

Abstract
Huntington’s disease (HD), a genetic neurodegenerative disorder, primarily affects the striatum and cortex with progressive loss of medium-sized spiny neurons (MSNs) and pyramidal neurons, disrupting cortico-striatal circuitry. A promising regenerative therapeutic strategy of transplanting human neural stem cells (hNSCs) is challenged by the need for long-term functional integration. We previously described that, with short-term hNSC transplantation into the striatum of HD R6/2 mice, human cells differentiated into electrophysiologically active immature neurons, improving behavior and biochemical deficits. Here, we show that long-term (8 months) implantation of hNSCs into the striatum of HD zQ175 mice ameliorates behavioral deficits, increases brain-derived neurotrophic factor (BDNF) levels, and reduces mutant huntingtin (mHTT) accumulation. Patch clamp recordings, immunohistochemistry, single-nucleus RNA sequencing (RNA-seq), and electron microscopy demonstrate that hNSCs differentiate into diverse neuronal populations, including MSN- and interneuron-like cells, and form connections. Single-nucleus RNA-seq analysis also shows restoration of several mHTT-mediated transcriptional changes of endogenous striatal HD mouse cells. Remarkably, engrafted cells receive synaptic inputs, innervate host neurons, and improve membrane and synaptic properties. Overall, the findings support hNSC transplantation for further evaluation and clinical development for HD.
Keywords: Huntington’s disease, stem cells, neural stem cells, zQ175 mice, BDNF, single-nucleus RNA-seq, electrophysiology, electron microscopy
Graphical abstract
Thompson and colleagues show that long-term implantation of human NSCs in Huntington’s disease (HD) mice ameliorates behavioral deficits and reduces mutant huntingtin accumulation. The human NSCs survive and differentiate into diverse neuronal populations, form connections, improve synaptic properties, and show restoration of several transcriptional changes in HD mouse cells.
Introduction
Huntington’s disease (HD) is a devastating neurodegenerative disorder that typically strikes individuals in midlife and average survival is 15–20 years after clinical motor onset.1 HD is caused by an autosomal dominant CAG (glutamine) repeat expansion in the huntingtin (HTT) gene.2 The most notable symptoms are hyperkinetic movement disorders such as chorea and dystonia that are prominent during early stages of the disease followed by parkinsonism later on. Other symptoms include difficulties with daily tasks, cognitive decline, memory loss, and eventually dementia, as well as psychiatric manifestations including depression.3,4 Neuropathologically, the disease affects the striatum early on, although eventually the entire brain (including white matter) becomes affected. Progressive loss of medium-sized spiny neurons (MSNs) and cortical pyramidal neurons, as well as loss of cortico-striatal synapses, leads to severe atrophy.5,6 At the molecular level, the disease is accompanied by progressive transcriptional dysregulation; loss of neuronal proteins, including brain-derived neurotrophic factor (BDNF), which supports survival of striatal neurons; and aberrant accumulation of aggregated huntingtin (HTT) protein species that correspond to impaired proteostasis and disease pathogenesis.7 There are currently no US Food and Drug Administration (FDA)-approved disease-modifying treatments for HD patients that either delay onset or modify disease progression.
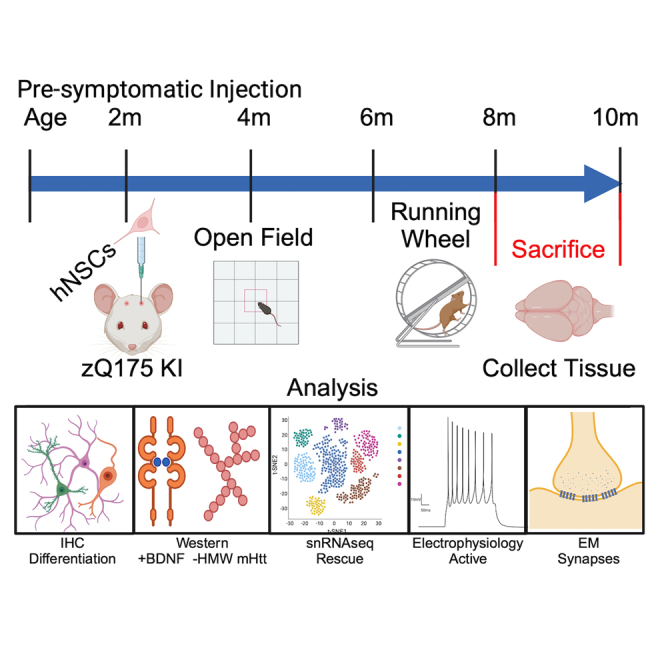
In recent years, there has been an explosion of studies in regenerative medicine for neurodegenerative and neurological diseases. Early trials of fetal cell transplantation for HD showed that the surgeries and treatments were safe, providing encouragement to pursue cell-based therapies. However, these human trials gave mixed to no signs of efficacy and variable survival of the grafts (for review, see Bachoud-Lévi et al.8), which may have been caused in part by the late stage of HD when patients were treated, immunosuppression regimens used, and the variability of the transplanted tissue or cells. Unlike the fetal cell products that were previously tested, current cell products moving into the clinic are defined cell types that can be reproducibly manufactured and show efficacy in preclinical studies. Clinical safety trials of stem cell-derived products are in various stages for Parkinson disease,9,10,11 amyotrophic lateral sclerosis,12 and epilepsy.13,14,15 The use of neural stem cells (NSCs) for the treatment of neurological disorders is in the early stages, but there is already information indicating that NSCs may offer a viable therapeutic avenue given their ability to differentiate into neurons and glia.16,17,18,19,20,21 Our previous study showed that short-term implants of embryonic stem cell-derived human NSCs (hNSCs) in HD transgenic R6/2 mice survive, are functional, and improve HD phenotypes.22,23 This work also included implants in the long-lived full-length homozygous knockin Q140 HD mouse model where we showed some behavioral improvements and reduced mutant HTT (mHTT) aggregation; however, characterization of cells was limited due to very low cell survival, perhaps caused by insufficient immunosuppression methods. Therefore, in the present study, we investigated whether implanted hNSCs could survive for an extended time period, the cell types they differentiate into in the host brain, whether cells are electrophysiologically active, whether they make connections with host cells, and whether neuroprotective effects persist. For these purposes, we used the heterozygous zQ175 mouse model, which recapitulates aspects of progressive human HD.24,25 Heterozygous zQ175 mice do not show overt behavioral symptoms until approximately 6 months of age and become fully symptomatic at 8–12 months.26 Alterations in striatal markers are observed by 12 weeks of age,25 and by 6 months of age there are extensive gene expression changes.27 Electrophysiological studies have demonstrated altered passive and active membrane properties of MSNs in symptomatic animals, as well as changes in synaptic activity.26,28,29,30,31 These functional alterations are associated with significant loss of neuronal dendritic spines. We tested viability, morphological, and electrophysiological properties of implanted ESI-017 hNSCs, hereafter designated as hNSCs, as well as their potential therapeutic benefits in zQ175 mice. The hNSCs were implanted in the striatum of pre-symptomatic mice (2.5 months), behavioral tests performed over 8 months, electrophysiological tests performed when the mice became fully symptomatic (10.5 months of age), and tissue was collected for immunohistochemical, biochemical, and morphological analyses. Finally, single-nucleus RNA sequencing (snRNA-seq) was used to determine host cell-type-specific changes elicited by hNSC transplantation 6 months post transplantation. Our data show that implanted hNSCs survive and a subset differentiate into mature MSNs and interneurons, establish connections with host neurons, rescue specific electrophysiological and behavioral phenotypes, and show restoration of several gene expression pathways across cell types.
Results
hNSCs transplanted long term in zQ175 HD model mice engraft and differentiate
We previously described the generation of a Good Medical Practice (GMP)-grade bank of hNSCs derived from embryonic stem cells that produced robust behavioral improvement in R6/2 mice, a rapidly progressing transgenic fragment mouse model of HD, and studies also showed beneficial effects in the long-lived full-length knockin Q140 HD mice.22,23 Results in Q140 mice23 used homozygous HD mice and an immunosuppression method that did not appear to allow long-term survival of hNSCs (Q140, 2 mg/kg cyclosporine [CSA] delivered by osmotic pumps vs. R6/2, 10 mg/kg CSA, daily intraperitoneally [i.p.] and CD4 Ab 10 mg/kg, weekly i.p.). Here, we sought to comprehensively evaluate whether hNSCs could survive for extended periods of time and could further differentiate. GMP-grade hNSCs were acquired as frozen aliquots (University of California, Davis [UC Davis]), thawed, and cultured as described.22,23 For immunosuppression, all mice received i.p. injections the day before surgery of CSA (10 mg/kg), daily thereafter, and mouse CD4 Ab (10 mg/kg), weekly thereafter. Mice were dosed by intrastriatal stereotactic delivery of 100,000 hNSCs per hemisphere at 2.5 months of age. To examine long-term survival of hNSCs, zQ175 mice were sacrificed at 10.5 months of age (8 months post implant). The fate of the implanted cells was determined using immunohistochemistry (IHC) with markers for human cells, neural progenitor cells, post-mitotic neurons, astrocytes, and oligodendrocytes. Implanted hNSCs survived and remained in the striatum with little migration away from the needle tract in most mice. Proliferative hNSCs implanted into mice are non-proliferative 8 months post implant as indicated by a lack of staining for the proliferation marker Ki67 or when analyzed for the incorporation of the nucleotide analog 5-ethynyl-2'-deoxyuridine (EdU) (Figures 1A and 1B). Implanted hNSCs no longer express the neural stem cell marker nestin (Figure 1C), indicating they have differentiated. A survey of cell markers revealed that the implanted hNSCs differentiated into lineages of immature neurons (doublecortin [DCX+]; Figures 1D and 1E), very few astrocytes (glial fibrillary acidic protein [GFAP+]; Figure 1C), and not into oligodendrocytes. Many cells appeared to differentiate into mature neurons (neuronal nuclei [NeuN+], Figures 1D, 1E, and 1I and BetaIII tubulin+, Figure 1G) or interneurons (calretinin [CR+], Figure 1F; glutamic acid decarboxylase 65/67 [GAD65/67+], Figure 1G; or ChAT+, Figure S1). We also detected glutamate transporter (vesicular glutamate transporter 1 [vGlut1+]) puncta surrounding implants potentially from cortical terminals (Figure 1F). Some hNSCs differentiated into MSNs (dopamine- and cyclic AMP [cAMP]-regulated neuronal phosphoprotein, DARPP-32+ and B cell lymphoma/leukemia 11B, Ctip2+, Figures 1H, 1I, and S2A). hNSCs also differentiated into cell types that exhibit inhibitory neuronal signals (gamma-aminobutyric acid [GABA+], Figure S2A). These longer-duration survival studies suggest that, given enough time, the engrafted cells are no longer proliferative and can differentiate into post-mitotic neurons typically found in striatum. The IHC data suggest that ∼58% of the human cells are NeuN positive within grafts and 7% of human cells in the implant are DARPP-32 positive.
Figure 1.

hNSCs implanted in zQ175 mice differentiate and do not proliferate
(A) Five-times magnification. hNSCs (human cytosolic marker SC121, red) in zQ175 mice do not express the proliferation marker Ki67 (green). Proliferating cells in the lateral ventricle are indicated by arrow. (B) Ten-times magnification, showing hNSCs (human nuclear marker Ku80, green) did not incorporate the nucleotide analog EdU 24 h post injection, indicating they are not dividing. (C) Twenty-times magnification shows hNSCs (Ku80, red) do not express the neuronal progenitor marker nestin (green) but some cells show expression of the astrocyte marker GFAP (blue, white arrow). The hNSC implant site is surrounded by a mouse glial cell scar (GFAP+ blue, yellow arrow). (D) hNSCs (Ku80, red) differentiate into both immature DCX+ (green) and more mature (NeuN, blue) neurons, shown at 63×. (E) Another image of hNSCs (Ku80, green) differentiating into both immature DCX+ (red) and more mature (NeuN, blue) neurons, shown at 63×. (F) Sixty-three-times magnification. hNSCs (SC121, red) differentiate into interneurons. Calretinin (green) and some vGlut1 (blue, white arrow) puncta can be observed in the implantation site. The inset image shows the entire implant at 10×. (G) Sixty-three-times magnification. hNSCs (human nuclear antigen [HNA], red) differentiate into a mixed population of cells that co-stain with GAD65/67 (green) or beta III-tubulin (blue). (H) Image shows hNSCs (HNA, green) differentiating into MSNs, DARPP-32+ (red) Ctip2+ (blue) at 60×. (I) Another 60× image showing hNSCs differentiating into MSNs using hDARPP-32+ (red) and hCtip2+ (blue) only, as well as some other hNSCs expressing the mature neuronal marker NeuN.
After analysis of multiple brain sections, we observed evidence of some cell migration on the white matter tracts between the striatum and cortex (half of the zQ175 but only three wild-type [WT] mice). In addition, a subset of the mice that displayed cell migration (one-third overall of implanted mice) exhibited nodules of cells that were positive for the human marker Ku80 adjacent to the cells in the implant site and in the ventricular space but not in the striatum. H&E stains on adjacent sections of tissue (Figures S2B and S2C) were performed and cytologically the cells in the nodules appeared to be mostly well-differentiated, mature-looking neurons. No evidence of proliferation was observed using Ki67 and nestin staining, suggesting these nodules did not have the potential for being or forming metastatic tumors. We performed IHC on another cohort of zQ175 and WT mice at 1, 2, and 5 months post implant in an attempt to evaluate the formation of nodules over time but did not observe any nodule formation. Between implantation of the original cohort and the nodule test mice, we made a minor improvement in mounting the mouse head during surgeries that may have reduced hNSC migration on white matter tracts.
hNSCs improve behavior in zQ175 HD mice
We previously established that engrafted hNSCs significantly improved multiple behavioral outcomes in R6/2 and homozygous Q140 HD model mice.23 Here, we determined the effect of long-term implantation of hNSCs on behavior of heterozygous zQ175 mice. We found strikingly significant improvements in the running wheel test in mice that were 7.5 months old (5 months post implant) in hNSC-implanted zQ175 mice compared to vehicle mice, suggesting significant prevention of the mutant HTT phenotypes and persistence of the effect (Figure 2A). The slope of motor learning was not significantly different among the three groups. In addition, we found significant improvements in distance traveled and velocity for hNSC-treated male and female mice combined compared to vehicle in the open field in mice that were 8 months of age (6 months post implant) (Figures 2B and 2C). Of note, one male and one female hNSC-implanted zQ175 mouse each performed well above average in the open field. All open field behavioral outcomes are provided in supplemental information (Figure S3).
Figure 2.

hNSCs implanted in zQ175 mice improve behavior, increase BDNF and pERK levels, and reduce levels of an HMW mHTT species
(A) Running wheel shows persistent improvement 5.5 months post treatment. Mean running wheel rotations per minute per night over 2 weeks in WT, zQ175 vehicle (Veh)-treated, or zQ175 hNSC and zQ175-Veh-treated male mice (five per group). Results are expressed as the mean ± SEM with one-way ANOVA Tukey HSD and Bonferroni post hoc tests. p values for zQ175-Veh treated to zQ175 hNSC-treated mice. Day 2 = 0.003, day 3 = 0.001, day 4 = 0.002, day 5 = 0.06, day 6 = 0.03, day 7 = 0.03, day 8 = 0.07, day 9 = 0.06, day 10 = 0.009, day 11 = 0.01, day 12 = 0.08, day 13 = 0.1, summarized as ∗p < 0.05, ∗∗p < 0.01, are shown below error bars. WT Veh to zQ175-Veh day 2 = 0.001, day 3 = 0.001, day 4 = 0.001, day 5 = 0.002, day 6 = 0.002, day 7 = 0.001, day 8 = 0.005, day 9 = 0.01, day 10 = 0.001, day 11 = 0.002, day 12 = 0.02, day 13 = 0.03, summarized as ∗∗p ≤ 0.03 at right. WT Veh to zQ175 hNSC was not significantly different at any time point. (B) Total distance traveled in the open field 6 months post implant. Mice were subjected to the open field and total distance (in centimeters) of their respective tracks were combined and statistically analyzed to visualize any differences in ambulation. The zQ175-Veh-treated mice traveled less distance than WT Veh and zQ175 hNSC-treated mice. (C) Velocity traveled in the open field 6 months post implant. Mice were subjected to the open field and velocity traveled (in centimeters per second) of their respective tracks were combined and statistically analyzed to visualize any differences in time of ambulation. The zQ175-Veh-treated mice traveled slower than WT Veh and zQ175 hNSC-treated mice. Groups for open field included seven male zQ175 Het hNSC, seven female Het hNSC, nine male zQ175 Het Veh, eight female zQ175 Het Veh, five male WT hNSC, five female WT hNSC, six male WT Veh, and six female WT Veh. Results are expressed as the mean ± SEM with one-way ANOVA Tukey HSD and Bonferroni post hoc tests: ∗p = 0.03 for genotype and p = 0.02 for hNSC treatment in zQ175. (D) Western blot analysis of whole-tissue mouse striatal lysates from zQ175 and WT mice. zQ175 mice exhibited non-significantly reduced levels of BDNF compared to WT mice and levels were significantly increased with hNSC treatment. zQ175 mice also showed significantly increased levels of pERK compared to vehicle controls. Quantitation of the relative protein expression for BDNF (E) and pERK (as the ratio of pERK to ERK) (F) is shown. (G) Western blot analysis of mouse striatal lysates separated into detergent-soluble and detergent-insoluble fractions. zQ175 mouse striatum is enriched in insoluble accumulated mHTT compared to WT mice. hNSC implantation in zQ175 mice results in a significant reduction of insoluble HMW accumulated HTT. Quantitation of the relative protein expression for mHTT is shown in (H) and hNSC implantation results in a significant decrease of insoluble HMW accumulated mHTT. Graph values represent means ± SEM and western blots were analyzed with ImageJ for quantification of BDNF (normalized to tubulin), ERK/pERK ratio or aggregate type per section. Data were analyzed by one-way ANOVA with Tukey HSD and Bonferroni post hoc tests (n = 3/group) ∗p = 0.03, ∗∗p = 0.005.
Engrafted hNSCs correlate with increased BDNF and decreased pathogenic accumulation of mHTT proteins
Increased levels of BDNF were demonstrated after hNSC implantation in the rapidly progressing R6/2 HD mouse model23; therefore, we evaluated whether this effect could be sustained following long-term engraftment in zQ175 mice. Striatal BDNF quantified by western blot analysis showed a slight but not significant genotype effect (decrease) in a subset of male zQ175 mice (n = 3/group) compared to WT. However, a significant increase in BDNF levels was observed in hNSC-treated zQ175 mice compared to vehicle with treated WT mice showing a non-significant increase (Figures 2D and 2E). We also observed an increase in the phosphorylation of extracellular signal-regulated kinase (ERK) protein suggesting potential activation of cellular trophic signaling cascades (Figures 2D and 2F), consistent with the increase in BDNF. Finally, our previous studies showed that hNSC treatment can reduce high-molecular-weight (HMW) mHTT species, a pathogenic marker for HD. Consistent with those results, we also observed persistent reduction in levels of an HMW mHTT species in hNSC-treated zQ175 mice (Figures 2G and 2H), suggesting prevention of aberrant protein homeostasis by the transplanted cells.
snRNA-seq of hNSC transplants reveals three distinct cell states of transplanted cells and rescue of HD genotype differentially expressed genes
snRNA-seq was performed on striatal tissue from female zQ175 mice implanted with hNSCs for 6 months to observe gene expression changes at a similar time frame as behavioral changes. Eight samples were analyzed with two biological replicates per condition; two genotypes, zQ175 and WT, with hNSC transplants or vehicle allowing comparison of treated and untreated mice. Single nuclei from frozen striatal samples were isolated (see section “materials and methods”) and then a Parse Biosciences kit used to fix the single-nucleus suspensions. Parse split and pool was performed, Illumina libraries prepared, and 3′-end sequencing performed. Parsebio proprietary pipeline was used for preprocessing and quality control (QC), and a gene by cell count matrix used as input for further analysis using Seurat. Cluster analysis of mouse nuclei revealed 13 unique cell clusters, which were annotated using known cell type markers as in Lim et al.32 (Figure S4). Figure 3A shows Uniform Manifold Approximation and Projections (UMAPs) of these 13 clusters with cell type labels colored by cluster (left) or genotype and treatment (right). Clear separation can be seen between WT and zQ175 vehicle-treated cells in each cell cluster; with a shift of zQ175 hNSC-treated cells compared to zQ175 vehicle treated (Figure 3A). When analyzing the human cell population, it first appeared that the isolation resulted in very few human nuclei, but, once the ratio of mouse cells to human-implanted cells was considered, the numbers were deemed to be appropriate. Three distinct clusters of human cells were indicated by cluster analysis (Figure 3B). Using cell marker analysis (Figure S5), we identified the largest cluster 0 as mainly immature or glial (astrocytes [SLC1A3, GFAP, S100B]) and neural progenitors (SOX2/5/6, PAX6, NES). Cluster 1 is mixed GABAergic, glutamatergic, MAP2 neurons, and immature neurons. Cluster 2 contained mainly neurons, most being positive for GAD1 and 2 and PVALB (GABAergic inhibitory interneurons) and some expression of DARPP-32 and DRD1, but at very low levels. UMAP of clusters showing levels of RBFOX3 encoding for the neuronal marker NeuN indicates that all clusters have some level of NeuN >0 (Figure S5). To integrate the human clusters derived from snRNA-seq with IHC described above and further define the clusters, we identified cells expressing a given IHC marker (e.g., NES, DCX, GFAP) on the three clusters (Figure S5B). Very few cells still expressed NES (identified primarily in cluster 0). Immature neurons (DCX) were primarily found within cluster 1 with some in cluster 0, consistent with staining. The relatively small number of GFAP-expressing cells are only found in cluster 0. GAD1, representing inhibitory GABAergic progenitor and mature neurons, is expressed in all clusters, consistent with staining. All three clusters express RBFOX3 (NeuN encoding), consistent with staining showing about half of the cells representing mature neurons. BDNF expression largely derives from cluster 1 (Figure S5C) and supports findings from western analysis. Summarizing the marker data in the clusters, we estimate that 0 represents 12.8% (immature or glia), 1 represents 92% (neuronal), and 2 represents 76% (primarily neuronal). These results support outcomes obtained from IHC. It is not clear why the cells tend to differentiate along a neuronal lineage over astrocytes and oligodendrocytes; possibilities include the HD mouse niche itself or differentiation propensity of the NSCs themselves derived from the ESI-017 cells.
Figure 3.

snRNA-seq of hNSC transplants reveals three distinct cell states of transplanted cells and rescue of HD genotype DEGs
(A) Uniform Manifold Approximation and Projection (UMAP) projection of 38,892 WT and zQ175 mouse nuclei from 13 major cell populations. UMAPs of the mouse cells forming 13 clusters with clusters colored by cell type annotation (left) or genotype and treatment combination for each cell (right). Within each cell type, gene expression differences were visible by shifts in shifts in cell populations of zQ175 and WT as well as treated and vehicle samples. (B) UMAP projection of transplanted human nuclei forming three clusters. Cell clusters were color coded by cluster ID. (C) Scatterplot showing per-cell-type Z score log2 fold change of genotype DEGs against treatment DEGs (het_treated versus het_vehicle). DEGs are color coded by cell type origin. Red line slope = 1 and blue line = Locally estimated scatterplot smoothing (LOESS) fit to data points.
Genotype differentially expressed genes (DEGs) were generated per cell type comparing zQ175 vehicle-treated cells against WT vehicle-treated cells (Table S1). These DEGs represent the HD genotype effect and showed dysregulation of genes known to be altered in this mouse model from multiple datasets.33 To assess how transplantation of the hNSCs may change the genotype effect on gene expression and whether the gene expression changes in the treated cells were rescuing or exacerbating the genotype changes, we generated DEGs for the effect of treatment on gene expression in the zQ175 Het mice (zQ175 hNSC-treated versus zQ175 vehicle Table S2) and compared changes that overlapped with genotype DEGs (e.g., genes that are altered in zQ175 vehicle mice compared to WT vehicle; Table S3). Figure 3C shows that, in most cases, the effect of treatment on overlapping genotype DEGs was to reverse/rescue the expression of that gene. We used Ingenuity Pathway Analysis (IPA) enrichment analysis to determine the functional impact of this rescue and found that the treatment effect also reverses/rescues the pathway activation Z score in most cases, showing rescue in pathways such as synaptogenesis, calcium transport, and axonal guidance signaling (Figure 4A). We also asked whether gene expression from the mouse and human clusters might inform the nature of cell-cell interactions between the host and grafted cells. We therefore carried out CellChat analysis34 to investigate ligand-receptor interactions. Chord plots were used to visualize the inferred interactions, which showed extensive communication between mouse and human cells. Two examples show interactions from both mouse and human cell clusters or primarily from human clusters (Figure 4B) and these cell cell interactions can be interactively explored using an R shiny based web interface at database: https://ucightf1.biochem.uci.edu/cellchatshiny/nsc/. For example, the neuronal growth regulator (NEGR) signaling pathway (Figure 4B, left panel), which functions as a trans-neural growth-promoting factor and is related to neuronal connectivity,35 shows that signals are coming from human cluster 1 (H1, primarily neurons) to mouse clusters 0, 1, 5, and 6 (D1, D2, and inhibitory neurons, blue lines) and in terms to H1 from mouse clusters M0, 1, 5, and 6, suggesting dynamic signaling. In contrast, the WNT signaling pathway, which has been implicated as dysregulated in HD,36,37,38,39 shows all three human clusters sending signals to most of the mouse clusters (except M3 and 4, which are oligos and astrocytes, respectively) (Figure 4B, right panel). Further, activation of WNT can restore neuronal circuits after synapse degeneration.40 Taken together, these results show that the transplanted hNSCs differentiate into a mixed population of cells mainly composed of neuronal-like cells, which can rescue some of the HD gene expression differences in zQ175 mouse cell types and communicate with the endogenous mouse cells.
Figure 4.

snRNA-seq of hNSC transplants reveals rescue of HD genotype DEGs
(A) Dot plot showing significant IPA pathways and predicted activation Z scores per cell type for genotype and treatment DEGs. (B) Chord diagrams from CellChat analysis showing all the significant ligand-receptor interactions in NEGR (left) and WNT (right) signaling pathway between the cell types from the two zQ175 mouse samples treated with hNSC. H0, H1, H2 are the three human cell clusters and M0–M12 are 13 mouse clusters identified from the corresponding UMAPs (Figures 3B and 3A, respectively). Edges are colored the same as source as senders. The inner thinner bar colors represent the targets that receive signal from the corresponding outer bar. A key is shown for mouse clusters.
Electrophysiological and morphological characterization of hNSCs
hNSCs transplanted for 4 weeks in R6/2 mice showed properties of immature, electrophysiologically active hNSCs23; however, we did not know whether this could be sustained or improved in long-term implanted mice. To perform electrophysiological studies, we used a subset of female zQ175 and WT mice (10.5 months of age, 8 months post implant) implanted at University of California, Irvine (UCI) and shipped live to University of California, Los Angeles (UCLA). Previous studies in zQ175 mice did not show significant differences in intrinsic or synaptic membrane properties between male and female mice.26,29 The hNSC grafts in zQ175 mice were easily identifiable under infrared illumination with differential interference contrast (IR-DIC) microscopy (Figures 5A1 and A2). In contrast to host tissue, which appeared darker due to myelin from fiber tracks, the graft appeared more translucent and densely populated by diverse cell types. In agreement with IHC findings, most cells (∼80%) within the graft sites were small (<15 μm in diameter), round or bipolar, and had few extended processes. These cells appeared to be visually similar to the immature neuronal cell types previously recorded in R6/2 mice 4–6 weeks after implantation.22,23 We also observed a number of cells that were larger in size (15–25 μm in diameter) with abundant and extensive processes that were visually different from host MSNs. As immature-looking engrafted cells were characterized extensively in R6/2 mice in our previous publication,23 in the present study we focused our recordings on these larger, more mature-looking cells. We found for all engrafted cell types that membrane properties were similar for cells recorded in either zQ175 or WT mice and data from recorded engrafted cells from both genotypes were pooled (Figures S6A and 5B).
Figure 5.

Two main types of hNSCs could be distinguished based on soma size and passive and active membrane properties
(A1) IR-DIC image from transplanted hNSCs in a zQ175 mouse revealed that most cells had small, round somata. (A2) A small percentage of hNSCs had larger somata and some dendritic branches. A patch pipette attached to the cell can be seen. Example cell is from a zQ175 mouse. (B) Bar graphs show mean ± SEM of cell membrane properties of host MSNs (WT and zQ175 pooled together) compared to implanted hNSCs. Capacitance, input resistance, and time constants are shown for recorded host MSNs and mature and immature hNSCs. Statistical significance was measured using one-way ANOVA tests followed by Bonferroni post hoc tests for pairwise comparisons; ∗∗∗p < 0.001. (C1) Recordings of intrinsic currents in response to step voltage commands (10-mV steps from −80 to +10 mV) in a host MSN (left) and in an MSN-like (center) and an interneuron-like (right) hNSC. Depolarizing voltage commands induced large Na+ currents followed by inactivating Ca2+ currents (arrows) of variable amplitudes. In the interneuron-like cell, repetitive spikes were observed but no prominent Ca2+ currents. (C2) Recordings of currents in response to a ramp voltage command (8 s, from −90 to +50 mV). Some hNSCs recorded (center) had properties similar to host MSNs, both displaying Ca2+ currents (black arrows) after membrane depolarization. Other large hNSCs recorded lacked Ca2+ currents but displayed repetitive Na+ spikes (right) and were probably interneurons. (C3) Spontaneous synaptic currents recorded at +10 mV in a host MSN and in MSN-like and interneuron-like hNSCs. These currents are mostly GABAergic. (C4) Spontaneous synaptic currents recorded at −70 mV in a host MSN and in an MSN-like and an interneuron-like hNSC. These currents are most likely glutamatergic. Traces in each column are from the same cell. Calibrations on the right apply to all traces in each row. (D) Upper panels show low-magnification images of several hNSCs. Arrows (yellow) indicate two cells that displayed MSN-like electrophysiological properties. Lower panels show processes from the same cells at higher magnification. Arrows (white) indicate possible dendritic spines. (E, A) Four medium to large hNSCs were recorded within the graft. (E, B) The same image showing staining for the human markers SC121 (cytoplasmic, green) and Ku80 (nuclear, blue). (E, C) Fluorescent image of one of the large biocytin-filled hNSCs. (E, D and E) Immunostaining of the same hNSC with human stem cell markers SC121 (arrow) and Ku80. (F) Merged image showing biocytin and the two human stem cell markers. The electrophysiology of this interneuron-like hNSC is shown in (C1) and (C2), right panels. (B) Capacitance: p = 2.0007e−05 for MSN versus hNSC Na+ < 1,000 pA and p = 6.1282e−08 for hNSC Na+ > 1,000 pA versus hNSC Na+ < 1,000 pA. Input resistance: p = 5.3000e−06 for MSN versus hNSC Na+ > 1,000 pA and p = 2.6645e−05 for MSN versus hNSC Na+ < 1,000 pA. Time constant: p = 8.9566e−05 for MSN versus hNSC Na+ < 1,000 pA and p = 1.7886e−05 for hNSC Na+ > 1,000 pA versus hNSC Na+ < 1,000 pA.
Recorded cells in the grafts appeared to differentiate into two groups: one population of cells with immature-like electrophysiological properties and one population with more mature-like properties (Figure S6A). In total, 49 engrafted cells were recorded (n = 18 in WT and 31 in zQ175 mice). The two groups of engrafted cells were recorded in voltage-clamp mode and separated based on Na+ current amplitudes. Passive membrane properties (cell membrane capacitance, input resistance, and decay time constant) differed between the two engrafted cell groups. Cells displaying Na+ current amplitudes <1 nA (n = 9; three in WT and six in zQ175 mice) were immature looking, had small membrane capacitances (mean ± SEM, 18.6 ± 2.2 pF; range 9–26 pF), high input resistances (847.3 ± 270.0 MΩ; range 92–2400 MΩ), and rapid time constants (0.51 ± 0.08 ms; range 0.25–0.9 ms) (Figure 5B). A number of these cells did not display Na+ currents and were likely very immature neuronal or glial cells (n = 2 in WT and n = 5 in zQ175 mice). In contrast, engrafted cells that displayed Na+ current amplitudes >1 nA (n = 40, 15 in WT and 25 in zQ175 mice) were mature looking had larger membrane capacitances (mean ± SEM, 128.5 ± 10.0 pF; range 37.8–340 pF), medium-high input resistances (625.1 ± 64.6 MΩ; range 142–1800 MΩ), and slower decay time constant (2.2 ± 0.2 ms; range 0.7–5.2 ms) (Figure 5B). Cell membrane properties of engrafted cells in WT or zQ175 mice are shown in Figure S6A.
To further characterize maturation and differentiation of hNSCs, we compared their basic membrane properties with those of MSNs recorded from the host (Figures 5B and 5C). Based on previous studies, the basic cell membrane properties (capacitance, input resistance, and time constant) of direct and indirect pathway MSNs are indistinguishable. Except for subtle differences in synaptic properties, overall it is very difficult to differentiate both types of neurons electrophysiologically. For this reason, in the present study we did not attempt to separate host MSNs into two classes.41,42 Cells with Na+ currents >1 nA had membrane capacitances similar to or even larger than that of host MSNs. In contrast, cells with small-amplitude Na+ current amplitudes of <1 nA had very small membrane capacitances, similar to recordings from immature neurons. Both types of engrafted cells had relatively high membrane input resistances compared with MSNs. In addition, differences in decay time constants were similar to those observed for cell capacitance. About 50% of recorded engrafted cells lacked inward Ca2+ currents (Figures 5C1 and C2 right panel; Figure S6B), usually visible with Cs+-based internal solution after depolarizing voltage commands, suggesting that these cells were not projection MSNs. However, the remaining recorded engrafted cells (n = 10 in WT mice and n = 14 in zQ175) displayed inward Ca2+ currents. Some of these cells displayed Ca2+ currents larger in amplitude and similar to those observed in host MSNs (n = 5 in WT mice and n = 6 in zQ175) (Figures 5C1 and C2 left and middle panels). The intrinsic membrane properties of these engrafted cells were not significantly different regardless of the mouse genotype (Figure S6C). These cells also displayed frequent spontaneous synaptic activity and could represent projection neurons with the potential to connect with other cells inside and outside the graft. Biocytin labeling revealed these cells had abundant dendritic processes and sparse spines (Figures 5C3 and C4 middle panel; Figures 5D and 5E).
The remainder of engrafted cells did not display Ca2+ currents. Instead, they displayed large inward Na+ and outward K+ currents and could fire repetitive action potentials. We were able to recover ∼50% of biocytin-filled hNSCs (9 out of 18 or 50% and 15 out of 31 or 48% recorded in WT and zQ175 mice, respectively). Importantly, over 80% of those recovered clearly co-immunostained with SC121 and Ku80 markers (8 out of 9 or 89% and 12 out of 15 or 80% in WT and zQ175 mice, respectively), thus confirming their human origin. Post-recording immunostaining with SC121 and Ku80 of biocytin-filled large, engrafted cells (visualized with streptavidin-Alexa 594) in fixed slices revealed engrafted cells with relatively large somata (compared to host MSNs) with cell diameters of up to ∼25 μm and extensively branched processes. Other visible engrafted cells were smaller in size and had either many or a small number of processes (Figures 5D and 5E). Another important difference was that, compared with host MSNs or MSN-like engrafted cells, many large, engrafted cells fired spontaneously. In cell-attached recording mode, cells fired rhythmically and received rhythmic GABAergic synaptic inputs (Figure S7A). We tentatively concluded these inputs were GABAergic because the GABA reversal potential was around −60 mV (Figure S7B). Due to this rhythmic input, one possibility is that the source of this GABA input is from other mature hNSCs, as engrafted cells remained in close proximity. In addition, host GABAergic interneurons, such as the somatostatin-expressing or neuropeptide Y (NPY)-expressing interneurons that fire spontaneously, may contribute to these rhythmic inhibitory events recorded in engrafted cells. Another type of recorded hNSC (n = 3) resembled low-threshold spiking (LTS) striatal interneurons (Figure S8). In current clamp mode, these cells displayed prominent delayed rectification at hyperpolarized membrane potentials (Figure S8B). When depolarized, they discharged in bursts of action potentials, seemingly riding on a low-threshold Ca2+ spike followed by a membrane hyperpolarization, which produced spontaneous oscillations and bursts of action potentials (Figure S8C). The last type of engrafted cell (n = 2) resembled cholinergic (ChAT-expressing) interneurons, also known as striatal tonically active neurons (TANs). They displayed rhythmic firing (2–3 Hz) and prominent delayed inward rectification. Electrophysiological identification of these interneurons was supported by IHC detection of appropriate markers (Figures S9A–S9C).
Synaptic properties of implanted hNSCs compared with host MSNs
Glutamatergic inputs onto hNSCs were examined by holding the membrane at −70 mV. GABAergic inputs were examined by holding the membrane at +10 mV. Immature hNSCs displayed very few synaptic inputs, whereas mature hNSCs had a wide range of synaptic inputs, some with frequencies as high as those recorded from MSNs (2.1 ± 0.2 Hz [range 0.2–6.9 Hz] for MSNs versus 1.5 ± 0.2 Hz [range 0.0–8.7 Hz] for hNSCs). Based on the frequency of spontaneous synaptic activity, cells displaying Na+ currents could be divided into those with high inhibitory postsynaptic current (IPSC) and high excitatory postsynaptic current (EPSC) frequencies, high IPSC and low EPSC frequencies, and low IPSC and high EPSC frequencies (Figure 6A). As expected, large, engrafted cells displayed higher frequencies than those of immature-looking engrafted cells (Figures 6B1 and 6B2). Some engrafted cells (n = 2) also were tested for their ability to respond to electrical stimulation in the vicinity of the graft. Both cells responded to the electrical stimulation displaying glutamatergic and GABAergic responses, as demonstrated by specific blockade with appropriate receptor antagonists (Figure 6C). Thus, these studies provided evidence that engrafted cells establish synaptic contacts with the host and probably among other implanted hNSCs as well.
Figure 6.

hNSCs in WT and Q175 mice display multiple synaptic inputs
(A) Sample traces of sIPSCs and sEPSCs recorded from hNSCs in a zQ175 mouse in ACSF. (B1 and B2) Summary of the sIPSC and sEPSC frequencies from MSNs and hNSCs categorized by the size of their Na+ currents. Statistical significance was measured between groups using Kruskal-Wallis one-way ANOVA on ranks followed by Holm-Sidak pairwise comparisons; ∗p = 0.024 for zQ175 Na+ > 1,000 pA versus WT Na+ < 1,000 pA. (C) Responses evoked by electrical stimulation (0.1–0.5 mA, 1-ms duration) of host striatal neurons (about 200 μm lateral to the graft) in a hNSC from a zQ175 mouse. Glutamatergic (Vh = −70 mV) and GABAergic (Vh = +10 mV) responses were reliably evoked by electrical stimulation. Responses were blocked by glutamate and GABAA receptor antagonists respectively. (D) Cell membrane properties recorded at a holding potential of −70 mV. (E) Fluorescent image of a recorded and biocytin-filled MSN (yellow arrow pointing to filled neuron [red]) near SC121-immunostained cells and processes (green). (F1 and F2) Effects on inhibitory (left) and excitatory (right) synaptic activity. Statistical significance was measured between groups using Kruskal-Wallis one-way ANOVA on ranks followed by Holm-Sidak pairwise comparisons, In (F), ∗∗p < 0.001 for WT versus zQ175-Veh and ∗p = 0.04 for zQ175-Veh versus zQ175-hNSC. In (G), ∗p = 0.047 for WT versus zQ175-Veh.
hNSCs improve some altered intrinsic and synaptic membrane properties of MSNs in host zQ175 mice
Given that we have previously shown that excitatory and inhibitory inputs to striatal MSNs and cortical pyramidal neurons in the zQ175 mouse model are altered,29 we obtained whole-cell voltage-clamp recordings to measure membrane and synaptic properties of neighboring host MSNs to determine whether transplanted hNSCs conferred modulatory outcomes (Figures 6D–6F). Data from WT mice implanted with hNSCs and injected with vehicle only were pooled as there were no consistent differences in measures from MSNs between the two groups (Figure S6D). We observed an improvement in the cell membrane properties of zQ175 MSNs from hNSC-treated mice compared to zQ175 MSNs from mice receiving the vehicle only (Figure 6D). Previously, we showed MSNs from symptomatic zQ175 mice have higher membrane input resistances than MSNs from WT mice.29 Compared with WT MSNs, zQ175 MSNs from vehicle-treated mice had significantly higher input resistances (WT 91.6 ± 10.1 versus zQ175-Veh 170.2 ± 19.7 MΩ, p < 0.001; Kruskal-Wallis ANOVA of ranks, followed by Holm-Sidak pairwise comparisons). Although input resistances were slightly higher in zQ175-hNSC MSNs (120.1 ± 13.1 MΩ) compared to WT MSNs, this difference was not statistically significant demonstrating improvement in this electrophysiological property. Input resistances were significantly lower in MSNs from hNSC-treated zQ175 mice compared to MSNs from vehicle-treated mice (p = 0.04). There were no significant differences in cell membrane capacitances or membrane time constants across all three groups (Figure 6D).
In terms of hNSCs’ effects on synaptic activity, as reported previously, in zQ175 MSNs29 the frequency of spontaneous IPSCs (sIPSCs) recorded at a holding potential of +10 mV was increased compared to WT MSNs (Figure 6F1). The increase was statistically significant in MSNs from zQ175 mice treated with vehicle (WT 1.3 ± 0.2 versus zQ175-Veh 2.2 ± 0.3 Hz, p = 0.047; Kruskal-Wallis ANOVA of ranks, followed by Holm-Sidak pairwise comparisons) and, although the sIPSC frequency was slightly higher in MSNs from hNSC-treated zQ175 mice (2.0 ± 0.3 Hz) compared to WT MSNs, it was not statistically significant (p = 0.114) (Figure 6F1), demonstrating that the transplant reduced the increase in sIPSCs in MSNs from the zQ175 mice compared to WT mice. From the same cells, we recorded spontaneous EPSCs (sEPSCs) at a holding potential of −70 mV and in the presence of a GABAA receptor antagonist (bicuculline methobromide [BIC], 10 μM). We observed a trend for decreased sEPSC frequency in MSNs from zQ175-Veh mice (1.8 ± 0.3 Hz) compared to WT MSNs (2.8 ± 0.5 Hz). Similarly, there was a trend for an increase in the frequency of sEPSCs in MSNs from the hNSC-treated zQ175 mice (2.4 ± 0.5 Hz) (Figure 6F2).
Taken together, these data show long-term survival and differentiation of hNSCs into mainly neuronal lineages including a subset of mature-like MSNs and interneurons. The engrafted human cells establish connections with the host neurons and rescue specific electrophysiological and behavioral pathologies.
Ultrastructural evidence that hNSCs establish synaptic contacts within and outside the graft
To examine the morphology of implanted hNSCs and determine whether synaptic contacts are present between the host and transplanted hNSCs, thus further supporting the electrophysiology results, we performed electron microscopy (EM). EM studies provided additional anatomical evidence that hNSCs in the graft received innervation from host neurons. A subset of the mice implanted at UCI (three females per group, zQ175 hNSC implanted, 10.5 months of age, 8 months post implant) were sent live to the Portland VA Medical Center where the mice were perfused with fixative and the brains were collected and fixed for EM tissue processing. Results indicate that mouse host cell nerve termini make both symmetrical and asymmetrical synaptic contacts with implanted hNSCs. As a proof of principle, in a subset of samples we performed double-immunostaining for vGlut1 (which labels cortical glutamate terminals) and SC121. There were mouse host cell nerve termini that were positively labeled with vGlut1 making asymmetrical synaptic contacts with engrafted cells (Figure 7A), suggesting that mouse (host) cortical neurons contribute to these connections. However, it is also possible that the other major host nerve terminal input to the hNSC-implanted neurons, where an asymmetrical synaptic contact is observed (Figures 7D and 7E), may be from the thalamus. Figures 7B and 7C shows unlabeled nerve termini making asymmetrical synaptic contacts with either an underlying engrafted cell-positive labeled dendrite (Figure 7B) or a labeled dendritic spine (Figure 7C). The unlabeled nerve terminal might originate from either the host striatum, thalamus, or cortex, while the labeled dendrite/dendritic spine originates from the implanted stem cells. Figure 7F shows a labeled nerve terminal making a symmetrical synaptic contact with an underlying engrafted cell-positive/labeled dendrite. The labeled nerve terminal and dendrite originate from the implanted stem cells. Within the implant, of the host (non-labeled) terminals contacting SC121+ dendrites/spines in the implant site, 60% of the asymmetrical contacts were on dendrites and 40% on spines (total of 35 observations), suggesting contacts with MSN-like engrafted cells. Of the host contacting SC121+ labeled dendrites, if the synaptic contact was symmetrical, 100% of those contacts were on the dendrite (22 observations). Of all the host (non-labeled) contacts onto SC121+ labeled cells in the implant area, 62.1% were asymmetrical while 37.9% of the contacts were symmetrical (total of 58 observations).
Figure 7.

EM studies revealed that the host tissue makes synaptic contacts with hNSCs in zQ175 mice within the implantation site
(A) hNSC/ diaminobenzidine (DAB)-labeled nerve terminal (L-NT) making a symmetrical synaptic contact (single white arrow) with an hNSC/DAB-labeled dendrite (L-DEND). vGlut1-L-NT (vGlut1 NT) making an asymmetrical synaptic contact with the head of a stubby spine that contains a perforated postsynaptic density (two white arrows pointing to synaptic contact). The vGlut1 NT (purple arrowheads pointing to VIP labeling) is distinguished from the adjacent nerve terminal (which is only DAB labeled). The primary origin of vGlut1-containing neurons is the cortex. (B) An unlabeled nerve terminal (U-NT) making an asymmetrical synaptic contact (arrow) with an underlying hNSC-positive labeled dendrite (L-DEND) (note the darkened DAB reaction product within the dendrite). The U-NT might originate from either the host striatum, thalamus, or cortex (see A), while the labeled dendrite originates from the implanted stem cells. (C) A U-NT is making an asymmetrical synaptic contact (arrow) with an underlying hNSC-positive/labeled dendritic spine (L-SP) (note the darkened DAB reaction product within the spine). As in (B), the U-NT might originate from either the host striatum, thalamus or cortex (see A), while the labeled dendritic spine originates from the implanted stem cells. (D) An L-NT is making an asymmetrical synaptic contact (arrow) with an underlying hNSC-positive/labeled dendrite (L-DEND) (note the darkened DAB reaction product within the nerve terminal and dendrite). The L-NT and dendrite originate from the implanted stem cells. (E) An L-NT is making an asymmetrical synaptic contact (arrow) with an underlying hNSC-positive/L-SP (note the darkened DAB reaction product within the nerve terminal and spine). The L-NT and spine originate from the implanted stem cells. (F) An L-NT is making a symmetrical synaptic contact (arrow) with an underlying hNSC-positive/L-DEND (note the darkened DAB reaction product within the nerve terminal and dendrite). The L-NT and dendrite originate from the implanted stem cells.
Within the implant, the percentage of SC121+ nerve terminals contacting SC121+ dendrites was determined. We found that, of those total synaptic contacts, 71.2% were making an asymmetrical synaptic contact, with 27.8% making a symmetrical contact (total of 115 observations). About 1% of the contacts could not be determined as to whether they were symmetrical or asymmetrical. Of those 71.2% of the contacts that were asymmetrical, 36.5% of those were on dendrites, while 34.7% were on spines. Of the 27.8% of the contacts that were symmetrical, 21.7% were on dendrites and 6.1% were on spines.
Investigating synaptic contacts outside the implant area (located ∼0.25 mm lateral of the implant site), of the percentage of SC121+ labeled nerve terminals contacting SC121 negatively labeled postsynaptic dendrites (i.e., from the host), 90.6% were making an asymmetrical synaptic contact while 9.4% were making a symmetrical contact (total of 32 observations). Of the 90.6% making an asymmetrical synaptic contact, 79.3% were contacting spines while 20.3% were contacting dendrites. There were also nerve terminals from the host striatum (i.e., SC121 negative) contacting SC121+ dendrites. Of those contacts, 52.6% were asymmetrical and 47.4% were making a symmetrical contact (1% could not be determined) (total of 19 observations) (Figure 8).
Figure 8.

Striatal synaptic contacts located outside the stem cell implantation zone
(A) An L-NT is making either an asymmetrical synaptic contact (black arrow) or a symmetrical contact (white arrow) with an underlying hNSC-negative/unlabeled dendritic spine (U-SP) (note the darkened DAB reaction product within the nerve terminals). The hNSC-L-NTs originate from the implanted stem cells, while the unlabeled dendritic spines originate from the host striatum. (B) U-NTs are making either an asymmetrical synaptic contact (white arrow) or a symmetrical contact (black arrow) with an underlying hNSC-positive/L-DEN (note the darkened DAB reaction product within the dendrite). The hNSC-L-DEN originates from the implanted stem cells, while the U-NTs originate from the host striatum. (C) U-NT making an asymmetrical synaptic contact (arrow) with an underlying hNSC-positive/L-DEND (note the darkened DAB reaction product within the dendrite). The hNSC-L-DEN originates from the implanted stem cells, while the U-NT originates from the host striatum. (D) An L-NT is making a symmetrical synaptic contact (black arrow) with an underlying hNSC-positive/ L-SP (note the darkened DAB reaction product within the nerve terminal and spine). The hNSC-L-NT and dendritic spine originate from the implanted stem cells.
Discussion
A major challenge in evaluating regenerative medicine approaches to treat neurodegenerative diseases, including HD, is enabling long-term assessment of cell fate, functional properties, and potential rescue of disease-associated phenotypes.43,44 Here, we evaluated whether hNSCs implanted in the striatum of zQ175 mice are viable and integrate into the host tissue after 8 months (about one-third of a 2-year lifespan). hNSCs survived, differentiating primarily along a neuronal lineage, including to resident neuronal populations in the striatum (MSNs and GABAergic and cholinergic interneurons). Implant cell fate appears to favor a neuronal developmental path in contrast to gliogenic, potentially due to the differentiation potential of the starting material or the transplantation niche.45,46,47 Most differentiated hNSCs had electrophysiological properties of immature neurons, but approximately 20% differentiated into electrophysiologically mature neurons with MSN- or interneuron-like properties. Transplanted hNSCs received synaptic inputs from neighboring human or host cells and innervated them. Notably, engrafted hNSCs increased striatal BDNF and pERK levels, reduced mHTT aggregated species, and ameliorated selected behavioral deficits of symptomatic zQ175 mice. Several HD genotype transcriptional changes were normalized to WT levels in mouse striatal cells when profiled using snRNA-seq. Further, grafted cells modified host MSNs and rescued some of the altered membrane and synaptic properties of symptomatic mice. Thus, the mechanisms whereby implanted hNSCs rescue HD alterations appear to involve improved striatal MSN membrane properties and circuit connectivity, BDNF production, and prevention of the accumulation of mHTT aggregating species. A comprehensive reconstruction of the striatal circuitry would require MSNs and a wide variety of interneurons, which also are significantly affected in HD.48,49,50,51,52 We speculate that using a progenitor population for implants offers the potential to populate areas of degeneration with more than one cell type, which may provide multiple benefits, and that even relatively immature cells have the potential to rescue some phenotypes, including through secretion of trophic factors.
EM and electrophysiology also suggest connections from the host originating in the cortex may support a potential reconnection of the cortico-striatal pathway, lost during HD progression, and reconnection could contribute to restoration of normal motor and cognitive functions. Data from snRNA-seq support potential connectivity showing rescue of synaptogenesis and axonal guidance pathways and cell-cell interactions between ligands and receptors. We hypothesize that the higher frequencies of synaptic inputs are due to the maturation and integration of hNSCs within the host tissue, as we did not observe engrafted cells with these synaptic properties in the shorter-term study of R6/2 hNSC-implanted mice.23
Electrophysiologically, ∼80% of engrafted cells remained immature, and it is unknown why these cells do not differentiate further. These cells could follow a human developmental time frame and with more time may mature.53 In the zQ175 mice, we found both the post-mitotic neuronal marker NeuN, as well as interneuron and MSN markers, but still observed clusters of implants to be DCX positive. Importantly, engrafted cells stop proliferating (no Ki67 or EdU incorporation) and are on a path to differentiate (e.g., loss of nestin). About 20% of the large, engrafted cells display electrophysiological properties of mature neurons, and evidence provided by IHC, snRNA-seq, and the presence of specific striatal neuronal markers supports this conclusion. MSN-like engrafted cells displayed Ca2+ currents typically observed in mature MSNs and IHC demonstrated the presence of DARPP-32 and Ctip2, which label striatal MSNs. Other engrafted cells fired rhythmically, had increased input resistances (compared to MSNs), lacked characteristic Ca2+ currents, and received rhythmic GABAergic synaptic events, suggesting that these cells could be GABAergic interneurons; IHC and RNA expression from these grafts indicated the presence of GAD and CR, specific interneuron markers. Some large engrafted cells also displayed rhythmic bursting and low-threshold spikes, reminiscent of the somatostatin-expressing (LTS) interneurons,51,54 significant in the context of HD because these two interneuron subtypes are spared during disease progression. In fact, CR+ interneurons appear increased in HD,55 suggesting neuroprotective properties or positive selection within the HD niche. Somatostatin and CR are neuroprotective56 and a study on grafted fetal striatal tissue demonstrated the presence of graft-derived neurons expressing DARPP-32, CR, and somatostatin.57 Some large engrafted cells may have evolved into NPY-expressing interneurons; in HD, MSNs expressing NPY are spared, and their numbers are even upregulated in HD patients.58 Based on passive and active membrane properties, some cells also differentiated into large cholinergic interneurons. Although no fast-spiking, parvalbumin-expressing interneurons were recorded electrophysiologically, at least in a mature state, this type of striatal GABAergic interneuron may also exist in the graft based on our snRNA-seq data.
Recent studies using a rat quinolinic acid model of HD showed that human embryonic stem cell-derived MSN progenitors differentiate in vitro, undergo maturation, improve behavior, integrate into host circuits, and display properties similar to those of the host striatum 2 and 6 months after transplantation.59,60 In addition, those studies showed that some cells were proliferative, the yield of DARPP-32/Ctip2 double-labeled MSNs was relatively low, and there was some degree of contamination from cortical neurons.60 Interestingly, like us, they also saw a low percentage of grafted cells expressing interneuronal markers calbindin and CR; however, the cells were not characterized electrophysiologically. It should be noted that excitotoxicity models may not reflect the same biochemical environment as the genetic mutation. Other strategies to generate striatal neurons for implant are in progress.61,62 A recent study reprogramed striatal astrocytes into GABAergic neurons through adenoassociated virus (AAV)-mediated ectopic expression of NeuroD1 and Dlx2 transcription factors.63 The striatal astrocyte-converted neurons showed action potentials and synaptic events and projected their axons to appropriate target regions in R6/2 mice with a significant extension of life span and improvement of motor deficits.63
We did not determine if grafted hNSCs send projections to the output nuclei of the basal ganglia (i.e., external globus pallidus and substantia nigra pars reticulata). However, improved behavior after transplantation suggests that connections to output nuclei may occur. A recent study supports that conclusion showing that hPSC-derived neural progenitor cells implanted in the mouse striatum establish synaptic connections onto MSN targets.64
It could be argued that our implantations during the pre-symptomatic stage, when the striatum is relatively healthy, diverge significantly from clinical scenarios. However, one of the objectives that drives cell therapies is to proactively prevent, stall, or decrease the emergence of deleterious symptoms through early intervention to protect cells from degeneration and premature death. Furthermore, the track record of therapeutic endeavors embarked upon during advanced symptomatic stages in HD have been largely negative. Given the gradual progression and hereditary nature of HD, the administration of treatments in the early stages could yield considerable benefit. There are a number of challenges inherent in human clinical development of cell-based therapies, including surgical and delivery methods, placement of implants to optimize coverage, advantages of different cell types for transplantation, and clinical trial design; these are discussed in a recent review of the challenges for cell-based therapies.65
In conclusion, our studies support future development of stem cell-based therapies. The present results show that implanted cells may provide trophic effects through enhanced BDNF levels and reduction of pathological mHTT, may compensate for transcriptional changes, and that engrafted cells establish synaptic contacts with themselves and host cells, differentiate into striatal resident cells, and thus form the building blocks for circuit regeneration. Given the electrophysiological and EM results, there is also promise that transplanted cells may make beneficial synaptic connections. Future studies will explore ways in which this approach may be utilized clinically.
Materials and methods
Cells
The use of hESCs and hNSCs was approved by Human Stem Cell Research Oversight Committees (hSCRO) at UCI, UCLA, and UC Davis. ESI-017 is one of the six clinical-grade hESC lines generated from supernumerary embryos by the Singapore Stem Cell Consortium.66 Their use for therapeutic application adheres to US FDA regulations for use of human cells. Of those lines, four (including ESI-017) were chosen for the generation of GMP hESC banks for preclinical research based on the absence of human and non-human pathogens.66,67 Subsequently, an hNSC line was differentiated from the GMP-grade hESC line ESI-017 as described previously.23 ESI-017 hNSCs were acquired as frozen aliquots, thawed, and then cultured for a minimal time out of thaw (2–3 days) using the same media reagents as the GMP facility prior to dose administration. The cells were not passaged.
Mice
All experimental procedures were in accordance with the Guide for the Care and Use of Laboratory Animals of the NIH and animal protocols were approved by Institutional Animal Care and Use Committees at UCI, UCLA, and the Portland VA Medical Center (Association for Assessment and Accreditation of Laboratory Animal Care [AAALAC]-accredited institutions). zQ175 heterozygous (Het) mice and their WT littermates were obtained from breeding colonies maintained at UCI (zQ175 Het mice had ∼163–199 CAG repeats, Laragen, Culver City, CA). All mice were housed on a 12/12-h light/dark schedule with ad libitum access to food and water. Mice were group housed as mixed treatment groups and only males were single housed for the running-wheel test. Groups included 10 male zQ175 hNSC, eight female zQ175 hNSC, nine male zQ175 vehicle, nine female zQ175 vehicle, seven male WT hNSC, seven female WT hNSC, six male WT vehicle, and six female WT vehicle.
Surgery
Two-and-a-half-month-old zQ175 Het mice and WT littermates were anesthetized, placed in a stereotaxic frame, and injected with either 100,000 hNSCs per side (2 μL/injection) or vehicle (2 μL Hank’s balanced salt solution [HBSS] with 20 ng/mL epidermal growth factor [STEMCELL Technologies, #78003] and human fibroblast growth factor [STEMCELL, #78006]) as a control treatment using a 5-μL Hamilton microsyringe (33G) and an injection rate of 0.5 μL/min. Coordinates relative to bregma were anterior-posterior (AP), 0.00; medial-lateral (ML), +/− 2.00; and dorsal-ventral (DV), −3.25 mm. For immunosuppression, all mice received i.p. injections of CSA (10 mg/kg, daily thereafter) and mouse CD4 Ab (10 mg/kg, weekly thereafter) the day before surgery and continued until mice were sacrificed (8 months after implantation).
Biochemical, molecular, and immunohistochemical analyses
Four male mice per group were given i.p. injections of EdU (Thermo Fisher Scientific) 24 h prior to sacrifice. Mice were euthanized by pentobarbital overdose and perfused with 0.01 M PBS. Striatum and cerebral cortex were dissected out of the left hemisphere and flash-frozen for biochemical analysis. The other halves were post-fixed in 4% paraformaldehyde (PFA), cryoprotected in 30% sucrose, and cut at 40 μm on a sliding vibratome for IHC. Sections were rinsed three times and placed in blocking buffer for 1 h (PBS, 0.02% Triton X-100, 5% goat serum), and primary antibodies placed in block overnight (ON) at 4°C. Sections were rinsed, incubated for 1 h in Alexa Fluor secondary antibodies, and mounted using Fluoromount G (Southern Biotechnology). Primary antibodies used include anti-Ki67 (Abcam, ab16667), anti-Ku80 (Abcam, ab80592), anti-nestin (Millipore Sigma, MAB5326), anti-GFAP (Abcam, ab4674), anti-DCX (Fisher Millipore, AB2253MI), anti-NeuN (Abcam, ab177487), anti-calretinin (Abcam, ab16694), anti-vGlut1 (Abcam, ab180188), anti-human nuclear antigen (HNA) (Abcam, ab191181), anti-GAD65/67 (Abcam, ab49832), anti-BetaIII tubulin (Abcam, ab78078), anti-DARPP-32 (Abcam, ab40802), and anti-Ctip2 (Abcam, ab233713). For IHC, a minimum of four mice per group were analyzed. For diaminobenzidine (DAB) staining for ChAT, sections were rinsed three times, then given 30 min in 3% H2O2 and 10% methanol, rinsed, and placed in blocking buffer for 1 h (TBS + 5% normal rabbit serum [NBS Vector S-5000] + 0.1% Triton X-100), then primary antibody (goat anti-ChAT 9 Millipore AB144P) placed in block overnight at 4°C. Sections were rinsed, incubated for 1 h in secondary antibody (rabbit anti-goat biotinylated secondary), incubated in ABC solution (Vector PK-6100) for 1 h at room temperature (RT) then 1–3 min in DAB, rinsed, and tissue mounted on slides. To quantify NeuN and DARPP-32-positive cells from the IHC data, Q175 male WT mice implanted with hNSCs for 8 months (four mice, four images per mouse, 20×) were analyzed for Ku80/NeuN positivity or human DARPP-32 positivity. Using Imaris analysis software to quantify the percentage of hNSC differentiated to neurons or MSNs, the 16 images for each group (average ± SD) determined NeuN positive = 58.94% ± 10.71% and DARPP-32 positive = 7% ± 1.41%. For confocal microscopy, sections were imaged with a Bio-Rad Radiance 2100 confocal system using lambda-strobing mode. Images represent either single confocal z slices or z stacks. Whole-cell tissue lysis was performed in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors (Complete Mini, Roche Applied Science), 0.1 mM phenylmethylsulfonyl fluoride (PMSF), 25 mM N-Ethylmaleimide (NEM), 1.5 mM aprotinin, and 23.4 mM leupeptin by douncing, then sonicated for 10 s, three times at 40% amplitude on ice. Samples were quantified using Lowry protein assay. For soluble/insoluble fractionation, striatal tissue was processed as described previously.68 For western analysis, RIPA lysates were resolved by reducing and running 60 μg of protein on 4%–12% bis-Tris poly-acrylamide gels (PAGE). Antibodies were anti-BDNF (Santa Cruz Biotechnology, clone N-20, for mature BDNF, catalog no. sc-546), anti-ERK (Cell Signaling Technology, catalog no. 9102), anti-pERK (Cell Signaling Technology, catalog no. 9106), and anti-alpha tubulin (Sigma-Aldrich, catalog no. T6074). Quantification of bands was performed using software from the NIH program ImageJ and densitometry application. Reduced, insoluble protein from insoluble fractions (50 μg) was resolved on 3%–8% Tris-acetate poly-acrylamide gels. Membranes were blocked in starting block (Invitrogen) for 20 min at RT and probed in primary antibody overnight at 4°C. Insoluble protein was quantified as relative protein abundance as previously.68 Antibody was anti-HTT (Millipore, #MAB5492).
Behavioral tests
Males and females were used except for the running wheel, where only males were used since estrus cycle influences running activity. Genotypes or treatments were unknown to the experimenter. All tests were done during the light phase except for the running wheel, where mice were allowed 24-h free access to the task. Running wheel data are only described for the dark phase. Slope of motor learning was calculated as mean nightly running wheel rotations per 3 min on night 5 minus night 2 divided by total number of nights (three) for initial and night 13 minus night 2 divided by total number of nights (11) for overall. Behavioral tasks, running wheel and open field, were performed in a manner to those previously described.23,69
snRNA-seq Parse Biosciences data analysis
Single nuclei were isolated from flash-frozen half-hemisphere full striatal in Nuclei EZ Lysis buffer (catalog #NUC101-1KT, Sigma-Aldrich) and incubated for 5 min. Samples were passed through a 70-μm filter and incubated in additional lysis buffer for 5 min and centrifuged at 500 × g for 5 min at 4°C before two washes in nuclei wash and resuspension buffer (1× PBS, 1% BSA, 0.2 U/μL RNase inhibitor). Nuclei were fixed using a Parse Biosciences WF100 kit and the manufacturers protocol (Parse Biosciences, Seattle, WA). Eight single-nucleus libraries were sequenced on an Illumina NovaSeq 6000 S2 100 flow cell. Illumina NovaSeq 6000 RTA (v3.4.4) software was used to perform basecalling, and FASTQ files were generated using the bcl2fastq Conversion software (v2.20) to convert BCL base call files outputted by the sequencing instrument into the FASTQ format. The data preprocessing and alignment were performed using the SPLiT-seq pipeline (https://github.com/yjzhang/split-seq-pipeline) from Parse Biosciences (split-pipe v0.9.6p) to convert raw FASTQ files into gene expression matrices. Specifically, raw reads were mapped to either mouse reference genome mm10 for samples with vehicle or a combined genome with human reference genome grch38 for the hNSC samples, using STAR aligner (https://github.com/alexdobin/STAR). Gene expression was quantified using raw counts.
The obtained count matrices for each sample were merged and filtered for nuclei with <500 genes, or >5%–10% mitochondrial genes. Prior to normalization, two mouse transcripts, AY036118 and Gm42418, were removed and disregarded in the downstream analysis due to potential rRNA contamination caused by sequence overlap with Rn45s.70,71,72 The filtered count matrix was then normalized and transformed using SCTransform (v1) in Seurat (v4.0.5). Downstream analyses with Seurat include dimension reduction by principal-component analysis (RunPCA), UMAP (RunUMAP), shared nearest neighbor (SNN) graph construction (FindNeighbors), and finally Louvain clustering (FindClusters) using the first 20 principal components and a resolution of 0.07 to determine cluster assignment. To identify cell type markers and DEGs between different conditions, Wilcoxon rank-sum test was used (FindMarkers) on the standard log-normalized counts.
CellChat analysis
We use CellChat software to gain insights into cell-to-cell interactions and intercellular networks.34 Specifically, the normalized expression matrices from the two samples treated with hNSCs and the corresponding cell type annotation generated from Seurat were passed to CellChat for ligand-receptor interaction analysis using the human database. Mouse genes were converted to orthologous human genes and only those with human orthologs were retained. Default parameters were used, except that min.cells were set to 10, which allows filtering out cell types with the total number of cells smaller than 10. Circle plot and chord plot were used to visualize the number of inferred communications between cell types by the netVisual_circle function in the CellChat package.
Electrophysiology
For electrophysiological studies, we used 12 female mice (10.5 months old) shipped live to UCLA from UCI. Groups included four zQ175 hNSC, four zQ175 vehicle, two WT hNSC, and two WT vehicle. Mice were anesthetized and transcardially perfused with high-sucrose-based slicing solution. Coronal slices (300 μm) were transferred to an incubating chamber containing standard artificial cerebrospinal fluid (ACSF). MSNs and hNSCs were visualized using IR-DIC optics. All recordings were performed in or around the injection site (recorded MSNs were adjacent to the graft, ∼150–250 μm). Biocytin (0.2%) was added to the patch pipette for cell visualization and location of recorded cells. Spontaneous postsynaptic currents were recorded in the whole-cell patch-clamp configuration in ACSF. Membrane currents were recorded in gap-free mode. Cells were voltage clamped at +10 mV and sIPSCs were recorded in ACSF. Spontaneous sEPSCs from grafted cells were recorded in ACSF at −70 mV (baseline). sEPSCs from MSNs were recorded in the presence of the GABAA receptor blocker, BIC (10 μM, Tocris, Minneapolis, MN) to better isolate glutamatergic events. Spontaneous synaptic currents were analyzed using the MiniAnalysis software (version 6.0, Synaptosoft, Fort Lee, NJ). To evoke responses in grafted cells, we used a monopolar glass electrode (impedance 1 MΩ), which was placed 200–300 μm lateral to the graft. Following recordings, slices were fixed with 4% PFA, then transferred to 30% sucrose at 4°C until IHC processing. To identify biocytin-filled recorded cells and hNSCs, fixed slices were washed, permeabilized with Triton (0.7%), and blocked for 4 h, followed by incubation with SC121 (1:1,000) and/or KU80 (1:500). After washing, slices were incubated in goat, anti-mouse Alexa 488 (1:1,000, Life Technologies, Carlsbad, CA, catalog #A-11001); goat, anti-rabbit Alexa 350 (1:1,000 Life Technologies, Carlsbad, CA; catalog # A-11046); and streptavidin conjugated with Alexa 594 (1:1,000, Life Technologies catalog # S11227). Slices were washed, mounted, and cells visualized with a Zeiss Apotome confocal microscope.
EM
Female zQ175 mice implanted with hNSCs for 8 months (n = 3 per group) at UCI were sent live to the Portland VA Medical Center. Mice were anesthetized and perfused with EM fixative (2.5% glutaraldehyde, 0.5% PFA, and 0.1% picric acid in 0.1 M phosphate buffer [pH 7.4]). Brains were then collected and further processed in a Pelco Biowave Pro+ (Ted Pella, Redding, CA), as previously reported,73 and then washed in PBS and stored overnight at 4°C. Striatum containing hNSCs (equivalent to +1.4 to +0.14 mm from bregma)74 was cut at 60 μm using a vibratome (Leica Microsystems). After pre-embed IHC of the striatum using diaminobenzidine (DAB) (Sigma, St Louis, MO) or ImmPACT VIP Substrate, horseradish peroxidase (HRP) (catalog # SK-4605) (Vector Labs, Burlingame, CA), hNSC antibody (SC121, 1:100; Takara: catalog # Y40410) and vGlut 1 (vGlut1) antibody (1:000, Synaptic Systems, Germany, catalog # 135-303), the tissue was processed for EM as previously described.23,73,75,76 Two striatal slices were selectively double labeled for SC121 (DAB) and vGlut1 (VIP) to determine if vGlut1-labeled terminals originating from the cortex77 innervated the implantation site. Striatal slices were embedded flat between two sheets of ACLAR (Electron Microscopy Sciences, Hatfield, PA) overnight in a 60°C oven to polymerize the resin. The area containing hNSCs was micro-dissected from the embedded slice and superglued onto a block for thin sectioning. Photographs were taken on a JEOL 1400 transmission electron microscope (JEOL, Peabody, MA) of DAB-labeled structures (i.e., SC121) and for a small number of sections, double labeled with DAB (SC121) and VIP (vGlut1)-labeled structures (i.e., hNSC-labeled cells, dendrites, nerve terminals). The DAB-labeled structures (i.e., SC121) were photographed both within and located ∼0.25 mm outside of the implantation zone, at a final magnification of 46,200 using a digital camera (AMT, Danvers, MA). Since the DAB and DAB/VIP labeling was restricted to the leading edge of the thin-sectioned tissue, only the area showing DAB and DAB/VIP labeling was photographed. The percentage of SC121/DAB-labeled asymmetrical and symmetrical synaptic contacts onto dendrites and spines within and outside the implant area was quantified.
Statistical analysis
Results are from a single cohort except for snRNA-seq, western blots for BDNF and pERK/ERK, and IHC for ChAT, which were from a different subset. Numbers were determined to have sufficient power using an analysis prior to the study. Assessment of differences in outcome were based upon previous experience and published results78,79 for HD models, and applying power analysis (G Power [http://www.psycho.uni-duesseldorf.de/abteilungen/aap/gpower3/]) led us to a minimal n = 5 for behavior and n = 3 for biochemical analysis. Statistical significance was achieved as described using rigorous analysis. All findings are reproducible. Experiments were performed at least three times using at least three different mice (biological replicates) and, in specific cases, tissue from one mouse was used multiple times (technical replicates); for example, in IHC at least three different mouse brains were used and multiple sections from each brain were examined to obtain data. Multiple statistical methods are further detailed above or in figure legends. Since the EM data are based on three implanted zQ175 mice, and not comparing them against the WT mice, the percentages reported are a comparison within a single group of three experimental mice; therefore, there was no statistical comparison. Statistical tests for behavioral tasks used one-way ANOVA followed by Tukey’s honestly significant difference (HSD) test with Scheffé, Bonferroni, and Holm multiple comparison post hoc methods. Data met the assumptions of the statistical tests used, and p values <0.05 were considered significant. All mice were randomly assigned and tasks performed in a random manner with individuals blinded to genotypes and treatment. Statistical comparisons of densitometry results were performed by one-way ANOVA followed by Tukey HSD and Bonferroni post hoc tests. For electrophysiology data, all statistical analyses were performed using SigmaPlot 13.0 software. Differences between multiple group means were assessed with appropriate one-way ANOVAs followed by Bonferroni post hoc tests, Kruskal-Wallis one-way ANOVA on ranks followed by Holm-Sidak post hoc tests, or Student’s t tests (unpaired) when only two groups were compared. Significance levels in the figures are given as specific p values and data are expressed as mean ± SEM.
Data and code availability
All data are available from the corresponding authors upon reasonable request. Single-cell and snRNA-seq data are deposited to database: NCBI Gene Expression Omnibus (GEO) at GSE245631. Code will be deposited in GitHub.
Acknowledgments
We thank Lexi Kopan and Sylvia Yeung for their technical contributions to the studies. Funding was provided by the California Institute for Regenerative Medicine (CIRM ETAII TR2-01841 and CLIN1-10953), and NIH grants NS096994 and U54HD087101 (M.S.L.). We thank BioTime, Inc., and AgeX for the ESI-017 cell line and the UC Davis Flow Cytometry Shared Resource, 2921 Stockton Blvd., Suite 1300/1670 Sacramento, CA 95817 for flow analysis. We also thank the UCI Institute for Memory Impairments and Neurological Disorders, the Sue and Bill Gross Stem Cell Center, the Optical Biology Shared Resource of the Cancer Center Support Grant (CA-62203), and the Genomics Research and Technology Hub Shared Resource of the Cancer Center Support Grant (CA-62203) at the University of California, Irvine for facilities and assistance in carrying out studies.
Author contributions
J.C.R., S.M.H., C.C., M.S.L., and L.M.T. designed the experiments and analyzed data. J.C.R., R.G.L., J.W., S.M.H., C.C., L.M.T., and M.S.L. wrote the manuscript. S.Y., A.L., and I.O. performed the experiments in mice. A.L. and M.N. did IHC. E.S.M. and J.C.R. performed analyses. R.G.L., J.W., and R.M. carried out snRNA-seq studies and bioinformatics analysis. S.M.H. and C.C. performed electrophysiology and analyzed data with guidance from M.S.L. C.M. performed EM with guidance from C.K.M. L.K. cultured hNSCs at UCI. B.F., D.C.-B., and G.B. supplied ESI-017 hNSCs and characterizations from the GMP facility at UC Davis.
Declaration of interests
L.M.T. and J.C.R. have greater than 5% ownership in NeuroAirmid, a newly launched company that has related interests in cell therapy treatments for neurodegenerative diseases.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.10.003.
Supplemental information
References
- 1.Ghosh R., Tabrizi S.J. Huntington disease. Handb. Clin. Neurol. 2018;147:255–278. doi: 10.1016/B978-0-444-63233-3.00017-8. [DOI] [PubMed] [Google Scholar]
- 2.Macdonald M. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 3.Bates G., Harper P.S., Jones L. 3rd edition. Oxford University Press; 2002. Huntington's Disease; p. 558. [Google Scholar]
- 4.Harper P.S., Jones L. In: Huntington's Disease. Third edition. Bates G.P., Harper P.S., Jones L., editors. Oxford University Press; 2002. Huntington's disease: Genetic and molecular studies; pp. 113–158. [Google Scholar]
- 5.Vonsattel J.P., DiFiglia M. Huntington disease. J. Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 6.Waldvogel H.J., Kim E.H., Tippett L.J., Vonsattel J.P.G., Faull R.L.M. The Neuropathology of Huntington's Disease. Curr. Top. Behav. Neurosci. 2015;22:33–80. doi: 10.1007/7854_2014_354. [DOI] [PubMed] [Google Scholar]
- 7.Saudou F., Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. doi: 10.1016/j.neuron.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Bachoud-Lévi A.C., Massart R., Rosser A. Cell therapy in Huntington's disease: Taking stock of past studies to move the field forward. Stem Cells. 2021;39:144–155. doi: 10.1002/stem.3300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barker R.A., TRANSEURO consortium TRANSEURO consortium. Designing stem-cell-based dopamine cell replacement trials for Parkinson's disease. Nat. Med. 2019;25:1045–1053. doi: 10.1038/s41591-019-0507-2. [DOI] [PubMed] [Google Scholar]
- 10.Jang S.E., Qiu L., Chan L.L., Tan E.K., Zeng L. Current Status of Stem Cell-Derived Therapies for Parkinson's Disease: From Cell Assessment and Imaging Modalities to Clinical Trials. Front. Neurosci. 2020;14 doi: 10.3389/fnins.2020.558532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barbuti P.A., Barker R.A., Brundin P., Przedborski S., Papa S.M., Kalia L.V., Mochizuki H., MDS Scientific Issues Committee Recent Advances in the Development of Stem-Cell-Derived Dopaminergic Neuronal Transplant Therapies for Parkinson's Disease. Mov. Disord. 2021;36:1772–1780. doi: 10.1002/mds.28628. [DOI] [PubMed] [Google Scholar]
- 12.Baloh R.H., Johnson J.P., Avalos P., Allred P., Svendsen S., Gowing G., Roxas K., Wu A., Donahue B., Osborne S., et al. Transplantation of human neural progenitor cells secreting GDNF into the spinal cord of patients with ALS: a phase 1/2a trial. Nat. Med. 2022;28:1813–1822. doi: 10.1038/s41591-022-01956-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Southwell D.G., Nicholas C.R., Basbaum A.I., Stryker M.P., Kriegstein A.R., Rubenstein J.L., Alvarez-Buylla A. Interneurons from embryonic development to cell-based therapy. Science. 2014;344 doi: 10.1126/science.1240622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fandel T.M., Trivedi A., Nicholas C.R., Zhang H., Chen J., Martinez A.F., Noble-Haeusslein L.J., Kriegstein A.R. Transplanted Human Stem Cell-Derived Interneuron Precursors Mitigate Mouse Bladder Dysfunction and Central Neuropathic Pain after Spinal Cord Injury. Cell Stem Cell. 2016;19:544–557. doi: 10.1016/j.stem.2016.08.020. [DOI] [PubMed] [Google Scholar]
- 15.Chang B.L., Chang K.H. Stem Cell Therapy in Treating Epilepsy. Front. Neurosci. 2022;16 doi: 10.3389/fnins.2022.934507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Akabawy G., Rattray I., Johansson S.M., Gale R., Bates G., Modo M. Implantation of undifferentiated and pre-differentiated human neural stem cells in the R6/2 transgenic mouse model of Huntington's disease. BMC Neurosci. 2012;13:97. doi: 10.1186/1471-2202-13-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi K.A., Hong S. Induced neural stem cells as a means of treatment in Huntington's disease. Expert Opin. Biol. Ther. 2017;17:1333–1343. doi: 10.1080/14712598.2017.1365133. [DOI] [PubMed] [Google Scholar]
- 18.Connor B. Concise Review: The Use of Stem Cells for Understanding and Treating Huntington's Disease. Stem Cells. 2018;36:146–160. doi: 10.1002/stem.2747. [DOI] [PubMed] [Google Scholar]
- 19.De Gioia R., Biella F., Citterio G., Rizzo F., Abati E., Nizzardo M., Bresolin N., Comi G.P., Corti S. Neural Stem Cell Transplantation for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020;21 doi: 10.3390/ijms21093103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han F., Bi J., Qiao L., Arancio O. Stem Cell Therapy for Alzheimer's Disease. Adv. Exp. Med. Biol. 2020;1266:39–55. doi: 10.1007/978-981-15-4370-8_4. [DOI] [PubMed] [Google Scholar]
- 21.Hayashi Y., Lin H.T., Lee C.C., Tsai K.J. Effects of neural stem cell transplantation in Alzheimer's disease models. J. Biomed. Sci. 2020;27:29. doi: 10.1186/s12929-020-0622-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holley S.M., Kamdjou T., Reidling J.C., Fury B., Coleal-Bergum D., Bauer G., Thompson L.M., Levine M.S., Cepeda C. Therapeutic effects of stem cells in rodent models of Huntington's disease: Review and electrophysiological findings. CNS Neurosci. Ther. 2018;24:329–342. doi: 10.1111/cns.12839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reidling J.C., Relaño-Ginés A., Holley S.M., Ochaba J., Moore C., Fury B., Lau A., Tran A.H., Yeung S., Salamati D., et al. Human Neural Stem Cell Transplantation Rescues Functional Deficits in R6/2 and Q140 Huntington's Disease Mice. Stem Cel Rep. 2018;10:58–72. doi: 10.1016/j.stemcr.2017.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farrar A.M., Murphy C.A., Paterson N.E., Oakeshott S., He D., Alosio W., McConnell K., Menalled L.B., Ramboz S., Park L.C., et al. Cognitive deficits in transgenic and knock-in HTT mice parallel those in Huntington's disease. J. Huntingtons Dis. 2014;3:145–158. doi: 10.3233/JHD-130061. [DOI] [PubMed] [Google Scholar]
- 25.Menalled L.B., Kudwa A.E., Miller S., Fitzpatrick J., Watson-Johnson J., Keating N., Ruiz M., Mushlin R., Alosio W., McConnell K., et al. Comprehensive behavioral and molecular characterization of a new knock-in mouse model of Huntington's disease: zQ175. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Heikkinen T., Lehtimäki K., Vartiainen N., Puoliväli J., Hendricks S.J., Glaser J.R., Bradaia A., Wadel K., Touller C., Kontkanen O., et al. Characterization of neurophysiological and behavioral changes, MRI brain volumetry and 1H MRS in zQ175 knock-in mouse model of Huntington's disease. PLoS One. 2012;7 doi: 10.1371/journal.pone.0050717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langfelder P., Cantle J.P., Chatzopoulou D., Wang N., Gao F., Al-Ramahi I., Lu X.H., Ramos E.M., El-Zein K., Zhao Y., et al. Integrated genomics and proteomics define huntingtin CAG length-dependent networks in mice. Nat. Neurosci. 2016;19:623–633. doi: 10.1038/nn.4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plotkin J.L., Day M., Peterson J.D., Xie Z., Kress G.J., Rafalovich I., Kondapalli J., Gertler T.S., Flajolet M., Greengard P., et al. Impaired TrkB receptor signaling underlies corticostriatal dysfunction in Huntington's disease. Neuron. 2014;83:178–188. doi: 10.1016/j.neuron.2014.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Indersmitten T., Tran C.H., Cepeda C., Levine M.S. Altered excitatory and inhibitory inputs to striatal medium-sized spiny neurons and cortical pyramidal neurons in the Q175 mouse model of Huntington's disease. J. Neurophysiol. 2015;113:2953–2966. doi: 10.1152/jn.01056.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Southwell A.L., Smith-Dijak A., Kay C., Sepers M., Villanueva E.B., Parsons M.P., Xie Y., Anderson L., Felczak B., Waltl S., et al. An enhanced Q175 knock-in mouse model of Huntington disease with higher mutant huntingtin levels and accelerated disease phenotypes. Hum. Mol. Genet. 2016;25:3654–3675. doi: 10.1093/hmg/ddw212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sepers M.D., Smith-Dijak A., LeDue J., Kolodziejczyk K., Mackie K., Raymond L.A. Endocannabinoid-Specific Impairment in Synaptic Plasticity in Striatum of Huntington's Disease Mouse Model. J. Neurosci. 2018;38:544–554. doi: 10.1523/JNEUROSCI.1739-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim R.G., Al-Dalahmah O., Wu J., Gold M.P., Reidling J.C., Tang G., Adam M., Dansu D.K., Park H.J., Casaccia P., et al. Huntington disease oligodendrocyte maturation deficits revealed by single-nucleus RNAseq are rescued by thiamine-biotin supplementation. Nat. Commun. 2022;13:7791. doi: 10.1038/s41467-022-35388-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Obenauer J.C., Chen J., Andreeva V., Aaronson J.S., Lee R., Caricasole A. Expression analysis of Huntington disease mouse models reveals robust striatum disease signatures. bioRxiv. 2022 doi: 10.1101/2022.02.04.479180. Preprint at. [DOI] [Google Scholar]
- 34.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singh K., Jayaram M., Kaare M., Leidmaa E., Jagomäe T., Heinla I., Hickey M.A., Kaasik A., Schäfer M.K., Innos J., et al. Neural cell adhesion molecule Negr1 deficiency in mouse results in structural brain endophenotypes and behavioral deviations related to psychiatric disorders. Sci. Rep. 2019;9:5457. doi: 10.1038/s41598-019-41991-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lim R.G., Quan C., Reyes-Ortiz A.M., Lutz S.E., Kedaigle A.J., Gipson T.A., Wu J., Vatine G.D., Stocksdale J., Casale M.S., et al. Huntington's Disease iPSC-Derived Brain Microvascular Endothelial Cells Reveal WNT-Mediated Angiogenic and Blood-Brain Barrier Deficits. Cell Rep. 2017;19:1365–1377. doi: 10.1016/j.celrep.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith-Geater C., Hernandez S.J., Lim R.G., Adam M., Wu J., Stocksdale J.T., Wassie B.T., Gold M.P., Wang K.Q., Miramontes R., et al. Aberrant Development Corrected in Adult-Onset Huntington's Disease iPSC-Derived Neuronal Cultures via WNT Signaling Modulation. Stem Cel Rep. 2020;14:406–419. doi: 10.1016/j.stemcr.2020.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Souza J.M., Abd-Elrahman K.S., Ribeiro F.M., Ferguson S.S.G. mGluR5 regulates REST/NRSF signaling through N-cadherin/beta-catenin complex in Huntington's disease. Mol. Brain. 2020;13:118. doi: 10.1186/s13041-020-00657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dupont P., Besson M.T., Devaux J., Liévens J.C. Reducing canonical Wingless/Wnt signaling pathway confers protection against mutant Huntingtin toxicity in Drosophila. Neurobiol. Dis. 2012;47:237–247. doi: 10.1016/j.nbd.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 40.Galli S., Stancheva S.H., Dufor T., Gibb A.J., Salinas P.C. Striatal Synapse Degeneration and Dysfunction Are Reversed by Reactivation of Wnt Signaling. Front. Synaptic Neurosci. 2021;13 doi: 10.3389/fnsyn.2021.670467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cepeda C., André V.M., Yamazaki I., Wu N., Kleiman-Weiner M., Levine M.S. Differential electrophysiological properties of dopamine D1 and D2 receptor-containing striatal medium-sized spiny neurons. Eur. J. Neurosci. 2008;27:671–682. doi: 10.1111/j.1460-9568.2008.06038.x. [DOI] [PubMed] [Google Scholar]
- 42.Gertler T.S., Chan C.S., Surmeier D.J. Dichotomous anatomical properties of adult striatal medium spiny neurons. J. Neurosci. 2008;28:10814–10824. doi: 10.1523/JNEUROSCI.2660-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jia N., Chong J., Sun L. Application of stem cell biology in treating neurodegenerative diseases. Int. J. Neurosci. 2020;132:815–825. doi: 10.1080/00207454.2020.1840376. [DOI] [PubMed] [Google Scholar]
- 44.Kim T.W., Koo S.Y., Studer L. Pluripotent Stem Cell Therapies for Parkinson Disease: Present Challenges and Future Opportunities. Front. Cel Dev. Biol. 2020;8:729. doi: 10.3389/fcell.2020.00729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Goldberg N.R.S., Marsh S.E., Ochaba J., Shelley B.C., Davtyan H., Thompson L.M., Steffan J.S., Svendsen C.N., Blurton-Jones M. Human Neural Progenitor Transplantation Rescues Behavior and Reduces alpha-Synuclein in a Transgenic Model of Dementia with Lewy Bodies. Stem Cell Transl. Med. 2017;6:1477–1490. doi: 10.1002/sctm.16-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Qian H., Kang X., Hu J., Zhang D., Liang Z., Meng F., Zhang X., Xue Y., Maimon R., Dowdy S.F., et al. Reversing a model of Parkinson's disease with in situ converted nigral neurons. Nature. 2020;582:550–556. doi: 10.1038/s41586-020-2388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoon Y., Kim H.S., Hong C.P., Li E., Jeon I., Park H.J., Lee N., Pei Z., Song J. Neural Transplants From Human Induced Pluripotent Stem Cells Rescue the Pathology and Behavioral Defects in a Rodent Model of Huntington's Disease. Front. Neurosci. 2020;14 doi: 10.3389/fnins.2020.558204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cepeda C., Galvan L., Holley S.M., Rao S.P., André V.M., Botelho E.P., Chen J.Y., Watson J.B., Deisseroth K., Levine M.S. Multiple sources of striatal inhibition are differentially affected in Huntington's disease mouse models. J. Neurosci. 2013;33:7393–7406. doi: 10.1523/JNEUROSCI.2137-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Reiner A., Shelby E., Wang H., Demarch Z., Deng Y., Guley N.H., Hogg V., Roxburgh R., Tippett L.J., Waldvogel H.J., Faull R.L.M. Striatal parvalbuminergic neurons are lost in Huntington's disease: implications for dystonia. Mov. Disord. 2013;28:1691–1699. doi: 10.1002/mds.25624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Holley S.M., Galvan L., Kamdjou T., Cepeda C., Levine M.S. Striatal GABAergic interneuron dysfunction in the Q175 mouse model of Huntington's disease. Eur. J. Neurosci. 2019;49:79–93. doi: 10.1111/ejn.14283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Holley S.M., Galvan L., Kamdjou T., Dong A., Levine M.S., Cepeda C. Major Contribution of Somatostatin-Expressing Interneurons and Cannabinoid Receptors to Increased GABA Synaptic Activity in the Striatum of Huntington's Disease Mice. Front. Synaptic Neurosci. 2019;11:14. doi: 10.3389/fnsyn.2019.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Holley S.M., Joshi P.R., Parievsky A., Galvan L., Chen J.Y., Fisher Y.E., Huynh M.N., Cepeda C., Levine M.S. Enhanced GABAergic Inputs Contribute to Functional Alterations of Cholinergic Interneurons in the R6/2 Mouse Model of Huntington’s Disease. ENEURO. 2015;2:0008–0014.2015. doi: 10.1523/ENEURO.0008-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Imitola J., Park K.I., Teng Y.D., Nisim S., Lachyankar M., Ourednik J., Mueller F.J., Yiou R., Atala A., Sidman R.L., et al. Stem cells: cross-talk and developmental programs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004;359:823–837. doi: 10.1098/rstb.2004.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tepper J.M., Koós T., Ibanez-Sandoval O., Tecuapetla F., Faust T.W., Assous M. Heterogeneity and Diversity of Striatal GABAergic Interneurons: Update 2018. Front. Neuroanat. 2018;12:91. doi: 10.3389/fnana.2018.00091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cicchetti F., Parent A. Striatal interneurons in Huntington's disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov. Disord. 1996;11:619–626. doi: 10.1002/mds.870110605. [DOI] [PubMed] [Google Scholar]
- 56.Kumar U. Somatostatin in medium-sized aspiny interneurons of striatum is responsible for their preservation in quinolinic acid and N-methyl-D-asparate-induced neurotoxicity. J. Mol. Neurosci. 2008;35:345–354. doi: 10.1007/s12031-008-9093-3. [DOI] [PubMed] [Google Scholar]
- 57.Capetian P., Knoth R., Maciaczyk J., Pantazis G., Ditter M., Bokla L., Landwehrmeyer G.B., Volk B., Nikkhah G. Histological findings on fetal striatal grafts in a Huntington's disease patient early after transplantation. Neuroscience. 2009;160:661–675. doi: 10.1016/j.neuroscience.2009.02.035. [DOI] [PubMed] [Google Scholar]
- 58.Wagner L., Björkqvist M., Lundh S.H., Wolf R., Börgel A., Schlenzig D., Ludwig H.H., Rahfeld J.U., Leavitt B., Demuth H.U., et al. Neuropeptide Y (NPY) in cerebrospinal fluid from patients with Huntington's Disease: increased NPY levels and differential degradation of the NPY1-30 fragment. J. Neurochem. 2016;137:820–837. doi: 10.1111/jnc.13624. [DOI] [PubMed] [Google Scholar]
- 59.Besusso D., Schellino R., Boido M., Belloli S., Parolisi R., Conforti P., Faedo A., Cernigoj M., Campus I., Laporta A., et al. Stem Cell-Derived Human Striatal Progenitors Innervate Striatal Targets and Alleviate Sensorimotor Deficit in a Rat Model of Huntington Disease. Stem Cel Rep. 2020;14:876–891. doi: 10.1016/j.stemcr.2020.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schellino R., Besusso D., Parolisi R., Gómez-González G.B., Dallere S., Scaramuzza L., Ribodino M., Campus I., Conforti P., Parmar M., et al. hESC-derived striatal progenitors grafted into a Huntington's disease rat model support long-term functional motor recovery by differentiating, self-organizing and connecting into the lesioned striatum. Stem Cel Res. Ther. 2023;14:189. doi: 10.1186/s13287-023-03422-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Straccia M., Garcia-Diaz Barriga G., Sanders P., Bombau G., Carrere J., Mairal P.B., Vinh N.N., Yung S., Kelly C.M., Svendsen C.N., et al. Quantitative high-throughput gene expression profiling of human striatal development to screen stem cell-derived medium spiny neurons. Mol. Ther. Methods Clin. Dev. 2015;2 doi: 10.1038/mtm.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Conforti P., Bocchi V.D., Campus I., Scaramuzza L., Galimberti M., Lischetti T., Talpo F., Pedrazzoli M., Murgia A., Ferrari I., et al. In vitro-derived medium spiny neurons recapitulate human striatal development and complexity at single-cell resolution. Cel Rep. Methods. 2022;2 doi: 10.1016/j.crmeth.2022.100367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu Z., Parry M., Hou X.Y., Liu M.H., Wang H., Cain R., Pei Z.F., Chen Y.C., Guo Z.Y., Abhijeet S., Chen G. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington's disease. Nat. Commun. 2020;11:1105. doi: 10.1038/s41467-020-14855-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Comella-Bolla A., Orlandi J.G., Miguez A., Straccia M., García-Bravo M., Bombau G., Galofré M., Sanders P., Carrere J., Segovia J.C., et al. Human Pluripotent Stem Cell-Derived Neurons Are Functionally Mature In Vitro and Integrate into the Mouse Striatum Following Transplantation. Mol. Neurobiol. 2020;57:2766–2798. doi: 10.1007/s12035-020-01907-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rosser A.E., Busse M.E., Gray W.P., Badin R.A., Perrier A.L., Wheelock V., Cozzi E., Martin U.P., Salado-Manzano C., Mills L.J., et al. Translating cell therapies for neurodegenerative diseases: Huntington's disease as a model disorder. Brain. 2022;145:1584–1597. doi: 10.1093/brain/awac086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crook J.M., Peura T.T., Kravets L., Bosman A.G., Buzzard J.J., Horne R., Hentze H., Dunn N.R., Zweigerdt R., Chua F., et al. The generation of six clinical-grade human embryonic stem cell lines. Cell Stem Cell. 2007;1:490–494. doi: 10.1016/j.stem.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 67.Sivarajah S., Raj G.S., Mathews A.J.V., Sahib N.B.E., Hwang W.S., Crook J.M. The generation of GLP-grade human embryonic stem cell banks from four clinical-grade cell lines for preclinical research. In Vitro Cel Dev. Biol. Anim. 2010;46:210–216. doi: 10.1007/s11626-010-9287-1. [DOI] [PubMed] [Google Scholar]
- 68.Ochaba J., Monteys A.M., O'Rourke J.G., Reidling J.C., Steffan J.S., Davidson B.L., Thompson L.M. PIAS1 Regulates Mutant Huntingtin Accumulation and Huntington's Disease-Associated Phenotypes In Vivo. Neuron. 2016;90:507–520. doi: 10.1016/j.neuron.2016.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hickey M.A., Kosmalska A., Enayati J., Cohen R., Zeitlin S., Levine M.S., Chesselet M.F. Extensive early motor and non-motor behavioral deficits are followed by striatal neuronal loss in knock-in Huntington's disease mice. Neuroscience. 2008;157:280–295. doi: 10.1016/j.neuroscience.2008.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu Y., Cook C., Sedgewick A.J., Zhang S., Fassett M.S., Ricardo-Gonzalez R.R., Harirchian P., Kashem S.W., Hanakawa S., Leistico J.R., et al. Single-Cell Profiling Reveals Divergent, Globally Patterned Immune Responses in Murine Skin Inflammation. iScience. 2020;23 doi: 10.1016/j.isci.2020.101582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nguyen H.T.T., Guevarra R.B., Magez S., Radwanska M. Single-cell transcriptome profiling and the use of AID deficient mice reveal that B cell activation combined with antibody class switch recombination and somatic hypermutation do not benefit the control of experimental trypanosomosis. Plos Pathog. 2021;17 doi: 10.1371/journal.ppat.1010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kimmel J.C., Penland L., Rubinstein N.D., Hendrickson D.G., Kelley D.R., Rosenthal A.Z. A murine aging cell atlas reveals cell identity and tissue-specific trajectories of aging. BioRxiv. 2019 doi: 10.1101/657726v1.full. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Moore C., Xu M., Bohlen J.K., Meshul C.K. Differential ultrastructural alterations in the Vglut2 glutamatergic input to the substantia nigra pars compacta/pars reticulata following nigrostriatal dopamine loss in a progressive mouse model of Parkinson's disease. Eur. J. Neurosci. 2021;53:2061–2077. doi: 10.1111/ejn.14894. [DOI] [PubMed] [Google Scholar]
- 74.Paxinos G., Franklin K.B. Third edition. Academic Press; 2008. The Mouse Brain in Stereotaxic Coordinates. [Google Scholar]
- 75.Walker R.H., Moore C., Davies G., Dirling L.B., Koch R.J., Meshul C.K. Effects of subthalamic nucleus lesions and stimulation upon corticostriatal afferents in the 6-hydroxydopamine-lesioned rat. PLoS One. 2012;7 doi: 10.1371/journal.pone.0032919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Parievsky A., Moore C., Kamdjou T., Cepeda C., Meshul C.K., Levine M.S. Differential electrophysiological and morphological alterations of thalamostriatal and corticostriatal projections in the R6/2 mouse model of Huntington's disease. Neurobiol. Dis. 2017;108:29–44. doi: 10.1016/j.nbd.2017.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaneko T., Fujiyama F., Hioki H. Immunohistochemical localization of candidates for vesicular glutamate transporters in the rat brain. J. Comp. Neurol. 2002;444:39–62. doi: 10.1002/cne.10129. [DOI] [PubMed] [Google Scholar]
- 78.Hockly E., Woodman B., Mahal A., Lewis C.M., Bates G. Standardization and statistical approaches to therapeutic trials in the R6/2 mouse. Brain Res. Bull. 2003;61:469–479. doi: 10.1016/s0361-9230(03)00185-0. [DOI] [PubMed] [Google Scholar]
- 79.Hickey M.A., Gallant K., Gross G.G., Levine M.S., Chesselet M.F. Early behavioral deficits in R6/2 mice suitable for use in preclinical drug testing. Neurobiol. Dis. 2005;20:1–11. doi: 10.1016/j.nbd.2005.01.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available from the corresponding authors upon reasonable request. Single-cell and snRNA-seq data are deposited to database: NCBI Gene Expression Omnibus (GEO) at GSE245631. Code will be deposited in GitHub.