

ABSTRACT
Eastern equine encephalitis virus (EEEV), Madariaga virus (MADV), and Venezuelan equine encephalitis virus complex (VEEV) are New World alphaviruses transmitted by mosquitoes. They cause febrile and sometimes severe neurological diseases in human and equine hosts. Detecting them during the acute phase is hindered by non-specific symptoms and limited diagnostic tools. We designed and clinically assessed real-time reverse transcription polymerase chain reaction assays (rRT-PCRs) for VEEV complex, MADV, and EEEV using whole-genome sequences. Validation involved 15 retrospective serum samples from 2015 to 2017 outbreaks, 150 mosquito pools from 2015, and 118 prospective samples from 2021 to 2022 surveillance in Panama. The rRT-PCRs detected VEEV complex RNA in 10 samples (66.7%) from outbreaks, with one having both VEEV complex and MADV RNAs. VEEV complex RNA was found in five suspected dengue cases from disease surveillance. The rRT-PCR assays identified VEEV complex RNA in three Culex (Melanoconion) vomerifer pools, leading to VEEV isolates in two. Phylogenetic analysis revealed the VEEV ID subtype in positive samples. Notably, 11.9% of dengue-like disease patients showed VEEV infections. Together, our rRT-PCR validation in human and mosquito samples suggests that this method can be incorporated into mosquito and human encephalitic alphavirus surveillance programs in endemic regions.
KEYWORDS: Venezuelan equine encephalitis, Madariaga virus, Eastern equine encephalitis virus, alphavirus, rRT-PCR
INTRODUCTION
New World alphaviruses (Togaviridae, genus Alphavirus) are a diverse group of mosquito-borne viruses that can cause severe disease in humans, including the Venezuelan equine encephalitis virus complex (VEEV complex), Madariaga virus (MADV), and Eastern equine encephalitis virus (1, 2). These persist in sylvatic-enzootic cycles throughout the Americas and are transmitted to humans by Aedes spp., Psorophora spp., and Culex spp. mosquitoes (2, 3).
Serologic and molecular evidence points to widespread VEEV complex infections in tropical Central and South America, indicating potential commonality yet significant underdiagnosis (2). At least 14 different viral subtypes within the VEEV complex have been identified to date (2), some associated with large equine and human outbreaks (VEEV subtypes IAB and IC) (1, 2). While most infections in humans are asymptomatic or subclinical, patients may develop acute febrile illness with headache, myalgias, arthralgias, nausea, and vomiting (4, 5). Cases can progress to encephalitis and result in long-term neurological effects (5, 6).
MADV, once considered a variant of EEEV, is an emerging virus that was first associated with large outbreaks in 2010 in the Darien province of Panama (5), where VEEV subtype ID has also been detected (7). MADV was primarily linked to equine disease, with a few human cases in Trinidad and Tobago and Brazil before the Panama outbreak (8, 9). This contrasts with North American EEEV, associated with severe and fatal human cases (3). MADV detection methods are limited, and its prevalence outside Darien province is not well understood (10). MADV’s geographic expansion to Northeast Brazil and Haiti highlights its potential for new areas (11, 12).
Accurate detection of VEEV complex, MADV, and EEEV during the acute phase is hindered by non-specific clinical signs and limited diagnostic tools. Antigen-based methods are unavailable, and serology requires paired samples to confirm diagnosis (1, 5). Current molecular tests lack optimal performance characteristics necessary for routine testing (13 – 19), and assay design is challenged by VEEV complex genetic variability (2). VEEV complex and MADV are often misdiagnosed as dengue virus due to similar symptoms during the acute phase (2). Common molecular tests involve pan-alphavirus primers amplifying a 400–500 nucleotide genome region, followed by sequencing or nested PCR for identification (5, 13, 14, 16, 18 – 20). These methods are labor-intensive and prone to contamination. Pan-alphavirus primers and conventional reverse transcription polymerase chain reaction (RT-PCR) chemistry may be less sensitive than real-time RT-PCR (rRT-PCR), with few reported rRT-PCR methods differentiating the VEEV complex and MADV (21).
The study aimed to design rRT-PCRs for the VEEV complex and MADV, with a secondary goal of developing a duplex MADV/EEEV rRT-PCR. These assays were evaluated using clinical samples from a Panama alphavirus outbreak and disease surveillance. Additionally, viral species, subtype, and genotype characterization were done using metagenomic sequencing on rRT-PCR-positive samples from humans and mosquitoes collected during the 2015 and 2022 outbreaks in Panama.
MATERIALS AND METHODS
VEEV complex, EEEV and MADV rRT-PCR design
Distinct alignments were established for the VEEV complex, EEEV, and MADV using comprehensive genome sequences from the NCBI GenBank (22) and aligned with MegAlign software (DNASTAR, Madison, WI, USA). The VEEV complex alignment encompassed complete genomes from Cabassou, Everglades, Mosso das Pedras, Mucambo, Pixuna, Rio Negro, Tonate, and VEEV subtypes (IAB, IC, ID, and IE). This compilation occurred in 2016 (n = 121 sequences), with a similar one for MADV in 2019 (n = 32). Employing Primer3 software (primer3.ut.ee), primers and probes were designed to contain ≤1 degenerate base and to align ≥95% with available sequences for each virus (Table 1). In silico validation details can be found in supplemental material.
TABLE 1.
Primers and probes in the VEEV and MADV/EEEV rRT-PCRs a
| Name | Sequence b | Concentration (nM) c | Location (5′−3′) d | Sequences fully matching e |
|---|---|---|---|---|
| VEEV | ||||
| VEEV forward 1 | GAAAGTTCACGTTGAYATCGAGGA | 200 | 44–67 | 156/159 (98) |
| VEEV forward 2 | GAAGGTTCACGTTGAYATCGAGGA | 200 | ||
| VEEV reverse 1 | GCTCTGGCRTTAGCATGGTC | 200 | 144–163 | 159/159 (100) |
| VEEV reverse 2 | GCTCTAGCRTTAGCATGGTC | 200 | ||
| VEEV probe | 5′-FAM-TTGAGGTAGAAGCHAAGCAGGTC-BHQ-1–3′ | 400 | 112–134 | 158/159 (99) |
| MADV/EEEV | ||||
| ME forward | GAGATAGAAGCMACGCAGGTC | 400 | 121–141; 99–119 | 31/32 (97); 1/449 (100) |
| ME reverse | TGYTTGGAATGCGTGTGC | 400 | 255–272; 233–250 | 32/32 (100); 9/449 (98) |
| MADV probe | 5′-FAM-CATCGAAAGCGAAGTGGACC-BHQ-1–3′ | 200 | 195–214 | 31/32 (97) |
| EEEV probe | 5′-CFO560-TGAGGGAGAAGTGGAYACAGACC-BHQ-1–3′ | 400 | 176–198 | 6/449 (99) |
BHQ, black hole quencher; CFO560, CAL Fluor Orange 560; and FAM, Fluorescein.
Probe sequences listed 5’′-fluorophore-sequence-quencher-3’′.
Concentration in the final reaction mixture.
Location in the following complete genome sequences: VEEV strain VEEV/Homo sapiens/GTM/69Z1/1969/IAB (Aaccession number KC344505.2); MADV strain Homo sapiens/Haiti-1901/2016 (MH359233.1); EEEV strain EEEV/Culiseta melanura/USA/SL13-0764-C/2013 (Aaccession number KX029319.1).
Displayed as number of complete genome sequences without a mismatch in the primer/probe sequence over all complete genome sequences aligned (%). Genomes downloaded on 22 Sept. 2021. Data shown for the combination of forward and reverse VEEV primers.
rRT-PCR assay performance and optimization
Primer and probe sets were evaluated in singleplex reactions containing 200 nM of each oligonucleotide and genomic RNA or quantified ssDNA containing the target region. Primer/probe sets were selected to generate the most sensitive detection based on cycle threshold (Ct) values, with preserved specificity. Primer and probe concentrations in the final reaction were then adjusted between 100 and 400 nM to optimize assay sensitivity. For VEEV, a total of four primers are mixed in a single reaction (Table 1). Additional validation, conditions, and lower limit of detection (LLOD) are given in the supplemental material (Fig. 1).
Fig 1.
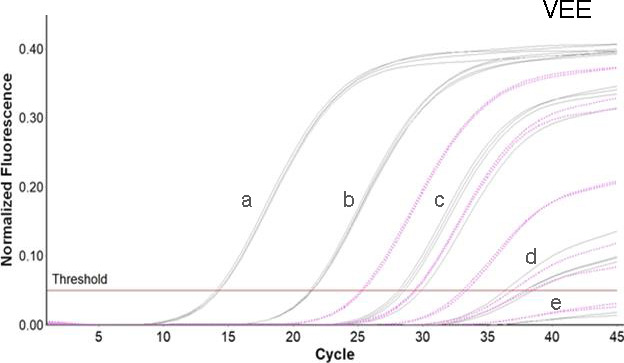
VEE amplification curves across a range of concentrations. Amplification curves are shown across a range of concentrations for the VEE complex rRT-PCR with ssDNA (gray curves, subtype IAB) and RNA (pink dotted curves, subtype IC). ssDNA was tested in quadruplicate at 8.0, 6.0, 4.0, 2.0, and 1.0 log10 copies/µL (labeled a–e, respectively). 10-fold dilutions of VEEV subtype IC RNA were tested in duplicate starting at the highest concentration available (5.0 log10 copies/µL).
Protocol validation with acute human samples
Acute human samples used in the protocol validation were collected in communities of Darien, the easternmost province in Panama, during three alphavirus outbreaks in 2015 and 2017. Cases identified in 2015 and 2017 were detected in the communities of Metetí, Cemaco, Tucutí, Yaviza, Nicanor, La Palma, and El Real de Santa María (Fig. 2A). The Darien province borders Colombia and encompasses the Darien Gap, and the Darien National Park, a UNESCO-designated World Heritage Site (23).
Fig 2.

Map with the distribution of VEEV human cases in Darien province in 2015 and 2017, and health centers in Panama and Darien provinces. (A) Distribution of VEEV cases used for protocol validation. Red dots represent the number of cases reported by locality. (B) Distribution of health centers used for prospective febrile surveillance in Panama and Darien provinces. The map was created with ArcGIS Desktop 10.6 using shapefiles from Esri [World Countries Generalized (2021); https://www.arcgis.com/home/item.html?id=2b93b06dc0dc4e809d3c8db5cb96ba69]. Data sources for the shapefiles include Esri, Garmin International Inc., U.S. Central Intelligence Agency, and National Geographic Society (24).
Patient recruitment in 2015 and 2017
Febrile patients were identified during an enhanced surveillance program by our outbreak response team using house-by-house visits during the 2015 and 2017 outbreaks. Blood samples were drawn from patients who met the case definition during the outbreak investigation.
Prospective acute disease surveillance in 2021 and 2022
In 2021, surveillance for emerging pathogens was established in Panama as part of the USA-National Institute of Allergy and Infectious Diseases, Centers for Research in Emerging Infectious Diseases Network initiative. The Coordinating Research on Emerging Arboviral Threats Encompassing the Neotropics (CREATE-NEO) in Panama undertakes acute febrile surveillance across 10 health centers in Panama and Darien Provinces (Fig. 2B) (https://www.utmb.edu/createneo/home/create-neo-home). Additional information of inclusion criteria is provided in the supplemental material.
Laboratory testing for acute disease surveillance
Acute samples (0–5 days) were first screened against DENV, CHIKV, and ZIKV virus using rRT-PCR as described previously (25), followed by testing with the newly designed MADV/VEEV rRT-PCR.
Mosquito collection
Mosquitoes were collected in a forested area (100 × 100 meters) in El Real de Santa María during the 2015 outbreak response. CDC light traps were employed over a 12-hour period (6:00 p.m. to 6:00 a.m.), positioned 1.5 meters above the ground level. These traps, equipped with octanol and CO2 as bait, were utilized for the encephalitis vector survey. Captured mosquitoes were anesthetized, identified to species using taxonomic keys (26), and preserved in liquid nitrogen. Specimens were grouped by species, with a maximum of 20 individuals per pool for subsequent analyses.
Viral isolation from mosquito pools
Mosquito pool homogenates were prepared with 20–50 mosquitoes in 2 mL of minimum essential medium supplemented with penicillin and streptomycin, and 20% fetal bovine serum (FBS), homogenized using a Tissue Lyser (Qiagen, Hidden, Germany) and centrifuged at 12,000 rpm for 10 min. A total of 200 µL of serum or mosquito homogenate was inoculated in each of two 12.5-cm2 flasks of Vero cells (Cercopithecus aethiops kidney normal cells, ATCC CCL-81). Vero cells were supplemented with 10% FBS for growth and maintained with 2% FBS and 1% penicillin/streptomycin at 37°C. Samples were passed twice and monitored for cytopathic effect. All viral isolations were undertaken in the biosafety level-3 containment laboratory at the Gorgas Memorial Institute in Panama City.
Generic alphavirus RT-PCR for human and mosquito samples
Viral RNA was extracted from human serum and mosquito pool homogenates using QIAamp RNA viral extraction kit (Qiagen, Valencia, CA, USA). Viral RNA from mosquitoes was also extracted using the Macherey-Nagel extraction kit (Düren, Germany). Volume for extraction was 160 and 200 µL for human serum and mosquito pool homogenates, whereas elution volume was 60 and 50 µL, respectively. Serum and mosquito homogenates were tested in 25 µL reactions for alphaviruses using a universal alphavirus RT-PCR, as previously described (19). Antibody response was assessed in all human serum samples from 2015 as described previously (27), further details are provided in the supplemental material.
Viral metagenomic sequencing
To confirm virus species, subtype, and genotype, we sequenced seven selected VEEV complex rRT-PCR positive mosquito and human samples from 2015 and 2022 using SMART-9N metagenomic sequencing as previously described (28). Additional information is provided in the supplemental material.
VEEV phylogenetic analysis
All available VEEV genome sequences, in GenBank, representing all antigenic complexes were selected to construct the alignment; duplicated sequences, partial sequences, and overlapping sequences were removed. Finally, the novel complete or near complete VEEV genome sequences (n = 7) were aligned with 132 representative VEEV genomes retrieved from NCBI GenBank using MAFFT version 7 (29). Selection of the best-fitting nucleotide substitution model and maximum likelihood phylogenetic reconstruction were performed with IQ-Tree v2.2.0.3 (30). Statistical robustness of the tree topology was assessed with 1,000 ultrafast bootstrap replicates.
RESULTS
rRT-PCR analytical evaluation
Primers and probes for the VEE complex singleplex and MADV/EEEV duplex rRT-PCRs are shown in Table 1 along with the optimized final reaction concentrations. The dynamic range for each assay extended from 2.0 to 8.0 log10 copies/µL (Fig. 1; Fig. S1). For the VEEV complex assay, the linear range was evaluated with ssDNA for subtypes IAB and IV and RNA from subtype IC (2.0–5.0 log10 copies/µL; Figure 2). The 95% LLODs, expressed in copies/µL, were: VEEV subtype IAB, 120; VEE subtype IV, 110; MADV, 19; and EEEV, 19. Assay exclusivity was evaluated by testing genomic RNA from VEEV subtype IC, EEEV, and a set of arboviruses, including flavi-, bunya-, and alphaviruses on a single run of the VEEV complex and MADV/EEEV rRT-PCRs. VEEV complex and EEEV only yielded signals in the respective assays for these viruses. None of the other tested viruses generated a signal in either assay. In addition, none of the 56 serum samples from Georgia, USA, or Asunción, Paraguay, tested positive in either assay.
Validation with clinical samples
A total of 15 febrile patients from the 2015 and 2017 alphavirus outbreaks who met the suspected or probable case definition were used to validate the new molecular assays. Previously, a total of eleven (11/15) acute serum samples collected during the 2015 and 2017 alphavirus outbreaks had tested positive using a generic alphavirus RT-PCR and were confirmed later by sequencing as VEEV-ID infections (17). In 2021, a second round of generic alphavirus RT-PCR using the same set of primers was run on these 15 stored samples, and all of them tested negative. Notably, using the newly designed rRT-PCR, we were able to detect 10 VEEV complex RNA positive samples (Ct range: 27–38), including two samples that had tested negative at the initial screening in 2017 (Table 2). Three of the VEEV complex rRT-PCR-positive samples were also anti-VEEV IgG and IgM positive, with only 0, 2, and 3 days since the onset of symptoms, respectively (Table 2). One sample was rRT-PCR positive for both VEEV and MADV viruses.
TABLE 2.
Characteristics and laboratory results of the samples used for protocol validation patients and clinical samples and laboratory results a
| Code | Township | Age b | Sex | Symptoms onset | Days of symptoms | RT-PCR-alpha (2015) | RT-PCR-alpha (2021) | rRT-PCR-VEE | Ct values | IgM-VEEV | IgM-MADV | IgG-VEEV | IgG-MADV | PRNT-veev c | PRNT-MADV c |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 258384 | El Real | 0–9 | M | Aug. 2015 | 0 | pos | neg | pos | 29.3 | neg | neg | neg | neg | <1:20 | <1:20 |
| 267738 | Cemaco | 0–9 | M | July 2017 | 3 | neg | neg | pos | 37.8 | pos | neg | neg | neg | <1:20 | <1:20 |
| 267411 | Tucuti | 0–9 | F | July 2017 | 5 | neg | neg | neg | – d | pos | neg | pos | pos | 1:40 | 1:40 |
| 258380 | El Real | 0–9 | F | Aug. 2015 | 1 | pos | neg | neg | – | neg | neg | neg | neg | <1:20 | <1:20 |
| 267410 | Yaviza | 0–9 | F | July 2017 | 2 | neg | neg | neg | – | pos | neg | pos | neg | <1:20 | <1:20 |
| 258657 | Yaviza | 10–19 | M | Sept. 2015 | 0 | pos | neg | pos | 28 | neg | neg | neg | neg | <1:20 | <1:20 |
| 258535 | Nicanor | 20–29 | F | Sept. 2015 | 2 | pos | neg | neg | – | neg | neg | neg | neg | <1:20 | <1:20 |
| 258401 | La Palma | 20–29 | M | Aug. 2015 | 2 | pos | neg | pos | 29 | neg | neg | neg | neg | <1:20 | <1:20 |
| 258395 | Metetí | 30–39 | M | Aug. 2015 | 2 | neg | neg | pos | 37 | pos | neg | neg | neg | <1:20 | <1:20 |
| 258399 | El Real | 30–39 | M | Aug. 2015 | 1 | pos | neg | pos | 26 | neg | neg | neg | neg | <1:20 | <1:20 |
| 258385 | El Real | 30–39 | M | Aug. 2015 | 2 | pos | neg | pos | 37 | neg | neg | neg | neg | <1:20 | <1:20 |
| 258398 | El Real | 30–39 | M | Aug. 2015 | 0 | pos | neg | pos | 27 | neg | neg | pos | neg | <1:20 | <1:20 |
| 258536 | Metetí | 30–39 | F | Sept. 2015 | 2 | pos | neg | neg | – | neg | neg | neg | neg | <1:20 | <1:20 |
| 258386 | El Real | 30–39 | M | Aug. 2015 | 5 | pos | neg | pos | 34 | neg | neg | neg | neg | ND | ND |
| 258379 | El Real | ≥40 | F | Aug. 2015 | 2 | pos | neg | pos | 31 | neg | neg | neg | neg | <1:20 | <1:20 |
Acute samples selected from the 2015 and 2017 alphavirus outbreaks in Darien Province. Ct, cycle threshold; neg, negative; pos, positive; and ND, not done.
Age categories in years.
Base on PRNT-80.
-, not done and postive results and higligthed in bold.
Prospective disease surveillance
A total of 118 febrile patients were recruited from 16 November 2021 to 1 December 2022. Of these, 84 (71.2%) were acute patients with the onset of symptoms ranging from 0 to 5 days. A total of 42 patients (50.0%) were DENV1 positive. We detected VEEV RNA (Ct range: 15–20) in five patients (11.9%; 95% CI: 4.0–25.6) with suspected dengue infection, one of which was from a fatal case in 2022. Details and results of disease surveillance are presented in Fig. 3.
Fig 3.
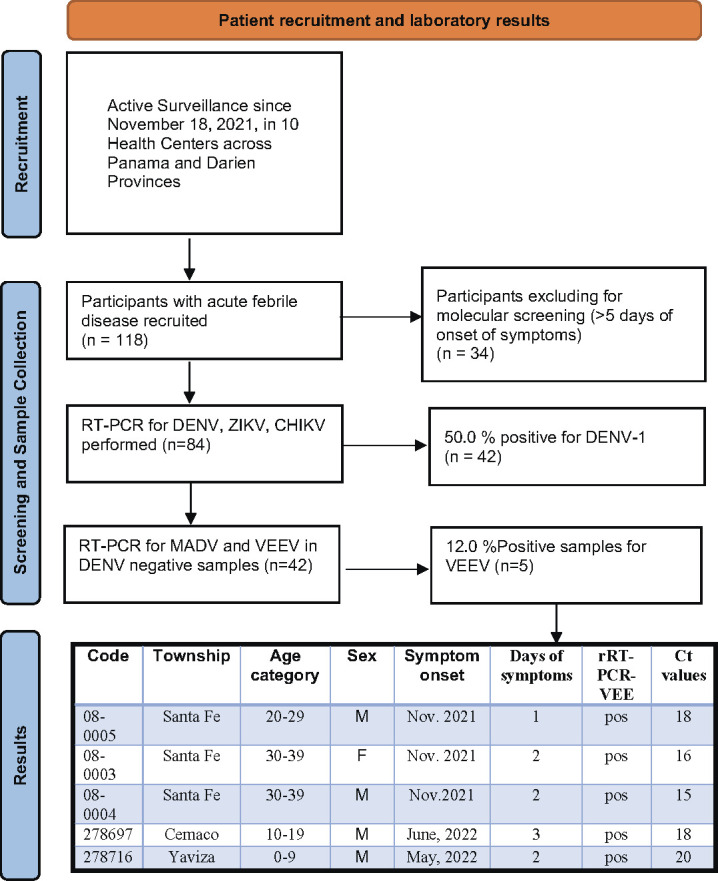
Flowchart of patient recruitment, characteristics, and RT-PCR results of febrile patients detected throughout disease surveillance. Febrile patients were recruited from 16 November 2021 to 1 December 2022, in 10 health care centers in Panama and Darien provinces.
Viral detection in mosquito pools
A total of 1,307 mosquitoes belonging to 35 species and 12 genera were collected in the community of El Real de Santa Maria, Panama, during a period of 5 days in 2015 (Table 3). The most abundant mosquito species was Coquilletidia venezualensis (37.5%, n = 490 of 1,307) and Culex Melanoconion vomerifer (34.4%, n = 450 of 1,307). Mosquito species, number of individuals, and pools are shown in Table 3. Of 150 mosquito pools, 3 Cx. (Mel.) vomerifer mosquito pools tested positive for VEEV by rRT-PCR (Ct range: 26–30). Two of these rRT-PCR-positive pools also yielded viral isolates.
TABLE 3.
Mosquito species collected during the 2015 outbreak in El Real de Santa Maria, Panama
| Mosquito species | N | (%) | Pools a | VEE-rRT-PCR positive |
MADV-rRT-PCR positive |
Viral isolates |
|---|---|---|---|---|---|---|
| Coquillettidia venezuelensis | 490 | 37.5 | 29 | 0 | 0 | 0 |
| Culex (Melanoconion) vomerifer | 450 | 34.4 | 27 | 3 | 0 | 2 |
| Culex (Melanoconion) pedroi | 32 | 2.4 | 4 | 0 | 0 | 0 |
| Aedes serratus | 31 | 2.4 | 7 | 0 | 0 | 0 |
| Aedes sp. | 30 | 2.3 | 5 | 0 | 0 | 0 |
| Culex (Melanoconion) sp. | 30 | 2.3 | 6 | 0 | 0 | 0 |
| Culex (Culex) interrogator | 27 | 2.1 | 5 | 0 | 0 | 0 |
| Anopheles trianulatus | 23 | 1.8 | 2 | 0 | 0 | 0 |
| Aedes eupoclamus | 14 | 1.1 | 4 | 0 | 0 | 0 |
| Culex (Culex) nigripalpus | 14 | 1.1 | 3 | 0 | 0 | 0 |
| Culex (Culex) sp. | 14 | 1.0 | 4 | 0 | 0 | 0 |
| Culex (Melanoconion) atratus | 14 | 1.0 | 1 | 0 | 0 | 0 |
| Culex (Melanoconion) adamesi | 13 | 1.0 | 3 | 0 | 0 | 0 |
| Others b | 125 | 9.6 | 50 | 0 | 0 | 0 |
| Total | 1307 | 100 | 150 | 3 | 0 | 2 |
Numbers of mosquito pools.
Species <1% abundance are listed as others.
VEEV subtype identification
Three mosquito pools and four human samples (including one from a fatal case in 2022), which tested positive for the new VEEV complex rRT-PCR, were sequenced using a virus-untargeted approach (28). The 20-fold genome coverage per base pair ranged from 45% to 100% (Table 4). The percentage of genome identity with the VEEV reference strain ranged from 87.7% to 90.0% (Table 4), while identity with the Panamanian VEEV ID subtype prototype strain 3880 ranged from 96% to 97% (Table 4). Maximum likelihood phylogenetic analysis indicated that the new viral genomes cluster together with historical Panamanian VEEV ID subtype strains within the Panama/Peru genotype (bootstrap statistical support = 100; Fig. 4).
TABLE 4.
Metadata and sequencing statistics for selected VEEV complex RNA positive samples c
| ID | Collection year | Location | Host species | Percent genome coverage 20× | Percent Nt identity with genome reference a , b | Percent identity with strain 3880 c | C values |
|---|---|---|---|---|---|---|---|
| 700677 | 2015 | Darien | Culex (Mel.) vomerifer | 100 | 89.8 | 92.1 | 27 |
| 700680 | 2015 | Darien | Culex (Mel.) vomerifer | 100 | 89.8 | 92.2 | 31 |
| 700732 | 2015 | Darien | Culex (Mel.) vomerifer | 100 | 90 | 92.3 | 26 |
| 258379 | 2015 | Darien | Human | 99.9 | 89.6 | 92 | 31 |
| 258398 | 2015 | Darien | Human | 70 | 88.7 | 90.7 | 27 |
| 258401 | 2015 | Darien | Human | 90.6 | 87.7 | 90 | 29 |
| 278716 | 2022 | Darien | Human | 45.98 | 88.1 | 90 | 20 |
Genbank accession no. NC_001449.1.
Nt = Nucleotide.
Panamanian VEEV ID subtype prototype strain 3880, GenBank accession no. L00930.1.
Fig 4.

VEEV complex maximum likelihood phylogenetic tree. Maximum likelihood phylogeny was estimated using 139 complete or near complete VEEV genomes. Publicly available Panamanian VEEV ID subtype strains are highlighted in gray (n = 15), and genomes generated in this study (n = 7) are highlighted in red. Bootstrap statistical support is shown for selected nodes. NCBI GenBank accession numbers for the new VEEV genomes are: OR644785, OR644786, OR644788, OR644801, OR644802, OR644803.
DISCUSSION
Encephalitic alphaviruses have been detected throughout the Americas and may account for a significant proportion of non-dengue acute febrile illness (1, 2, 5, 9). Assays for their molecular detection, although existing (14 – 20), are often time-consuming, involving multiple PCR rounds or subsequent genome sequencing limited to well-equipped facilities (14 – 20). Co-circulation and the potential for co-infection with these viruses further complicate their identification, especially when clinical presentations are similar, and convenient methods for detecting VEEV complex and MADV are lacking (19). In Panama, for instance, both VEEV subtype ID and MADV have been identified, with co-circulation detected along the Colombian border (5 – 7). Typically, cases are identified during the neurological phase of the disease (5, 31), where the virus is cleared from serum, necessitating reliance on serological testing. Given that alphaviruses can induce IgM responses lasting 2–3 months, anti-VEEV or anti-MADV IgM detection alone could lead to misdiagnosis without seroconversion (5, 31).
We have developed singleplex and duplex rRT-PCRs for detecting VEEV complex, MADV, and EEEV viral RNA in clinical and mosquito samples. These assays identified VEEV ID subtype and MADV in samples previously negative using a reference RT-PCR (19). We also identified a VEEV ID subtype—MADV co-infection, highlighting an advantage of our VEEV complex and MADV/EEEV rRT-PCRs over prior methods. Co-infection cases are epidemiologically significant and may have clinical relevance if associated with more severe disease (5). Our rRT-PCR assays can be rapidly integrated into testing algorithms in endemic regions. The current rRT-PCR detects VEEV ID subtype RNA within the initial 5 days of symptoms, preceding IgM and IgG antibody responses which usually manifest after 5–7 days following symptom onset (32). Intriguingly, three patients with detectable VEEV complex RNA were also VEEV IgM and IgG-reactive, suggesting possible VEEV re-infections with potential implications for vaccine development. However, early IgM responses cannot be ruled out, necessitating further research on alphavirus humoral immunity.
Through our prospective disease surveillance in Panama, we have demonstrated a notable prevalence of alphavirus detection. About 11.9% of individuals exhibiting symptoms similar to dengue have been found to have VEEV infections. These findings align with earlier assessments indicating that roughly 10% of clinical dengue cases in endemic countries can be attributed to VEEV infection (2). Moreover, this suggests a co-circulation of alphaviruses alongside other endemic arboviral infections, including dengue. Given the clinical similarities between VEEV complex infections and dengue, there exists the potential for underestimating the true burden of VEEV-related disease (2).
VEEV ID subtype RNA was found in Cx. (Mel.) vomerifer mosquito pools trapped during the 2015 outbreak in El Real de Santamaria. These mosquitoes were previously implicated as VEEV ID subtype vectors (2). Two pools yielded viral isolates. Notably, pan-alphavirus conventional RT-PCRs failed to detect viral RNA in these pools, suggesting the new rRT-PCR’s heightened sensitivity for VEEV complex RNA detection in mosquitoes. Neither MADV nor EEEV infections were detected in mosquitoes using various methods. A similar pattern emerged from past outbreak investigations by our group in Panama (27, 33). Interestingly, MADV detection frequency in Culex (Mel.) spp. mosquitos is low in Panama (34, 35), unlike the endemic region of Iquitos, Peru, where MADV in the enzootic vector Culex (Mel.) pedroi is frequent (9, 36). Reasons for this variation in MADV and VEEV ID subtype frequency in Panama and MADV and VEEV in Panama vs Iquitos remain uncertain, possibly involving vector competence or viral competition, even enhanced VEEV ID subtype transmission via insect-specific viruses (37).
While our assays were validated with a limited number of human and mosquito samples, prospective surveillance allowed further validation. Unlike previous methods relying on plasmids, viral isolates, or a few human serum samples (13 – 21), we validated with human serum, mosquitoes, and post-mortem tissue samples. Our approach failed to detect two samples previously positive using standard alphavirus generic primers (19). Interestingly, a subsequent generic alphavirus RT-PCR in 2017 also failed to reamplify the former positives, possibly due to viral RNA degradation over time (38)
An rRT-PCR based on 33 VEEV sequences was reported by Vina-Rodriguez et al. but excluded other VEEV complex species and lacked clinical evaluation (21). Our assays used more complete genome sequences, with in silico primer and probe alignment to contemporary sequences. Untargeted metagenomic sequencing confirmed VEEV ID subtype detection using VEEV complex primers; this subtype has been detected in central and eastern Panama regions (7). These findings highlight molecular and genomic approaches’ potential to enhance the detection of acute encephalitis alphavirus infections, even in archived samples.
Further prospective testing is necessary for comprehensive clinical performance characterization, including quantitative diagnostics and challenging assays with interfering substances. Limitations include the design requiring two separate assays for three viruses due to overlapping optimal design targets. However, the two rRT-PCRs can be executed simultaneously, improving lab workflow. The VEEV complex assay can also be multiplexed with rRT-PCRs for other neurotropic arboviruses without performance loss (Jesse J. Waggone, unpublished data).
We developed sensitive and specific VEEV complex, MADV, and EEEV rRT-PCRs, surpassing available molecular methods. These assays detect VEEV-MADV co-infections, VEEV human infections, potential VEEV reinfections, and active VEEV viral circulation in mosquitoes during alphavirus outbreaks. Implementing these assays in endemic regions may enhance neurotropic alphavirus identification and characterization.
ACKNOWLEDGMENTS
We thank Xacdiel Rodriguez, Yelissa Rios, Yaneth Pittí, Oriel Lezcano, Eddier Rivera, and Mileika Santos for technical support with sample processing and mosquito classification and Alberto Cumbrera for the map construction. We also thank Leyda Abrego for providing reagents for the rRT-PCR, and Milena Gomez, Thais M. Coletti, Esmenia Rocha, Geovana Maria Pererira, and Erika R. Manuli for technical support with metagenomic sequencing.
J.-P.C. was funded by the Clarendon Scholarship from the University of Oxford and the Lincoln-Kingsgate Scholarship from Lincoln College, University of Oxford (grant number SFF1920_CB2_MPLS_1293647).
This work was supported by SENACYT, through the grants numbers FID-16-201 and FID-2021-96 grant to J-.P.C.; the National Institute of Allergy and Infectious Diseases, National Institutes of Health (grant K08AI110528 to J.J.W.), and the Centers for Research in Emerging Infectious Diseases (CREID) Coordinating Research on Emerging Arboviral Threats Encompassing the Neotropics (CREATE-NEO) 1U01AI151807 grant awarded to N.V./K.A.H. by the National Institutes of Health (NIH); and by the Medical Research Council‐São Paulo Research Foundation CADDE partnership award (MR/S0195/1 and FAPESP18/14389‐0 to N.R.F.) (https://caddecentre.org). C.A.D. was supported by the NIHR HPRU in Emerging and Zoonotic Infections, a partnership between PHE, the University of Oxford, the University of Liverpool, and the Liverpool School of Tropical Medicine (grant no. NIHR200907).
Contributor Information
Jean-Paul Carrera, Email: jpcarrera@gorgas.gob.pa.
Nuno R. Faria, Email: nfaria@ic.ac.uk.
Jesse J. Waggoner, Email: jjwaggo@emory.edu.
Elitza S. Theel, Mayo Clinic, Rochester, Minnesota, USA
ETHICS STATEMENT
The use of human samples for protocol validation was approved by the Panamanian Ministry of Health (protocol number 2077), Gorgas’s Institutional Review Board (IRB) (protocol: 335/CBI/ICGES/21), Emory University IRB (IRB00097089), and the Ethics Committee of the Instituto de Investigaciones en Ciencias de la Salud, Universidad Nacional de Asunción (P06/2017). Prospective disease surveillance was approved by Gorgas’s IRB (protocol: 073/CBI/ICGES/21).
DATA AVAILABILITY
All the data used for human and mosquito validation are contained within the manuscript. Accession numbers for the newly generated genomes are OR644785, OR644786, OR644788, OR644801, OR644802, OR644803. Accession numbers and strain information of sequences used for primer design are shown in the supplemental material.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jcm.00152-23.
Addicional information of recruitment and methods used in the study.
Genome accession numbers used in primer design.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Navarro JC, Carrera JP, Liria J, Auguste AJ, Weaver SC. 2017. Alphaviruses in Latin America and the introduction of chikungunya virus, p 169–192. In Human virology in Latin America: from biology to control. Springer International, Publishing, Cham, Switzerland. doi: 10.1007/978-3-319-54567-7 [DOI] [Google Scholar]
- 2. Aguilar PV, Estrada-Franco JG, Navarro-Lopez R, Ferro C, Haddow AD, Weaver SC. 2011. Endemic venezuelan equine encephalitis in the Americas: hidden under the dengue umbrella. Future Virol 6:721–740. doi: 10.2217/FVL.11.5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arrigo NC, Adams AP, Weaver SC. 2010. Evolutionary patterns of Eastern equine encephalitis virus in North versus South America suggest ecological differences and taxonomic revision. J Virol 84:1014–1025. doi: 10.1128/JVI.01586-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Forshey BM, Guevara C, Laguna-Torres VA, Cespedes M, Vargas J, Gianella A, Vallejo E, Madrid C, Aguayo N, Gotuzzo E, Suarez V, Morales AM, Beingolea L, Reyes N, Perez J, Negrete M, Rocha C, Morrison AC, Russell KL, Blair PJ, Olson JG, Kochel TJ, NMRCD Febrile Surveillance Working Group . 2010. Arboviral etiologies of acute febrile illnesses in Western South America, 2000-2007. PLoS Negl Trop Dis 4:e787. doi: 10.1371/journal.pntd.0000787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Carrera J-P, Forrester N, Wang E, Vittor AY, Haddow AD, López-Vergès S, Abadía I, Castaño E, Sosa N, Báez C, Estripeaut D, Díaz Y, Beltrán D, Cisneros J, Cedeño HG, Travassos da Rosa AP, Hernandez H, Martínez-Torres AO, Tesh RB, Weaver SC. 2013. Eastern equine encephalitis in Latin America. N Engl J Med 369:732–744. doi: 10.1056/NEJMoa1212628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carrera JP, Pittí Y, Molares-Martínez JC, Casal E, Pereyra-Elias R, Saenz L, Guerrero I, Galué J, Rodriguez-Alvarez F, Jackman C, Pascale JM, Armien B, Weaver SC, Donnelly CA, Vittor AY. 2020. Clinical and serological findings of madariaga and venezuelan equine encephalitis viral infections: a follow-up study 5 years after an outbreak in Panama. Open Forum Infect Dis 7:faa359. doi: 10.1093/ofid/ofaa359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Quiroz E, Aguilar PV, Cisneros J, Tesh RB, Weaver SC, Halstead SB. 2009. Venezuelan equine encephalitis in Panama: fatal endemic disease and genetic diversity of etiologic viral strains. PLoS Negl Trop Dis 3:e472. doi: 10.1371/journal.pntd.0000472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Corniou B, Ardoin P, Bartholomew C, Ince W, Massiah V. 1972. First isolation of a South American strain of Eastern equine virus from a case of encephalitis in Trinidad. Trop Geogr Med 24:162–167. [PubMed] [Google Scholar]
- 9. Aguilar PV, Robich RM, Turell MJ, O’Guinn ML, Klein TA, Huaman A, Guevara C, Rios Z, Tesh RB, Watts DM, Olson J, Weaver SC. 2007. Endemic Eastern equine encephalitis in the Amazon region of Peru. Am J Trop Med Hyg 76:293–298. doi: 10.4269/ajtmh.2007.76.293 [DOI] [PubMed] [Google Scholar]
- 10. Vittor AY, Armien B, Gonzalez P, Carrera J-P, Dominguez C, Valderrama A, Glass GE, Beltran D, Cisneros J, Wang E, Castillo A, Moreno B, Weaver SC. 2016. Epidemiology of emergent madariaga encephalitis in a region with endemic venezuelan equine encephalitis: initial host studies and human cross-sectional study in Darien, Panama. PLoS Negl Trop Dis 10:e0004554. doi: 10.1371/journal.pntd.0004554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gil L, Magalhaes T, Santos B, Oliveira LV, Oliveira-Filho EF, Cunha JLR, Fraiha ALS, Rocha BMM, Longo BC, Ecco R, Faria GC, Furtini R, Drumond SRM, Maranhão RPA, Lobato ZIP, Guedes M, Teixeira RBC, Costa EA. 2021. Active circulation of madariaga virus, a member of the Eastern equine encephalitis virus complex, in northeast Brazil. Pathogens 10:983. doi: 10.3390/pathogens10080983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lednicky JA, White SK, Mavian CN, El Badry MA, Telisma T, Salemi M, OKech BA, Beau De Rochars VM, Morris JG. 2019. Emergence of madariaga virus as a cause of acute febrile illness in children, Haiti, 2015-2016. PLoS Negl Trop Dis 13:e0006972. doi: 10.1371/journal.pntd.0006972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pfeffer M, Proebster B, Kinney RM, Kaaden OR. 1997. Genus-specific detection of alphaviruses by a semi-nested reverse transcription-polymerase chain reaction. Am J Trop Med Hyg 57:709–718. doi: 10.4269/ajtmh.1997.57.709 [DOI] [PubMed] [Google Scholar]
- 14. Brightwell G, Brown JM, Coates DM. 1998. Genetic targets for the detection and identification of Venezuelan equine encephalitis viruses. Arch Virol 143:731–742. doi: 10.1007/s007050050326 [DOI] [PubMed] [Google Scholar]
- 15. Romeiro MF, de Souza WM, Tolardo AL, Vieira LC, Henriques DA, de Araujo J, Siqueira CEH, Colombo TE, Aquino VH, da Fonseca BAL, de Morais Bronzoni RV, Nogueira ML, Durigon EL, Figueiredo LTM. 2016. A real-time RT-PCR for rapid detection and quantification of mosquito-borne alphaviruses. Arch Virol 161:3171–3177. doi: 10.1007/s00705-016-3019-0 [DOI] [PubMed] [Google Scholar]
- 16. Linssen B, Kinney RM, Aguilar P, Russell KL, Watts DM, Kaaden OR, Pfeffer M. 2000. Development of reverse transcription-PCR assays specific for detection of equine encephalitis viruses. J Clin Microbiol 38:1527–1535. doi: 10.1128/JCM.38.4.1527-1535.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang E, Paessler S, Aguilar PV, Carrara A-S, Ni H, Greene IP, Weaver SC. 2006. Reverse transcription-PCR-enzyme-linked immunosorbent assay for rapid detection and differentiation of alphavirus infections. J Clin Microbiol 44:4000–4008. doi: 10.1128/JCM.00175-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pisano MB, Seco MPS, Ré VE, Farías AA, Contigiani MS, Tenorio A. 2012. Specific detection of all members of the Venezuelan equine encephalitis complex: development of a RT-nested PCR. J Virol Methods 186:203–206. doi: 10.1016/j.jviromet.2012.05.009 [DOI] [PubMed] [Google Scholar]
- 19. Sánchez-Seco MP, Rosario D, Quiroz E, Guzmán G, Tenorio A. 2001. A generic nested-RT-PCR followed by sequencing for detection and identification of members of the alphavirus genus. J Virol Methods 95:153–161. doi: 10.1016/s0166-0934(01)00306-8 [DOI] [PubMed] [Google Scholar]
- 20. Bronzoni RVM, Moreli ML, Cruz ACR, Figueiredo LTM. 2004. Multiplex nested PCR for Brazilian alphavirus diagnosis. Trans R Soc Trop Med Hyg 98:456–461. doi: 10.1016/j.trstmh.2003.09.002 [DOI] [PubMed] [Google Scholar]
- 21. Vina-Rodriguez A, Eiden M, Keller M, Hinrichs W, Groschup MH. 2016. A quantitative real-time RT-PCR assay for the detection of Venezuelan equine encephalitis virus utilizing a universal alphavirus control RNA. Biomed Res Int 2016:8543204. doi: 10.1155/2016/8543204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Ostell J, Pruitt KD, Sayers EW. 2018. GenBank. Nucleic Acids Res 46:D41–D47. doi: 10.1093/nar/gkx1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Darien National Park. 2023. Available from: https://whc.unesco.org/en/list/159
- 24. World Countries (Generalized): Esri . 2023. https://www.arcgis.com/home/item.html?id= 2b93b06dc0dc4e809d3c8db5cb96ba69.
- 25. Santiago GA, Vázquez J, Courtney S, Matías KY, Andersen LE, Colón C, Butler AE, Roulo R, Bowzard J, Villanueva JM, Muñoz-Jordan JL. 2018. Performance of the trioplex real-time RT-PCR assay for detection of Zika, dengue, and chikungunya viruses. Nat Commun 9:1391. doi: 10.1038/s41467-018-03772-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sallum MA, Forattini OP. 1996. Revision of the spissipes section of culex (melanoconion) (diptera:culicidae). J Am Mosq Control Assoc 12:517–600. [PubMed] [Google Scholar]
- 27. Carrera J-P, Cucunubá ZM, Neira K, Lambert B, Pittí Y, Liscano J, Garzón JL, Beltran D, Collado-Mariscal L, Saenz L, Sosa N, Rodriguez-Guzman LD, González P, Lescano AG, Pereyra-Elías R, Valderrama A, Weaver SC, Vittor AY, Armién B, Pascale J-M, Donnelly CA. 2020. Endemic and epidemic human alphavirus infections in Eastern Panama: an analysis of population-based cross-sectional surveys. Am J Trop Med Hyg 103:2429–2437. doi: 10.4269/ajtmh.20-0408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Claro IM, Ramundo MS, Coletti TM, da Silva CAM, Valenca IN, Candido DS, Sales FCS, Manuli ER, de Jesus JG, de Paula A, Felix AC, Andrade PDS, Pinho MC, Souza WM, Amorim MR, Proenca-Modena JL, Kallas EG, Levi JE, Faria NR, Sabino EC, Loman NJ, Quick J. 2021. Rapid viral metagenomics using SMART-9N amplification and nanopore sequencing. Wellcome Open Res 6:241. doi: 10.12688/wellcomeopenres.17170.2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, Lanfear R. 2020. Corrigendum to: IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol 37:2461. doi: 10.1093/molbev/msaa131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Luciani K, Abadía I, Martínez-Torres AO, Cisneros J, Guerra I, García M, Estripeaut D, Carrera J-P. 2015. Case report: madariaga virus infection associated with a case of acute disseminated encephalomyelitis. Am J Trop Med Hyg 92:1130–1132. doi: 10.4269/ajtmh.14-0845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Torres-Ruesta A, Chee R-L, Ng LFP. 2021. Insights into antibody-mediated alphavirus immunity and vaccine development landscape. Microorganisms 9:899. doi: 10.3390/microorganisms9050899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Torres R, Samudio R, Carrera J-P, Young J, Márquez R, Hurtado L, Weaver S, Chaves LF, Tesh R, Cáceres L, Coffey LL. 2017. Enzootic mosquito vector species at equine encephalitis transmission foci in the República de Panama. PLoS One 12:e0185491. doi: 10.1371/journal.pone.0185491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dietz WH, Galindo P, Johnson KM. 1980. Eastern equine encephalomyelitis in Panama: the epidemiology of the 1973 epizootic. Am J Trop Med Hyg 29:133–140. doi: 10.4269/ajtmh.1980.29.133 [DOI] [PubMed] [Google Scholar]
- 35. Srihongse S, Galindo P. 1967. The isolation of Eastern equine encephalitis virus from culex (melanoconion) taeniopus Dyar and Knab in Panama. Mosq News 27:74–76. [Google Scholar]
- 36. Turell MJ, O’guinn ML, Jones JW, Sardelis MR, Dohm DJ, Watts DM, Fernandez R, da Rosa AT, Guzman H, Tesh R, Rossi CA, Ludwig GV, Mangiafico JA, Kondig J, Wasieloski LP, Pecor J, Zyzak M, Schoeler G, Mores CN, Calampa C, Lee JS, Klein TA. 2005. Isolation of viruses from mosquitoes (Diptera: Culicidae) collected in the Amazon basin region of Peru. J Med Entomol 42:891–898. doi: 10.1093/jmedent/42.5.891 [DOI] [PubMed] [Google Scholar]
- 37. Olmo RP, Todjro YMH, Aguiar ERGR, de Almeida JPP, Ferreira FV, Armache JN, de Faria IJS, Ferreira AGA, Amadou SCG, Silva ATS, de Souza KPR, Vilela APP, Babarit A, Tan CH, Diallo M, Gaye A, Paupy C, Obame-Nkoghe J, Visser TM, Koenraadt CJM, Wongsokarijo MA, Cruz ALC, Prieto MT, Parra MCP, Nogueira ML, Avelino-Silva V, Mota RN, Borges MAZ, Drumond BP, Kroon EG, Recker M, Sedda L, Marois E, Imler J-L, Marques JT. 2023. Mosquito vector competence for dengue is modulated by insect-specific viruses. Nat Microbiol 8:135–149. doi: 10.1038/s41564-022-01289-4 [DOI] [PubMed] [Google Scholar]
- 38. Relova D, Rios L, Acevedo AM, Coronado L, Perera CL, Pérez LJ. 2018. Impact of RNA degradation on viral diagnosis: an understated but essential step for the successful establishment of a diagnosis network. Vet Sci 5:19. doi: 10.3390/vetsci5010019 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Addicional information of recruitment and methods used in the study.
Genome accession numbers used in primer design.
Data Availability Statement
All the data used for human and mosquito validation are contained within the manuscript. Accession numbers for the newly generated genomes are OR644785, OR644786, OR644788, OR644801, OR644802, OR644803. Accession numbers and strain information of sequences used for primer design are shown in the supplemental material.