

Abstract

The development of selective catalysts for direct conversion of ammonia into nitrous oxide, N2O, will circumvent the conventional five-step manufacturing process and enable its wider utilization in oxidation catalysis. Deviating from commonly accepted catalyst design principles for this reaction, reliant on manganese oxide, we herein report an efficient system comprised of isolated chromium atoms (1 wt %) stabilized in the ceria lattice by coprecipitation. The latter, in contrast to a simple impregnation approach, ensures firm metal anchoring and results in stable and selective N2O production over 100 h on stream up to 79% N2O selectivity at full NH3 conversion. Raman, electron paramagnetic resonance, and in situ UV–vis spectroscopies reveal that chromium incorporation enhances the density of oxygen vacancies and the rate of their generation and healing. Accordingly, temporal analysis of products, kinetic studies, and atomistic simulations show lattice oxygen of ceria to directly participate in the reaction, establishing the cocatalytic role of the carrier. Coupled with the dynamic restructuring of chromium sites to stabilize intermediates of N2O formation, these factors enable catalytic performance on par with or exceeding benchmark systems. These findings demonstrate how nanoscale engineering can elevate a previously overlooked metal into a highly competitive catalyst for selective ammonia oxidation to N2O, paving the way toward industrial implementation.
Keywords: single-atom catalysis, ammonia oxidation, nitrous oxide, chromium, ceria
Introduction
Selective oxidations of hydrocarbons represent a key challenge to meet the modern standards of sustainability.1−6 Nitrous oxide, N2O, can aid in resolving it, being a highly selective oxidant able to donate a single oxygen atom while generating inert N2 as the sole byproduct.7,8 Accordingly, the issue of substrate overoxidation, frequently encountered when using O2, is avoided, and numerous industrially relevant reactions have been shown to proceed in fewer steps and with increased selectivity.9−12 However, despite the momentum garnered in the early 2000s to utilize N2O for chemical synthesis, its widespread adoption is still hindered by the high cost of N2O manufacturing, comprising five steps starting from NH3. Instead, propelled by the recent advances in the field of blue and green NH3 production, direct NH3 oxidation to N2O has emerged as a more efficient and sustainable synthesis method but requires the design of a suitable catalyst.13−15 In this endeavor, manganese oxide-based materials have been most widely investigated, leading to the discovery of a promising Mn–Bi–O/α-Al2O3 system at the Boreskov Institute of Catalysis.16 Still, issues of suboptimal N2O selectivity, catalyst deactivation, and the necessity to work in excess of O2, incurring significant downstream costs, have precluded commercialization.17,18 Recently, our group has reported CeO2-supported gold nanoparticles and low-valent manganese single atoms as highly efficient catalysts for NH3 oxidation to N2O.19,20 Leveraging the redox properties of CeO2 has enabled operation under stoichiometric conditions achieving N2O selectivity above 80% and 3- to 4-fold higher productivity per gram of catalyst than Mn–Bi–O/α-Al2O3. This has demonstrated that precise control over the nanostructure of the metal and the properties of the carrier are key to maximizing the catalytic effect21,22 and could be translated to other underexplored materials. In this respect, chromium presents an intriguing avenue of research. Owing to its capacity to assume a range of oxidation states, it has a prominent role in oxidation catalysis, such as oxidative alkane dehydrogenation23−25 and methane oxidation26,27 processes, among others. Furthermore, several recent works have reported the superior catalytic activity of chromium single-atom catalysts (SACs), such as Cr/TiO2 for CH4 oxidation26 or Cr/MWCNT for oxidative desulfurization.28 Still, very little work has been done on investigating the potential of Cr in catalyzing NH3 oxidation, reflected by only a few reports, primarily focused on NO formation in the high temperature (>1073 K) regime over bulk and supported chromium oxides.29−31 The relatively lower catalytic activity compared to manganese oxides and challenges of deactivation via irreversible reduction have led to its abandonment. However, nanoscale engineering may allow the subversion of previously encountered issues and the emergence of new catalytic synergies between the metal and the support.
Herein, we report the first effective application of chromium as a selective catalyst for NH3 oxidation to N2O, used in the form of isolated atoms embedded within the ceria lattice by coprecipitation (CP) and rivaling the performance of the state-of-the-art systems. The incorporation of a low amount of the metal into the carrier, as opposed to simple deposition on the surface, is shown to both (i) robustly anchor Cr sites, ensuring stability for 100 h on stream and (ii) facilitate the generation of oxygen vacancies, increasing their density. The Cr-induced modification of the CeO2 carrier further extends to the kinetics of associated redox processes, accelerating vacancy healing, as demonstrated by in situ UV–vis spectroscopy. This is complemented by the transient kinetic, spectroscopic, and computational analyses, showing the lattice oxygen of CeO2 to directly participate in N2O formation. Thus, the superior oxygen buffer capacity, coupled with the ability of Cr sites to dynamically restructure, stabilizing key intermediates of N2O synthesis, is identified as the central element governing catalytic performance. Our results highlight the importance of atomic precision in catalyst nanostructuring to unlock the potential of commonly overlooked metals and bring NH3 oxidation to N2O one step closer to practical application.
Experimental Section
Catalyst Preparation
Supported chromium catalysts on various carriers (Cr/support, support = CeO2, ZrO2, Al2O3, and Nb2O5) were synthesized via the incipient wetness impregnation (IWI) method with a nominal Cr content of 1 wt %. Accordingly, the metal precursor, Cr(NO3)3·9H2O (Sigma-Aldrich, 99%), was dissolved in deionized water and the resulting solution was added dropwise to the respective support, including CeO2 (Sigma-Aldrich, nanopowder <25 nm), m-ZrO2 (Alfa Aesar, catalyst support), α-Al2O3 (Alfa Aesar, 99.98%), and Nb2O5 (Sigma-Aldrich, 99.99%). After impregnation, all of the samples were dried overnight under vacuum at 353 K and then calcined in static air at 823 K (heating rate = 3 K min–1, hold time = 5 h).
CrCeOx-T catalysts (T = calcination temperature, i.e., 673, 873, or 1073 K) were synthesized via the CP method with a nominal Cr content of 1 wt %. Accordingly, the metal precursors, Cr(NO3)3·9H2O (Sigma-Aldrich, 99%) and Ce(NO3)3·6H2O (Sigma-Aldrich, 99%) were dissolved in 80 cm3 water. H2O2 (Sigma-Aldrich, 50 wt % in H2O) was added to oxidize the Ce3+ to the more easily hydrolyzable Ce4+ with a molar ratio n(H2O2)/(n(Ce) + n(Cr)) = 3 as reported elsewhere.19,32 NH3 (VWR Chemicals, 25%) was added dropwise under vigorous stirring until a pH of 9.5 was reached. The suspension was further stirred for 24 h and subsequently filtered. The residue was washed with at least 1 L of water and dried overnight in a vacuum oven at 353 K. The dried powder was calcined in static air at 673, 873, or 1073 K (heating rate of 3 K min–1, hold time of 5 h).
Catalyst Characterization
Inductively coupled plasma optical emission spectrometry was conducted by using a Horiba Ultra 2 instrument equipped with a photomultiplier tube detector. The sample was dissolved in an Anton-Paar Multiwave 7000 microwave digestion system, using a 1:3 mixture of HCl (VWR Chemicals, 37%) and HNO3 (Sigma-Aldrich, 65%) at 513 K and 80 bar of Ar.
Powder X-ray diffraction (XRD) measurements were conducted on a Rigaku SmartLab diffractometer using Cu–Kα radiation (λ = 0.1541 nm). The data were recorded in the 2θ range of 10–70° with an angular step size of 0.017° and a counting time of 0.26 s per step.
N2 sorption was measured at 77 K in a Micrometrics TriStar II instrument. The sample (mcat = 0.2 g, particle size <0.2 mm) was degassed at 473 K for 3 h prior to the analysis.
Volumetric chemisorption of O2 was performed at 673 K in a Micromeritics 3Flex Chemi instrument. Prior to the measurement, the sample (mcat = 0.1 g, particle size <0.2 mm) was loaded into a U-shaped quartz microreactor, dried in He at 393 K (heating rate = 10 K min–1, hold time = 1 h), evacuated (hold time = 30 min), and then cooled down to 303 K under vacuum. The sample was then heated to 673 K (heating rate = 10 K min–1) and the O2 adsorption isotherms were measured.
High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was conducted on an aberration-corrected Hitachi HD2700CS microscope operated at 200 kV. Samples were prepared by dipping the copper grid supporting a perforated carbon foil in a suspension of the solid in ethanol and drying in air. Energy-dispersive X-ray spectroscopy (EDXS) was performed on a Thermo Fisher Scientific Talos F200X microscope with a high-brightness field emission gun operated at an acceleration potential of 200 kV. The EDXS system of this microscope is composed of 4 silicon drift detectors that enable the recording of EDXS maps with a proper signal-to-noise ratio in a relatively short collection time (5–15 min). HAADF-STEM, high-resolution scanning transmission electron microscopy (HRSTEM) images, and EDXS maps of as-prepared and used CrCeOx-673 and Cr/CeO2 catalysts, presented in Figure S12 of the manuscript, were acquired with a probe aberration-corrected Titan Themis operated at 300 kV, equipped with a Super-X EDX detector. The sample was prepared by dipping the copper grid supporting a perforated carbon foil in a suspension of the solid in methanol and drying it in air. The sample was cleaned gently with argon–oxygen plasma to reduce sample contamination.
X-ray photoelectron spectroscopy (XPS) was performed on a Physical Electronics Quantum 2000 spectrometer using monochromatic Al–Kα radiation, generated by an electron beam operated at 15 kV, and equipped with a hemispherical capacitor electron-energy analyzer. The sample was analyzed at an electron take-off angle of 45° and a constant analyzer pass energy of 46.95 eV with a spectra resolution step width of 0.2 eV. All XPS signals were referenced using the C 1s photoemission of adventitious carbon, which was set at 284.8 eV. Fitting of the acquired Cr 2p3/2 spectra was performed based on the parameters reported elsewhere, using CasaXPS software.33
Temperature-programmed reduction with hydrogen (H2-TPR) was acquired in a Micromeritics Autochem HP unit equipped with a thermal conductivity detector. The sample (mcat = 0.3 g, particle size <0.2 mm) was loaded into a U-shaped quartz microreactor, dried in Ar at 473 K (total flow rate, FT = 20 cm3 min–1, heating rate = 20 K min–1, hold time = 30 min), and then cooled to 303 K. The sample was subsequently heated to 1073 K in flowing 5 vol % H2 in Ar (FT = 20 cm3 min–1, heating rate = 10 K min–1).
Raman spectroscopy was performed on a Horiba LabRAM HR Evolution UV–vis–NIR confocal Raman system using a Cobolt Samba Nd/YAG laser with a wavelength of 532 nm, a power of 3.2 mW and a 50× Olympus LMPlanFLN objective. Spectra were collected with an acquisition time of 10 s and an accumulation number of 5.
In situ ultraviolet–visible (UV–vis) spectroscopy was performed by using an Avantes AVASPEC fiber optical spectrometer equipped with an AvaLight-DH-S-BAL deuterium-halogen light source and a CCD array detector. The details of the UV–vis setup were described elsewhere.34 BaSO4 was used as a white reference material. The catalyst (mcat = 0.2 g, particle size = 0.25–0.35 mm) was loaded into a quartz reactor (inner diameter = 6 mm). The catalyst bed was fixed between two layers of quartz wool. A high-temperature reflection probe, including six light fibers and one reading fiber, was positioned perpendicular to the reactor. The catalyst was heated to 673 K in flowing 10 vol % O2 in Ar (FT = 20 cm3 min–1, heating rate = 10 K min–1, hold time = 15 min). The time-resolved UV–vis spectra (λ = 200–800 nm) were recorded every 30 s. Subsequently, the sample was flushed with Ar (hold time = 20 min), while the spectral collection was continued every 60 s. The reduction of the sample was then performed in flowing 1 vol % NH3 in He (FT = 20 cm3 min–1, hold time = 30 min), followed by flushing with Ar (hold time = 20 min) and reoxidation in flowing 3 vol % O2 in Ar (FT = 20 cm3 min–1, hold time = 30 min). During the reduction and reoxidation treatments, UV–vis spectra in the range of 250–800 nm and the Kubelka–Munk (KM) function at 700 nm were recorded every 5 and 1 s, respectively. Rrel was defined as a ratio of reflectance of the reduced sample, Rreduced, to that of the fully oxidized one, Roxidized (eq 1). It was vice versa for Rrel during reoxidation. From this reflectance, the relative KM function F(Rrel) was calculated according to eq 2. The KM function was determined from the ratio of the reflectance of the oxidized sample to that of BaSO4:
| 1 |
| 2 |
Temporal analysis of products (TAP) experiment was performed in the TAP-2 reactor system operating in pulse mode with a time resolution of ca. 0.100 ms.35,36 The catalyst (mcat = 0.38 g, particle size = 0.15–0.4 mm) was placed between two layers of quartz particles (particle size = 0.25–0.35 mm) within the isothermal zone of an in-house-developed quartz reactor (inner diameter = 6 mm, length = 40 mm). Prior to pulse experiments, the catalyst was heated to 773 K in flowing O2 (FT = 4 cm3 min–1) and kept in this flow for 0.5 h. After that it was cooled to 473 K and exposed to vacuum of ca. 10–5 Pa. Single-pulse experiments with 18O2/NH3/Ar = 1:1:1 were performed at 673 K. The total pulse size was between 7 × 1015 and 1.4 × 1016 molecules. Multipulse experiments with an NH3/Ar = 1:1 mixture were carried out at 673 K with the total pulse size of 7–9 × 1015 molecules. The feed mixtures were prepared by using NH3 (Messer Griesheim, 3.8), 18O2 (Campro Scientific, 97% 18O2), and Ar (Air Liquide, 5.0) without additional purification. Feed components and reaction products were quantified by an online quadrupole mass spectrometer (HAL RC 301, Hiden Analytics) at the following m/z values: 46 (N218O), 44.0 (N216O), 36.0 (18O2), 34.0 (18O16O), 32.0 (16O2, N18O), 30.0 (N16O), 28.0 (N2), 19.0 (H218O), 17.0 (H216O), 15.0 (NH3), 2.0 (H2), and 40.0 (Ar). During experiments with the 18O2/NH3/Ar = 1:1:1 mixture, the pulses were repeated 10 times for each m/z and averaged to improve the signal-to-noise ratio. In the multipulse experiments, the pulses were repeated 30 times for each m/z and treated separately without averaging. The contribution of the compounds to the respective m/z values was estimated by using standard fragmentation patterns determined in separate experiments.
Continuous-wave electron paramagnetic resonance (CW-EPR) spectroscopy experiments were conducted on a Bruker Elexsys E500 spectrometer operating at X-band frequencies, using an ER4102ST microwave resonator and equipped with an Oxford helium (ESR900) cryostat. All CW-EPR spectra were acquired at room temperature or 10 K with the following spectrometer parameters: microwave frequency = 9.8 GHz, sweep width = 590 mT, center field = 300 mT, modulation frequency = 100 kHz, modulation amplitude = 5 G, microwave power = 2.012 mW, power attenuation = 20 dB, conversion time = 1310.72 ms, and time constant = 327.68 ms. All measured g-factors were offset-corrected against a known standard (i.e., free radical 1,1-diphenyl-2-picrylhydrazyl).
Catalytic Evaluation
The catalytic performance in ammonia oxidation was evaluated at atmospheric pressure in a fixed-bed microreactor (Figure S1).19,20 The gases, He (PanGas, purity 4.6, diluent), NH3 (PanGas, purity 3.8), O2 (PanGas, purity 5.0), and Ar (PanGas, purity 5.0, internal standard), were fed using thermal mass-flow controllers (Bronkhorst), connected to a mixing unit equipped with a pressure gauge. The catalyst [particle size = 0.15–0.4 mm, mcat = 0.002–0.2 g for kinetic tests, and 0.05 g for stability tests; for tests at elevated GHSV (>15,000 cm3 h–1 gcat–1) the catalyst bed was diluted with SiC (particle size = 0.5–0.6 mm) to minimize the formation of hot spots] was loaded into a quartz microreactor (inner diameter = 8 mm for mcat > 0.03 g or 2 mm for mcat < 0.03 g), containing a bed made of quartz wool and placed in an electrical oven. The temperature of the catalyst bed was monitored and controlled using a K-type thermocouple placed in a coaxial quartz thermowell. Prior to testing, the catalyst was heated in a He flow (Tbed = 473 K, FT = 40 cm3 min–1) for 30 min, subsequently heated to the desired temperature (Tbed = 473–723 K) and allowed to stabilize for at least 30 min before the reaction mixture (8 vol % NH3, 8 vol % O2, 4 vol % Ar, and 80 vol % He) was fed at a total volumetric flow of FT = 50–250 cm3 min–1.
Nitrogen-containing compounds (NH3, N2, N2O, NO2, and NO), as well as O2 and Ar were quantified via an online gas chromatograph equipped with a GS CP-Volamine column coupled to a mass spectrometer (GC–MS, Agilent, GC 7890B, MSD 5977A). Upon acquisition of the full chromatogram, individual ion chromatograms at m/z 17, 28, 30, 32, 40, 44, and 46 were extracted. A single peak was observed on chromatograms at m/z 32, 40, and 44, allowing one to directly quantify O2, Ar, and N2O, respectively. Sufficiently different retention times of N2O (t = 1.966 s), N2 (t = 1.887 s), and NO (t = 1.892 s) allowed resolution of the peaks attributed to N2O fragments, N2 and NO in the product stream at m/z 28 and 30, so that subsequent quantification of N2 and NO could be performed. Sufficiently different retention times of NH3 (t = 1.985 s) and H2O (t = 2.065 s) allowed the resolution of their respective peaks at m/z 17 and quantification of NH3. No peaks were observed at m/z 46 for any of the investigated catalysts. The conversion of NH3 and O2 was calculated according to eq 3:
| 3 |
where ṅini and ṅouti denote the molar flows of NH3 or O2 at the reactor inlet and outlet, respectively. Selectivity toward individual products was determined according to eq 4:
| 4 |
where v is the number of N atoms in the product molecule (i.e., v = 2 for N2O or N2, and v = 1 for NO). The space-time yield (STY) of N2O was calculated according to eq 5:
| 5 |
where mcat denotes the catalyst mass. Nitrogen (BN) and oxygen (BO) balances were evaluated for each catalytic test according to eqs 6 and 7, respectively:
| 6 |
| 7 |
The error of BO was less than 5% in all experiments. Due to the nonlinear response of the NH3 MS signal at low concentrations, it was adjusted accordingly to achieve a comparable value of BN to BO. After the tests, the reactor was quenched to room temperature in a He flow and the catalyst samples were retrieved for further characterizations.
Computational Methods
To gain insights into the possible structural configurations of isolated chromium atoms on different surfaces of CeO2, and their corresponding reactivity, DFT modeling was conducted with the Vienna Ab Initio Simulation Package (VASP, versions 5.4.4 and 6.3.0),37 using the Perdew–Burke–Ernzerhof (PBE) functional,38 and the HSE03 hybrid functional with 13% exact exchange (HSE03-13).38,39 The valence electrons were extended in plane waves with a basis set cutoff of 500 eV.40,41 PBE + U framework was employed to carry out the structural relaxations. On the Ce 4f manifolds, an additional Hubbard U term (Ueff = 4.5 V) was applied, following previous literature reports.42−44 To avoid nonphysical charge transfer caused by favored electron localization on the Ce centers due to Hubbard correction, for the substitutional structures, we also applied a value of Ueff = 4.0 V on Cr 3d. Ideally, both values should be optimized and obtained self-consistently (for instance via density functional perturbation theory),45 however, as they further depend on the adapted oxidation state and chemical environment of the respective center, a consistent description throughout is challenging. Thus, we deem the selected choice of parameters as a reasonable compromise between an accurate description of the complex electronic structure, and computational feasibility, in line with previous theoretical studies.46 Projector augmented wave (PAW) method was applied to the core electron, utilizing appropriate PAW–PBE pseudopotentials. Simulations were performed spin unrestricted, applying dipole corrections where appropriate. The threshold for electronic convergence was set at 1 × 10–6 eV and the positions of atoms were relaxed until residual forces reached 0.015 eV Å–1. For selected structures, the HSE03-13 hybrid functional was also used to refine the energies, fixing the atomic coordinates at the PBE + U optimized positions. In view of the significantly higher computational cost of HSE03-13, the threshold for electronic convergence was lowered to 1 × 10–4 eV. The “automated interactive infrastructure and database for computational science” (AiiDA) was used to drive the simulations on the high-performance computing facilities.47−49 Dipole corrections and spin polarization were applied throughout, while sampling of the Brillouin zone was restricted to the Gamma point.
For the (111), (110), and (100) facets of CeO2 slab models were constructed as (3 × 3), (2 × 2) and (3 × 3) supercells, extending 9, 6, and 9 atomic layers along the z-direction, of which the bottom 4, 3, and 4 layers were fixed at the optimized bulk positions, respectively. At least 10 Å of vacuum was added on top of the surfaces to minimize interactions of vertically repeated slab images under periodic boundary conditions. Formal oxidation states of chromium single-atoms (SAs) were assigned using the localized magnetic moments of reduced Ce3+ centers, where a threshold of 0.8 μB was applied.50 SA chromium adsorption and substitution energies were calculated following eqs 8 and 9, where EadsSAC and EsubSAC are the energies of the respective SACs, Epris is the energy of the corresponding pristine ceria slab, ECr is the energy of SA chromium, using bulk Cr as reference (Ecoh = −9.49 (−10.32) eV atom–1 with PBE + U (HSE03)), and ECe is the energy of the substituted cerium atom, evaluated via eq 10. Adsorption energies of reactants and reaction intermediates were evaluated using eq 8, accordingly:
| 8 |
| 9 |
| 10 |
Results and Discussion
Synthesis–Performance Relationships
Aiming at
developing efficient Cr-based catalysts for selective NH3 oxidation, chromium was first deposited onto various support materials
by the IWI method with a nominal Cr content of 1 wt % (Table S1, Cr/support). The catalytic performance
of as-prepared materials in NH3 oxidation was evaluated
in a fixed-bed reactor (Figure S1). First,
the temperature of the catalyst bed was varied in the range of 473–723
K to investigate its effect on product selectivity and NH3 conversion (Figure S2). Cr/CeO2 displayed the highest activity, as reflected by the relatively lower
temperature at which 50% NH3 conversion was attained (T50, Figure 1a). It also showed the highest N2O selectivity,
reaching 64% at 623 K. To investigate whether this promotional effect
of CeO2 could be further enhanced through closer interaction
of the metal with the carrier, a set of chromium–cerium mixed
oxide catalysts was prepared via a CP method, maintaining the nominal
Cr content at 1 wt % and varying the calcination temperature (CrCeOx-T, T =
673, 873, or 1073 K). Notably, all of the CP samples showed similar
or superior N2O selectivity compared to Cr/CeO2, with CrCeOx-673 achieving a value of
79% at 673 K (Figure 1a). The synergy between Cr and CeO2 was further punctuated
by the fact that CeO2 alone is catalytically inactive.
As all of the temperature ramp experiments were performed employing
a low gas-hourly space velocity (GHSV = 15,000 cm3 h–1 gcat–1), it resulted
in complete NH3 conversion at temperatures relevant for
selective N2O formation (623–673 K). Accordingly,
to assess the intrinsic catalytic activity of each system, the applied
GHSV was varied (Figure S3), and the corresponding
data acquired at 20% NH3 conversion, including the STY
of N2O ( ), is shown in Figure 1a. It is immediately
apparent that the product
distribution of all catalysts was strongly affected by the decrease
in contact time, leading to a drastic reduction in N2O
selectivity in favor of N2. Nevertheless, the previously
observed trend holds with CrCeOx-673 clearly
standing out as the most intrinsically selective system and maintaining
this feature at varying degrees of NH3 conversion (Figure S4). Furthermore, CrCeOx-673 also achieved the highest
), is shown in Figure 1a. It is immediately
apparent that the product
distribution of all catalysts was strongly affected by the decrease
in contact time, leading to a drastic reduction in N2O
selectivity in favor of N2. Nevertheless, the previously
observed trend holds with CrCeOx-673 clearly
standing out as the most intrinsically selective system and maintaining
this feature at varying degrees of NH3 conversion (Figure S4). Furthermore, CrCeOx-673 also achieved the highest  of 687 mmol h–1 gcat–1, more than double that of its impregnated
counterpart, underscoring the importance of a strong interaction between
Cr and CeO2. Finally, the stability of each catalyst was
evaluated for 50 h on stream (Figure 1b). All materials prepared by IWI, as well as CrCeOx-1073 experienced deactivation, losing between
20 and 80% of their initial activity. Conversely, the two mixed oxide
catalysts calcined at a lower temperature exhibited stable performance,
emphasizing the importance of additional stabilization achieved when
embedding Cr within the ceria lattice. In fact, the duration of the
test for CrCeOx-673 was extended to 100
h, during which both the rate of NH3 conversion and the
corresponding product distribution remained essentially constant,
firmly establishing it as a stable, selective, and highly productive
material for N2O production via NH3 oxidation
(Figure 1c).
of 687 mmol h–1 gcat–1, more than double that of its impregnated
counterpart, underscoring the importance of a strong interaction between
Cr and CeO2. Finally, the stability of each catalyst was
evaluated for 50 h on stream (Figure 1b). All materials prepared by IWI, as well as CrCeOx-1073 experienced deactivation, losing between
20 and 80% of their initial activity. Conversely, the two mixed oxide
catalysts calcined at a lower temperature exhibited stable performance,
emphasizing the importance of additional stabilization achieved when
embedding Cr within the ceria lattice. In fact, the duration of the
test for CrCeOx-673 was extended to 100
h, during which both the rate of NH3 conversion and the
corresponding product distribution remained essentially constant,
firmly establishing it as a stable, selective, and highly productive
material for N2O production via NH3 oxidation
(Figure 1c).
Figure 1.

(a) Catalytic performance in NH3 oxidation of chromium supported on different supports (Cr/MOx), coprecipitated chromium–cerium oxides (CrCeOx-T), and CeO2 represented by the temperature at which 50% NH3 conversion is achieved (T50), the space-time yield (STY) of N2O (top panel), NH3 conversion and product selectivity (bottom panel). For each catalyst, the results acquired at two different gas-hourly space velocity (GHSV) values are shown, with the one on the right adjusted such that 20% NH3 conversion could be attained. (b) Stability test of selected catalysts expressed in terms of the normalized rate of N2O formation (r/r0), and (c) conversion and selectivity profile of CrCeOx-673 over 100 h on stream. Reaction conditions: Tbed = 673 K; mcat = 0.002–0.2 g; GHSV = 15,000–3,000,000 cm3 h–1 gcat–1 (a), 120,000 cm3 h–1 gcat–1 (b, c); feed composition = 8 vol % NH3, 8 vol % O2, 4 vol % Ar, 80 vol % He; P = 1 bar.
Oxygen Availability as a Performance Descriptor
Having observed stark differences between Cr-based materials in their ability to catalyze N2O production via NH3 oxidation, which appears to largely be a result of the support effect, we sought to identify the carrier property governing this behavior. Given the nature of the reaction and the variable reducibility of supports employed, redox properties were first assessed. To this end, H2-TPR of the catalysts and respective supports was performed (Figure S5). No reduction peak was observed for Cr/Nb2O5, which was tentatively attributed to the formation of a stable CrNbO4 phase.51 Conversely, a single reduction peak was observed for Cr/Al2O3 and Cr/ZrO2, at 520 and 550 K, respectively. In the former case, it likely originates from the reduction of Cr2O3 to Cr, whereas in the latter, considering the larger peak area, H2 likely also reacted with the surface oxygen of ZrO2. Similarly, in Cr/CeO2 a characteristic peak due to surface reduction of CeO2 was seen at 580 K, occurring at a lower temperature and with higher intensity than for the pristine support, suggesting that the introduction of Cr improves reducibility of CeO2. Among the three mixed oxide catalysts (CrCeOx-T), the presence of Cr also has a pronounced promotional effect on surface reducibility, with increasing calcination temperature leading to reduced H2 consumption and onset of reduction at a lower temperature. This is in line with variations in the surface area (SBET,Table S1), as the sample with the highest SBET value (i.e., CrCeOx-673) has a correspondingly large reduction peak, further validating that it is attributable to the removal of surface O atoms. Furthermore, the higher reduction temperature over CrCeOx-673 serves as a fingerprint of stronger interaction between CeO2 and Cr in comparison with those over CrCeOx-873 and CrCeOx-1073. This is likely the result of differences in CeO2 crystallite size (Figure S6), which was reported to influence the metal–support interactions.52 Indeed, increasing the calcination temperature is expected to lead to sintering, hence resulting in larger CeO2 particles and lower surface area.
To complement the H2-TPR measurements and evaluate the ability of the catalysts to interact with gas-phase oxygen, we performed O2 chemisorption experiments at 673 K (Figure 2a). The corresponding amount of chemisorbed O2, herein referred to as oxygen uptake, was found to be negligible in the case of nonreducible Nb2O5 and Al2O3, while ZrO2, commercial and lab-synthesized CeO2, all exhibited appreciable interaction with O2. It was found to be consistently enhanced upon the introduction of Cr, while the overall ranking of catalysts remained largely unaffected, with CrCeOx-673 having the largest oxygen uptake value. It should be noted that during pretreatment of the sample, it is exposed to a vacuum, which is known to strip labile surface oxygen atoms.53 Thus, the measured amount of chemisorbed O2 also takes into account lattice oxygen that had to be replenished. Accordingly, there is good agreement between hydrogen consumption in H2-TPR tests and the oxygen uptake values of the catalysts (Figure S7a). Finally, to verify whether the observed differences in oxygen uptake can indeed be attributed to differences in the density of oxygen vacancies, CeO2-based catalysts were investigated by means of Raman spectroscopy (Figure S8a). In all samples, the Raman feature at 595 cm–1 could be attributed to the defect band (D-band) caused by oxygen vacancies of CeO2.54,55 The relative intensity of the D-band decreased with a rising calcination temperature for CrCeOx-T catalysts. The peak intensity of CrCeOx-673 and CrCeOx-873 also indicates a higher density of oxygen vacancies than that of Cr/CeO2, prepared by IWI. Furthermore, the D-band of Cr-free CeO2 materials was consistently weaker than that of their Cr-containing counterparts (Figure S8b), indicating that the introduction of Cr species induces the formation of oxygen vacancies on CeO2 and therefore prompts O2 activation. These findings are all in agreement with the above-discussed results of O2 chemisorption measurements. Therefore, the oxygen uptake value can serve as a robust and quantitative measure of the availability of oxygen.
Figure 2.

(a) Oxygen uptake of carriers (open symbols),
and corresponding
Cr-based catalysts (solid symbols), determined by volumetric O2 chemisorption, (b) N2O selectivity and (c)  as a function of oxygen uptake, and (d)
as a function of oxygen uptake, and (d) 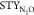 as a function of the partial reaction order
with respect to oxygen, n(O2). Reaction
conditions: Tbed = 673 K, mcat = 0.002–0.2 g; GHSV = 15,000–3,000,000
cm3 h–1 gcat–1; feed composition = 8 vol % NH3, 8 vol % O2, 4 vol % Ar, 80 vol % He; P = 1 bar.
as a function of the partial reaction order
with respect to oxygen, n(O2). Reaction
conditions: Tbed = 673 K, mcat = 0.002–0.2 g; GHSV = 15,000–3,000,000
cm3 h–1 gcat–1; feed composition = 8 vol % NH3, 8 vol % O2, 4 vol % Ar, 80 vol % He; P = 1 bar.
When the oxygen uptake values are plotted against
N2O selectivity, a clear trend emerges (Figure 2b). This can be understood
by considering
the stoichiometry of NH3 oxidation, which requires increasingly
more oxygen to form N2, N2O, and NO. Hence,
the ability to form the more oxidized species (i.e., N2O) is contingent upon the ample supply of oxygen, which is enabled
by the use of redox-active supports, primarily CeO2. This
point is further reinforced by the correlation between oxygen uptake
and 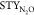 , which suggests that oxygen availability
also impacts the overall rate of NH3 conversion—an
expected outcome given that dehydrogenation of NH3 must
be driven by the elimination of H2O (Figure 2c). To validate the material’s ability
to modulate the oxygen supply as a key property governing catalytic
performance, macrokinetic analysis was performed. Accordingly, the
reaction rate was measured while varying the partial pressure of reactants
and reaction temperature to extract the corresponding reaction orders
(Figure S9) and the apparent activation
energy (Figure S10), respectively. The
obtained reaction orders with respect to O2 were found
to have an inverse relation with
, which suggests that oxygen availability
also impacts the overall rate of NH3 conversion—an
expected outcome given that dehydrogenation of NH3 must
be driven by the elimination of H2O (Figure 2c). To validate the material’s ability
to modulate the oxygen supply as a key property governing catalytic
performance, macrokinetic analysis was performed. Accordingly, the
reaction rate was measured while varying the partial pressure of reactants
and reaction temperature to extract the corresponding reaction orders
(Figure S9) and the apparent activation
energy (Figure S10), respectively. The
obtained reaction orders with respect to O2 were found
to have an inverse relation with 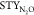 , indicating that the less dependent a material
is on the partial pressure of the O2, the higher its N2O productivity (Figure 2d). In fact, the best catalytic system (i.e., CrCeOx-673) exhibits a slightly negative reaction order
of −0.05, which is indicative of its ability to allow the reaction
to proceed via a Mars-van Krevelen (MvK) type mechanism,56 with the involvement of lattice oxygen, while
gas-phase O2 could be very easily activated and has a slightly
inhibiting effect. O2 poisoning has also been reported
for Pd single atoms on defect-rich CeO2 nanocrystals (<8
nm) in CO oxidation.52 Furthermore, a similar
inverse relation between
, indicating that the less dependent a material
is on the partial pressure of the O2, the higher its N2O productivity (Figure 2d). In fact, the best catalytic system (i.e., CrCeOx-673) exhibits a slightly negative reaction order
of −0.05, which is indicative of its ability to allow the reaction
to proceed via a Mars-van Krevelen (MvK) type mechanism,56 with the involvement of lattice oxygen, while
gas-phase O2 could be very easily activated and has a slightly
inhibiting effect. O2 poisoning has also been reported
for Pd single atoms on defect-rich CeO2 nanocrystals (<8
nm) in CO oxidation.52 Furthermore, a similar
inverse relation between 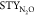 and the apparent activation energy of NH3 oxidation was identified (Figure S7b).
and the apparent activation energy of NH3 oxidation was identified (Figure S7b).
Chromium Speciation
Having established the material’s ability to facilitate the oxygen supply as a governing descriptor of catalytic ability, we endeavored to gain a deeper understanding of underlying structural features. To shed light on the speciation of Cr, HAADF-STEM coupled to EDXS of as-prepared catalysts was performed (Figures 3a and S11). The analysis revealed that Cr is homogeneously distributed on the ZrO2 and CeO2-based systems. In fact, elemental mappings of Cr/CeO2 and CrCeOx-673 acquired at the nanometer scale suggest atomic dispersion of Cr. Although high-resolution HAADF-(HR)STEM could not provide direct observations of single Cr atoms, due to the relatively much larger mass of Ce, it refuted the presence of clusters or nanoparticles. Thus, since CeO2 (111) is the most stable facet,57 and was commonly observed (Figure S12), single Cr atoms are expected to predominantly reside on CeO2 (111). In contrast, Cr-rich regions were detected over the Al2O3 and Nb2O5 supports. Notably, despite clear evidence of nanometer-sized Cr-containing particles being present in the latter, XRD patterns of pristine supports and as-prepared Cr catalysts evidenced no differences (Figure S13). This indicates that they are either poorly crystalline or too few in number to produce characteristic reflections. To understand the origin of distinct deactivation patterns that Cr catalysts exhibited, the samples were also studied by HAADF-STEM and EDXS after the stability test (Figure 3a; Figures S11 and S12). Evidence of Cr agglomeration was found in all samples, except for CrCeOx-673 and CrCeOx-873, which are incidentally the only two to remain stable during the catalytic test. This points to Cr dispersion having a central role in ensuring and maintaining high catalytic activity. Furthermore, the contrasting behavior of Cr/CeO2 and CrCeOx-673 highlighted the importance of employing a suitable synthetic technique to ensure the sufficiently strong anchoring of Cr atoms and thus catalytic stability. It is also notable that while the incorporation of Cr into the lattice of CeO2 effectively stabilized it and prevented its agglomeration during the reaction for the samples calcined at 673 and 873 K, some agglomeration was still observed in CrCeOx-1073. A possible explanation for this behavior is the fact that this sample also showed the weakest strength of the metal–support interaction and the highest reducibility, as shown by the H2-TPR analysis (Figure S5). This, in tandem with a large concentration of NH3 in the feed and elevated reaction temperature (673 K), could have induced the migration of Cr atoms, particularly from the bulk of the catalyst, via an ex-solution-like process.58,59 The tendency of solid solutions to undergo such transformations is associated with the ease of surface reduction,60,61 which could explain why CrCeOx-673 and CrCeOx-873 did not experience a similar transformation.
Figure 3.

(a) STEM-EDX maps, (b) Cr 2p3/2 XPS spectra, and (c) EPR spectra acquired at room temperature of selected as-prepared and used Cr-based catalysts.
Cr 2p3/2 XPS was subsequently employed to probe the electronic structure and discern the oxidation states of Cr in selected representative catalysts.33 The presence of both Cr6+ and Cr3+ species on the as-prepared Cr/CeO2 was identified, albeit with the former comprising only ∼20% of the total Cr content (Figure 3b). In contrast, only Cr3+ was observed on Cr/Al2O3, with Cr likely being present as the stable Cr2O3 phase (Figure 3a). Therefore, the type of support strongly affects the dispersion degree and oxidation state of the Cr species. The oxidation state of Cr in CrCeOx-673 was found to be predominantly Cr3+. This is likely the result of Cr occupying the positions of Ce4+ in the ceria lattice, forming Cr–O–Ce linkages, which inhibits further oxidation of Cr,62,63 and inducing the formation of oxygen vacancies, which is well in line with the high oxygen uptake of CrCeOx-673. Analysis of the used catalysts by XPS also revealed clear differences. After the reaction, Cr/CeO2 was comprised solely of Cr3+, which suggests that highly dispersed Cr6+ species were reduced and likely experienced agglomeration as a result, as seen in the corresponding micrographs. In contrast, the XPS spectra of as-prepared and used Cr/Al2O3 are hardly distinguishable. Similarly, the speciation of Cr in CrCeOx-673 is largely unchanged after 100 h on stream, with only a minor increase in Cr6+, perhaps as a result of the formation of a surface chromate species.
To deepen the understanding of Cr speciation, electron paramagnetic resonance (EPR) spectroscopy was utilized. The continuous-wave (CW) EPR spectrum of the as-prepared CrCeOx-673 acquired at room temperature was found to mainly consist of a narrow signal around g = 2, characterized by an anisotropic g tensor (Figure 3c). This signal could be attributed to low-spin Cr3+ (d3, S = 1/2) centers, corresponding to magnetically isolated ions in a highly distorted orthorhombic coordination. This indicates that Cr is highly dispersed and likely incorporated inside the ceria lattice. Additional minor signals are present and partially overlap with the main signal. These can be attributed to Cr5+ and/or Ce3+. Measurements at 10 K showed the presence of an additional weak low-field signal around g = 4 (Figure S14), characteristic of strongly axially distorted S = 3/2 centers with zero-field splitting, higher than the Zeeman interaction. This signal could be attributed to high-spin Cr3+ (d3, S = 3/2) in a more symmetric, tetragonally distorted environment, possibly coordinated on the surface. The Cr/CeO2 sample showed the same, yet significantly weaker, low-spin Cr3+ “single-atom” signal that was already observed for the CP sample (Figure 3c). The narrow Ce3+/Cr5+ signal is also observed. In addition, partially overlapping with the latter, a broader unstructured signal is present, which is most likely due to dipolar or exchange-coupled Cr3+ ions and may be attributed to small Cr2O3 clusters and aggregates. Furthermore, the overall intensity of the Cr3+ signals is significantly lower compared to CrCeOx-673, suggesting that a large amount of Cr3+ is present as EPR-silent antiferromagnetically coupled dimers. The low-temperature spectrum (Figure S14) also showed a g = 4 feature, attributed to high-spin Cr3+ in tetragonal coordination. Moreover, a series of broad peaks could be observed, centered around g = 2 and with an average spacing of approximately 800 G (Figure S14). This pattern could be attributed to the fine splitting due to spin–orbit coupling in a high-spin S = 3/2 system with zero-field splitting, comparable to the Zeeman interaction (and consequently lower than for the CP sample). The local geometry of surface-coordinated Cr3+ in Cr/CeO2 is therefore very different from CrCeOx-673, being less axially distorted and closer to orthorhombic. Finally, the Cr/Al2O3 sample exhibited a distinct, unstructured cluster signal, similar to the one observed for the Cr/CeO2 sample, showing that Cr is highly aggregated on the Al2O3 surface, most likely in the form of Cr2O3 clusters and nanoparticles.
When considering the room-temperature spectra of the used samples (Figure 3c), the low-spin Cr3+ signal in CrCeOx-673 decreased by about 60%, while its line shape remained unchanged. This could be partially attributed to Cr3+ oxidation to the EPR-silent Cr6+, evidenced by XPS. At the same time, the intensity of the high-spin Cr3+, appearing at low field and observable at low temperature, is virtually unchanged. In addition, the spectrum shows extremely broad and strongly anisotropic signals, covering the whole experimental field sweep. This signal exhibits the typical characteristics of extended ferro/antiferromagnetically coupled systems, closely resembling the signals attributed to exchange-coupled oxygen vacancy-bound polarons, previously observed in oxygen-depleted catalysts, based on In2O3 and ZrO2.64 Similarly, the spectrum of used Cr/CeO2 exhibited a broad oxygen vacancy-related signal. However, its intensity is significantly lower, which agrees with its lower oxygen uptake value. Furthermore, the intensity of the sharp “single-atom” signal is drastically reduced, which is in line with the agglomeration of Cr observed in EDXS mappings (Figure 3a). Any signal from Cr3+ ions in formed Cr2O3 agglomerates is expected to be broad as well and cannot be separated from the broad oxygen vacancy-related signal. Conversely, the spectrum of the used Cr/Al2O3 still evidenced the signal characteristic of Cr2O3 clusters and nanoparticles, showing little difference from the spectrum of the as-prepared material (Figure 3c). No oxygen vacancy-related signals were observed, indicating that lattice oxygen did not participate in the reaction, which is consistent with the nonreducible nature of Al2O3.
Catalytic Role of Lattice Oxygen
With availability of lattice oxygen established as a performance descriptor, its direct participation in NH3 oxidation to N2O was studied by using a TAP reactor.36,65 Initially, pulse experiments with a mixture of 18O2/NH3/Ar = 1:1:1 at 673 K were performed. The use of isotopically labeled 18O2 enabled us to discern the origin of O in N2O from either lattice of CeO2 or adsorbed O2 from the gas phase. The transient responses of the products are shown in Figure 4a. Quantitative analysis revealed that the abundance of 16O-containing products depends strongly on the catalyst used (Figure 4b). In the case of CrCeOx-T, the ratio of 16O- and 18O-containing products for both N2O (N216O/N218O) and NO (N16O/N18O) decreased with the increasing of calcination temperature of the catalyst and hence diminishing density of oxygen vacancies. Remarkably, these two ratios over CrCeOx-673 exceed 20 and 40, respectively, indicating that N216O and N16O constitute over 95% of O-containing products. Cr/CeO2 also showed a N216O/N218O and N16O/N18O ratio exceeding 1 (ca. 3.8). These results clearly prove the direct participation of lattice oxygen (16O) of CeO2 in the formation of N2O and NO, which is in line with the MvK mechanism proposed earlier. Conversely, gas-phase O2 is primarily responsible for the replenishment of oxygen vacancies. This is supported by the identical shape of transient responses of N216O and N218O, as well as of N16O and N18O. In contrast, only minor fractions of N216O and N16O were detected over Cr/Al2O3 (Figure 4b). This can be attributed to the nonreducible nature of Al2O3. To investigate the effect of CeO2 reduction degree on product formation, a multipulse experiment with a mixture of NH3/Ar = 1:1 was performed at 673 K over CrCeOx-673. With the increase in the number of NH3 pulses, the progressive depletion of lattice oxygen in the catalyst could be detected (Figure 4c). Moreover, the change in the reduction degree was found to influence the product selectivity (Figure 4d). It was essentially constant until 20% of available oxygen was depleted, producing almost exclusively N2O and NO. This buffer region is indicative of the ample supply of oxygen provided by the catalyst. With the gradual consumption of lattice oxygen, the selectivity to N2O and NO decreased, whereas the selectivity to less oxidized N2 increased. These results complement the conclusion drawn from the single-pulse experiments (Figure 4a,b) and clearly demonstrate that NH3 oxidation proceeds via a MvK mechanism over CeO2-based catalysts.
Figure 4.

Temporal analysis of products (TAP) of NH3 oxidation over selected catalysts. (a) Height-normalized transient responses in NH3 oxidation, and (b) relative contribution of lattice (16O) and gas-phase (18O) oxygen to N2O and NO formation, expressed as molar ratio of products, after pulsing 18O2/NH3/Ar = 1:1:1 at 673 K. The transient response of N18O over the CrCeOx-673 catalyst is not shown due to its small quantity and strong noise. (c) Amount of lattice oxygen removed from CrCeOx-673, and (d) corresponding product selectivity upon successive pulsing of NH3/Ar = 1:1 at 673 K over this catalyst.
The dynamic redox cycle involved in the MvK mechanism, comprising the consumption of lattice oxygen and healing of generated vacancies, requires facile activation of gas-phase O2 to sustain the availability of active oxygen species. To study the kinetics of the oxidation and reduction processes, time-resolved in situ UV–vis spectroscopy was used. For this purpose, the as-prepared Cr/CeO2 and CrCeOx-673 were first fully oxidized in 10 vol % O2/Ar, then reduced by 1 vol % NH3/He and consecutively reoxidized by 3 vol % O2/Ar at 673 K. At the same time, UV–vis spectra were recorded to monitor the catalyst state (Figure S15). Upon exposure to NH3, the intensity in the UV–vis spectrum above 500 nm increased due to the reduction of the catalyst.66 When no further changes to the spectrum were observed, the feed composition was changed, and the UV–vis spectrum was acquired during the reoxidation process, until the initial state of the catalyst was recovered, signifying that the redox cycle was reversible. The normalized temporal changes in the KM function at 700 nm were subsequently used to evaluate the redox behavior of the catalyst during reduction and reoxidation (Figure 5). As indicated, the rate of surface reduction was found to be about four-fold higher over Cr/CeO2 than over CrCeOx-673, whereas reoxidation proceeded twice as fast over CrCeOx-673 (Figure 5). These observations suggest that under the typical conditions employed for NH3 oxidation, where the reducing (i.e., NH3) and oxidizing (i.e., O2) agents are present in stoichiometric amounts, CrCeOx-673 should generally remain in a more oxidized state. Thus, the fast redox kinetics coupled to the larger pool of available lattice oxygen significantly enhances the oxygen buffer ability of CrCeOx-673, which we put forward as the reason for the superior N2O selectivity and productivity of this catalyst.
Figure 5.

Normalized temporal changes in the relative Kubelka–Munk function at 700 nm during in situ (a) reduction and (b) reoxidation treatments of CrCeOx-673 and Cr/CeO2. The corresponding UV–vis spectra are shown in Figure S15. The rate constants of reduction and reoxidation were acquired by linear fitting in the region of steepest increase, only considering the data points that yield a linear correlation with R2 > 0.9. Reaction conditions: T = 673 K; mcat = 0.2 g; FT = 20 cm3 min–1; feed composition: 1 vol % NH3 in He (reduction); 3 vol % O2 in Ar (reoxidation); and P = 1 bar.
Experimentally Guided Modeling of N2O Formation
Having established that the CeO2 carrier is the primary source of active oxygen species for the reaction, we aimed to expand our understanding of the mechanism by density functional theory (DFT) simulations. As a starting point, suitable models for the impregnated Cr/CeO2 and coprecipitated CrCeOx catalysts had to be developed. It should be noted that although EPR analysis suggested that some Cr in the Cr/CeO2 sample might be present in the form of antiferromagnetically coupled Cr3+ dimers, as well as a small amount of Cr2O3 aggregates, the metal still appears to primarily occur as isolated Cr3+ sites. Thus, we constructed a catalyst library of possible adsorbed and substitutional Cr SA structures based on the CeO2 low-index facets to model Cr/CeO2 and CrCeOx-673 catalysts, respectively (Figure S16). In line with the observation that Cr/CeO2 is not stable under the reaction conditions, we identify adsorbed SA Cr on the CeO2 (111) facet as only a metastable state (Figure S16 and Table S2), contrary to the favorable formation of substitutional CrCeOx (Table S3). Here, replacing a surface Ce center by Cr leads to an initial threefold surface oxygen coordination, which restructures during optimization due to the much smaller size of Cr compared to Ce. In the resulting structure, the symmetry is lowered and the Cr center adopts a tetrahedral coordination sphere, reminiscent of the chromate ion, CrO4,2 as shown in Figures 6 and S16. Importantly, despite one of the oxygen ligands of Cr being removed from its original lattice position, breaking two Ce–O bonds, and introducing a formal vacancy in the process, Cr retains the 4+ oxidation state of the replaced Ce4+ (Table S3). However, when an actual surface oxygen vacancy of the ceria lattice, away from the SA center, is introduced (Figure S17), the Cr atom gets reduced to a 3+ state, in line with previous reports for the bulk substitutional position.63 Thus, under vacuum conditions present during XPS measurements, oxygen expulsion and surface reduction likely lead to substitutional Cr3+ as the major species.
Figure 6.

Feasible reaction pathway proceeding via the proposed nitroxyl, HNO, and hyponitrous acid-like, H2N2O2, intermediates en route to N2O formation is shown for a representative CrCeOx catalyst model based on the majority CeO2 (111) facet. Energies (in eV) were evaluated with PBE + U and the HSE03-13 hybrid functionals (also see Table S4).
We also found that upon removal of the dangling Cr-bound oxygen ligand (which might, for instance, be consumed during initial NH3 dehydrogenation), another restructuring occurs, in which Cr restores its chromate-like tetrahedral coordination sphere by moving further down into the lattice and forming bonds with subsurface oxygen centers (Figure S17). Accordingly, this restructuring leads to a second surface oxygen vacancy in the lattice with a formation energy of 2.11 eV (0.72 eV) when evaluated with HSE03-13 (PBE + U), comparable with (significantly lower than) vacancy formation energies previously reported for the pristine CeO2 surface.67 Thus, integration of Cr into the lattice not only facilitates vacancy formation of the CeO2 support but also unlocks an alternate mechanism of providing active surface oxygen species.
Next, we evaluated the adsorption of the reactants, namely, O2 and NH3, and the formation of the HNO and H2N2O2 intermediates, considering Cr of the (111)-based adsorbed Cr/CeO2 catalyst, as well as Cr and Ce centers of the adjacent vacancy of the restructured, substitutional CrCeOx catalyst as possible adsorption sites. We began our exploration of the reactivity landscape with the mechanistic scheme proposed for the previously reported CeO2-based Mn catalyst,20 where the reaction sequence begins by the adsorption and activation of gas-phase O2, followed by dehydrogenation of NH3 to yield nitroxyl, HNO, a key intermediate en route to N2O formation. Then, a second HNO is generated accordingly, and N–N bond formation is achieved by the dimerization of both fragments mediated by the metal center. This results in a hyponitrous acid-like intermediate, H2N2O2, which is stabilized via ring formation with the low-valent Mn atom. Its dissociation finally leads to the elimination of N2O and H2O, recovering the catalyst.
Interestingly, in the substitutional (111)-based CrCeOx system, we found that the Cr atom cannot bind the reactants due to its coordinately saturated state. Instead, adsorption on Ce of the adjacent vacancy is strongly favored (Table S4). We therefore deem it likely to be the reactive site of the catalyst. The structures of the main intermediates along with the energies of the reaction sequence based on this site are presented in Figure 6. Initial adsorption of O2 is more favorable than adsorption of NH3 (Table S4), thus it is assumed to occur first (and be responsible for the slightly negative reaction order with respect to O2). Importantly, the second Ce atom remains accessible for the coordination of NH3, bringing both reactants into the proximity and facilitating the dehydrogenation of NH3. The resulting HNO fragment is then stabilized in the oxygen vacancy. After the second dehydrogenation, however, the proposed hyponitrous acid-like ring intermediate is not stable in its fully protonated state. Proton transfer to the dangling Cr-bound oxygen atom, as shown in Figure 6 (or, alternatively, the CeO2 surface), instead allows for a stable ring intermediate. The elimination of N2O and H2O finally ended the cycle. The analogous reaction sequence for the Cr/CeO2 system (Figure S18) rationalizes that adsorbed low-valent Cr can initially show reactivity similar to CrCeOx. However, as the isolated Cr is not stable in this structure, catalytic activity is lost over time on stream due to agglomeration.
To summarize, the integration of Cr into the lattice via CP naturally facilitates the formation of oxygen vacancies, in line with the increased oxygen uptake of the system, while the incurred structural distortions activate the surrounding CeO2 surface, allowing for the reaction to proceed. Thus, our computational investigation again points to the fundamental role of the availability of reactive oxygen for the formation of N2O. Lastly, as seen in the hyponitrous acid-like intermediate, the Cr-bound oxygen can also participate in surface acid–base equilibria, possibly assisting reactivity.
Catalyst Benchmarking
Having developed promising catalytic
systems (i.e., Cr/CeO2 and CrCeOx-673), we sought to evaluate how they compare to previously
reported benchmark systems, comprising Au nanoparticles supported
on CeO2 (Au/CeO2), Mn single atoms stabilized
on the surface of CeO2 (Mn/CeO2) and a mixed
manganese–bismuth oxide supported on alumina (Mn–Bi–O/α-Al2O3). The comparison was made based on several performance
metrics, namely, the highest achieved N2O selectivity,
STYN2O, and catalyst stability (Figure 7). Although CrCeOx-673 was slightly inferior to Au/CeO2 in terms of N2O selectivity (Figure 7a), it was comparable with Mn/CeO2 and superior
to all others. Furthermore, it displayed N2O productivity
second only to that of Mn/CeO2, which exceeded it by a
narrow margin (Figure 7b). Finally, the excellent stability of CrCeOx-673 also made it the sole competitor to Mn/CeO2, whereas all other catalysts experienced deactivation (Figure 7c). In contrast to
CrCeOx-673, Cr/CeO2 ranks last
in all categories except for  , which further highlights the benefits
of establishing a strong interaction between the metal and CeO2 by adopting a suitable synthetic technique. Based on this
evaluation, CrCeOx-673 stands out as a
highly competitive catalytic system for selective and stable N2O production via NH3 oxidation.
, which further highlights the benefits
of establishing a strong interaction between the metal and CeO2 by adopting a suitable synthetic technique. Based on this
evaluation, CrCeOx-673 stands out as a
highly competitive catalytic system for selective and stable N2O production via NH3 oxidation.
Figure 7.

Performance comparison of the best catalysts presented in this work with the benchmark systems, in terms of (a) highest achieved N2O selectivity, (b) STY of N2O, and (c) deactivation constant (kd). kd value was obtained by fitting the normalized rate of N2O production over 50+ h on stream to a power function of the form y = ax–kd. Reaction conditions: Tbed = 673 K; mcat = 0.01–0.2 g; GHSV = 15,000 (a) or 450,000 cm3 h–1 gcat–1(b, c); feed composition = 8 vol % NH3, 8 vol % O2, 4 vol % Ar, 80 vol % He; P = 1 bar. The N2O selectivity of Au/CeO2 and Mn–Bi–O/α-Al2O3 was evaluated at 573 and 623 K, respectively.
Conclusions
Contrary to the conventional wisdom of using manganese as the main component of catalysts for selective NH3 oxidation to N2O, in this work, we demonstrate the viability of a previously overlooked metal, Cr, for this purpose. We utilized atomic scale engineering to exploit the properties of the metal and the carrier by incorporating a small amount of isolated Cr atoms (1 wt %) within the CeO2 crystal through CP. The choice of the synthetic technique was shown to be crucial, as simple impregnation resulted in poorly stabilized atoms of Cr, which agglomerated during the reaction, whereas introducing Cr in the carrier allowed atomic dispersion and the stable rate of N2O formation to be maintained for 100 h on stream. The calcination temperature was also found to regulate the extent of the Cr-induced increase in the density of oxygen vacancies in CeO2, which, in turn, was identified as a performance descriptor. In addition, the rate of associated surface reduction and reoxidation processes was enhanced, thus resulting in a comprehensive improvement of catalyst oxygen buffer ability. The latter is of particular note, as the combination of kinetic and TAP studies evidenced direct participation of lattice oxygen of CeO2 in N2O formation. Further mechanistic insights were obtained by DFT simulations, revealing that it is the ability of Cr atoms to dynamically change oxidation state and coordinatively restructure that enables facile oxygen vacancy creation and stabilization of nitroxyl and nitrous acid-like reaction intermediates. Furthermore, reactant adsorption was found to partially occur over Ce atoms, thus bringing us to the conclusion that isolated Cr atoms form a catalytic ensemble with the proximal CeO2. Thus, this catalyst serves as an example of isolated metal atoms inducing carrier modification and causing the latter to take on a cocatalytic role, a phenomenon that has only recently begun to be recognized in the SAC community.68−70 In fact, the curated partnership of Cr and CeO2 has allowed the attainment of catalytic performance that superseded nearly all benchmark systems and was on par with that of the state-of-the-art Mn/CeO2. The understanding of the catalyst design principles acquired in this work, whose viability has been demonstrated for an underexplored and generally disregarded metal, will aid in this endeavor and bring us one step closer to the implementation of a more economic and sustainable method of N2O synthesis.
Acknowledgments
This publication was created as part of NCCR Catalysis (Grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation. The Scientific Center for Optical and Electron Microscopy (ScopeM) at the ETH Zurich is thanked for access to their facilities. The work of J.G. and N.L. was supported by the Generalitat de Catalunya and the European Union under Grant 2020-FI-B-00266, the Ministerio de Ciencia e Innovación, with ref no. PID2021-122516OB-I00, and the Severo Ochoa Grant (MCIN/AEI/10.13039/501100011033 CEX2019-000925-S). The authors also thank the Barcelona Supercomputing Center (BSC-RES) for providing computational resources. R.E. and H.E. acknowledge the financial support from the Swiss National Science Foundation (Project no. 200021_196381).
Data Availability Statement
The experimental and computational data presented in the main figures of the manuscript are publicly available through the Zenodo (10.5281/zenodo.8285840) and ioChem-BD (https://iochem-bd.iciq.es/browse/review-collection/100/64626/648b674425655ee479759631) repositories, respectively. Further data supporting the findings of this study are available in the Supporting Information. All other relevant source data are available from the corresponding author upon request.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c04463.
Summary of results of elemental analysis and N2 sorption experiments; simulated single-atom Cr adsorption and substitution energies on different facets of CeO2; simulated adsorption energies of reactants and key intermediates on the substitutional (111)-based CrCeOx catalyst model; schematic of the experimental setup for NH3 oxidation; additional catalytic tests of Cr-based materials; H2-TPR profiles of Cr-based catalysts and corresponding carriers; HAADF-STEM micrographs with corresponding particle size distributions of CrCeOx-T catalysts; the scatter plots of H2 consumption in H2-TPR tests vs oxygen uptake and N2O space-time yield vs apparent activation energy of NH3 conversion for Cr-based catalysts; Raman spectra of CeO2-based materials; results of the kinetic investigation; HAADF-STEM micrographs and EDX mappings of as-prepared and used Cr-based catalysts; XRD patterns and low-temperature EPR spectra of Cr-based materials; UV–vis spectra of CeO2-based catalysts acquired over the course of reduction and reoxidation treatment; library of simulated structures of adsorbed and lattice-substituted single Cr atoms on CeO2; simulated process of oxygen cycling enabled by the vacancy in the (111) based CrCeOx catalyst; and simulated reaction sequence of N2O formation over an adsorbed isolated Cr atom (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Q.Y. and I.S. are contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Pérez-Ramírez J.; Mondelli C.; Schmidt T.; Schlüter O. F. K.; Wolf A.; Mleczko L.; Dreier T. Sustainable chlorine recycling via catalysed HCl oxidation: from fundamentals to implementation. Energy Environ. Sci. 2011, 4, 4786–4799. 10.1039/c1ee02190g. [DOI] [Google Scholar]
- Agarwal N.; Freakley S. J.; McVicker R. U.; Althahban S. M.; Dimitratos N.; He Q.; Morgan D. J.; Jenkins R. L.; Willock D. J.; Taylor S. H.; Kiely C. J.; Hutchings G. J. Aqueous Au-Pd colloids catalyze selective CH4 oxidation to CH3OH with O2 under mild conditions. Science 2017, 358, 223–227. 10.1126/science.aan6515. [DOI] [PubMed] [Google Scholar]
- Grant J. T.; Venegas J. M.; McDermott W. P.; Hermans I. Aerobic oxidations of light alkanes over solid metal oxide catalysts. Chem. Rev. 2018, 118, 2769–2815. 10.1021/acs.chemrev.7b00236. [DOI] [PubMed] [Google Scholar]
- Szécsényi Á.; Li G.; Gascon J.; Pidko E. A. Mechanistic complexity of methane oxidation with H2O2 by single-site Fe/ZSM-5 catalyst. ACS Catal. 2018, 8, 7961–7972. 10.1021/acscatal.8b01672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Pei C.; Gong J. Shale gas revolution: Catalytic conversion of C1–C3 light alkanes to value-added chemicals. Chem. 2021, 7, 1755–1801. 10.1016/j.chempr.2021.02.002. [DOI] [Google Scholar]
- Yan H.; Liu B.; Zhou X.; Meng F.; Zhao M.; Pan Y.; Li J.; Wu Y.; Zhao H.; Liu Y.; Chen X.; Li L.; Feng X.; Chen D.; Shan H.; Yang C.; Yan N. Enhancing polyol/sugar cascade oxidation to formic acid with defect rich MnO2 catalysts. Nat. Commun. 2023, 14, 4509. 10.1038/s41467-023-40306-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severin K. Synthetic chemistry with nitrous oxide. Chem. Soc. Rev. 2015, 44, 6375–6386. 10.1039/C5CS00339C. [DOI] [PubMed] [Google Scholar]
- Arinaga A. M.; Ziegelski M. C.; Marks T. J. Alternative oxidants for the catalytic oxidative coupling of methane. Angew. Chem., Int. Ed. 2021, 60, 10502–10515. 10.1002/anie.202012862. [DOI] [PubMed] [Google Scholar]
- Hermans I.; Janssen K.; Moens B.; Philippaerts A.; Van Berlo B.; Peeters J.; Jacobs P. A.; Sels B. F. Solvent- and metal-free ketonization of fatty acid methyl esters and triacylglycerols with nitrous oxide. Adv. Synth. Catal. 2007, 349, 1604–1608. 10.1002/adsc.200600645. [DOI] [Google Scholar]
- Xiao D. J.; Bloch E. D.; Mason J. A.; Queen W. L.; Hudson M. R.; Planas N.; Borycz J.; Dzubak A. L.; Verma P.; Lee K.; Bonino F.; Crocellà V.; Yano J.; Bordiga S.; Truhlar D. G.; Gagliardi L.; Brown C. M.; Long J. R. Oxidation of ethane to ethanol by N2O in a metal–organic framework with coordinatively unsaturated iron(II) sites. Nat. Chem. 2014, 6, 590–595. 10.1038/nchem.1956. [DOI] [PubMed] [Google Scholar]
- Held A.; Kowalska-Kuś J.; Nowińska K.; Góra-Marek K. Potassium-modified silica-supported vanadium oxide catalysts applied for propene epoxidation. J. Catal. 2017, 347, 21–35. 10.1016/j.jcat.2016.12.001. [DOI] [Google Scholar]
- Aydin Z.; Zanina A.; Kondratenko V. A.; Rabeah J.; Li J.; Chen J.; Li Y.; Jiang G.; Lund H.; Bartling S.; Linke D.; Kondratenko E. V. Effects of N2O and water on activity and selectivity in the oxidative coupling of methane over Mn–Na2WO4/SiO2: role of oxygen species. ACS Catal. 2022, 12, 1298–1309. 10.1021/acscatal.1c04915. [DOI] [Google Scholar]
- Ye D.; Tsang S. C. E. Prospects and challenges of green ammonia synthesis. Nat. Synth. 2023, 2, 612–623. 10.1038/s44160-023-00321-7. [DOI] [Google Scholar]
- Pérez-Ramírez J.; Kondratenko E. V.; Kondratenko V. A.; Baerns M. Selectivity-directing factors of ammonia oxidation over PGM gauzes in the Temporal Analysis of Products reactor: primary interactions of NH3 and O2. J. Catal. 2004, 227, 90–100. 10.1016/j.jcat.2004.06.023. [DOI] [Google Scholar]
- Baerns M.; Imbihl R.; Kondratenko V. A.; Kraehnert R.; Offermans W. K.; van Santen R. A.; Scheibe A. Bridging the pressure and material gap in the catalytic ammonia oxidation: structural and catalytic properties of different platinum catalysts. J. Catal. 2005, 232, 226–238. 10.1016/j.jcat.2005.03.002. [DOI] [Google Scholar]
- Slavinskaya E. M.; Veniaminov S. A.; Notté P.; Ivanova A. S.; Boronin A. I.; Chesalov Y. A.; Polukhina I. A.; Noskov A. S. Studies of the mechanism of ammonia oxidation into nitrous oxide over Mn-Bi-O/α-Al2O3 catalyst. J. Catal. 2004, 222, 129–142. 10.1016/j.jcat.2003.09.029. [DOI] [Google Scholar]
- Ivanova A. S.; Slavinskaya E. M.; Mokrinskii V. V.; Polukhina I. A.; Tsybulya S. V.; Prosvirin I. P.; Bukhtiyarov V. I.; Rogov V. A.; Zaikovskii V. I.; Noskov A. S. The role of support in formation of the manganese–bismuth oxide catalyst for synthesis of nitrous oxide through oxidation of ammonia with oxygen. J. Catal. 2004, 221, 213–224. 10.1016/j.jcat.2003.06.001. [DOI] [Google Scholar]
- Parmon V. N.; Panov G. I.; Uriarte A.; Noskov A. S. Nitrous oxide in oxidation chemistry and catalysis: application and production. Catal. Today 2005, 100, 115–131. 10.1016/j.cattod.2004.12.012. [DOI] [Google Scholar]
- Tang Z.; Surin I.; Rasmussen A.; Krumeich F.; Kondratenko E. V.; Kondratenko V. A.; Pérez-Ramírez J. Ceria-supported gold nanoparticles as a superior catalyst for nitrous oxide production via ammonia oxidation. Angew. Chem., Int. Ed. 2022, 61, e202200772 10.1002/anie.202200772. [DOI] [PubMed] [Google Scholar]
- Surin I.; Tang Z.; Geiger J.; Damir S.; Eliasson H.; Agrachev M.; Krumeich F.; Mitchell S.; Kondratenko V. A.; Kondratenko E. V.; Jeschke G.; Erni R.; López N.; Pérez-Ramírez J. Low-valent manganese atoms stabilized on ceria for nitrous oxide synthesis. Adv. Mater. 2023, 35, 2211260 10.1002/adma.202211260. [DOI] [PubMed] [Google Scholar]
- Montini T.; Melchionna M.; Monai M.; Fornasiero P. Fundamentals and catalytic applications of CeO2-based materials. Chem. Rev. 2016, 116, 5987–6041. 10.1021/acs.chemrev.5b00603. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Zhao S.; Feng J.; Song S.; Shi W.; Wang D.; Zhang H. Unraveling the physical chemistry and materials science of CeO2-based nanostructures. Chem. 2021, 7, 2022–2059. 10.1016/j.chempr.2021.02.015. [DOI] [Google Scholar]
- Baek J.; Yun H. J.; Yun D.; Choi Y.; Yi J. Preparation of highly dispersed chromium oxide catalysts supported on mesoporous silica for the oxidative dehydrogenation of propane using CO2: insight into the nature of catalytically active chromium sites. ACS Catal. 2012, 2, 1893–1903. 10.1021/cs300198u. [DOI] [Google Scholar]
- Wang J.; Zhu M.-L.; Song Y.-H.; Liu Z.-T.; Wang L.; Liu Z.-W. Molecular-level investigation on supported CrOx catalyst for oxidative dehydrogenation of propane with carbon dioxide. J. Catal. 2022, 409, 87–97. 10.1016/j.jcat.2022.03.027. [DOI] [Google Scholar]
- Otroshchenko T.; Jiang G.; Kondratenko V. A.; Rodemerck U.; Kondratenko E. V. Current status and perspectives in oxidative, non-oxidative and CO2-mediated dehydrogenation of propane and isobutane over metal oxide catalysts. Chem. Soc. Rev. 2021, 50, 473–527. 10.1039/D0CS01140A. [DOI] [PubMed] [Google Scholar]
- Shen Q.; Cao C.; Huang R.; Zhu L.; Zhou X.; Zhang Q.; Gu L.; Song W. Single chromium atoms supported on titanium dioxide nanoparticles for synergic catalytic methane conversion under mild conditions. Angew. Chem., Int. Ed. 2020, 59, 1216–1219. 10.1002/anie.201913309. [DOI] [PubMed] [Google Scholar]
- Zeng M.; Cheng L.; Gu Q.; Yang B.; Yu B.; Xu J.; Zhang Y.; Pan C.; Cao X.-M.; Lou Y.; Zhu Y. ZSM-5-confined Cr1–O4 active sites boost methane direct oxidation to C1 oxygenates under mild conditions. EES. Catal. 2023, 1, 153–161. 10.1039/D2EY00080F. [DOI] [Google Scholar]
- Jiang W.; An X.; Xiao J.; Yang Z.; Liu J.; Chen H.; Li H.; Zhu W.; Li H.; Dai S. Enhanced oxygen activation achieved by robust single chromium atom-derived catalysts in aerobic oxidative desulfurization. ACS Catal. 2022, 12, 8623–8631. 10.1021/acscatal.2c01329. [DOI] [Google Scholar]
- Amores J. M. G.; Escribano V. S.; Ramis G.; Busca G. An FT-IR study of ammonia adsorption and oxidation over anatase-supported metal oxides. Appl. Catal. B: Environ. 1997, 13, 45–58. 10.1016/S0926-3373(96)00092-6. [DOI] [Google Scholar]
- Sadykov V. A.; Isupova L. A.; Zolotarskii I. A.; Bobrova L. N.; Noskov A. S.; Parmon V. N.; Brushtein E. A.; Telyatnikova T. V.; Chernyshev V. I.; Lunin V. V. Oxide catalysts for ammonia oxidation in nitric acid production: properties and perspectives. Appl. Catal. A: Gen. 2000, 204, 59–87. 10.1016/S0926-860X(00)00506-8. [DOI] [Google Scholar]
- Pérez-Ramírez J.; Kondratenko E. V. Mechanism of ammonia oxidation over oxides studied by temporal analysis of products. J. Catal. 2007, 250, 240–246. 10.1016/j.jcat.2007.06.014. [DOI] [Google Scholar]
- Moser M.; Vilé G.; Colussi S.; Krumeich F.; Teschner D.; Szentmiklósi L.; Trovarelli A.; Pérez-Ramírez J. Structure and reactivity of ceria–zirconia catalysts for bromine and chlorine production via the oxidation of hydrogen halides. J. Catal. 2015, 331, 128–137. 10.1016/j.jcat.2015.08.024. [DOI] [Google Scholar]
- Biesinger M. C.; Payne B. P.; Grosvenor A. P.; Lau L. W. M.; Gerson A. R.; Smart R. S. C. Resolving surface chemical states in XPS analysis of first row transition metals, oxides and hydroxides: Cr, Mn, Fe. Co and Ni. Appl. Surf. Sci. 2011, 257, 2717–2730. 10.1016/j.apsusc.2010.10.051. [DOI] [Google Scholar]
- Ovsitser O.; Cherian M.; Brückner A.; Kondratenko E. V. Dynamics of redox behavior of nano-sized VOx species over Ti–Si-MCM-41 from time-resolved in situ UV/Vis analysis. J. Catal. 2009, 265, 8–18. 10.1016/j.jcat.2009.04.005. [DOI] [Google Scholar]
- Gleaves J. T.; Yablonskii G. S.; Phanawadee P.; Schuurman Y. TAP-2: an interrogative kinetics approach. Appl. Catal. A: Gen. 1997, 160, 55–88. 10.1016/S0926-860X(97)00124-5. [DOI] [Google Scholar]
- Pérez-Ramírez J.; Kondratenko E. V. Evolution, achievements, and perspectives of the TAP technique. Catal. Today 2007, 121, 160–169. 10.1016/j.cattod.2007.01.001. [DOI] [Google Scholar]
- Kresse G.; Furthmüller J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15. 10.1016/0927-0256(96)00008-0. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Hegner F. S.; Herraiz-Cardona I.; Cardenas-Morcoso D.; Lopez N.; Galan-Mascaros J. R.; Gimenez S. Cobalt hexacyanoferrate on BiVO4 photoanodes for robust water splitting. ACS Appl. Mater. Interfaces 2017, 9, 37671. 10.1021/acsami.7b09449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. 10.1103/PhysRevB.59.1758. [DOI] [Google Scholar]
- Blochl P. E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. 10.1103/PhysRevB.50.17953. [DOI] [PubMed] [Google Scholar]
- Hubbard J. Electron correlations in narrow energy bands. Proc. R. Soc. A 1997, 276, 238. 10.1098/rspa.1963.0204. [DOI] [Google Scholar]
- Dudarev S. L.; Botton G. A.; Savrasov S. Y.; Humphreys C. J.; Sutton A. P. Electron-energy-loss spectra and the structural stability of nickel oxide: an LSDA+U study. Phys. Rev. B 1998, 57, 1505. 10.1103/PhysRevB.57.1505. [DOI] [Google Scholar]
- Fabris S.; de Gironcoli S.; Baroni S.; Vicario G.; Balducci G. Taming multiple valency with density functionals: a case study of defective ceria. Phys. Rev. B 2005, 71, 041102(R) 10.1103/PhysRevB.71.041102. [DOI] [Google Scholar]
- Timrov I.; Marzari N.; Cococcioni M. Hubbard parameters from density-functional perturbation theory. Phys. Rev. B 2018, 98, 085127 10.1103/PhysRevB.98.085127. [DOI] [Google Scholar]
- Wang J.; Gong X.-Q. A DFT+U study of V, Cr and Mn doped CeO2(111). Appl. Surf. Sci. 2018, 428, 377–384. 10.1016/j.apsusc.2017.09.120. [DOI] [Google Scholar]
- Pizzi G.; Cepellotti A.; Sabatini R.; Marzari N.; Kozinsky B. AiiDA: automated interactive infrastructure and database for computational science. Comput. Mater. Sci. 2016, 111, 218–230. 10.1016/j.commatsci.2015.09.013. [DOI] [Google Scholar]
- Huber S. P.; Zoupanos S.; Uhrin M.; Talirz L.; Kahle L.; Häuselmann R.; Gresch D.; Müller T.; Yakutovich A. V.; Andersen C. W.; Ramirez F. F.; Adorf C. S.; Gargiulo F.; Kumbhar S.; Passaro E.; Johnston C.; Merkys A.; Cepellotti A.; Mounet N.; Marzari N.; Kozinsky B.; Pizzi G. AiiDA 1.0, a scalable computational infrastructure for automated reproducible workflows and data provenance. Sci. Data 2020, 7, 300. 10.1038/s41597-020-00638-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrin M.; Huber S. P.; Yu J.; Marzari N.; Pizzi G. Workflows in AiiDA: engineering a high-throughput, event-based engine for robust and modular computational workflows. Comput. Mater. Sci. 2021, 187, 110086 10.1016/j.commatsci.2020.110086. [DOI] [Google Scholar]
- Daelman N.; Capdevila-Cortada M.; Lopez N. Dynamic charge and oxidation state of Pt/CeO2 single-atom catalysts. Nat. Mater. 2019, 18, 1215. 10.1038/s41563-019-0444-y. [DOI] [PubMed] [Google Scholar]
- Pérez Flores J. C.; García-Alvarado F. Electrical conductivity of the oxygen-deficient rutile CrNbO4−δ. Solid State Sci. 2009, 11, 207–213. 10.1016/j.solidstatesciences.2008.05.009. [DOI] [Google Scholar]
- Muravev V.; Parastaev A.; van den Bosch Y.; Ligt B.; Claes N.; Bals S.; Kosinov N.; Hensen E. J. M. Size of cerium dioxide support nanocrystals dictates reactivity of highly dispersed palladium catalysts. Science 2023, 380, 1174–1179. 10.1126/science.adf9082. [DOI] [PubMed] [Google Scholar]
- Fierro J. L. G.; Soria J.; Sanz J.; Rojo J. M. Induced changes in ceria by thermal treatments under vacuum or hydrogen. J. Solid State Chem. 1987, 66, 154–162. 10.1016/0022-4596(87)90230-1. [DOI] [Google Scholar]
- Loridant S. Raman spectroscopy as a powerful tool to characterize ceria-based catalysts. Catal. Today 2021, 373, 98–111. 10.1016/j.cattod.2020.03.044. [DOI] [Google Scholar]
- Wu Z.; Li M.; Howe J.; Meyer H. M. III; Overbury S. H. Probing defect sites on CeO2 nanocrystals with well-defined surface planes by Raman spectroscopy and O2 adsorption. Langmuir 2010, 26, 16595–16606. 10.1021/la101723w. [DOI] [PubMed] [Google Scholar]
- Muravev V.; Spezzati G.; Su Y.-Q.; Parastaev A.; Chiang F.-K.; Longo A.; Escudero C.; Kosinov N.; Hensen E. J. M. Interface dynamics of Pd–CeO2 single-atom catalysts during CO oxidation. Nat. Catal. 2021, 4, 469–478. 10.1038/s41929-021-00621-1. [DOI] [Google Scholar]
- Skorodumova N. V.; Baudin M.; Hermansson K. Surface properties of CeO2 from first principles. Phys. Rev. B 2004, 69, 075401 10.1103/PhysRevB.69.075401. [DOI] [Google Scholar]
- Naeem M. A.; Abdala P. M.; Armutlulu A.; Kim S. M.; Fedorov A.; Müller C. R. Exsolution of metallic Ru nanoparticles from defective, fluorite-type solid solutions Sm2RuxCe2–xO7 to impart stability on dry reforming catalysts. ACS Catal. 2020, 10, 1923–1937. 10.1021/acscatal.9b04555. [DOI] [Google Scholar]
- Xiao Y.; Xie K. Active exsolved metal–oxide interfaces in porous single-crystalline ceria monoliths for efficient and durable CH4/CO2 reforming. Angew. Chem., Int. Ed. 2022, 61, e202113079 10.1002/anie.202113079. [DOI] [PubMed] [Google Scholar]
- Kim M.; Kwon G.; Jung W.-G.; Choi Y.; Kim B.-J.; Lee H. Direct observation of rhodium ex-solution from a ceria nanodomain and its use for hydrogen production via propane steam reforming. ACS Appl. Mater. Interfaces 2021, 13, 48508–48515. 10.1021/acsami.1c09799. [DOI] [PubMed] [Google Scholar]
- Kwon O.; Sengodan S.; Kim K.; Kim G.; Jeong H. Y.; Shin J.; Ju Y.-W.; Han J. W.; Kim G. Exsolution trends and co-segregation aspects of self-grown catalyst nanoparticles in perovskites. Nat. Commun. 2017, 8, 15967. 10.1038/ncomms15967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing G. Z.; Yi J. B.; Wang D. D.; Liao L.; Yu T.; Shen Z. X.; Huan C. H. A.; Sum T. C.; Ding J.; Wu T. Strong correlation between ferromagnetism and oxygen deficiency in Cr-doped In2O3−δ nanostructures. Phys. Rev. B 2009, 79, 174406 10.1103/PhysRevB.79.174406. [DOI] [Google Scholar]
- Hao X.; Yoko A.; Inoue K.; Xu Y.; Saito M.; Chen C.; Seong G.; Tomai T.; Takami S.; Shluger A. L.; Xu B.; Adschiri T.; Ikuhara Y. Atomistic origin of high-concentration Ce3+ in {100}-faceted Cr-substituted CeO2 nanocrystals. Acta Mater. 2021, 203, 116473. 10.1016/j.actamat.2020.11.015. [DOI] [Google Scholar]
- Pinheiro Araujo T.; Mondelli C.; Agrachev M.; Zou T.; Willi P. O.; Engel K. M.; Grass R. N.; Stark W. J.; Safonova O. V.; Jeschke G.; Mitchell S.; Perez-Ramirez J. Flame-made ternary Pd-In2O3-ZrO2 catalyst with enhanced oxygen vacancy generation for CO2 hydrogenation to methanol. Nat. Commun. 2022, 13, 5610. 10.1038/s41467-022-33391-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan K.; Maguire N.; Fushimi R.; Gleaves J. T.; Goguet A.; Harold M. P.; Kondratenko E. V.; Menon U.; Schuurman Y.; Yablonsky G. S. Forty years of temporal analysis of products. Catal. Sci. Technol. 2017, 7, 2416–2439. 10.1039/C7CY00678K. [DOI] [Google Scholar]
- Kondratenko E. V.; Sakamoto Y.; Okumura K.; Shinjoh H. Time-resolved in situ UV/vis spectroscopy for analyzing the dynamics of surface and bulk redox processes in materials used as oxygen storage capacitors. Catal. Today 2011, 164, 46–51. 10.1016/j.cattod.2010.10.015. [DOI] [Google Scholar]
- Pérez-Bailac P.; Lustemberg P. G.; Ganduglia-Pirovano M. V. Facet-dependent stability of near-surface oxygen vacancies and excess charge localization at CeO2 surfaces. J. Phys.: Condens. Matter 2021, 33, 504003. 10.1088/1361-648X/ac238b. [DOI] [PubMed] [Google Scholar]
- Kaiser S. K.; Chen Z.; Faust Akl D.; Mitchell S.; Pérez-Ramírez J. Single-atom catalysts across the periodic table. Chem. Rev. 2020, 120, 11703–11809. 10.1021/acs.chemrev.0c00576. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Wang M.; Zhu Q.; Xiao D.; Ma D. Ensemble effect for single-atom, small cluster and nanoparticle catalysts. Nat. Catal. 2022, 5, 766–776. 10.1038/s41929-022-00839-7. [DOI] [Google Scholar]
- Giulimondi V.; Mitchell S.; Pérez-Ramírez J. Challenges and opportunities in engineering the electronic structure of single-atom catalysts. ACS Catal. 2023, 13, 2981–2997. 10.1021/acscatal.2c05992. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental and computational data presented in the main figures of the manuscript are publicly available through the Zenodo (10.5281/zenodo.8285840) and ioChem-BD (https://iochem-bd.iciq.es/browse/review-collection/100/64626/648b674425655ee479759631) repositories, respectively. Further data supporting the findings of this study are available in the Supporting Information. All other relevant source data are available from the corresponding author upon request.