

SUMMARY
Development is regulated by various factors, including protein methylation status. While PRMT5 is well known for its roles in oncogenesis by mediating symmetric di-methylation of arginine, its role in normal development remains elusive. Using Myod1Cre to drive Prmt5 knockout in embryonic myoblasts (Prmt5MKO), we dissected the role of PRMT5 in myogenesis. The Prmt5MKO mice are born normally but exhibit progressive muscle atrophy and premature death. Prmt5MKO inhibits proliferation and promotes premature differentiation of embryonic myoblasts, reducing the number and regenerative function of satellite cells in postnatal mice. Mechanistically, PRMT5 methylates and destabilizes FoxO1. Prmt5MKO increases the total FoxO1 level and promotes its cytoplasmic accumulation, leading to activation of autophagy and depletion of lipid droplets (LDs). Systemic inhibition of autophagy in Prmt5MKO mice restores LDs in myoblasts and moderately improves muscle regeneration. Together, PRMT5 is essential for muscle development and regeneration at least partially through mediating FoxO1 methylation and LD turnover.
In brief
Kim et al. report the role of PRMT5 in muscle development using knockout (KO) mice. Myoblast-specific Prmt5-KO impairs myogenesis and causes premature death. PRMT5 regulates subcellular localization of FoxO1, which influences autophagy of lipid droplets. The work uncovers an interplay between PRMT5, FoxO1, and lipophagy essential for muscle development.
Graphical Abstract
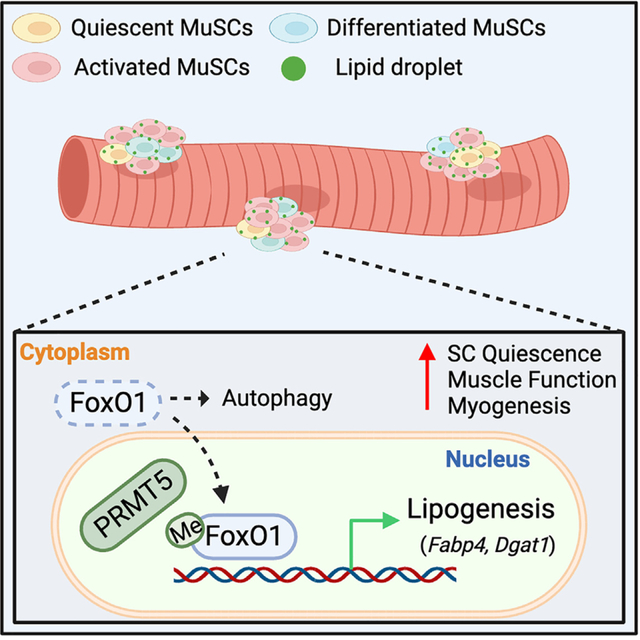
INTRODUCTION
Embryonic myogenic progenitor cells (also called myoblasts) and adult satellite cells (SCs) play an essential role in muscle development and regeneration, respectively.1,2 Embryonic and fetal myogenesis involves two stages of muscle fiber (myofiber) formation.3,4 The first phase, referred to as primary myogenesis, takes place at embryonic day (E) 9.5–14.5, encompassing the differentiation of myoblasts into primary myofibers.3,4 Subsequently, secondary myogenesis occurs at E14.5–17.5, during which myoblasts continue to proliferate and differentiate to form secondary myofibers, as well as deposit SCs at the peripheral of myofibers.3,4 These secondary myofibers are composed of a heterogeneous cell population with different contractile, physiological, and biochemical properties, thereby contributing to the overall morphogenesis and function of a complex musculature.3,4 In the neonatal mice, SCs account for up to 30% of muscle nuclei and undergo extensive proliferation to contribute nuclei to growing myofibers.5 This process of myonuclear accretion through SC proliferation and fusion is crucial for postnatal muscle growth. SCs become quiescent in adult, but they can be activated by muscle injury or other cues to rapidly proliferate and differentiate to repair the injured muscles. Together, proliferation of myoblasts and SCs is critical for muscle development, growth, and regeneration.
Macroautophagy (hereafter referred to as autophagy) serves as an intracellular adaptive mechanism, sustaining quality control and recycling of misfolded proteins and dysfunctional organelles under normal and stress conditions.6 Substantial evidence suggests that proper autophagic activity is necessary for myoblast proliferation, muscle development, and regeneration, as it regulates protein turnover and provides the essential components and energy for new protein synthesis.7–10 Autophagy is triggered to provide bioenergetic support when quiescent SCs become activated, but excessive autophagy activation has been implicated in a plethora of muscular diseases, including muscle atrophy and degeneration.6,11–13 Accumulating evidence has demonstrated that autophagy is indispensable for the breakdown of lipid droplets (LDs) (a process called lipophagy) in skeletal muscle tissues, and the dynamic turnover of LDs is vital in determining the fate of SCs.14,15 Despite considerable understanding on the regulation of LD degradation through autophagic flux, including lipophagy,15 the molecular mechanisms regulating lipophagy in SCs remain poorly characterized.
FoxO1, a member of the FoxO protein family, primarily functions as a transcription factor that regulates gene expression. In this regard, we have shown that nuclear FoxO1 interacts with Notch Intracellular Domain (NICD) to enforce SC quiescence.16 In addition, FoxO1 has also been reported to regulate various physiological processes (such as autophagy) beyond its nuclear functions.17 Accumulating evidence has demonstrated that cytosolic FoxO1, regulated by post-translational modifications (PTMs), triggers autophagy.18,19 Specifically, acetylation of FoxO1 promotes its binding to ATG7, the autophagic initiation protein, thereby stimulating autophagy in cancer cells and podocytes under nutrient-deprived conditions.20,21 Similarly, phosphorylated FoxO1 leads to its cytoplasmic retention, initiating autophagy through interaction with ATG7.22,23 Moreover, FoxO1 has also been implicated in regulating autophagy in skeletal muscle to maintain muscle mass and function, particularly during nutrient deprivation.24–26 However, the mechanisms governing subcellular localization of FoxO1 in myogenic progenitors are not well understood.
Protein arginine methyltransferases (PRMT1–9) are enzymes that mediate the transfer of a methyl group to an arginine (R) residue in substrate proteins. PRMTs regulate a wide range of biological processes, including gene expression, protein localization, and signal transduction, in skeletal muscles through regulating the methylation status of key proteins responsible for the homeostasis of SCs.27,28 Emerging studies have demonstrated that arginine methylation, mediated by PRMTs, also regulates SC function in vivo.27 For example, PRMT1 methylates SIX1 to control MyoD expression, thereby promoting SC proliferation and enhancing muscle regeneration.29 Similarly, PRMT4, also known as CARM1, has been reported to modulate the asymmetric division of SCs and autophagy through Pax7 methylation.30 PRMT7 is recognized not only as an epigenetic regulator that maintains the regenerative capacity of SCs through DNMT3b/p21 axis but also as a promoter of SC differentiation through p38MAPK methylation.31,32 A recent study reported that PRMT5 plays a critical role in SC proliferation and adult muscle regeneration by regulating postnatal day (P) 21 via an unknown mechanism.33 However, it remains unknown whether and how PRMT5 plays a role in embryonic myoblasts and muscle development.
To gain insight into the physiological function of PRMT5 in myogenesis, we generated a conditional knockout (KO) mouse using MyoDCre to drive the deletion of the Prmt5 allele in embryonic myoblasts (referred to as Prmt5MKO). Our investigation revealed that the Prmt5MKO mice exhibited a significant reduction in skeletal muscle mass and contractile function, accompanied by immune cell infiltration. Further analysis uncovered a link between muscle atrophy and early developmental defects, attributed to diminished myoblast proliferation and premature differentiation. Notably, the Prmt5-null myoblasts had elevated cytoplasmic FoxO1 levels, associated with autophagy activation and subsequent depletion of lipid droplets within the myoblasts. Accordingly, pharmacological inhibition of autophagy normalized lipid content and partially restored muscle regeneration in the Prmt5MKO mice. Collectively, our study establishes the role of PRMT5 as a regulator of autophagy in SCs, which subsequently affects muscle development and repair.
RESULTS
Adult Prmt5MKO mice exhibit muscle atrophy and early lethality
To investigate the roles of PRMT5 in myogenic progenitors in vivo, we generated a myogenic lineage-specific conditional Prmt5 KO mice (abbreviated as Prmt5MKO) by crossing Prmt5flox/flox mice with MyoDCre mice expressing Cre recombinase under the control of the Myod1 gene promoter. In this mouse model, the deletion of exon 7 in the Prmt5 gene causes a frameshift and premature stop codon, resulting in a truncated protein without any essential functional domains.34 Real-time qPCR validated the efficient depletion of Prmt5 in skeletal muscles in Prmt5MKO mice (Figure S1A).
Morphologically, the Prmt5MKO mice appeared smaller than wild-type (WT) littermates, as indicated by significantly lower body weights than WT siblings at all examined time points (Figures 1A and S1B). The body weight differences between WT and Prmt5MKO mice became more prominent with age (Figure 1B). As a consequence, the Prmt5MKO mice failed to thrive after 4 months, with all dying before 18 weeks of age (Figure 1C). EchoMRI analysis confirmed that the Prmt5MKO mice exhibited a significantly lower average lean mass than their WT littermates, without significant alternation in fat mass (Figure 1D). The size and weight of various muscle groups, including soleus (SOL), extensor digitorum longus (EDL), tibialis anterior (TA), gastrocnemius (GAS), quadriceps (QU), and diaphragm (Di) were noticeably reduced in the Prmt5MKO mice compared to their WT littermates (Figures 1E and S1C). We further analyzed TA muscle cross sections with myofiber boundaries (sarcolemma) marked by an antibody against dystrophin. The myofiber cross-sectional area (CSA) distribution showed an increased proportion of small myofibers and diminished large myofibers in the Prmt5MKO TA, resulting in a significantly smaller average myofiber size (Figures 1F–1H). Total numbers of myofiber per TA muscle were also reduced in the Prmt5MKO mice (Figure 1I). To assess the impact of Prmt5 deletion on motor function, we conducted treadmill and grip strength tests. The Prmt5MKO mice showed significantly diminished running time, maximum speed, and running distance, as well as reduced grip strength, compared to their WT littermates (Figures 1J–1M). We also directly evaluated muscle force of EDL and SOL muscles from WT and Prmt5MKO mice and observed that Prmt5MKO muscles exhibited lower absolute force (Figures 1L and 1M) and specific force (Figures 1N and 1O).
Figure 1. Conditional KO of Prmt5 in embryonic myoblasts (Prmt5MKO) reduces muscle mass, impairs muscle function, and leads to pre-term death in adult mice.
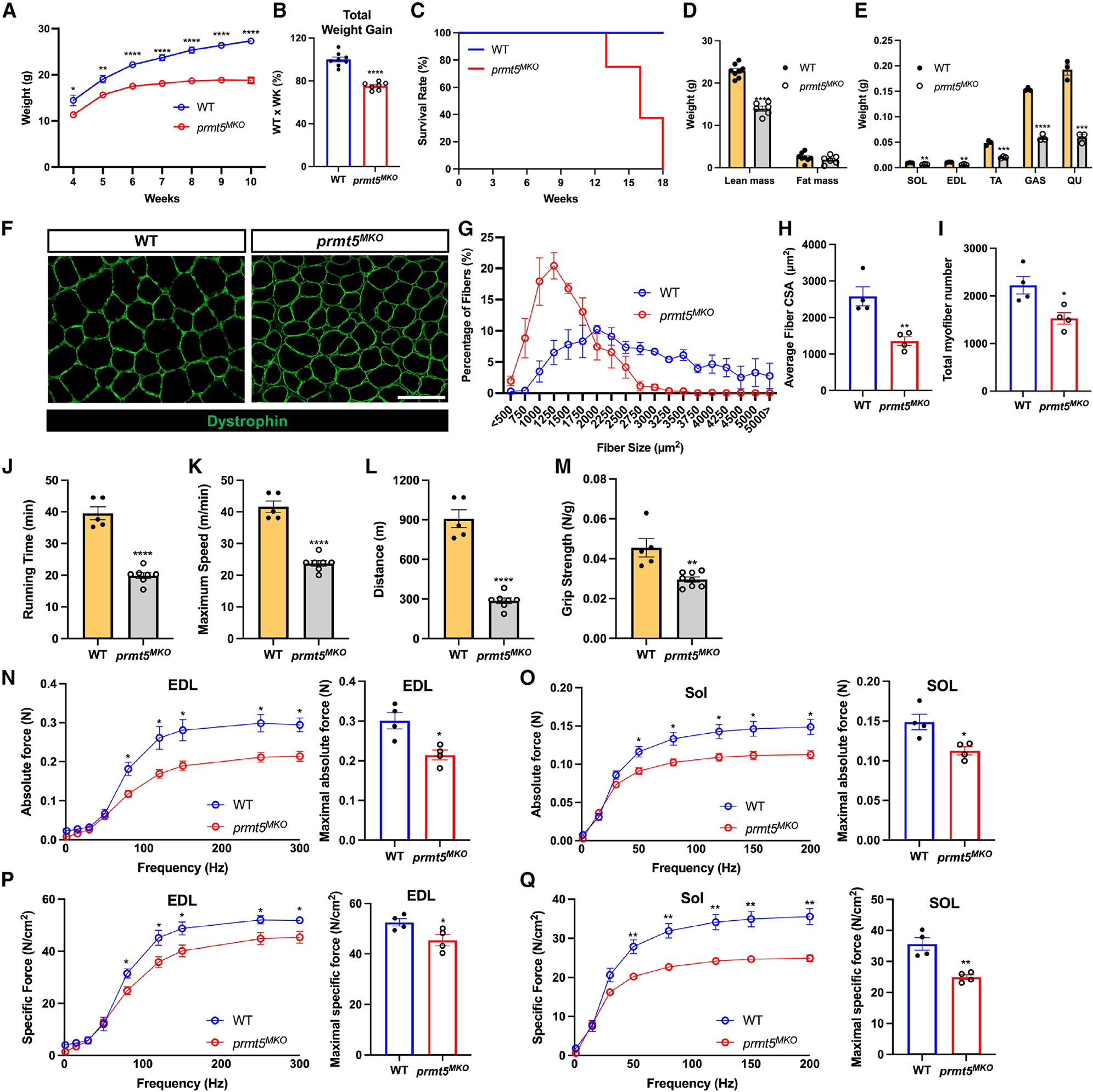
(A and B) Growth curve (A) and total body weight gain over 6 weeks (B) in 4-week-old WT and Prmt5MKO mice (n = 7/group).
(C) Survival of adult WT and Prmt5MKO mice; all KO mice died before reaching 18 weeks old (n = 15).
(D) Lean and fat mass of 8-week-old WT and Prmt5MKO mice (n = 6) measured by EchoMRI body composition analyzer.
(E) Weight of soleus (SOL), extensor digitorum longus (EDL), tibialis anterior (TA), gastrocnemius (GAS), and quadriceps (QU) muscles in adult WT and Prmt5MKO mice (n = 3).
(F) Myofiber morphology outlined by sarcolemma dystrophin immunofluorescence of TA muscle sections from WT and Prmt5MKO mice (n = 4). Scale bar, 100 μm.
(G) Frequency distribution of TA muscle myofiber cross-sectional area (CSA) in adult WT and Prmt5MKO mice (n = 4).
(H and I) Average myofiber CSA (H) and total myofiber number (I) per TA muscle of WT and Prmt5MKO mice (n = 4).
(J–L) Running time (J), maximum speed (K), and running distance (L) of adult WT and Prmt5MKO mice measured by treadmill exhaustion test (n = 5–7).
(M) Grip strength test normalized to body weight in adult WT and Prmt5MKO mice (n = 5–8).
(N and O) Absolute contractile force as a function of frequency of electric stimulation (left) and maximal absolute force (right) measured in the EDL (N) and SOL (O) muscles isolated from adult WT and Prmt5MKO mice (n = 4).
(P and Q) Specific force as a function of frequency (left) and maximal specific force (right) of EDL (P) and SOL (Q) muscles isolated from adult WT and Prmt5MKO mice (n = 4).
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by t tests. See also Figure S1.
We also performed pathological analysis of skeletal muscles from both Prmt5MKO and littermate control mice. H&E staining revealed that the Prmt5MKO TA muscles contained numerous centrally nucleated myofibers (CNFs) (Figure S1D, asterisks), a defining characteristic of myopathy. In addition, the Prmt5MKO Di muscles exhibited an accumulation of interstitial mononuclear cells and were visibly thinner compared to WT muscles (Figure S1E). Furthermore, an elevated number of IgM+ myofibers and CD68+ cells, markers for degenerating myofibers and infiltrated macrophages/monocytes, respectively, were detected in the Prmt5MKO muscles, indicative of inflammation (Figures S1F and S1G). To further substantiate our findings, we quantified several immune cell markers in mononuclear cells dissociated from hindlimb muscles using fluorescence-activated cell sorting (FACS). The Prmt5MKO muscles contained a significantly higher population of immune cells (CD45+, CD11b+, MHCII+, F4/80+) compared to WT muscles (Figures S1H and S1I). Collectively, these results demonstrate that myogenic line-age-specific ablation of Prmt5 leads to age-dependent decline in muscle size and contractile function, accompanied by pathogenic myopathy and chronic inflammation.
Prmt5MKO diminishes SCs and their regenerative capacity in adult mice
As SCs are derived from embryonic myoblasts, we further explored whether the deletion of Prmt5MKO affects the generation of SCs.35 Using FACS, we isolated SCs from the hindlimb muscles of WT and Prmt5MKO mice, revealing a significantly lower abundance of SCs in the Prmt5MKO mice (Figure S2A). RT-qPCR analysis demonstrated reduced Pax7 and elevated Myog mRNA levels in the Prmt5MKO muscles (Figure S2B). Consistently, immunofluorescence staining confirmed that the Prmt5MKO muscles had significantly lower numbers of Pax7+ SCs and an increased numbers of MyoG+ differentiating cells (Figures S2C and S2D). Similarly, staining for Pax7 in freshly isolated myofibers revealed fewer Pax7+ cells and myonuclei per myofiber in the Prmt5MKO myofibers (Figures S2E–S2G). In contrast, a higher proportion of myofiber-associated SCs expressed MyoG in Prmt5MKO mice (Figure S2H), and the Prmt5-deficient myoblasts fused poorly despite normal differentiation (Figure S2I). These results demonstrate an essential role of PRMT5 in maintaining SCs, as the Prmt5MKO SCs undergo spontaneous differentiation in adult muscles.
To assess the functionality of the remaining Prmt5-null SCs, we examined the regeneration of TA muscles following cardiotoxin (CTX)-induced injury (Figures S3A and S3B). Histological analyses revealed that the Prmt5MKO muscles displayed severe inflammatory infiltration, resulting in 21% and 39% reductions in the recovery rate compared to WT muscles at 5 and 21 days post injury (DPI), respectively (Figure S3C). Immunohistochemistry also demonstrated a significant reduction in the number of Pax7+ cells, and MyoG+ cells, as well as in the newly regenerated myofibers surrounded by laminin in the Prmt5MKO mice at 5 DPI (Figures S3D and S3E). At 21 DPI, a reduced number of Pax7+ cells and an increase in MyoG+ cells were detected in the Prmt5MKO muscles, but regenerated laminin-surrounded myofibers were observed in both WT and KO muscles (Figures S3F and S3G). The initial lack of regenerated myofibers at 5 DPI followed by eventual catch-up at 21 DPI suggest a delay in the regeneration process. Taken together, these results indicate that PRMT5 in embryonic myoblasts plays a critical role in the generation of SCs, and continued expression of PRMT5 is necessary for the efficient regenerative capacity of the SCs.
Prmt5MKO impairs SC proliferation and delays muscle growth and maturation
Given the reduced number of SCs and myonuclei in Prmt5MKO mice (Figures S2F and S2G), we hypothesized that PRMT5 in SCs may be crucial for myonuclei accretion during postnatal growth. To test this hypothesis, we examined WT and Prmt5MKO mice during early postnatal muscle growth (P7, P14). We observed fewer Pax7+ cells and more MyoG+ cells at P7 and P14 in Prmt5MKO muscles (Figures 2A and 2B), suggesting reduced proliferation and premature differentiation of these Prmt5-null myogenic progenitors. Interestingly, we also noticed numerous nascent eMyHC+ myofibers in the Prmt5MKO muscles, which were rarely detectable in WT muscles at P7 (Figure 2C), indicating a delayed postnatal maturation or formation of myofibers. Immunoblotting analysis not only confirmed the robust upregulation of eMyHC proteins in the Prmt5MKO muscles at P7 but also revealed diminished protein contents of MyoD in the Prmt5MKO muscles at both P7 and P14 (Figures 2D and 2E). As MyoD marks proliferating myoblasts (non-quiescent SCs) in growing muscles, the diminished MyoD level demonstrates proliferation defects of the Prmt5MKO SCs. These findings collectively suggest that PRMT5 is indispensable for SC proliferation and early postnatal muscle growth and maturation, resulting in a significant reduction in body weight in the Prmt5MKO relative to WT mice (Figure 2F).
Figure 2. Prmt5 KO in embryonic myoblasts (Prmt5MKO) delay postnatal myogenesis and muscle growth.
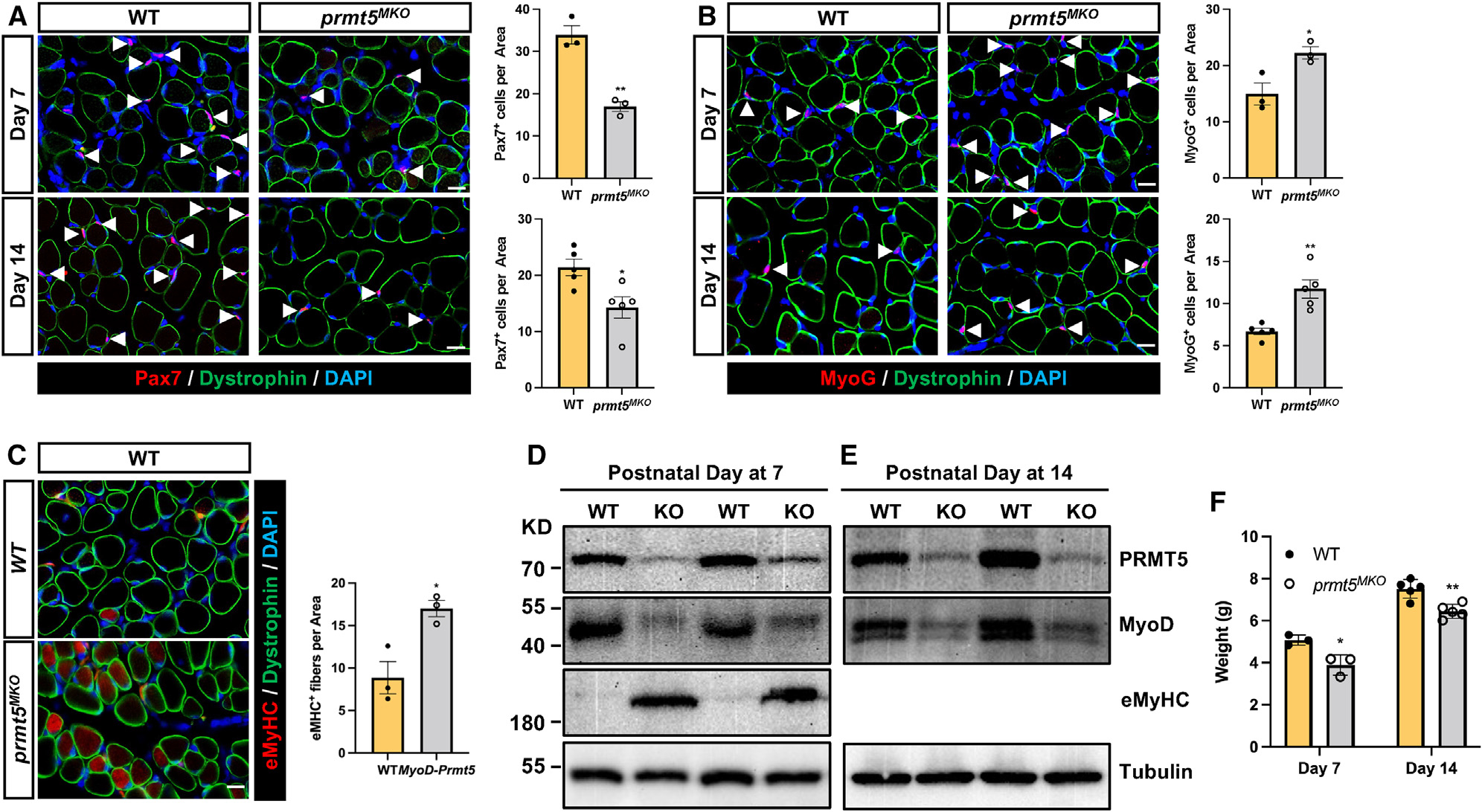
(A and B) immunofluorescence of Pax7 (A) and MyoG (B) in the left panels and corresponding quantification of cells, in right panels, of WT and Prmt5MKO TA muscle sections at P7 (n = 3) and P14 (n = 5). Myofiber membranes are stained with dystrophin and nuclei are stained by DAPI sarcolemma. Scale bar, 10 μm.
(C) eMyHC immunofluorescence (left) and quantification of eMyHC+ myofibers (right) in TA muscle of WT and Prmt5MKO mice at P7 (n = 4–5). Scale bar, 10 μm.
(D and E) immunoblotting analysis showing the protein contents of PRMT5, MyoD, eMyHC, and tubulin at P7 (D) and P14 (E) in skeletal muscles of WT and Prmt5MKO mice. KD, kilodalton size marker.
(F) Body weights of P7 and P14 WT and Prmt5MKO mice (P7, n = 3; P14, n = 5).
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01 by t test. See also Figure S3.
PRMT5 is essential for embryonic and perianal myogenesis
A reduced number of Pax7+ SCs at P7 suggests a defect in the generation of myoblasts and SCs during embryonic myogenesis in the Prmt5MKO mice. To explore the involvement of PRMT5 in the early stages of myogenesis, we analyzed hindlimb muscles of WT and Prmt5MKO mice during embryonic and perinatal myogenesis. We observed a significant decrease in body size and body weight of Prmt5MKO newborn mice (P1) compared to WT littermates (Figure 3A). Immunofluorescence analysis revealed reduced proliferating SCs (calculated by proportions of Pax7+Ki67+ cells among all Pax7+ cells) and increased MyoG+ cells in KO muscles (Figures 3B and 3C). Additionally, hindlimb muscles of E18.5 Prmt5MKO mice were smaller with fewer Pax7+ cells and more MyoG+ cells compared to their WT littermates (Figures 3D–3G).
Figure 3. Prmt5 KO in embryonic myoblast (Prmt5MKO) impedes cell proliferation and promotes premature differentiation, hampering embryonic and fetal myogenesis.
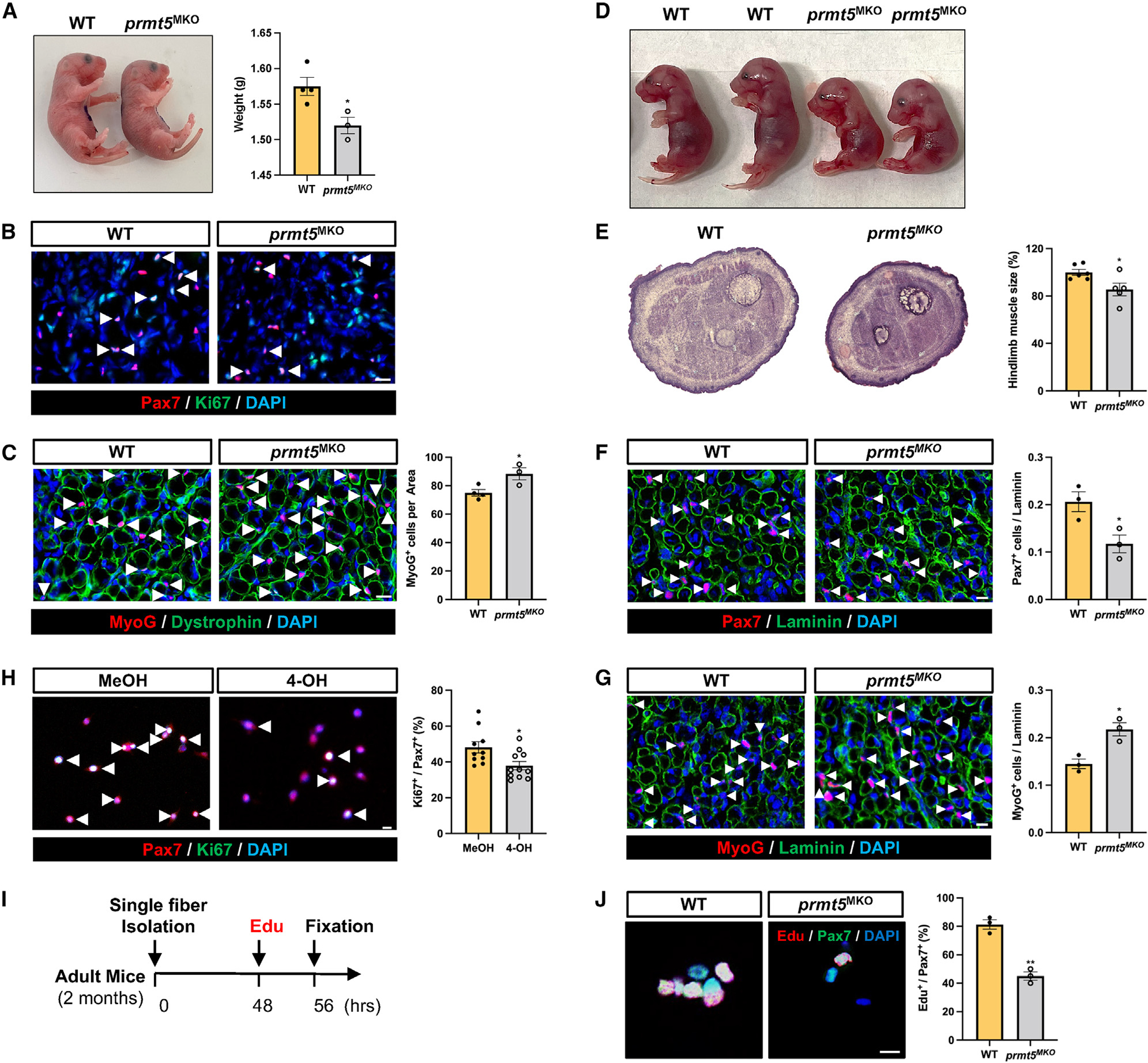
(A) Gross morphology (left) and body weight (right) of newborn mice (P1) in WT and Prmt5MKO mice (n = 3–4).
(B) Pax7+ and Ki67+ immunofluorescence (left) and quantification (right) in hindlimb cross sections of newborn WT and Prmt5MKO mice (n = 3–4). Scale bar, 10 μm.
(C) MyoG immunofluorescence (left) and quantification of MyoG+ cells per area (right) in hindlimb muscle sections of P1 WT and Prmt5MKO mice (n = 3–4). Scale bar, 10 μm.
(D) Gross morphology of E18.5 WT and Prmt5MKO embryos.
(E) H&E staining (left) and quantification of E18.5 hindlimb (right) of WT and Prmt5MKO mice.
(F and G) Pax7 (F) and MyoG (G) immunofluorescence images (left) and corresponding quantification of positive cells per myofiber (right) in WT and Prmt5MKO E18.5 hindlimb sections (n = 3). Scale bar, 10 μm.
(H) Pax7 and Ki67 immunofluorescence (left) and percentage of proliferating (Ki67+) cells among Pax7+ myoblasts (right). Primary myoblasts were treated with vehicle or 4-OH-TMX (4-hydroxyl-tamoxifen) (to induce deletion of Prmt5). Scale bar, 10 μm.
(I) Schematic outline of EdU incorporation assay to assess proliferating SCs on single myofibers isolated from adult WT and Prmt5MKO mice.
(J) Pax7 immunofluorescence cells and EdU click labeling (left), along with the quantification (percentages of EdU+ cells among Pax7+ cells) grown on WT and Prmt5MKO myofibers (right) (n = 3). Scale bar, 10 μm.
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01 by t test. See also Figure S2.
To further investigate the effects of PRMT5 on SC proliferation, we established myoblasts from tamoxifen (TAM)-inducible Prmt5flox/flox/RosaCreER mice and treated the cells with 4-hydroxy-tamoxifen (4-OH) to induce Prmt5 deletion, or with methanol (MeOH) as vehicle control. We found a significant reduction in Ki67+ cells in 4-OH treated (Prmt5-KO) myoblasts compared to MeOH-treated control cells (Figure 3H). Furthermore, EdU (5-ethynyl-2′-deoxyuridine) labeling of proliferating cells on cultured single myofibers revealed significantly lower percentages of EdU+Pax7+ SCs on Prmt5MKO myofibers relative to WT myofibers (Figures 3I and 3J). Taken together, these results demonstrate that PRMT5 plays an important role in maintaining embryonic and perinatal myogenesis, and its deletion hinders proliferative capacity of myoblasts and impairs muscle development.
Prmt5MKO alters lipid metabolism and impairs SC fate homeostasis
PRMT5 has been shown to regulate lipid metabolism in adipocytes and skeletal muscles.34,36 We have recently identified LD as an important metabolic regulator of SC fate,14 we therefore explored whether PRMT5 regulates SCs through LDs. We utilized oil red O (ORO) staining to visualize and quantify LD contents in myoblasts. The Prmt5-null myoblasts exhibited a remarkably reduced intensity of ORO+ LDs (Figure 4A). Immunofluorescent staining of LDs by BODIPY also revealed a decreased intensity of LDs in Prmt5-KO compared to WT myoblasts (Figure 4B). Additionally, using the myofiber explant model, we observed that SCs on 72-h-cultured single myofibers isolated from Prmt5MKO mice failed to accumulate LDs, while WT SCs accumulated many LDs (Figure 4C). Diminished LD content was associated with upregulation of lipolysis gene (Pnpla2) and downregulated genes related to lipid synthesis (Fabp4, Dgat1) in Prmt5-null myoblasts (Figure 4D).
Figure 4. Prmt5 KO diminishes LD abundance and oxidative respiration of SCs, disrupting their cell fate homeostasis.
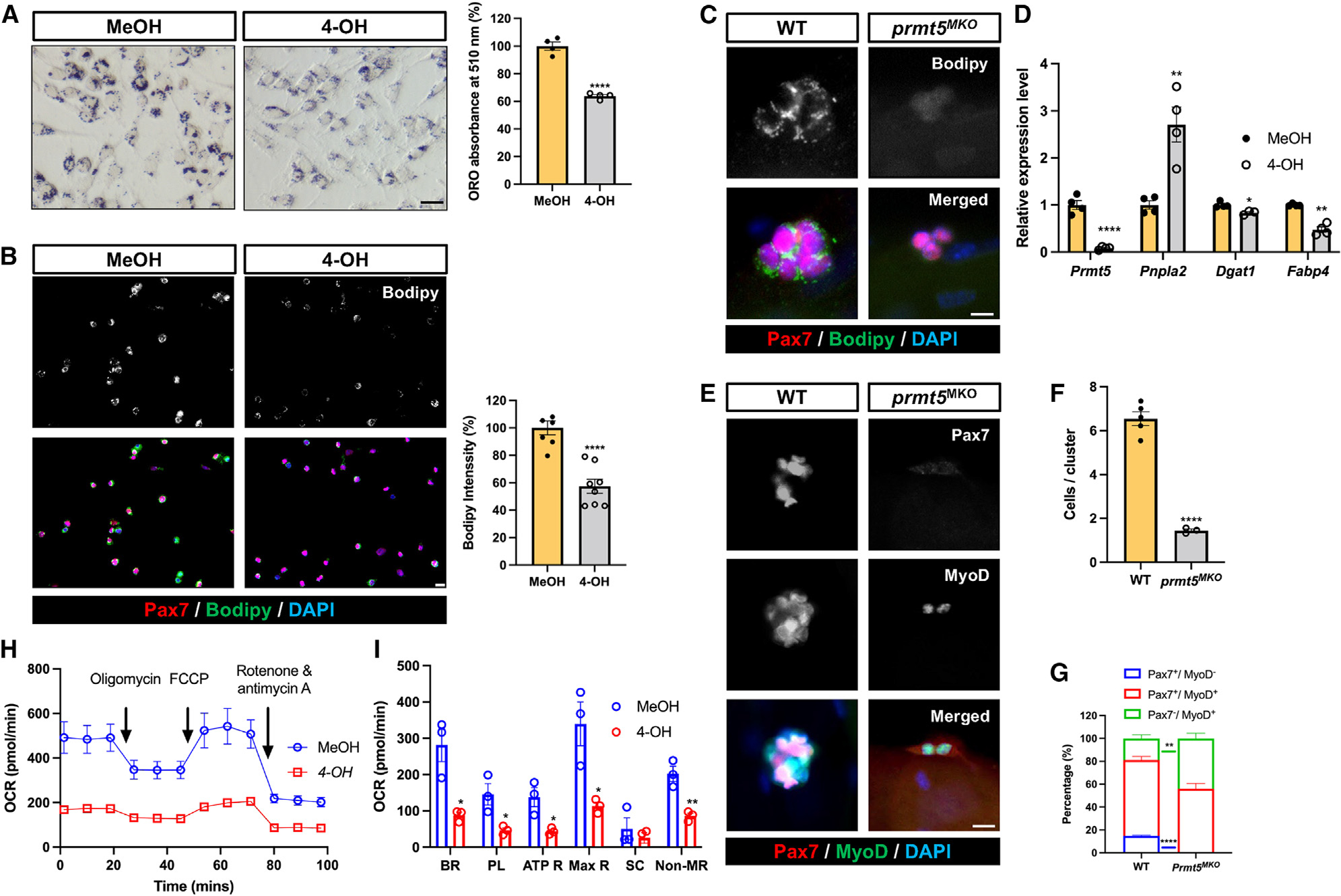
(A) Oil red O staining (left) and quantification (right) of LD content in control (MeOH) and Prmt5 KO (4-OH) myoblasts. Scale bar, 50 μm.
(B) Pax7 and Bodipy immunofluorescence (left) and quantification of Bodipy intensity (right) in control (MeOH) and Prmt5 KO (4-OH) myoblasts. Scale bar, 10 mm.
(C) Pax7 and Bodipy immunofluorescence (left) and quantification of Bodipy intensity (right) in SCs after proliferating for 72 h on cultured single myofibers isolated from WT and Prmt5MKO mice. Scale bar, 10 μm.
(D) qPCR analysis of lipogenic marker (Pnpla2) and adipogenesis markers (Dgat1, Fabp4) in control (MeOH) and Prmt5 KO (4-OH) myoblasts (n = 4).
(E–G) Pax7 and MyoD fluorescence to distinguish cell fate status in WT and Prmt5MKO SCs (E), quantification of SCs per cluster (F), and proportions of self-renewing (Pax7+MyoD−), proliferating (Pax7+MyoD+), and differentiating (Pax7−MyoD+) cells (G) after culturing on single myofibers for 72 h (n = 3). Scale bar, 10 μm.
(H) Seahorse measurement of oxygen consumption rate (OCR) in control (MeOH) and Prmt5 KO (4-OH) myoblasts in response to sequential addition of oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and rotenone/antimycin A (n = 3).
(I) Average OCR for basal respiration (BR), proton leak (PL), ATP respiration (ATP R), maximal respiration (Max R), spare capacity, and non-mitochondrial respiration (NR) based on H (n = 3). SC, spare capacity.
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ****p < 0.0001 by t test. See also Figure S4.
To investigate whether LD reduction in the Prmt5-null cells alters SC fate, the SCs were stained with Pax7 and MyoD antibodies (Figure 4E). In Prmt5-null myofibers, there was not only a reduction in the number of cells per cluster but also a significant decrease in Pax7+/MyoD− (self-renewing) cells, accompanied by an increase in the proportion of Pax7−/MyoD+ (differentiating) cells (Figures 4F and 4G). To evaluate the energetic consequence of LD reduction in Prmt5-null myoblasts, we used Seahorse bioanalyzer to determine the oxygen consumption rate (OCR) (Figure 4H). The results showed a significant decrease in OCR for basal respiration, proton leak, ATP respiration, maximal respiration, and non-mitochondrial respiration in Prmt5-null myoblasts compared to WT cells (Figures 4H and 4I). These findings indicate that Prmt5MKO alters lipid metabolism and oxygen-dependent cellular bioenergetics in SCs, disrupting the homeostasis of SC fate.
Prmt5MKO myoblasts exhibit elevated autophagy
We next explored the molecular mechanism responsible for the depletion of LDs and spontaneous differentiation of Prmt5MKO SCs. Previous work indicated that autophagy plays a critical role in both lipid catabolism and SC activation from quiescence.12,37 To investigate the potential role of PRMT5 in regulating myoblast autophagy, we exposed both WT (MeOH treated) and KO (4-OH treated) Rosa26CreER/Prmt5flox/flox myoblasts to a 3-h serum withdrawal to induce autophagy in the presence or absence of the autophagy inhibitor, hydroxychloroquine (HCQ). Compared to WT myoblasts, the Prmt5-KO myoblasts exhibited a notable increase in LC3-lipidation, indicated by the LC3 II/LC3 I ratios (Figure 5A). Notably, the elevated LC3 II/I ratio in Prmt5-KO myoblasts was normalized by HCQ (Figure 5A), indicating that Prmt5 KO promotes autophagy. To exclude the possibility that autophagy and LD depletion are induced by the 4-OH treatment, we treated myoblasts derived from Rosa26CreER/Prmt5flox/+ mice with MeOH or 4-OH. No significant differences were observed in the autophagic response (LC3BII/I levels) or LD content (Bodipy intensity) in myoblasts, excluding a role of 4-OH (Figures S4A and S4B). The Prmt5MKO myoblasts also showed higher mRNA levels of autophagy-related genes (LC3B, Lamp1, Ulk1, Beclin1, Atg7) compared to WT myoblasts (Figure 5B). Moreover, the average intensity of LC3B immunofluorescence in Prmt5-null SCs was higher than that of WT SCs attached to freshly isolated single myofibers (Figure 5C). These results illustrate that Prmt5MKO activates autophagy in SCs.
Figure 5. Prmt5 KO activates autophagy in satellite cells.
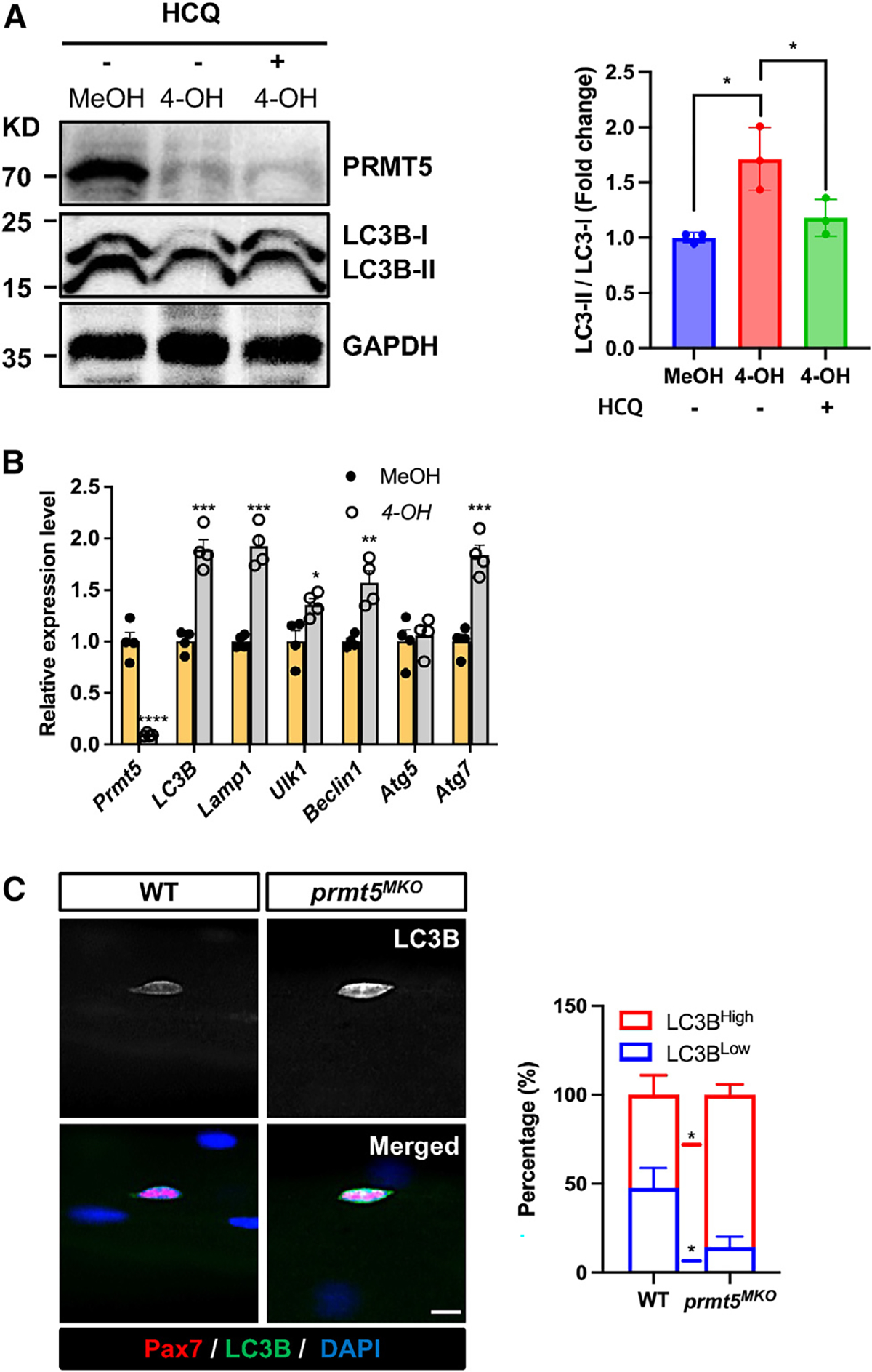
(A) Western blotting showing levels of PRMT5, LC3B, and GAPDH in control (MeOH) and Prmt5 KO (4-OH) myoblasts in the absence or presence of hydroxychloroquine (HCQ), along with quantified levels (normalized to GAPDH) of LC-II/I, as an indicator of autophagy (right). KD, kilodalton size marker.
(B) qPCR analysis of autophagy makers in control (MeOH) and Prmt5 KO (4-OH) myoblasts (n = 4).
(C) Pax7 and LC3B immunofluorescence (left) and the relative intensity of LC3B (right) in Pax7+ cells grown on freshly isolated single myofibers from WT and Prmt5MKO mice (n = 4). Scale bar, 10 μm.
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by t test. See also Figure S5.
PRMT5 mediates FoxO1 methylation to modulate its subcellular localization
We next sought to understand how PRMT5 regulates autophagy. Forkhead box protein O1 (FoxO1) is a transcriptional factor that, when sequestrated in the cytoplasm, induces autophagy.38 Interestingly, we observed that Prmt5MKO resulted in increased levels of cytoplasmic FoxO1 (Figure 6A). Additionally, we performed immunofluorescence staining of FoxO1 in SCs on freshly isolated myofibers (Figure 6B). While FoxO1 expression was predominantly localized to the nucleus in WT SCs (nearly 100%), approximately 65% of Prmt5MKO SCs exhibited nuclear FoxO1, with 35% of SCs showing cytoplasmic FoxO1 distribution (Figure 6B). These results provide an unexplored function of PRMT5 in regulating the subcellular distribution of FoxO1 in SCs.
Figure 6. PRMT5 mediates methylation of FoxO1 to alter its subcellular localization.
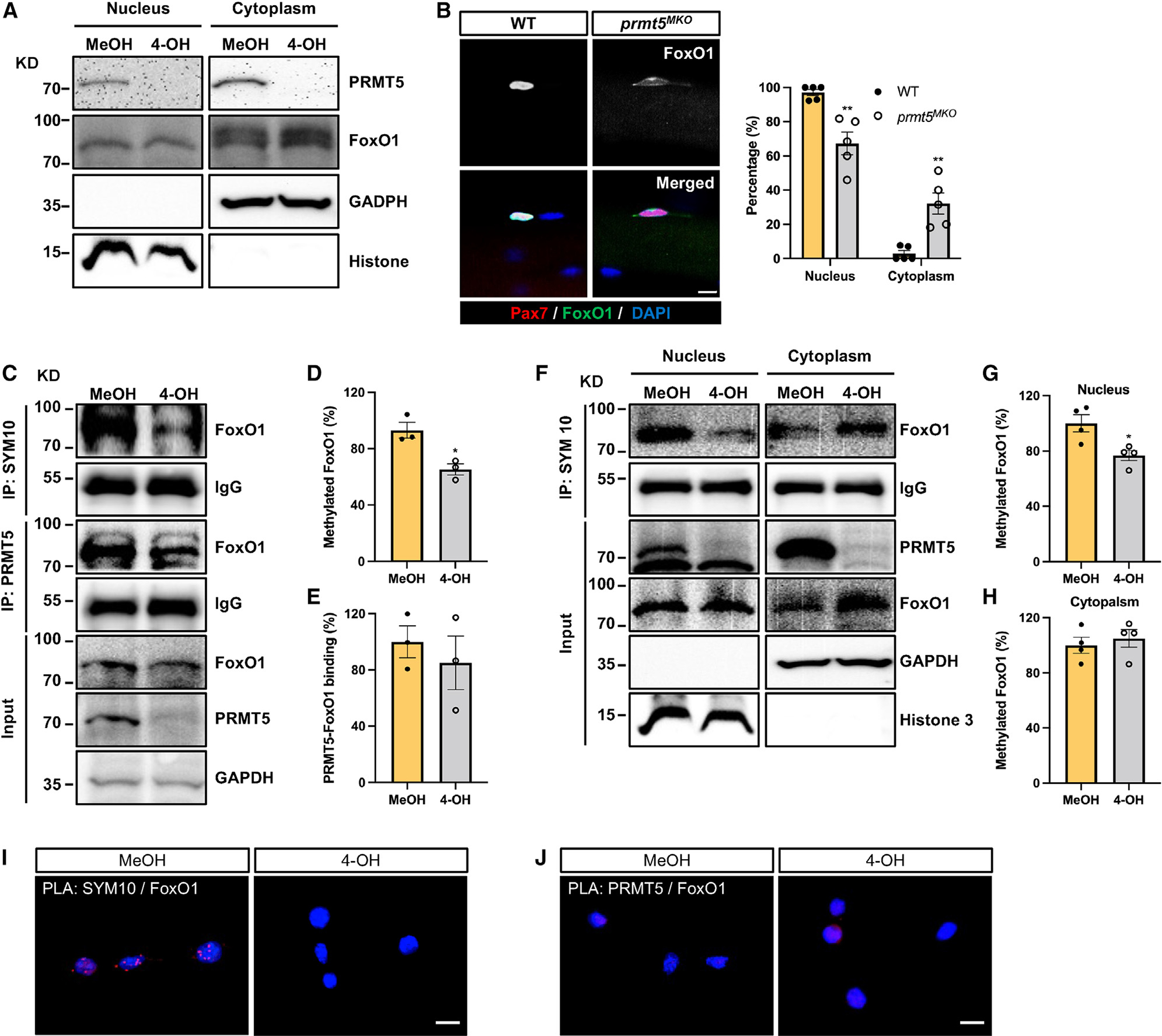
(A) Western blot images showing subcellular fraction of control (MeOH) and Prmt5 KO (4-OH) myoblasts and cytoplasmic retention of FoxO1 in Prmt5 KO cells. KD, kilodalton size marker.
(B) Pax7 and FoxO1 immunofluorescence (left) of SCs on freshly isolated myofibers from WT and Prmt5MKO mice, along with quantification of FoxO1 subcellular location (right) (n = 5). Scale bar, 10 μm.
(C) Control (MeOH) and Prmt5 KO (4-OH) myoblasts were immunoprecipitated with SYM10 and PRMT5 antibody and blotted with FoxO1, PRMT5, and GAPDH antibodies. KD, kilodalton size marker.
(D and E) Quantification of methylated FoxO1 (D) and PRMT5-FoxO1 binding (E) (n = 3).
(F) Control (MeOH) and Prmt5 KO (4-OH) myoblasts were immunoprecipitated with SYM10 after subcellular fractionation and blotted with PRMT5, FoxO1, GAPDH, and histone 3 antibodies. KD, kilodalton size marker.
(G and H) Quantification of methylated FoxO1 in the nucleus (G) and cytoplasm (H) (n = 4).
(I and J) Proximity ligation assay (PLA) on control (MeOH) and Prmt5 KO (4-OH) myoblasts using SYM10 and FoxO1 (I), or PRMT5 and FoxO1 (J) antibody pairs.
The presence of red punta indicates potential protein modification or interaction. Scale bar, 10 μm. Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01 by t test. See also Figure S6.
We then performed a series of biochemical analyses to further understand how PRMT5 regulates subcellular localization of FoxO1 in SCs. We investigated the potential protein interaction between FoxO1 and PRMT5 and whether or not PRMT5 mediates FoxO1 methylation to alter its subcellular localization. A co-immunoprecipitation (coIP) experiment revealed that, compared to the control (MeOH) group, Prmt5 deletion (4-OH) significantly reduced the dimethylation of FoxO1, marked by the SYM10 antibody (Figures 6C and 6D). However, a direct interaction between PRMT5 and FoxO1 was not detected (Figures 6C and 6E). To explore the effect of Prmt5 KO on the intracellular localization of FoxO1, we conducted coIP following cellular fractionation. Remarkably, Prmt5 KO only reduced the levels of methylated FoxO1 in the nucleus, without significantly altering FoxO1 methylation in the cytoplasm, resulting in a higher level of total cytoplasmic FoxO1 (Figures 6F–6H). Conversely, the overexpression of Prmt5 led to enhanced FoxO1 methylation, and pharmacological inhibition of PRMT5 using BLL3.3 decreased FoxO1 methylation in C2C12 myoblasts (Figures S5A and S5B). To further verify the involvement of PRMT5 in FoxO1 methylation and its potential interaction with FoxO1, we employed in situ proximity ligation assay (PLA), a technique that enables the visualization of colocalized proteins within 40 nm as puncta signal. The results confirmed colocalization of symmetric demethylated protein and FoxO1 in the nucleus of WT myoblasts but not in KO myoblasts (Figure 6I). However, we did not observe PLA signals of PRMT5 and FoxO1 in either WT or KO myoblasts (Figure 6J) To investigate the biological significance of PRMT5-mediated FoxO1 methylation, we conducted FoxO1 protein stability assay after the addition of cycloheximide (CHX) in control (MeOH) and KO (4-OH) C2C12 myoblasts. The results revealed that FoxO1 stability was increased over time in the KO group (Figure S5C), indicating that PRMT5-mediated methylation destabilizes FoxO. Overall, these findings suggest that PRMT5 governs the methylation of nuclear FoxO1 and Prmt5-KO leads to the accumulation of FoxO1 in the cytoplasm.
To directly examine the role of cytoplasmic FoxO1 in autophagy, we transfected C2C12 myoblasts with different constructs, including an empty vector (EV), WT FoxO1, a cytoplasmic FoxO1 mutant lacking the DNA binding domain (ΔDB), and a variant of ΔDB with mutations in the three AKT phosphorylation sites (ΔDB3A) that promote nuclear localization (Figure S6A). Immunoprecipitation-western blot analysis revealed that myoblasts transfected with WT or ΔDB had elevated autophagy, as indicated by LC3II/LC1 ratio, and increased association between FoxO1 and autophagy initiation factor ATG7, as compared to ΔDB3A-transfected myoblasts (Figures S6B and S6C). Subsequently, we only detected the interaction between FoxO1 and ATG7 in the cytoplasm of primary myoblasts, but not in the nucleus (Figure S6D). Collectively, these findings provide substantial evidence that cytoplasmic FoxO1 associates with ATG 7 to modulate autophagic activity in muscle cells.
Pharmacological inhibition of autophagy partially rescues Prmt5MKO myoblasts
As inhibition of autophagy increases lipid content,37,39 we tested whether inhibition of autophagic flux with HCQ could rescue the lipid content and function of Prmt5-null myoblasts (Figure 7A). This treatment led to an increased intensity of LDs in both WT and Prmt5-null myoblasts, with a more significantly robust increase in KO cells (Figure 7B). Furthermore, HCQ treatment remarkably upregulated the expression of the lipid metabolism marker (Dgat1), while simultaneously suppressing the autophagic marker LC3B in Prmt5-null SCs (Figure 7C).
Figure 7. Pharmacological inhibition of autophagy alleviates defects of Prmt5-KO myoblasts.
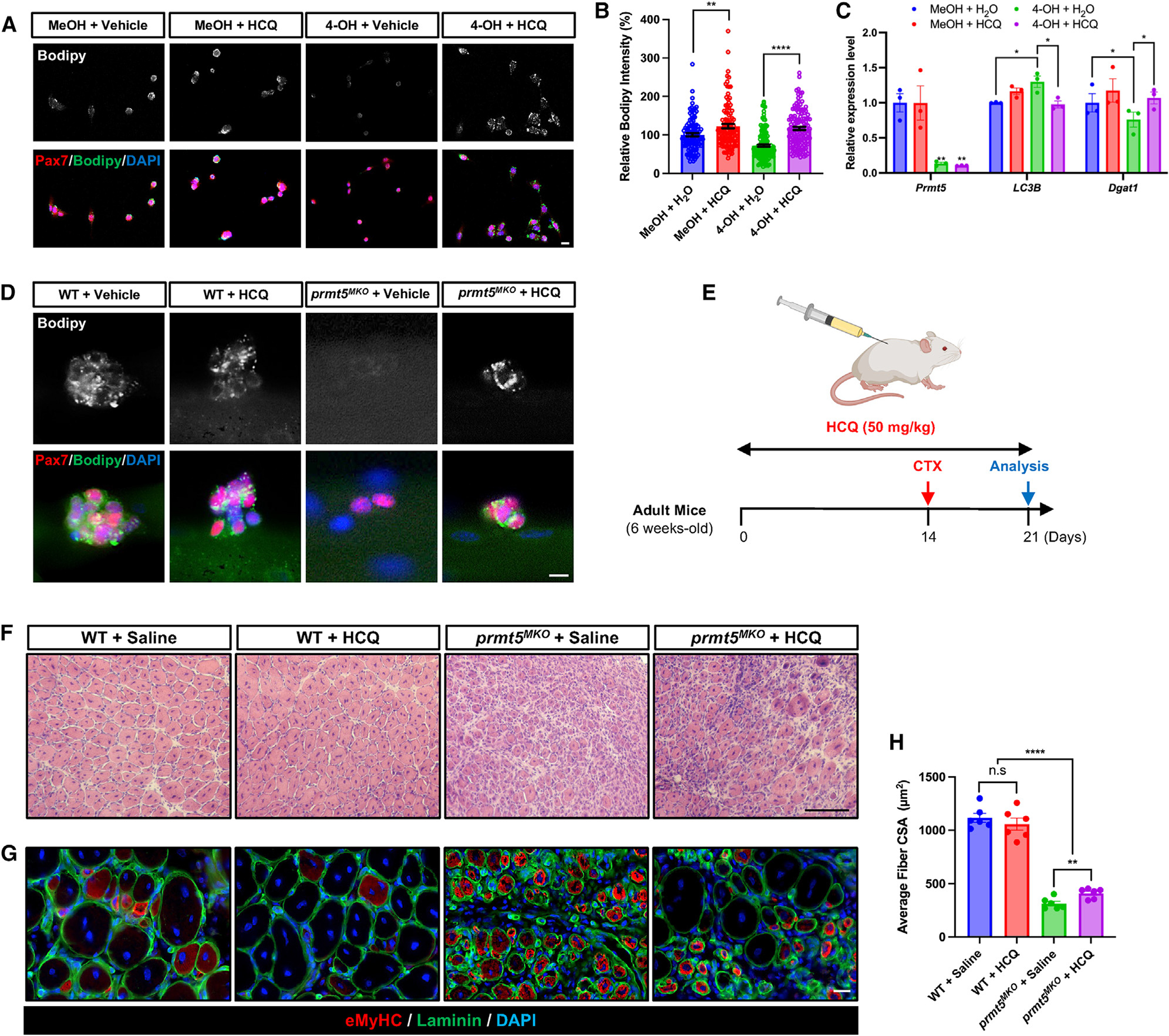
(A and B) Pax7 and Bodipy immunofluorescence (A), along with quantification of Bodipy intensity (B) in control (MeOH) and Prmt5 KO (4-OH) myoblasts treated with or without HCQ. Scale bar, 10 μm.
(C) qPCR analysis of autophagy marker (LC3B) and lipid metabolism markers (Dgat1) in control (MeOH) and Prmt5 KO (4-OH) myoblasts treated with or without HCQ (n = 3).
(D) Immunostaining of Pax7 and Bodipy in SCs after being cultured for 72 h on myofibers from WT and Prmt5MKO treated with or without HCQ (n = 3). Scale bar, 10 μm.
(E) Experimental design for vehicle or HCQ treatment in adult WT and Prmt5MKO mice to examine muscle regeneration.
(F) H&E images of WT and Prmt5MKO TA muscle cross sections at 7 DPI after vehicle or HCQ treatment. Scale bar, 100 μm.
(G and H) Immunostaining of eMyHC and laminin (G) along with quantification of myofiber size (H) in WT and Prmt5MKO TA muscle cross sections at 7 DPI after vehicle or HCQ treatment (n = 6). Scale bar, 10 μm.
Values are expressed as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 by t test.
We also examined the effect of autophagy inhibition on SCs attached on myofibers isolated from WT and Prmt5MKO mice. Immunostaining with Bodipy no significant changes in LD abundance in SCs of HCQ-treated WT myofibers, while Prmt5-deficient SCs exhibited increased LD abundance in response to HCQ (Figure 7D). These results suggest that the inhibition of autophagy flux by HCQ rescues lipid contents in Prmt5-null SCs. To assess whether HCQ rescues Prmt5-null SCs in vivo, we treated WT and Prmt5MKO mice with HCQ via daily intraperitoneal injections for 14 days. We then performed CTX-induced muscle injury, followed by 7 days of recovery, during which HCQ was continuously injected (Figure 7E). While HCQ treatment did not result in significant differences in muscle morphology of WT mice, noticeable improvement in muscle regeneration were observed in Prmt5MKO mice. Specifically, HCQ treatment led to a 30% increase in the average size of laminin+ myofibers in the Prmt5MKO mice (Figures 7F–7H). However, the overall recovery of the HCQ-treated Prmt5MKO muscles was still worse than the WT muscles (Figures 7F–7H), indicating that HCQ only partially restores muscle regeneration in the Prmt5MKO mice.
DISCUSSION
Our study delineates a physiological role of PRMT5 in myoblasts during muscle development, growth, and regeneration. We provide compelling evidence that PRMT5 mediates FoxO1 methylation and controls subcellular localization to regulate lipophagy in myoblasts. Our findings suggest that PRMT5-mediated methylation of FoxO1 in the nucleus promotes nuclear retention and transcriptional activity of FoxO1, influencing the expression of genes involved in lipogenesis. In the absence of PRMT5, FoxO1 is sequestered in the cytoplasm, which triggers autophagy along with the spontaneous differentiation of SC and results in impaired muscle regeneration. Subsequently, the pharmacological inhibition of autophagy using HCQ treatment effectively restores lipid content and partially enhances muscle regeneration in Prmt5MKO SCs.
Our study employed MyoDCre mice, genetically engineered to specifically target embryonic myoblasts, to investigate the impact of Prmt5 deletion on muscle development. A previous study has shown that loss of Prmt5 using Pax7Cre mice did not affect embryonic myogenesis but leads to premature death, presumably due to off-target deletion of Prmt5 in the central nervous system.33,40,41 Consistently, PRMT5-mediated histone modifications are crucial for neurogenesis in neural stem cells (NSCs) and conditional deletion of Prmt5 in NSCs and oligodendrocytes leads to premature death.42–44 The lack of effect on myogenesis in the Pax7Cre-driven Prmt5 KO mice may be explained by the compensatory role of Pax3-lineage myogenic cells in myogenesis. The Pax genes, including Pax3 and its paralog Pax7, are known to play crucial roles in the myogenesis process by activating the transcription of myogenic regulatory factors.45,46 Previous mouse studies have shown that Pax3 expression (E8) is initiated earlier than Pax7 expression (E9) during embryonic myogenesis, and the majority of myogenic cells in the limb are derived from Pax3+ cells.47,48 While Pax7 has been recognized as essential for adult myogenesis and regeneration, its role in fetal myogenesis is limited or may be compensated by other proteins.49–51 Utilizing MyoDCre-mediated deletion of Prmt5 provides distinct advantages to investigate myogenesis of muscle progenitors, as MyoD is intimately involved in the developmental pathway during fetal myogenesis.45,46 Moreover, PRMT5 is required for the interaction between Brg1 and MyoD, which plays a crucial role in regulating muscle differentiation.52 Our finding suggests that leveraging the MyoDCre-mediated ablation of PRMT5 is a valuable in vivo tool for elucidating the role of PRMT5 in myogenesis. This approach led to the discovery of a key role of PRMT5 in embryonic myogenesis.
Emerging studies have highlighted the critical role of LDs in the behavior and fate of SCs, and the availability of LDs through de novo lipogenesis is implicated in NSC proliferation.14,53,54 PRMTs have been reported to modulate lipid metabolism and influence cellular activity in various tissues. For instance, PRMT1 has been shown to facilitate proliferation through its interaction with C/EBPβ during adipocyte differentiation, and PRMT3-mediated methylation of LDHA (lactate dehydrogenase A) promotes glycolysis, a key process in lipid metabolism, thereby influencing tumor growth.55,56 Moreover, PRMT5 has been implicated in facilitating fatty acid biogenesis by methylating mSREP1a and enhancing LD formation through SPT methylation, which subsequently interacts with Bscl2 in adipocytes.34 Despite the established role of PRMT5 in SC proliferation,33 the molecular mechanisms underlying the defective proliferation of Prmt5-KO SCs are not well understood. In our study, we observed that PRMT5 deletion led to a reduction in LD content, which resulted in impaired SC proliferation, but promoted spontaneous SC differentiation, accompanied by compromised energy production. Rhabdomyosarcoma, a solid tumor originating in muscle tissues, is associated with high frequencies of Pax7+ SCs.57,58 Our study suggests that targeting PRMT5 may diminish the population of proliferating SCs, thereby inhibiting the growth of rhabdomyosarcoma.
Autophagy is a key player in regulating the homeostasis of skeletal muscles, and it triggers the proliferation of SCs by boosting energy supply.59 In vivo studies involving the pharmacological and genetic inactivation of Sirt1 have revealed that autophagy is induced during SC activation.12 Moreover, several studies have highlighted the role of autophagy in modulating intracellular lipid reserves through a process known as lipophagy.8,15,37 A recent study has indicated that autophagy is critical for removing intramyocellular triglycerides in myocytes.15 Our study supports the finding that the loss of PRMT5 triggers autophagy, accompanied by LD clearance. Moreover, we observed an elevation in LD contents in Prmt5-null SCs upon treatment with an autophagy inhibitor, which could potentially explain the augmented muscle repair capacity observed in Prmt5MKO mice. Dysfunctions in autophagy have been observed in various myopathies.26,59 The use of a small molecule to promote autophagy activation has shown promise in enhancing muscle regeneration and ameliorating a spectrum of myopathy and DMD (Duchenne muscular dystrophy) conditions.13,60,61 On the other hand, the impaired muscle regeneration observed in Islr-null muscle was effectively rescued by genetic deletion of Dcat1/Islr, which antagonized autophagy activation.62 Furthermore, an in vivo study has demonstrated that the inhibition of the autophagy pathway remarkably ameliorates pathology associated with congenital muscular dystrophy caused by laminin α2 chain deficiency (MDC1A).63 Muscle regeneration is an intricate process involving coordinated action of multiple cell types.35 Circulating cytokines/chemokines secreted from immune cells can facilitate the activation of SCs to replace damaged tissue, and autophagy serves as a crucial regulator to either enhance or suppress the immune response.64,65 Hence, it is essential to have a dedicated mechanism to control autophagy in SCs at an appropriate level. Our results identify PRMT5 as a negative regulator (or inhibitor) of autophagy in myoblasts.
The protein FoxO1 plays a critical role in regulating autophagy by promoting the expression of several key genes to maintain cellular homeostasis.66,67 Emerging studies have shown that FoxO1 activity and localization within cells are tightly controlled by PTMs.22,68 Phosphorylation of specific residues on FoxO1 by kinases such as AKT and serum/glucocorticoid regulated kinase 1 (SGK1) leads to its retention in the cytoplasm, inhibiting its transcriptional activity.69,70 Conversely, dephosphorylation of FoxO1 by PP2a promotes its nuclear translocation, inducing the expression of its target genes.71,72 Furthermore, under stress conditions, acetylated FoxO1 associates with ATG7 in the cytoplasm, initiating the autophagic process.20 These findings highlight the significance of PTMs in regulating function and localization of FoxO1. Besides phosphorylation and acetylation, FoxO1 also undergoes lysine methylation mediated by G9a, which controls its protein stability.73 Furthermore, PRMT1 was reported to methylate FoxO1 on arginine residues 248 and 250, which subsequently inhibits the phosphorylation of FoxO1 at serine 253.38 Our study revealed that PRMT5-mediated methylation subsequently prevents the translocation of FoxO1 to the cytoplasm. In the absence of PRMT5, FoxO1 remains sequestered in the cytoplasm, leading to changes in the cellular function of SCs. This is supported by another study reporting that SC-specific Pten-KO resulted in nuclear exclusion of FoxO1, indicating that cytoplasmic FoxO1 promotes exhaustion of quiescent SCs.16
Our study sheds light on the significant function of PRMT5 in regulating autophagy as a means of lipid catabolism (lipophagy), an area of growing interest due to its potential implications in metabolic syndrome.74,75 Specifically, our findings elucidated the importance of PRMT5-mediated methylation of FoxO1 in the nucleus, which is essential for maintaining muscle development. Nevertheless, the precise molecular mechanism underlying the downstream targets of FoxO1 in SCs remains uncertain. A previous study has reported that FoxO1 binds to the promoter of PGC1α, modulating fatty acid catabolism in the liver under stress conditions.25 Moreover, the activation of FoxO1 can bind to the promoter region of the Pnpla2 gene (encoded by ATGL protein), resulting in elevated triglyceride breakdown for energy production.76 These findings suggest that there may be multiple potential regulatory factors involved in the PRMT5-FoxO1 axis signaling pathway, which could affect lipophagy in SCs. Understanding these mechanisms is essential for exploring the role of PRMT5 in the context of SC function for muscle development and regeneration.
Limitations of the study
While our myogenic-specific conditional KO mice reveal a physiological role of PRMT5 in myogenesis, the underlying mechanisms remain to be fully elucidated. We provided evidence that Prmt5 KO induces cytoplasmic accumulation of FoxO1, which is associated with the autophagy initiation factor ATG7, elevates autophagic activities, and reduces LD content. However, pharmacological inhibition of autophagy using HCQ only partially restored muscle regeneration in the Prmt5MKO mice following muscle injury. This may represent a key limitation of the systemic delivery of HCQ, which could influence many other cell types beyond SCs. For example, HCQ has been shown to modulate the function of both innate and adaptive immune cells,77,78 which have been reported to play key roles in muscle regeneration.79 In this context, it is not surprising that HCQ treatment has been shown to promote muscle atrophy in humans.80,81 In our study, while the systemic HCQ treatment restored LD contents and perhaps the regenerative function of SCs, its off-target effect on other cell types may have largely negated the positive effects on SCs, resulting in only partial rescue of overall regeneration. Indeed, our recent study has demonstrated that myofiber-specific restoration of LDs rescues the phenotype of myofiber-specific Prmt5-KO mice very effectively.82 Future studies should explore the possibility of targeted delivery of HCQ into muscle cells using for example nanoparticle delivery tools.83,84
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information an inquiries regarding resources and reagents should be directed to the Lead Contact, Shihuan Kuang, Ph.D (skuang@purdue.edu)
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contract upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
All mouse procedures were approved by the Purdue University Animal Care and Use committee. Prmt5flox/flox mice were generously provided by Chengdeng Hu (Purdue University), and other mice strains were acquired from Jackson Laboratory using the following stock numbers: MyoDCre (stock# 014140) and Rosa26CreER (stock# 008463). The mice were housed in the animal facility with free access to standard rodent chow food and water. Unless otherwise noted, 8- to10-week-old mice of both sexes (male and female) were used in this study.
Primary myoblast isolation and culture
Primary myoblast derived from hindlimb muscles of 4-week-old WT and RosaCreER/Prmt5f/f mice (male and female) were minced and then digested using a type II collagenase and Dispase B mixture (Roche).85 Digestion was neutralized by adding growth media containing F-10 Ham’s medium (Thermo Fisher Scientific), 20% fetal bovine serum (FBS), 1% penicillin, 4 ng/mL basic fibroblast growth factor (Thermo Fisher Scientific), and the cells were cultured on collagen-coated plates. Primary myoblasts were then purified using pre-plating, which was performed 2–3 times. For in vitro genetic deletion, the myoblasts were treated with 4-hydroxyl Tamoxifen (4-OH, 0.4 μM) for 4 days to induce Cre-mediated deletion, while methanol (MeOH) was used as a vehicle control.
Single myofiber isolation and culture
Single myofibers were extracted from EDL muscles of WT and Prmt5MKO mice. briefly, EDL muscles were meticulously dissected and placed in a digestion medium containing 1.5 mg/mL collagenase I in Dulbecco’s Modified Eagle’s Medium (Sigma, DMEM) and incubated for a specific time (WT for 50 min and Prmt5MKO for 80 min) at 37°C subsequently, the EDL muscles were carefully transferred to a pre-heated plate containing 5 mL of DMEM, and single myofibers were released by flushing the muscles gently with a large pore glass pipette. The released single myofibers were then transferred, cultured in culture plate (60 mm) coated with horse serum and incubated in DMEM supplemented with 20% FBS (HyClone), 4 ng/mL basic FGF (Promega), and 1% penicillin-streptomycin (Hyclone) at 37°C. The myofibers were collected at specific time points for further analysis.
METHOD DETAILS
Muscle injury and drug treatment
Muscle injury was induced by injecting 50 μL of CTX (Sigma, 10 μM) dissolved in saline into TA muscle. Muscles were harvested at the indicated days post injection to evaluate the completion of regeneration. For preparation of Hydroxychloroquine (HCQ, TCI, H1306), it was dissolved in saline at 10 mg/mL. Prior to CTX injury, a dose of 50 mg/kg (50 mpk) was administrated by intraperitoneal (IP) injection daily for a period of two weeks. Recovery rate is determined by calculating the ratio of the injured muscle weight to the uninjured muscle weight.
Histology and immunofluorescence staining
Whole muscle tissues or TA muscle from WT and Prmt5MKO mice were frozen in an optimum cutting temperature (OCT) compound. A Leica CM 1850 cryostat was employed to cross-section the frozen muscles into 10 μm sections. These sections were then subjected to H&E staining or immunofluorescence staining. For immunofluorescent staining, briefly, samples including cross-sections, single myofibers, and primary myoblasts were fixed in 4% paraformaldehyde (PFA) for 15 min, followed by quenching with 100 mM glycine for 10 min. Samples were then incubated in blocking buffer, which contained 5% goat serum, 2% BSA, 0.2% Triton C-100 and 0.1% sodium azide, for 2 h. The samples were then incubated with primary antibodies at the indicated ratio, followed by incubation with secondary antibody.
Body composition analysis measurement
The body composition of live mice was measured using the Echo-MRI 130 analyzer (EchoMRI LLC, Houston, Tx, USA). The instrument was calibrated with corn oil prior to use. The mouse was then gently placed into a cylindrical holder, which was inserted into the EchoMRI system to measure lean mass.
Measurement of treadmill, grip strength test and muscle contractile function
Prior to the treadmill exercise test, WT and Prmt5MKO mice were trained on a treadmill (Eco3/6; Columbus Instruments, Columbus, OH) set at a constant speed of 10 m/min for 5 min per day for 3 consecutive days. On the day of the exercise test, the mice were subjected to the treadmill at 10 m/min for 5 min, after which the speed was increased by 2 m/min every 2 min until the mice reached exhaustion. Excise capacity including running time, running distance, and maximum speed were then analyzed.
To measure the gripping strength, a digital grip strength meter was employed (Columbus Instruments). The mouse grasped the grid, and its tail was gently pulled along the axis the grid to record peak tension at the time of release. The grip strength was assessed in three trials, each repeated fifteen times, and the average strength was normalized to body weight (N/g).
For the assessment of the contractile properties of slow-twitch Soleus and fast-twitch EDL muscles, we employed an ex vivo muscle test system (Aurora Scientific, 1200A intact Muscle Test System). The hindlimb of mice (anesthetized with isoflurane) was excised and placed in a bicarbonate-buffered solution. Braided silk suture thread (Fine Science Tools, 4–0) was tied around each end of the muscle tendons, the muscles were mounted between two platinum electrodes in a tissue bath apparatus containing a bicarbonate-buffered solution at room temperature. To generate the force-frequency relationship, we select frequencies between 1 and 300 Hz for the EDL muscle and 1–200 Hz for the Soleus muscle. Once the contractile properties of the muscles were measured, the muscles were removed from the organ bath, freed from the connective tissue, dried with a blotting paper, and weighed. We determined the muscle cross-sectional area (CSA) by dividing the wet muscle mass by the product of Lo and muscle-specific density (1.056 g/cm3). To calculate the specific force (N/cm2), we divided the muscle force (N) by the CSA (cm2).
Primary myoblast isolation, culture, and flow cytometry analysis
Primary myoblasts derived from hindlimb muscles of 4-week-old WT and RosaCreER/Prmt5f/f mice were minced and then digested using a type II collagenase and Dispase B mixture (Roche).85 Digestion was neutralized by adding growth media containing F-10 Ham’s medium (Thermo Fisher Scientific), 20% fetal bovine serum (FBS), 1% penicillin, 4 ng/mL basic fibroblast growth factor (Thermo Fisher Scientific), and the cells were cultured on collagen-coated plates. Primary myoblasts were then purified using pre-plating, which was performed 2–3 times. For in vitro genetic deletion, the myoblasts were treated with 4-hydroxyl Tamoxifen (4-OH, Sigma, 0.4 μM) for 4 days to induce Cre-mediated deletion, while methanol (MeOH) was used as a vehicle control.
For flow cytometry, primary myoblast isolation was conducted.86 In brief, hindlimb muscles from both WT and Prmt5MKO mice were minced and digested with type II collagenase (700 U/ml) at 37 for 60 min. The samples were then centrifuged, and additional digestion solution (100 U/ml type II collagenase and 1 U/ml Dispase II) was added, followed byincubation at 37°C for 30 min. The resulting samples were filtered through 40-μm cell strainers and washed with F-10 HAM’s medium containing 10% horse serum and 1% penicillin-streptomycin (Hyclone). The suspension cells were then centrifuged, treated with red blood cell lysis solution (Promega, Cat#Z3141), and washed with autoclaved PBS. To quantify the satellite cell population, cells from both WT and Prmt5MKO mice were stained with antibody cocktail consisting of CD31-PE (BioLegend, Cat#102408), CD45-PE (BioLegend, Cat#103106), Sca1-Pacific Blue (BioLegend, Cat#108120), and VCAM-APC (BioLegend, Cat#105718) for 30 min at 4°C in the dark. Satellite cells were identified and purified by gating with CD31−CD45−Sca1−VCAM1+ using a BD-FACS Aria III FACS system (BD Biosciences). For the quantification of immune cell population, CD45(Invitrogen, Cat#12-0451-82), CD11b (Invitrogen, Cat#25-0112-82), MHCII (Invitrogen, Cat#11-5322-82), and F4/80 (Invitrogen, Cat#11-5322-82) antibodies were used for analysis.
Immunoblotting analysis
Protein extraction from homogenized muscle tissues and primary myoblasts was carried out using RIPA buffer, which contained 25 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, and 0.1% SDS), supplemented with proteinase inhibitor and phenylmethylsulphonyl fluoride (PMSF). The quantification of supernatant proteins was performed using Peirce BCA Protein Assay Reagent (Pierce Biotechnology). The proteins were separated by electrophoresis, and then transferred onto a polyvinylidene fluoride (PVDF) membrane. After blocking the membrane with 5% fat-free milk for 1 h at room temperature, primary antibodies were incubated with the membrane overnight at 4°C. Detection of immunoreactivity was performed using an enhanced chemiluminescence western blotting substrate (Santa Cruz Biotechnology) on a FluorChem R system (Proteinsimple). For Subcellular fractions, trypsinized cells were incubated in a hypotonic buffer (0.1% NP40, 20 mM Tris-HCL (pH 7.4), 10 mM KCL, 2 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 5 mM PMSF) for 3 min and were subjected to centrifugation at 2,000 rpm. The resulting supernatant was further centrifuged at 15,000 rpm to separate and collect only supernatant (cytosolic fraction). Subsequently, the pellet was washed twice using isotonic buffer (0.1% NP40, 20 mM Tris-HCl (pH 7.4), 150 mM KCL, 2 mM MgCl2, 1 mM EGTA, 0.5 mM DTT, 0.5 mM PMSF) for 3 min each. These washes were performed to eliminate any cytosolic proteins, and the resultant pellt was designated as the nuclear fraction.87
Total RNA extraction and qRT-PCR
Cell and tissues were subjected to RNA extraction using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer’s instruction. The extracted 2 mg of total RNA was reverse transcribed using random primers, M-MLV reverse transcriptase, and DTT. Real-time qPCR was performed using SYBR green master mix and gene-specific primers, listed in Table S1. Changes in relative expression of mRNA level were analyzed by employing the 2−ΔΔCT method and normalized to β-actin.
Sea horse
Primary myoblasts cells (1×105 cells) extracted from hindlimb muscles of WT and RosaCreER/Prmt5f/f mice were seeded into XF24 microplates (SeaHorse, Bioscience). On the day of the measurements, the primary myoblasts were thoroughly washed three times using XF medium (supplemented with SeaHorse XF RPMI medium, 5 mM glucose, 2 mM pyruvate, 1 mM glutamine, pH 7.4) and were then pre-incubated for 1 h at 37°C in a non-CO2 incubator using the same medium. Subsequently, oligomycin (3 μM), FCCP (3 μM), Rotenone (1.5 μM), and Antimycin A (1.5 μM) were preloaded in a sequential manner into the cartridges, which were injected into XF wells to monitor oxygen consumption rate (OCR, pmol/min). The SeaHorse Wave software was used to measure the respiration rates of all mitochondria and normalize them based on the cellular protein contents.
Oil Red O (ORO) staining
Primary myoblasts were fixed with 4% PFA for 10 min, followed by staining with ORO working solution for 30 min at room temperature. After rinsing with PBs to remove excess stain, the ORO-stained myoblasts were imaged under bright field microscopy. Once imaging was completed, the cells were allowed to air dry for 1 h. The stained lipids were eluted from the myoblasts by adding isopropanol to each well and incubating for 20 min with gentle shaking. The eluted solution was transferred to a new microplate, and the absorbance was measured at a suitable wavelength (e.g., 510 nm) using a microplate reader.
Cell cycle assay
The myofibers from both WT and Prmt5MKO mice were isolated and cultured in a Petri dish (60 mm) coated with horse serum in DMEM supplemented with 20% FBS (HyClone), 4 ng/ml basic FGF (Promega), and 1% Penicillin-streptomycin (HyClone) at 37°C. EdU (Cayman Chemical) was added to the culture medium at a concentration of 1mM. Cultured single myofibers were sampled at the designated time points for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
A minimum of three biological replicates were utilized for performing experiments in vivo. The muscle histological analysis encompassing measurements including myofiber sectional area (CSA), the number of central nuclei, and myogenic cells per area were quantified using Fiji-imageJ software or Adobe PhotoShop software. The determination of the fusion index involved assessing the proportion of nuclei within MF20+ myotubes to the total nuclei number per image. The intensity of Bodipy in primary myoblasts was assessed using the Fiji-ImageJ software. The sample size or number of replicates (denoted as ‘n’) for each experiment is indicated in the figure legends. The data were presented as mean ± standard error of the mean (S.E.M). All quantitative analyses were conducted using Student’s t-test with a two-tail distribution, calculated using GraphPad Prism. The significance levels are denoted by p < 0.05, p < 0.01, p < 0.001, and p < 0.0001. A p value <0.05 was defined as statistically significant, whereas a p value greater than 0.05 was considered not significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rabbit Polyclonal anti-PRMT5 | Proteintech | Cat# 18436–1-AP, RRID: AB_2171798 |
| Rabbit Polyclonal anti-Laminin | Sigma | Cat #L9393, RRID: AB_477163 |
| Rabbit Polyclonal anti-Dystrophin | Abcam | Cat# ab15277, RRID: AB_301813 |
| Rabbit Polyclonal anti-Tubulin | Abcam | Cat# ab6046, RRID: AB_2210370 |
| Mouse Monoclonal anti-GAPDH | Santacruz | Cat# sc-32233, RRID: AB_627679 |
| Rabbit Polyclonal anti-LC3B | Invitrogen | Ca# PA5–32254, RRID: AB_2549727 |
| Mouse Monoclonal anti-FoxO1 | Cell signaling | Cat# 14952, RRID: AB_2722487 |
| Rabbit Polyclonal anti-FoxO1 | Cell signaling | Cat# 2880, RRID: AB_2106495 |
| Mouse Monoclonal anti-ATG7 | Proteintech | Cat# 67341–1-Ig, RRID: AB_2882599 |
| Rabbit Polyclonal anti-SYM10 | Millipore | Cat# 07–412, RRID: AB_310594 |
| Mouse Monoclonal anti-CD68 | Abcam | Cat# Ab31630, RRID: AB_1141557 |
| Mouse Monoclonal anti-Pax7 | DSHB | Cat# Pax7, RRID: AB_2299243 |
| Mouse Monoclonal anti-MyoD | Santa Cruz Biotechnology | Cat# sc-377460, RRID: AB_2813984 |
| Mouse Monoclonal anti-MyoG | DSHB | Cat# F5D, RRID: AB_2146602 |
| Mouse Monoclonal anti-MyHC | DSHB | Cat# MF20, RRID: AB_2147781 |
| Mouse Monoclonal anti-eMyHC | DSHB | Cat# F1.652, RRID: AB_528358 |
| Rabbit Polyclonal anti-Ki67 | Abcam | Cat# ab15580, RRID: AB_443209 |
| Mouse Monoclonal anti-Flag | Sigma | Cat# F1804, RRID: AB_262044 |
| PE Rat anti-mouse CD31 antibody | BD Biosciences | Cat# 553373, RRID: AB_394819 |
| PE anti-mouse CD45 antibody | eBioscience | Cat# 12–0451-82,RRID: AB_465668 |
| Pacific Blue anti-mouse Ly-6A/E (Sca-1) antibody | BioLegned | Cat# 122520, RRID: AB_2143237 |
| APC anti-mouse CD106 antibody | Biolegend | Cat# 105718, RRID: AB_1877141 |
| PE-Cy7 anti-mouse CD11b | eBioscience | Cat# 25–0112-82, RRID: AB_469588 |
| FITC anti-mouse MHC II | eBioscience | Cat# 11–5322-82, RRID: AB465235 |
| FITC anti-mouse F4/80 | eBioscience | Cat# 11–4801-81, RRID AB_2735037 |
| Alexa 568 goat anti-mouse IgG1 | Thermo Fisher Scientiic | Cat# A-21124, RRID: AB_2535766 |
| Alexa 647 goat anti-mouse IgG2b | Thermo Fisher Scientiic | Cat# A-21242, RRID: AB_2535811 |
| Alexa 488 goat-anti mouse IGM | Thermo Fisher Scientiic | Cat# A21042, RRID: AB_141357 |
| Alexa 488 goat-anti rabbit IgG | Thermo Fisher Scientiic | Cat# A-11034, RRID: AB_2576217 |
| HRP AffiniPure goat anti-mouse IgG | Jackson ImmunoResearch | Cat# 115–035-003, RRID: AB_10015289 |
| HRP AffiniPure goat anti-rebbit IgG | Jackson ImmunoResearch | Cat# 111–035-003, RRID: AB_2313567 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| 4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | H6278 |
| Hydroxychloroquine | Tcichemicals | H1306 |
| Cardiotoxin | Sigma-Aldrich | 217503 |
| Collagenase, Type I | Worthington | LS004197 |
| Collagenase, Type II | Worthington | LS004179 |
| Dispase II | Sigma-Aldrich | 04942078001 |
| Red blood cell lysis solution | Promega | Z3141 |
| Dulbecco’s Modified Eagle Medium | Gibco | 11995065 |
| Ham’s F10 Nutrient Mix | Gibco | 11550043 |
| Fetal bovine serum | HyClone | SH30080.03 |
| Donor Hose Serum | Corning | MT35030CV |
| Opti-MEM | Gibco | 31985070 |
| Penicillin-Streptomycin | Sigma-Aldrich | P4333 |
| Seahorse XF base medium | Agilent Technologies | 103334–100 |
| Phosphate-buffered saline | Gibco | 21600–069 |
| Bodipy 493/503 | Invitrogen | D3922 |
| M.O.M (Mouse on Mouse) blocking buffer | Vector lab | MKB-2213 |
| DAPI | Invitrogen | D1306 |
| O.C.T. Compound | Fisher Scientific | 23–730-571 |
| bFGF (Fibroblast growth factor, basic) | Promega | 9PIG507 |
| BD Matrigel Matrix | BD Science | 356235 |
| Collagen from rat tail | Sigma-Aldrich | C7661 |
| 5-Ethynyl-2'-deoxyuridine (EdU) | Cayman Chemical | 20518 |
| Lipofectamin 2000 | Invitrogen | 11668030 |
| Xfect transection Reagent | Takara | 631318 |
| Goat serum | MP Biomedicals | 08642921 |
| TRIzol Reagent | Sigma-Aldrich | T9424 |
| Chloroform | VWR Chemicals | BDH1109 |
| Methanol | Fisher Scientific | A412–20 |
| NP-40 | Thermo Scientific | 85124 |
| SDS (Sodium Dodecyl Sulfrate) | Fisher Scientific | 02–004-080 |
| Ammonium acetate | Fisher Scientific | A637–500 |
| PMSF | Calbiochem | 7110-OP |
| Sodium pyruvate | Sigma-Aldrich | P5280 |
| Paraformaldehyde | Sigma-Aldrich | P6148 |
| Oil Red O (ORO) | Sigma-Aldrich | O0625 |
| Sodium pyruvate | Sigma-Aldrich | P5280 |
| Cycloheximide | Thermo Fisher Scientific | J66901–03 |
|
| ||
| Critical commercial assays | ||
|
| ||
| M-MLV reverse transcriptase | Invitrogen | 28025021 |
| Pierce BCA Protein Assay Reagent | Thermo Scientific | 232225 |
| Western Blotting Chemiluminescence Luminol Reagent | Santa Cruz Biotechnology | sc-2048 |
| Seahorse XF Cell Mito Stress Test | Agilent Technologies | 103015–100 |
| Duolink in situ red Detection Reagent Red | Sigma | DUO92008 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse: C2C12 | ATCC | Cat# CRL-1772 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: FVB.Cg-Myod1tm2.1(icre)Glh/J | The Jackson Laboratory | JAX Stock: #14140 |
| Mouse: B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J | The Jackson Laboratory | JAX Stock: #008463 |
| Mouse: Prmt5flox/flox | Kim et al.36, Jia et al.34 | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 | This paper | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pcDNA3 Flag FKHR | Addgene | 13507 |
| 1354 pcDNA3 flag FKHR ΔDB | Addgene | 10694 |
| 1357 pcDNA3 flag FKHR AAA ΔDB | Addgene | 10696 |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo 10 | FLOWJO, LLC | RRID: SCR_008520 |
| Fiji-ImageJ | RRID:SCR_002285 | |
| Seahorse Wave | Agilent Technologies | RRID: SCR_014526 |
| PRISM 6.0 | GraphPad Prism | RRID:SCR_002798 |
| Adobe Photoshop | Adobe Inc. | RRID: SCR_014199 |
Highlights.
PRMT5 is essential for muscle development and function in vivo
PRMT5 regulates fate of muscle stem cells and muscle regenerative capacity
PRMT5 governs autophagy through FoxO1 methylation, influencing its subcellular localization
Autophagy inhibition partially improves muscle regeneration in Prmt5 KO mice
ACKNOWLEDGMENTS
This work was supported by grants from the US National Institutes of Health (R01AR078695, R01DK132819, R01AR079235) to S.K. and (F31AR077424) to S.O. We thank Dr. Changdeng Hu (Purdue University, USA) for kindly providing the Prmt5flox/flox mice. We extend our gratitude to Jun Wu and Mary Larimore for their valuable contributions in maintaining the mouse colony and the members of the Kuang laboratory for their valuable insights and critical comments.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113329.
REFERENCES
- 1.Blau HM, Cosgrove BD, and Ho ATV( (2015). The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 21, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yin H, Price F, and Rudnicki MA (2013). Satellite cells and the muscle stem cell niche. Physiol. Rev. 93, 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez-Outeiriño L, Hernandez-Torres F, Ramírez-de Acuña F, Matías-Valiente L, Sanchez-Fernandez C, Franco D, and Aranega AE (2021). Muscle satellite cell heterogeneity: does embryonic origin matter? Front. Cell Dev. Biol. 9, 750534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Relaix F, Bencze M, Borok MJ, Der Vartanian A, Gattazzo F, Mademtzoglou D, Perez-Diaz S, Prola A, Reyes-Fernandez PC, Rotini A, and Taglietti 5th.. (2021). Perspectives on skeletal muscle stem cells. Nat. Commun. 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pallafacchina G, Blaauw B, and Schiaffino S (2013). Role of satellite cells in muscle growth and maintenance of muscle mass. Nutr. Metabol. Cardiovasc. Dis. 23, S12–S18. [DOI] [PubMed] [Google Scholar]
- 6.Xia Q, Huang X, Huang J, Zheng Y, March ME, Li J, and Wei Y (2021). The role of autophagy in skeletal muscle diseases. Front. Physiol. 12, 638983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang W, Huang J, Wu H, Wang Y, Du Z, Ling Y, Wang W, Wu Q, and Gao W (2020). Molecular mechanisms of cancer cachexia-induced muscle atrophy. Mol. Med. Rep. 22, 4967–4980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang S, Peng X, Yang S, Li X, Huang M, Wei S, Liu J, He G, Zheng H, Yang L, et al. (2022). The regulation, function, and role of lipophagy, a form of selective autophagy, in metabolic disorders. Cell Death Dis. 13, 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegué E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boya P, Codogno P, and Rodriguez-Muela N (2018). Autophagy in stem cells: repair, remodelling and metabolic reprogramming. Development 145, dev146506. [DOI] [PubMed] [Google Scholar]
- 11.Chang NC (2020). Autophagy and stem cells: self-eating for self-renewal. Front. Cell Dev. Biol. 8, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tang AH, and Rando TA (2014). Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 33, 2782–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiacco E, Castagnetti F, Bianconi V, Madaro L, De Bardi M, Nazio F, D’Amico A, Bertini E, Cecconi F, Puri PL, and Latella L (2016). Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ. 23, 1839–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yue F, Oprescu SN, Qiu J, Gu L, Zhang L, Chen J, Narayanan N, Deng M, and Kuang S (2022). Lipid droplet dynamics regulate adult muscle stem cell fate. Cell Rep. 38, 110267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lam T, Harmancey R, Vasquez H, Gilbert B, Patel N, Hariharan V, Lee A, Covey M, and Taegtmeyer H (2016). Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Dis. 2, 16061–16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yue F, Bi P, Wang C, Shan T, Nie Y, Ratliff TL, Gavin TP, and Kuang S (2017). Pten is necessary for the quiescence and maintenance of adult muscle stem cells. Nat. Commun. 8, 14328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aman Y, Schmauck-Medina T, Hansen M, Morimoto RI, Simon AK, Bjedov I, Palikaras K, Simonsen A, Johansen T, Tavernarakis N, et al. (2021). Autophagy in healthy aging and disease. Nat. Aging 1, 634–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Farhan M, Silva M, Li S, Yan F, Fang J, Peng T, Hu J, Tsao MS, Little P, and Zheng W (2020). The role of FOXOs and autophagy in cancer and metastasis—Implications in therapeutic development. Med. Res. Rev. 40, 2089–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yue J, Aobulikasimu A, Sun W, Liu S, Xie W, and Sun W (2022). Targeted regulation of FoxO1 in chondrocytes prevents age-related osteoarthritis via autophagy mechanism. J. Cell Mol. Med. 26, 3075–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhao Y, Yang J, Liao W, Liu X, Zhang H, Wang S, Wang D, Feng J, Yu L, and Zhu W-G (2010). Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol. 12, 665–675. [DOI] [PubMed] [Google Scholar]
- 21.Wang B, Ding W, Zhang M, Li H, Guo H, Lin L, Chen J, and Gu Y (2016). Role of FOXO1 in aldosterone-induced autophagy: A compensatory protective mechanism related to podocyte injury. Oncotarget 7, 45331–45351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang S, Xia P, Huang G, Zhu P, Liu J, Ye B, Du Y, and Fan Z (2016). FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 7, 11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou J, Liao W, Yang J, Ma K, Li X, Wang Y, Wang D, Wang L, Zhang Y, Yin Y, et al. (2012). FOXO3 induces FOXO1-dependent autophagy by activating the AKT1 signaling pathway. Autophagy 8, 1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kamei Y, Miura S, Suzuki M, Kai Y, Mizukami J, Taniguchi T, Mochida K, Hata T, Matsuda J, Aburatani H, et al. (2004). Skeletal Muscle FOXO1 (FKHR) Transgenic Mice Have Less Skeletal Muscle Mass, Down-regulated Type I (Slow Twitch/Red Muscle) Fiber Genes, and Impaired Glycemic Control*[boxs]. J. Biol. Chem. 279, 41114–41123. [DOI] [PubMed] [Google Scholar]
- 25.Xu M, Chen X, Chen D, Yu B, and Huang Z (2017). FoxO1: a novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 8, 10662–10674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leduc-Gaudet J-P, Franco-Romero A, Cefis M, Moamer A, Broering FE, Milan G, Sartori R, Chaffer TJ, Dulac M, Marcangeli V, et al. (2023). MYTHO is a novel regulator of skeletal muscle autophagy and integrity. Nat. Commun. 14, 1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.So H-K, Kim S, Kang J-S, and Lee S-J (2021). Role of protein arginine methyltransferases and inflammation in muscle pathophysiology. Front. Physiol. 12, 712389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stouth DW, VanLieshout TL, Shen NY, and Ljubicic V (2017). Regulation of skeletal muscle plasticity by protein arginine methyltransferases and their potential roles in neuromuscular disorders. Front. Physiol. 8, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanc RS, Vogel G, Li X, Yu Z, Li S, and Richard S (2017). Arginine methylation by PRMT1 regulates muscle stem cell fate. Mol. Cell Biol. 37, e00457–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawabe YI, Wang YX, McKinnell IW, Bedford MT, and Rudnicki MA (2012). Carm1 regulates Pax7 transcriptional activity through MLL1/2 recruitment during asymmetric satellite stem cell divisions. Cell Stem Cell 11, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blanc RS, Vogel G, Chen T, Crist C, and Richard S (2016). PRMT7 preserves satellite cell regenerative capacity. Cell Rep. 14, 1528–1539. [DOI] [PubMed] [Google Scholar]
- 32.Jeong H-J, Lee S-J, Lee H-J, Kim H-B, Anh Vuong T, Cho H, Bae G-U, and Kang J-S (2020). Prmt7 promotes myoblast differentiation via methylation of p38MAPK on arginine residue 70. Cell Death Differ. 27, 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang T, Gu€nther S, Looso M, Künne C, Krüger M, Kim J, Zhou Y, and Braun T (2015). Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun. 6, 7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jia Z, Yue F, Chen X, Narayanan N, Qiu J, Syed SA, Imbalzano AN, Deng M, Yu P, Hu C, and Kuang S (2020). Protein arginine methyltransferase PRMT5 regulates fatty acid metabolism and lipid droplet biogenesis in white adipose tissues. Adv. Sci. 7, 2002602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chargé SBP, and Rudnicki MA (2004). Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 84, 209–238. [DOI] [PubMed] [Google Scholar]
- 36.Kim KH, Jia Z, Snyder M, Chen J, Qiu J, Oprescu SN, Chen X, Syed SA, Yue F, Roseguini BT, et al. (2023). PRMT5 links lipid metabolism to contractile function of skeletal muscles. EMBO Rep. 24, e57306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, and Czaja MJ (2009). Autophagy regulates lipid metabolism. Nature 458, 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, Mukai H, Kasuya Y, and Fukamizu A (2008). Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol. Cell 32, 221–231. [DOI] [PubMed] [Google Scholar]
- 39.Bik E, Mateuszuk L, Orleanska J, Baranska M, Chlopicki S, and Majzner K (2021). Chloroquine-induced accumulation of autophagosomes and lipids in the endothelium. Int. J. Mol. Sci. 22, 2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim K, Orvis J, and Stolfi A (2022). Pax3/7 regulates neural tube closure and patterning in a non-vertebrate chordate. Front. Cell Dev. Biol. 10, 999511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jostes B, Walther C, and Gruss P (1990). The murine paired box gene, Pax7, is expressed specifically during the development of the nervous and muscular system. Mech. Dev. 33, 27–37. [DOI] [PubMed] [Google Scholar]
- 42.Chittka A, Nitarska J, Grazini U, and Richardson WD (2012). Transcription factor positive regulatory domain 4 (PRDM4) recruits protein arginine methyltransferase 5 (PRMT5) to mediate histone arginine methylation and control neural stem cell proliferation and differentiation. J. Biol. Chem. 287, 42995–43006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Calabretta S, Vogel G, Yu Z, Choquet K, Darbelli L, Nicholson TB, Kleinman CL, and Richard S (2018). Loss of PRMT5 promotes PDGFRα degradation during oligodendrocyte differentiation and myelination. Dev. Cell 46, 426–440.e5. [DOI] [PubMed] [Google Scholar]
- 44.Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, and Guccione E (2013). Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev. 27, 1903–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Relaix F, Rocancourt D, Mansouri A, and Buckingham M (2004). Divergent functions of murine Pax3 and Pax7 in limb muscle development. Genes Dev. 18, 1088–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Messina G, and Cossu G (2009). The origin of embryonic and fetal myoblasts: a role of Pax3 and Pax7. Genes Dev. 23, 902–905. [DOI] [PubMed] [Google Scholar]
- 47.Buckingham M, and Relaix F (2007). The role of Pax genes in the development of tissues and organs: Pax3 and Pax7 regulate muscle progenitor cell functions. Annu. Rev. Cell Dev. Biol. 23, 645–673. [DOI] [PubMed] [Google Scholar]
- 48.Hutcheson DA, Zhao J, Merrell A, Haldar M, and Kardon G (2009). Embryonic and fetal limb myogenic cells are derived from developmentally distinct progenitors and have different requirements for β-catenin. Genes Dev. 23, 997–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuang S, Chargé SB, Seale P, Huh M, and Rudnicki MA (2006). Distinct roles for Pax7 and Pax3 in adult regenerative myogenesis. J. Cell Biol. 172, 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Seale P, Sabourin LA, Girgis-Gabardo A, Mansouri A, Gruss P, and Rudnicki MA (2000). Pax7 is required for the specification of myogenic satellite cells. Cell 102, 777–786. [DOI] [PubMed] [Google Scholar]
- 51.Oustanina S, Hause G, and Braun T (2004). Pax7 directs postnatal renewal and propagation of myogenic satellite cells but not their specification. EMBO J. 23, 3430–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dacwag CS, Ohkawa Y, Pal S, Sif S, and Imbalzano AN (2007). The protein arginine methyltransferase Prmt5 is required for myogenesis because it facilitates ATP-dependent chromatin remodeling. Mol. Cell Biol. 27, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knobloch M, Braun SMG, Zurkirchen L, Von Schoultz C, Zamboni N, Araúzo-Bravo MJ, Kovacs WJ, Karalay Ö, Suter U, Machado RAC, et al. (2013). Metabolic control of adult neural stem cell activity by Fasn-dependent lipogenesis. Nature 493, 226–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ramosaj M, Madsen S, Maillard V, Scandella V, Sudria-Lopez D, Yuizumi N, Telley L, and Knobloch M (2021). Lipid droplet availability affects neural stem/progenitor cell metabolism and proliferation. Nat. Commun. 12, 7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu Q, Wang D, Liang F, Tong X, Liang Z, Wang X, Chen Y, and Mo D (2022). Protein arginine methyltransferase PRMT1 promotes adipogenesis by modulating transcription factors C/EBPb and PPARg. J. Biol. Chem. 298, 102309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lei Y, Han P, Chen Y, Wang H, Wang S, Wang M, Liu J, Yan W, Tian D, and Liu M (2022). Protein arginine methyltransferase 3 promotes glycolysis and hepatocellular carcinoma growth by enhancing arginine methylation of lactate dehydrogenase A. Clin. Transl. Med. 12, e686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soleimani VD, and Rudnicki MA (2011). New insights into the origin and the genetic basis of rhabdomyosarcomas. Cancer Cell 19, 157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boscolo Sesillo F, Fox D, and Sacco A (2019). Muscle stem cells give rise to rhabdomyosarcomas in a severe mouse model of Duchenne muscular dystrophy. Cell Rep. 26, 689–701.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee DE, Bareja A, Bartlett DB, and White JP (2019). Autophagy as a therapeutic target to enhance aged muscle regeneration. Cells 8, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Civiletto G, Dogan SA, Cerutti R, Fagiolari G, Moggio M, Lamperti C, Benincá C, Viscomi C, and Zeviani M (2018). Rapamycin rescues mitochondrial myopathy via coordinated activation of autophagy and lysosomal biogenesis. EMBO Mol. Med. 10, e8799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Masiero E, and Sandri M (2010). Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy 6, 307–309. [DOI] [PubMed] [Google Scholar]
- 62.Zhang K, Zhang Y, Gu L, Lan M, Liu C, Wang M, Su Y, Ge M, Wang T, Yu Y, et al. (2018). Islr regulates canonical Wnt signaling-mediated skeletal muscle regeneration by stabilizing Dishevelled-2 and preventing autophagy. Nat. Commun. 9, 5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carmignac V, Svensson M, Körner Z, Elowsson L, Matsumura C, Gawlik KI, Allamand V, and Durbeej M (2011). Autophagy is increased in laminin α2 chain-deficient muscle and its inhibition improves muscle morphology in a mouse model of MDC1A. Hum. Mol. Genet. 20, 4891–4902. [DOI] [PubMed] [Google Scholar]
- 64.Fu X, Wang H, and Hu P (2015). Stem cell activation in skeletal muscle regeneration. Cell. Mol. Life Sci. 72, 1663–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van de Vyver M, and Myburgh KH (2012). Cytokine and satellite cell responses to muscle damage: interpretation and possible confounding factors in human studies. J. Muscle Res. Cell Motil. 33, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lettieri Barbato D, Tatulli G, Aquilano K, and Ciriolo MR (2013). FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 4, e861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Xiong X, Tao R, DePinho RA, and Dong XC (2012). The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J. Biol. Chem. 287, 39107–39114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Horst A, and Burgering BMT (2007). Stressing the role of FoxO proteins in lifespan and disease. Nat. Rev. Mol. Cell Biol. 8, 440–450. [DOI] [PubMed] [Google Scholar]
- 69.Pan CW, Jin X, Zhao Y, Pan Y, Yang J, Karnes RJ, Zhang J, Wang L, and Huang H (2017). AKT-phosphorylated FOXO 1 suppresses ERK activation and chemoresistance by disrupting IQGAP 1-MAPK interaction. EMBO J. 36, 995–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Du Y-N, Tang X-F, Xu L, Chen W-D, Gao P-J, and Han W-Q (2018). SGK1-FoxO1 signaling pathway mediates Th17/Treg imbalance and target organ inflammation in angiotensin II-induced hypertension. Front. Physiol. 9, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schachter TN, Shen T, Liu Y, and Schneider MF (2012). Kinetics of nuclear-cytoplasmic translocation of Foxo1 and Foxo3A in adult skeletal muscle fibers. Am. J. Physiol. Cell Physiol. 303, C977–C990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yan L, Lavin VA, Moser LR, Cui Q, Kanies C, and Yang E (2008). PP2A regulates the pro-apoptotic activity of FOXO1. J. Biol. Chem. 283, 7411–7420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chae Y-C, Kim J-Y, Park JW, Kim K-B, Oh H, Lee K-H, and Seo S-B (2019). FOXO1 degradation via G9a-mediated methylation promotes cell proliferation in colon cancer. Nucleic Acids Res. 47, 1692–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sebastián D, and Zorzano A (2020). Self-eating for muscle fitness: autophagy in the control of energy metabolism. Dev. Cell 54, 268–281. [DOI] [PubMed] [Google Scholar]
- 75.Zhang X, Evans TD, Jeong S-J, and Razani B (2018). Classical and alternative roles for autophagy in lipid metabolism. Curr. Opin. Lipidol. 29, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, Luo Z, Farmer SR, and Kandror KV( (2011). SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 52, 1693–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gies V, Bekaddour N, Dieudonné Y, Guffroy A, Frenger Q, Gros F, Rodero MP, Herbeuval JP, and Korganow AS (2020). Beyond Antiviral Effects of Chloroquine/Hydroxychloroquine. Front Immunol 11, 1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Devarajan A, and Vaseghi M (2021). Hydroxychloroquine can potentially interfere with immune function in COVID-19 patients: Mechanisms and insights. Redox Biol 38, 101810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ziemkiewicz N, Hilliard G, Pullen NA, and Garg K (2021). The Role of Innate and Adaptive Immune Cells in Skeletal Muscle Regeneration. Int J Mol Sci 22, 3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Naddaf E, Paul P, and AbouEzzeddine OF (2021). Chloroquine and Hydroxychloroquine Myopathy: Clinical Spectrum and Treatment Outcomes. Front Neurol 11, 616075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Biguetti CC, Santiago JF Jr., Fiedler MW, Marrelli MT, and Brotto M (2021). The toxic effects of chloroquine and hydroxychloroquine on skeletal muscle: a systematic review and meta-analysis. Sci Rep 11, 6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim KH, Jia Z, Snyder M, Chen J, Qiu J, Oprescu SN, Chen X, Syed SA, Yue F, Roseguini BT, Imbalzano AN, Hu C, and Kuang S (2023). PRMT5 links lipid metabolism to contractile function of skeletal muscles. EMBO Rep 24, e57306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Li Y, Cho MH, Lee SS, Lee DE, Cheong H, and Choi Y (2020). Hydroxychloroquine-loaded hollow mesoporous silica nanoparticles for enhanced autophagy inhibition and radiation therapy. J Control Release 325, 100–110. [DOI] [PubMed] [Google Scholar]
- 84.Huang D, Yue F, Qiu J, Deng M, and Kuang S (2020). Polymeric nanoparticles functionalized with muscle-homing peptides for targeted delivery of phosphatase and tensin homolog inhibitor to skeletal muscle. Acta Biomater 118, 196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim KH, Qiu J, and Kuang S (2020). Isolation, Culture, and Differentiation of Primary Myoblasts Derived from Muscle Satellite Cells. Bio. Protoc. 10, e3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu L, Cheung TH, Charville GW, and Rando TA (2015). Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat. Protoc. 10, 1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Senichkin VV, Prokhorova EA, Zhivotovsky B, and Kopeina GS (2021). Simple and efficient protocol for subcellular fractionation of normal and apoptotic cells. Cells 10, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contract upon request.