

Abstract
Autosomal recessive congenital ichthyosis (ARCI) is a group of diseases presenting as collodion baby at birth. ARCI is categorized as Harlequin ichthyosis, lamellar ichthyosis, and non-bullous congenital ichthyosiform erythroderma (NBCIE), bathing suit icthyosis (BSI) and others. We describe the case of a male newborn with NBCIE whose whole exome sequencing revealed two variants of TGM1 gene (NM_000359.3) in a compound heterozygous state: c.790C>T (p.Arg264Trp) in exon 5 and c.2060G>A (p.Arg687His) in exon 13. In the literature, the Arg264Trp variant has been reported as homozygous or compound heterozygous with other variants in patients with BSI. In contrast, the Arg687His variant has been reported only as homozygous in patients with BSI. To the best of our knowledge, this is the first case whose two compound heterozygous variants, exhibiting the NBCIE phenotype, instead of the BSI.
Keywords: Ichthyosis, Infant, newborn, Transglutaminase 1
INTRODUCTION
ARCI (autosomal recessive congenital ichthyosis) is a heterogeneous group of disorders characterized by a deficiency in skin barrier function. ARCI refers to a group of nonsyndromic phenotypes. Harlequin ichthyosis (HI), lamellar ichthyosis (LI), non-bullous congenital ichthyosiform erythroderma (NBCIE), bathing suit ichthyosis (BSI), self-healing collodion baby (SHCB), and others are subtypes of ARCI1.
The term “collodion baby” describes the newborn covered by a shiny, taut, cellophane-like membrane at birth. Its incidence has been estimated to be ~1 in 300,000 births. Ectropion (turning the lower eyelids outward) and eclabium (eversion of the lips) are the other manifestations. These neonates have an increased risk for infections, dehydration, electrolyte imbalances, respiratory problems, feeding difficulty, and ocular issues, whose management require a multidisciplinary approach2.
Systemic retinoids play a crucial role in managing inherited disorders of keratinization and might be life-saving in HI2,3,4. Herein, we report a term neonate presented as collodion baby and diagnosed with NBCIE.
CASE REPORT
The patient, a male neonate of a non-consanguineous couple, was born at 39 weeks 6 days of gestational age by spontaneous vaginal delivery without any complication. The Apgar scores were 9 and 10. There was no relevant family history. He was referred to our Neonatal Intensive Care Unit at the 18th hour of postnatal age due to his red, stretched, shiny skin appearance for further investigation and treatment.
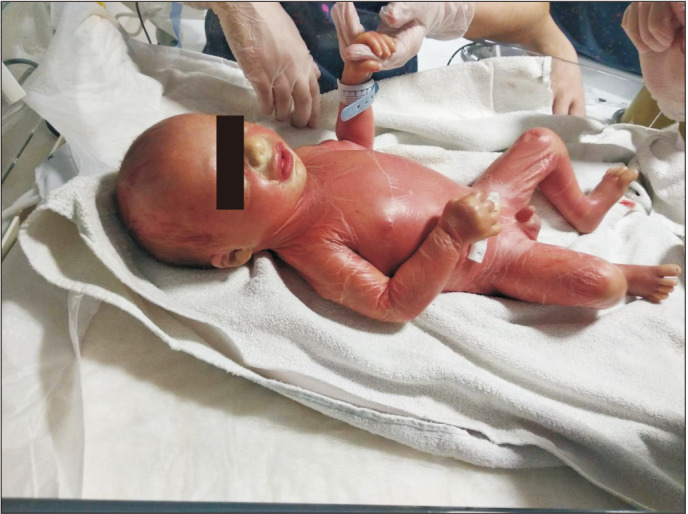
On his first physical examination, the patient was evaluated as critically ill. There was a generalized thick translucent desquamation, including bilateral palmoplantar surfaces. Desquamation was more prominent in the skin folds. Ectropion and eclabium were all present (Fig. 1). There was no mucosal involvement, nail abnormalities, or bullae. He had minimal contractures on his upper extremities.
Fig. 1. Collodion membrane, generalized scaling, prominent eclabium, and bilateral ectropion were present on the first day of life.
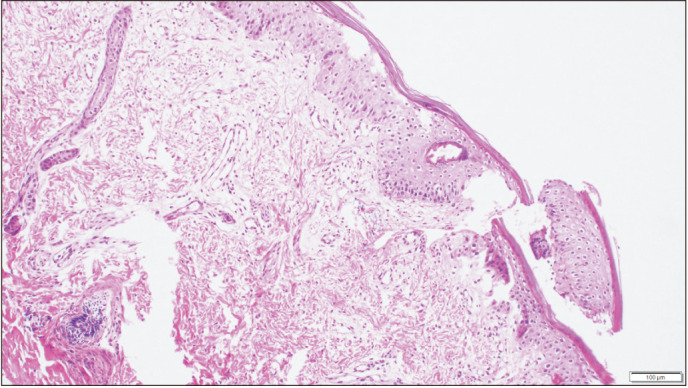
Due to the severe ectropion and eclabium, erythroderma and diffused thick membrane covering body, and the rapid onset sepsis and resulting increased neonatal mortality, oral acitretin 1 mg/kg/day was initiated at the 39th hour of hospitalization. An incisional skin biopsy was performed at the 60th hour of hospitalization. In skin biopsy, compact mild hyperkeratosis and parakeratosis were present in the epidermis. There was a significant loss in the granular layer, intercellular edema in the epidermis, and was compatible with icththyosis histopathologically (Fig. 2).
Fig. 2. Incisional biopsy, light microscopy finding: compact mild hyperkeratosis and parakeratosis in the epidermis and a major loss in the granular layer, intercellular edema in the epidermis (H&E, ×400).
During the first 4 days, parenteral nutrition via central venous catheterization were administered. Since methicillin-resistant Staphylococcus epidermidis was isolated in the catheter culture, antimicrobial therapy was switched to vancomycin.
His health improved after restoring the skin barrier. He got full enteral feeding on day 10. All antibiotics were discontinued on day 13. The incubator's humidity were gradually lowered. Extremity contractures required physiotherapy.
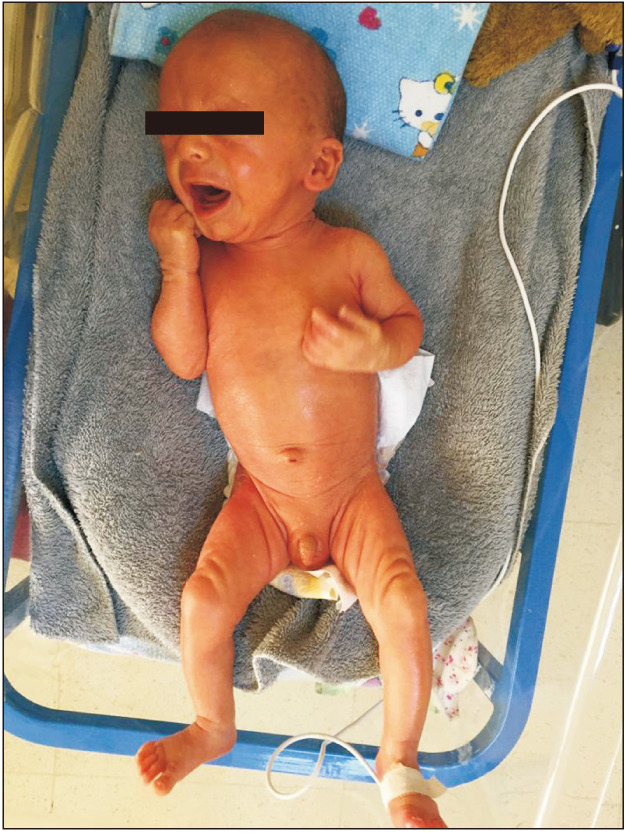
Acitretin treatment was interrupted on day 8. The collodion membrane and desquamation improved significantly. His skin was almost normal (Fig. 3). The ectropion and eclabium were also resolved. His ophthalmologic and audiologic examination was normal. Counseling and multidisciplinary follow-up by all departments were planned before the discharge.
Fig. 3. After 7 days of acitretin treatment, the findings of generalized scaling, ectropion, and eclabium were resolved.
In the virtual gene panel analysis (Sophia CES_v2 solution; Illumina Nexseq 500 system) containing 102 genes associated with ichthyosis, two heterozygous variants in the TGM1 gene were detected in the patient; NM_000359.3: c.790C>T (p.Arg264Trp) and NM_000359.3: c.2060G>A (p. Arg687His). Both variants have been associated with autosomal recessive ichthyosis in the literature and were evaluated according to the American College of Medical Genetics and Genomics 2015 criteria5 and interpreted as pathogenic. Parental testing by Sanger sequencing revealed that the variants were in trans. Mothers’ and fathers’ analysis revealed respectively; NM_000359.3: c.2060G>A (p. Arg687His) and NM_000359.3: c.790C>T (p.Arg264Trp).
At 3rd, 6th months, 1st year, and 18 months follow-up of the patient, white and thin scales in body and mild erythema in the face, extremities were seen (Supplementary Fig. 1). All anthropometric measurements were in the 50th percentiles. The neurocognitive development was within normal limits.
DISCUSSION
ARCI refers to HI, LI, NBCIE, and BSI and rare variants1,4. Our case had a collodion membrane at birth, a typical feature of NBCIE, LI, and BSI; and presented with generalized large white scales, prominent erythema, ectropion, and eclabium. Progressive course without regression and the generalized distribution of the skin lesions enabled us to exclude the diagnoses of BSI and SHCB. In BSI, scales are visible, especially on the trunk, but the face or extremities are usually unaffected4. In SHCB, after the membrane desquamates, the skin of extremities heals; but the axillary region, scalp, mid-trunk skin areas remain involved until 3 months of age1,4. The most striking form of collodion appears in HI, in which almost all parts of the skin is covered with large thick scales that split apart. Our patient did not present nose or ear abnormalities and armor-like collodion membrane; thus, HI diagnosis was excluded.
In addition to the clinical differences, histopathologic findings helped us distinguish NBCIE from LI. NBCIE shows only mild thickening of the stratum corneum with foci of parakeratosis, whereas LI has a markedly thickened stratum corneum without areas of parakeratosis1,4,6. Our findings pointed out the NBCIE diagnosis.
As recommended in the guidelines, oral retinoids could be considered in cases with prominent erythroderma7.
Mutations in 13 genes are known to cause ARCI: ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, LIPN, NIPAL4, PNPLA1, SDR9C7, SLC27A4, SULT2B1, ST14, and TGM17. Some phenotypic features may associate with certain gene mutations, but paradigms for genotype-phenotype correlation need refining7.
The transglutaminase-1 enzyme, involved in forming the keratinized cell envelope, is coded by TGM18.The most frequent TGM1 mutation was c.877–2A>G (IVS5–2A>G) in 9 patients. Six patients with this mutation had more erythema and less scale, categorized as NBCIE, while 3 others were classified as having LI8. TGM1 mutation presents in the SHCB, BSI, NBCIE, LI, overlap NBCIE-LI and severe LI8.
ABCA12, ALOX12B, ALOXE3, CERS3, CYP4F22, LIPN, NIPAL4/ICHTHYIN, PNPLA1, and TGM1 have been reported associated with NBCIE3. It was summarized all mutations led to NBCIE and NBCIE/LI in Table 1.
Table 1. Mutations in TGM1 gene in NBCIE patients.
| Phenotype | Gene | Location | Variant | Zygosity | Reference |
|---|---|---|---|---|---|
| NBCIE | TGM1 | Exon 3 | p.Arg143Cys | Compound heterozygous | 10 |
| Exon 4 | p.Ser212Phe | ||||
| NBCIE | TGM1 | Exon 11 | p.Tyr503Ter (Y503X) | Compound heterozygous | 8 |
| Exon 13 | p.Ser669Ter (S669X) | ||||
| NBCIE-LI overlap | TGM1 | Intron 5 | IVS5-2A>G | Compound heterozygous | 7 |
| Intron 7 | IVS7+1G>T | ||||
| Lamellar/NBCIE overlap | TGM1 | Intron 5 | IVS5-2A>G | Compound heterozygous | 7 |
| Exon 3 | p.Arg126His | ||||
| Lamellar /NBCIE overlap | TGM1 | Intron 2 | IVS2-10delGTinsAGAGC ATGCTACAG; | Compound heterozygous | 7 |
| Intron 5 | IVS5-2A>G | ||||
| Lamellar/NBCIE overlap | TGM1 | Intron 5 | IVS5-2A>G | Homozygous | 7 |
| Lamellar/NBCIE overlap | TGM1 | Intron 14 | IVS14-2A>G | Homozygous | 7 |
| NBCIE/lamellar overlap | TGM1 | Exon7 | c.1083delC, (p.Ile361Ilefs*23) | Homozygous | 7 |
| LI/NBCIE overlap | TGM1 | Exon7 | p.Gly375Asp | Homozygous | 7 |
| Lamellar/NBCIE overlap | TGM1 | Exon3 | p.Arg143Leu | Compound heterozygous | 7 |
| Intron 7 | IVS7-1G>A | ||||
| Lamellar/NBCIE overlap | TGM1 | Exon 6 | p.Arg323Trp | Compound heterozygous | 7 |
| Exon 10 | p.Met487Arg | ||||
| Lamellar/NBCIE overlap | TGM1 | Exon4 | p.Trp193* | Compound heterozygous | 7 |
| Exon7 | p.Val379Leu | ||||
| NBCIE | TGM1 (NM_000359) | Exon 5 | p.Arg264Trp | Compound heterozygous | Our patient |
| Exon 13 | p.Arg687His |
NBCIE: non-bullous congenital ichthyosiform erythroderma, LI: lamellar ichthyosis.
In our patient compound heterozygous Arg687His and Arg264Trp variants were detected in the TGM1 gene. The Arg687His variant is located between the ß-Barrel 1 and 2 domains of the transglutaminase-1 enzyme and is expected to cause decreased cytosolic and membranous transglutaminase-1 activity9. This variant has been reported as homozygous in a patient with BSI and LI9,10. The Arg264Trp variant is located in the core domain of the transglutaminase-1 enzyme and is expected to decrease membranous enzyme function9. This variant has also been reported as homozygous in patients with BSI9,11. In most BSI patients, mutations have been reported in the core domain9,11,12. In our patient, whose two variants were compound heterozygous, the clinical findings were consistent with the NBCIE type. This reveals that mutations with a particular type in TGM1-associated ARCI may cause different phenotypes in different combinations. In addition, other modifier variants may also affect the genotype-phenotype relationship.
Prenatal diagnosis plays a vital role in the prevention. For families with an affected child with ARCI, in which ARCI-causing mutations were detected in the parents or a sibling, prenatal diagnosis is indicated. Chorionic villus or amniotic fluid sampling procedures enable DNA based prenatal diagnosis13. Prenatal ultrasonography could aid in diagnosing HI via the physical examination findings such as eclabium, ectropion, rudimentary ears, contractures, and dense floating particles in amniotic fluid (snowflake sign)14.
Consanguinity could be expected in the family history due to the autosomal recessive inheritance pattern. However, our case was a child of a non-consanguineous couple, highlighting the importance of the prenatal ultrasonographic diagnosis.
Knowing the different types of ichthyosis and how to manage them in the newborn contributes to the long-term survival of the infant. Skin biopsy supports the clinical diagnosis. Molecular diagnosis is a crucial diagnostic tool and has become the gold standard for the diagnosis of the ichthyoses. There is a broad clinical spectrum and diversity of genotype-phenotype correlation, and new cases with different genotypic variants may make it possible to provide a more complete picture of the ARCI group of diseases.
ACKNOWLEDGMENT
Written informed consent for publication of their details was obtained from the parents.
Footnotes
CONFLICTS OF INTEREST: The authors have nothing to disclose.
FUNDING SOURCE: None.
SUPPLEMENTARY MATERIALS
Supplementary data can be found via http://anndermatol.org/src/sm/ad-21-134-s001.pdf.
(A) Fine, white scales, erythroderma, and eclabium are observed on the body, extremities, and full face. Thin scales are white to light grey on the extremities, forehead, ears, neck at 3rd months. (B) Light Brown and platelike scales on the vertex of the scalp at 3rd months. (C) Thin scales are white to gray and beige on the inguinal area, extremities, and hyper-linearity of the toes at 6th months. (D) Thin scales are white on the whole back, body, neck, and head, with mild erythroderma on the neck and head and alopecia. (E) Dry, scaly skin and erythroderma on the trunk, axillar region, neck, and extremities at 1st year follow-up. (F) Dry, scaly skin on the arm and palms at 18th months follow-up.
References
- 1.Oji V, Tadini G, Akiyama M, Blanchet Bardon C, Bodemer C, Bourrat E, et al. Revised nomenclature and classification of inherited ichthyoses: results of the First Ichthyosis Consensus Conference in Sorèze 2009. J Am Acad Dermatol. 2010;63:607–641. doi: 10.1016/j.jaad.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 2.Glick JB, Craiglow BG, Choate KA, Kato H, Fleming RE, Siegfried E, et al. Improved management of Harlequin ichthyosis with advances in neonatal intensive care. Pediatrics. 2017;139:e20161003. doi: 10.1542/peds.2016-1003. [DOI] [PubMed] [Google Scholar]
- 3.Takeichi T, Akiyama M. Inherited ichthyosis: non-syndromic forms. J Dermatol. 2016;43:242–251. doi: 10.1111/1346-8138.13243. [DOI] [PubMed] [Google Scholar]
- 4.Akiyama M. Harlequin ichthyosis and other autosomal recessive congenital ichthyoses: the underlying genetic defects and pathomechanisms. J Dermatol Sci. 2006;42:83–89. doi: 10.1016/j.jdermsci.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mazereeuw-Hautier J, Hernández-Martín A, O'Toole EA, Bygum A, Amaro C, Aldwin M, et al. Management of congenital ichthyoses: European guidelines of care, part two. Br J Dermatol. 2019;180:484–495. doi: 10.1111/bjd.16882. [DOI] [PubMed] [Google Scholar]
- 7.Simpson JK, Martinez-Queipo M, Onoufriadis A, Tso S, Glass E, Liu L, et al. Genotype-phenotype correlation in a large English cohort of patients with autosomal recessive ichthyosis. Br J Dermatol. 2020;182:729–737. doi: 10.1111/bjd.18211. [DOI] [PubMed] [Google Scholar]
- 8.Herman ML, Farasat S, Steinbach PJ, Wei MH, Toure O, Fleckman P, et al. Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat. 2009;30:537–547. doi: 10.1002/humu.20952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oji V, Hautier JM, Ahvazi B, Hausser I, Aufenvenne K, Walker T, et al. Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: evidence for a temperature-sensitive phenotype. Hum Mol Genet. 2006;15:3083–3097. doi: 10.1093/hmg/ddl249. [DOI] [PubMed] [Google Scholar]
- 10.Liu JJ, Yuan YY, Zhang XQ, Li ZM, Xu YS, Gao SM, et al. Mutations of transglutaminase-1 in Chinese patients with autosomal recessive congenital ichthyosis: a case report with clinical and genetic analysis of Chinese cases reported in literature. Clin Exp Dermatol. 2015;40:56–62. doi: 10.1111/ced.12410. [DOI] [PubMed] [Google Scholar]
- 11.Marukian NV, Hu RH, Craiglow BG, Milstone LM, Zhou J, Theos A, et al. Expanding the genotypic spectrum of bathing suit ichthyosis. JAMA Dermatol. 2017;153:537–543. doi: 10.1001/jamadermatol.2017.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marukian NV. Ichthyosis: assessing severity and genotype-phenotype correlations [thesis] New Haven (CT): Yale University; 2017. [Google Scholar]
- 13.Akiyama M, Titeux M, Sakai K, McMillan JR, Tonasso L, Calvas P, et al. DNA-based prenatal diagnosis of harlequin ichthyosis and characterization of ABCA12 mutation consequences. J Invest Dermatol. 2007;127:568–573. doi: 10.1038/sj.jid.5700617. [DOI] [PubMed] [Google Scholar]
- 14.Bongain A, Benoit B, Ejnes L, Lambert JC, Gillet JY. Harlequin fetus: three-dimensional sonographic findings and new diagnostic approach. Ultrasound Obstet Gynecol. 2002;20:82–85. doi: 10.1046/j.1469-0705.2002.00708.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Fine, white scales, erythroderma, and eclabium are observed on the body, extremities, and full face. Thin scales are white to light grey on the extremities, forehead, ears, neck at 3rd months. (B) Light Brown and platelike scales on the vertex of the scalp at 3rd months. (C) Thin scales are white to gray and beige on the inguinal area, extremities, and hyper-linearity of the toes at 6th months. (D) Thin scales are white on the whole back, body, neck, and head, with mild erythroderma on the neck and head and alopecia. (E) Dry, scaly skin and erythroderma on the trunk, axillar region, neck, and extremities at 1st year follow-up. (F) Dry, scaly skin on the arm and palms at 18th months follow-up.