

ABSTRACT
Anabolic treatment options for osteoporosis remain limited. One approach to discovering novel anabolic drug targets is to identify genetic causes of extreme high bone mass (HBM). We investigated a pedigree with unexplained HBM within the UK HBM study, a national cohort of probands with HBM and their relatives. Whole exome sequencing (WES) in a family with HBM identified a rare heterozygous missense variant (NM_004482.4:c.1657C > T, p.Arg553Trp) in GALNT3, segregating appropriately. Interrogation of data from the UK HBM study and the Anglo‐Australasian Osteoporosis Genetics Consortium (AOGC) revealed an unrelated individual with HBM with another rare heterozygous variant (NM_004482.4:c.831 T > A, p.Asp277Glu) within the same gene. In silico protein modeling predicted that p.Arg553Trp would disrupt salt‐bridge interactions, causing instability of GALNT3, and that p.Asp277Glu would disrupt manganese binding and consequently GALNT3 catalytic function. Bi‐allelic loss‐of‐function GALNT3 mutations alter FGF23 metabolism, resulting in hyperphosphatemia and causing familial tumoral calcinosis (FTC). However, bone mineral density (BMD) in FTC cases, when reported, has been either normal or low. Common variants in the GALNT3 locus show genome‐wide significant associations with lumbar, femoral neck, and total body BMD. However, no significant associations with BMD are observed at loci coding for FGF23, its receptor FGFR1, or coreceptor klotho. Mendelian randomization analysis, using expression quantitative trait loci (eQTL) data from primary human osteoblasts and genome‐wide association studies data from UK Biobank, suggested increased expression of GALNT3 reduces total body, lumbar spine, and femoral neck BMD but has no effect on phosphate concentrations. In conclusion, rare heterozygous loss‐of‐function variants in GALNT3 may cause HBM without altering phosphate concentration. These findings suggest that GALNT3 may affect BMD through pathways other than FGF23 regulation, the identification of which may yield novel anabolic drug targets for osteoporosis. © 2023 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: HIGH BONE MASS, EXOME SEQUENCING, MONOGENIC, GALNT3, PHOSPHATE
Introduction
Osteoporotic fractures affect one in three women and one in five men over the age of 50 worldwide, causing substantial morbidity and mortality, with annual healthcare costs in the USA alone of over $20 billion.( 1 ) Osteoporotic fractures are the leading cause of hospitalization in women over the age of 45 in the UK, occupying more bed‐days than any other condition including breast cancer, diabetes, and cardiovascular disease.( 2 ) Antiresorptive drugs, such as bisphosphonates or denosumab, currently form the mainstay of osteoporosis treatment. Long‐term antiresorptive use is limited by concern regarding rare side effects, including atypical femoral fractures and osteonecrosis of the jaw. In contrast, anabolic treatments actively stimulate new bone formation. Until recently, parathyroid hormone (PTH) derivatives and analogs such as teriparatide (PTH1‐34) and abaloparatide (PTH‐related protein analog) were the only anabolic treatment options available but the requirement for daily subcutaneous injections and, in many countries, restrictions on prescribing limit their use.( 3 ) Romosozumab was recently approved as an anabolic treatment in the USA and Europe, with once‐monthly subcutaneous administration, stimulating bone formation while also reducing bone resorption.( 4 ) Despite clinical efficacy, concerns regarding cardiovascular side effects restricted approval by the European Medicines Agency to selected women with severe osteoporosis. Thus, there remains a need for safe, and preferably orally administered, anabolic treatments for osteoporosis.
Romosozumab is an excellent example of where data from human genetic studies can be used to identify novel drug targets. This agent, which consists of an anti‐sclerostin antibody, was developed after the discovery that mutations in and around SOST,( 5 , 6 ) the gene encoding sclerostin, underlie the rare diseases sclerosteosis( 5 ) and van Buchem's disease( 7 ); common variants at this locus are also associated with bone mineral density (BMD) (reported in multiple genome‐wide association studies [GWASs]).( 8 ) Thus, a highly successful approach to anabolic drug discovery is to identify genetic causes underlying skeletal dysplasias associated with high bone mass (HBM), specifically HBM resulting from excessive osteoblastic bone formation. Both sclerosteosis and van Buchem's disease are associated with pathological features arising from bone overgrowth, such as nerve compression.( 9 ) In contrast, several other monogenic disorders have been identified where HBM due to excessive osteoblast activity is an incidental finding, caused by mutations in other Wnt signaling proteins, namely LRP5 and LRP6, usually without sinister consequences of bony overgrowth.( 10 )
The UK HBM study was established to identify new monogenic causes for HBM.( 11 ) Pathological variants in known HBM genes (SOST/LRP5) were identified in a small minority, leaving most with unexplained HBM.( 12 ) Although enrichment of common variant “high‐BMD” alleles in known BMD‐associated loci was evident,( 13 ) it is likely that further monogenic causes exist that are yet to be discovered. To examine this question, we undertook whole exome sequencing (WES) of our HBM population and analyzed the data using a bespoke pipeline developed to identify underlying monogenic variants in individuals and/or families with phenotypes of interest.( 14 ) Using this approach, we recently identified a rare missense pathogenic variant in SMAD9, segregating with HBM in an autosomal dominant pattern within a UK HBM pedigree.( 15 ) The same variant was identified in two additional unrelated individuals with HBM sequenced as part of the Anglo‐Australian Osteoporosis Genetics Consortium (AOGC).( 16 ) Moreover, common variants at the same locus are associated with BMD in the general population.( 15 ) These findings led to the identification of the SMAD‐dependent BMP signaling pathway as a potential anabolic target for osteoporosis treatment.
Both SOST and SMAD9 gene discoveries highlight the value of investigating rare monogenic HBM in identifying novel anabolic drug targets. We sought to identify additional novel monogenic causes of HBM by analyzing WES data from further kindreds in our UK HBM study.
Methods
The UK HBM study
The HBM study is a UK‐based multicenter observational study of adults with unexplained HBM, identified incidentally on routine clinical dual‐energy X‐ray absorptiometry (DXA) scanning. Briefly, DXA databases containing 335,115 DXA scans across 13 UK centers were searched; all scans explained by artifact or known causes of high BMD were excluded, principally degenerative disease/osteoarthritis (OA). Unexplained HBM was defined as (1) first lumbar vertebra (L1) Z‐score of ≥+3.2 plus total hip (TH) Z‐score of ≥+1.2 and/or (2) TH Z‐score ≥+3.2 plus L1 Z‐score of ≥+1.2. Full details of DXA database screening and participant recruitment were previously reported together with the clinical, biochemical, and imaging assessments performed.( 11 , 15 ) Baseline recruitment took place between 2005 and 2010 and a follow‐up study was performed between 2016 and 2018.( 17 ) The study recruited 337 individuals with unexplained HBM (240 probands and 97 affected relatives).
Anglo‐Australasian Osteoporosis Genetics Consortium (AOGC) HBM and LBM cases
The AOGC extreme truncate population comprises 1128 Australian, 74 New Zealand, and 753 British unrelated women of white Caucasian ancestry, at age 55 to 85 years, ≥5 years postmenopausal, with either HBM (TH BMD Z‐scores +1.5 to +4.0, n = 1055) or low bone mass (LBM) (Z‐scores −4.0 to −1.5, n = 900), with no known secondary cause for altered BMD in either group.( 16 )
Whole exome sequencing and analysis
HBM study probands (excluding individuals with pathogenic variants in known HBM genes) underwent WES. Following review of each HBM pedigree, family members were also selected who were related to the index case and had a phenotype and DNA collected. Where the pedigree spanned three generations, the most distant relatives were sequenced. Additionally, WES was performed in 126 HBM and 493 LBM individuals from AOGC, as well as 240 from the HBM study. Library creation, sequencing, base calling, sequence alignment, and variant calling were performed as previously described.( 18 ) After quality filtering, data were analyzed for carriage of at least one rare (either novel or population‐based minor allele frequency [MAF] <0.005) using the Genome Aggregation Database (gnomAD) version 2.1.1,( 19 ) coding, nonsynonymous single nucleotide variant (SNV), or indel in a highly conserved region (Genomic Evolutionary Rate Profiling [GERP] score >1.5). Data were then filtered based on functional prediction of SNVs using Polymorphism Phenotyping version 2 (Polyphen‐2),( 20 ) Sorting Intolerant from Tolerant (SIFT) 4G,( 21 ) PMut,( 22 ) and Mutation Taster.( 23 )
HBM pedigrees were analyzed for variants segregating appropriately for autosomal dominant inheritance (i.e., carried by affected individuals, not carried by unaffected individuals); compound heterozygous (i.e., two different mutation within the same gene) and homozygous inheritances were also considered. Replication was sought by scrutinizing similarly filtered data from all UK HBM and AOGC extreme HBM individuals (seeking carriage of the same variant or other variant within the same gene). For this analysis, a threshold TH or lumbar spine (LS) Z‐score ≥ +2.5 was used to select the most extreme high‐BMD individuals from both cohorts. Data from 493 AOGC LBM individuals (with threshold LS Z‐scores of <0.5 and TH Z‐score <1.5) were similarly scrutinized as negative controls. Variants of interest were validated using Sanger sequencing.
Measurement of intact and C‐terminal FGF23 concentrations
Human intact (iFGF23) and C‐terminal (cFGF23) forms of FGF23 were analyzed by the Immutopics second‐generation Enzyme‐Linked Immunosorbent Assay (ELISA) kits (Immutopics, San Clemente, CA, USA). The iFGF23 assay employs a murine monoclonal antibody and an affinity purified goat polyclonal antibody to detect epitopes within the amino terminal and carboxyl‐terminal portions of FGF‐23, whereas the antibodies in the cFGF23 assay bind to both the intact molecule and large carboxyl terminal fragments of human FGF‐23. Each sample was analyzed in duplicate as per manufacturer's instruction, with interassay coefficient of variation (CV%) <10% across the assay range of 1.5–2200 pg/mL for iFGF23 and 1.5–1400 RU/mL for cFGF23.
Protein structural modeling
Using the crystal structure of T. guttata GalNAc‐T3 in complex with uridine diphosphate (UDP), manganese, and FGF23 (PDB ID: 6S22)( 24 ) as the template (80% identity to human GalNAc‐T3), a homology model was built with SWISS‐MODEL.( 25 ) Point mutations were introduced and visualized in silico using PyMOL version 2.4 (www.pymol.org/2/). This highly homologous structure, solved at <2 Å resolution with well‐resolved binding of the Mn2+ cofactor and the UDP and FGF23 substrates, allows for confident prediction of the functionally relevant structure of human GalNAc‐T3, for which no crystal structure is available.
Literature review and mapping of all reported GALNT3 mutations
PubMed (www.pubmed.ncbi.nlm.nih.gov) was searched using the terms “GALNT3,” “GalNac‐T3,” “familial tumoral calcinosis,” “FTC,” “hyperostosis‐hyperphosphataemia syndrome,” and “hyperostosis‐hyperphosphatemia syndrome.” Abstracts were reviewed to identify case reports of individuals with GALNT3 variants. Clinical details (including BMD) were reviewed; identified GALNT3 variants were collated.
Identification of common GALNT3 variants associated with BMD
GWASs assessing LS and femoral neck (FN) (UK10K BMD( 26 ) and GEFOS LS‐ FN‐ BMD( 8 )), total body (TB) (life‐course TB BMD GWAS( 8 )), and forearm BMD (UK10K BMD) and BMD estimated from heel ultrasound (eBMD) (UK Biobank eBMD( 27 , 28 )) were interrogated for associations within ±50 kb of the coding sequence of GALNT3, FGF23, FGFR1, and klotho using the Musculoskeletal Knowledge Portal (MSKKP; https://msk.hugeamp.org), a data‐mining platform that includes genomic data relating to 291 musculoskeletal traits from 269 datasets.( 29 ) A publicly available GWAS of serum phosphate concentration, conducted in UK Biobank (http://www.nealelab.is/uk-biobank/), was also interrogated for associations with the same four loci.
A PheWAS was conducted using the MSKKP (https://msk.hugeamp.org/) and GWASATLAS (https://atlas.ctglab.nl), of which the latter is an online database of publicly available summary results statistics from 4,756 GWASs from 473 unique studies across 3,302 unique traits and 28 domains.( 30 ) Significance for pleiotropic associations used the traditional genome‐wide significance threshold for SNP‐trait PheWAS (p < 5 × 10−8).
Mendelian randomization (MR) using osteoblast eQTL data
We performed a two‐sample MR. The exposure (GALNT3‐expression quantitative trait loci [eQTLs]) was identified by searching eQTL data from primary human osteoblasts( 31 ) to identify SNPs within 0.5 kB of GALNT3 associated with GALNT3 expression levels (cis‐eQTLs). Other phosphate‐handling genes (FGF23, FGFR1, and klotho) were similarly evaluated. F‐statistics were calculated to evaluate instrument strength. The outcome comprised BMD as assessed in the GWAS datasets described earlier.( 26 , 28 , 32 ) We also examined possible associations of these eQTLs with serum phosphate concentration based on the UK Biobank Phosphate GWAS (http://www.nealelab.is/uk-biobank/). MR analyses were based on Wald ratios,( 33 ) where a single eQTL SNP was identified, or inverse variance weighted (IVW)( 34 ) and generalized IVW,( 35 ) where multiple SNPs were available.
Results
A HBM pedigree with a segregating GALNT3 c.1657C > T p.Arg553Trp variant
A pedigree with unexplained HBM, segregating in an autosomal dominant pattern, was identified within the UK HBM population (Figure 1A). The clinical and biochemical characteristics of each individual are summarized in Table 1, with further details below. All recruited individuals from this pedigree had high BMD Z‐scores and high body mass index (BMI). OA and dental overcrowding was common, with all individuals having multiple tooth extractions. There was no history of headaches, nerve compression, or low‐trauma fractures. No individuals had received antiresorptive or anabolic drugs. Neither parent had a history of fracture.
Fig. 1.
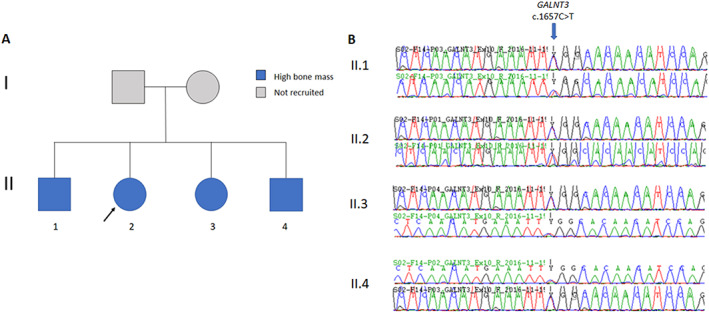
(A) HBM pedigree. (B) Electropherograms with GALNT3 c.1657C > T variant.
Table 1.
Clinical and biochemical characteristics of HBM individuals with GALNT3 c.1657C > T, p.R553W mutations, and a further unrelated HBM individual with a GALNT3 c.831 T > A, p.D277E mutation
| HBM pedigree GALNT3 c.1657C > T, p.Arg553Trp | Additional isolated HBM case GALNT3 c.831 T > A, p.Asp277Glu | |||||
|---|---|---|---|---|---|---|
| II.1 brother | II.2 Proband | II.3 sister | II.4 brother | S1 | ||
| Age | 67 | 64 | 59 | 58 | 61 | |
| Sex | Male | Female | Female | Male | Female | |
| Ethnicity | British (white) | British (white) | British (white) | British (whte) | British (white) | |
| Height (cm) | 178 | 162 | 160 | 182 | 162 | |
| Weight (kg) | 86 | 89 | 74 | 94 | 69 | |
| BMI (kg/m2) | 27.1 | 33.9 | 28.9 | 28.4 | 26.3 | |
| DXA L1 Z‐score | +3.4 | +5.2 | +3.3 | +2.9 | +5.2 | |
| DXA TH Z‐score | +1.8 | +5.1 | +2.4 | +2.0 | +3.2 | |
| Adult fracture | Yes (only digits) | No | No | No | No | |
| Dental overcrowding | Yes | Yes | Yes | Yes | No | |
| Torus | Yes | No | No | No | No | |
| Nerve compression | No | No | No | No | No | |
| Difficulties with swimming | Not known | No | No | No | Yes | |
| Blood tests a | ||||||
| Adjusted calcium (mmol/L) | – | 2.43 | – | 2.28 | – | |
| Phosphate (mmol/L) | – | 1.2 | – | 0.9 | 1.3 | |
| iFGF23 (pg/mL) | 43.4 | 52.5 | – | 22.8 | – | |
| cFGF23 (RU/mL) | 25.2 | 59.5 | – | 28.0 | – | |
| ALP (IU/L) | 50 | 78 | – | 70 | 65 | |
| P1NP (μg/L) | 28 | 27 | – | 16 | 15 | |
| CTX (μg/L) | 0.05 | 0.09 | – | 0.09 | 0.09 | |
| Osteocalcin (μg/L) | 12.2 | 12.0 | – | 12.0 | 6.0 | |
Abbreviations: ALP, alkaline phosphatase; BMI, body mass index; cFGF23, C‐terminal fragment fibroblast growth factor‐23; CTX, C‐terminal telopeptide of type I collagen; DXA, dual energy X‐ray absorptiometry; iFGF23, intact fibroblast growth factor‐23; P1NP, procollagen type 1 N propeptide.
Reference ranges: adjusted calcium 2.25–2.70; phosphate 0.8–1.5; iFGF23 28–121; cFGF23 < 100; ALP 20–120; P1NP: postmenopausal female 26–110, mle 20–76; CTX 0.1–0.5; osteocalcin 6.8–32.2.
Clinical features
Proband
The proband was a 64‐year‐old female with BMD Z‐scores of +5.2 at first lumbar vertebra (L1) and + 5.1 at TH. She had no difficulties swimming. She did not have any joint or limb pain and had never fractured. She had required wisdom tooth extractions and was noted to have minimal unilateral hearing loss on a recent hearing test (type of hearing loss not known). She had hypertension (diagnosed at age 34 years) and hypercholesterolaemia (diagnosed at age 64 years). She received menopausal hormone therapy between ages 50 and 57 years and had a hysterectomy and bilateral salpingo‐oophorectomy for stage 1 uterine cancer at age 62 years.
On examination, her height was 1.62 m and weight was 89 kg (BMI 33.9 kg/m2). She had mandibular enlargement but no mandibular or maxillary tori and no cranial nerve impingement. She had signs of OA (palpable crepitus in both knees and Heberden's nodes bilaterally) but normal range of movement throughout all joints, including her spine. Radiographs demonstrated degenerative changes in the hands (carpometacarpal joints and several distal interphalangeal joints), knees (medial and lateral compartments), and LS (joint space narrowing and osteophytosis).
Brother of proband
The proband's 67‐year‐old brother had BMD Z scores +3.4 at L1 and + 1.8 at TH. He had no difficulties swimming and denied any joint or limb pain. He had fractured his fingers playing cricket at age 16 years and his thumb playing hockey at age 22 years. He had four teeth removed due to overcrowding. He reported gradual bilateral hearing loss (type not known) over the preceding 10 years. He had hypercholesterolemia and had undergone a prostatectomy for prostate cancer at age 61 years.
On examination, his height was 1.78 m and weight was 86 kg (BMI 27.1 kg/m2). He had a small torus arising from the lower left mandible and lower jaw dental overcrowding. He had Heberden's nodes affecting multiple fingers. He had reduced forward flexion in his LS but otherwise normal range of joint movement. Radiographs revealed joint space narrowing and osteophytosis affecting multiple distal interphalangeal joints.
Sister of proband
The proband's 59‐year‐old sister resided outside of the UK, so history was obtained over the telephone, physical examination was not performed, and DXA scanning was performed in her home country.
The sister had BMD Z‐scores of +3.3 at L1 and + 2.4 at TH. She had no swimming difficulties. She had no joint or limb pain and had never fractured. Tooth extractions were performed due to dental overcrowding, and she required an orthodontic brace for 3 years. She had hypertension (diagnosed at age 52 years). She was taking regular calcium/vitamin D supplements. Her height was 1.6 m and weight was 74 kg (BMI 28.9 kg/m2).
Brother of proband
The proband's 58‐year‐old brother had BMD Z‐scores of +2.9 at L1 and + 2.0 at TH. He had no swimming difficulties. He had bilateral ankle pain for over 30 years (i.e., since approximately age 25 years). He had bilateral hip pain and had bilateral hip replacement for OA during the study follow‐up period. Baseline radiographs revealed severe joint space loss at both hips. He had never fractured. He had wisdom teeth removed at age 27 years. He had hypertension (diagnosed at age 50 years), asthma, and depression.
His height was 1.82 m and weight was 94 kg (BMI 28.4 kg/m2). He had a broad frame. He had no tori and no cranial nerve impingement. He had bilateral knee crepitus.
Sequencing results
A heterozygous missense variant in GALNT3 (NM_004482:exon 4:c.1657C > T, p.Arg553Trp) was identified, present in all affected siblings (Figure 1). The variant (rs764878326) is rare (GnomAD MAF 0.000016 in European non‐Finnish populations), with a GERP score of 4.47, and predicted to be pathogenic by multiple protein‐prediction algorithms (probably damaging by PolyPhen, damaging by SIFT, and disease‐causing by PMut [pathology score 0.81] and Mutation Taster). No other rare pathogenic variants with appropriate segregation were identified. Sanger sequencing confirmed the variant in each sequenced individual (Figure 1B).
A further HBM case with a rare heterozygous GALNT3 variant
Scrutiny of other UK HBM cases, and the HBM AOGC arm, identified an unrelated individual with a different rare heterozygous variant in GALNT3 (c.831 T > A, p.Asp277Glu).
Clinical features
Proband
This 61‐year‐old female had a BMD Z‐score of +5.2 at L1 and + 3.2 at TH. She was able to swim but had difficulty floating. She had never fractured. She had OA and fibromyalgia and reported widespread pain in her joints and back with muscle spasms. She reported multiple teeth “breaking” when younger, and she required multiple fillings throughout her 20s and 30s. She had a total hysterectomy at age 40 years and a cholecystectomy at age 60 years.
On examination, her height was 1.62 m and weight was 69 kg (BMI 26.3 kg/m2). She had no tori. Cranial nerve examination was normal. She had osteoarthritic changes in her hands and knees. She had reduced range of movement in her cervical spine, but range of movement was normal elsewhere. Radiographs revealed degenerative changes within the hands (joint space narrowing, osteophytosis, and subchondral cysts) and knees (joint space narrowing and osteophytosis).
Her mother had a fragility fracture of her hip. Other family information was limited.
Sequencing results
A rare, heterozygous missense variant in GALNT3 (NM_004482.4:c.831 T > A, p.Asp277Glu) was identified in the proband. This variant (rs139397826) is rare (MAF 0.000025), with a GERP score of 2.59, and is predicted to be pathogenic by multiple protein‐prediction algorithms (probably damaging by PolyPhen, damaging by SIFT, and disease‐causing by PMut [pathology score 0.92] and Mutation Taster).
LBM controls
GALNT3 variants in the LBM cohort
Neither variant (c.1657C > T p.Arg553Trp or c.831 T > A p.Asp.277Glu) was observed on interrogation of the AOGC LBM cohort. No other rare pathogenic GALNT3 variants were identified in this cohort when the same filtering parameters were used.
Protein structural modeling predicts that p.Arg553Trp and p.Asp277Glu disrupt GALNT3 function
GALNT3 encodes the 633 amino acid Golgi‐associated glycosyltransferase, polypeptide N‐acetylgalactosaminyltransferase‐3 (GALNT3). GALNT3 belongs to a large family of Golgi‐associated glycosyltransferases (GalNAc‐Ts) that initiate mucin‐type O‐glycosylation, catalyzing the transfer of N‐acetyl‐D‐galactosamine (GalNAc) from uridine‐diphosphate‐GalNAc (UDP‐GalNAc) to a serine or threonine residue on protein substrates, with relatively broad acceptor substrate specificity.( 24 ) GALNT3 consists of a short N‐terminal cytoplasmic tail, a hydrophobic transmembrane domain, a central catalytic (glycosyltransferase) domain, and a C‐terminal ricin B‐type lectin (carbohydrate binding) domain with a β‐trefoil fold. There are two conserved domains within the glycosyltransferase region: the N‐terminal domain (domain A, also known as the GT1 motif) and the C‐terminal domain (domain B, also known as the Gal/GalNAc‐T motif). The ricin B‐type lectin domain binds to GalNAc and contributes to glycopeptide specificity.
The c.1657C > T p.Arg553Trp variant in the first pedigree is located within the carbohydrate‐binding lectin domain. Substitution of arginine with tryptophan is predicted to disrupt salt‐bridge interactions, leading to instability of the protein (Figure 2).
Fig. 2.
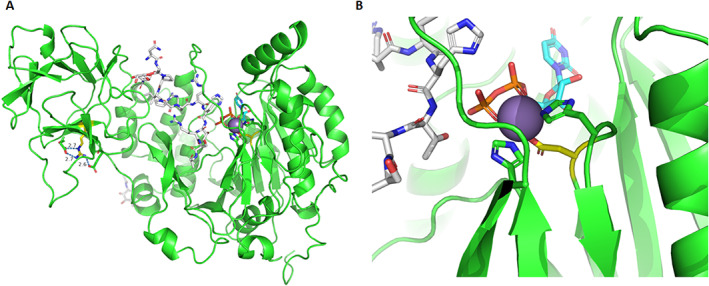
(A) Overview of GALNT3 protein structure with R553 indicated by yellow carbons, with salt‐bridge hydrogen‐bond interactions shown as blue dotted lines. R553W will result in the disruption of these salt‐bridge interactions, causing instability. (B) D277 (Asp277) is shown with yellow carbons and Mn2+ in purple, with the other residues coordinating to Mn2+ (H279 & H415). The D277E mutation is predicted to affect Mn2+ coordination/binding. The UDP substrate, whose binding will depend on Mn2+, is shown with cyan carbons and FGF23c with white carbons. Images generated using PyMOL version 2.4.
The c.831 T > A p.Asp277Glu variant is located within domain A of the glycosyltransferase domain and is part of the D277XH279 motif involved in Mn2+ (manganese) coordination. Mn2+ is key for binding the UDP pyrophosphate group; substitution of aspartate with glutamate is predicted to disrupt Mn2+ coordination and, consequently, UDP binding and, thus, the catalytic function of GALNT3 (Figure 2).
GALNT3 variants and bone mass
Rare variants
The gnomAD database indicates that the loss‐of‐function (LoF) oe (ratio of observed to expected number of LoF variants) in GALNT3 is 0.58 (90% CI 0.4–0.86), that is, only 58% of the expected LoF variants are observed. This suggests that GALNT3 is likely to be under a degree of selection against LoF variants.
Biallelic LoF GALNT3 variants are known to be associated with the extremely rare autosomal recessive conditions of hyperphosphatemic familial tumoral calcinosis (FTC) and hyperostosis‐hyperphosphatemia syndrome (HHS) (OMIM: 211900). Mice homozygous for a LoF Galnt3 mutation (Trp589Arg), a model for FTC and HHS, have higher areal BMD compared to wild‐type mice, while heterozygous mice had intermediate BMD.( 16 ) Mice homozygous for the mutation also had high cortical bone volume and trabecular number on micro–computed tomography (CT).( 36 )
A literature review identified 38 novel/rare LoF GALNT3 variants in humans, all reported in association with FTC and/or HHS (41 homozygotes, 21 compound heterozygotes) (Table 2). DXA‐measured BMD Z‐scores (at TH and/or LS) were reported in only four individuals with GALNT3‐associated FTC and HHS (all at age <25 years). Three individuals had normal BMD and one had “extremely reduced” BMD.( 48 ) One case series of seven individuals with GALNT3‐associated FTC and/or HHS reported that Z‐ and T‐scores were “within two standard deviations of the mean” for all individuals, apart from one individual who had low T‐scores in the context of chronic untreated systemic inflammation.( 46 ) GALNT3 variants associated with FTC/HHS showed no obvious clustering within the structure of the GALNT3 protein or relative to the two variants described in our HBM individuals (Figure 3).
Table 2.
Summary of published case reports of GALNT3 variants
| Variant | Protein consequence | Clinical consequence | BMD Z‐scores | Reference |
|---|---|---|---|---|
| c.2 T > A | Loss of start codon | HHS | NR | Gok et al.( 37 ) |
| c.41_58 del | p.Arg14Serfs*8 | FTC | NR | Garringer et al.( 38 ) |
| c.254_255delCT | p.Pro85Argfs*6 | FTC | Two females, homozygote, skeletal site not specified. Z‐scores of +0.7 (age 10) and 0.0 (age 9) | Kisla et al.( 39 ) |
| c.260‐266delGGCAAA | p Arg87Thrfs*19 | FTC | Two females (compound heterozygote, ages 6 and 8) –Z‐scores reported as “within 2 SD of mean” | Ramnitz et al. |
| c.484C > T | p.Arg162* | FTC | NR | Topaz et al.,( 40 ) Ichikawa et al.,( 41 ) Demellawy et al.,( 42 ) Carmichael et al.( 43 , 44 ) |
| c.485G > A | p.Arg162Gln | FTC | NR | Ichikawa et al.( 45 ) |
| c.516‐2A > T | p.Cys173Leu fs176* | FTC | Two females (compound heterozygote, ages 6 and 8) –Z‐scores reported as “within 2 SD of mean”( 46 ) | Laleye et al.,( 47 ) Ichikawa et al.,( 41 ) Ramnitz et al.( 46 ) |
| c.516‐2A > G | p.Cys173 fs176* | FTC/HHS | One female (age 16) LS −5.1 | Masi et al.( 48 ) |
| c.539G > A | p.Arg180His | FTC | NR | Sun et al.( 49 ) |
| c.659 T > A | p.Ile220Asn | FTC | NR | Sun et al.( 49 ) |
| c.677delC | p.Ala226Valfs*3 | FTC/HHS | NR | Ichikawa al.,( 45 ) Dauchez et al.( 50 ) |
| c.746_749delTCAG | p.Val249Aspfs*8 | FTC | One report of one female (compound heterozygote, age 6) with Z‐scores reported as “within 2 SD of mean” | Ramnitz et al.( 46 ) Guerra et al.( 51 ) |
| c.767G > T | p.Gly256Val | FTC/HHS | NR | Rafaelson et al.( 52 ) |
| c.782G > A | p.Arg261Gln | FTC | NR | Mahjoubi et al.( 53 ) |
| c.803‐804insC | p.Thr269Asn fs281* | HHS | NR | Ichikawa et al.( 54 ) |
| c.815C > A | p.Thr272Lys | FTC | NR | Ichikawa et al.( 55 ) |
| c.839G > A | p.Cys280Tyr | HHS | NR | Gok et al.( 37 ) |
| c.842A > G | p.Glu281Gly | FTC/HHS | NR | Joseph et al.( 56 ) |
| c.892delT* | p.Tyr298Thrfs*5 | FTC | One report of one female (compound heterozygote, age 6) with Z‐scores reported as “within 2 SD of mean” | Ramnitz et al.( 46 ) |
| c.966 T > G | p.Tyr322* | FTC | NR | Barbieri et al.( 57 ) |
| c.1076C > A | p.Thr359Lys | FTC | NR | Ichikawa et al.( 55 ) |
| c.1097 T > G | p.Leu366Arg | FTC/HHS | NR | Joseph et al.( 56 ) |
| c.1102_1103 insT | p.Ser368Phefs*8 | FTC | NR | Garringer et al.( 38 ) |
| c.1245 T > A | p.His415Gln | FTC | NR | Yancovitch et al.( 58 ) |
| c.1312C > T | p.Arg438Cys | FTC/HHS | One female (compound heterozygote, age 22)( 59 ) | Yancovitch et al.( 58 ) Dumitrescu et al.( 59 ) Ramnitz et al.( 46 ) |
| TH +1.9 | ||||
| LS +0.3 | ||||
| Forearm +1.0 | ||||
| One report of one female (age 36) with T‐ and Z‐scores reported as “within 2 SD of mean”( 46 ) | ||||
| c.1313G > A | p.Arg438His | HHS | NR | Olauson et al.( 60 ) |
| c.1319C > A | p.Ala440Glu | FTC | NR | Finer et al.( 44 ) |
| c.1387A > T | p.Arg463* | FTC | NR | Campagnoli et al.( 61 ) |
| c.1392 + 1G > A | Splicing error | HHS | NR | Ichikawa et al.( 45 ) |
| c.1441C > T | p.Gln481* | FTC | NR | Barbieri et al.( 57 ) |
| c.1460G > A | p.Trp487* | FTC | NR | Garringer et al.( 38 ) |
| c.1524 + 1G > A – skip exon 7 | Deletion of 44 amino acids from codons 464–508 | FTC/HHS | NR | Topaz et al.( 40 ) Frishberg et al.,( 62 ) Frishberg et al.( 63 ) |
| c.1524 + 5G > A – skip exon 7 | Deletion of 44 amino acids from codons 464–508 | FTC | One report of one female (compound heterozygote, age 36) with T and Z‐scores reported as “within 2 SD of mean”( 46 ) | Topaz et al.( 40 ) Ramnitz et al.( 46 ) |
| c.1584_1585insA | p.Pro529Thrfs*17 | FTC | 1 male (homozygote, age 29) | Ramnitz et al.( 46 ) |
| No Z‐scores but T‐score reported as | ||||
| AP spine −3.9 | ||||
| TH −3.2. Male sibling (homozygote, age 19 with same variant) reported as having T‐ and Z‐scores “within 2 SD of mean” | ||||
| c.1626 + 1G > A – skip exon 8 | Deletion of 34 amino acids from codons 509–542 | HHS | NR | Ichikawa et al.( 54 ) |
| c.1681 T > A | p.Cys561Ser | FTC | NR | Dayal et al.( 64 ) |
| c.1720 T > G | p.Cys574Gly | FTC/HHS | NR | Ichikawa et al.( 45 ) |
| c.1774C > T | p.Gln592X | FTC | One female (compound heterozygote, age 22)( 59 ) | Specktor et al.( 65 ) Dumitrescu et al.( 59 ) Ramnitz et al.( 46 ) |
| TH +1.9 | ||||
| LS +0.3 | ||||
| Forearm +1.0 | ||||
| One female (compound heterozygote, age 36) with T‐ and Z‐scores reported as “within 2 SD of mean”( 46 ) |
Note: Reported as per the sequence variant nomenclature guidelines from the Human Genome Variation Society (http://varnomen.hgvs.org/).
Abbreviations: BMD, Bone mineral density; FTC, Familial tumoral calcinosis; HHS, Hyperostosis‐hyperphosphatemia syndrome; LS, Lumbar spine; NR, Not reported; SD, standard deviation; TH, Total hip.
Fig. 3.
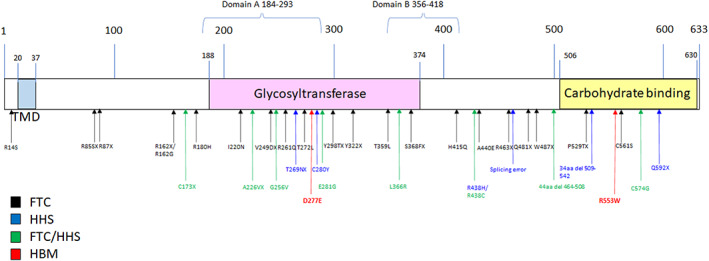
Schematic representation of GALNT3 protein structure. Arrows at sites of known FTC (black) and HHS (blue). Mutations in cases with features of both FTC and HHS are represented in green. HBM mutations are shown in red.
We sought to clarify the effect of heterozygous or homozygous GALNT3 rare LoF variants on bone mass by assessing BMD in a family in whom two individuals with FTC were homozygous for a GALNT3 variant affecting a splice site (c.1524 + 1G > A) (Figure 4). This variant was previously shown to result in disruption of the intron 7 donor splice site consensus sequence.( 40 ) Both homozygous individuals had normal BMD at both LS and TH; in contrast, both carrier parents and their heterozygous sister had Z‐scores <1.0 at LS but normal BMD at TH (Figure 4).
Fig. 4.
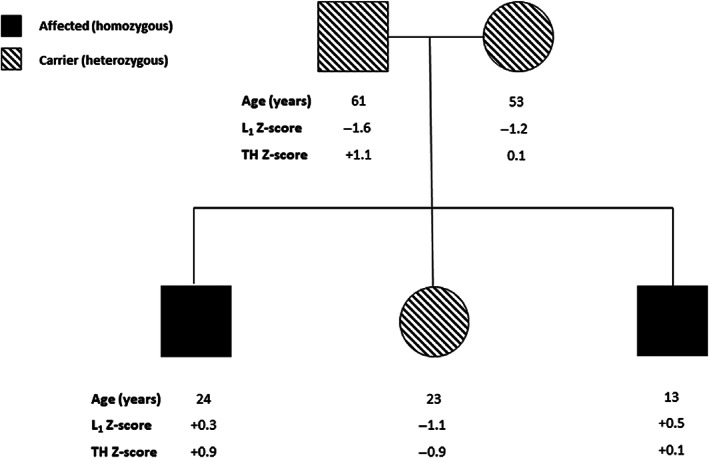
BMD measurements in Italian pedigree with FTC and GALNT3 c.1524 + 1G > A variant.
Common variants
Common variants at the GALNT3 locus are associated with BMD, as reported in an earlier GWAS in the AOGC cohort (lead SNP rs1863196 with suggestive association with BMD at TH [p = 2.3 × 10−5] and LS [p = 0.037]).( 16 )
Interrogation of subsequent BMD GWASs using MSKKP confirmed the previous suggestive results from AOGC. Significant (p < 5 × 10−8) associations were observed between SNPs in the GALNT3 locus (chr2:166,554,098 to 166,701,169) and BMD at TB (lead SNP rs7586085, p = 8.64 × 10−21),( 32 ) femoral neck (lead SNP rs10170839, p = 1.2 × 10−14),( 26 ) and LS (lead SNP rs11680288, p = 3.12 × 10−9) (Table 3); no significant association was found with forearm BMD,( 26 ) or eBMD( 28 ) (locus zoom plots for each result: Supplemental Materials Figures S1–S5). Phenome screening revealed an additional genome‐wide significant association with type 2 diabetes (lead SNP rs11686403, p = 9.68 × 10−11), although not of genome‐wide significance when adjusted for BMI (lead SNP rs6710518, p = 2.86 × 10−5) (no significant pleiotropic associations were found using GWASATLAS).
Table 3.
Genome‐wide significant associations of SNPs in the GALNT3 genomic region (p < 5 × 10−8) in order of significance
| Trait | Lead SNP | MAF | p value | β | 95% CI low | 95% CI high | Odds ratio |
|---|---|---|---|---|---|---|---|
| Total body BMD( 32 ) | rs7586085 | 0.41 | 8.64 × 10−21 | −0.0532 | −0.064 | −0.042 | |
| Femoral neck BMD( 26 ) | rs10170839 | 0.42 | 1.2 × 10−14 | −0.0594 | −0.074 | −0.045 | |
| Type 2 diabetes( 66 , 67 , 68 , 69 , 70 , 71 , 72 , 73 , 74 , 75 , 76 , 77 , 78 , 79 , 80 , 81 , 82 , 83 , 84 , 85 ) | rs11686403 | 0.41 | 9.68 × 10−11 | −0.022 | −0.028 | −0.017 | 0.098 |
| Lumbar spine BMD( 26 ) | rs11680288 | 0.42 | 3.12 × 10−9 | −0.0542 | −0.072 | −0.037 | |
| Phosphate | rs777356 | 0.48 | 2.73 × 10−5 | −0.01019 | −0.015 | −0.0054 | |
| eBMD( 28 ) | rs12692777 | 0.40 | 7.20 × 10−4 | −0.0117 | −0.018 | −0.005 | |
| Forearm BMD( 26 ) | rs34100816 | 0.27 | 1.78 × 10−3 | −0.0527 | −0.085 | −0.02 |
Note: Lead SNPs for estimated BMD from heel ultrasound (eBMD), forearm BMD, and phosphate are also shown, although these did not reach genome‐wide significance.
Abbreviations: BMD, bone mineral density; CI, confidence interval; eBMD, bone mineral density estimated from heel ultrasound; LDL, low‐density lipoprotein; MAF, minor allele frequency; SNP, single nucleotide polymorphism; β, beta value.
Given the effect of GALNT3 on phosphate metabolism via FGF23 (discussed below), we examined whether common variants in genes coding for other members of the FGF23 pathway (FGF23, its receptor FGFR1, and coreceptor klotho) were also associated with BMD. No associations reached genome‐wide significance (Table S1).
GALNT3 variants and phosphate homeostasis
Rare variants
Biallelic loss‐of‐function variants in GALNT3 cause disorders of phosphate homeostasis, specifically hyperphosphatemic FTC and HHS,( 86 ) acting through the FGF23 pathway.( 63 , 87 ) Although heterozygous carriers of FTC/HHS‐associated GALNT3 variants do not show any clinical features of FTC or HHS,( 61 ) subtle biochemical abnormalities, including slightly elevated phosphate concentrations, have been reported.( 41 , 56 )
Thus, in our HBM cases with heterozygous GALNT3 variants, we measured phosphate (II.2, II.4, S1), intact FGF23 (iFGF23) (II.1, II.2, II.4), and C‐terminal FGF23 (cFGF23) (II.1, II.2, II.4) concentrations where possible. These results were all in the normal reference range (Table 1).
Common variants
Interrogating the UK Biobank GWAS of phosphate concentrations identified strong associations with variants in the FGF23 locus (lead SNP rs2970818 p = 3.23 × 10−230) and klotho locus (lead SNP rs7324259, p = 2.99 × 10−10). However, associations with loci containing GALNT3 and FGFR1 did not reach genome‐wide significance (Table 3, Table S1).
Mendelian randomization studies suggest that GALNT3 expression reduces BMD
We used MR to examine whether GALNT3, FGF23, FGFR1, or klotho expression affected BMD. A single cis‐SNP, rs13427694, was associated with GALNT3 mRNA levels in primary human osteoblasts at p < 0.05, providing a weak genetic instrument as reflected by an F‐statistic of 7.8 (Table S2). Three cis‐SNPs were associated with FGFR1 mRNA (F‐statistic 7.0). One cis‐SNP was associated with klotho mRNA, which provided an insufficiently strong instrument for further analysis (F‐statistic 5.0), whereas no cis‐SNP was identified for FGF23.
As shown in Figure 5, subsequent MR analysis suggested that the GALNT3 osteoblast eQTL was inversely related to FN, LS, and TB BMD, with no associations seen within forearm BMD or heel eBMD, or with serum phosphate levels. While the FGFR1 eQTL was unrelated to FN, LS, or TB BMD, generalized IVW analysis suggested a decrease in forearm BMD, though IVW estimates crossed the null. Notably, osteoblast FGFR1 expression was related to an increase in phosphate, particularly in generalized IVW analyses.
Fig. 5.
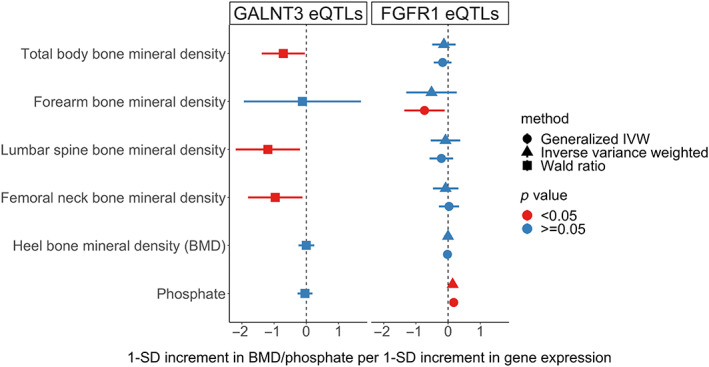
Association of GALNT3 and FGFR1 eQTLs with BMD and phosphate.
Discussion
We reported two rare heterozygous missense GALNT3 variants underlying HBM in two independent kindreds. In silico protein modeling predicts that these two variants are likely to result in loss of GALNT3 function, whether through structural instability (c.1657C > T p.Arg553Trp), which may lead to misfolding and more rapid degradation of the protein, or impairment of manganese binding and, consequently, loss of catalytic function (c.831 T > A p.Asp277Glu).
Analysis of common genetic variants from GWASs supports the suggestion from our HBM studies that GALNT3 plays an important role in regulating bone mass, given loci within this gene show associations at genome‐wide significance with LS, FN, and TB BMD. Though genetic associations do not demonstrate a causal role for a given locus, our MR analysis using cis‐eQTL data from primary human osteoblasts suggests that GALNT3 expression is causally associated with BMD. The eQTL analysis showed that increased expression of GALNT3 was associated with reduced BMD, consistent with our finding of two likely LoF GALNT3 variants causing an increase in BMD.
Our study demonstrates that the association between GALNT3 and BMD is supported by both rare and common variants and is consistent with findings from mouse models. As shown previously, drug targets with support from monogenic and population genetic studies are more likely to proceed successfully through the drug development pipeline and be approved.( 88 , 89 ) It is possible that targeting GALNT3‐dependent BMD pathways may open a new avenue in the search for new anabolic treatments for osteoporosis.
It is important to consider potential unwanted effects of targeting GALNT3. Three of the four HBM individuals from the index pedigree had OA; artifactual increases in BMD due to OA were excluded by use of L1 BMD measurements and careful inspection of DXA images. This predisposition to OA is consistent with previous findings from the UK HBM study where individuals with generalized HBM were shown to be at increased risk of developing OA and requiring joint replacement.( 17 , 90 ) “Bone‐forming” features of OA have previously been reported in association with HBM,( 91 ) perhaps reflecting an underlying tendency to increased osteoblast activity and excess bone formation, thereby contributing to the osteophytosis observed in several individuals. Dental overcrowding also appeared to be common, but a torus was only seen in one individual. Individuals with these two rare heterozygous GALNT3 variants appeared to display an otherwise benign clinical phenotype, with an absence of the nerve compression seen with other skeletal dysplasias.( 5 , 92 )
It is known that biallelic loss‐of‐function GALNT3 mutations cause the rare autosomal recessive disorder FTC and HHS in which deficient GALNT3‐mediated O‐glycosylation results in enhanced cleavage and inactivation of the phosphaturic hormone FGF23, leading to increased/normal inactive C terminal FG23, low intact FGF23, and hyperphosphatemia.( 87 ) However, whether these rare variants alter BMD has not been assessed in most cases. Two cases of FTC caused by novel variants in GALNT3 were reported to have very low BMD.( 46 , 48 ) However, both individuals had other clinical risk factors that could have led to low bone mass. The first individual had hypogonadism and reduced physical activity due to extraskeletal masses,( 48 ) while the second individual had untreated chronic systemic inflammation.( 46 ) It remains unclear whether these FTC‐associated GALNT3 variants had any role in directly causing this low BMD alongside disturbing phosphate metabolism.
Acknowledging that we were only able to assess these parameters in a few individuals, the normal phosphate, iFGF23, and cFGF23 concentrations in our heterozygous HBM GALNT3 cases raised the possibility that GALNT3 was able to alter BMD independently of the FGF23 pathway. Interrogation of the UK Biobank phosphate GWASs and our MR analysis further supported this hypothesis. In contrast to TB, lumbar, and spinal BMD, our GALNT3 eQTL was unrelated to circulating phosphate, and though there was a suggestive association between the GALNT3 genomic region and phosphate, this failed to reach genome‐wide significance.
In contrast to GALNT3, no SNPs within the FGF23 genomic region were associated with any BMD parameter at genome‐wide significance. The same applies to FGFR1 and klotho, the downstream receptor and coreceptor for FGF23, respectively, defects in which are associated with phosphate abnormalities, likely due to loss of FGF23 activity. This further supports the separation of this pathway from effects on BMD. As expected, FGF23 and klotho were strongly associated with phosphate on GWAS, although the association for FGFR1 did not reach genome‐wide significance in the UK Biobank dataset. FGFR1 cis‐eQTLs identified in osteoblast cultures were unrelated to LS, FN, and TB BMD. That said, FGFR1 expression was associated with reduced forearm BMD in generalized IVW analyses, though no effect was seen in IVW analyses, possibly as the latter incorporated fewer SNPs. Whereas FGFR1 expression had a smaller effect on BMD compared to GALNT3 expression, unlike the latter, FGFR1 expression had a strong effect on serum phosphate levels, providing further evidence of dissociation of GALNT3 and FGF23 function.
Taken together, studies of common genetic variants and gene expression suggest important functional differences between GALNT3 and FGF23/FGFR1/klotho, with common variants in the former predominantly affecting BMD and the latter phosphate homeostasis. Previous studies of GALNT3 glycosylation activity focused on FGF23 as its substrate. Since the effects of GALNT3 on BMD appear to be independent of phosphate homeostasis, these could be mediated by glycosylation of distinct protein(s) primarily involved in regulating bone mass, which are yet to be identified. HBM caused by heterozygous p.Arg553Trp and p.Asp277Glu GALNT3 variants may result from the disruption of interactions between GALNT3 and these bone regulatory protein(s).
Bone turnover markers (CTX, osteocalcin, and P1NP) in our HBM individuals were either low or toward the lower limit of normal, indicating that bone turnover overall may be suppressed. This is in keeping with previous findings from the UK HBM cohort, where bone turnover markers generally tended to be lower than those in controls.( 93 ) This was also previously found to be the case with some individuals with LRP5 mutations,( 94 ) although others reported normal or high turnover.( 95 , 96 , 97 ) One possible explanation for this is that generalized HBM may be caused by mechanisms that increase osteoblast activity and bone formation when the HBM phenotype is being developed (i.e., during puberty), and these same mechanisms then suppress bone turnover during later life to maintain the increased BMD.( 93 ) Another possible explanation is that these HBM variants lead to wnt activation (similar to HBM variants in LRP5 and SOST), causing suppression of bone resorption as a consequence of the known inhibitory action of wnts on osteoclastogenesis. This mechanism underlies the suppression of bone turnover markers that is seen following the use of romosozumab (which leads to wnt pathway activation)( 4 ).
The mechanism by which GALNT3 affects BMD without disturbing phosphate homeostasis requires further investigation to avoid precipitating features of FTC or HHS, particularly because there did not appear to be any obvious clustering or segregation of HBM‐associated GALNT3 variants compared to FTC/HHS‐associated GALNT3 variants within the structure of the protein. Furthermore, male mice with W589R‐induced FTC also had infertility, a feature not widely reported in humans with FTC or HHS. GALNT3 is highly expressed in the testis; one case report described a boy with testicular microlithiasis associated with oligozoospermia.( 61 ) The finding that common variants in GALNT3 on GWAS appear to be protective against type 2 diabetes also raises the concern that inactivating GALNT3 may increase the risk of developing this condition.
There are several limitations to our study. Since phenotyping was only performed in a limited number of family members, information about comorbidities was not consistently available in the kindreds used for this study. Phosphate and FGF23 measurements were found to be normal but were only tested at one time point in some of the HBM cases. Serum phosphate concentration is regulated by several hormones, including PTH, and is heavily influenced by food intake, neither of which was quantified or measured. It has been shown that dietary phosphate intake can affect the development of ectopic calcification in FTC mouse models. High dietary intake of phosphate in humans has also been associated with larger FTC lesions. Dietary phosphate intake in studied individuals was not documented, so it is unclear whether this had any effect on the biochemical phenotype observed in these HBM cases. A further limitation is that the eQTL data used in our study were derived from human osteoblasts obtained from a small sample of 95 donors, so the derived genetic instruments were relatively weak, risking weak instrument bias.
In conclusion, we report two rare heterozygous GALNT3 variants as putative causes of HBM. Our study indicates that c.1657C > T p.Arg553Trp and c.831 T > A p.Asp277Glu variants are likely to cause loss of function GALNT3. We hypothesize that these GALNT3 variants may affect BMD through mechanisms that are independent of phosphate and FGF23 regulation. This is supported by evidence from population genetic studies where common variants in GALNT3, including a GALNT3 cis‐eQTL, are associated with BMD more strongly than with phosphate levels. Further investigation of the mechanism by which this occurs may yield novel anabolic drug targets for osteoporosis treatment.
Disclosures
The authors have no potential conflicts of interest.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4795.
Supporting information
Table S1. Top genome‐wide associations for SNPs in genomic regions of FGF23, FGFR1, and KL with BMD parameters and phosphate. Abbreviations: BMD: bone mineral density; CI: confidence interval; eBMD: estimated BMD from heel ultrasound; FGF23: fibroblast growth factor‐23; FGFR1: fibroblast growth factor receptor 1; FN: femoral neck; KL: klotho; LS: lumbar spine; MAF: minor allele frequency; SNP: single nucleotide polymorphism; β: beta coefficient.
Table S2. GALNT3 and FGFR1 osteoblast‐derived eQTLs used as genetic instruments for the exposure in two‐sample MR. The F‐statistic for each genetic instrument is shown together with the method of MR analysis used. Abbreviations: eQTL, expressive quantitative trait loci; FGFR1, fibroblast growth factor receptor 1, GIVW, generalized inverse variance weighted; IVW, inverse variance weighted; MR, Mendelian randomization; SNP, single nucleotide polymorphism; β, beta coefficient.
Figure S1. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with total body BMD (generated using Musculoskeletal Knowledge Portal)
Figure S2. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with femoral neck mineral density (generated using Musculoskeletal Knowledge Portal)
Figure S3. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with lumbar spine BMD (generated using Musculoskeletal Knowledge Portal)
Figure S4. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with type 2 diabetes (generated using Musculoskeletal Knowledge Portal)
Figure S5. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with type 2 diabetes, adjusted for BMI (generated using Musculoskeletal Knowledge Portal)
Acknowledgments
AOGC investigators include Eugene McCloskey,1 Geoffrey C. Nicholson,2 Richard Eastell,1 Richard L. Prince,3 John A. Eisman,4,5 Graeme Jones,6 Philip Sambrook7 (deceased), Ian R. Reid,8 Elaine M. Dennison,9 and John Wark.2 NH was supported by the Medical Research Council (MR/V00199X/1), Elizabeth Blackwell Institute for Health Research, University of Bristol, and the Wellcome Trust Institutional Strategic Support Fund (8064/Hassan/WT ISSF 3). CLG was funded by the Wellcome Trust (080280/Z/06/Z), the EU 7th Framework Programme Ref. 247642 (GEoCoDE), a British Geriatric Society travel grant, and Versus Arthritis (formerly Arthritis Research UK) (Grant Ref. 20000). MAB was funded by a National Health and Medical Research Council (Australia) Principal Research Fellowship. ELD was funded by a National Health and Medical Research Council (Australia) Career Development Award (569807). AML was funded by a National Health and Medical Research Council Early Career Fellowship (APP1158111). The AOGC was funded by the National Health and Medical Research Council (Australia) (Grant Ref. 511132). Funding was also received from the Australian Cancer Research Foundation and Rebecca Cooper Foundation (Australia).
1University of Sheffield, UK, 2University of Melbourne, Geelong, Australia, 3University of Western Australia, Perth, Australia, 4St Vincent's Clinical School, University of New South Wales, 5Menzies Research Institute, University of Tasmania, Hobart, Australia, 6Kolling Institute, Royal North Shore Hospital, University of Sydney, Australia, 7University of Auckland, New Zealand, 8Medical Research Council Resource Centre, Southampton, UK.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
References
- 1. Burge R, Dawson‐Hughes B, Solomon DH, Wong JB, King A, Tosteson A. Incidence and economic burden of osteoporosis‐related fractures in the United States, 2005‐2025. J Bone Miner Res. 2007;22:465–475. [DOI] [PubMed] [Google Scholar]
- 2. Kanis JA, Delmas P, Burckhardt P, Cooper C, Torgerson D. Guidelines for diagnosis and management of osteoporosis. The European Foundation for Osteoporosis and Bone Disease. Osteoporos Int. 1997;7:390–406. [DOI] [PubMed] [Google Scholar]
- 3. Koller G, Goetz V, Vandermeer B, et al. Persistence and adherence to parenteral osteoporosis therapies: a systematic review. Osteoporos Int. 2020;31:2093–2102. [DOI] [PubMed] [Google Scholar]
- 4. McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370:412–420. [DOI] [PubMed] [Google Scholar]
- 5. Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot‐containing protein. Am J Hum Genet. 2001;68:577–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Staehling‐Hampton K, Proll S, Paeper BW, et al. A 52‐kb deletion in the SOST‐MEOX1 intergenic region on 17q12‐q21 is associated with van Buchem disease in the Dutch population. Am J Med Genet. 2002;110:144–152. [DOI] [PubMed] [Google Scholar]
- 7. Balemans W, Patel N, Ebeling M, et al. Identification of a 52 kb deletion downstream of the SOST gene in patients with van Buchem disease. J Med Genet. 2002;39:91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Estrada K, Styrkarsdottir U, Evangelou E, et al. Genome‐wide meta‐analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat Genet. 2012;44:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hamersma H, Gardner J, Beighton P. The natural history of sclerosteosis. Clin Genet. 2003;63:192–197. [DOI] [PubMed] [Google Scholar]
- 10. Gregson CL, Duncan EL. The genetic architecture of high Bone mass. Front Endocrinol (Lausanne). 2020;11:595653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gregson CL, Steel SA, O'Rourke KP, et al. 'Sink or swim': an evaluation of the clinical characteristics of individuals with high bone mass. Osteoporos Int. 2012;23:643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gregson CL, Wheeler L, Hardcastle SA, et al. Mutations in known monogenic high Bone mass loci only explain a Small proportion of high Bone mass cases. J Bone Miner Res. 2016;31:640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gregson CL, Newell F, Leo PJ, et al. Genome‐wide association study of extreme high bone mass: contribution of common genetic variation to extreme BMD phenotypes and potential novel BMD‐associated genes. Bone. 2018;114:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zankl A, Duncan EL, Leo PJ, et al. Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino‐terminal transcriptional activation domain of MAFB. Am J Hum Genet. 2012;90:494–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gregson CL, Bergen DJM, Leo P, et al. A rare mutation in SMAD9 associated with high Bone mass identifies the SMAD‐dependent BMP signaling pathway as a potential anabolic target for osteoporosis. J Bone Miner Res. 2020;35:92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Duncan EL, Danoy P, Kemp JP, et al. Genome‐wide association study using extreme truncate selection identifies novel genes affecting bone mineral density and fracture risk. PLoS Genet. 2011;7:e1001372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hartley A, Hardcastle SA, Frysz M, et al. Increased development of radiographic hip osteoarthritis in individuals with high bone mass: a prospective cohort study. Arthritis Res Ther. 2021;23:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McInerney‐Leo AM, Schmidts M, Cortes CR, et al. Short‐rib polydactyly and Jeune syndromes are caused by mutations in WDR60. Am J Hum Genet. 2013;93:515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012;40:W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lopez‐Ferrando V, Gazzo A, de la Cruz X, Orozco M, Gelpi JL. PMut: a web‐based tool for the annotation of pathological variants on proteins, 2017 update. Nucleic Acids Res. 2017;45:W222–W228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods. 2014;11:361–362. [DOI] [PubMed] [Google Scholar]
- 24. de Las Rivas M, Paul Daniel EJ, Narimatsu Y, et al. Molecular basis for fibroblast growth factor 23 O‐glycosylation by GalNAc‐T3. Nat Chem Biol. 2020;16:351–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Waterhouse A, Bertoni M, Bienert S, et al. SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 2018;46:W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng HF, Forgetta V, Hsu YH, et al. Whole‐genome sequencing identifies EN1 as a determinant of bone density and fracture. Nature. 2015;526:112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kemp JP, Morris JA, Medina‐Gomez C, et al. Identification of 153 new loci associated with heel bone mineral density and functional involvement of GPC6 in osteoporosis. Nat Genet. 2017;49:1468–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morris JA, Kemp JP, Youlten SE, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat Genet. 2019;51:258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kiel DP, Kemp JP, Rivadeneira F, et al. The musculoskeletal knowledge portal: making omics data useful to the broader scientific community. J Bone Miner Res. 2020;35:1626–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Watanabe K, Stringer S, Frei O, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51:1339–1348. [DOI] [PubMed] [Google Scholar]
- 31. Grundberg E, Kwan T, Ge B, et al. Population genomics in a disease targeted primary cell model. Genome Res. 2009;19:1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Medina‐Gomez C, Kemp JP, Trajanoska K, et al. Life‐course genome‐wide association study Meta‐analysis of Total body BMD and assessment of age‐specific effects. Am J Hum Genet. 2018;102:88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2017;26:2333–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bowden J, Del Greco MF, Minelli C, Davey Smith G, Sheehan N, Thompson J. A framework for the investigation of pleiotropy in two‐sample summary data Mendelian randomization. Stat Med. 2017;36:1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu Z, Zheng Z, Zhang F, et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat Commun. 2018;9:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Esapa CT, Head RA, Jeyabalan J, et al. A mouse with an N‐ethyl‐N‐nitrosourea (ENU) induced Trp589Arg Galnt3 mutation represents a model for hyperphosphataemic familial tumoural calcinosis. PLoS One. 2012;7:e43205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gok F, Chefetz I, Indelman M, Kocaoglu M, Sprecher E. Newly discovered mutations in the GALNT3 gene causing autosomal recessive hyperostosis‐hyperphosphatemia syndrome. Acta Orthop. 2009;80:131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garringer HJ, Mortazavi SM, Esteghamat F, et al. Two novel GALNT3 mutations in familial tumoral calcinosis. Am J Med Genet A. 2007;143A:2390–2396. [DOI] [PubMed] [Google Scholar]
- 39. Kisla Ekinci RM, Gurbuz F, Balci S, et al. Hyperphosphatemic familial Tumoral calcinosis in two siblings with a novel mutation in GALNT3 gene: experience from southern Turkey. J Clin Res Pediatr Endocrinol. 2019;11:94–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Topaz O, Shurman DL, Bergman R, et al. Mutations in GALNT3, encoding a protein involved in O‐linked glycosylation, cause familial tumoral calcinosis. Nat Genet. 2004;36:579–581. [DOI] [PubMed] [Google Scholar]
- 41. Ichikawa S, Lyles KW, Econs MJ. A novel GALNT3 mutation in a pseudoautosomal dominant form of tumoral calcinosis: evidence that the disorder is autosomal recessive. J Clin Endocrinol Metab. 2005;90:2420–2423. [DOI] [PubMed] [Google Scholar]
- 42. Demellawy DE, Chang N, de Nanassy J, Nasr A. GALNT3 gene mutation‐associated chronic recurrent multifocal osteomyelitis and familial hyperphosphatemic familial tumoral calcinosis. Scand J Rheumatol. 2015;44:170–172. [DOI] [PubMed] [Google Scholar]
- 43. Carmichael KD, Bynum JA, Evans EB. Familial tumoral calcinosis: a forty‐year follow‐up on one family. J Bone Joint Surg Am. 2009;91:664–671. [DOI] [PubMed] [Google Scholar]
- 44. Finer G, Price HE, Shore RM, White KE, Langman CB. Hyperphosphatemic familial tumoral calcinosis: response to acetazolamide and postulated mechanisms. Am J Med Genet A. 2014;164A:1545–1549. [DOI] [PubMed] [Google Scholar]
- 45. Ichikawa S, Baujat G, Seyahi A, et al. Clinical variability of familial tumoral calcinosis caused by novel GALNT3 mutations. Am J Med Genet A. 2010;152A:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramnitz MS, Gourh P, Goldbach‐Mansky R, et al. Phenotypic and genotypic characterization and treatment of a cohort with familial Tumoral calcinosis/hyperostosis‐hyperphosphatemia syndrome. J Bone Miner Res. 2016;31:1845–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laleye A, Alao MJ, Gbessi G, et al. Tumoral calcinosis due to GALNT3 C.516‐2A >T mutation in a black African family. Genet Couns. 2008;19:183–192. [PubMed] [Google Scholar]
- 48. Masi L, Beltrami G, Ottanelli S, et al. Human Preosteoblastic cell culture from a patient with Severe Tumoral calcinosis‐hyperphosphatemia due to a new GALNT3 gene mutation: study of In vitro mineralization. Calcif Tissue Int. 2015;96:438–452. [DOI] [PubMed] [Google Scholar]
- 49. Sun L, Zhao L, Du L, et al. Identification of two novel mutations in the GALNT3 gene in a Chinese family with hyperphosphatemic familial tumoral calcinosis. Bone Res. 2016;4:16038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dauchez A, Souffir C, Quartier P, Baujat G, Briot K, Roux C. Hyperphosphatemic familial Tumoral calcinosis with Galnt3 mutation: transient response to anti‐Interleukin‐1 treatments. JBMR Plus. 2019;3:e10185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guerra MG, Videira T, de Fonseca D, Vieira R, Dos Santos J, Pinto S. Hyperphosphataemic familial tumoral calcinosis: case report of a rare and challenging disease. Scand J Rheumatol. 2020;49:80–81. [DOI] [PubMed] [Google Scholar]
- 52. Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Long‐term clinical outcome and phenotypic variability in hyperphosphatemic familial tumoral calcinosis and hyperphosphatemic hyperostosis syndrome caused by a novel GALNT3 mutation; case report and review of the literature. BMC Genet. 2014;15:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mahjoubi F, Ghadir M, Samanian S, Heydari I, Honardoost M. Hyperphosphatemic familial tumoral calcinosis caused by a novel variant in the GALNT3 gene. J Endocrinol Invest. 2020;43:1125–1130. [DOI] [PubMed] [Google Scholar]
- 54. Ichikawa S, Guigonis V, Imel EA, et al. Novel GALNT3 mutations causing hyperostosis‐hyperphosphatemia syndrome result in low intact fibroblast growth factor 23 concentrations. J Clin Endocrinol Metab. 2007;92:1943–1947. [DOI] [PubMed] [Google Scholar]
- 55. Ichikawa S, Imel EA, Sorenson AH, et al. Tumoral calcinosis presenting with eyelid calcifications due to novel missense mutations in the glycosyl transferase domain of the GALNT3 gene. J Clin Endocrinol Metab. 2006;91:4472–4475. [DOI] [PubMed] [Google Scholar]
- 56. Joseph L, Hing SN, Presneau N, et al. Familial tumoral calcinosis and hyperostosis‐hyperphosphataemia syndrome are different manifestations of the same disease: novel missense mutations in GALNT3. Skeletal Radiol. 2010;39:63–68. [DOI] [PubMed] [Google Scholar]
- 57. Barbieri AM, Filopanti M, Bua G, Beck‐Peccoz P. Two novel nonsense mutations in GALNT3 gene are responsible for familial tumoral calcinosis. J Hum Genet. 2007;52:464–468. [DOI] [PubMed] [Google Scholar]
- 58. Yancovitch A, Hershkovitz D, Indelman M, et al. Novel mutations in GALNT3 causing hyperphosphatemic familial tumoral calcinosis. J Bone Miner Metab. 2011;29:621–625. [DOI] [PubMed] [Google Scholar]
- 59. Dumitrescu CE, Kelly MH, Khosravi A, et al. A case of familial tumoral calcinosis/hyperostosis‐hyperphosphatemia syndrome due to a compound heterozygous mutation in GALNT3 demonstrating new phenotypic features. Osteoporos Int. 2009;20:1273–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Olauson H, Krajisnik T, Larsson C, Lindberg B, Larsson TE. A novel missense mutation in GALNT3 causing hyperostosis‐hyperphosphataemia syndrome. Eur J Endocrinol. 2008;158:929–934. [DOI] [PubMed] [Google Scholar]
- 61. Campagnoli MF, Pucci A, Garelli E, et al. Familial tumoral calcinosis and testicular microlithiasis associated with a new mutation of GALNT3 in a white family. J Clin Pathol. 2006;59:440–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frishberg Y, Topaz O, Bergman R, et al. Identification of a recurrent mutation in GALNT3 demonstrates that hyperostosis‐hyperphosphatemia syndrome and familial tumoral calcinosis are allelic disorders. J Mol Med (Berl). 2005;83:33–38. [DOI] [PubMed] [Google Scholar]
- 63. Frishberg Y, Ito N, Rinat C, et al. Hyperostosis‐hyperphosphatemia syndrome: a congenital disorder of O‐glycosylation associated with augmented processing of fibroblast growth factor 23. J Bone Miner Res. 2007;22:235–242. [DOI] [PubMed] [Google Scholar]
- 64. Dayal D, Gupta S, Kumar R, Srinivasan R, Lorenz‐Depiereux B, Strom TM. A novel homozygous variant in exon 10 of the GALNT3 gene causing hyperphosphatemic familial tumoral calcinosis in a family from North India. Intractable Rare Dis Res. 2021;10:55–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Specktor P, Cooper JG, Indelman M, Sprecher E. Hyperphosphatemic familial tumoral calcinosis caused by a mutation in GALNT3 in a European kindred. J Hum Genet. 2006;51:487–490. [DOI] [PubMed] [Google Scholar]
- 66. Bonas‐Guarch S, Guindo‐Martinez M, Miguel‐Escalada I, et al. Re‐analysis of public genetic data reveals a rare X‐chromosomal variant associated with type 2 diabetes. Nat Commun. 2018;9:321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen J, Sun M, Adeyemo A, et al. Genome‐wide association study of type 2 diabetes in Africa. Diabetologia. 2019;62:1204–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen P, Ong RT, Tay WT, et al. A study assessing the association of glycated hemoglobin A1C (HbA1C) associated variants with HbA1C, chronic kidney disease and diabetic retinopathy in populations of Asian ancestry. PLoS One. 2013;8:e79767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Consortium , Williams AL, Jacobs SB, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506:97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mercader JM, Liao RG, Bell AD, et al. A loss‐of‐function splice acceptor variant in IGF2 is protective for type 2 diabetes. Diabetes. 2017;66:2903–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Flannick J, Fuchsberger C, Mahajan A, et al. Sequence data and association statistics from 12,940 type 2 diabetes cases and controls. Sci Data. 2017;4:170179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fuchsberger C, Flannick J, Teslovich TM, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536:41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gaulton KJ, Ferreira T, Lee Y, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. 2015;47:1415–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mahajan A, Wessel J, Willems SM, et al. Refining the accuracy of validated target identification through coding variant fine‐mapping in type 2 diabetes. Nat Genet. 2018;50:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Guindo‐Martinez M, Amela R, Bonas‐Guarch S, et al. The impact of non‐additive genetic associations on age‐related complex diseases. Nat Commun. 2021;12:2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hebert HL, Shepherd B, Milburn K, et al. Cohort profile: genetics of diabetes audit and Research in Tayside Scotland (GoDARTS). Int J Epidemiol. 2018;47:380–381j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kanai M, Akiyama M, Takahashi A, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50:390–400. [DOI] [PubMed] [Google Scholar]
- 78. Laakso M, Kuusisto J, Stancakova A, et al. The metabolic syndrome in men study: a resource for studies of metabolic and cardiovascular diseases. J Lipid Res. 2017;58:481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Morris AP, Voight BF, Teslovich TM, et al. Large‐scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Ng MC, Shriner D, Chen BH, et al. Meta‐analysis of genome‐wide association studies in African Americans provides insights into the genetic architecture of type 2 diabetes. PLoS Genet. 2014;10:e1004517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Replication, D. I. G., Meta‐analysis, C., Asian Genetic Epidemiology Network Type 2 Diabetes, C., South Asian Type 2 Diabetes, C., Mexican American Type 2 Diabetes, C., Type 2 Diabetes Genetic Exploration by Nex‐generation sequencing in muylti‐Ethnic Samples, C , Mahajan A, Go MJ, et al. Genome‐wide trans‐ancestry meta‐analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46:234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Scott RA, Scott LJ, Magi R, et al. An expanded genome‐wide association study of type 2 diabetes in Europeans. Diabetes. 2017;66:2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Spracklen CN, Horikoshi M, Kim YJ, et al. Identification of type 2 diabetes loci in 433,540 east Asian individuals. Nature. 2020;582:240–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wessel J, Majarian TD, Highland HM, et al. Rare non‐coding variation identified by large scale whole genome sequencing reveals unexplained heritability of type 2 diabetes. medRxiv. 2020:20221812. [Google Scholar]
- 85. Zhao W, Rasheed A, Tikkanen E, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49:1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Kato K, Hansen L, Clausen H. Polypeptide N‐acetylgalactosaminyltransferase‐associated phenotypes in mammals. Molecules. 2021;26:5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ito N, Fukumoto S. Congenital Hyperphosphatemic conditions caused by the deficient activity of FGF23. Calcif Tissue Int. 2021;108:104–115. [DOI] [PubMed] [Google Scholar]
- 88. King EA, Davis JW, Degner JF. Are drug targets with genetic support twice as likely to be approved? Revised estimates of the impact of genetic support for drug mechanisms on the probability of drug approval. PLoS Genet. 2019;15:e1008489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Nelson MR, Tipney H, Painter JL, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–860. [DOI] [PubMed] [Google Scholar]
- 90. Hartley A, Hardcastle SA, Paternoster L, et al. Individuals with high bone mass have increased progression of radiographic and clinical features of knee osteoarthritis. Osteoarthr Cartil. 2020;28:1180–1190. [DOI] [PubMed] [Google Scholar]
- 91. Hardcastle SA, Dieppe P, Gregson CL, et al. Prevalence of radiographic hip osteoarthritis is increased in high bone mass. Osteoarthr Cartil. 2014;22:1120–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Leupin O, Piters E, Halleux C, et al. Bone overgrowth‐associated mutations in the LRP4 gene impair sclerostin facilitator function. J Biol Chem. 2011;286:19489–19500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Gregson CL, Paggiosi MA, Crabtree N, et al. Analysis of body composition in individuals with high bone mass reveals a marked increase in fat mass in women but not men. J Clin Endocrinol Metab. 2013;98:818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Frost M, Andersen T, Gossiel F, et al. Levels of serotonin, sclerostin, bone turnover markers as well as bone density and microarchitecture in patients with high‐bone‐mass phenotype due to a mutation in Lrp5. J Bone Miner Res. 2011;26:1721–1728. [DOI] [PubMed] [Google Scholar]
- 95. Balemans W, Devogelaer JP, Cleiren E, Piters E, Caussin E, Van Hul W. Novel LRP5 missense mutation in a patient with a high bone mass phenotype results in decreased DKK1‐mediated inhibition of Wnt signaling. J Bone Miner Res. 2007;22:708–716. [DOI] [PubMed] [Google Scholar]
- 96. Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL‐receptor‐related protein 5. N Engl J Med. 2002;346:1513–1521. [DOI] [PubMed] [Google Scholar]
- 97. Whyte MP, Reinus WH, Mumm S. High‐bone‐mass disease and LRP5. N Engl J Med. 2004;350:2096–2099 author reply 2096‐2099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Top genome‐wide associations for SNPs in genomic regions of FGF23, FGFR1, and KL with BMD parameters and phosphate. Abbreviations: BMD: bone mineral density; CI: confidence interval; eBMD: estimated BMD from heel ultrasound; FGF23: fibroblast growth factor‐23; FGFR1: fibroblast growth factor receptor 1; FN: femoral neck; KL: klotho; LS: lumbar spine; MAF: minor allele frequency; SNP: single nucleotide polymorphism; β: beta coefficient.
Table S2. GALNT3 and FGFR1 osteoblast‐derived eQTLs used as genetic instruments for the exposure in two‐sample MR. The F‐statistic for each genetic instrument is shown together with the method of MR analysis used. Abbreviations: eQTL, expressive quantitative trait loci; FGFR1, fibroblast growth factor receptor 1, GIVW, generalized inverse variance weighted; IVW, inverse variance weighted; MR, Mendelian randomization; SNP, single nucleotide polymorphism; β, beta coefficient.
Figure S1. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with total body BMD (generated using Musculoskeletal Knowledge Portal)
Figure S2. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with femoral neck mineral density (generated using Musculoskeletal Knowledge Portal)
Figure S3. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with lumbar spine BMD (generated using Musculoskeletal Knowledge Portal)
Figure S4. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with type 2 diabetes (generated using Musculoskeletal Knowledge Portal)
Figure S5. Locus zoom plot for genome‐wide significant associations in GALNT3 locus with type 2 diabetes, adjusted for BMI (generated using Musculoskeletal Knowledge Portal)
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.