

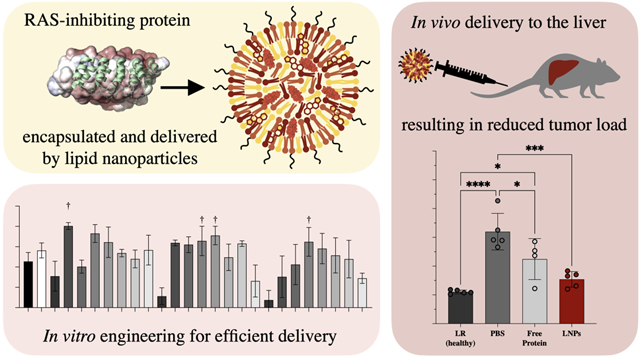
Abstract
Mutated RAS proteins are potent oncogenic drivers and have long been considered “undruggable”. While RAS-targeting therapies have recently shown promise, there remains a clinical need for RAS inhibitors with more diverse targets. Small proteins represent a potential new therapeutic option, including K27, a Designed Ankyrin Repeat Protein (DARPin) engineered to inhibit RAS. However, K27 functions intracellularly, and is incapable of entering the cytosol on its own, currently limiting its utility. To overcome this barrier, we have engineered a lipid nanoparticle (LNP) platform for potent delivery of functional K27-D30—a charge modified version of the protein—intracellularly in vitro and in vivo. This system efficiently encapsulates charge-modified proteins, facilitates delivery in up to 90% of cells in vitro, and maintains potency after at least 45 days of storage. In vivo, these LNPs deliver K27-D30 to the cytosol of cancerous cells in the liver, inhibiting RAS-driven growth and ultimately reducing tumor load in an HTVI-induced mouse model of hepatocellular carcinoma. This work shows that K27 holds promise as a new cancer therapeutic when delivered using this LNP platform. Furthermore, this technology has the potential to broaden the use of LNPs to include new cargo types—beyond RNA—for diverse therapeutic applications.
Keywords: lipid nanoparticles, intracellular delivery, protein engineering, small proteins, cancer
Graphical Abstract
Among the many targets in cancer that have been considered “undruggable”, one of the most widely researched is the RAS family of proteins (KRAS, NRAS, and HRAS).1 RAS mutations are found in ~30% of all human cancers, most notably in pancreatic (~98%) and colorectal (~52%) cancers, which are also among the most lethal.2 Due to interactions with both the mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K) pathways, mutated RAS proteins are potent oncogenic drivers, and thus are highly desirable as a target for cancer therapy. Recently, inhibitors have been developed which target the most common KRAS mutation in non-small-cell lung cancer: KRASG12C.3 The drug sotorasib (known previously as AMG510), is the first direct KRAS-targeting drug to be FDA approved, and is indicated for treatment of non-small cell lung carcinoma.4,5 However, the specific point mutation targeted by this drug is found only in a small subpopulation of RAS-mutant cancers, making the usage of this therapeutic scientifically important, but clinically limited.
Although the connection between KRAS and cancer has been known for decades, the direct targeting of RAS proteins has previously been stated as a difficult, if not impossible, task. This is due to the lack of hydrophobic binding pockets available on the protein surface, which are utilized by traditional small-molecule inhibitors. The current KRASG12C-targeting drugs are exceptional due to the biological differences present in the KRASG12C mutant.6 These slight differences allow for structural changes to the protein, creating opportunity for covalent small molecule binding to the mutated site. While there have been extensive efforts to develop other RAS-targeting small molecules, and there are drugs recently in development targeting the KRASG12D mutation (MRTX1133)7, many other compounds developed in the past have been largely ineffectual or even toxic, leading researchers to focus on alternative therapeutics such as protein and gene therapies, which may have more widespread applicability.2,8
One such alternate therapeutic is K27, a Designed Ankyrin Repeat Protein (DARPin), an antibody mimetic engineered to potently bind to and inhibit RAS activity.9 DARPins are small proteins which have regions that can be selected through display mechanisms to bind to their target proteins with high affinity and specificity. K27 has been shown to reduce the amount of active RAS and inhibit downstream signaling when expressed intracellularly, in turn causing a reduction in cancer cell growth in vitro.9 However, K27 is incapable of crossing the plasma membrane on its own and must be present in the cytosol to inhibit RAS, currently limiting its usage in vivo.
To overcome these barriers, this work details the development and optimization of a lipid nanoparticle (LNP) system that utilizes ionizable lipids to deliver K27 intracellularly. LNPs have been recently propelled into the public eye with the development and widespread administration of Moderna and BioNTech/Pfizer’s COVID-19 mRNA vaccines.10–12 However, LNPs have long been used in research applications due to their ease of production, good cyto-compatibility, and efficient intracellular delivery; as well as in clinical applications, with the FDA approval of Alnylam’s Onpattro.13 The incorporation of ionizable lipids allows LNPs to remain neutral at physiological pH, but become positively changed in acidic environments, such as the endosome—facilitating endosomal escape and cytosolic delivery of the cargo.14,15
While LNPs have been more commonly used for nucleic acid cargo (DNA, siRNA, mRNA), there have been recent successes with the usage of LNPs for alternate cargos, including proteins, often for gene editing applications.16–18 In these applications, the complexation of negatively charged nucleic acids—such as guide RNA—with proteins allows lipid and charge interactions without necessitating alterations to the protein itself. This method works well but requires co-encapsulation and delivery of these nucleic acids. To deliver proteins alone using LNPs, some sort of charge modification is required. In this work, recent developments in protein engineering are utilized to modify proteins with a negatively charged peptide sequence, to enable LNP encapsulation. Similar charge modifications have been previously explored for lipid-mediated delivery, fusing proteins to supercharged sequences, domains that bind nucleic acids, or oligonucleotides.19–21 However, these are also typically in gene-editing applications, where the function of the protein delivered does not depend on high delivery concentration. The modular nature of LNP components allows for facile adjustment of physiochemical properties, making it possible to re-optimize for alternate cargos that have not yet been tested. This optimization can be used to achieve high delivery efficiency, potentially allowing the delivery of cargos which have been previously under-investigated due to the relatively higher intracellular concentrations required for inhibitory effect.
Here, we developed a LNP platform for potent systemic in vivo delivery of DARPins with therapeutic applications in an HTVI-induced mouse model of hepatocellular carcinoma. First, a permanent negative charge was introduced to K2722,23, as the addition of a thirty aspartic acid (D30) anionic polypeptide allows for charge interaction and complexation with positively charged lipids. Further, the addition of split-GFP or split-luciferase (S11 or HiBiT) amino acid sequences allow for quantification of intracellular delivery when a reporter cell line is used that expresses the complimentary subunit. For delivery of this modified DARPin cargo, a library of 24 ionizable lipids were synthesized and screened in the split-GFP engineered cell line HCT116, a colorectal cancer model.24 The top-scoring lipids were included in a second library which also included alternate excipient and cargo ratios. From these two screens, a top performer was chosen to move forward for testing in vivo. This optimized LNP formulation contains an ionizable lipid (C14–4), a cationic lipid (DOTAP), a neutral/helper lipid (DOPE), cholesterol, and a lipid-anchored PEG polymer, and was formulated by microfluidic mixing (Figure 1).24–26 This formulation was able to consistently deliver functional K27 intracellularly both in vitro and in vivo. Here, LNPs are introduced as a promising delivery platform for K27 and demonstrated anti-cancer activity, inhibiting RAS-driven growth and reducing tumor load. Further, this work develops a formulation of LNPs for the small protein K27, expanding the cargo types amenable to delivery.
Figure 1.
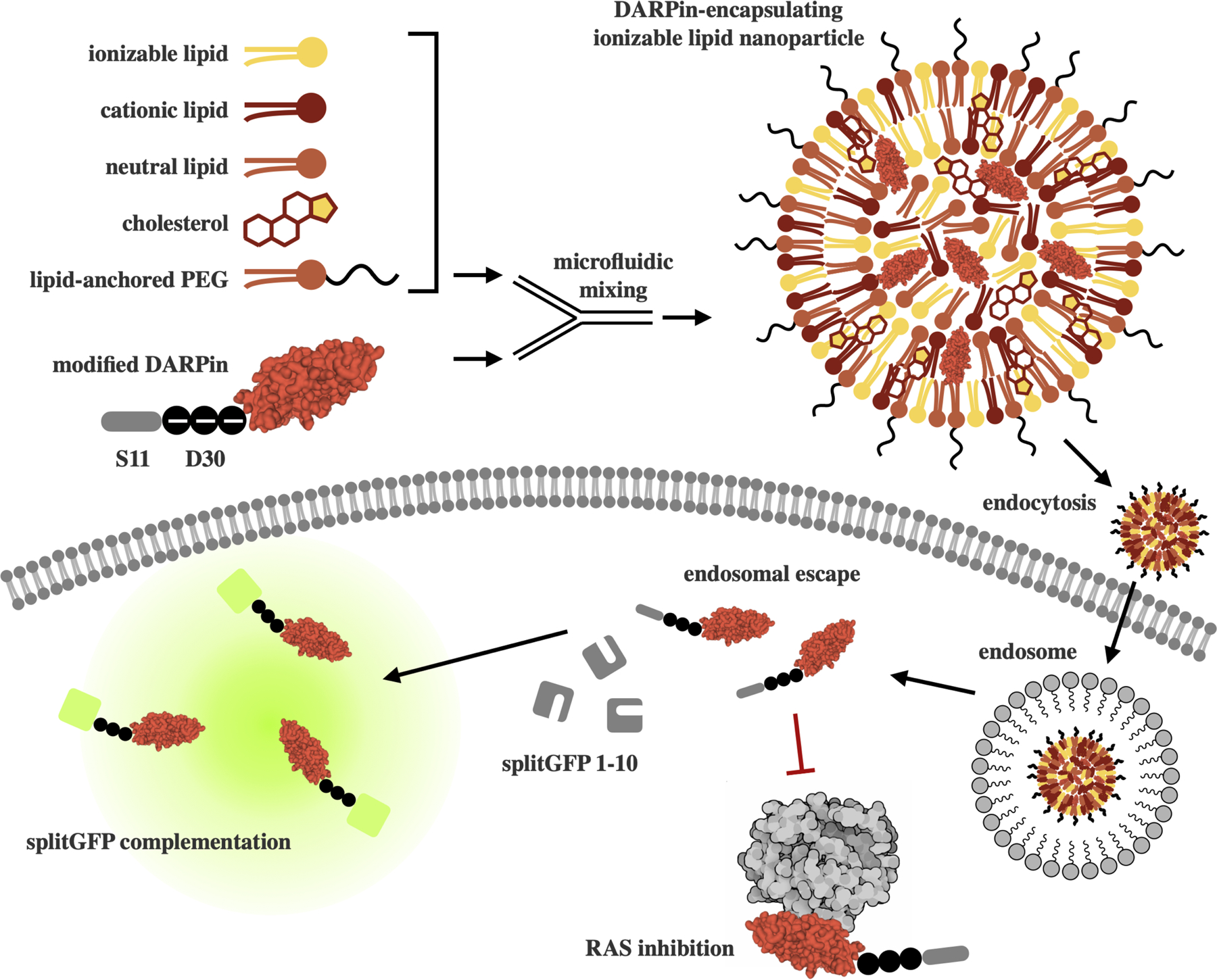
A LNP platform for small protein DARPin delivery was engineered using library screening to identify (i) ionizable lipids for potent DARPin delivery, and (ii) excipient and cargo molar ratios tailored for DARPin delivery. Using this LNP, a K27 DARPin—modified with a 30-repeat of negatively charged aspartic acid (D30)—can be delivered intracellularly, undergoing endosomal escape to become available in the cytosol. In screening applications, K27 modified with S11 complexes with GFP (1–10) in the reporter cell line to produce fluorescence. In functional applications, K27 binds to and inhibits RAS.
Results and Discussion
Identification of Key Formulation Parameters for Small Protein Delivery
The delivery of DARPins—or any similarly structured small proteins—using LNPs is largely unexplored. Therefore, before using traditional methods for LNP engineering and development, it was important to first evaluate the key LNP formulation parameters that influence K27 encapsulation and delivery. Without these parameters, it is unknown which components are best suited for modulation when designing LNP libraries. As the foundation for this work, a previously described optimized LNP formulation for siRNA encapsulation and delivery was used.27
For initial formulations, the molar ratios between lipid components were based on this siRNA-optimized formulation. The ionizable lipid C12–200 and the helper lipid 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) were also kept consistent. The molar ratio of cargo to lipid for these initial tests was held at 1:50, based on initial charge calculations. Based on LNP formulations for larger protein delivery applications,17 the modulation of the aqueous component of mixing and introduction of the permanently positively charged lipid 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were identified and shown to be major influencers of K27 encapsulation and delivery, using the engineered K27-S11 protein and split-GFP reporter assay (Figure 1). In this assay, K27 modified with S11 complexes with GFP (1–10) in the reporter cell line to produce fluorescence. For this work, HCT116 colorectal cancer cells were engineered to express GFP (1–10)22 and the percentage of GFP+ cells—correlating to cells which are receiving K27 into the cytosol—is reported.
While nucleic acids are typically introduced to the LNP in a citrate buffer, proteins may not always tolerate the extreme pH (pH 3) of this buffer. DARPins are comparatively robust proteins, but it was still important to test the influence of aqueous phase pH on LNP formulation and ultimately intracellular delivery (Figure 2.A). To more clearly illustrate these differences, the data shown here were collected using the finalized formulation resulting from this work (B6).
Figure 2.
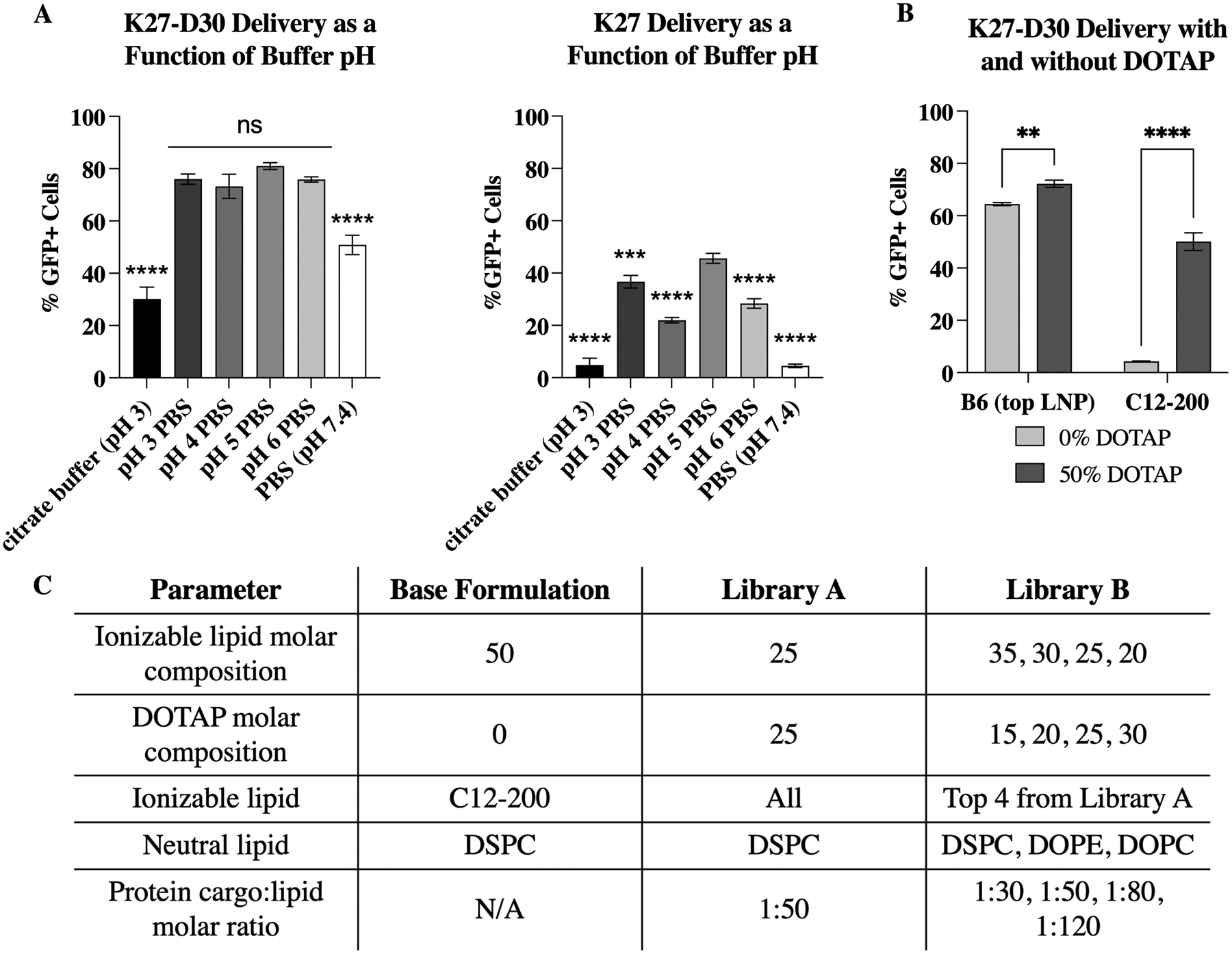
Identification of optimal buffer pH and inclusion of DOTAP as key parameters for K27 delivery and the design of LNP screening libraries used throughout this work. (A) Usage of pH 5 PBS as the aqueous component results in the best K27 delivery in both charge-modified (left) and non-charge modified (right) DARPin. When charge modified proteins are used (left), citrate buffer (pH 3) and PBS (pH 7.4) are significantly different than all other buffers tested. All shifted PBS buffers perform similarly and are non-significant from each other. When non-charge modified proteins are used (right), all pHs perform significantly different, except for citrate buffer (pH 3) and non-shifted PBS (7.4). Statistics shown are comparison to pH 5 PBS, which is used in formulations moving forward as it performs better than all other buffers tested. n ≧ 3, ***: p<0.001, ****: p <0.0001. (B) The introduction of the cationic lipid DOTAP improves K27 delivery in both base (C12–200) and optimized (B6) formulations. n = 3; **: p <0.01 and ****: p <0.0001. (C) LNP formulations and libraries designed based on these results and used in this work. Library A aims to identify top ionizable lipids, while library B aims to optimize excipients and cargo ratios. For these formulations, molar compositions are equal to molar percentages.
Interestingly, LNPs formulated using citrate buffer (pH 3) performed worse than PBS shifted to the same pH. This could be due to multiple factors including the negative charge on citrate, which may be outcompeting the anionic polypeptide charge on K27. While most of the pH values tested (pH 3, 4, 5, and 6) resulted in delivery performances that were not statistically different from each other, they were all statistical improvements when compared to citrate buffer and non-shifted (neutral, pH 7.4) PBS (p < 0.0001, not shown on graph). However, when non-charge modified proteins were used (K27), the differences became more pronounced, with pH 5 PBS performing significantly better than all other buffers tested (p ≦ 0.001, not shown on graph). It is likely that a pH of 5 performs best as it is under these conditions that the balance between the positively-charged ionizable lipid (pKa < 6.5) and negatively-charged protein—with a theoretical isoelectric point (pI) of 3.99—is optimized (Figure S1). At pH 5, it appears that there is some of the improved positive charge that occurs at lower pHs (due to the ionizability of the lipid) without reducing charge and stability of DARPins too much. Overall, charge modified proteins (K27-D30) resulted in improved intracellular delivery, regardless of aqueous phase buffer.
Another way to influence charge balances is with the introduction of charged lipids. Introduction of DOTAP at an equimolar amount to the ionizable lipid component significantly improved delivery when applied to the base C12–200 formulation (Figure 2.B). Interestingly, this increase was less dramatic when applied to our final top formulation (B6), although still significant. With the reduced and more localized charge of the modified K27-D30, as compared to traditional nucleic acid cargo, it is likely that the introduction of additional permanent positive charge—in the form of DOTAP—helps improve charge interactions and stability of the particle.
Using these identified parameters, two libraries were designed to screen LNP formulations for efficient K27 encapsulation and delivery (Figure 2.C). Library A aims to identify top ionizable lipids for potent DARPin delivery, while holding all other components standard. Namely, (i) pH 5 shifted PBS as the aqueous component, (ii) DOTAP at an equimolar amount to the ionizable lipid, (iii) DSPC as the helper lipid, and (iv) a cargo to lipid molar ratio of 1:50 were used in all formulations in library A. Library B was designed to identify excipient and cargo molar ratios tailored for DARPin delivery. Specifically, ionizable lipid and helper lipid type, as well as cargo to lipid and ionizable lipid to DOTAP molar ratios. Both LNP libraries were formulated using microfluidic mixing of the two phases: the ethanol phase, which contains lipid components, and the aqueous phase (pH 5 PBS), which contains the cargo to be encapsulated. The usage of microfluidic devices, as opposed to bulk mixing, creates smaller and more consistent particle sizes. These devices also have the potential to scale up for clinical use.25,26
Screening an Ionizable Lipid Library
Library A was designed to evaluate the usage of 24 previously designed24 ionizable lipids which are structural analogues to C12–200. These lipids were synthesized in the lab using nucleophilic addition/SN2 reactions, combining eight polyamine cores with one of three epoxide-terminated alkyl chains (Figure 3.A).24 They were additionally characterized to confirm structure (Table S2). These lipids—as well as C12–200 and C12–113, as comparison controls—were used to formulate K27-encapsulating LNPs via microfluidic mixing. This library incorporates the parameters identified as important for protein encapsulation: the inclusion of DOTAP and the usage of pH 5 PBS as the aqueous component. After formulation, Library A was characterized for size, polydispersity, surface zeta potential, and protein concentration (Figure S2.A).
Figure 3.
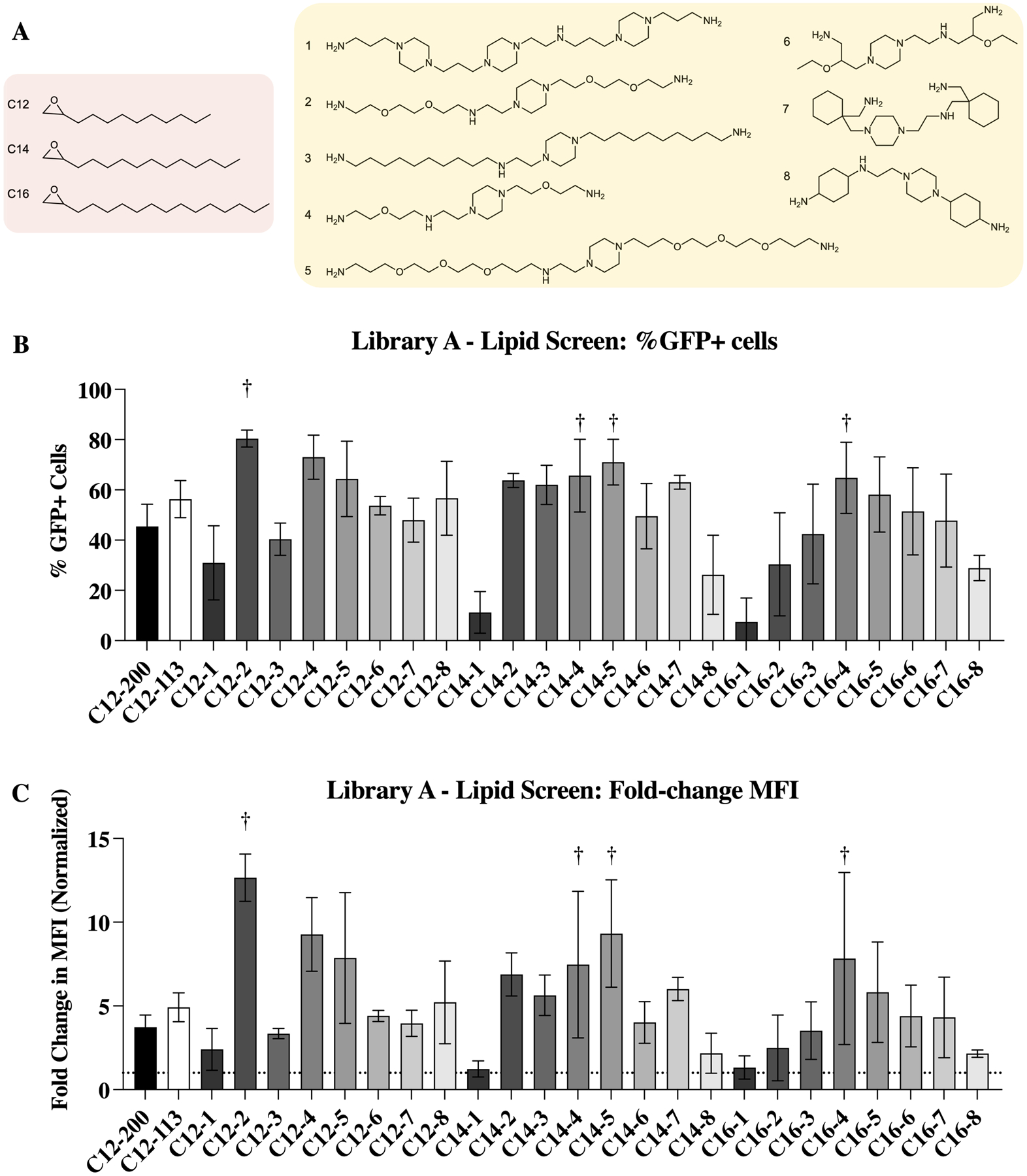
Identification of ionizable lipids for potent DARPin delivery. (A) Ionizable lipid tail and core structures were reacted to form a library of 24 ionizable lipids. (B) Intracellular delivery (%GFP+ cells) of K27 by LNPs formulated with each ionizable lipid. Top performers (C12–2, C14–4, C14–5, and C16–4) are highlighted here. (C) Same library screen represented instead by the change in mean fluorescence intensity. The same top performers are again identified. n = 4.
To evaluate the ability of these LNPs to deliver K27 intracellularly, a previously described split-GFP reporter assay was used (Figure 1).22,23,28 In this assay, K27 modified with S11 complexes with GFP (1–10) in the reporter cell line to produce fluorescence. For this work, HCT116 colorectal cancer cells were engineered to express GFP (1–10)22 and the percentage of GFP+ cells—correlating to cells which are receiving K27 into the cytosol—is reported in Figure 3.B. Further, median fluorescence intensity, which additionally evaluates the intensity of GFP—correlating to the number of K27 proteins successfully delivered per cell—is reported in Figure 3.C. From this split-GFP assay, four ionizable lipids were identified as top performers: C12–2, C14–4, C14–5, and C16–4.
To evaluate possible structure/function relationships between the ionizable lipids used and the resulting LNPs formed, multiple linear regression was used to identified parameters which may influence characteristics of the LNPs. From this analysis, it was found that delivery efficiency—measured by reconstituted GFP fluorescence—was influenced by tail length as well as the number of Nitrogens in the lipid structure. While C12–2 exhibited the highest fluorescence overall, lipids with C14 tails had the highest mean fluorescence, with C16 tails performing the worst overall. The one polyamine core tested that contains greater than 5 Nitrogens (core 1) created statistically worse performing LNPs, regardless of tail length (Figure S2.C).
Size (z-average) was found to be influenced primarily by tail length and the number of Oxygens in the lipid core. Both an increase in tail length and an increase in the number of Oxygens was found to correlate to an increase in mean diameter. LNPs with C12 tails were significantly smaller than both C14 and C16-containing lipids (Figure S2.D). PDI also appeared to be influenced by tail length, with LNPs containing C16 tails having the highest mean PDI values, significantly higher than their C12 counterparts, which have the lowest mean PDI (Figure S2.E).
Finally, zeta potential appears to be influenced by the number of rings present in the lipid core, with cores containing only one ring (2, 3, 4, 5, and 6) being more negatively charged than those with three rings (1, 7, and 8) (Figure S2.F). While these possible structure/function findings are specific to K27 DARPin cargo, they may be interesting the context of larger LNP structure/function studies. It has been described in other applications that the structural characteristics of lipids can greatly inform cargo delivery.29,30 For example, chemical changes which effect packing—such as lipid tail saturation or branching—can influence membrane destabilization and endosomal escape.
Optimizing a Lipid Nanoparticle Formulation
Library B was designed to further optimize LNP excipients to accommodate the DARPin cargo. The top four ionizable lipids from library A (C12–2, C14–4, C14–5, and C16–4) were included in this library at various molar ratios. The total molar percent of charged lipid—including ionizable lipids and cationic lipids—remained constant, but within that fixed amount the ratio of ionizable lipid to cationic lipid was varied (Figure 2.A). This was, in part, to screen for formulations which remained potent at low DOTAP percentages, as cationic lipids have been recently noted as having significant toxicity in vivo. While DSPC is the most used neutral (helper) lipid for siRNA delivery, other lipids such as DOPE and DOPC have been used to improve encapsulation of alternate nucleic acid cargos, such as mRNA. In this library, all three neutral lipids were evaluated. Finally, the ratio of protein cargo (K27) to total lipids was varied, to optimize charge balances and minimize the amount of free protein lost to formulation. To evaluate the effects of these changes, library B was designed using a design of experiments (DOE) approach, similar to previous work.31 This method allows a design space of 256 potential LNP formulations to be evaluated using 16 representative LNP formulations (Figure 4.A).
Figure 4.
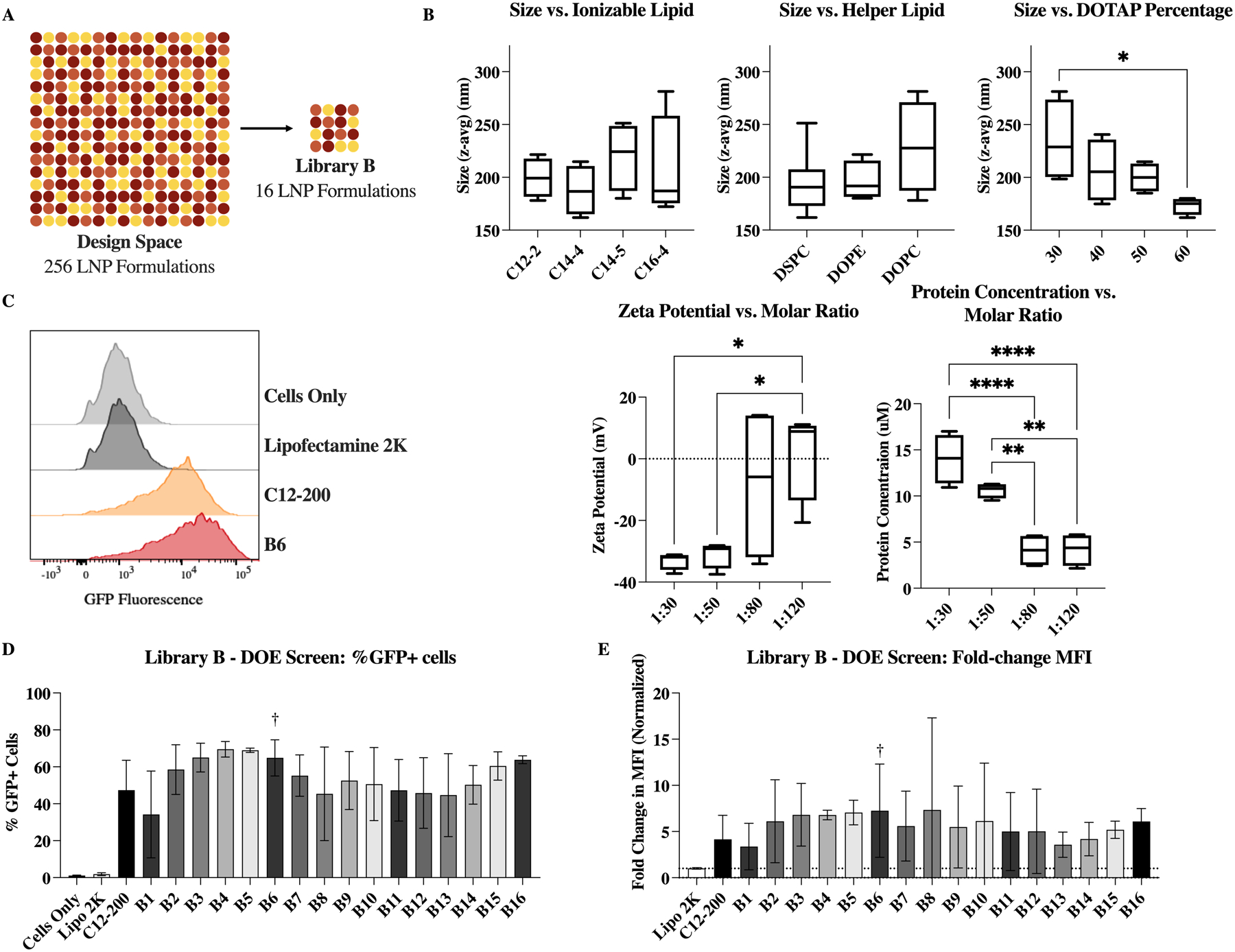
Optimizing LNP excipient components for DARPin delivery using a Design of Experiments (DOE) approach. (A) DOE allows for a design space of 256 LNPs to be analyzed by testing only 16 LNPs (B) Variables which were found to have impact on LNP physiochemical properties, as identified by multiple linear regression; *: p <0.1, **: p <0.01, and ****: p <0.0001. (C) Representative flow plots of negative (untreated) control, commercial control Lipofectamine 2000, C12–200 comparison formulation, and identified top performer, B6. (D) Intracellular delivery (%GFP+ cells) of K27 by each LNP formulation. Top performer B6 is highlighted here. (E) Same library screen represented instead by the change in mean fluorescence intensity (normalized to commercial control). n = 4.
In general, characterization of the LNPs formulated for library B was more variable than library A, especially in the measured surface zeta potential and protein concentration values (Figure S3). Again, multiple linear regression was used to identify key parameters which may influence characteristics of the resulting LNPs (Figure 4.B). Size was found to be potentially influenced by the choice of lipids. Specifically, increased amounts of DOTAP caused a reduction in LNP size. When DOPC was used as the neutral lipid, size appeared slightly increased and more variable, while DSPC and DOPE produced smaller particles. Similarly, different ionizable lipids resulted in slightly different LNP sizes—although none of these differences were statistically significant.
As the molar ratio of protein cargo to total lipids was increased—increasing the number of lipids per K27 molecule—the zeta potential became more neutral (Figure 4.B). This could be caused by a reduction in free or surface bound protein, which has been modified with the D30 negative charge. In fact, at the lower molar ratios (1:30 and 1:50), the measured zeta potential was around −30 mV (average of −33 and −31, respectively). Finally, as the molar ratio of protein cargo to total lipids was increased, the total protein concentration of the LNPs decreased. This is an unsurprising result, as the protein quantity was the attribute which was varied—either increased or decreased—to evaluate the various molar ratios, and the total lipids used in each formulation remains constant. Interestingly, increasing the amount of protein in a formulation did not increase delivery, possibly due to an upper limit on encapsulation.
All LNPs in library B performed well in the split-GFP reporter assay, outperforming the commercial control lipofectamine 2000 which showed negligible delivery in HCT116 cells (Figure 4.D and E). Both the percentage of GFP+ cells (Figure 4.D) and the mean fluorescence intensity—normalized to lipofectamine 2000—(Figure 4.E) are shown. From this delivery and characterization data, B6 was identified as a top performer, as it delivered K27 intracellularly in an effective manner with reduced DOTAP content and minimal protein loss.
in vitro Lipid Nanoparticle Mediated DARPin Delivery
Once B6 was identified as a top performer, it was tested further for delivery efficiency, toxicity, stability, and in vitro activity. From TEM images, the B6 LNP formulation was of consistent size and shape, with lamellar structure (Figure 5.A). In addition to performing well in the library B screen, B6 shows a clear dose-responsive delivery curve, with minimal but significant delivery at the lowest concentration tested (7.81 nM), ultimately achieving intracellular delivery to >90% of cells at the highest dosages (≧125nM) (Figure 5.B). Minimal in vitro toxicity as measured by an LDH assay was seen at these higher concentrations when LNPs were incubated with cells for 24h. Toxicity was not observed until the highest dosage (500nM) for 8h. In vivo, it is unlikely that LNPs will be present at such high concentrations for such long periods of time, indicating that the B6 formulation holds promise for in vivo testing.
Figure 5.
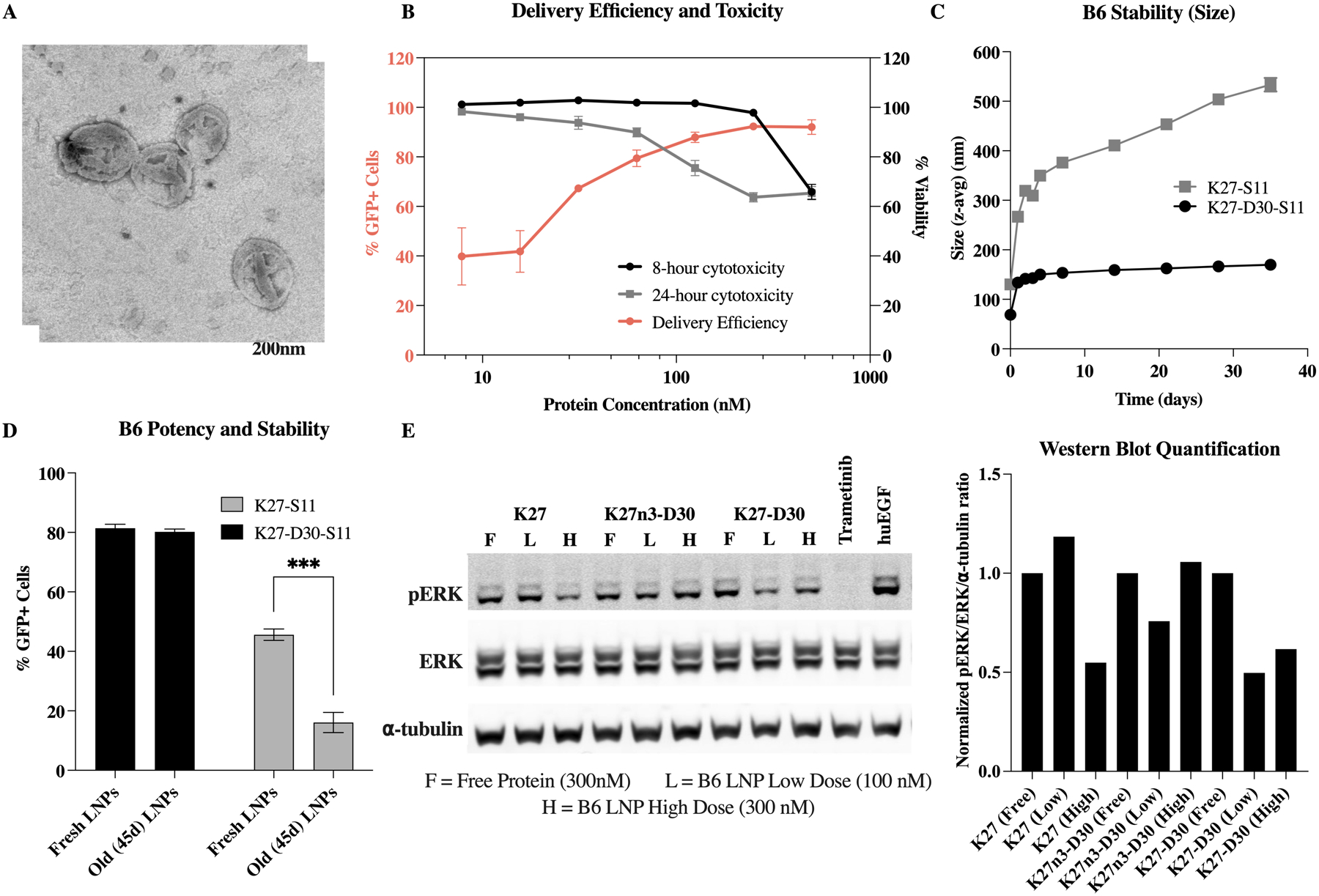
Top LNP (B6) delivers functional K27 DARPin in vitro. (A) TEM image of top LNP formulation, B6, showing multilamellar structure. Image supports DLS-determined size of 198.4 ± 4.4 nm. (B) Delivery efficiency, as measured by %GFP+ cells, and toxicity, represented as % viability. Delivery efficiency plateaus around 90% above 100 nM. (C, D) D30 modification of K27 allows for LNP stability out to at least 45 days post formulation. K27 without D30 modification shows increase in size and decrease in delivery efficacy after being stored. ***: p<0.001. (E) Western blot shows decrease in downstream phosphorylated ERK due to RAS inhibition when treated with B6 LNPs encapsulating K27 at high dose and B6 LNPs encapsulating K27-D30 at both low and high dose.
For clinical translatability, it is also important that these LNPs retain functionality when stored. To test stability, B6 LNPs were formulated, and intracellular delivery was tested using the split-GFP reporter assay. LNPs stored at 2–4°C retained their original size when formulated using K27 DARPins modified with the D30 negative charge (K27-D30-S11) (Figure 5.C). These LNPs also retained their potency, with no statistical change in delivery efficiency seen after 45 days of storage (Figure 5.D). However, B6 LNPs formulated with non-charge modified K27 (K27-S11) displayed increasing hydrodynamic radius over time, suggesting particle instability and aggregation. Additionally, their already lower delivery efficacy was further reduced after storage (Figure 5.C and D). The D30 charge modification appears to be necessary for the formation of stable and efficient particles using these methods. Further, the D30 charge modification significantly improves encapsulation efficiency, with 68% of charge modified proteins (K27-D30-S11) being encapsulated by or associated with LNPs in our top formulation. In contrast, 95% of non-charge modified proteins (K27-S11) remain free in solution, with only 5% being encapsulated by or otherwise associated with LNPs (Figure S4.B).
The split-GFP reporter assay is a powerful and stringent tool to measure intracellular delivery of K27 with decent throughput. However, it does not measure the activity of the K27 DARPin once delivered to the cytosol. Thus, we assessed functional K27 delivery and RAS inhibition by measuring RAS downstream signaling—in this case, ERK phosphorylation—by western blotting (Figure 5.E). A non-functional negative control variant of DARPin K27 (K27n3-D30) was purified and compared against both charge modified (K27-D30) and non-charge modified (K27) functional proteins. When cells were incubated with free protein, none of the DARPin proteins caused a reduction in pERK levels. Non-charged modified K27—delivered via B6 LNP—shows a slight reduction in pERK only at the higher dosage (300nM), while charge modified K27-D30 shows pERK reduction at both the higher and the lower dosage (100nM). Trametinib—a small molecule MEK inhibitor—was used here as a positive control. These results show that B6 can successfully and stably encapsulate and deliver K27-D30 intracellularly in vitro, at levels which allow for endogenous Ras inhibition.
Lipid Nanoparticles deliver DARPin to alternate cell lines in vitro
LNPs are traditionally used for the encapsulation and delivery of nucleic acid cargos, and while formulations have been optimized for each nucleic acid type, the field tends to assume that all cargos within the same type will encapsulate and deliver the same. For example, formulations are optimized for delivery of reporter mRNAs—such as luciferase or GFP-encoding—and then tested for functionality by switching to functional-encoding mRNAs, without changing any of the LNP parameters. This makes LNP platforms widely applicable and generalizable. Proteins, however, are much more heterogenous macromolecules than mRNAs or siRNAs, and it was therefore important to test the platform nature of the B6 formulation, to evaluate if it can be used as a universal delivery system for small proteins.
Even LNP formulations for nucleic acid cargos must be re-optimized for delivery to different cell types or tissues of interest. Therefore, we first tested the delivery of the K27 DARPin to alternate GFP (1–10) reporter cell lines using the top B6 formulation. HT1080 (human fibrosarcoma), A549 (human lung adenocarcinoma), and HEK293T (embryonic kidney) all showed similar K27 delivery to the HCT116 colorectal line used in previous studies—with delivery to 77–87% of cells (Figure 6.A). MDA-MB-231 (triple negative human breast adenocarcinoma) and CT26 (murine colorectal carcinoma) showed moderate and low delivery of K27, respectively. Variable performance based on cell type is not uncommon for LNPs, or delivery vehicles in general. This difference in delivery may be due to differences in uptake mechanisms, cytoplasmic trafficking, or general cellular activity.32 All cell lines tested here are considered “difficult-to-transfect” when talking about cationic lipids and commercially available transfection reagents.33,34 Regardless of these differences, it appears from this data that the B6 formulation generally delivers well to other cell lines.
Figure 6.
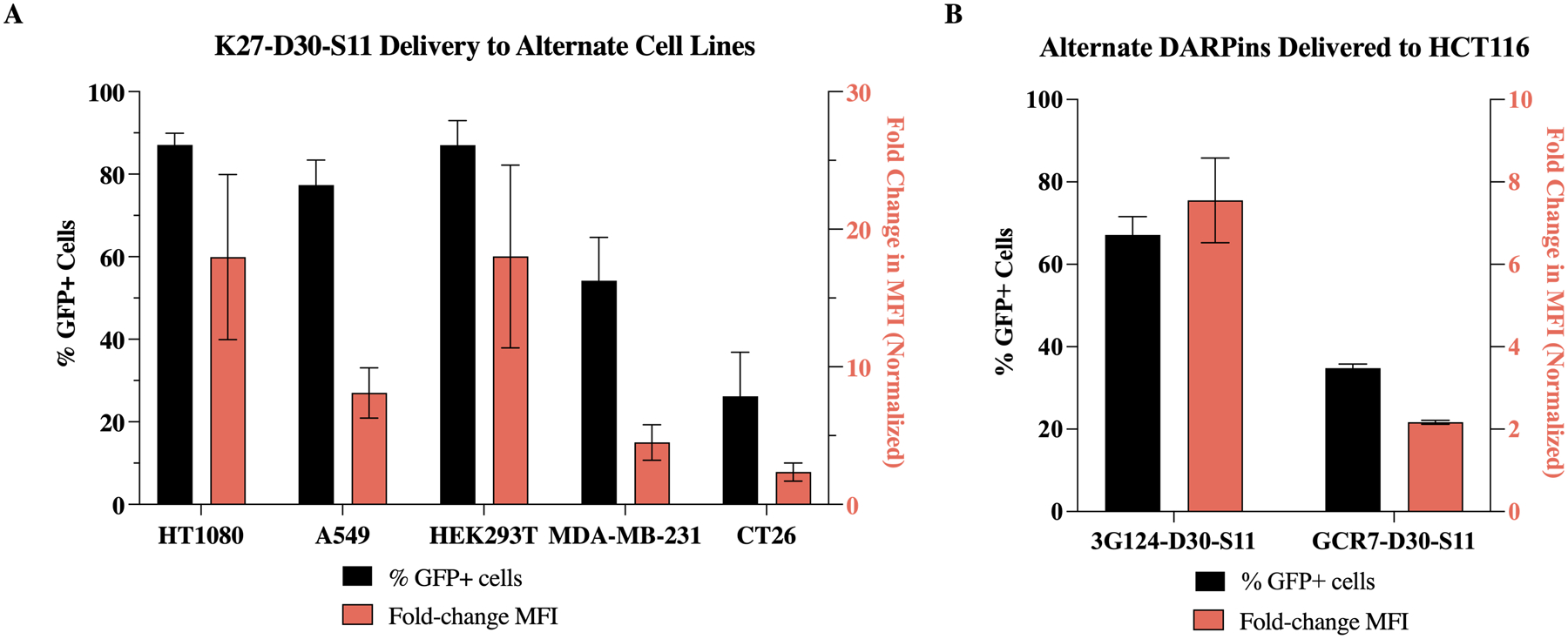
LNP B6 enables potent intracellular delivery of K27-D30—and other DARPins—to multiple cancer cell lines in vitro. (A) Intracellular delivery—%GFP+ cells (left axis) and mean fluorescence intensity (right axis, normalized to untreated cells)—of K27-D30 to multiple cancer cell lines in vitro. Delivery efficiency of B6 is dependent on cell line. (B) Intracellular delivery of multiple DARPin cargos to HCT116 cells in vitro. Delivery efficiency of B6 is dependent on macromolecular cargo. n ≧ 4.
To determine if the top B6 formulation was generalizable to protein cargos based off the same scaffold, two alternate DARPin cargos were expressed and purified. The first is an anti-GFP DARPin (3G12435). This protein, like K27, contains three internal repeats and stabilizing N and C caps. The second protein purified was a GFP “clamp” comprising two non-competing anti-GFP DARPins fused together (gc_R736). When delivered to HCT116 cells using B6 LNPs, charge-modified 3G124 delivered at similar efficiency to K27, while gc_R7 delivered about half as efficiently—67% and 35% of cells, respectively (Figure 6.B). This suggests that lipid nanoparticles optimized for delivery of one class of proteins—in this case, DARPins—can be applied to proteins with similar structure. Currently, there are many different DARPins available which target intracellular proteins.37–39 They can be developed relatively easily and produced in high yields but are currently limited in use due to poor delivery methods. For other DARPins, this technology could be immediately applicable. However, for other protein scaffolds that are more structurally distinct—such as nanobodies and affibodies—delivery efficiency was found to be less reliable (Figure S5.A). LNPs may have to be re-optimized to accommodate changes in tertiary structure, surface charge distribution, and molecular weight (Figure S5.B).
Biodistribution of K27-encapsulating LNPs in an in vivo HTVI tumor model
Most LNP formulations—encapsulating nucleic acids—distribute primarily to the liver when administered intravenously (IV).40 This phenomenon has been largely attributed to the resulting protein corona which forms upon LNP administration. Lipid structures which have affinity for certain serum proteins may alter corona composition and resulting biodistribution. Specifically, ApoE affinity has been linked to liver delivery.41 Recent work in the field has shown that the inclusion of charged lipids can influence LNP trafficking, redirecting distribution to the spleen or lungs based on negative or positive charge additions, respectively.42 Similarly, overall apparent charge of LNPs may also influence biodistribution profiles. Since the formulations used in this work incorporate DOTAP, a positively charged lipid, it is important to determine whether this causes trafficking to the lungs. This is especially important given that the top performer, B6, has a measured surface zeta potential of 9.5 mV, making it slightly positively charged.
To evaluate biodistribution in a therapeutically relevant in vivo tumor model, hydrodynamic tail vein injection (HTVI) was used to induce tumor formation in the liver of C57BL/6 mice.43 This HTVI hepatocellular carcinoma model was identified as a therapeutically relevant model as it more closely mimics a metastatic cancer than a primary liver tumor, due to the large number of small tumors, as opposed to a single large primary tumor. Colorectal cancer specifically is known to metastasize mainly to the liver, causing tumor growth across the entire organ. It is at this stage that patients would typically be receiving IV chemotherapeutics as a primary treatment, as opposed to opting for surgical removal of the primary tumor. As the in vitro studies were completed primarily using a colorectal cancer line, we decided to focus on the treatment of liver-metastatic colon cancer as an initial proof-of concept for this delivery system. While the HTVI model is not a perfect metastasis model—as the cells in question are still hepatocytes, making it a hepatocellular carcinoma and not an adenocarcinoma—from a delivery perspective it is still a much better model than a subcutaneous xenograph tumor model.
Plasmid DNA encoding MYC and NRASG12V oncogenes (CaMIN, Sleeping Beauty transposon system, Figure S6)44 induced hepatocarcinogenesis following HTVI, and once tumors were fully formed (7 weeks after HTVI), mice were injected IV with carboxytetramethylrhodamine (TAMRA) fluorophore-labelled K27-D30, either free in solution or encapsulated using the B6 LNP formulation, at 3 mg/kg. TAMRA labelled K27-D30 was produced by site-specific labelling of protein with a single TAMRA fluorophore via sortase tag expressed protein ligation.45
At 6h post-injection, fluorescence signal in the free protein group was found primarily in the kidneys (Figure 7.A). Since the free protein does not efficiently enter cells on its own, it is likely present in the kidneys after being filtered from the blood. With a size of ~25kDa, free K27-D30 protein should pass easily through the glomerular filtration barrier, while LNPs—with diameters above 100nm—should be retained in the body.46 Fluorescence signal from the K27-D30 LNP group was found in both the kidneys and liver (Figure 7.A). Since this formulation is not processed to remove free protein—as we have determined encapsulation efficiency to be high, around 70% (Figure S4.B)—the signal from the kidneys is likely due to residual free protein. Other than the kidneys, signal is seen predominantly in the liver, indicating that LNP-mediated protein delivery is occurring primarily in the liver, and that the inclusion of DOTAP is not significantly shifting biodistribution to the lungs.
Figure 7.
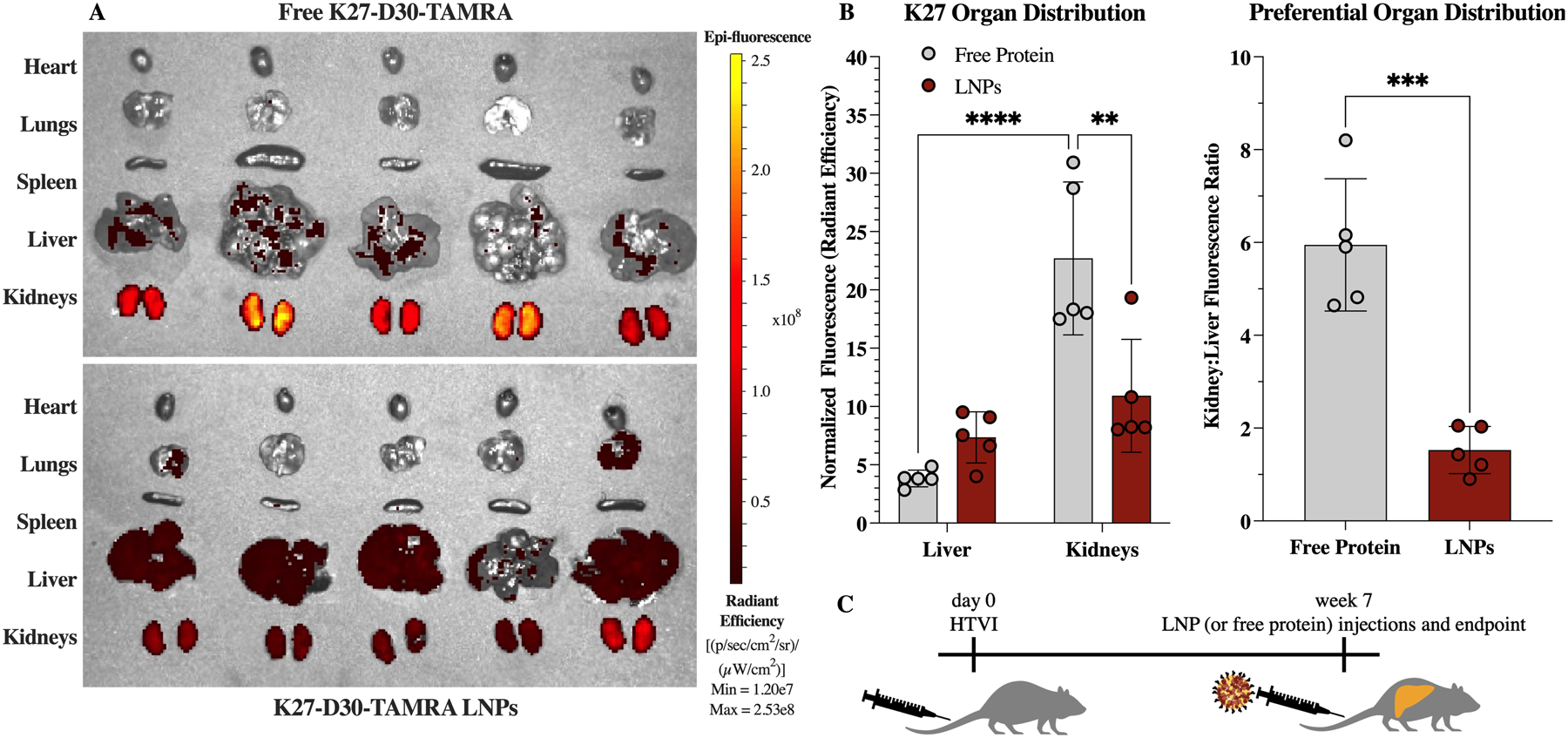
Biodistribution in an HTVI-induced mouse model of hepatocellular carcinoma, after IV administration of K27 protein at 3 mg/kg. (A) IVIS images showing fluorescence of TAMRA label on K27-D30 protein. Free protein (top) was found primarily in the kidneys, while administration of K27-encapsulating LNPs resulted in protein distribution to the kidneys and the liver. (B) Quantification of fluorescence intensity, normalized to background. Free protein administration resulted in a statistically greater signal in the kidneys, compared to free protein signal in the liver and LNP-delivered protein signal in the kidneys. n = 5; **: p <0.01, ***: p<0.001, and ****: p <0.0001.
Overall, fluorescence in the liver was increased and fluorescence in the kidneys was significantly decreased when K27-D30 was delivered via LNPs, as compared to free protein (Figure 7.B). This causes a significant shift in preferential organ distribution, with LNPs having a lower kidney:liver fluorescence ratio. Some liver distribution is still seen with the free protein, and it is possible that the cancerous tissue and leaky vasculature in the liver is contributing to free protein and LNP accumulation at that site.
in vivo intracellular delivery of K27-D30 to HTVI modified cells
Beyond distribution to organs of interest, it is also important to determine if K27-D30 protein can be delivered intracellularly to cancerous cells in the liver. A similar HTVI model was used to induce tumor growth, this time with the addition of a split nanoLuc luciferase reporter (LgBiT, Figure S6) to evaluate intracellular delivery of protein. We chose to switch to a split-luciferase complementation system owing to high signal to noise ratio in vivo and to avoid autofluorescence issues that may arise from the split-GFP system used for in vitro studies. In this model, HTVI-transduced cells intracellularly express LgBiT, an 18kD fragment of NanoLuc luciferase.47 LgBiT was stably integrated into mouse hepatocytes via the Sleeping Beauty system in which co-injection of a Sleeping Beauty transposase encoding plasmid catalyzed gene transfer of LgBiT. The S11 peptide used in vitro was replaced with HiBiT, an 11 amino acid peptide which complements with LgBiT to generate a luminescent reporter with similar performance to full-length NanoLuc.48
After IV delivery of free or LNP-encapsulated K27-D30 (tagged with the split-luciferase peptide HiBiT) significant luminescent signal could be seen only in livers that received K27-D30-HiBiT LNPs (Figure 8.A). This luminescence was about 60x higher than background, and 12x higher than free protein signal (Figure 8.B). No significant signal was observed in livers taken from mice that received free K27-D30-HiBiT protein or PBS IV injections (Figure 8.C).
Figure 8.
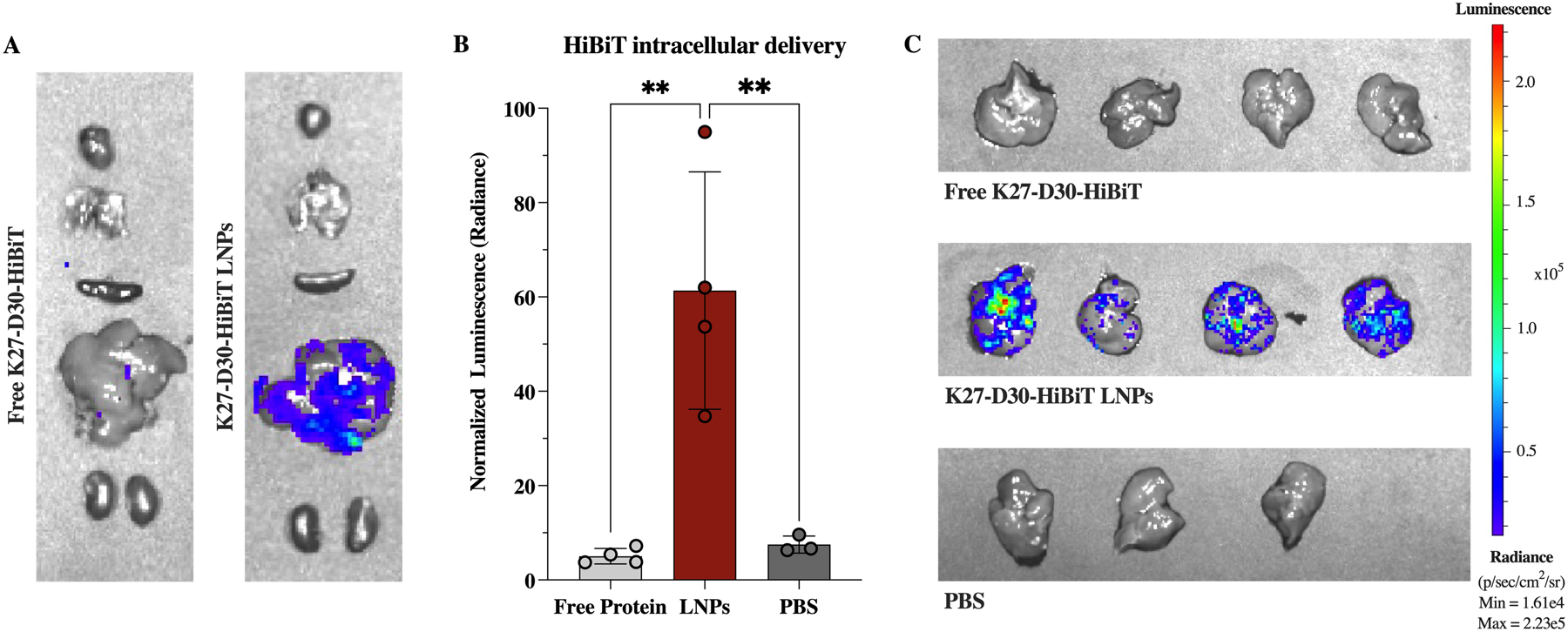
In vivo intracellular delivery of K27 protein in an HTVI-induced mouse model of hepatocellular carcinoma. (A) Representative IVIS images of murine organs after IV administration of 3 mg/kg K27-D30-HiBiT protein either free (left) or delivered via LNPs (right). (B) Quantification of luminescent signal—normalized to background—shows significant increase in signal when protein is delivered via LNPs, indicating intracellular delivery of protein in the liver. n ≧ 3; **: p <0.01. (C) IVIS images of livers after IV administration of free K27-D30-HiBiT protein (top), K27-D30-HiBiT LNPs (middle), or PBS (bottom).
This data supports what was determined in vitro, that free protein is not capable of crossing cell membranes and cannot act intracellularly without a delivery vehicle. From the biodistribution data, it appears that some free protein is present locally in the liver. However, that free protein does not enter the intracellular environment to interact with the LgBiT fragment expressed in the cytosol. In contrast, the LNP-delivered protein is consistently delivered intracellularly to HTVI-modified hepatocytes, indicating that this system holds promise for delivering K27-D30 as a treatment for cancer in the liver.
Therapeutic Efficacy of K27-D30 encapsulating LNPs
With confirmation that IV-administered LNPs could deliver K27-D30 intracellularly to a model of hepatocellular carcinoma, the therapeutic potential of LNP-delivered DARPin K27 was investigated. Another HTVI model was used to induce tumor growth and evaluate therapeutic efficacy. This time, all mice received CaMiN plasmids to induce hepatocarcinogenesis, and half additionally received firefly Luciferase plasmids, as a method to track the growth of tumor cells created by HTVI (Figure S6). Both CaMiN and Luciferase genes were integrated via Sleeping Beauty. Tumors were allowed to grow for 5 weeks before treatment, to allow initial small tumors to form (Figure 9.A). Treatment consisted of 4.5 mg/kg of protein administered IV twice a week, either free or encapsulated using the B6 LNP. Mice were weighed every week and tolerated this dosing scheme without any significant change in body weight (Figure S7.C). This same dose was administrated IV to healthy mice as well to further evaluate biotoxicity—specifically liver toxicity—as this is the main location of delivery. After 24h, serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were found to, on average, increase slightly, but with no statistical difference from PBS treated mice (Figure S7.D).
Figure 9.
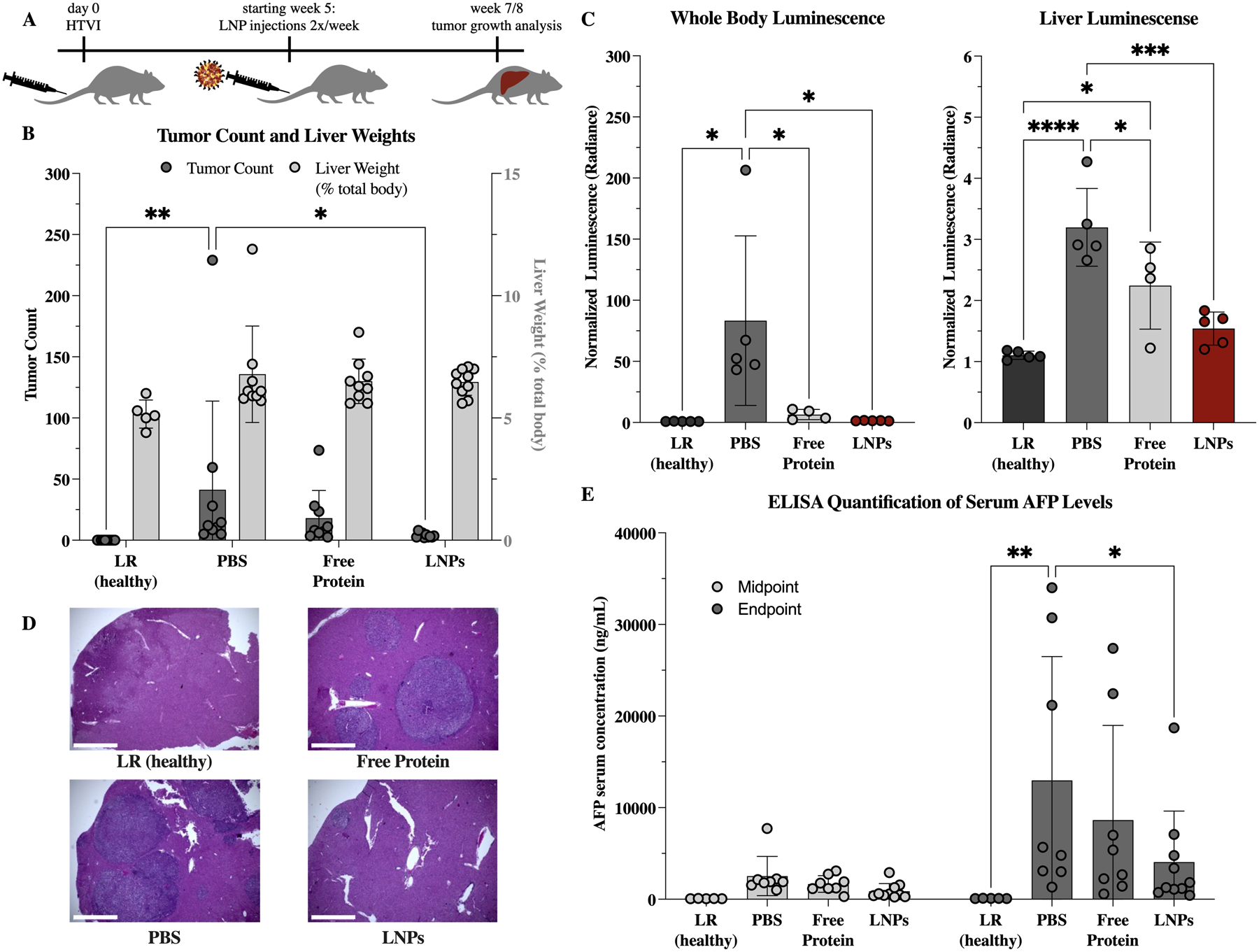
Therapeutic effect of K27-D30 encapsulating LNPs on tumor growth in an HTVI-induced mouse model of hepatocellular carcinoma. (A) Experimental scheme. Mice received HTVI on day 0, began receiving treatment week 5, and were evaluated for tumor growth at an endpoint of 7 or 8 weeks for the non-luciferase and luciferase expressing subsets, respectively. (B) Tumor counts and liver weights (as a percentage of total body weight) at endpoint for: healthy (received LR only during HTVI at day 0), PBS (received PBS injections starting week 5), free protein (received 4.5 mg/kg of K27-D30 2x/week starting week 5), and LNP (received 4.5 mg/kg of LNP-encapsulated K27-D30 2x/week starting week 5) groups. n = 5 for LR and n ≧ 9 for others; *: p <0.05, **: p <0.01. Group receiving LNPs shows significantly reduced tumor counts as compared to PBS control. (C) Luminescence (collected using IVIS) of whole body and extracted livers of mice that received an additional luciferase reporter gene during HTVI. Both free protein and LNP groups show reduction in luciferase signal over PBS control, indicating reduced luciferase production in these tumors. n ≧ 4; *: p <0.05, ***: p <0.001, and ****: p <0.0001. (D) Representative images of H&E stained liver sections showing tumors in LR (healthy), PBS, free protein, and LNP groups. Fewer, smaller, and more dispersed tumors—visible here by change in color—were seen in the LNP group, when compared to free protein and PBS. Scale bar = 1000μm. (E) Serum levels of alpha-fetoprotein (AFP), which indicate the presence of primary liver cancer. At midpoint (week 5), all groups are statistically similar. At endpoint (week 7/8), PBS group is elevated compared to healthy control, and LNP group is significantly reduced, when compared to PBS control. n = 5 for LR and n ≧ 8 for others; *: p <0.05, **: p <0.01
At 7 or 8 weeks after HTVI—for the non-luciferase and luciferase-expressing groups, respectively—mice were evaluated for tumor burden using a number of metrics. The liver weight was non-significantly increased in all mice that received functional HTVI as compared to the healthy Lactated Ringer’s (LR) solution injection group (Figure 9.B). The PBS control-treated group had significantly more tumors than the K27-D30-LNP group (Figure 9.B). Overall, the livers from the LNP group had smaller, more dispersed tumors as compared to the PBS and free protein groups, although the nature of this model is highly heterogeneous (Figure S7.A). Whole body in vivo imaging system (IVIS) images showed only background luminescence in the healthy group, and similar levels in the LNP groups, with only the PBS group having significantly increased luminescence (Figure 9.C). However, in excised livers, more differences can be seen within groups, with PBS, free protein, LNPs, and healthy livers having the most to the least luminescent signal, respectively. Interestingly, while luminescence is elevated above background in the LNP group, it is not statistically different from the healthy control. After tumors were counted, representative liver lobes were processed for histology. Hematoxylin and eosin staining illustrated the differences in tumor burden between treatments (Figure 9.D, Figure S7.B). In both the PBS and free protein groups, it was easy to find regions with large tumor masses or numbers. In the LNP group, tumors were much smaller when located and there were fewer areas found with multiple close tumor sites.
Finally, blood was taken from all mice at the midpoint (week 5) and endpoints (week 7/8) of the study to evaluate the level of the tumor biomarker alpha-fetoprotein (AFP) in the serum.49 At the midpoint, all but the healthy group show slightly elevated AFP levels, indicating the beginning of tumor formation (Figure 9.E). At the endpoint, AFP levels were further elevated, with the PBS control rising to levels significantly higher than both the healthy control and the K27-D30 LNP treated group. Combined, these tumor growth metrics indicate that treatment with K27-D30 encapsulating LNPs led to fewer, smaller tumors as compared to no treatment or free protein.
Overall, the LNP group showed the lowest whole body and liver luminescence, tumor counts, and serum AFP levels, significantly lower than the PBS groups in all metrics tested. Interestingly, the free protein group had the next lowest average levels, although this group was not significantly different than the PBS group in many of the metrics tested. It is possible that the administration of free protein has some effect on tumor growth, possibly due to a non-specific immune response to the non-endogenous protein. However, if the free protein can alter tumor progression, it does not seem a potent enough effect to significantly impact tumor burden.
Conclusion
This work demonstrates that the small protein K27 holds potential promise as a new cancer therapeutic when delivered using the LNP platform developed herein. Through extensive ionizable lipid screening and design of experiments optimization, a lipid nanoparticle platform was developed which encapsulates K27 at ~70% efficiency and delivers K27 to upwards of 90% of cells in vitro with minimal toxicity. Here, uniform LNPs were produced reliably and rapidly using a microfluidic platform, with similar components to those used in FDA-approved systems.10–12 In comparison, most existing protein encapsulation systems suffer from poor encapsulation efficiencies, complex synthesis methods, and poor scalability.50,51 The B6 LNP system is stable at 2–4°C for at least 45 days and shows applicability to alternate cell lines and alternate types of DARPins. In vivo, this system delivers K27 intracellularly to cancerous cells in the liver and can deliver K27 to a therapeutic level, resulting in reduced tumor counts and serum levels of the biomarker AFP.
As an anti-cancer therapy, the delivery of K27 using this engineered LNP system shows promise for the treatment of liver cancers, or cancers that have metastasized to the liver. With a high percentage of liver tumors being RAS-driven, there are several potential clinical applications for a novel treatment and delivery system such as this one. While the differences in tumor growth shown here are minimal—possibly because the pCaMIN-induced tumors are driven by MYC in addition to RAS—this treatment may be synergistic with existing chemotherapeutics or targeted cancer therapies and may help to tip the scales in the favor of many patients with aggressive RAS-driven cancers. The HTVI based model used for tracking in vivo cytosolic delivery was developed for this work. This system pairs Sleeping Beauty and NanoBiT (split nanoLuc) technologies, and can be used in future studies that investigate intracellular protein delivery. To our knowledge, this is the first demonstration of a LNP platform for potent systemic in vivo delivery of RAS-targeting K27 DARPin proteins, with therapeutic applications in an HTVI-induced mouse model of hepatocellular carcinoma
Overall, this work is not only an investigation into the therapeutic applicability of K27, but also serves as an early step towards the expansion of LNPs for the delivery of alternate cargo types, beyond nucleic acids such as RNA. With the recent expansion of LNP use in the clinic, it is our hope that such a platform is highly translational and may inspire further work utilizing LNPs for macromolecular delivery for a number of diverse therapeutic applications.
Experimental Methods
Protein Cloning and Expression
For all cloning, gBlocks encoding recombinant proteins were synthesized by Integrated DNA Technologies (Coralville, IA). All proteins were expressed and purified in either previously described sortase tag expressed protein ligation (STEPL)52 or proximity-based sortase-mediated ligation (PBSL)53 one-step purification/ligation systems. Purified protein concentration was determined by BCA assay (ThermoFisher; Waltham, MA). Constructs were cloned with flexible GS-rich linkers between binding protein, D25/D30, S11, and HiBiT sequences.
To generate 3G124-D30-S11 and gc_R7-D30-S11, gBlocks for the DARPins were inserted into a pSTEPL backbone between NdeI and XhoI already containing C-terminal D30 and S11 peptide sequences by In-fusion cloning (Takara Bio USA; Mountain View, CA). To generate HiBiT-tagged proteins, pSTEPL DARPinK27-D30-S11 and pSTEPL DARPinK27n3-D30-S11 were first double-digested with Bsu36I and AgeI, and the larger DNA fragment was gel extracted to remove the S11 tag. Then, a gBlock encoding both HiBiT and a sequence reconstructing the sortase recognition motif (LPETG) was inserted into the vector by In-fusion cloning. Successful cloning was confirmed by sanger sequencing (Azenta Life Sciences; South Plainfield, New Jersey). Plasmids were transformed into T7 Express competent E. coli (New England Biolabs; Ipswich, MA). For STEPL purification of gc_R7 and all DARPins, transformed T7 Express were grown for 20–24 hours at 37°C in 2YT autoinduction media.
Expression cultures of aGFPnb-S11, aGFPnb-D25-S11, aTaqAffi-S11, aTaqAffi-D30-S11 were grown at 37°C for 24 hours in 2YT autoinduction media, while Omomyc-S11 and Omomyc-E30-S11 were grown at 37°C for 16–20 hours. Nanobody, affibody and Omomyc proteins were all purified by PBSL.
Fluorescently tagged proteins—used for encapsulation efficiency and biodistribution studies—were created by labelling K27 with a C-terminal carboxytetramethyl rhodamine (GGGSK-TAMRA peptide, LifeTein; Somerset, NJ) using STEPL. Proteins for animal studies were further processed using Pierce™ Endotoxin Removal kits according to manufacturer’s instructions with a modified equilibration buffer containing 400mM NaCl.
Cell Culture
A549, HT1080 and HEK293T cells were obtained from our own stocks. MDA-MB-231 cells were gifted by Erle Robertson. These four cell lines were maintained in complete DMEM media containing 10% FBS and 1% penicillin/streptomycin. GFP (1–10) engineered versions of A549, HT1080, and HEK293T, which we previously described,23 and MDA-MB-231 GFP (1–10), which was made for this study, were maintained in the same media supplemented with 2 μg/mL puromycin (Takara Bio).
CT26 cells were obtained from Celeste Simon and maintained in RPMI containing 10% FBS and 1% P/S. CT26 GFP (1–10) cells were maintained in the same media supplemented with 8 μg/mL puromycin. HCT116 cells were gifted by Michael Farwell and were maintained in McCoy 5A media with 10% FBS and 1% penicillin/streptomycin. HCT116 GFP (1–10) cells, which we previously described,22 were maintained in the same media supplemented with 2 μg/mL puromycin. All cells were maintained in a 5% CO2, 37°C humidified incubator.
Generating GFP (1–10) Cells
Concentrated VSV-G pseudotyped lentivirus containing CMV-driven GFP (1–10)-IRES-PuroR was generously gifted by Philip Zoltick. CT26 and MDA-MB-231 cells were incubated overnight with different volumes of lentivirus in complete DMEM medium supplemented with 8 μg/mL polybrene. The following day, media was replaced with complete DMEM without polybrene, and cells were grown to confluence. MDA-MB-231 cells expressing GFP (1–10) were selected with media containing 2 μg/mL puromycin and CT26 cells were selected with media containing 8 μg/mL puromycin. To confirm GFP (1–10) expression, transduced cells were pelleted, lysed in cell lysis buffer (Cell Signaling Technology 9803; Danvers, MA) with added protease inhibitor (Cell Signaling Technology 5871) and centrifuged to remove debris. Clarified lysates were incubated with purified recombinant S11-containing protein in TNG buffer (100 mM Tris-HCl, 150 mM NaCl, 10% v/v glycerol, pH 7.4) for at least 1 hour at 37°C, and reconstituted GFP fluorescence was analyzed on a BioTek Synergy H1 (Winooski, VT) microplate reader in black-bottom 96-well plates (λexcitation/λemission = 488nm/530nm). Polyclonal GFP (1–10) cells with high GFP complementation were frozen for further use.
Sleeping Beauty Transposons
The Sleeping Beauty transposon, pSBbi (Addgene #60523; Watertown, MA) was digested with NcoI and HindIII (New England Biolabs; Ipswich, MA) and the resulting backbone was gel-purified for further cloning. To generate pSBbi-LgBiT, a gBlock encoding LgBiT containing a Kozak sequence and 5’ and 3’ overhangs homologous to the backbone was incorporated into pSBbi by In-fusion cloning. To clone pSBbi-Luc2, overhang PCR was performed on pGL4.54 Luc2-TK (Promega; Madison, WI) to amplify the Luc2 gene with appropriate homologous ends and a 5’ Kozak sequence. The resulting PCR product was cloned into the pSBbi backbone by In-fusion cloning. Both pSBbi-LgBiT and pSBbi-Luc2 plasmids were prepared using an endotoxin-free maxiprep kit (Qiagen; Hilden, Germany).
Ionizable Lipid Synthesis
The ionizable lipids used in this study were synthesized by reacting epoxide-terminated alkyl chains (Avanti Polar Lipids; Alabaster, AL) with polyamine cores (Enamine; Monmouth Jct, NJ) using Michael addition chemistry, as previously described.24,54 Components were combined with a 7-fold excess of alkyl chains and mixed with a magnetic stir bar for 48 h at 80°C. The crude product was then transferred to a Rotavapor R-300 (BUCHI; Newark, DE) for solvent evaporation, and the lipids were suspended in ethanol for use in formulation without further purification.
Ionizable Lipid Characterization
To confirm structures of the 24 ionizable lipids synthesized, 1H-NMR and LC-MS were used. 1H NMR spectra were acquired in d-chloroform using an Avance Neo 400 MHz spectrometer (Bruker; Billerica, MA). LC-MS spectra were acquired in ethanol using an SQD equipped with an Acquity UPLC (Milford, MA), using a C8 column with a 2 min wash followed by a gradient mobile phase from 50% water (1% trifluoroacetic acid) and 50% acetonitrile (1% trifluoroacetic acid) to 100% acetonitrile (1% trifluoroacetic acid).
LNP Formulation
To synthesize LNPs, an aqueous phase containing the small protein of interest (typically K27 DARPin) and an ethanol phase containing lipid and cholesterol components were mixed using a microfluidic device as previously described.24–26 The aqueous phase was prepared using PBS, shifted to desired pH (typically 5). To prepare the ethanol phase, ionizable lipid, 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), or 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), lipid-anchored polyethylene glycol (PEG) (Avanti Polar Lipids; Birmingham, AL), and cholesterol (Sigma; St. Louis, MO) components were combined. Pump 33 DS syringe pumps (Harvard Apparatus; Holliston, MA) were used to mix the ethanol and aqueous phases at a 3:1 v/v ratio in a microfluidic device produced as previously described.26 After mixing, LNPs were dialyzed against 1x PBS for 1 hour to remove ethanol.
LNP Characterization
To determine protein concentration, a micro-BCA protein assay kit (ThermoFisher; Waltham, MA) was used. LNPs were diluted in PBS with 2% SDS, as per manufacturer instructions, to accommodate the presence of lipids in the sample. BCA working reagent was added to each sample, and samples were incubated for 2 hours at 37°C in a sonicating bath, to allow for quantification of encapsulated, as well as free or surface-anchored protein. Samples were plated in triplicate in 96-well plates and resulting absorbance was measured using an Infinite M Plex plate reader (Tecan; Morrisville, NC), alongside a standard curve of DARPinK27 used to quantify protein concentration. Size and zeta potential were determined by diluting LNPs in PBS and water respectively, and measuring in a Zetasizer (Malvern Panalytical; Malvern, UK). A JEOL JEM-1010 transmission electron microscope (TEM, JEOL; Akishima, Tokyo) was used to image top performing LNPs at 60,000x magnification. Encapsulation efficiency was evaluated as previously described,22 by separating free and encapsulated TAMRA-labelled K27 by size exclusion chromatography and measuring fluorescence in resulting fractions.
Protein Delivery
In a typical delivery assay 35,000 cells were used for a 48-well plate format. For Lipofectamine delivery in 48-well plates, 2 μL Lipofectamine 2000 (Invitrogen; Waltham, MA) was mixed with 8 μL of Opti-MEM Reduced Serum Medium (ThermoFisher; Waltham, MA), and protein was separately diluted to 10 μM in 10 μL of Opti-MEM. The diluted Lipofectamine and protein solutions were mixed by pipetting 5–10 times. Proteins were incubated for 15 minutes at room temperature to promote complexation. Then, Lipofectamine:Protein complexes were added to cells in 180 μL of antibiotic-free media so that the total protein concentration per well was 500nM. For LNP delivery, indicated amounts of LNP:Protein formulations were added directly to each well. Cells were incubated with proteins for 6 hours at 37°C before flow cytometry analysis and 8 hours for western blot analysis.
Flow Cytometry
Following protein delivery in GFP (1–10) cells in a 48-well plate, cells were washed once with cold PBS, detached with 0.25% trypsin, and pelleted in a 4°C table-top centrifuge at 600xg. Cell pellets were resuspended in flow buffer (PBS, 1% w/v BSA, 1 mM EDTA) and analyzed on a BD Accuri C6 analyzer (BD Biosciences; Franklin Lakes, NJ). For screening assays, cells were pelleted in 96-well U bottom plates and analyzed by CytoFLEX (Beckman Coulter; Brea, CA) or BD LSR Fortessa (BD Biosciences). At least 8,000 total events were collected. Data were analyzed with BD Accuri CFlow 6 Software or FlowJo v10. The gate for GFP positivity is defined based on the negative control: corresponding GFP (1–10) cells only (without treatment, resulting in no fluorescence). The positive gate is defined such that only 1% of the negative control sample would fall within that positive gate. Representative flow histograms were generated in FlowJo v10.
In vitro cytotoxicity
LDH Cytotoxicity Assay. HCT116 cells were plated overnight in 96-well plates (20,000 cells/well). The following day, media was replaced with 100 μL of antibiotic-free media with dilutions of B6:K27-D30-S11, and cells were incubated for an additional 8 h or 24 h. Cytotoxicity was measured using a lactate dehydrogenase (LDH) detection kit according to the manufacturer’s instructions (Dojindo Molecular Technologies CK12; Rockville, MD) and normalized to both live and dead controls.
Western Blotting
First, B6:DARPinK27 proteins were delivered in 6-well plates for 8 hours in HCT116 cells. As a positive control, cells were treated with 100nM of the MEK inhibitor Trametinib for 1 hour. Following delivery, cells were lysed in plate using cell lysis buffer (Cell Signaling Technology 9803; Danvers, MA) with added protease/phosphatase inhibitor (Cell Signaling Technology 5872S; Danvers, MA), and centrifuged at 15,000g. Approximately 30μg of protein was boiled in LiCor loading buffer (LiCor 928–40004; Lincoln, NE), resolved on a Bolt™ 4–12%, Bis-Tris gel (ThermoFisher; Waltham, MA) and transferred onto a PVDF membrane for 1 hour at 20V. Membranes were blotted with mouse anti-pErk 1/2 (1:2000 dilution, Cell Signaling Technology 9106S; Danvers, MA) and rabbit anti-Erk 1/2 (1:2000 dilution, Cell Signaling Technology 9102S; Danvers, MA) primary antibodies. Membranes were then incubated with goat anti-rabbit 680RD (LiCor 925–68071; Lincoln, NE) and donkey anti-mouse 800CW (LiCor 925–32212; Lincoln, NE) IR-functionalized secondary antibodies (1:15,000 dilution). Imaging was performed on a LiCor Odyssey system. Membranes were stripped with NewBlot™ buffer and re-probed for α-tubulin as a loading control (Cell Signaling Technology 2144S; Danvers, MA).
Band intensities were calculated using ImageJ, and the pERK signal was divided by both total ERK and loading control signals. The pERK/ERK/α-tubulin ratio in samples with LNP-delivered proteins was normalized to the corresponding free protein control.
In vivo biotoxicity
To evaluate biotoxicity, serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured 24h after IV injection of 4.5 mg/kg K27, encapsulated using LNPs. Mouse serum was diluted and assayed using ELISA kits (Abcam; Cambridge, UK) following manufacturer’s protocols.
Hydrodynamic Tail Vein Injection (HTVI)
To evaluate delivery in a therapeutically relevant in vivo tumor model, hydrodynamic tail vein injection (HTVI) was used to induce tumor formation in the liver, as previously described.43,55,56 Mice were weighed, and plasmids of interest (pPGK-SB13, pCaMIN, pSBbi-LgBiT, and/or pSB-Luc2)44 were diluted into lactated ringer’s solution up to a volume that is 10% of animal body weight. This volume was administered into the tail vein—via a catheter (Terumo Medical SV*27EL; Shibuya City, Tokyo)—in under 10 seconds. By administering plasmid DNA in this high speed and volume fashion, about 40% of hepatocytes can take up the transgenes of interest and more than 95% of mice exhibit expression after injection.55 pPGK-SB13 and pCaMIN were both generously gifted by Daniel Dauch, University of Tuebingen.
The Sleeping Beauty transposon system was used to insert different plasmids for different parts of this work. For biodistribution, pPGK-SB13, and pCaMIN were administered. pCaMIN is used to express the oncogenes MYC and NRASG12V, inducing tumor growth. For intracellular delivery, pSBbi-LgBiT was additionally administered as a split-luciferase reporter. For therapeutic studies, either pCaMIN alone or pCaMNN and pSBbi-Luc2 (luciferase)—as a constitutive reporter—were delivered. In all cases, each mouse received 10 μg of each transposon sequence administered (pCaMIN, pSBbi-LgBiT, and/or pSB-Luc2) and 2 μg of Sleeping Beauty transposase plasmid (pPGK-SB13), and tumors formed 6–8 weeks after HTVI.
In Vivo Biodistribution and Intracellular Delivery
Male C57BL/6 mice underwent HTVI to induce expression of CaMIN for biodistribution studies or CaMIN, and LgBiT for intracellular delivery studies. 7 weeks after HTVI, mice were given 3 mg/kg of free K27 protein or LNPs encapsulating K27 (or PBS as a control) via tail vein injection. For biodistribution, K27 was modified with the D30 negative charge and the fluorescent TAMRA tag (K27-D30-TAMRA). For intracellular delivery, K27 was modified with the D30 negative charge repeat and HiBiT (K27-D30-HiBiT). HiBiT is a small 11 amino acid peptide that binds with high affinity (KD = 0.7nM) to LgBiT. Once bound, the NanoBIT complex has luciferase activity and will produce luminescent signal when Nano-Glo in vivo Substrate is added (Fluorofuramizine FFz, Promega Nano-Glo; Madison, WI). Tumors in the intracellular delivery group should express LgBiT, and luminescent signal should only be seen if K27-D30-HiBiT is delivered intracellularly.
6 h after protein/LNP injections, mice in the intracellular delivery group received IP injections of Nano-Glo substrate, and all mice were imaged, sacrificed, and organs were excised for additional imaging. IVIS was used to collect fluorescent (TAMRA) and luminescent (NanoBIT and/or Luciferase) whole body and organ images. Images were analyzed using Living Image (PerkinElmer; Waltham, MA) software.
In vivo Therapeutic Model
Female C57BL/6 mice underwent HTVI to induce expression of CaMIN alone or CaMIN and luciferase for therapeutic studies. One week after HTVI, mice with luciferase expression received IP injections of luciferin, and were imaged using IVIS to confirm successful HTVI and luciferase expression. 5 weeks after HTVI, when initial small tumors should be beginning to form, mice began to receive 2x/week tail vein injections of 4.5 mg/kg free K27 protein or LNPs encapsulating K27. Blood was also collected to evaluate serum cytokine levels. 7 weeks after HTVI (or 8 weeks, in the case of CaMIN/Luc mice), mice with luciferase expression received IP injections of luciferin and blood was also collected from all mice to evaluate serum cytokine levels. Mice were imaged, sacrificed, and organs were excised for additional imaging using IVIS.
Livers were weighed and tumors were counted by eye. Two separate counts were averaged to determine tumor count per liver, and a third count was added if the first two significantly disagreed. Blood was allowed to sit for 2h at room temperature to promote clotting, and then samples were spun at 2,000xg for 10 minutes to separate serum. Serum was stored at −20°C until analysis, which was done using a mouse alpha-Fetoprotein/AFP Quantikine ELISA Kit (Bio-Techne; Minneapolis, MN).
After weighing and counting, livers of interest were fixed in 10% neutral buffered formalin for at least 48h and changed into 70% ethanol for at least 24h. Fixed tissue was processed and embedded by the histotechnology facility at the Wister Institute, to produce H&E-stained tissue slides. Images were collected on an EVOS FL Auto 2 (Invitrogen; Waltham, MA) at 4x and 20x magnification.
Statistical Analysis
Statistical analyses were performed on GraphPad Prism (v9) software. If otherwise unspecified, ANOVA was applied as appropriate. Statistical significance was defined at α = 0.05. Multiple batches of proteins and LNPs were used throughout this study.
Supplementary Material
Acknowledgements
R.M.H. and A.C. were supported by the National Science Foundation Graduate Research Fellowship Program (NSF-GRFP) under Grant No. DGE-1845298. M.S.P was supported by the National Institute of Dental & Craniofacial Research (NIDCR) of the National Institutes of Health (NIH) under Award Number T90DE030854 and the Center for Innovation & Precision Dentistry (CiPD) at the University of Pennsylvania. D.Y. and K.J.W are supported by the Arnold and Mabel Beckman Foundation (Beckman Young Investigator Award) and the NIH National Cancer Institute (R01 CA259201). M.J.M. was supported by a National Institutes of Health (NIH) Director’s New Innovator Award (DP2 TR002776), a Burroughs Wellcome Fund Career Award at the Scientific Interface (CASI), additional funding from the NIH (NCI R01 CA241661, NCI R37 CA244911, NIDDK R01 DK123049), and an NSF CAREER Award (CBET-2145491).
We are grateful to the Wistar Institute’s Histotechnology Facility for providing technical support. Funding support for The Wistar Institute core facilities was provided by Cancer Center Support Grant P30 CA010815. The NSF Major Research Instrumentation Program (award NSF CHE-1827457) and Vagelos Institute for Energy, Science and Technology supported the purchase of the NMR used in this study. We are grateful to Joshua Glover at the Pennsylvania University Perelman School of Medicine Stem Cell and Xenograft core for assistance with preliminary in vivo studies.
We also acknowledge Christian Figueroa-Espada for assistance with manuscript revisions and David Mai for assistance with initial flow cytometry studies.
Footnotes
Supporting Information Available: diagram of estimated charge balance, additional LNP characterization and analysis, alternative small protein encapsulation and delivery, diagram of Sleeping Beauty transposon system used, additional in vivo therapeutic data, amino acid sequences of proteins used in this work, and lipid characterization (DOC)
References
- (1).Moore AR; Rosenberg SC; McCormick F; Malek S RAS-Targeted Therapies: Is the Undruggable Drugged? Nat Rev Drug Discov 2020, 19 (8), 533–552. 10.1038/s41573-020-0068-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Cox AD; Fesik SW; Kimmelman AC; Luo J; Der CJ Drugging the Undruggable RAS: Mission Possible? Nat Rev Drug Discov 2014, 13 (11), 828–851. 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM K-Ras(G12C) Inhibitors Allosterically Control GTP Affinity and Effector Interactions. Nature 2013, 503 (7477), 548–551. 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kwan AK; Piazza GA; Keeton AB; Leite CA The Path to the Clinic: A Comprehensive Review on Direct KRAS(G12C) Inhibitors. J Exp Clin Cancer Res 2022, 41 (1), 27. 10.1186/s13046-021-02225-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Sheridan C Oncologists Greet Lumakras: The World’s First KRAS Inhibitor. Nat Biotechnol 2021, 39 (9), 1032–1034. 10.1038/s41587-021-01053-9. [DOI] [PubMed] [Google Scholar]
- (6).Ostrem JML; Shokat KM Direct Small-Molecule Inhibitors of KRAS: From Structural Insights to Mechanism-Based Design. Nat Rev Drug Discov 2016, 15 (11), 771–785. 10.1038/nrd.2016.139. [DOI] [PubMed] [Google Scholar]
- (7).The KRASG12D Inhibitor MRTX1133 Elucidates KRAS-Mediated Oncogenesis. Nat Med 2022, 28 (10), 2017–2018. 10.1038/s41591-022-02008-6. [DOI] [PubMed] [Google Scholar]
- (8).Shin S-M; Choi D-K; Jung K; Bae J; Kim J; Park S; Song K-H; Kim Y-S Antibody Targeting Intracellular Oncogenic Ras Mutants Exerts Anti-Tumour Effects after Systemic Administration. Nat Commun 2017, 8 (1), 15090. 10.1038/ncomms15090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Guillard S; Kolasinska-Zwierz P; Debreczeni J; Breed J; Zhang J; Bery N; Marwood R; Tart J; Overman R; Stocki P; Mistry B; Phillips C; Rabbitts T; Jackson R; Minter R Structural and Functional Characterization of a DARPin Which Inhibits Ras Nucleotide Exchange. Nat Commun 2017, 8 (1), 16111. 10.1038/ncomms16111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Vogel AB; Kanevsky I; Che Y; Swanson KA; Muik A; Vormehr M; Kranz LM; Walzer KC; Hein S; Güler A; Loschko J; Maddur MS; Ota-Setlik A; Tompkins K; Cole J; Lui BG; Ziegenhals T; Plaschke A; Eisel D; Dany SC; Fesser S; Erbar S; Bates F; Schneider D; Jesionek B; Sänger B; Wallisch A-K; Feuchter Y; Junginger H; Krumm SA; Heinen AP; Adams-Quack P; Schlereth J; Schille S; Kröner C; de la Caridad Güimil Garcia R; Hiller T; Fischer L; Sellers RS; Choudhary S; Gonzalez O; Vascotto F; Gutman MR; Fontenot JA; Hall-Ursone S; Brasky K; Griffor MC; Han S; Su AAH; Lees JA; Nedoma NL; Mashalidis EH; Sahasrabudhe P. v; Tan CY; Pavliakova D; Singh G; Fontes-Garfias C; Pride M; Scully IL; Ciolino T; Obregon J; Gazi M; Carrion R; Alfson KJ; Kalina W. v; Kaushal D; Shi P-Y; Klamp T; Rosenbaum C; Kuhn AN; Türeci Ö; Dormitzer PR; Jansen KU; Sahin U BNT162b Vaccines Protect Rhesus Macaques from SARS-CoV-2. Nature 2021, 592 (7853), 283–289. 10.1038/s41586-021-03275-y. [DOI] [PubMed] [Google Scholar]
- (11).Jackson LA; Anderson EJ; Rouphael NG; Roberts PC; Makhene M; Coler RN; McCullough MP; Chappell JD; Denison MR; Stevens LJ; Pruijssers AJ; McDermott A; Flach B; Doria-Rose NA; Corbett KS; Morabito KM; O’Dell S; Schmidt SD; Swanson PA; Padilla M; Mascola JR; Neuzil KM; Bennett H; Sun W; Peters E; Makowski M; Albert J; Cross K; Buchanan W; Pikaart-Tautges R; Ledgerwood JE; Graham BS; Beigel JH An MRNA Vaccine against SARS-CoV-2 — Preliminary Report. New England Journal of Medicine 2020, 383 (20), 1920–1931. 10.1056/NEJMoa2022483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cavalcanti E; Isgrò M; Rea D; Capua L; Trillò G; Russo L; Botti G; Miscio L; Buonaguro F; Bianchi A Vaccination Strategy and Anti - SARS-CoV-2 S Titers in Healthcare Workers of the INT – IRCCS “Fondazione Pascale” Cancer Center (Naples, Italy). Infect Agent Cancer 2021, 16. 10.1186/s13027-021-00375-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Garber K Alnylam Launches Era of RNAi Drugs. Nat Biotechnol 2018, 36 (9), 777–778. 10.1038/nbt0918-777. [DOI] [PubMed] [Google Scholar]
- (14).Maugeri M; Nawaz M; Papadimitriou A; Angerfors A; Camponeschi A; Na M; Hölttä M; Skantze P; Johansson S; Sundqvist M; Lindquist J; Kjellman T; Mårtensson I-L; Jin T; Sunnerhagen P; Östman S; Lindfors L; Valadi H Linkage between Endosomal Escape of LNP-MRNA and Loading into EVs for Transport to Other Cells. Nat Commun 2019, 10 (1), 4333. 10.1038/s41467-019-12275-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Schlich M; Palomba R; Costabile G; Mizrahy S; Pannuzzo M; Peer D; Decuzzi P Cytosolic Delivery of Nucleic Acids: The Case of Ionizable Lipid Nanoparticles. Bioeng Transl Med 2021, 6 (2), e10213–e10213. 10.1002/btm2.10213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Li Y; Bolinger J; Yu Y; Glass Z; Shi N; Yang L; Wang M; Xu Q Intracellular Delivery and Biodistribution Study of CRISPR/Cas9 Ribonucleoprotein Loaded Bioreducible Lipidoid Nanoparticles. Biomater Sci 2019, 7 (2), 596–606. 10.1039/C8BM00637G. [DOI] [PubMed] [Google Scholar]
- (17).Wei T; Cheng Q; Min Y-L; Olson EN; Siegwart DJ Systemic Nanoparticle Delivery of CRISPR-Cas9 Ribonucleoproteins for Effective Tissue Specific Genome Editing. Nat Commun 2020, 11 (1), 3232. 10.1038/s41467-020-17029-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chang J; Chen X; Glass Z; Gao F; Mao L; Wang M; Xu Q Integrating Combinatorial Lipid Nanoparticle and Chemically Modified Protein for Intracellular Delivery and Genome Editing. Acc Chem Res 2019, 52 (3), 665–675. 10.1021/acs.accounts.8b00493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kim YB; Zhao KT; Thompson DB; Liu DR An Anionic Human Protein Mediates Cationic Liposome Delivery of Genome Editing Proteins into Mammalian Cells. Nat Commun 2019, 10 (1), 2905. 10.1038/s41467-019-10828-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Zuris JA; Thompson DB; Shu Y; Guilinger JP; Bessen JL; Hu JH; Maeder ML; Joung JK; Chen Z-Y; Liu DR Cationic Lipid-Mediated Delivery of Proteins Enables Efficient Protein-Based Genome Editing in Vitro and in Vivo. Nat Biotechnol 2015, 33 (1), 73–80. 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eltoukhy AA; Chen D; Veiseh O; Pelet JM; Yin H; Dong Y; Anderson DG Nucleic Acid-Mediated Intracellular Protein Delivery by Lipid-like Nanoparticles. Biomaterials 2014, 35 (24), 6454–6461. https://doi.org/ 10.1016/j.biomaterials.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chan A; Wang HH; Haley RM; Song C; Gonzalez-Martinez D; Bugaj L; Mitchell MJ; Tsourkas A Cytosolic Delivery of Small Protein Scaffolds Enables Efficient Inhibition of Ras and Myc. Mol Pharm 2022, 19 (4), 1104–1116. 10.1021/acs.molpharmaceut.1c00798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wang HH; Tsourkas A Cytosolic Delivery of Inhibitory Antibodies with Cationic Lipids. Proc Natl Acad Sci U S A 2019, 116 (44), 22132–22139. 10.1073/pnas.1913973116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Billingsley MM; Singh N; Ravikumar P; Zhang R; June CH; Mitchell MJ Ionizable Lipid Nanoparticle-Mediated MRNA Delivery for Human CAR T Cell Engineering. Nano Lett 2020, 20 (3), 1578–1589. 10.1021/acs.nanolett.9b04246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Shepherd SJ; Issadore D; Mitchell MJ Microfluidic Formulation of Nanoparticles for Biomedical Applications. Biomaterials 2021, 274, 120826. https://doi.org/ 10.1016/j.biomaterials.2021.120826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shepherd SJ; Warzecha CC; Yadavali S; El-Mayta R; Alameh M-G; Wang L; Weissman D; Wilson JM; Issadore D; Mitchell MJ Scalable MRNA and SiRNA Lipid Nanoparticle Production Using a Parallelized Microfluidic Device. Nano Lett 2021, 21 (13), 5671–5680. 10.1021/acs.nanolett.1c01353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ball RL; Hajj KA; Vizelman J; Bajaj P; Whitehead KA Lipid Nanoparticle Formulations for Enhanced Co-Delivery of SiRNA and MRNA. Nano Lett 2018, 18 (6), 3814–3822. 10.1021/acs.nanolett.8b01101. [DOI] [PubMed] [Google Scholar]
- (28).Cabantous S; Terwilliger TC; Waldo GS Protein Tagging and Detection with Engineered Self-Assembling Fragments of Green Fluorescent Protein. Nat Biotechnol 2005, 23 (1), 102–107. 10.1038/nbt1044. [DOI] [PubMed] [Google Scholar]
- (29).Cheng X; Lee RJ The Role of Helper Lipids in Lipid Nanoparticles (LNPs) Designed for Oligonucleotide Delivery. Adv Drug Deliv Rev 2016, 99, 129–137. https://doi.org/ 10.1016/j.addr.2016.01.022. [DOI] [PubMed] [Google Scholar]
- (30).Eygeris Y; Gupta M; Kim J; Sahay G Chemistry of Lipid Nanoparticles for RNA Delivery. Acc Chem Res 2022, 55 (1), 2–12. 10.1021/acs.accounts.1c00544. [DOI] [PubMed] [Google Scholar]
- (31).Billingsley MM; Hamilton AG; Mai D; Patel SK; Swingle KL; Sheppard NC; June CH; Mitchell MJ Orthogonal Design of Experiments for Optimization of Lipid Nanoparticles for MRNA Engineering of CAR T Cells. Nano Lett 2022, 22 (1), 533–542. 10.1021/acs.nanolett.1c02503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Figueroa E; Bugga P; Asthana V; Chen AL; Stephen Yan J; Evans ER; Drezek RA A Mechanistic Investigation Exploring the Differential Transfection Efficiencies between the Easy-to-Transfect SK-BR3 and Difficult-to-Transfect CT26 Cell Lines. J Nanobiotechnology 2017, 15 (1), 36. 10.1186/s12951-017-0271-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Maurisse R; De Semir D; Emamekhoo H; Bedayat B; Abdolmohammadi A; Parsi H; Gruenert DC Comparative Transfection of DNA into Primary and Transformed Mammalian Cells from Different Lineages. BMC Biotechnol 2010, 10 (1), 9. 10.1186/1472-6750-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Yamano S; Dai J; Moursi AM Comparison of Transfection Efficiency of Nonviral Gene Transfer Reagents. Mol Biotechnol 2010, 46 (3), 287–300. 10.1007/s12033-010-9302-5. [DOI] [PubMed] [Google Scholar]
- (35).Brauchle M; Hansen S; Caussinus E; Lenard A; Ochoa-Espinosa A; Scholz O; Sprecher SG; Plückthun A; Affolter M Protein Interference Applications in Cellular and Developmental Biology Using DARPins That Recognize GFP and MCherry. Biol Open 2014, 3 (12), 1252–1261. 10.1242/bio.201410041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hansen S; Stüber JC; Ernst P; Koch A; Bojar D; Batyuk A; Plückthun A Design and Applications of a Clamp for Green Fluorescent Protein with Picomolar Affinity. Sci Rep 2017, 7 (1), 16292. 10.1038/s41598-017-15711-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bery N; Legg S; Debreczeni J; Breed J; Embrey K; Stubbs C; Kolasinska-Zwierz P; Barrett N; Marwood R; Watson J; Tart J; Overman R; Miller A; Phillips C; Minter R; Rabbitts TH KRAS-Specific Inhibition Using a DARPin Binding to a Site in the Allosteric Lobe. Nat Commun 2019, 10 (1), 2607. 10.1038/s41467-019-10419-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Parizek P; Kummer L; Rube P; Prinz A; Herberg FW; Plückthun A Designed Ankyrin Repeat Proteins (DARPins) as Novel Isoform-Specific Intracellular Inhibitors of c-Jun N-Terminal Kinases. ACS Chem Biol 2012, 7 (8), 1356–1366. 10.1021/cb3001167. [DOI] [PubMed] [Google Scholar]
- (39).Kummer L; Parizek P; Rube P; Millgramm B; Prinz A; Mittl PRE; Kaufholz M; Zimmermann B; Herberg FW; Plückthun A Structural and Functional Analysis of Phosphorylation-Specific Binders of the Kinase ERK from Designed Ankyrin Repeat Protein Libraries. Proc Natl Acad Sci U S A 2012, 109 (34), E2248–E2257. 10.1073/pnas.1205399109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Dilliard SA; Cheng Q; Siegwart DJ On the Mechanism of Tissue-Specific MRNA Delivery by Selective Organ Targeting Nanoparticles. Proc Natl Acad Sci U S A 2021, 118 (52), e2109256118. 10.1073/pnas.2109256118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Qiu M; Tang Y; Chen J; Muriph R; Ye Z; Huang C; Evans J; Henske EP; Xu Q Lung-Selective MRNA Delivery of Synthetic Lipid Nanoparticles for the Treatment of Pulmonary Lymphangioleiomyomatosis. Proceedings of the National Academy of Sciences 2022, 119 (8), e2116271119. 10.1073/pnas.2116271119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Cheng Q; Wei T; Farbiak L; Johnson LT; Dilliard SA; Siegwart DJ Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR-Cas Gene Editing. Nat Nanotechnol 2020. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kieckhaefer JE; Maina F; Wells RG; Wangensteen KJ Liver Cancer Gene Discovery Using Gene Targeting, Sleeping Beauty, and CRISPR/Cas9. Semin Liver Dis 2019, 39 (2), 261–274. 10.1055/s-0039-1678725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Seehawer M; Heinzmann F; D’Artista L; Harbig J; Roux P-F; Hoenicke L; Dang H; Klotz S; Robinson L; Doré G; Rozenblum N; Kang T-W; Chawla R; Buch T; Vucur M; Roth M; Zuber J; Luedde T; Sipos B; Longerich T; Heikenwälder M; Wang XW; Bischof O; Zender L Necroptosis Microenvironment Directs Lineage Commitment in Liver Cancer. Nature 2018, 562 (7725), 69–75. 10.1038/s41586-018-0519-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Warden-Rothman R; Caturegli I; Popik V; Tsourkas A Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal Chem 2013, 85 (22), 11090–11097. 10.1021/ac402871k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Du B; Jiang X; Das A; Zhou Q; Yu M; Jin R; Zheng J Glomerular Barrier Behaves as an Atomically Precise Bandpass Filter in a Sub-Nanometre Regime. Nat Nanotechnol 2017, 12 (11), 1096–1102. 10.1038/nnano.2017.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Dixon AS; Schwinn MK; Hall MP; Zimmerman K; Otto P; Lubben TH; Butler BL; Binkowski BF; Machleidt T; Kirkland TA; Wood MG; Eggers CT; Encell LP; Wood K. v. NanoLuc Complementation Reporter Optimized for Accurate Measurement of Protein Interactions in Cells. ACS Chem Biol 2016, 11 (2), 400–408. 10.1021/acschembio.5b00753. [DOI] [PubMed] [Google Scholar]
- (48).Schwinn MK; Machleidt T; Zimmerman K; Eggers CT; Dixon AS; Hurst R; Hall MP; Encell LP; Binkowski BF; Wood K. v. CRISPR-Mediated Tagging of Endogenous Proteins with a Luminescent Peptide. ACS Chem Biol 2018, 13 (2), 467–474. 10.1021/acschembio.7b00549. [DOI] [PubMed] [Google Scholar]
- (49).Sell S Alpha-Fetoprotein, Stem Cells and Cancer: How Study of the Production of Alpha-Fetoprotein during Chemical Hepatocarcinogenesis Led to Reaffirmation of the Stem Cell Theory of Cancer. Tumour Biol 2008, 29 (3), 161–180. 10.1159/000143402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Fu A; Tang R; Hardie J; Farkas ME; Rotello VM Promises and Pitfalls of Intracellular Delivery of Proteins. Bioconjug Chem 2014, 25 (9), 1602–1608. 10.1021/bc500320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Yu M; Wu J; Shi J; Farokhzad OC Nanotechnology for Protein Delivery: Overview and Perspectives. Journal of Controlled Release 2016, 240, 24–37. https://doi.org/ 10.1016/j.jconrel.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Warden-Rothman R; Caturegli I; Popik V; Tsourkas A Sortase-Tag Expressed Protein Ligation: Combining Protein Purification and Site-Specific Bioconjugation into a Single Step. Anal Chem 2013, 85 (22), 11090–11097. 10.1021/AC402871K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang HH; Altun B; Nwe K; Tsourkas A Proximity-Based Sortase-Mediated Ligation. Angewandte Chemie International Edition 2017, 56 (19), 5349–5352. 10.1002/ANIE.201701419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Riley RS; Kashyap M. v; Billingsley MM; White B; Alameh M-G; Bose SK; Zoltick PW; Li H; Zhang R; Cheng AY; Weissman D; Peranteau WH; Mitchell MJ Ionizable Lipid Nanoparticles for in Utero MRNA Delivery. Sci Adv 2021, 7 (3), eaba1028. 10.1126/sciadv.aba1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Chiu AP; Keng VW Liver-Specific Delivery of Sleeping Beauty Transposon System by Hydrodynamic Injection for Cancer Gene Validation. In Cancer Driver Genes: Methods and Protocols; Starr TK, Ed.; Springer New York: New York, NY, 2019; pp 185–196. 10.1007/978-1-4939-8967-6_15. [DOI] [PubMed] [Google Scholar]
- (56).Preziosi ME; Zahm AM; Vázquez-Salgado AM; Ackerman D; Gade TP; Kaestner KH; Wangensteen KJ In Vivo Screen Identifies LXR Agonism Potentiates Sorafenib Killing of Hepatocellular Carcinoma. bioRxiv 2019, 668350. 10.1101/668350. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.