

Abstract
In vivo apoptosis of human mesenchymal stromal cells (MSCs) plays a critical role in delivering immunomodulation. Yet, caspase activity not only mediates the dying process but also death-independent functions that may shape the immunogenicity of apoptotic cells. Therefore, a better characterization of the immunological profile of apoptotic MSCs (ApoMSCs) could shed light on their mechanistic action and therapeutic applications. We analyzed the transcriptomes of MSCs undergoing apoptosis and identified several immunomodulatory factors and chemokines dependent on caspase activation following Fas stimulation. The ApoMSC secretome inhibited human T cell proliferation and activation, and chemoattracted monocytes in vitro. Both immunomodulatory activities were dependent on the cyclooxygenase2 (COX2)/prostaglandin E2 (PGE2) axis. To assess the clinical relevance of ApoMSC signature, we used the peripheral blood mononuclear cells (PBMCs) from a cohort of fistulizing Crohn’s disease (CD) patients who had undergone MSC treatment (ADMIRE-CD). Compared with healthy donors, MSCs exposed to patients’ PBMCs underwent apoptosis and released PGE2 in a caspase-dependent manner. Both PGE2 and apoptosis were significantly associated with clinical responses to MSCs. Our findings identify a new mechanism whereby caspase activation delivers ApoMSC immunosuppression. Remarkably, such molecular signatures could implicate translational tools for predicting patients’ clinical responses to MSC therapy in CD.
Keywords: mesenchymal stromal cells, apoptosis, fistulizing Crohn’s disease, prostaglandin E2, biomarker, NFκB
Graphical abstract
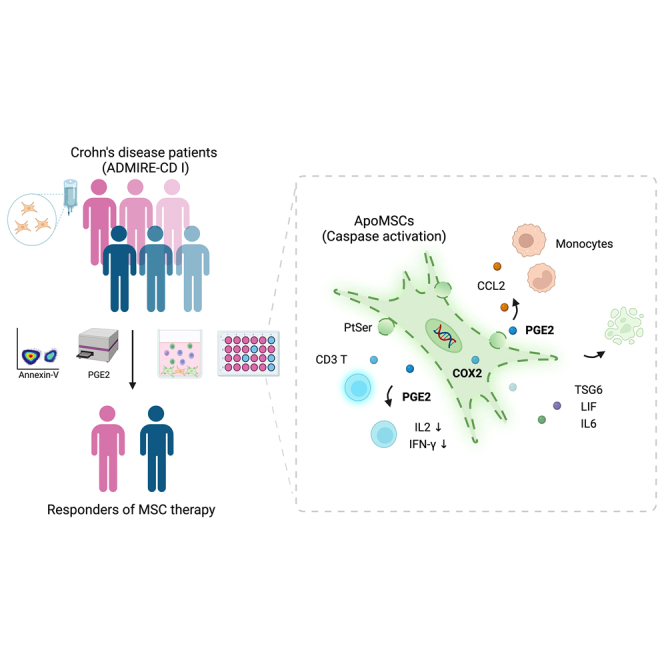
Dazzi and colleagues identify a new mechanism whereby caspase activation in MSCs induces a NFκB-dependent secretome that actively effects immunomodulation via COX2/PGE2, corroborating apoptosis is critical for MSC immunosuppression. The levels of PGE2 and apoptosis induced in MSC by patients’ immune cells predicts clinical responses to MSC in Crohn’s disease.
Introduction
In vivo apoptosis of mesenchymal stromal cells (MSCs) has recently been recognized as a critical mechanism to deliver immunosuppression.1,2,3 In graft-versus-host disease (GvHD), MSC apoptosis is induced by recipient activated cytotoxic cells through a contact-dependent bystander effect that is not antigen-specific yet mediated through Fas stimulation and granzyme B.1 Apoptotic MSCs (ApoMSCs) are efferocytosed by recipient mononuclear phagocyte system (MPS) cells, which are then reprogrammed to immunosuppressive cells by upregulating molecules such as indoleamine 2,3-dioxygenase (IDO) and prostaglandin E2 (PGE2) to alleviate pathogenic inflammation in vivo.1,4,5 A similar observation has been reported in a subsequent study using an asthma model, where researchers found that MSC apoptosis and efferocytosis polarized lung alveolar macrophages to effect immunosuppression.2 Remarkably, in both models, prevention of MSC apoptosis in vivo compromised the therapeutic effects of MSCs after infusion.1,2 Despite being a relatively underappreciated mechanism of MSC immunomodulation, the notion that apoptotic cell clearance initiates immunosuppression via efferocytosis has been previously documented.6,7
However, caspase activity not only mediates the dying process but also upregulates a variety of death-independent functions that may shape the immunogenic properties of apoptotic cells.8,9 Emerging evidence has documented that, after caspase activation, cells undergoing apoptosis release a variety of metabolites10 and cytokines11,12 that can have an impact on the surrounding environment. Therefore, considering the critical role of MSC apoptosis in the generation of clinical immunosuppression, it is important to better understand whether additional mechanisms involving caspase activation can directly influence immune effector cells regardless of efferocytosis in the MSC recipient.
Several commercial MSC products like Ryoncil/remestemcel-L (Mesoblast) and Alofisel/darvadstrocel (Takeda) have already gained the approval for treating patients for acute GvHD13 and perianal fistulizing Crohn’s disease (CD)14 respectively. Despite the encouraging response rates, only a proportion of patients respond to therapy.15 Therefore, a reliable method for patient screening and stratification is urgently needed to ultimately improve the overall response rate and cost-effectiveness of MSC treatment.16
In order to address these questions, we have profiled the transcriptome of MSCs undergoing apoptosis and characterized the resulting immunological activity of the ApoMSC secretome. Building on our in vitro findings, we addressed the significance of MSC apoptosis and death-associated secretome using samples from the phase III clinical study ADMIRE-CD, which showed that allogeneic MSCs (Alofisel/darvadstrocel) were effective and safe in treating perianal fistulizing CD patients.15
Results
Activation of apoptotic caspases in MSCs induces an NF-κB-dependent secretome that is independent of the dying process
Activated CD8+ and CD56+ cytotoxic cells induced apoptosis in bone marrow-derived MSCs via granzyme B and Fas receptor.1 Modeling the pro-apoptotic stimuli generated by cytotoxic cells, we exposed bone marrow-derived MSCs to anti-Fas activating antibody and recombinant granzyme B, and assessed their gene transcription profile. The gene set enrichment analysis (GSEA)17,18 revealed that there was a list of gene sets upregulated in the ApoMSCs compared with the control. Among them, the gene set of tumor necrosis factor (TNF)-α signaling via NF-κB was the most significant followed by the gene set of apoptosis (Figure 1A). Within its core enriched genes, we could identify several genes encoding for immunomodulatory molecules (IL-6, LIF, TNFAIP6, and cyclooxygenase2/COX2 encoding gene PTGS2) and chemokines (CCL20, CXCL3, CXCL1, and CCL2) (Figure 1B). According to the role of Fas stimulation in eliciting NF-κB signaling,19 we observed that anti-Fas treatment alone without granzyme B was sufficient to induce the most differentially upregulated genes such as LIF, TNFAIP6, PTGS2, and IL-6 (Figure 1C). Therefore, we decided to continue characterizing the apoptotic signaling by using only Fas stimulation. The gene expression profile was later validated at the protein level with TSG-6, COX2/PGE2, LIF, and interleukin (IL)-6 found proportionally increased in the ApoMSCs and their conditioned medium (Figures 1D and 1E). The chemokine protein array further confirmed the presence of high levels of CCL20 and CCL2, and also detected high levels of IL-8 (Figure S1A).
Figure 1.

Fas stimulation induces the non-death signaling in bone marrow MSCs during apoptosis
(A) Bone marrow MSCs from different batches (5 × 105 per 96 well) were treated with 5 μg/mL recombinant granzyme B and 10 μg/mL anti-Fas clone CH11 in complete RPMI for 24 h at 37°C, 5% CO2. A total of 10 samples from five independent groups were prepared for the human microarray analysis. GSEA used the hallmark gene set to analyze differently expressed genes in ApoMSCs compared with MSCs. Other parameters are set as default. Gene sets with FDR less than 0.25 are considered as significant. (B) Clustered heatmap was generated using the heatmap.2 function from R.Studio. Within the gene set of TNF-α signaling via NF-κB, 76 genes were identified as core genes that contributed most to the enrichment result. Within those 76 genes, there is a list of immunomodulatory factors and chemokines. (C) RNA was isolated from untreated MSCs and Fas-stimulated MSCs, which were treated with 10 μg/mL anti-Fas (clone CH-11) for 24 h at 37°C, 5% CO2. Expression levels of LIF, TNFAIP6, PTGS2, and IL-6 were determined by qRT-PCR. The bar chart showed the relative fold changes of markers in comparison with untreated MSC after normalization with HPRT-1. (D) Increased expression of TSG-6 (35 kDa) and COX2 (70 kDa) in Fas-stimulated MSCs was confirmed also at the protein level by immunoblotting. α-Tubulin (55 kDa) was used as loading control. ImageJ software was used to quantify the intensity of bands. The expression level of TSG-6 and COX2 in each experiment was normalized to the α-Tubulin. (E) ELISA analysis also confirmed the increased level of LIF, IL-6, and PGE2 in the supernatant of Fas-stimulated MSCs compared with the untreated. (D and E) Experimental data were expressed as mean ± SD. Unpaired t test was used to compare the mean difference between the two groups. (F) Dot plots illustrating the distribution of COX2 expression among the Annexin-V+ or Annexin-V− population. (G) The intracellular COX2 was stained using human anti-COX2 (clone AS67) and the COX2+ population was gated according to the isotype control. The level of PGE2, LIF, and IL-6 in the cell culture supernatant were assessed using human PGE2, LIF, and IL-6 ELISA kits, respectively. (F and G) All results shown are representative of least three independent experiments. One-way ANOVA and post hoc Tukey test were used to compare the mean differences among the samples. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗,### p values <0.001; ∗∗∗∗, ####, ˆˆˆˆ p values <0.0001; n.s, no significance.
To ensure that the molecules identified were specifically originating from MSCs undergoing apoptosis, we co-stained the cells with COX2, as representative of the enriched markers, together with Annexin-V, and confirmed that COX2 was expressed by about half of the apoptotic (Annexin-V+) MSCs (Figure 1F). Next, we investigated the impact of caspase activation on the secretome. Overall, most of the molecules induced by Fas stimulation were dependent on caspase activation. Caspase inhibition prevented the upregulation of the immunoregulatory factors COX2 and PGE2, LIF, and chemokines like CCL2, IL-8, and CCL20 (Figures 1G and S1A). IL-6 was an exception as its increment could not be reversed (Figure 1G). As expected, caspase inhibition completely prevented the apoptotic cell death as almost all MSCs remained Annexin-V− and 7AAD− under Fas stimulation (Figure S1B). These data suggested that no other forms of cell death were initiated.
We next investigated whether other apoptotic stimuli were able to induce an apoptotic secretome similar to Fas stimulation. MSCs were exposed to recombinant TNF-related apoptosis-inducing ligand (TRAIL) to stimulate their TRAIL-Receptors (TRAIL-Rs). PGE2, selected as representative marker among the secreted molecules, was analyzed in the conditioned medium. A much lower degree of apoptosis and PGE2 levels were detected compared with Fas stimulation (Figure S1C). Nonetheless, caspase inhibition completely prevented both the TRAIL-induced apoptotic cell death and PGE2 production, suggesting that the non-death signaling may be shared among the death receptors. On the other hand, another common apoptotic stimulus granzyme B alone failed to induce PGE2 despite triggering MSC apoptosis (Figure S1D).
Since the transcriptional profile clearly indicated a prominent activation of the NF-κB signaling pathway in the Fas-stimulated MSCs, we attempted to validate the role of NF-κB activation in the induction of the apoptotic secretome by knocking down the expression of RelA (also known as p65, a subunit of the NF-kB transcription factor complex) in the bone marrow-derived MSCs using short hairpin RNA (shRNA) (Figure 2A). Compared with the scrambled control (Sh-Ctl), RelA knockdown had no impact on the percentages of apoptotic cell death induced by Fas stimulation (Figure 2B). Yet, RelA knockdown totally prevented the upregulation of COX2 as well as the increase in PGE2 and LIF (Figures 2C–2E). The level of IL-6 was less affected, implying that additional pathways might contribute to its regulation (Figure 2F). To confirm the RelA knockdown data, we used the selective NF-κB inhibitor BAY11-7085.20 BAY11-7085 efficiently blocked COX2 upregulation in ApoMSCs but had no effects on the proportion of apoptotic cell death (Figure 2G). Consistent with the previous results, BAY11-7085 fully prevented the increase of PGE2 and LIF in the supernatant while producing a minor effect on IL-6 levels (Figure 2H). In contrast to caspase inhibition, which stopped both the apoptotic death and the upregulation of the Fas-induced secretome (Figures 1F and 1G), NF-κB inhibition selectively interferes with Fas-induced secretome, but not apoptosis.
Figure 2.

NF-κB activation in the apoptotic MSCs produces an immunosuppressive secretome independent of the dying process
(A) To knock down the RelA in the bone marrow MSCs, MSCs were infected by lentivirus vector using pLentiLox.3.7 with the shRNA control sequence (Sh-Ctl) and RelA sequence (Sh-RelA) for 48 h at 37°C, 5% CO2. Expression of RelA (65 kDa) was assessed by immunoblotting. β-Tubulin (43 kDa) was used as the loading control. (B) After 24-h treatment of anti-Fas, the percentage of apoptosis in the MSCs was assessed using Annexin-V and 7AAD assay. (C) The intracellular COX2 level was assessed using flow cytometry as described before. (D–F and H) The level of PGE2, LIF, and IL-6 in the cell culture supernatant were assessed using human PGE2, LIF, and IL-6 ELISA kits, respectively. One-way ANOVA and post hoc Tukey test were used to compare the mean differences among the samples. (G) Dot plots illustrating the distribution of COX2 expression among the Annexin-V+ or Annexin-V− population. Thirty micromolar BAY 11–7085 was added to MSCs during the Fas stimulation to prevent NF-κB activation. Two-way ANOVA and post hoc Tukey test were used to compared the mean differences between groups. ∗ p values < 0.05; ∗∗,## p values < 0.01; ∗∗∗, ˆˆˆ p values < 0.001; ∗∗∗∗, ####, ˆˆˆˆ p values <0.0001; n.s, no significance. All results shown are representative of at least three independent experiments. Experimental data were expressed as mean ± SD.
Fas-induced MSC secretome mediates immunosuppression and monocyte chemotaxis via COX2/PGE2 activity
We then characterized the immunological function of the Fas-induced MSC secretome by testing the conditioned media generated from Fas-stimulated MSCs (Fas-CM) and untreated controls (Ut-CM) (Figures S2A and S2B). To minimize the likelihood of any anti-Fas antibody remaining in the conditioned medium, the bone marrow-derived MSCs were only exposed to the treatment for 15-min followed by washing and resuspension in fresh media. We first tested the functional impacts of Fas-CM on CD3/CD28-induced T cell proliferation and activation in vitro. Despite the presence of pro-inflammatory cytokines like IL-6 and IL-8, Fas-CM significantly reduced CD3 T cell proliferation in a dose-dependent manner compared with controls (Figure 3A). The secretion of IL-2 and interferon (IFN)-γ from the T cells was also markedly suppressed in the presence of Fas-CM (Figures 3B and 3C).
Figure 3.

Fas-induced secretome mediates immunosuppression and monocyte chemotaxis via PGE2 activity
(A) CellTrace-stained CD3 T cells were treated with varying amount of Fas-CM or Ut-CM at 37°C, 5% CO2 for 3 days. CD3/28 Dynabeads were used to stimulated CD3 T cells. Percentage of relative proliferation was calculated by the following formula: (% of V450low population of samples/% of V450low population of positive control: CD3 with beads only) × 100%. The amount of (B) IL-2 and (C) IFN-γ in the cell culture medium was assessed using ELISA kits. (D–F) Different amounts of anti-PGE2 neutralizing antibodies (clone 2B5) were added to the Fas-CM and incubated for 30 min at 37°C, 5% CO2 before adding to stimulated CD3 T cells. Likewise, the amount of (E) IL-2 and (F) IFN-γ in the cell culture medium was assessed. (G) Isolated human monocytes were first resuspended in transwells and incubated for 10 min at 37°C, 5% CO2. Fas-CM and Ut-CM was diluted 50-fold with complete RPMI and then then added to the 24-well for another 2-h incubation at 37°C, 5% CO2. Migrated monocyte concentration (cell/μL) was then calculated based on the manufacturer’s manual of CountBright Absolute Counting Beads. Different amount of anti-CCL2/MCP-1, anti-CCL20, and anti-CXCL8/IL-8 neutralizing antibodies (all mouse IgG1) were added to the culture medium and incubated for 20 min at 37°C, 5% CO2 before adding to the 24-well plates. The same amount of PBS was added as vehicle controls. (H) MSCs were stimulated with anti-Fas CH11 in the presence of caspase inhibitor Z-VAD-FMK (50μM), NF-κB inhibitor BAY 11–7085 (30 μM), or selective COX2 inhibitor NS-398 (100 μM). The level of CCL2 in the cell culture medium was assessed after 24 h treatment using ELISA kit. All results shown are representative of at least three independent experiments. Experimental data were expressed as mean ± SD. One-way ANOVA and post hoc Tukey test were used to compare the mean differences among the samples (∗p values <0.05, ∗∗p values <0.01, ∗∗∗p values <0.001, ∗∗∗∗p values <0.0001, n.s, not significant).
Since PGE2 levels were found to be particularly high compared with other molecules in the ApoMSC secretome and PGE2 had previously been identified as a critical molecule modulating T cell function,21,22 we evaluated whether the PGE2 from Fas-CM could account for the immunosuppressive activity. We added escalating doses of neutralizing anti-PGE2 antibody23 to the Fas-CM and observed that they proportionally reversed the inhibitory effects of Fas-CM on CD3+ T cell proliferation and IL-2/IFN-γ production (Figures 3D–3F). We ruled out the role of ApoMSC-derived extracellular vesicles on the CD3 T cells (Figures S2C–S2F). Therefore, PGE2 is a critical mediator of ApoMSC immunosuppressive activity.
We next assessed the chemotactic activity of the Fas-CM because of the upregulation of multiple chemokines in the supernatant. Furthermore, CCL2 was proposed as a novel “find-me” signal for the apoptotic cells to recruit THP-1 monocytes.24 It was plausible to hypothesize the ApoMSC secretome would contain “find-me” signals to help recruit phagocytes for dead cell clearance. Our prior study demonstrated that human monocytes polarized into immunosuppressive cells through the efferocytosis of ApoMSCs.5 Hence, we assessed the chemotactic activity of the Fas-CM using primary human monocytes. Fas-CM demonstrated potent chemotactic activity for human monocytes compared with the Ut-CM (Figure 3G). To identify the chemokine responsible for such activity, we individually neutralized CCL2, IL-8, and CCL20, which were the most upregulated, in the Fas-CM (Figure S1B). Only the neutralization of CCL-2 but not IL-8 nor CCL-20 reversed the numbers of migrated monocytes (Figure 3G). The full reversion of migration suggested that CCL-2 was solely responsible for the monocyte chemotaxis among the molecules in the Fas-CM. Although it was expected to see that the CCL-2 released by the Fas-stimulated MSCs was dependent on the caspase activity and NF-κB activation, we found that CCL-2 production was also dependent on the COX2/PGE2 activity as selective COX2 inhibitor NS-398 could prevent its increase (Figure 3H). Similarly in a mouse model of glioma, COX2 inhibitors reduced the CCL2 production in the tumor to reduce myeloid-derived suppressor cell accumulation in the tumor microenvironment.25
In addition to its chemotactic property toward monocytes, we observed that Fas-CM reprogrammed monocytes to acquire an immunosuppressive phenotype. Monocytes exposed to Fas-CM (Fas-CM-Mφ) exhibited M2-like markers including downregulation of CD86 and HLA-DR, and upregulation of CD206 (Figure S3A). Remarkably, Fas-CM-Mφ exhibited a potent antiproliferative action on CD3/CD28-stimulated T cells (Figure S3B).
PBMCs from CD patients induce apoptosis and caspase-dependent COX2/PGE2 in MSCs
We have previously proposed that MSC apoptosis could be used as a potential biomarker for identifying acute GvHD patients responding to MSC therapy.1 In this study, we uncovered a range of molecules that are upregulated during MSC apoptosis. Among them, we have identified that PGE2 in the ApoMSC secretome could in part inhibit T cell functions. In order to functionally validate the impact of ApoMSC-derived PGE2 on MSC therapeutic activity, we exposed adipose tissue-derived MSCs (Alofisel/darvadstrocel) used in the clinical trial ADMIRE-CD to PBMCs obtained from perianal fistulizing CD patients enrolled in the same study15 and assessed the induction of MSC apoptosis and the release of PGE2. In line with the bone marrow MSCs in GvHD, we observed that PBMCs isolated from CD patients induced much higher levels of MSC (Alofisel/darvadstrocel) apoptosis compared with the healthy donors or MSCs alone (Figure 4A).
Figure 4.

MSCs (Alofisel/darvadstrocel) undergo apoptosis when in contact with CD patients’ PBMCs and secrete PGE2 in a caspase-dependent manner
(A) 5 × 104 adipose tissue-derived MSCs (Alofisel/darvadstrocel) were cultured for 4 h with 1 × 106 PBMCs and apoptosis was evaluated by assessing Annexin-V/7AAD staining by flow cytometry. MSC alone and MSC co-cultured with PBMCs from healthy donors were used as controls. (B) 5 × 104 MSCs were cultured for 4 h with 1 × 106 PBMCs either in direct contact or in transwell and apoptosis was evaluated by assessing Annexin-V/7AAD staining by flow cytometry. (C) Levels of PGE2 were detected in the supernatant by ELISA. MSCs alone (n = 8), PBMCs alone (n = 3), co-culture of MSC:PBMC obtained from healthy controls (HC, n = 4) and Crohn’s disease patients (CD, n = 24). (D) PGE2 levels detected by ELISA after 4 h MSC:PBMC co-culture either in straight contact or in a transwell system. (E) Expression levels of PGE2 were measured in the co-culture supernatant by ELISA after treatment with the pan-caspase inhibitor Z-VAD-FMK (50 μM) and compared with the untreated control. Data are expressed as mean ± SD. Unpaired t test was used to analyze mean differences between the samples in (A) and (B). Nonparametric Wilcoxon signed rank test and Kruskal-Wallis test followed by Dunn’s post-test were used to compare the differences between two or more groups in (C), (D), and (E) (∗p values <0.05, ∗∗p values <0.01, ∗∗∗p values <0.001, ∗∗∗∗p values <0.0001, n.s, not significant).
Cell-cell contact was essential to trigger apoptosis in MSCs since cell death was prevented when effector cells and target cells were physically separated (Figure 4B). We observed that the PBMC:MSC co-culture supernatant contained much higher PGE2 levels in comparison with the amount detected in either MSCs or PBMCs alone, or in healthy donor samples (Figure 4C). PGE2 production was dependent on cell-cell contact (Figure 4D), thus excluding the contribution of soluble factors “licensing” MSCs in inducing PGE2. To confirm the role of caspase activation in mediating the PGE2 upregulation, caspase inhibitor was added during the co-culture, and it significantly reduced the increase of PGE2 (Figure 4E).
To reassure whether PGE2 was secreted by MSCs, we analyzed PBMC:MSC cultures for COX2. We found COX2 to be selectively upregulated in cell tracer-labeled MSCs and completely reversed after caspase inhibition. Due to limited availability of samples from the ADMIRE-CD trial, we selected two different CD patients who were chosen because their PBMCs exhibited high cytotoxicity against MSCs (Figure S4A).
PBMC cytotoxicity against MSCs and increment of PGE2 are associated with clinical response to MSC therapy in CD patients
Finally, we asked whether MSC apoptosis and PGE2 increment induced by patients’ PBMCs could predict clinical responses to MSC therapy (Alofisel/darvadstrocel). We found that the proportion of MSC apoptosis and PGE2 levels were significantly higher when PBMCs were obtained from responders compared with non-responders (Figures 5A and 5D). We also assessed whether, in addition to PGE2, other molecules detected in the Fas-stimulated transcriptome (Figure 1B) could exhibit a similar predictive potential for MSC therapy. The levels of IL-8, CCL2, CXCL6, CXCL10, LIF, and IL-6 were measured and compared between responders and non-responders. The molecular profile detected in the co-culture supernatant largely recapitulates the one induced in ApoMSC secretome by Fas stimulation. However, except for PGE2 and IL-6, none of the tested molecules were differentially expressed in the two groups (Figures S5A–S5F). Yet we decided to proceed with the downstream analysis with only PGE2 as an apoptosis-dependent molecule because the pathway regulating IL-6 during MSC apoptosis was less understood.
Figure 5.

PBMC cytotoxicity against MSCs (Alofisel/darvadstrocel) and increment of PGE2 are associated with clinical response to MSC therapy
MSC (Alofisel/darvadstrocel) apoptosis (A) and PGE2 levels (D) were compared in the MSC arm between patients who never responded to treatment and patients who responded either 6, 12, or 24 weeks after treatment. (B and E) ROC curve analysis for ApoMSC and PGE biomarkers. (B) ApoMSC yielded an AUC of 0.8 (95% CI: 0.61–0.99, p = 0.01) with 78% sensitivity and 75% specificity selecting a cutoff value of 9.79. (E) PGE2 yielded an AUC of 0.84 (95% CI: 0.68–1, p = 0.007). A PGE2 value > 2,008 pg/mL detected clinical response with 81% sensitivity and 75% specificity. (C and F) MSC apoptosis (C) and PGE2 (F) were assessed in patients who maintained remission and patients who relapsed. Unpaired t test or Mann-Whitney test was used to analyze mean differences between the groups (∗p values <0.05, ∗∗p values <0.01, ns, not significant).
Using receiver operating characteristic (ROC) curve analysis, we demonstrated that MSC apoptosis (Figure 5B) and PGE2 (Figure 5E) were significantly associated with clinical responses. By selecting the cutoff value of 9.79%, MSC apoptosis was able to predict response with 78% sensitivity and 75% specificity; and with positive and negative predictive values (PPV and NPV) of 87% and 60%, respectively. On the other hand, by selecting a cutoff of 2008 pg/mL, PGE2 predicted clinical responses with 83% sensitivity, 75% specificity, 86.7% PPV, and 66.7% NPV.
Another concern about MSC therapy in CD patients is that not all patients who achieve a clinical response remain in remission. Among our available samples, four patients experienced a relapse 6 (n = 1), 12 (n = 1), or 18 (n = 2) weeks after remission. Relapses were recorded 12 (n = 1) or 24 (n = 3) weeks after the infusion. We observed that PBMCs from durable remitters induced significantly higher PGE2 levels compared with those from relapsing patients. Although two out of four relapsing patients exhibited moderate levels of PGE2, those levels were comparable to the range of non-responders. On the other hand, we failed to demonstrate a difference in the two groups when we compared the levels of apoptosis (Figures 5C and 5F).
Finally, we reported no difference in terms of apoptosis and PGE2 between the MSC and placebo arms (Figures S5G and S5H), nor among the responders, long-term remitters, and relapsed patients belonging to the placebo arm (Figures S5I and S5J).
Discussion
Described in 1972 by Kerr et al, apoptosis was first characterized as a form of regulated cell death proceeding in an immunologically silent manner.26 Despite being stated as pro-inflammatory under some circumstances,9 here we show that apoptotic cell death in MSCs under Fas receptor stimulation is actively immunosuppressive. The caspase activation in MSCs not only triggers apoptosis but also induces an immunosuppressive secretome that appears to be partly distinct from the dying process (Figures 1, 2, and S1). Our findings shed light on a novel molecular signature whereby caspase activation induces a list of immunosuppressive molecules in ApoMSCs under the regulation of the NF-κB pathway (Figure 2). Although NF-κB is required to produce the apoptotic secretome, its activation does not affect the fate of cell death. The molecules upregulated include COX2/PGE2, CCL2, LIF, TSG-6, and IL-6 (Figure 1), which share many similarities with the immune molecular repertoire of MSCs exposed to the conventional cytokine “licensing.”27,28,29 We speculate this could explain why the role of apoptosis in vivo has been unappreciated until very recently.1,2,3,30
Our study demonstrates that PGE2 is critical for apoptotic MSCs to directly suppress T cell activation independently of the already described mechanism mediated by efferocytosis (Figures 3A–3F). PGE2 is known to inhibit T cell activation via the IL-2 blockade.21,22 Previous studies have reported that the production of PGE2 from apoptotic cells occurs via the caspase-dependent iPLA2,31,32 whose function is to increase the synthesis of arachidonic acid for COX1/2 activity. Here we showed that PGE2 from ApoMSCs is dependent on COX2 activity under the NF-κB (Figure 2). Collectively, this indicates that caspase activation might contribute to PGE2 production by regulating multiple molecular checkpoints throughout the pathway. Of note, the extracellular vehicles isolated from the same amount of Fas-CM (12.5% v/v) had no impact on the T cell proliferation in vitro (Figure S2F). Although PGE2 neutralization did only partly reverse the inhibition of T cell proliferation, its effect was more significant on IL-2 and IFN-γ suppression (Figures 3E and 3F). Caspase activation in MSCs also induces the upregulation of CCL2, which was found to predominately facilitate monocyte migration (Figures 3G and 3H). Monocytes are the frontline innate phagocytes during the onset of inflammation33 and they polarize into immunosuppressive cells following efferocytosis of apoptotic cells including the ApoMSCs.5,34 Therefore, it is plausible that caspase-dependent CCL2 might serve as a “find-me” signal, an idea previously proposed by Cullen and colleagues,24 thereby facilitating additional efferocytosis and immune-silencing clearance of apoptotic cells. On the other hand, CCL2 could also be involved in polarizing immunosuppressive macrophages.35 In a murine model of colitis, CCL2 has been found to be required for bone marrow MSC-induced IL-10+ polarization of intestinal and peritoneal macrophages in vivo, which was essential for alleviating gut injury.35
After decades of pre-clinical and clinal studies about MSCs in treating CD,36,37 an adipose tissue-derived MSC product (Alofisel/darvadstrocel, Takeda) has been officially approved for treating the perianal fistulizing CD patients in the European Union and Japan.14 The phase III clinical study ADMIRE-CD (NCT01541579), published in 2016, has shown local administration of allogeneic MSCs are an effective and safe treatment for complex perianal fistulas in patients with CD,15 and clinical responses were sustained for at least 1 year after the treatment in a follow-up study.38 Despite its efficacy, the lack of criteria to stratify patients remains a great challenge. Prognostic tools or biomarkers in the field are required to identify responders to improve the overall responses and cost-effectiveness of MSC therapy.16 In GvHD, clinical responses to MSC treatment can be predicted based on the ability of patients to induce apoptosis in bone marrow MSCs.1 Remarkably, we observed similar levels of cytotoxicity from CD patients’ PBMCs on adipose tissue-derived MSCs (Alofisel/darvadstrocel) whereby elevated MSC apoptosis was associated with clinical responses in ADMIRE-CD patients (Figure 4). Despite the different sources of MSCs, PBMC cytotoxicity was seemingly applicable in both GvHD and CD patients. Advancing from our prior GvHD studies,1,39 here we have retrospectively assessed the clinical significance of the ApoMSC secretome signature in the ADMIRE-CD patients. We found that patients inducing high levels of PGE2 in MSCs (Alofisel/darvadstrocel) achieved clinical improvement and healing of the fistula following MSC infusion, whereas patients displaying lower PGE2 levels failed to respond (Figure 5). Altogether, these findings imply that measuring the levels of MSC apoptosis and PGE2 can be effective tools to predict clinical responses and stratify patients for treatment. We also observed that the levels of PGE2 could identify relapsing patients as their PGE2 levels dropped back to levels comparable to the non-responders (Figure 5). A larger scale of clinical studies in the future is unquestionably warranted to validate these clinical findings. Interestingly, MSC apoptosis was not associated with long-term remission. This discrepancy could result from a defective MPS, as already hypothesized in previous studies,40 impairing the ability to clear apoptotic cells and sustain long-term immunosuppression. Alternatively, some of the MSC batches received by the patients may have a different ability to undergo apoptosis and produce PGE2.
Although we characterized the immunological impacts of the ApoMSC secretome, myeloid cell efferocytosis of apoptotic MSCs is an essential mechanistic pathway to alleviate inflammation.1,2,5,39,41 It remains to be determined whether the immunosuppressive activity of the ApoMSC secretome is as effective as the one produced by phagocytes following ApoMSC efferocytosis. Nonetheless, it is reasonable to hypothesize that the apoptotic secretome would at least provide an additional mechanism to facilitate efferocytosis and dampen inflammation in situ. In previous murine colitis models, PGE2 secretion from the infused MSCs was found indispensable to alleviate colitis inflammation.42,43 Therefore, the ApoMSC secretome is likely to play a complementary role to the ApoMSC efferocytosis process, thus demonstrating the multifaceted functions of apoptosis to achieve immune silencing. Moreover, our data indicate that caspase activation is critical to provide a further death-independent signal for the ApoMSCs in vivo. Our conclusions are supported by Pang et al.,2 who found that apoptotic cells exhibit a better activity compared with heat-inactivated, fixed MSCs or MSCs with incomplete apoptotic stimulation.
The new mechanism that we have described may provide a new tool for the selection of MSC batches, whereby those more susceptible to undergo apoptosis and release of PGE2 could be preferred for treatment with potential implication on the cells’ dose. Although we have only used one donor’s bone marrow MSCs for the in vitro secretome characterization, we observed comparable levels of caspase-dependent apoptosis and PGE2 release in MSC from another donor’s bone marrow (Figure S1E). Since the MSCs infused in the ADMIRE-CD were expanded from only one donor (later become Alofisel/darvadstrocel), we have used the same MSCs to address the significance of the biomarker. However, we did not look into adipose tissue-derived MSCs from further donors and we acknowledge this as a limitation. Due to restricted availability of ADMIRE-CD samples, we could not answer this question. More studies are needed to understand if the tissue origin of MSCs differently affects their susceptibility to undergo apoptosis when exposed to PBCMs isolated from patients.
Our study, strengthened by many others, continues to corroborate the notion that MSC apoptosis is an important step contributing to their mechanistic action in vivo. Notably, here we report that caspase activation induces an immunosuppressive secretome with added chemoattractive properties for phagocytes, which might benefit in situ efferocytosis. The impact of the mechanism was validated as a biomarker to predict clinical responses to MSCs in fistulizing CD patients. Our results pave the way to improve the design of MSC-based clinical trials and provide new information on how to develop the next generation of MSC products.
Materials and methods
Isolation and culture of human MSCs
Human bone marrow-derived MSCs were isolated from the bone marrow aspirate taken from the iliac crest of a healthy donor and sub-cultured as previously described.1,5 The human samples are obtained from the Imperial College Healthcare Tissue Bank (ICHTB, Human Tissue Authority [HTA] license 12275) under the approval of UK National Research Ethics Service to release human material for research (12/WA/0196). MSCs at passage 3 to 7 (all from the same MSC donor) were used for experiments. Different batches of MSCs were used to obtain independent experimental repeats. MSCs were previously checked for positivity of CD90, 105, CD106, CD73, human leukocyte antigen class I, and the lack of expression of CD14, CD31, and CD45. Human adipose tissue-derived MSCs (Alofisel/darvadstrocel) were kindly provided and used as part of a collaboration with TiGenix company.15
Microarray and gene set enrichment analysis
Bone marrow MSCs from different batches (5 × 105 per 96 well) were treated with 5 μg/mL recombinant granzyme B (Enzo Life Sciences, UK) and 10 μg/mL anti-Fas activating monoclonal antibody (clone CH11, Merck Millipore, UK) in complete RPMI 1640 medium containing 10% (v/v) FBS (Labtech.com, UK) and 1% (v/v) P-S (Thermo Fisher Scientific, UK) for 24 h at 37°C, 5% CO2. For each treatment, the untreated MSCs were prepared as the control group. A total of 10 samples from five independent groups were prepared for the human microarray analysis. RNA samples were extracted using TRIzol with chloroform and purified using RNeasy Mini Kit (QIAGEN, UK). Purified RNA was resuspended in RNase-free water. To evaluate the quality, the RNA was examined using Agilent RNA 6000 Nano kits (Agilent Technologies, UK) according to the manufacturer’s protocol and measured by the Agilent 2100 Bioanalyser. For the microarray, 100 ng of selected RNA samples was input and processed using GeneChip WT PLUS Reagent Kit (Affymetrix, UK). Samples were processed and converted at King’s Genomic Center. Gene set enrichment analysis (GSEA) (version 2.2.3) was used to determine the significant gene sets or signaling pathways differently expressed between the MSCs and ApoMSCs. The hallmark gene sets were selected to perform the analysis with other parameters kept as default. Statistics with false discovery rate (FDR) below 0.25 (25%) were considered as significant. Heatmaps are generated using Rstudio via the heatmap.2 function to demonstrate the expression difference from the core enriched genes contributing to the enrichment results. Among the top genes, the immunomodulatory factors and chemokines are further selected to create another heatmap.
Real-time qPCR
Gene expression was further validated using TaqMan RNA-to-CTM 1-Step Kit (Thermo Fisher Scientific, UK); 20 ng of RNA sample was used in each reaction. The primer sequences and the gene product sizes are listed in Table S2. The threshold cycle (CT) was analyzed by StepOne and StepOnePlus Software version 2.1. The expression level of the target gene was normalized to that of hypoxanthine phosphoribosyltransferase 1 (HPRT1). Relative gene expression was calculated using the 2–ΔΔ CT formula.
Protein extraction and immunoblotting
Cell lysates were homogenized using 100 μL RIPA buffer with phenylmethanesulfonyl fluoride and protease inhibitors. After vortexing several times and incubation on ice for 30 min, the protein extracts were obtained after centrifuging at 10,000 × g for 15 min at 4°C. The amount of protein was determined by bicinchoninic acid protein assay kit. The amount of protein in samples was measured by NanoDrop 1000 Spectrophotometer. Ten to 20 μg protein per sample was prepared in laemmli buffer (Thermo Fisher Scientific, UK) and boiled at 100°C for 5 min before running the SDS-PAGE. Protein samples were later transferred onto activated polyvinylidene difluoride (PVDF) membranes using electroblotting.
Tris-buffered saline-Tween20
Five percent (w/v) non-fat milk in Tris-buffered saline-Tween20 (TBS-T) was used to block the membrane for 1 h at room temperature before overnight probing of primary antibody in 3% BSA (w/v) TBS-T at 4°C. The next day, membrane was washed three times by TBS-T at room temperature and proceeded to secondary antibody (with horseradish peroxidase) probing in 5% non-fat milk TBS-T for 1-h incubation at room temperature. Chemiluminescence was developed using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, UK). Last, the membrane was imaged using Azure c300 Chemiluminescent Western Blot Imaging System (Azure Biosystems, USA) and quantified using ImageJ software. A full list of antibodies used is stated in Table S3 a-Tubulin was used as the protein loading control.
ELISA and Luminex assay
For ELISA and Luminex, cell culture supernatant was collected and the components were analyzed using different ELISA kits for LIF, IL-6, CCL-2, IL-2, and IFN-γ (Thermo Fisher Scientific, UK), PGE2 and CCL-2 (Cayman, USA), and Human Magnetic Luminex Assay (R&D systems, UK).
Flow cytometry analysis
For human specific molecules, the following antibodies were used: anti-COX2 (PE, clone AS67), anti-CD14 (APC-eFluor 780, clone 61D3), anti-CD3 (APC, clone UCHT1), anti-CD80 (APC-H7, clone L307.4), anti-CD86 (PE, clone IT2.2), anti-HLA-DR (APC, clone G46-6), anti-CD163 (APC, clone GHI/61), anti-CD206 (eFluor450, clone 19.2), anti-CD68 (PerCPCy5.5, clone Y1/82A), CD95/Fas (APC, clone DX2). The status of cell death was determined using PE-Annexin-V and 7-Aminoactinomycin D (7-AAD) double staining kit (BD Biosciences, UK). For intracellular staining, cells are fixed by Cytofix/Cytoperm Fixation and Permeabilization Solution (BD Biosciences, UK) and permeabilized using 1X permeabilization buffer (Thermo Fisher Scientific, UK). For co-culture assays, Celltrace fluorescence dye was used to identify certain populations (Thermo Fisher Scientific, UK).
Lentiviral transduction
Lentiviral infection was performed using pLentiLox.3.7 as described before.44 Both vector, RelA shRNA and control sequence were purchased from Sigma-Aldrich, UK. Knockdown efficiency was verified using western blot. β-Actin was used as the protein loading control.
Use of inhibitors
One hundred micromolar selective COX2 inhibitor NS-398 (MedChemExpress Europe, Sweden) was used to inactivate COX2. Thirty micromolar selective NF-κB inhibitor BAY 11–7082 (Sigma-Aldrich, UK) was used to prevent the activation of the NF-κB pathway. Fifty micromolar pan-caspases inhibitor Z-VAD-FMK (R&D Systems, UK) was used to prevent the activation of apoptotic caspases.
Isolation of human primary mononuclear cells and specific immune cell types
Human blood samples of healthy donors were purchased from the National Health Service Blood and Transplant with the non-clinical account number (P243) for research purposes. For this project being conducted in England, no ethical approval was needed from the NHS Research Ethics Committee. Storage of isolated human cell samples is under the HTA license (#11023). PBMCs were isolated from the blood samples of heathy donors and inflammatory bowel disease patients using Ficoll-Paque (Sigma-Aldrich, UK) following 1,800 rpm centrifugation for 32 min at 20°C. Isolated PBMCs were washed and resuspended in ACK lysis buffer (Thermo Fisher Scientific, UK) for 5 min to remove the red blood cells. Certain immune cell types were isolated from healthy donor PBMCs. To isolate monocytes, PBMCs were subjected to Pan Monocyte Isolation Kit II (Miltenyi Biotec, UK). To isolate CD3+ lymphocytes, CD3 MicroBeads (Miltenyi Biotec, UK) were used.
CellTrace proliferation assay
CellTrace-stained human CD3 T cells were treated with varying amounts of conditioned medium. Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher Scientific, UK) in 1:1 cell-to-bead ratio was used to stimulate the proliferation of PBMCs and CD3 T cells, respectively. The proliferating population of T cells was measured by flow cytometry according to the loss of fluorescence intensity due to cell division. The percentage of proliferation is relative to the proliferation from the CD3 T cells with Dynabeads alone. To assess the impact of CM, different proportions of CM (v/v%) were added during the stimulation. To confirm the functional relevance of PGE2, selective anti-PGE2 monoclonal antibody 2B5 (Cayman, USA)23 was added to the CM and incubated for 30 min at 37°C, 5% CO2 before adding to the reaction. The same volume of PBS was also added as vehicle control.
Monocyte chemotaxis assay
Isolated human monocytes (3 × 105 per transwell) were first resuspended in 100 μL complete RPMI medium (10% FBS, 1% P-S) and then transferred to 8-μm pore transwell chambers (Sigma-Aldrich, UK) sitting on an empty 24 well. The monocytes were incubated in the transwell chambers for 10 min at 37°C, 5% CO2. The assay quantifies how many monocytes can migrate in a vertical direction through the porous membrane into the lower compartment, in which RPMI containing chemo-attractants or simply a higher concentration of FBS is present (positive control: 20% FBS RPMI). To prepare the samples for the lower compartment, CM was harvested from different MSCs. They were 50-fold diluted using complete RPMI to a total volume of 600 μL. For the neutralization, specific antibodies were added to the CM and incubated for 20 min at 37°C, 5% CO2 (all monoclonal mouse immunoglobulin [Ig]G1, R&D Systems, UK). After incubation, transwells were removed carefully and the supernatant in the 24-well was collected in a flow cytometry tube. Two washes of PBS were followed to ensure all the cells were collected. Collected cells were centrifuged and resuspended in 300 μL buffer (PBS 2% FBS, 2 mM EDTA). Fifteen microliters of CountBright Absolute Counting Beads (Thermo Fisher Scientific, UK) was added to each tube before FACS analysis. Migrated monocyte concentration (cell/μL) was then calculated based on the manufacturer’s manual.
Cytotoxic assay with patients’ PBMCs
PBMCs were collected from patients enrolled in the clinical trial ADMIRE-CD (https://clinicaltrials.gov/show/NCT01541579)15 during randomization visits (at least 2 weeks before treatment administration) and stored in liquid nitrogen until use. Forty-two available frozen PBMC samples (MSC-treated and placebo arms) were thawed and incubated overnight in complete RPMI medium (10% FBS, 1% Pen-Strep) in ultra-low attachment plates. The viability of thawed PBMCs was checked before subjecting them to the co-culture assay and the majority of samples had viability >85%. MSCs (Alofisel/darvadstrocel), which were the same source as those administrated into patients during the trial, were stained with 2.5 μM CellTraceTM Violet (Thermo Fisher Scientific, UK) and plated (5 × 104 cells/well) in a 24-well plate overnight in a total volume of 500 μL. The day after, PBMCs were harvested and added to the MSC monolayer at a PBMC:MSC ratio of 20:1. After 4 h of co-culture, supernatant was removed, and cells were trypsinized and collected for flow cytometry analysis. In some conditions, transwell insert (pore size 0.4 μm) (Corning, UK) was used to separate the physical contact between PBMCs and MSCs. Status of apoptosis was determined using PE-Annexin-V and 7-Aminoactinomycin D (7AAD) double staining kit (BD Biosciences, UK) and flow cytometry. MSCs were identified as Violet+ cells. The percentage of Annexin-V+7AAD-MSC population was evaluated and referred as apoptotic MSC (ApoMSCs).
Patient information
Patients’ characteristics are summarized in Table 1. Among the 42 patient samples available for this study, 27 patients were in the MSC-treated arm and 15 in the placebo arm. Age was 37.5 ± 11 (mean ± SD) with no statistically significant difference observed between the two groups. For all experiments, the operator was blind to patients’ clinical details. Patient consent was given before the disclosure of their status of clinical responses to MSC therapy, which were defined as closure of all external openings that were draining before treatment despite gentle finger compression, and absence of collections larger than 2 cm of the fistulas confirmed by magnetic resonance imaging. Clinical assessment was evaluated 6, 12, and 24 weeks after MSC or placebo treatment. We indicated responders as those patients who exhibited a response at either week 6, 12, or 24 after treatment and maintained clinical remission at week 24, and non-responders were defined as the patients who did not show a clinical response at any time point. Patients who showed relapse were responders at week 6 or 12 but relapsed at week 24.
Table 1.
Patient characteristics
| Alofisel®/darvadstrocel (n = 27) | Placebo (n = 15) | |
|---|---|---|
| Age (years) | 35 | 42 |
| Male | 12 (44%) | 6 (40%) |
| Female | 15 (56%) | 9 (60%) |
| Perianal Crohn’s Disease Activity Index Score | 6.1 | 8.6 |
| C-reactive protein (nmol/L) | 8.6 | 11.8 |
| Treatments | ||
| Anti-TNF | 10 (37%) | 8 (54%) |
| Immunomodulators | 6 (22%) | 3 (20%) |
| anti-TNF and immunomodulators | 5 (19%) | 2 (13%) |
| Neither | 6 (22%) | 2 (13%) |
Statistical analysis
Statistical analysis was performed using PRISM version 6.0 (GraphPad, USA). Experimental data were expressed as mean ± SD from at least three technical repeats. One-way ANOVA and post hoc Tukey test were used to compare the mean differences when there were more than two samples. Two-way ANOVA and post hoc Tukey test were used to compare the mean differences between groups. Unpaired t test was used to compare the mean differences between two samples. p values less than 0.05 are considered as statistically significant.
Handling the patient data, normality of distribution of experimental data was first assessed with the Kolmogrov-Smirnov test and Shapiro-Wilk test. When data were normally distributed, unpaired and paired Student’s t tests were performed to compare the differences between two groups. When data were not normally distributed, Mann-Whitney U test and Wilcoxon rank test were used to compare the differences between unpaired and paired groups, respectively. p values less than 0.05 are considered as statistically significant. ROC curve analysis was used to assess the predictive ability of the biomarkers.
Data and code availability
The data generated in this study are available within the article and its supplementary data files. All raw data are available upon request.
Acknowledgments
This work was supported by King’s Commercialisation Institute and in part by Cancer Research UK grant C26587, NIHR Imperial Biomedical Research Centre (BRC) grant P81135, and Medical Research Council (MRC) grant MR/V027581/1 to G.F. Infrastructure support for this research was provided in part by the NIHR Imperial BRC. T.S.C. was a recipient of the Hong Kong Scholarships by King's College London Hong Kong Foundation Ltd and the Chinese Student Awards by the Great Britain-China Educational Trust. C.G. was a recipient of the Health School Studentship funded by King’s College London. G.M.B. was funded by a CAPES (Coordination for the Improvement of Higher Education Personnel - Brazil) scholarship. We are grateful to the patients who have provided samples for this study. We thank King’s Genomic Center for collecting the microarray data. We would also like to thank Dr Valeria Zuliani for the technical support.
Author contributions
T.S.C., C.G., A.G., and F.D. conceived the study. O.R., A.A., and R.C. provided patient samples. T.S.C. and A.G. prepared the samples for microarray analysis. T.S.C. characterized the molecular profile of ApoMSC secretome and validated their functions in vitro. C.G. generated the data with patients’ samples with the help of A.A. T.S.C. and C.G. performed the Luminex assay. D.C. provided essential support for lentivirus production and infection. G.M.B. generated data of extracellular vesicles. G.F. provided resources for the lentiviral transduction and supervision. M.C. and F.C. supervised the bioinformatic analysis. T.S.C., C.G., and F.D. wrote the final manuscript with input from R.H. All authors reviewed the final manuscript.
Declaration of interests
A.A. and O.R. are employees of Takeda and hold Takeda stock/stock options. R.H. and F.D. are employees of AstraZeneca. R.C. is a member of the Advisory Board of Takeda on the use of MSC in fistulizing Crohn’s disease. This study did not receive any funding from Takeda or any other pharmaceutical companies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.10.004.
Supplemental information
References
- 1.Galleu A., Riffo-Vasquez Y., Trento C., Lomas C., Dolcetti L., Cheung T.S., Von Bonin M., Barbieri L., Halai K., Ward S., et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017;9:eaam7828. doi: 10.1126/scitranslmed.aam7828. [DOI] [PubMed] [Google Scholar]
- 2.Pang S.H.M., D’Rozario J., Mendonca S., Bhuvan T., Payne N.L., Zheng D., Hisana A., Wallis G., Barugahare A., Powell D., et al. Mesenchymal stromal cell apoptosis is required for their therapeutic function. Nat. Commun. 2021;12:6495. doi: 10.1038/s41467-021-26834-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiss A.R.R., Dahlke M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of action of living, apoptotic, and dead MSCs. Front. Immunol. 2019;10:1191. doi: 10.3389/fimmu.2019.01191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de Witte S.F.H., Luk F., Sierra Parraga J.M., Gargesha M., Merino A., Korevaar S.S., Shankar A.S., O’Flynn L., Elliman S.J., Roy D., et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells. 2018;36:602–615. doi: 10.1002/stem.2779. [DOI] [PubMed] [Google Scholar]
- 5.Cheung T.S., Galleu A., von Bonin M., Bornhäuser M., Dazzi F. Apoptotic mesenchymal stromal cells induce prostaglandin E2 in monocytes: implications for the monitoring of mesenchymal stromal cells activity. Haematologica. 2019;104:e438–e441. doi: 10.3324/haematol.2018.214767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mudter J., Neurath M.F. Apoptosis of T cells and the control of inflammatory bowel disease: Therapeutic implications. Gut. 2007;56:293–303. doi: 10.1136/gut.2005.090464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saas P., Daguindau E., Perruche S. Concise Review: Apoptotic Cell-Based Therapies-Rationale, Preclinical Results and Future Clinical Developments. Stem Cells. 2016;34:1464–1473. doi: 10.1002/stem.2361. [DOI] [PubMed] [Google Scholar]
- 8.McArthur K., Kile B.T. Apoptotic Caspases: Multiple or Mistaken Identities? Trends Cell Biol. 2018;28:475–493. doi: 10.1016/j.tcb.2018.02.003. [DOI] [PubMed] [Google Scholar]
- 9.Galluzzi L., Vitale I., Aaronson S.A., Abrams J.M., Adam D., Agostinis P., Alnemri E.S., Altucci L., Amelio I., Andrews D.W., et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Medina C.B., Mehrotra P., Arandjelovic S., Perry J.S.A., Guo Y., Morioka S., Barron B., Walk S.F., Ghesquière B., Krupnick A.S., et al. Metabolites released from apoptotic cells act as tissue messengers. Nature. 2020;580:130–135. doi: 10.1038/s41586-020-2121-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao Y., Herndon J.M., Zhang H., Griffith T.S., Ferguson T.A. Antiinflammatory effects of CD95 ligand (FasL)-induced apoptosis. J. Exp. Med. 1998;188:887–896. doi: 10.1084/jem.188.5.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W., Frank M.E., Jin W., Wahl S.M. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 2001;14:715–725. doi: 10.1016/s1074-7613(01)00147-9. [DOI] [PubMed] [Google Scholar]
- 13.Murata M., Teshima T. Treatment of Steroid-Refractory Acute Graft-Versus-Host Disease Using Commercial Mesenchymal Stem Cell Products. Front. Immunol. 2021;12:724380–724388. doi: 10.3389/fimmu.2021.724380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levy O., Kuai R., Siren E.M.J., Bhere D., Milton Y., Nissar N., De Biasio M., Heinelt M., Reeve B., Abdi R., et al. Shattering barriers toward clinically meaningful MSC therapies. Sci. Adv. 2020;6:eaba6884. doi: 10.1126/sciadv.aba6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Panés J., García-Olmo D., Van Assche G., Colombel J.F., Reinisch W., Baumgart D.C., Dignass A., Nachury M., Ferrante M., Kazemi-Shirazi L., et al. Expanded allogeneic adipose-derived mesenchymal stem cells (Cx601) for complex perianal fistulas in Crohn’s disease: a phase 3 randomised, double-blind controlled trial. Lancet. 2016;388:1281–1290. doi: 10.1016/S0140-6736(16)31203-X. [DOI] [PubMed] [Google Scholar]
- 16.Martin I., Galipeau J., Kessler C., Le Blanc K., Dazzi F. Challenges for mesenchymal stromal cell therapies. Sci. Transl. Med. 2019;11:eaat2189. doi: 10.1126/scitranslmed.aat2189. [DOI] [PubMed] [Google Scholar]
- 17.Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S., Mesirov J.P. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Packham G., Lahti J.M., Fee B.E., Gawn J.M., Coustan-Smith E., Campana D., Douglas I., Kidd V.J., Ghosh S., Cleveland J.L. Fas activates NF-κB and induces apoptosis in T-cell lines by signaling pathways distinct from those induced by TNF-α. Cell Death Differ. 1997;4:130–139. doi: 10.1038/sj.cdd.4400217. [DOI] [PubMed] [Google Scholar]
- 20.Lawrence T., Gilroy D.W., Colville-Nash P.R., Willoughby D.A. Possible new role for NF-κB in the resolution of inflammation. Nat. Med. 2001;7:1291–1297. doi: 10.1038/nm1201-1291. [DOI] [PubMed] [Google Scholar]
- 21.Walker C., Kristensen F., Bettens F., DeWeck A.L. Lymphokine regulation of activated (G1) lymphocytes. I. Prostaglandin E2-induced inhibition of interleukin 2 production. J. Immunol. 1983;130:1770–1773. [PubMed] [Google Scholar]
- 22.Kalinski P. Regulation of Immune Responses by Prostaglandin E2. J. Immunol. 2012;188:21–28. doi: 10.4049/jimmunol.1101029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mnich S.J., Veenhuizen A.W., Monahan J.B., Sheehan K.C., Lynch K.R., Isakson P.C., Portanova J.P. Characterization of a monoclonal antibody that neutralizes the activity of prostaglandin E2. J. Immunol. 1995;155:4437–4444. [PubMed] [Google Scholar]
- 24.Cullen S.P., Henry C.M., Kearney C.J., Logue S.E., Feoktistova M., Tynan G.A., Lavelle E.C., Leverkus M., Martin S.J. Fas/CD95-Induced Chemokines Can Serve as “ Find-Me” Signals for Apoptotic Cells. Mol. Cell. 2013;49:1034–1048. doi: 10.1016/j.molcel.2013.01.025. [DOI] [PubMed] [Google Scholar]
- 25.Fujita M., Kohanbash G., Fellows-Mayle W., Hamilton R.L., Komohara Y., Decker S.A., Ohlfest J.R., Okada H. COX-2 blockade suppresses gliomagenesis by inhibiting myeloid-derived suppressor cells. Cancer Res. 2011;71:2664–2674. doi: 10.1158/0008-5472.CAN-10-3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kerr J.F., Wyllie A.H., Currie A.R. Apoptosis : a Basic Biological Phenomenon With Wide- Br. J. Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung T.S., Dazzi F. Mesenchymal-myeloid interaction in the regulation of immunity. Semin. Immunol. 2018;35:59–68. doi: 10.1016/j.smim.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Shi Y., Wang Y., Li Q., Liu K., Hou J., Shao C., Wang Y. Immunoregulatory mechanisms of mesenchymal stem and stromal cells in inflammatory diseases. Nat. Rev. Nephrol. 2018;14:493–507. doi: 10.1038/s41581-018-0023-5. [DOI] [PubMed] [Google Scholar]
- 29.Marigo I., Dazzi F. The immunomodulatory properties of mesenchymal stem cells. Semin. Immunopathol. 2011;33:593–602. doi: 10.1007/s00281-011-0267-7. [DOI] [PubMed] [Google Scholar]
- 30.Moll G., Hoogduijn M.J., Ankrum J.A. Editorial: Safety, Efficacy and Mechanisms of Action of Mesenchymal Stem Cell Therapies. Front. Immunol. 2020;11:243–244. doi: 10.3389/fimmu.2020.00243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bell C.R., Pelly V.S., Moeini A., Chiang S.-C., Flanagan E., Bromley C.P., Clark C., Earnshaw C.H., Koufaki M.A., Bonavita E., Zelenay S. Chemotherapy-induced COX-2 upregulation by cancer cells defines their inflammatory properties and limits the efficacy of chemoimmunotherapy combinations. Nat. Commun. 2022;13:2063. doi: 10.1038/s41467-022-29606-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li F., Huang Q., Chen J., Peng Y., Roop D.R., Bedford J.S., Li C.Y. Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 2010;3:ra13. doi: 10.1126/scisignal.2000634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shi C., Pamer E.G. Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 2011;11:762–774. doi: 10.1038/nri3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boada-Romero E., Martinez J., Heckmann B.L., Green D.R. The clearance of dead cells by efferocytosis. Nat. Rev. Mol. Cell Biol. 2020;21:398–414. doi: 10.1038/s41580-020-0232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giri J., Das R., Nylen E., Chinnadurai R., Galipeau J. CCL2 and CXCL12 Derived from Mesenchymal Stromal Cells Cooperatively Polarize IL-10+ Tissue Macrophages to Mitigate Gut Injury. Cell Rep. 2020;30:1923–1934.e4. doi: 10.1016/j.celrep.2020.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ibraheim H., Giacomini C., Kassam Z., Dazzi F., Powell N. Advances in mesenchymal stromal cell therapy in the management of Crohn’s disease. Expert Rev. Gastroenterol. Hepatol. 2018;12:141–153. doi: 10.1080/17474124.2018.1393332. [DOI] [PubMed] [Google Scholar]
- 37.Ciccocioppo R., Fioravante M., Frulloni L. Mesenchymal stromal cell therapy in intestinal diseases. Curr. Opin. Organ Transpl. 2018;23:679–689. doi: 10.1097/MOT.0000000000000586. [DOI] [PubMed] [Google Scholar]
- 38.Panés J., García-Olmo D., Van Assche G., Colombel J.F., Reinisch W., Baumgart D.C., Dignass A., Nachury M., Ferrante M., Kazemi-Shirazi L., et al. Long-term Efficacy and Safety of Stem Cell Therapy (Cx601) for Complex Perianal Fistulas in Patients With Crohn’s Disease. Gastroenterology. 2018;154:1334–1342.e4. doi: 10.1053/j.gastro.2017.12.020. [DOI] [PubMed] [Google Scholar]
- 39.Cheung T.S., Bertolino G.M., Giacomini C., Bornhäuser M., Dazzi F., Galleu A. Mesenchymal Stromal Cells for Graft Versus Host Disease: Mechanism-Based Biomarkers. Front. Immunol. 2020;11:1338. doi: 10.3389/fimmu.2020.01338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marks D.J.B., Harbord M.W.N., MacAllister R., Rahman F.Z., Young J., Al-Lazikani B., Lees W., Novelli M., Bloom S., Segal A.W. Defective acute inflammation in Crohn’s disease: A clinical investigation. Lancet. 2006;367:668–678. doi: 10.1016/S0140-6736(06)68265-2. [DOI] [PubMed] [Google Scholar]
- 41.Giacomini C., Granéli C., Hicks R., Dazzi F. The critical role of apoptosis in mesenchymal stromal cell therapeutics and implications in homeostasis and normal tissue repair. Cell. Mol. Immunol. 2023;20:570–582. doi: 10.1038/s41423-023-01018-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Song W.J., Li Q., Ryu M.O., Nam A., An J.H., Jung Y.C., Ahn J.O., Youn H.Y. Canine adipose tissue-derived mesenchymal stem cells pre-treated with TNF-alpha enhance immunomodulatory effects in inflammatory bowel disease in mice. Res. Vet. Sci. 2019;125:176–184. doi: 10.1016/j.rvsc.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shin T.H., Friedrich S., Brat G.A., Rudolph M.I., Sein V., Munoz-Acuna R., Houle T.T., Ferrone C.R., Eikermann M., Kim H.S. Tnf-α priming elicits robust immunomodulatory potential of human tonsil-derived mesenchymal stem cells to alleviate murine colitis. Surg. Endosc. 2020;34:1–12. [Google Scholar]
- 44.Mauro C., Leow S.C., Anso E., Rocha S., Thotakura A.K., Tornatore L., Moretti M., De Smaele E., Beg A.A., Tergaonkar V., et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011;13:1272–1279. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files. All raw data are available upon request.