

Abstract
Despite advanced therapeutics, esophageal squamous cell carcinoma (ESCC) remains one of the deadliest cancers. Here, we propose a novel therapeutic strategy based on synthetic lethality combining trifluridine/tipiracil and MK1775 (WEE1 inhibitor) as a treatment for ESCC. This study demonstrates that trifluridine induces single‐strand DNA damage in ESCC cells, as evidenced by phosphorylated replication protein 32. The DNA damage response includes cyclin‐dependent kinase 1 (CDK1) (Tyr15) phosphorylation as CDK1 inhibition and a decrease of the proportion of phospho‐histone H3 (p‐hH3)‐positive cells, indicating cell cycle arrest at the G2 phase before mitosis entry. The WEE1 inhibitor remarkedly suppressed CDK1 phosphorylation (Try15) and reactivated CDK1, and also increased the proportion of p‐hH3‐positive cells, which indicates an increase of the number of cells into mitosis. Trifluridine combined with a WEE1 inhibitor increased trifluridine‐mediated DNA damage, namely DNA double‐strand breaks, as shown by increased γ‐H2AX expression. Moreover, the combination treatment with trifluridine/tipiracil and a WEE1 inhibitor significantly suppressed tumor growth of ESCC‐derived xenograft models. Hence, our novel combination treatment with trifluridine/tipiracil and a WEE1 inhibitor is considered a candidate treatment strategy for ESCC.
Keywords: DNA damage response, esophageal squamous cell carcinoma, synthetic lethality, trifluridine, WEE1 inhibitor
We suggest combining trifluridine/tipiracil and a WEE1 inhibitor as a novel therapeutic strategy against esophageal squamous cell carcinoma. This approach has demonstrated efficacy by combining the induction of DNA damage with the suppression of the DNA damage response.
Abbreviations
- ATR
ataxia telangiectasia and Rad3‐related protein
- ATM
ataxia telangiectasia mutated
- CDK1
cyclin‐dependent kinase 1
- CHK1
checkpoint kinase 1
- CHK2
checkpoint kinase 2
- DDR
DNA damage response
- ESCC
esophageal squamous cell carcinoma
- FTD/TPI
trifluridine/tipiracil
- hH3
histone H3
- MDM2
mouse double minute 2
- PDX
patient‐derived xenograft
- PFS
progression‐free survival
- RPA32
replication protein A32
- TP53
tumor suppressor protein P53
1. INTRODUCTION
Esophageal squamous cell carcinoma (ESCC) is the major histological type of esophageal cancer 1 , 2 and the sixth leading cause of cancer‐related mortality worldwide. 3 Unfortunately, despite recent progress in therapeutics, the prognosis for patients with ESCC remains poor. 4 , 5 , 6 , 7 , 8 TAS‐102 is an orally administered drug that is a fixed combination (1:0.5) of trifluridine (FTD) and tipiracil (TPI). It is currently in use against refractory metastatic gastric cancer 9 and colorectal cancer. 10 FTD is incorporated into DNA as a thymidine analog that induces DNA damage, whereas the TPI component inhibits the degradation of FTD and maintains the blood concentration of FTD. 11 Previously, we conducted a phase II study of FTD/TPI for advanced/recurrent esophageal cancer resistant/intolerable to standard therapies. We reported that FTD/TPI had good tolerability for heavily pretreated patients with ESCC and efficacy in controlling the primary esophageal lesion. However, overall, it failed to maintain progression‐free survival (PFS), 12 indicating that FTD/TPI needs additional cotreatment. Therefore, we aimed to develop a combination therapy with FTD/TPI and another small molecule to achieve better efficacy against ESCC.
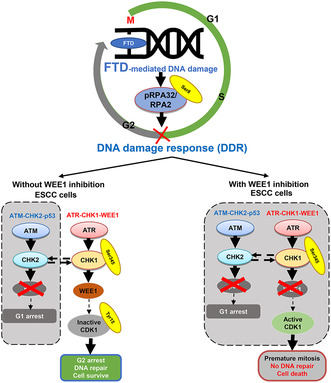
Recently, the development of therapeutic strategies based on synthetic lethality (a phenomenon in which the mutation of either gene alone is compatible with viability, but the simultaneous mutation of the two genes causes death) has received increasing attention. 13 The DNA damage response (DDR) is an important factor for synthetic lethality and a critical mechanism for maintaining genome stability. 14 DDR is coordinated by two distinct kinase signaling cascades, the ataxia telangiectasia Rad3‐related (ATR)‐checkpoint kinase 1 (CHK1)‐WEE1 (ATR‐CHK1‐WEE1) and the ataxia telangiectasia mutated (ATM)‐checkpoint kinase 2 (CHK2)‐p53 (ATM‐CHK2‐p53) pathways. 15 , 16 These pathways regulate specific cell cycle checkpoints: The ATR‐CHK1‐WEE1 pathway regulates the S‐phase and G2 checkpoint, 17 , 18 whereas the ATM‐CHK2‐p53 pathway regulates the G1 checkpoint. 17 Therefore, a synthetic lethality strategy perturbating these two pathways makes cells vulnerable to DNA damage.
In ESCC, the tumor suppressor protein p53 (encoded by the TP53 gene), a part of the ATM‐CHK2‐p53 pathway, 19 is frequently (83%–92%) mutated in ESCC 20 , 21 ; therefore, ESCC is considered to depend on the ATR‐CHK1‐WEE1 pathway to maintain DNA integrity. We recently reported synthetic lethality from a combination of genetic loss of function of p53 and a chemical inhibition with a CHK1 inhibitor in p53‐mutant ESCC cells with DNA damage induced by FTD/TPI. 22 However, CHK1 inhibitors are not clinically available for further clinical development. Here, we explored the concept of synthetic lethality with CHK1 perturbation to the whole ATR‐CHK1‐WEE1 pathway. Following the ATR‐CHK1‐WEE1 pathway, the cyclin‐dependent kinase 1 (CDK1, also known as cell division control protein 2 [CDC2]), a cell cycle regulator, is inactivated during S‐G2 phases through the phosphorylation on Thr14 and Tyr15 by WEE1. 23 , 24 , 25 In the late stage of G2, active CDK1 interacts with cyclin B1 to form the mitosis‐promoting factor that can lead a cell to the M (mitotic) phase. 26 , 27 Unscheduled CDK1 activation during incomplete S‐G2 phase can result in premature mitosis with unrepaired DNA damage and cell death, which is called “mitotic catastrophe.” 24 Therefore, the inhibition of WEE1 may induce entry into unscheduled mitosis triggered by the activation of CDK1, which can lead to cell death.
Based on these findings, we investigated whether combination treatment with FTD/TPI and a WEE1 inhibitor is an effective therapeutic strategy for ESCC.
2. MATERIALS AND METHODS
2.1. Cell culture
Human ESCC cell lines (TE‐1, TE‐5, TE‐8, TE‐10, TE‐11) were obtained from the RIKEN Bioresource Center. All cells were cultured as described previously. 22 In addition, the cells were all cultured in RPMI 1640 (Life Technologies Corp.) supplemented with 10% FBS and 0.1% penicillin‐streptomycin (Nacalai Tesque). All cells were grown at 37°C in a humidified 5% CO2 atmosphere.
2.2. Small molecule compounds
MK1775 (WEE1 inhibitor) was purchased from MedChemExpress. FTD/TPI (TAS‐102) was provided by Taiho Pharmaceutical Co., Ltd. PD0166285 was purchased from AdooQ BioScience. LY2606368 (CHK1 inhibitor) was purchased from Selleck Chemicals. Nutlin‐3a (MDM2 inhibitor) was purchased from MedChemExpress.
2.3. Western blot analysis
Western blotting was performed as reported previously. 22 In brief, cells were washed twice with ice‐cold PBS and lysed with RIPA Buffer (Nacalai Tesque). The protein samples were separated on Mini‐PROTEAN TGX Gels (Bio‐Rad Laboratories, Inc.) and transferred to a polyvinylidene difluoride membrane (Trans‐Blot Turbo Transfer Pack, Bio‐Rad Laboratories, Inc.).
The list of primary antibodies is provided in Table S1.
2.4. WST‐1 assay
WST‐1 assays were performed as previously described. 22 , 28 In brief, 100 μL of ready‐to‐use WST‐1 reagent (Roche Applied Science) was added directly into 96‐well plates. After 30 min of incubation, the plate was read at 450 nm with a reference reading at 630 nm by a multiwell plate reader (Infinite 200 Pro; Tecan Group Ltd.).
2.5. Flow cytometry assay
Cells were cultured and treated with FTD (10 μM) and/or MK1775 (100 nM) in the designated time courses. Then, cells were harvested, fixed by 100 μL 4% formaldehyde, and permeabilized by cold 100% methanol slowly while gently vortexing to a final concentration of 90% methanol. After washing by PBS, cells were resuspended in 100 μL of diluted phospho‐histone H3 (Ser10) (D2C8) XP Rabbit mAb (Alexa Fluor® 647 Conjugate) (Cell Signaling Technology (CST), #3458) prepared in antibody dilution buffer at a ratio of 1:50. After 30 min at room temperature, cells then were analyzed.
2.6. Clonogenic assay
Cells were plated at 1–2 × 103 cells per well in six‐well plates and incubated for 24 h. Then, FTD (0.5 and 1 μM) and/or MK1775 (30 and 100 nM) were added and incubated for a week. During this time, the medium was changed every 3 days with the same concentration of drug. Next, cells were fixed with 20% glutaraldehyde for 15 min, stained with 0.05% crystal violet, and left to dry overnight. Digital images of the colonies were obtained using a scanning device (EPSON GY‐X830).
2.7. Cell cycle analysis
Esophageal squamous cell carcinoma cells cultured in the presence of FTD following time courses were then harvested and fixed with 70% ice‐cold ethanol. To determine DNA content, cells were treated with ribonuclease, stained with 25 μL of propidium iodide solution (BioLegend # 421301), and finally analyzed by BD FACS flow cytometry.
2.8. CellTiter‐Glo assay
Organoids were prepared in 96‐well plates and incubated and exposed to Nutlin‐3a for 24 h as designed. The plates were equilibrated at room temperature for approximately 30 min. A volume of CellTiter‐Glo 3D Reagent (Promega # G9682) equal to the volume of the culture medium present in each well was added, and the contents were mixed for 2 min on an orbital. The plates were then incubated at room temperature for 10 min to stabilize the luminescent signal.
2.9. CytoTox‐Glo cytotoxicity assay
Cells were plated as 2 × 103 cells per well in black flat‐bottom 96‐well plates and then incubated overnight. After 72 h of treating with FTD 10 μM and/or MK1775 100 nM, each well was added 50 μL assay reagent, shaken for 1 min, and incubated for 15 min at room temperature. Then, we measured dead cell luminescence by a GloMax‐Multi detection system. The procedure was repeated with lysis reagent, and all‐cell luminescence was measured.
2.10. In vivo experiment
TE‐8 cells were cultured and then prepared in RPMI medium (1 × 106 cells per mouse) at a 1:1 ratio and injected subcutaneously into the flanks of BALB/cSlc‐nu/nu male mice (6–8 weeks old) purchased from Japan SLC, Inc. To establish patient‐derived xenograft (PDX) tumors, biopsy specimens, which were generated in our lab after obtaining written patient consent, from a 60‐year‐old male with a poorly differentiated cT2N0M0 ESCC were placed in a subcutaneous pocket created by a 5‐mm incision in the left flank of 6‐week‐old male BALB/cSlc‐nu/nu mice, then closed by suturing. Calipers were used for measurements twice a week, and tumor volumes were calculated (volume = length × width2 × 0.5) until tumors reached a volume of about 150–300 mm3. The mice were randomly assigned to groups (n = 5 or 6 each) and treated with FTD/TPI (200 mg/kg/day, orally) and/or MK1775 (30 mg/kg/day, orally) for 3 weeks (TE‐8) or 4 weeks (PDX). As a control for FTD/TPI and MK1775, 0.5% hydroxypropyl methylcellulose (10 mL/kg) and/or 20% captisol (CyDex Pharmaceuticals, Inc.) were used, respectively. Mice were sacrificed when tumors reached an end point. Dissected tumors were snap‐frozen and stored at −80°C or fixed in 10% buffered formalin for the histopathologic process.
2.11. Histologic and immunohistochemical (IHC) staining
Tissue samples were fixed in 4% paraformaldehyde phosphate buffer solution (Wako Pure Chemical Industries, Ltd.) overnight at 4°C, embedded in paraffin, and cut into 4‐μm sections for standard hematoxylin and eosin (H&E) staining and IHC. Deparaffinization and antigen retrieval by incubation in protease solution (Nichirei Biosciences) were carried out for 5 min. The glass slides were washed in PBS (six times, 5 min each) and mounted with 1% normal serum in PBS for 30 min. 29 The primary antibody, rabbit monoclonal anti‐γ‐H2AX (#9718, CST) at 1:100 dilution was subsequently applied for 40 min, followed by PBS washes (three times, 5 min each). Slides were incubated with a secondary antibody solution in a Histofine Simple Stain MAX PO (R) Kit (Nichirei Biosciences) for 30 min, which was followed by PBS washes. Coloring reaction was carried out with diaminobenzidine, and nuclei were counterstained with hematoxylin. Immuno‐stained tissues were assessed using a BIOREVO BZ‐9000 Microscope (Keyence) for γ‐H2AX staining.
2.12. Radiotherapy
Radiation treatment (RT, 2 grays [Gy]) was conducted in a single fraction of tumors, as reported previously. 30 Briefly, mice were positioned in a modified 50‐mL conical plastic tube to allow irradiation of the tumor area while keeping the rest of the body outside the RT field using a collimator. The tumors were locally irradiated with 2 Gy of 137Cs γ‐rays using a Gammacell 40 Exactor (MDS Nordion International).
2.13. Statistical analysis
Unless otherwise stated, data are presented as the means ± SD of triplicate experiments. Correlations between the proportion of mitotic cells assessed by flow cytometry and IC50 values obtained using WST‐1 assay between two groups, FTD and combination FTD‐MK1775, were evaluated for statistical significance using a t‐test. In addition, the interaction between FTD/TPI and WEE1 inhibitor treatments for TE‐1 CytoTox‐Glo assay and ESCC TE‐8 tumor and PDX tumor xenografts were assessed using two‐way analysis of variance (ANOVA). When significant interactions were noted, more than two groups were analyzed using Tukey's honestly significant difference test. A p‐value <0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 9 for Windows.
3. RESULTS
3.1. FTD activates DDR processes and induces a G2 arrest
We exposed various ESCC cell lines to FTD for different durations to observe the activation of DDR. We used a clinically relevant dose (10 μM) of FTD based on phase I clinical trial data. 31 In Figure 1A, the protein levels of phospho‐replication protein A 32 (RPA32), which is a marker for single‐strand DNA damage, 32 were enhanced by FTD 10 μM and remarkedly increased after 48 h compared with the control at 0 h. We also investigated whether FTD‐induced DDR could inhibit CDK1. CDK1 has several phosphorylation sites, including the Tyrosine 15 residue, which, when phosphorylated, deactivates CDK1. 33 Therefore, we examined the p‐CDK1 status at the Tyr15 site after treatment by FTD. Along the ATR‐CHK1‐WEE1 axis, CHK1 was activated by phosphorylation of this protein at Ser345 (Figure 1B), followed by downstream inactivation of CDK1, as the blots of phospho‐CDK1 at Tyr15 increased over time and peaked after 48 h, showing a sequential effect of FTD on the DDR pathway. Next, we observed whether the inactivation of CDK1 had negatively regulated the cell cycle in FTD‐treated ESCC cells.
FIGURE 1.
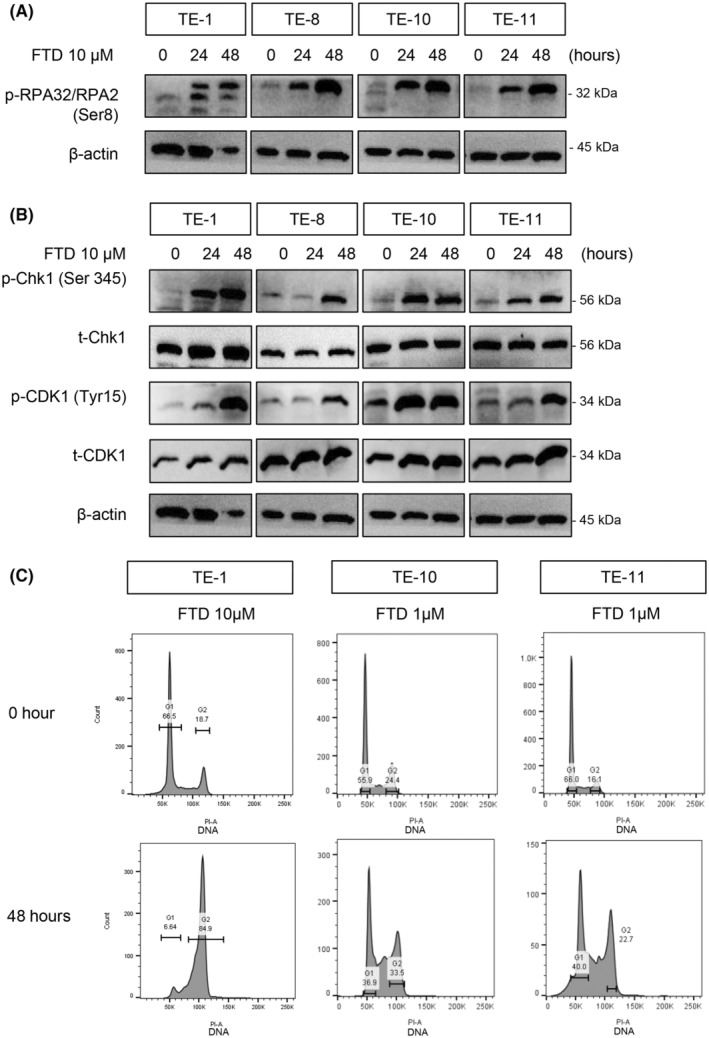
Trifluridine (FTD) activates DNA damage response (DDR), deactivates cyclin‐dependent kinase 1 (CDK1), and arrests cells at the G2 phase. (A, B) Immunoblots showing DDRs caused by FTD at different time points. (A) Single‐strand DNA damage, shown by phosphorylation of replication protein A32 (RPA32)/RPA2 induced by FTD, which increased over time. (B) FTD activated the ataxia telangiectasia and Rad3‐related protein (ATR)‐CHK1 pathway (indicated by phosphorylation) and deactivated CDK1 (indicated by phosphorylation at Tyr15). β‐Actin bands, as loading control, were used for both panels (A, B) since these results were from the same protein samples. (C) Cell cycle distribution of TE‐1, TE‐10, and TE‐11 cells exposed to FTD 1 μM and 10 μM for 0 and 48 h. Cells were stained with propidium iodide and analyzed by flow cytometry.
To investigate whether FTD could induce the arrest at the G2 phase in ESCC cells, we treated them with FTD and analyzed the cell cycle distribution at several time points, varying from 0 to 48 h (Figure 1C). We excluded TE‐8 from this analysis because of multiple peaks at the baseline due to aneuploidy. Consistent with the elevation of p‐CHK1 S345 and p‐CDK1 Tyr15, after 48 h of FTD treatment (Figure 1A), flow cytometry data showed that 4N DNA reached the highest level at 48 h after FTD treatment, suggesting a G2 arrest to give the cell time for DDR. Hence, FTD is able to induce DDR and inactivate CDK1, which triggers a G2 arrest in ESCC cells.
3.2. A WEE1 inhibitor suppresses FTD‐induced DDR and induces DNA double‐strand breaks
Next, we tested whether adding a WEE1 inhibitor to the FTD‐exposed ESCC cells could inhibit the inactivation of CDK1 and lead cells into mitosis. Before cell‐based assays, we investigated DepMap gene dependency data with the shRNA library (DEMETER2 v6, DepMap, Broad Institute). We found that CHK1 gene dependency correlated more with WEE1 than with ATR or ATM‐CHK2‐TP53 pathway genes in all cell line data sets and squamous cell carcinoma data sets, including ESCC (Figure 2A, Figure S1A–C), which supports our idea to use the WEE1 inhibitor, not an ATR inhibitor. After this result, we continued to observe a relevant suppression of phospho‐CDK1 by the CHK1 inhibitor (Prexasertib, LY2606368) (Figure 2B) and WEE1 inhibitor (Adavosertib, MK1775) (Figure 2C) following increasing doses after 24 h of administration. When the cells were treated with a CHK1 inhibitor or a WEE1 inhibitor (MK1775 or PD0166285) alone, CHK1/WEE1 inhibitors dephosphorylated Tyr15 residues of CDK1, indicating their function to inhibit DDRs, which attenuate cell cycles (Figure 2B,C, Figure S1D,E), although the effect size is different among the cell lines.
FIGURE 2.
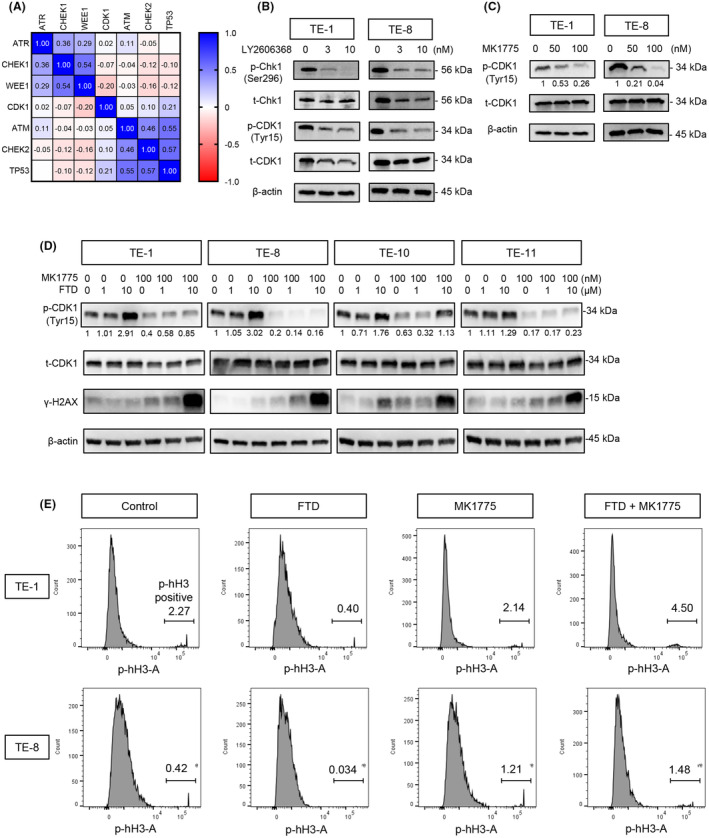
MK1775 suppresses trifluridine (FTD)‐induced DNA damage responses (DDRs) by cyclin‐dependent kinase 1 (CDK1) activation. (A) Gene dependency correlation among DNA damage repair pathway genes and CDK1. Matrix of Pearson r values calculated from all cell lines included in the data set (712 cell lines). Data set: DEMETER2_v6 combined RNAi (DepMap: https://depmap.org/portal/, Broad Institute). (B, C) Immunoblots of DDR checkpoints, which were suppressed after exposing the cells to a Chk1 inhibitor (LY2606368) and WEE1 inhibitor (MK1775) with increasing concentrations. (D) With FTD, MK1775 not only activated CDK1 but also induced DNA double‐strand breaks, indicated by g‐H2AX. (E) Flow cytometry showing the percentage of mitotic cells, which is indicated by p‐hH3 following the treatment of FTD, MK1775, and a combination of them.
Addition of a WEE1 inhibitor (MK1775) to FTD‐treated cells dephosphorylated CDK1 (activated CDK1) in all cells (TE‐1, TE‐8, TE‐10, TE‐11; Figure 2D), suggesting the potential to activate CDK1. Next, we monitored how the accumulation of mitotic cells varied by treatment with those reagents in ESCC cells. We observed the proportion of mitotic cells in the M phase by measuring the phosphorylation of histone H3 34 and found a decrease in mitotic cells treated by FTD only, as a blocked mitotic entry in the presence of FTD. However, the proportion of mitotic cells was remarkably increased after 24 h of cotreatment with MK1775 and FTD (p < 0.0001), suggesting that the WEE1 inhibitor had blocked FTD‐induced DDR and led more cells into the mitosis phase (Figure 2E, Figure S1F). Furthermore, combination of a WEE1 inhibitor (MK1775 or PD0166285) and FTD induced γ‐H2AX, indicating double‐strand breaks of DNA due to a forced cell cycle, even with DNA damage caused by FTD increased the toxicity in ESCC cells (Figure 2D, Figure S1D,E).
3.3. The WEE1 inhibitor enhances the cytotoxic effect of FTD on various ESCC cell lines
Next, we examined whether the combination therapy has effective cytotoxicity against ESCC cells in vitro. First, we examined cell viability with or without MK1775 and/or FTD. In the WST‐1 assay, the addition of MK1775 (100 nM) sensitized FTD‐treated TE‐1 and TE‐8 cells and remarkedly reduced the IC50 concentration of FTD in both cell lines (p < 0.0001 and p = 0.0028, respectively) (Figure 3A). Next, to determine whether the combination was cytotoxic or just cytostatic, we performed a dead cell assay (CytoTox‐Glo). As shown in Figure 3B, adding 100 nM MK1775 to FTD resulted in a significant increase in the dead cell ratio compared with every single treatment and control group, indicating that the effect of the combination treatment was cytotoxic. Furthermore, a significant interaction between FTD and MK1775 was observed when ESCC cells were treated by the combination (p‐values for interaction in two‐way ANOVA: 0.003, 0.0152, 0.0108, and 0.0164 for TE‐1, TE‐8, TE‐10, and TE‐11, respectively), suggesting a synergistic effect to kill the cells with FTD and MK1775.
FIGURE 3.
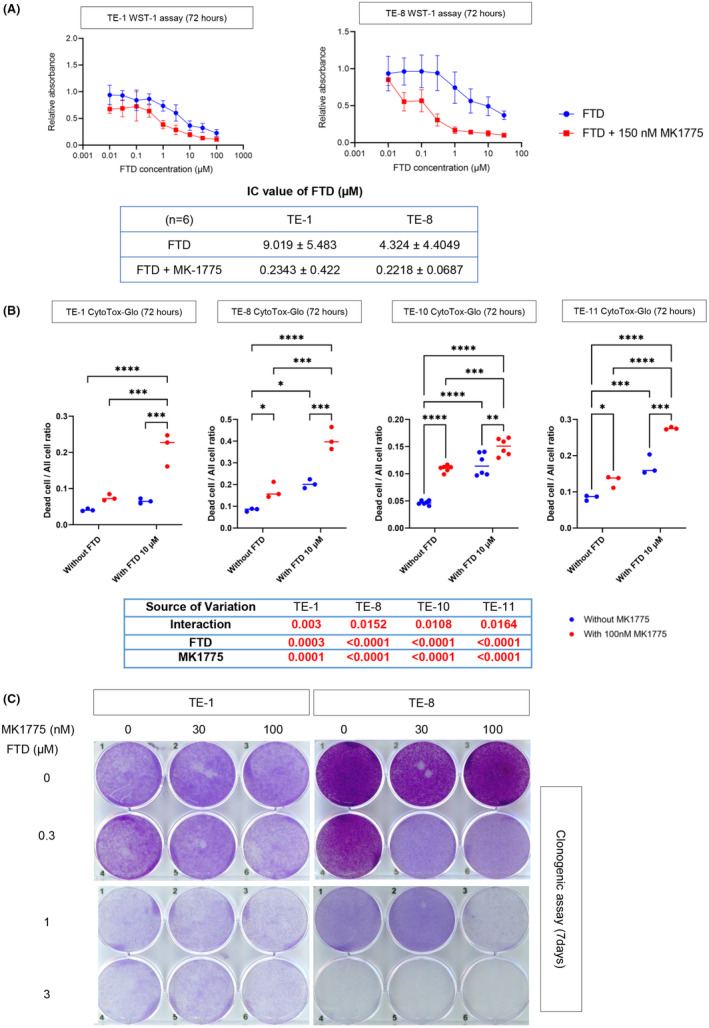
The combination of trifluridine (FTD) and MK1775 shows cytotoxicity to esophageal squamous cell carcinoma (ESCC) cells. (A) Cell survival curve and IC50 after 72 h of treatment with FTD only and combination treatment of FTD and MK1775 with TE‐1 and TE‐8. (B) Comparison of ESCC cells’ live/dead ratio between control and every single treatment and combination. Two‐way analysis of variance (ANOVA) was used for the statistical analysis. (C) Clonogenic assay of TE‐1 and TE‐8 after treatment with the indicated doses of FTD and MK1775 for 1 week. In each well, 4 × 103 cells were plated, which were then collected and stained with crystal violet. After washing, pictures were taken using a scanner (EPSON GT‐X830). *p < 0.05; **p < 0.005; ***p <0.0005; ****p < 0.0001.
Furthermore, we performed a clonogenic assay to examine the long‐term (7 days) combined therapeutic effects of FTD and MK‐1775 on ESCC cell lines (TE‐1 and TE‐8). This combination has shown stronger cytotoxicity in ESCC cells than the monotherapy of each drug. Altogether, MK1775 had a synergistic effect of increasing the cytotoxicity of FTD on ESCC cell lines (Figure 3C).
3.4. FTD/TPI and WEE1 inhibitor combination shows potent antitumor effect in ESCC xenograft models
Given the promising in vitro data, we next tested the combination treatment with FTD/TPI and MK1775 in ESCC xenografted tumors. In xenografted tumors from the TE‐8 cell line, the tumor growth curves were attenuated by each monotherapy MK1775 or FTD/TPI compared with control mice (Figure 4A). Moreover, tumor growth was significantly more suppressed in the combination group of FTD/TPI and MK‐1775 than under other conditions (tumor growth compression was approximately 86% in TE‐8 cell xenograft models) (Figure 4A).
FIGURE 4.
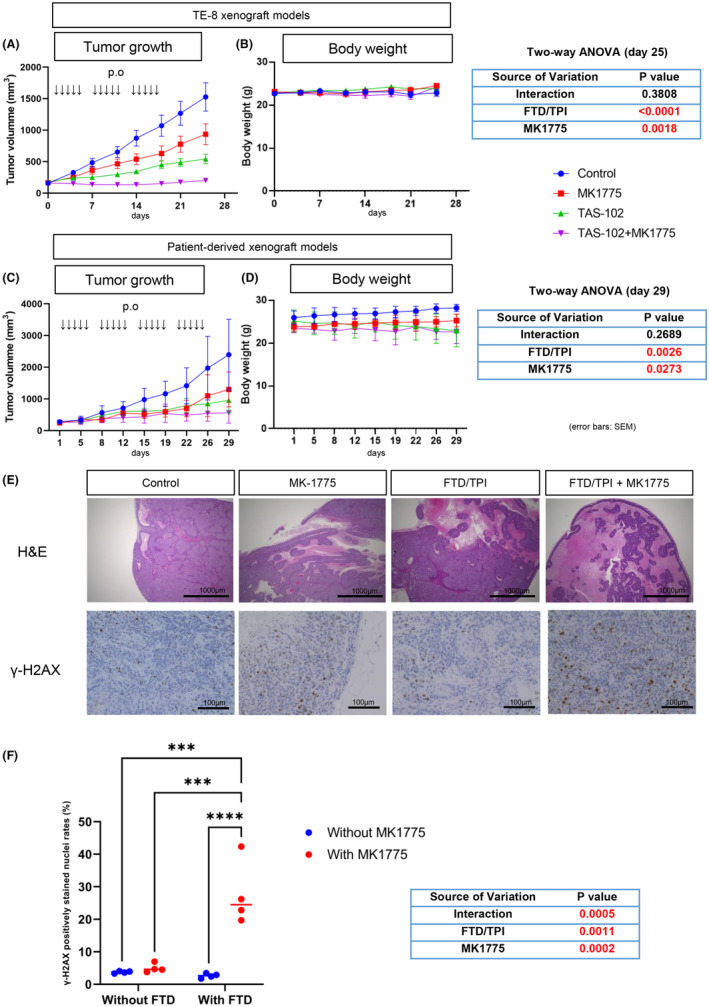
TAS‐102 and MK1775 show potent antitumor effects on esophageal squamous cell carcinoma xenograft tumors. Nude mice bearing TE‐8 xenograft (A) and patient‐derived xenograft (PDX) (C) were administered a vehicle (●), MK‐1775 at 30 mg/kg (■), trifluridine (FTD)/tipiracil (TPI) at 200 mg/kg (▲), FTD/TPI + MK‐1775 (▼), and oral gavage, five times weekly, n = 12–13 per arm. Mean relative tumor volumes +/− SE are shown. Two‐way analysis of variance (ANOVA) was used for the statistical analysis. (B) Body weight from the same group as shown in (A) was monitored during the study. Mean relative tumor volumes +/− SE are shown. (D) Body weight from the same groups as shown in (C). (E) Hematoxylin and eosin (H&E) images of PDX#5 tumor tissues in each treatment group on day 29 (scale bar, 1000 μm) and immunohistochemical (IHC) staining images for γ‐H2AX on day 29 (scale bar, 100 μm). (F) γ‐H2AX positively stained nuclei were counted in eight random fields from two representative slides in each group. ****p < 0.0001.
We also used PDX tumors to evaluate the efficacy of the combination of FTD/TPI and MK1775. Before using them for xenograft experiments, we ran whole‐exome sequencing and detected various gene mutations, including TP53, with allele frequencies above 99% (Table S2). We further confirmed the functional loss of TP53 by checking the growth of organoids with an MDM2 inhibitor Nutlin‐3a (Figure S2). PDX#5‐derived organoids did not show a significant loss of viability after exposure to 10 μM Nutlin‐3a, indicating that PDX#5 has a loss‐of‐function TP53 mutation. With PDX#5, the FTD/TPI and MK‐1775 combination showed better suppression of tumor growth than either FTD/TPI alone or MK1775 alone or vehicle control (tumor growth suppression was nearly 71.3%) (Figure 4C).
Hematoxylin and eosin images showed that combination therapy tumors had more necrosis regions and fewer growing tissues than the single‐treatment and control groups (Figure 4E). We also found that the combination treatment of FTD/TPI and MK1775 induced γ‐H2AX in the PDX#5 tissues (Figure 4E,F). Although a synergistic effect was not observed in terms of tumor size suppression (Figure 4C), there was a significant interaction of γ‐H2AX induction between FTD and MK1775 (Figure 4F), suggesting a potential synergistic effect of these two compounds in cytotoxicity. As for safety, no remarkable alteration was noticed during the treatment course in the TE‐8 and PDX experiments, except for minor body weight loss in FTD/TPI‐treated mice in the PDX#5 experiment (Figure 4B–D). Overall, the combination of FTD/TPI and MK1775 showed a potent antitumor effect in ESCC mouse models.
Considering the importance of local control with radiotherapy in ESCC treatment, we further assessed the radiosensitizing effect of FTD/TPI and MK1775 by in vivo models. We conducted an experiment with four groups: vehicle as control, radiotherapy, chemotherapy (FTD/TPI and MK1775), and a combination of chemotherapy and radiotherapy. Chemotherapy showed an additive effect on radiotherapy to suppress tumor growth, although radiotherapy's effect was not statistically significant in this xenograft model (Figure S3A). No severe complications were observed in this experiment, and no significant body weight loss was observed on day 25 in all groups (Figure S3B). In addition, more γ‐H2AX‐positive cells were observed in the combination treatment than in other groups (Figure S3C,D), indicating that radiotherapy might be able to enhance the cytotoxicity together with our novel chemotherapy with FTD/TPI and a WEE1 inhibitor, although in our experiment we could not see significant synergistic effects.
Hence, the combination of FTD/TPI and MK1775 showed a potent antitumor effect as chemotherapy or chemoradiotherapy against ESCC.
4. DISCUSSION
This study showed that the combination of FTD/TPI and a WEE1 inhibitor had potent cytotoxicity and antitumor effects against ESCC. Based on our previous report of synthetic lethality with coinhibition of ATR‐CHK1‐WEE1 (chemically) and ATM‐CHK2‐p53 (genetically), we chose WEE1 as our target because it is the closest node to the cell cycle regulation in concert with FTD. 25 Furthermore, as supportive data, the dependency on genes in the ATR‐CHK1‐WEE1 pathway in the DepMap data set showed that the dependency on CHK1 in ESCC cells was the most correlated with that on WEE1, rather than the upstream ATR (Figure S1A,B). Therefore, this drug in combination with FTD/TPI and a WEE1 inhibitor is an effective therapeutic strategy for ESCC.
In the present study, FTD activated the ATR‐CHK1‐WEE1 pathway and decreased mitotic cells in ESCC cells. These cell responses are suggested to be the DDR to FTD‐derived DNA damage. Nonetheless, at the same time point, G2 peak elevated in TE‐1 cells treated with FTD 10 μM, while TE‐10 and TE‐11 only required 1 μM of FTD to achieve this phase. TE‐8 was also observed with high G2 peak after 48 h of 1 μM FTD treatment; however, this cell line exhibited multiple peaks on histogram, not appropriate to be shown. As well as when evaluating the cytotoxicity, a difference in interaction effect between two drugs could be seen among the cell lines, especially TE‐1 versus the others. These results suggest that different cell lines would have different sensitivity to FTD treatment. Furthermore, the ATM‐CHK2‐P53 pathway is impaired in ESCC cells because of highly frequent TP53 mutations. Therefore, inhibition of the ATR‐CHK1‐WEE1 pathway by a WEE1 inhibitor is considered to fail DNA damage repair function in ESCC cells severely. Indeed, the combination of FTD and a WEE1 inhibitor increased the percentage of mitotic cells compared with monotherapy with FTD. Accordingly, premature mitosis proceeds in cells that have not completed DNA damage repair, leading to severe DNA damage and cell death (Figure 5).
FIGURE 5.
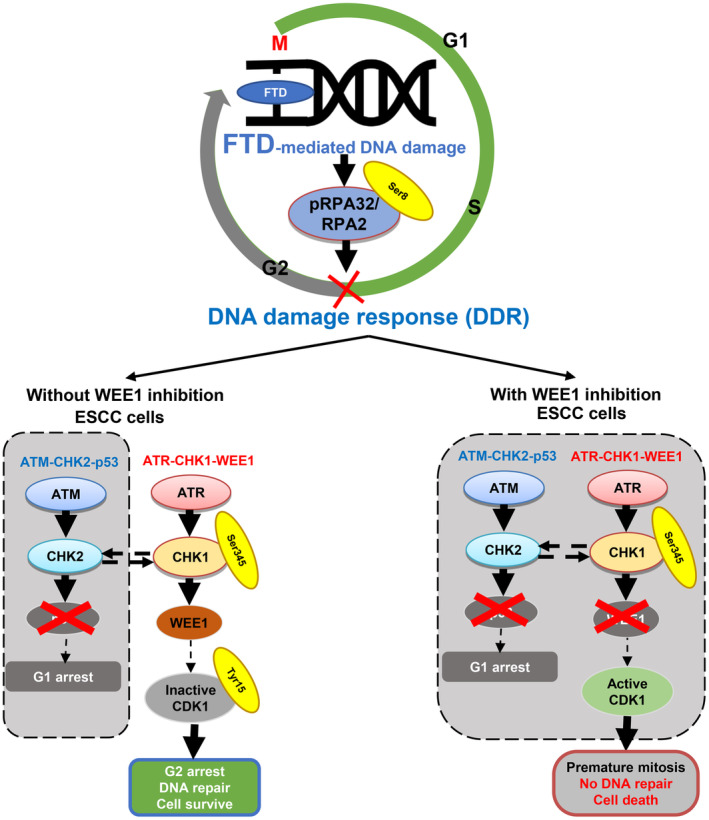
Proposed combination treatment strategy to tackle p53‐mutant esophageal squamous cell carcinoma (ESCC) tumors with a WEE1 inhibitor. Cotreatment with trifluridine (FTD) and MK1775 inhibited ataxia telangiectasia and Rad3‐related protein (ATR)‐checkpoint kinase 1 (CHK1)‐WEE1, a compensation DNA damage response pathway in p53‐mutant ESCC cells, resulting in cytotoxicity via synthetic lethality.
In this study, the antitumor effect of combination treatments with FTD/TPI and MK1775 was stronger than that with either treatment alone. Because we could not show any significant interaction (synergistic effect) between the two compounds, FTD/TPI and MK1775, in both TE‐8 and PDX xenograft tumors, a combination of this treatment is considered to exert an additive antitumor effect on ESCC. This result is inconsistent with our previous report, 22 which showed a synergistic effect of FTD/TPI and prexasertib (CHK1 inhibitor). This discrepancy may be due to the difference between the WEE1 and CHK1 inhibitors. As WEE1 is a downstream target of CHK1, it may take time for the WEE1 inhibitor to work properly. Our in vitro data showing that FTD needs time to activate downstream of the DDR pathway, phosphorylate CDK1, and arrest cell cycle appear to support this idea. However, the variation in number of treatment cycles between two xenografts may be the result of the difference in the background of mutation between cancer cells and actual patients. On the other hand, a remarkable percentage of γ‐H2AX induced by combination and a good synergy were observed, which has encouraged us for further investigation in the future to fully comprehend the effectiveness and impact of this combination.
In addition, we also showed that chemoradiotherapy with FTD/TPI, MK1775, and radiation had a good antitumor effect in vivo. FTD/TPI monotherapy has shown some good positive but moderate effect on sensitivity to radiotherapy in metastatic colon cancer. 35 Together with that, MK1775 was also proven to enhance the sensitivity to radiotherapy in ESCC. 36 Therefore, we believe that this combination of FTD/TPI and MK1775 can be a good candidate to improve the outcome of radiotherapy effectiveness. To our knowledge, this is the first study reporting the radiosensitizing effect of the combination of MK1775 and FTD/TPI. Therefore, a triple‐treatment plan combining FTD/TPI, MK1775, and radiation therapy might be a novel, powerful therapeutic strategy for patients with ESCC who need strong loco‐regional control at the primary site. However, we did not see tumor shrinkage or remission in our in vivo experiments, probably because of the suboptimal radiation dose for the TE‐8 model we used.
Although there have been upgrades in identifying genomic drivers, clinically approved targeted therapies are limited. 37 Recent advances in treatment against unresectable ESCC include the application of anti‐PD‐1 inhibitors and an anti‐CTLA4 inhibitor that improves the overall survival (OS) of patients with ESCC. 38 , 39 However, given the modest response rate of immunotherapy, improving chemotherapy against ESCC using molecular‐targeted therapy such as our novel FTD/TPI and MK1775 combination is still fundamental. The FTD/TPI and MK1775 combination could be a good candidate strategy as the last line of treatment after taxanes, given the good tolerability of FTD/TPI shown in our previous phase II trial. 12 Several trials are already evaluating WEE1 inhibitors in non‐small cell lung, ovarian, colon, and squamous cells of head and neck cancer when using monotherapy or combined with other cytotoxic agents. In a phase II trial conducted in 718 patients with RAS/TP53 mutant metastatic colorectal cancer (FOCUS4‐C), monotherapy of MK1775 increased PFS with good tolerance but no improvement in OS. 40
On the other hand, many preclinical and clinical studies have demonstrated the antitumor effect of MK1775 in combination with chemotherapy targeting the S and G2 phases. Therefore, a phase II study evaluated MK1775 in a single dose and combined it with different chemotherapies (gemcitabine, carboplatin, paclitaxel, or pegylated liposomal doxorubicin) in patients with primary platinum‐resistant ovarian, fallopian tube, or peritoneal cancer. 41 The highest efficacy of MK1775 was obtained in combination with carboplatin with a response rate of 66.7% and a median PFS of 12 months. 42 The results of this trial to achieve good efficacy using the combination of an anti‐DDR inhibitor and a cytotoxic agent were consistent with our study.
Our study has several limitations. First, the safety profiles of WEE1 inhibitor and FTD/TPI have not been investigated, although no apparent adverse events were found in mice. We expect good tolerability of our combination strategy in patients because the mechanism of action of the WEE1 inhibitor depends on P53 mutation 43 , 44 ; however, the phase I clinical trial must be conducted carefully to determine tolerable doses of both drugs. Second, the efficacy of the WEE1 inhibitor may differ among the patients considering the different sensitivity to MK1775 in different cell lines. Although all the cell lines and PDX we used in this report had TP53 mutation, there might be some genetic or epigenetic factors which affect the efficacy of the combination therapy.
In conclusion, we demonstrated the cytotoxicity and antitumor effects of FTD and WEE1 inhibition in ESCC cells and their mechanism to desuppress CDK1 by WEE1 inhibition to run cell division, thereby suggesting a novel therapeutic strategy for patients with ESCC. Furthermore, we have shown two possibilities for this strategy, namely chemotherapy and chemoradiotherapy. Further validation studies in clinical trials are needed.
FUNDING INFORMATION
Taiho Pharmaceutical Co., Ltd. provided partial funding for this project. T. Nguyen is supported by grants from the Japan Material Foundation, Support for Pioneering Graduate Students presented by the Kyoto University Graduate Division. This work was funded in part by a Grant‐in‐Aid for Scientific Research (grant No. 22K07960: Shinya Ohashi).
CONFLICT OF INTEREST STATEMENT
S. Ohashi and M. Muto report receiving other commercial research support from Taiho Pharmaceutical Co. Ltd. O. Kikuchi reports receiving other commercial research support from Taiho and AstraZeneca and honoraria from Taiho Pharmaceutical Co. Ltd. No potential conflicts of interest were disclosed by the other authors.
ETHICS STATEMENTS
Approval of the research protocol by an Institutional Review Board: The Ethics Committee of Kyoto University (Kyoto, Japan, G0770‐4).
Informed Consent: Yes.
Registry and the Registration No. of the study/trial: N/A.
Animal Studies: Approved by Institutional Animal Care and Use Committee at Kyoto University (MedKyo21505).
Supporting information
Figure S1.
Figure S2.
Figure S3.
Table S1.
Table S2.
ACKNOWLEDGMENTS
The authors would like to thank the Medical Research Support Center, Graduate School of Medicine, Kyoto University for flow cytometry, supported by the Platform for Drug Discovery, Informatics, and Structural Life Science from the Ministry of Education, Culture, Sports, Science and Technology, Japan. In addition, the authors thank the Center for Anatomical, Pathological, and Forensic Medical Research, Kyoto University Graduate School of Medicine for preparing the microscope slides.
Nguyen Vu TH, Kikuchi O, Ohashi S, et al. Combination therapy with WEE1 inhibition and trifluridine/tipiracil against esophageal squamous cell carcinoma. Cancer Sci. 2023;114:4664‐4676. doi: 10.1111/cas.15966
REFERENCES
- 1. Ohashi S, Miyamoto S, Kikuchi O, Goto T, Amanuma Y, Muto M. Recent advances from basic and clinical studies of esophageal squamous cell carcinoma. Gastroenterology. 2015;149:1700‐1715. [DOI] [PubMed] [Google Scholar]
- 2. Sung H, Ferlay J, Siegel RL, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209‐249. [DOI] [PubMed] [Google Scholar]
- 3. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 4. Mariette C, Piessen G, Triboulet JP. Therapeutic strategies in oesophageal carcinoma: role of surgery and other modalities. Lancet Oncol. 2007;8:545‐553. [DOI] [PubMed] [Google Scholar]
- 5. van Hagen P, Hulshof MC, van Lanschot JJ, et al. Preoperative chemoradiotherapy for esophageal or junctional cancer. N Engl J Med. 2012;366:2074‐2084. [DOI] [PubMed] [Google Scholar]
- 6. Ando N, Kato H, Igaki H, et al. A randomized trial comparing postoperative adjuvant chemotherapy with cisplatin and 5‐fluorouracil versus preoperative chemotherapy for localized advanced squamous cell carcinoma of the thoracic esophagus (JCOG9907). Ann Surg Oncol. 2012;19:68‐74. [DOI] [PubMed] [Google Scholar]
- 7. Shapiro J, van Lanschot JJB, Hulshof M, et al. Neoadjuvant chemoradiotherapy plus surgery versus surgery alone for oesophageal or junctional cancer (CROSS): long‐term results of a randomised controlled trial. Lancet Oncol. 2015;16:1090‐1098. [DOI] [PubMed] [Google Scholar]
- 8. Tachimori Y, Ozawa S, Numasaki H, et al. Comprehensive registry of esophageal cancer in Japan, 2011. Esophagus. 2018;15:127‐152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shitara K, Doi T, Dvorkin M, et al. Trifluridine/tipiracil versus placebo in patients with heavily pretreated metastatic gastric cancer (TAGS): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet Oncol. 2018;19:1437‐1448. [DOI] [PubMed] [Google Scholar]
- 10. Zaniboni A, Bertocchi P, Barni S, Petrelli F. TAS‐102 (Lonsurf) for the treatment of metastatic colorectal cancer. A concise review. Clin Colorectal Cancer. 2016;15:292‐297. [DOI] [PubMed] [Google Scholar]
- 11. Lenz HJ, Stintzing S, Loupakis F. TAS‐102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev. 2015;41:777‐783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mori Y, Kikuchi O, Horimatsu T, et al. Multicenter phase II study of trifluridine/tipiracil for esophageal squamous carcinoma refractory/intolerant to 5‐fluorouracil, platinum compounds, and taxanes: the ECTAS study. Esophagus. 2022;19:444‐451. [DOI] [PubMed] [Google Scholar]
- 13. Kaelin WG. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689‐698. [DOI] [PubMed] [Google Scholar]
- 14. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179‐204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996;274:1664‐1672. [DOI] [PubMed] [Google Scholar]
- 16. Zhou B‐BS, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature. 2000;408:433‐439. [DOI] [PubMed] [Google Scholar]
- 17. O'Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547‐560. [DOI] [PubMed] [Google Scholar]
- 18. Xiao Z, Chen Z, Gunasekera AH, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA‐damaging agents. J Biol Chem. 2003;278:21767‐21773. [DOI] [PubMed] [Google Scholar]
- 19. Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177‐2196. [DOI] [PubMed] [Google Scholar]
- 20. Kim J, Bowlby R, Mungall AJ, et al. Integrated genomic characterization of oesophageal carcinoma. Nature. 2017;541:169‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Song Y, Li L, Ou Y, et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature. 2014;509:91‐95. [DOI] [PubMed] [Google Scholar]
- 22. Ohashi S, Kikuchi O, Nakai Y, et al. Synthetic lethality with trifluridine/tipiracil and checkpoint kinase 1 inhibitor for esophageal squamous cell carcinoma. Mol Cancer Ther. 2020;19:1363‐1372. [DOI] [PubMed] [Google Scholar]
- 23. Morgan DO. Principles of CDK regulation. Nature. 1995;374:131‐134. [DOI] [PubMed] [Google Scholar]
- 24. Castedo M, Perfettini JL, Roumier T, Kroemer G. Cyclin‐dependent kinase‐1: linking apoptosis to cell cycle and mitotic catastrophe. Cell Death Differ. 2002;9:1287‐1293. [DOI] [PubMed] [Google Scholar]
- 25. Parker LL, Piwnica‐Worms H. Inactivation of the p34cdc2‐cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992;257:1955‐1957. [DOI] [PubMed] [Google Scholar]
- 26. Fisher DL, Nurse P. A single fission yeast mitotic cyclin B p34cdc2 kinase promotes both S‐phase and mitosis in the absence of G1 cyclins. EMBO J. 1996;15:850‐860. [PMC free article] [PubMed] [Google Scholar]
- 27. Nigg EA. Mitotic kinases as regulators of cell division and its checkpoints. Nat Rev Mol Cell Biol. 2001;2:21‐32. [DOI] [PubMed] [Google Scholar]
- 28. Kikuchi O, Ohashi S, Horibe T, et al. Novel EGFR‐targeted strategy with hybrid peptide against oesophageal squamous cell carcinoma. Sci Rep. 2016;6:22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Toda Y, Kono K, Abiru H, et al. Application of tyramide signal amplification system to immunohistochemistry: a potent method to localize antigens that are not detectable by ordinary method. Pathol Int. 1999;49:479‐483. [DOI] [PubMed] [Google Scholar]
- 30. Baba K, Nomura M, Ohashi S, et al. Experimental model for the irradiation‐mediated abscopal effect and factors influencing this effect. Am J Cancer Res. 2020;10:440‐453. [PMC free article] [PubMed] [Google Scholar]
- 31. Doi T, Ohtsu A, Yoshino T, et al. Phase I study of TAS‐102 treatment in Japanese patients with advanced solid tumours. Br J Cancer. 2012;107:429‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fotedar R, Roberts JM. Cell cycle regulated phosphorylation of RPA‐32 occurs within the replication initiation complex. EMBO J. 1992;11:2177‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. McGowan CH, Russell P. Human Wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12:75‐85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hendzel MJ, Wei Y, Mancini MA, et al. Mitosis‐specific phosphorylation of histone H3 initiates primarily within pericentromeric heterochromatin during G2 and spreads in an ordered fashion coincident with mitotic chromosome condensation. Chromosoma. 1997;106:348‐360. [DOI] [PubMed] [Google Scholar]
- 35. Rothkamm K, Christiansen S, Rieckmann T, et al. Radiosensitisation and enhanced tumour growth delay of colorectal cancer cells by sustained treatment with trifluridine/tipiracil and X‐rays. Cancer Lett. 2020;493:179‐188. [DOI] [PubMed] [Google Scholar]
- 36. Yang L, Shen C, Pettit CJ, et al. Wee1 kinase inhibitor AZD1775 effectively sensitizes esophageal cancer to radiotherapy. Clin Cancer Res. 2020;26:3740‐3750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fatehi Hassanabad A, Chehade R, Breadner D, Raphael J. Esophageal carcinoma: towards targeted therapies. Cell Oncol. 2020;43:195‐209. [DOI] [PubMed] [Google Scholar]
- 38. Janjigian YY, Shitara K, Moehler M, et al. First‐line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro‐oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open‐label, phase 3 trial. Lancet. 2021;398:27‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kato K, Cho BC, Takahashi M, et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION‐3): a multicentre, randomised, open‐label, phase 3 trial. Lancet Oncol. 2019;20:1506‐1517. [DOI] [PubMed] [Google Scholar]
- 40. Seligmann JF, Fisher DJ, Brown LC, et al. Inhibition of WEE1 is effective in TP53‐ and RAS‐mutant metastatic colorectal cancer: a randomized trial (FOCUS4‐C) comparing adavosertib (AZD1775) with active monitoring. J Clin Oncol. 2021;39:3705‐3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rajeshkumar NV, De Oliveira E, Ottenhof N, et al. MK‐1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53‐deficient pancreatic cancer xenografts. Clin Cancer Res. 2011;17:2799‐2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore KN, Chambers SK, Hamilton EP, et al. Adavosertib with chemotherapy in patients with primary platinum‐resistant ovarian, fallopian tube, or peritoneal cancer: an open‐label, four‐arm, phase II study. Clin Cancer Res. 2022;28:36‐44. [DOI] [PubMed] [Google Scholar]
- 43. Ku BM, Bae YH, Koh J, et al. Mutational status of TP53 defines the efficacy of Wee1 inhibitor AZD1775 in KRAS‐mutant non‐small cell lung cancer. Oncotarget. 2017;8:67526‐67537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bridges KA, Hirai H, Buser CA, et al. MK‐1775, a novel Wee1 kinase inhibitor, radiosensitizes p53‐defective human tumor cells. Clin Cancer Res. 2011;17:5638‐5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Figure S2.
Figure S3.
Table S1.
Table S2.