

Abstract
Down syndrome (DS) is the most prevalent chromosomal disorder associated with a higher incidence of pulmonary arterial hypertension (PAH). The dysfunction of vascular endothelial cells (ECs) is known to cause pulmonary arterial remodeling in PAH, although the physiological characteristics of ECs harboring trisomy 21 (T21) are still unknown. In this study, we analyzed the human vascular ECs by utilizing the isogenic pairs of T21-induced pluripotent stem cells (iPSCs) and corrected disomy 21 (cDi21)-iPSCs. In T21-iPSC-derived ECs, apoptosis and mitochondrial reactive oxygen species (mROS) were significantly increased, and angiogenesis and oxygen consumption rate (OCR) were significantly impaired as compared with cDi21-iPSC-derived ECs. The RNA-sequencing identified that EGR1 on chromosome 5 was significantly upregulated in T21-ECs. Both EGR1 suppression by siRNA and pharmacological inhibitor could recover the apoptosis, mROS, angiogenesis, and OCR in T21-ECs. Alternately, the study also revealed that DYRK1A was responsible to increase EGR1 expression via PPARG suppression, and that chemical inhibition of DYRK1A could restore the apoptosis, mROS, angiogenesis, and OCR in T21-ECs. Finally, we demonstrated that EGR1 was significantly upregulated in the pulmonary arterial ECs from lung specimens of a patient with DS and PAH. In conclusion, DYRK1A/PPARG/EGR1 pathway could play a central role for the pulmonary EC functions and thus be associated with the pathogenesis of PAH in DS.
Keywords: induced pluripotent stem cell, Down syndrome, endothelial cell, EGR1, pulmonary hypertension
Graphical abstract
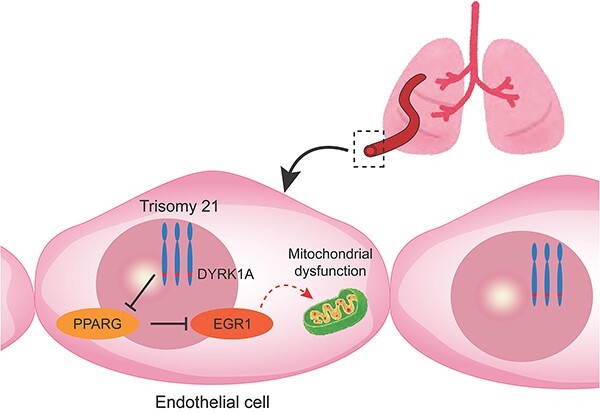
Introduction
Down syndrome (DS) caused by the trisomy of human chromosome 21 (T21) is the most frequent chromosomal disorder with an estimated prevalence of one in every 700 births each year [1]. It has been known that DS is associated with a higher incidence of pulmonary arterial hypertension (PAH), especially in patients with congenital heart defects [2, 3]. A recent large cohort study revealed that the prevalence of PAH in DS was estimated at 28%, far higher than in general population [4]. The major risk factors associated with PAH in DS have been linked to respiratory diseases such as upper airway obstruction, hypoventilation, and recurrent respiratory infections [5]. However, the pulmonary histology showed that early progression of pulmonary arterial remodeling could occur in patients with DS [6, 7]. Previous studies regarding the pathogenesis of idiopathic or hereditary PAH (I/HPAH) demonstrated that pulmonary vascular remodeling was involved in the vascular endothelial cell (EC) dysfunction [8, 9]. In contrast, the cellular characteristics and behaviors of vascular ECs with T21 were not elucidated till present, mainly due to the lack of appropriate cellular and animal models for DS that harbor a complete T21.
Recently, patient-specific induced pluripotent stem cells (iPSCs) became a useful technique for exploring the cellular and molecular pathogeneses of various genetic disorders. In this context, Gu et al. reported that iPSC-derived ECs from patients with HPAH and BMPR2 gene mutation showed a significant EC dysfunction, consequently, a specific molecular pathway for the protection of EC functions was identified [10]. This study suggested that iPSC-derived ECs could be a reliable method for the investigation of the molecular pathogenesis of PAH, despite the incomplete illustration of the pulmonary or systemic arterial ECs in iPSC-derived ECs.
In this study, we mainly utilized the isogenic pairs of human iPSC-derived ECs which were established by patients with DS and the genetically corrected disomy 21 (cDi21) line to investigate the cellular physiology of ECs harboring T21.
Results
All lines of human iPSCs established from patients with DS and healthy controls can efficiently differentiate into ECs
We used MiraCell iPS Cell to Endothelial Cell Differentiation Kit for the differentiation of our iPSC lines into vascular ECs. EC morphology showed a typical cobblestone appearance, similar in T21-, T21’-, cDi21-, and two healthy controls-iPSC-derived ECs (HC_1 and HC_2). We confirmed that almost all cells were CD31-positive ECs by immunocytochemistry, and we estimated the purity of ECs by flow cytometry for CD31 as 97.3 ± 1.4%, 97.2 ± 0.97%, 98.7 ± 0.54%, 92.4 ± 3.4%, and 97.1 ± 0.082%, respectively (Supplementary Fig. 1).
The ECs differentiated from cDi21-iPSCs show equivalent cellular physiological functions as those from healthy control-iPSCs
In order to exclude the possible inter-individual errors of iPSC lines, we generated the isogenic pairs of T21-iPSCs and cDi21-iPSCs [11]. Before comparing cellular functions of ECs between the isogenic pairs of iPSC-derived ECs, we confirmed that ECs, derived from cDi21-iPSCs harboring experimentally corrected normal karyotype, were physiologically equivalent to those derived from HC-iPSCs. In this framework, we carried out the following five fundamental cellular functional assays: cellular proliferation, migration, adhesion, apoptosis, and angiogenesis. We did not find a significant difference among the vascular ECs differentiated from cDi21-iPSCs and the two independent lines of HC-iPSCs (Supplementary Fig. 2). Therefore, cDi21-ECs could be considered as an isogenic normal control of T21-ECs hereafter.
T21-ECs show increased apoptosis, impaired angiogenesis, and mitochondrial dysfunction
Afterwards, we compared cellular physiological functions between the vascular ECs derived from isogenic pairs of T21- and cDi21-iPSCs, and another DS patient derived (T21’)-iPSCs. No significant difference was detected among T21-, T21’-, and cDi21-ECs in the properties of proliferation (cDi21, 23.8 ± 2.99%; T21, 23.6 ± 1.74%; T21’, 23.7 ± 2.17%; P = 0.997; Fig. 1A), migration (cDi21, 1.00 ± 0.076; T21, 0.96 ± 0.079; T21’, 0.96 ± 0.038; P = 0.872; Fig. 1B), and adhesion (cDi21, 560.2 ± 40.08/mm2; T21, 416.0 ± 68.70/mm2; T21’, 506.4 ± 52.57/mm2; P = 0.251; Fig. 1C). In contrast, T21- and T21’-ECs exhibited significantly more apoptotic cells as assessed by flow cytometry for annexin V and propidium iodide (cDi21, 1.85 ± 0.287%; T21, 6.65 ± 0.794%; T21’, 7.41 ± 0.248%; P < 0.01 vs cDi21; Fig. 1, D and E). Alternately, we analyzed angiogenesis abilities by tube formation assay, and found that T21- and T21’-ECs were significantly impaired in angiogenesis (cDi21, 114.8 ± 4.52; T21, 44.67 ± 4.26; T21’, 48.5 ± 3.25; P < 0.001 vs cDi21; Fig. 1, F and G).
Figure 1.

Cellular physiology and mitochondrial respiratory function of T21-ECs and cDi21-ECs. (A) Proliferation assay evaluating the percentages of EdU-positive cells. There is no significant difference among cDi21-, T21-, and T21’-ECs. (B) Relative migration distance 12 h after the wound scratch of the cells. There is no significant difference among cDi21-, T21-, and T21’-ECs. (C) Cell adhesion assay by counting the number of attached cells 2 h after seeding. There is no significant difference among cDi21-, T21-, and T21’-ECs. (D) Representative images of apoptosis assay by flow cytometry for annexin V and propidium iodide (PI). (E) The quantitative analyses of annexin V-positive and PI-negative cell populations show that apoptotic cells are significantly increased in T21- and T21’-ECs as compared with cDi21-ECs. (F) Representative images of phase contrast microscopy in tube formation assay. Note that lots of cellular tubes are formed in cDi21-ECs. Scale bar: 500 μm. (G) The quantitative assessments tube formation show that ability of angiogenesis is impaired in T21- and T21’-ECs as compared with cDi21-ECs. (H) Representative fluorescent images of MitoSOX red staining for mitochondrial superoxide production assay. Scale bar: 50 μm. (I) The quantitative measurements of MitoSOX fluorescence intensity show that mitochondrial superoxide production is significantly increased in T21- and T21’-ECs. (J) Oxygen consumption rate (OCR) at baseline and after sequential injection of oligomycin, carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and Rotenone/Antimycin A (R/AA). (K) Basal respiration and maximal respiration are significantly impaired in T21- and T21’-ECs. Three independent experiments were conducted each in A, B, C, E, G, and I. Five independent experiments were conducted in J and K. Data are presented as mean ± SEM. Data were analyzed by the Tukey–Kramer multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05 as compared to cDi21-ECs.
Assuming that mitochondrial dysfunction is involved in cellular apoptosis, we investigated mitochondrial respiratory activity, and found that T21- and T21’-ECs exhibited higher production of mitochondrial reactive oxygen species (mROS) as compared with cDi21-ECs (cDi21, 142.0 ± 1.25; T21, 240.1 ± 22.1; T21’, 209.6 ± 14.3; P < 0.05 vs cDi21; Fig. 1, H and I). Next, we evaluated mitochondrial respiratory function by oxygen consumption rate (OCR) assay. In this context, T21- and T21’-ECs showed a significant decrease in normalized rate where basal respiration and maximal respiration were significantly impaired (basal: cDi21, 100.0 ± 3.34; T21, 51.8 ± 10.31; T21’, 42.1 ± 0.045; P < 0.01 vs cDi21; maximal: cDi21, 200.0 ± 20.19; T21, 89.72 ± 15.53; T21’, 79.34 ± 11.81; P < 0.01 vs cDi21; Fig. 1, J and K). Consequently, DS specific ECs were more apoptotic and less angiogenic compared with cDi21-ECs, which could be linked to the mitochondrial respiratory dysfunction and oxidative stress. These results regarding cellular features of DS-ECs are consistent with the previous reports using pulmonary ECs-derived from patients with I/HPAH [12–14].
RNA-sequencing shows that gene expression profiles are altered in T21-ECs and identifies EGR1 as a candidate for mitochondrial dysfunction in DS
To clarify the molecular mechanisms that underlie T21-EC dysfunctions, we carried out an RNA-sequencing analysis using two independent iPSC-derived EC lines of patients with DS (T21 and T21’), two independent iPSC-derived EC lines of HC_1 and HC_2, and an isogenic iPSC-derived EC line of cDi21. T-distributed stochastic neighbor embedding (tSNE) showed distinctive expression patterns among each of the two lines of T21-ECs, as well as the two lines of HC-ECs and cDi21-ECs. Noting that, the expression pattern of cDi21-ECs was similar to HC-ECs (Fig. 2A). The pathway analyses demonstrated that both the mitochondria related genes and endothelial cell related genes were dysregulated in T21-ECs (Fig. 2, B and C). To identify the causative genes of T21-EC mitochondrial and endothelial dysfunction, we focused on the differential expression genes among T21- and control-lines of ECs. As a result, we identified significantly upregulated and downregulated genes, where none was located on the chromosome 21 (Fig. 2D). Furthermore, we searched for candidate genes which could result in EC dysfunctions associated with PAH, apoptosis, and mitochondrial dysfunction, and found EGR1 as a potent candidate for T21-EC dysfunction. Our results confirmed that EGR1 expression was significantly increased approximately 1.9-fold in both T21-ECs and T21’-ECs compared to cDi21-ECs by quantitative polymerase chain reaction (PCR) analysis (Fig. 2E). Noting that, EGR1, which is a transcriptional factor encoded on chromosome 5, is known to promote apoptosis via mitochondria-dependent manner [15]. In addition, EGR1 is reported to be involved in the vascular remodeling of PAH [16].
Figure 2.

The RNA-sequencing analyses of T21-, cDi21-, and healthy control-ECs. RNAs were extracted from two independent iPSC-derived EC lines of patients with Down syndrome (T21 and T21’), two independent iPSC-derived EC lines of healthy controls (HC_1 and HC_2), and an isogenic iPSC-derived EC line of corrected to disomy 21 (cDi21). (A) T-distributed stochastic neighbor embedding (tSNE) plots of RNA-sequencing. The comprehensive expression patterns of T21- and T21’-ECs are separated from cDi21-ECs and HC-ECs. (B) The pathway analysis of RNA-sequencing reveals that the mitochondria related genes are dysregulated in T21- and T21’-ECs as compared to HC-ECs and cDi21-ECs. (C) The RNA-sequencing reveals that the endothelial cell related genes are dysregulated in T21- and T21’-ECs. (D) Differential gene expression (DEG) analysis reveals significantly upregulated and downregulated genes in T21-ECs. (E) Quantitative PCR analysis of EGR1 in T21-, T21’-, cDi21-, and HC-ECs. Data are presented as mean ± SEM. Data were analyzed using the Tukey–Kramer multiple comparison test. **P < 0.01.
Suppression of EGR1 expression restores angiogenesis and mitochondrial dysfunctions of T21-ECs
To determine whether the increase in EGR1 expression is truly responsible for endothelial and mitochondrial dysfunctions of T21-ECs, we analyzed angiogenesis and OCR subsequently after siEGR1 treatment. First, we checked the suppression of EGR1 expression in T21-ECs following the treatment of two different targets of siEGR1; both could effectively reduce EGR1 expression in T21- ECs (Supplementary Fig. 3A). As the 24 h treatment of lipofection reagent disturbed the cellular viability and angiogenesis of iPSC-derived ECs, we selected 6 h treatment of siEGR1. We confirmed EGR1 expression levels in T21-, T21’-, cDi21-, and HC-ECs under 6 h treatment of siEGR1, and found that the relative mRNA expressions of EGR1 in all lines were suppressed at similar levels. Although EGR1 expression level is known to rapidly change depending on the cellular status, we confirmed that suppression of EGR1 expression was persistent for at least 6 h after the retrieval of siEGR1 from culture media, meaning that there was no time effect during the angiogenesis and OCR experimental assays (Supplementary Fig. 3B). Afterward, we found that the reduction of EGR1 expression could significantly restore the angiogenesis property, mitochondrial OCR, the ratio of apoptotic cells, and mROS production both in T21- and T21’-ECs (Fig. 3), suggesting that the elevation of EGR1 expression could be substantially responsible for DS-associate endothelial and mitochondrial dysfunctions.
Figure 3.

Knockdown of EGR1 expression in T21-EC restores angiogenesis and mitochondrial respiratory function. (A) Representative phase contrast images of tube formation assay in scrambled siRNA-treated or siEGR1-treated ECs. Scale bar: 500 μm. (B) The quantitative analyses of tube formation assays reveal that angiogenesis is significantly recovered in siEGR1 treated T21- and T21’-ECs as same level as that of cDi21-ECs. (C) OCR of each cell line and condition at baseline and after sequential supplementation of oligomycin (Oligo), FCCP, and Rotenone/Antimycin A (R/AA). (D) Quantitative analyses of basal respiration and maximal respiration show significant improvement of OCR in siEGR1 treated T21- and T21’-ECs. (E) Quantitative analyses of annexin V-positive and PI-negative cell populations show that apoptotic cells are significantly decreased in siEGR1-treated T21- and T21’-ECs, similar to cDi21-ECs. (F) Quantitative measurements of MitoSOX fluorescence intensity show that the level of mitochondrial superoxide production is significantly recovered in siEGR1-treated T21- and T21’-ECs, which is similar to the level in cDi21-ECs.Three to four independent experiments were conducted in each. Data are presented as mean ± SEM. Data were analyzed by Tukey–Kramer multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05.
Next, to confirm whether EGR1 suppression by the chemical inhibitor was also effective to restore the T21-EC dysfunction, we administered pioglitazone to the culture media of T21-, T21’- and cDi21-ECs. Pioglitazone is a PPARγ activator that reduces EGR1 expression [17]. Alternatively, we confirmed that a supplementation of 10 μM pioglitazone could efficiently increase PPARG expression and reduce EGR1 expression in T21-ECs (Fig. 4, A and B). Therefore, we clearly verified that angiogenesis property, mitochondrial OCR, the ratio of apoptotic cells, and mROS production could be significantly recovered in T21- and T21’-ECs after pioglitazone treatment (Fig. 4, C–H). This result suggested that pioglitazone could be a candidate for drug treatment to improve the endothelial function in patients with DS.
Figure 4.

Pioglitazone suppresses EGR1 mRNA expression in DS-ECs and can restore angiogenesis and mitochondrial respiratory function. (A and B) Relative mRNA expression of PPARG and EGR1 in T21-ECs treated with DMSO or pioglitazone (PIO). PIO treatment significantly increases PPARG expression and suppresses EGR1 expression. (C) Representative phase contrast images of tube formation assays in each cell line and condition. Scale bar: 500 μm. (D) The quantitative analyses of tube formation assays demonstrate that angiogenesis is significantly recovered in PIO-treated T21- and T21’-ECs as same level as that of cDi21-ECs. (E) OCR of each cell line and condition at baseline and after sequential treatment of oligomycin (Oligo), FCCP, and Rotenone/Antimycin A (R/AA). (F) Quantitative analyses of basal respiration and maximal respiration show significant improvement of OCR in PIO-treated T21- and T21’-ECs. (G) Quantitative analyses of annexin V-positive and PI-negative cell populations show that apoptotic cells are significantly decreased in PIO-treated T21- and T21’-ECs, similar to cDi21-ECs. (H) Quantitative measurements of MitoSOX fluorescence intensity show that the level of mitochondrial superoxide production is significantly restored in pioglitazone-treated T21- and T21’-ECs, which is similar to the level in cDi21-ECs. Three independent experiments were conducted in each. Data are presented as mean ± SEM. Data were analyzed by unpaired t-test and Tukey–Kramer multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05.
Chemical inhibition of DYRK1A can modulate PPARG and EGR1 expressions leading to recovery of EC function in DS
In order to investigate how EGR1 expression is altered in T21-ECs, we examined the candidates of responsible genes on chromosome 21. DYRK1A is located within the DS critical region of the chromosome 21, and any elevation in its expression is reported to impair the angiogenesis, which is related to the suppression of solid tumor growth in DS through ECs inactivation [18]. Moreover, overexpression of DYRK1A was reported to suppress PPARG expression [19]. Given that PPARG was reported to inhibit EGR1 expression [20], we hypothesized that an upregulation of DYRK1A might increase EGR1 expression via suppressing PPARG pathway in T21-ECs. On the other side, to examine whether DYRK1A was responsible for the endothelial dysfunction in DS, we administered harmine, a potent DYRK1A inhibitor, to the culture media of T21-, T21’-, and cDi21-ECs. Consequently, we confirmed that DYRK1A expression level was almost 1.5-fold higher in our T21- and T21’-ECs by RNA-sequencing and quantitative PCR analyses (Fig. 5A). Harmine treatment could effectively increase PPARG expression and suppress EGR1 expression in T21- and T21’-ECs with similar levels of cDi21-ECs (Fig. 5, B and C). Therefore, we demonstrated that harmine treatment could significantly improve the angiogenesis property, the mitochondrial respiratory function, apoptosis, and mROS production in T21- and T21’-ECs (Fig. 5, D–I).
Figure 5.

DYRK1A inhibitor, harmine reduces EGR1 mRNA expression in T21-EC via PPARG pathway and restores angiogenesis and mitochondrial respiratory function. (A) Relative mRNA expressions of DYRK1A in T21- and T21’-ECs, evaluated by quantitative PCR. Approximately 1.5-fold higher expression is observed both in T21- and T21’-ECs as compared with cDi21-ECs. (B) Relative mRNA expression of PPARG in cDi21-ECs, T21-, and T21’-ECs treated with DMSO or harmine; the latter significantly increases PPARG expression in T21- and T21’-ECs, but not in cDi21-ECs. (C) Relative mRNA expression of EGR1 treated with DMSO or harmine; the latter significantly reduces EGR1 expression in T21- and T21’-ECs. (D) Representative phase contrast images of tube formation assays in each cell line and condition. Scale bar: 500 μm. (E) Quantitative analyses of tube formation assays reveal that angiogenesis is significantly recovered in harmine-treated T21- and T21’-ECs as same level as that of cDi21-ECs. (F) OCR of each cell line and condition at baseline and after sequential treatment of oligomycin (Oligo), FCCP, and Rotenone/Antimycin A (R/AA). (G) Quantitative analyses of basal respiration and maximal respiration show significant improvement of OCR in harmine-treated T21- and T21’-ECs. (H) Quantitative analyses of annexin V-positive and PI-negative cell populations show that apoptotic cells are significantly decreased in harmine-treated T21- and T21’-ECs, similar to cDi21-ECs. (I) Quantitative measurements of MitoSOX fluorescence intensity show that the level of mitochondrial superoxide production is significantly decreased in harmine-treated T21- and T21’-ECs, which is similar to the level in cDi21-ECs. Three to four independent experiments were conducted in each. Data are presented as mean ± SEM. Data were analyzed by Tukey–Kramer multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05.
DYRK1A overexpression deteriorates endothelial and mitochondrial function in control ECs
We transfected a vector expressing DYRK1A into cDi21-ECs and two healthy control (HC)-ECs to investigate whether DYRK1A is solely responsible for endothelial and mitochondrial dysfunctions in DS-ECs. First, we examined the relative expression levels of DYRK1A in cDi21-ECs. Additionally, we found that DYRK1A is 20-fold more abundant in DYRK1A expression vector-cDi21- than in empty vector (EV) transfected-cDi21-ECs (Fig. 6A). DYRK1A overexpression significantly downregulated and upregulated the expression of PPARG and EGR1, respectively (Fig. 6A). DYRK1A overexpression impaired tube formation abilities and OCR in cDi21- and HC-ECs (Fig. 6, B–E). Then, the experiment revealed significant apoptosis of ECs and increased mROS levels in DYRK1A-overexpressed-cDi21- and HC-ECs (Fig. 6, F and G). Overall, our results suggest that DYRK1A upregulation could affect the expression levels of PPARG and EGR1 in DS-ECs, leading to EC dysfunction in patients with DS.
Figure 6.

DYRK1A overexpression impairs angiogenesis and mitochondrial respiratory function in cDi21- and HC-ECs. (A) Transfection of the DYRK1A-expression vector induced significant higher mRNA level of DYRK1A in cDi21-ECs as compared with that of empty vector (EV). Overexpression of DYRK1A results in a significant decrease of PPARG and an increase of EGR1 levels in cDi21-ECs compared with EV-treated-ECs. (B) Representative images of the tube formation assays in cDi21- and two HC-EC lines treated with EV and DYRK1A expression vector, visualized by phase contrast microscopy. Scale bar: 500 μm. (C) Quantitative analyses of the tube formation assays show that angiogenesis is significantly impaired in DYRK1A-overexpressed cDi21- and HC-ECs. (D) OCR at baseline and after sequential treatment of oligomycin (Oligo), FCCP, and Rotenone/Antimycin A (R/AA) in each line treated with EV and DRYK1A expression vector. (E) Quantitative analyses of basal respiration and maximal respiration show significant impairment of OCR in cells treated with DYRK1A-expression vector. (F) Quantitative analyses of annexin V-positive and PI-negative cell populations demonstrate that the proportion of apoptotic cells significantly increased in DYRK1A-overexpressed ECs. (G) Quantitative measurements of MitoSOX fluorescence intensity show a significant increase in the level of mitochondrial superoxide production in DYRK1A-overexpressed ECs. Three independent experiments were conducted in each. Data are presented as mean ± SEM. Data were analyzed by unpaired t-test and Tukey–Kramer multiple comparison test. ***P < 0.001, **P < 0.01, *P < 0.05.
EGR1 is upregulated in the pulmonary ECs of a human patient with DS and PAH
Finally, to evaluate whether EGR1 plays an important role in the pathogenesis of PAH in human patients with DS, we analyzed the lung specimens of a patient with DS and PAH, whose upper lobe of his left lung was partly resected due to a massive hemoptysis with severe PAH subsequent to a cardiac surgery for a ventricular septal defect. Therefore, we conducted an immunohistochemistry of EGR1 in the lung tissues and counted the percentages of EGR1-positive nuclei of his pulmonary arterial ECs (Fig. 7, A and B). The quantitative analysis showed that the number of EGR1- positive pulmonary arterial ECs was significantly greater as compared with a healthy control (healthy control, 21.5 ± 4.37%; Down syndrome, 81.5 ± 3.89%; P < 0.001; Fig. 7C). This outcome suggested that EGR1 is upregulated in human patients with DS and PAH, indicating that EGR1 is a key regulator of pulmonary EC dysfunction in DS as well as in iPSC-derived ECs.
Figure 7.

Immunohistochemistry of EGR1 in pulmonary arteries of patients with Down syndrome and PAH. (A) Representative images of immunohistochemistry of EGR1 in the lung tissues of healthy control (upper panels) and patients with DS and PAH (lower panels). Scale bar: 200 μm. (B) Representative images of quantitative evaluation for EGR1 positive pulmonary ECs. The parentages of 3,3′-Diaminobenzidine (DAB) positive nuclei of pulmonary arterial ECs were counted. The black arrowheads indicate EGR1-positive nuclei and the white arrowhead indicates EGR1-negative nucleus. Scale bar: 50 μm. (C) EGR1-positive cell ratio of pulmonary ECs in DS is significantly higher than that of healthy control. Seven arteries and nine arteries were analyzed in healthy control and in DS, respectively. Data are presented as mean ± SEM. Data were analyzed by an unpaired Student’s t test. ***P < 0.001.
Discussion
In this study, we originally proved that vascular EC function is impaired in DS by using isogenic pairs of iPSCs derived from DS patients. Recently, patient-specific iPSC technology facilitates the investigation of pathogenic mechanisms in many genetic disorders including PAH. Nevertheless, to reduce the inter-individual error of iPSC lines, isogenic pairs of iPSCs have been considered as an appropriate in in vitro model [21, 22].
First of all, we analyzed the five cellular physiological functions of T21-ECs, including proliferation, adhesion, migration, apoptosis, and angiogenesis abilities. According to previous studies utilizing the ECs from patients with I/HPAH or animal models, these cellular properties were impaired in PAH [23, 24]. In fact, we demonstrated that T21-ECs had an impaired angiogenesis and an increased apoptosis, which is compatible with the previous studies of IPAH both in vitro and in vivo [25]. In contrast, we could not find any significant difference in cellular proliferation, migration, and adhesion in T21-ECs, although many previous studies revealed that the latter properties of cellular function were impaired in pulmonary ECs of PAH [23]. This discrepancy might be due to the difference of pathogenesis between I/HPAH and PAH associated with DS. Afterward, we focused on the mitochondrial respiratory function of T21-ECs as mitochondrial dysfunction was reported in DS; a previous study proved that fetal dermal fibroblasts from patients with DS exhibited the impairment of mitochondrial respiration and elevation of ROS [26]. On the other hand, the impairment of mitochondrial respiratory function and metabolism have been thoroughly examined in patients with PAH and model animals [12–14, 24]. Several studies showed that ROS increased in the lung tissues of patients with PAH [12], and that OCR of pulmonary ECs in patients with IPAH significantly decreased [24]. Although we were unable to assess the metabolic shift of glycolysis and alteration of mitochondrial membrane potential, our results suggest that mitochondrial dysfunction could be associated with the pathogenesis of PAH in DS.
Subsequently, we found that the comprehensive gene expression patterns in T21-ECs changed compared with healthy control-ECs. In addition, we identified EGR1 as a candidate gene for mitochondrial dysfunction in DS. EGR1, which is a Cys2-His2 type zinc finger transcription factor, resides on human chromosome 5 [27]. Its expression is induced by various stimuli including hypoxia, shear stress, and pro-inflammatory cytokines [28–30]. EGR1 binds to GC-rich domain of genes such as TGFB [31], PDGFA [29], PDGFB [32], and p53 [33], all of which are involved in the pathogenesis of PAH in animal models and human patients [34–36]. Additionally, EGR1 is also involved in the mitochondrial function [15, 37]. Later, we showed that treatment with siEGR1 and pioglitazone could restore the mitochondrial function and angiogenesis in DS-ECs, suggesting that EGR1 is a key regulator of the mitochondrial function and plays an important role in PAH pathogenesis of patients with DS.
Next, we focused on DYRK1A and PPARG pathways. DYRK1A is one of the most important genes in human chromosome 21, which is closely associated with the pathogenesis of DS mainly in neurological disorders [38]. Dyrk1a transgenic mouse was reported to decrease PPARG expression, and PPARG was described as an important upstream regulator of EGR1 [17, 19]. Moreover, PPARG pathway has attracted attention for its involvement in the pathogenesis of PAH [39, 40]. We demonstrated that a chemical inhibition of DYRK1A could restore the mitochondrial function and angiogenesis in T21-ECs. On the contrary, DYRK1A overexpression might deteriorate the mitochondrial and endothelial functions in control ECs. Taken together, we strongly suggest that DYRK1A/PPARG/EGR1 pathway would play a central role in the pathogenesis of PAH associated with DS.
Finally, to confirm whether our in vitro experiments using patient-specific iPSC lines truly recapitulate the pathogenesis of PAH in DS, we analyzed the lung specimen of a patient with DS with severe PAH and congenital heart disease; the results showed that EGR1 expression in the pulmonary arterial ECs was significantly elevated in the patient with DS with PAH, which indicates that EGR1 is a potent key regulator of the pathogenesis of PAH in human patients with DS, and that the DYRK1A/PPARG/EGR1 pathway might be a candidate therapeutic target of DS with PAH.
Nevertheless, there are several limitations in this study; the most important one is that the differentiated ECs from iPSCs are not specific to pulmonary ECs. However, a previous study using iPSCs from patients with HPAH and a BMPR2 mutation could also successfully recapitulate the pathological phenotype of EC dysfunction [10]. Another limitation resides on the number of patients in both in vitro and in vivo experiments. We cannot exclude the possibility of inter-individual variability. We utilized the isogenic pairs of iPSCs: T21- and cDi21-iPSCs; therefore, we considered that the possible errors of iPSC lines could be reduced in this study. Moreover, we analyzed the lung specimen from only one patient with DS, due to the fact that human lung specimens from patients with DS are hard to obtain. Since EGR1 expression is associated with cell shear stress, we assessed the response of T21-ECs to shear stress using a liquid flow pump system. However, T21- and T21’-ECs detached from the cell culture plates even under weak stimuli (2 to 5 dyne). Therefore, we could not determine the shear stress response of these cells in vitro. Consequently, further studies are necessary to reveal whether the suppression of EGR1 is effective for DS-associated PAH in human.
In conclusion, we identified EGR1 as a key regulator of mitochondrial dysfunction of pulmonary ECs in DS. Moreover, we suggested that DYRK1A/PPARG/EGR1 could be a central pathway for pulmonary EC dysfunction in patients with DS.
Materials and Methods
Generation and culture of iPSCs from patients with DS and healthy controls
T21-iPSC lines were generated as previously described [11, 41]. Briefly, the dermal fibroblasts cultured from two patients with DS and two healthy infants were transfected by a Sendai virus vector encoding human OCT3/4, SOX2, KLF4, and CMYC. Noting that, no patient with DS had congenital heart disease. We generated a cDi21-iPSC line by introducing a chromosome elimination cassette into the extra maternal chromosome by TALENs, obtained after Cre-loxP mediated recombination and FIAU selection [11]. The iPSC lines were characterized by immunostaining for pluripotency markers, karyotyping, and teratoma formation. We maintained the cells by Cellartis DEF-CS 500 Basal Medium (Takara bio, Kusatsu, Japan) and passaged following the manufacturer’s recommendations.
Differentiation to vascular ECs
We differentiated ECs from each iPSC line by using MiraCell iPS Cell to Endothelial Cell Differentiation Kit (Takara bio) following the manufacturer’s protocol, and maintained on fibronectin-coated cell-culture dishes with MiraCell EC Culture Medium (Takara bio). ECs were used for all experiments between passages 2 and 6.
Immunofluorescence and flow cytometry analyses
For immunocytochemical analysis, we fixed the cells with 4% paraformaldehyde for 10 min, and permeabilized it with a PBS solution containing 0.1% of Triton-X 100 for 10 min. After blocking with 5% FBS, we incubated the cells with anti-CD31 (1/30; 14-0319-82, Thermo Fisher Scientific, Waltham, MA, USA) primary antibody at 4°C overnight. Afterward, we incubated the cells with secondary Goat anti-Mouse Alexa Fluor 488 antibody (1/400; A-11001, Thermo Fisher Scientific) for 60 min at room temperature, and we stained the nuclei with Hoechst 33 342 (H342, Dojindo Molecular Technologies, Kumamoto, Japan). We developed the images by IN Cell Analyzer 6000 (GE Healthcare, Chicago, IL, USA). For flow cytometry, we suspended the cells in HBSS containing 3% FBS and 0.3 mM EDTA, fixed with 4% paraformaldehyde, and stained with the same antibodies used in immunofluorescence. Mouse IgG isotype control (401 401, BioLegend, San Diego, CA, USA) was applied as a negative control. We analyzed the cells via a BD FACS Canto II (BD Biosciences, Franklin Lakes, NJ, USA), and performed the data analysis using FlowJo v10 software (BD Biosciences).
Proliferation assay
We cultured ECs at 15 000 cells per well in 96-well plates for one day, then, we applied 10 μM EdU (Click-iT EdU Cell Proliferation Kit for Imaging, Thermo Fisher Scientific) and incubated the plates for 24 h. Afterward, we stained the cells with AlexaFluor 488 (Thermo Fisher Scientific) and the nuclei with Hoechst 33 342. We analyzed the cells at a × 400 magnification by IN Cell Analyzer 6000, and we calculated the percentages of EdU-positive nuclei.
Apoptosis assay
Prior to the apoptosis assay, we cultured the cells at 100 000 cells per well in 6-well plates for 24 h. Next, we washed the cells with Cell Staining Buffer (Biolegend) and stained them with FITC-Annexin V and propidium iodide (PI) (Biolegend) at 4°C for 10 min, and analyzed them by BD FACS Canto II. Data analysis was performed using FlowJo v10 software.
Migration assay
We modified the migration assay from the previous publication [42]; we cultured the cells at 60 000 per well in 24-well plates 24 h prior to the assay. After confirming the confluence, we scratched the cells with a vertical wound through the cell monolayer using a 200 μl pipette tip. We washed the cellular debris with PBS twice and added MiraCell EC Culture Medium to the cells. We obtained the phase contrast microscopic images and measured the distance of one side to the other side of the wound. We analyzed the ratio of shortening from the start to 12 h compared with a control using ImageJ software (https://imagej.nih.gov/ij/index.html).
Adhesion assay
We seeded the cells at 20 000 cells per well in 96-well plates coated with fibronectin. Later, after 2 h incubation at 37°C, we washed the cells twice with PBS and stained them with Hoechst 33 342. We analyzed the number of nuclei by IN Cell Analyzer 6000.
Tube formation assay
We modified the tube formation assay from the previous publication [43]; we merged growth factor reduced Matrigel (Merck Millipore, Burlington, MA, USA) into 96-well plates (40 μl/well) and incubated them at 37°C for 30 min. We seeded the cells at 15 000 per well for 6 h. We captured the images with a BZ-X700 microscope (Keyence, Osaka, Japan). We quantified the tube formation by the number of branching points.
Measurement of mitochondrial reactive oxygen species (mROS)
We measured mROS using MitoSOX red (Thermo Fisher Scientific) following the manufacture’s protocol. We seeded the cells briefly at 15 000 per well in 96-well black plates. We loaded the cells with 5 μM MitoSOX red and 50 nM MitoTracker Green (Thermo Fisher Scientific) for 15 min, washed twice in PBS, and analyzed them by IN Cell Analyzer 6000. We indexed the level of mROS by mean red fluorescence intensity on the mitochondria stained in green.
Measurement of OCR
We measured OCR via a Seahorse XFe96 Analyzer (Agilent Technologies, Santa Clara, CA, USA). We seeded the cells at 10 000 cells per well in 96 well Seahorse assay plates coated with fibronectin. We studied the cells in Seahorse XF DMEM (Agilent Technologies) supplemented with 10 mM glucose, 2 mM glutamine and 1 mM pyruvate. We determined OCR in the basal state or after sequentially adding 1.0 μM oligomycin, 2.0 μM carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP), and 0.5 μM rotenone/antimycin A. We corrected the values of the OCR measurement for cell numbers and expressed the data as a percentage of the basal OCR of control cell lines.
RNA isolation and RNA-sequence
We extracted total RNA using NucleoSpin (Takara Bio) following the manufacturer’s procedure; we measured the RNA concentration by NanoDrop 2000c (Thermo Fisher Scientific), and conducted an RNA-sequencing by next generation sequencer as previously described [44]. Sequencing was performed on an Illumina HiSeq 2500 platform in 75-base single-end mode (Illumina, San Diego, CA, USA). The transcriptomes of T21-ECs, healthy-ECs, and cDi21-ECs were analyzed with Subio platform (https://www.subioplatform.com/ja/). All sequence data sets are available at Gene Expression Omnibus, GSE203257.
siRNA gene silencing
Knockdown of EGR1 was carried out using Silencer Select siRNA with Lipofectamine RNAiMAX (Thermo Fisher Scientific) following the manufacturer’s protocol. Before performing tube formation assay and measuring OCR, we transfected ECs with siRNA (5 nM) for 6 h. The sequences of siRNAs were as follows; siEGR1_1 sense: 5′-CAACGACAGCAGUCCCAUUtt-3′, antisense: 5′-AAUGGGACUGCUGUCGUUGga-3′; siEGR1_2 sense: 5′-GCAGAGUCUUUUCCU GACAtt-3′, antisense: 5′-UGUCAGGAAAAGACUCUGCgg-3′.
Plasmid transfection
The plasmids pRP-Vec (pRP[Exp]-EGFP-CAG > ORF_Stuffer, Cat. VB900122-4426sjs) and pRP-DYRK1A (pRP[Exp]-EGFP-CAG > hDYRK1A[NM_001396.5], Cat. VB900135-9088zng) were purchased from VectorBuilder (Chicago, IL, USA). Differentiated ECs were transfected with the plasmids for 6 h using Lipofectamine 2000 reagent (Thermo Fisher Scientific). Subsequently, the medium was replaced with MiraCell EC Culture Medium overnight. ECs were provided for all experiments 24 h following the plasmid transfection.
Quantitative real-time PCR
We extracted total RNA using NucleoSpin RNA column kit (Takara Bio). After RNA isolation, we performed reverse transcription through the use of ReverTra ACE (TOYOBO, Osaka, Japan) following the manufacturer’s instructions. Afterward, we conducted quantitative real-time PCR with QuantStudio 7 system by using THUDERBIRD SYBR qPCR Mix kit (TOYOBO). We analyzed each sample in technical triplicates with the following protocol: 10 min at 95°C, followed by 40 cycles of 15 s at 95°C, and 1 min at 60°C. Each PCR analysis was completed by a melt curve cycle of 15 s at 95°C, 60 s at 60°C, and 15 s at 95°C. We analyzed all the data by using the standard curve method, and evaluated the relative amount of mRNAs compared to that of the house-keeping gene ACTB. We calculated the cycle threshold under the default settings by real-time sequence detection software (Applied Biosystems).
We used the following primer pairs for qPCR; EGR1 forward: 5′-AGCAGCACCTTCAACCCTCAGG-3′, reverse 5′-GAGTGGTTTGGCTGGGGTAACT-3′; PPARG forward: 5′-TGGTGACTTTATGGAGCCCAA-3′, reverse 5′-GGCAAACAGCTGTGAGGACTCAG-3′; DYRK1A forward: 5′-CCTTGCCATTGATATGTGGTCCC-3′, reverse 5′-GCAGGTGGAATACCCAGAACTTC-3′; ACTB forward: 5′-TCAAGATCATTGCTCCTCCTGAG-3′, reverse 5′-ACATCTGCTGGAAGGTGGACA-3′.
Chemical inhibition of DYRK1A/PPARG/EGR1 pathway
We seeded the cells at 80 000 per well in 24-well plates for RNA extraction, at 100 000 per well in 6-well plates for tube formation assay, and 10 000 per well in 96 well Seahorse assay plates coated with fibronectin for OCR measurement, 24 h prior to drug treatment. We applied a 24 h treatment with 10 μM of pioglitazone (Wako, Osaka, Japan) and 2.5 μM of harmine (Thermo Fisher Scientific) for inhibiting EGR1 and DYRK1A, respectively.
Immunohistochemistry for EGR1 in the lungs of a patient with DS
We performed immunohistochemistry using ImmPRESS HRP Horse Anti-Rabbit IgG PLUS Polymer Kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer’s instructions. We deparaffinized the paraffin-embedded slides in xylene and rehydrated them in a graded series of ethanol. We performed antigen retrieval by autoclaving the slides in 10 mM citrate buffer, at pH 6.0. We blocked the activity of the endogenous peroxidase with 3% hydrogen peroxide for 30 min. After blocking, we incubated the slides with a 1:200 dilution of rabbit monoclonal anti–EGR1 antibody (ab194357, Abcam, Cambridge, UK) at 4°C overnight in a humidified chamber. Next, we incubated the slides with ImmPRESS HRP horse anti-rabbit IgG polymer reagent (Vector Laboratories) for 30 min, followed by 3,3′-diaminobenzidine (DAB) staining for 40 s. We counterstained the slides with Mayer hematoxylin, dehydrated in a graded series of ethanol followed by xylene, and mounted with Mount Quick (Daido Sangyo, Saitama, Japan). Paraffin-embedded normal lung sections were purchased from Novus Biologicals (62-years old, male, NBP2-30182).
Data availability
The RNA-sequencing raw fastq files, gene by cell transcript counts matrix, and metadata files were available in Gene Expression Omnibus database (GSE203257).
Statistics
All data were expressed as means ± standard error. We performed all statistical analyses through R version 4.0.4 and RStudio 1.4.1106. We used an unpaired t-test for comparison of two groups, and performed a multiple group comparison by Tukey–Kramer test. P value < 0.05 was considered to indicate a statistical significance.
Study approval
All studies using iPSCs and human lung samples are approved by the Ethics Committee of Osaka University Graduate School of Medicine (approval no. 13123-823 and no. 15118-4). Written informed consents were obtained from the parents of the patients prior to participation.
Conflict of interest statement
None declared.
Supplementary Material
Contributor Information
Hidehiro Suginobe, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Hidekazu Ishida, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Yoichiro Ishii, Department of Pediatric Cardiology, Osaka Children’s and Women’s Hospital, 840 Murodohcho, Izumi, Osaka 594-1101, Japan.
Kazutoshi Ueda, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Chika Yoshihara, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Atsuko Ueyama, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Renjie Wang, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Hirofumi Tsuru, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan; Department of Pediatrics, Niigata University School of Medicine, 1-757 Asahimachi-dori, chuo-ku, Niigata 951-8510, Japan.
Kazuhisa Hashimoto, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Masaki Hirose, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Ryo Ishii, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Jun Narita, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Yasuji Kitabatake, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Keiichi Ozono, Department of Pediatrics, Osaka University Graduate School of Medicine, 2-2 Yamadaoka, Suita, Osaka 565-0871, Japan.
Funding
This study was supported by grants from the Ministry of Education, Science, Sports, and Culture of Japan [no. 17K16261 and 20K16854].
Author contributions
H.S, K.U., C.Y., A.U., R.W., H.T., conducted the cell culture and differentiation, and contributed to all experimental measurements. H.I., R.I., J.N., K.O. designed all the study. K.H. and M.H. corrected the sample of the patients and controls. H.S. and Y.I. analyzed the RNA-sequence data. Y.K. established the T21- and cDi21-, and healthy-iPSC lines. H.S. and H.I. wrote the manuscript. H.I., Y.K., K.O. revised the data and the manuscript. All authors approved the final version of the manuscript.
References
- 1. Presson AP, Partyka G, Jensen KM. et al. Current estimate of Down syndrome population prevalence in the United States. J Pediatr 2013;163:1163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hals J, Hagemo PS, Thaulow E. et al. Pulmonary vascular resistance in complete atrioventricular septal defect. A comparison between children with and without Down’s syndrome. Acta Paediatr 1993;82:595–8. [DOI] [PubMed] [Google Scholar]
- 3. Colvin KL, Yeager ME. What people with Down syndrome can teach us about cardiopulmonary disease. Eur Respir Rev 2017;26:160098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bush D, Galambos C, Ivy DD. et al. Clinical characteristics and risk factors for developing pulmonary hypertension in children with Down syndrome. J Pediatr 2018;202:212–219.e2. [DOI] [PubMed] [Google Scholar]
- 5. Bush D, Galambos C, Dunbar Ivy D. Pulmonary hypertension in children with Down syndrome. Pediatr Pulmonol 2021;56:621–9. [DOI] [PubMed] [Google Scholar]
- 6. Suzuki K, Yamaki S, Mimori S. et al. Pulmonary vascular disease in Down’s syndrome with complete atrioventricular septal defect. Am J Cardiol 2000;86:434–7. [DOI] [PubMed] [Google Scholar]
- 7. Clapp S, Perry BL, Farooki ZQ. et al. Down’s syndrome, complete atrioventricular canal, and pulmonary vascular obstructive disease. J Thorac Cardiovasc Surg 1990;100:115–21. [PubMed] [Google Scholar]
- 8. Tuder RM, Archer SL, Dorfmüller P. et al. Relevant issues in the pathology and pathobiology of pulmonary hypertension. J Am Coll Cardiol 2013;62:D4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leopold JA, Maron BA. Molecular mechanisms of pulmonary vascular remodeling in pulmonary arterial hypertension. Int J Mol Sci 2016;17:761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gu M, Shao N-Y, Sa S. et al. Patient-specific iPSC-derived endothelial cells uncover pathways that protect against pulmonary hypertension in BMPR2 mutation carriers. Cell Stem Cell 2017;20:490–504.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Omori S, Tanabe H, Banno K. et al. A pair of maternal chromosomes derived from meiotic nondisjunction in trisomy 21 affects nuclear architecture and transcriptional regulation. Sci Rep 2017;7:764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bowers R, Cool C, Murphy RC. et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 2004;169:764–9. [DOI] [PubMed] [Google Scholar]
- 13. Dorfmüller P, Chaumais M-C, Giannakouli M. et al. Increased oxidative stress and severe arterial remodeling induced by permanent high-flow challenge in experimental pulmonary hypertension. Respir Res 2011;12:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wedgwood S, Black SM. Role of reactive oxygen species in vascular remodeling associated with pulmonary hypertension. Antioxid Redox Signal 2003;5:759–69. [DOI] [PubMed] [Google Scholar]
- 15. Yuan S, Wen J, Cheng J. et al. Age-associated up-regulation of EGR1 promotes granulosa cell apoptosis during follicle atresia in mice through the NF-κB pathway. Cell Cycle 2016;15:2895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van der Feen DE, Dickinson MG, Bartelds B. et al. Egr-1 identifies neointimal remodeling and relates to progression in human pulmonary arterial hypertension. J Heart Lung Transplant 2016;35:481–90. [DOI] [PubMed] [Google Scholar]
- 17. Wan H, Yuan Y, Liu J. et al. Pioglitazone, a PPAR-γ activator, attenuates the severity of cerulein-induced acute pancreatitis by modulating early growth response-1 transcription factor. Transl Res 2012;160:153–61. [DOI] [PubMed] [Google Scholar]
- 18. Baek K-H, Zaslavsky A, Lynch RC. et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009;459:1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Song W-J, Song E-AC, Jung M-S. et al. Phosphorylation and inactivation of glycogen synthase kinase 3β (GSK3β) by dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). J Biol Chem 2015;290:2321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan C, Chen J, Ding Y. et al. The crucial role of PPARγ-Egr-1-pro-inflammatory mediators Axis in IgG immune complex-induced acute lung injury. Front Immunol 2021;12:634889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Soldner F, Laganière J, Cheng AW. et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 2011;146:318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim HS, Bernitz JM, Lee D-F. et al. Genomic editing tools to model human diseases with isogenic pluripotent stem cells. Stem Cells Dev 2014;23:2673–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Humbert M, Guignabert C, Bonnet S. et al. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 2019;53:1801887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xu W, Koeck T, Lara AR. et al. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A 2007;104:1342–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xu W, Erzurum SC. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. Compr Physiol 2011;1:357–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Izzo A, Mollo N, Nitti M. et al. Mitochondrial dysfunction in Down syndrome: molecular mechanisms and therapeutic targets. Mol Med 2018;24:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol 2002;193:287–92. [DOI] [PubMed] [Google Scholar]
- 28. Lo LW, Cheng JJ, Chiu JJ. et al. Endothelial exposure to hypoxia induces Egr-1 expression involving PKCalpha-mediated Ras/Raf-1/ERK1/2 pathway. J Cell Physiol 2001;188:304–12. [DOI] [PubMed] [Google Scholar]
- 29. Khachigian LM, Anderson KR, Halnon NJ. et al. Egr-1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress-response element in the PDGF A-chain promoter. Arterioscler Thromb Vasc Biol 1997;17:2280–6. [DOI] [PubMed] [Google Scholar]
- 30. Goetze S, Kintscher U, Kaneshiro K. et al. TNFalpha induces expression of transcription factors c-fos, Egr-1, and Ets-1 in vascular lesions through extracellular signal-regulated kinases 1/2. Atherosclerosis 2001;159:93–101. [DOI] [PubMed] [Google Scholar]
- 31. Baron V, Adamson ED, Calogero A. et al. The transcription factor Egr1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53, and fibronectin. Cancer Gene Ther 2006;13:115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khachigian LM, Lindner V, Williams AJ. et al. Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science 1996;271:1427–31. [DOI] [PubMed] [Google Scholar]
- 33. Boone DN, Qi Y, Li Z. et al. Egr1 mediates p53-independent c-Myc-induced apoptosis via a noncanonical ARF-dependent transcriptional mechanism. Proc Natl Acad Sci U S A 2011;108:632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Botney MD, Bahadori L, Gold LI. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol 1994;144:286–95. [PMC free article] [PubMed] [Google Scholar]
- 35. Perros F, Montani D, Dorfmüller P. et al. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med 2008;178:81–8. [DOI] [PubMed] [Google Scholar]
- 36. Wang Z, Yang K, Zheng Q. et al. Divergent changes of p53 in pulmonary arterial endothelial and smooth muscle cells involved in the development of pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2019;316:L216–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nemani N, Dong Z, Daw CC. et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci Signal 2020;13:eaaz6206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Duchon A, Herault Y. DYRK1A, a dosage-sensitive gene involved in neurodevelopmental disorders, is a target for drug development in Down syndrome. Front Behav Neurosci 2016;10:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ameshima S, Golpon H, Cool CD. et al. Peroxisome proliferator-activated receptor gamma (PPARgamma) expression is decreased in pulmonary hypertension and affects endothelial cell growth. Circ Res 2003;92:1162–9. [DOI] [PubMed] [Google Scholar]
- 40. Hansmann G, Wagner RA, Schellong S. et al. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation 2007;115:1275–84. [DOI] [PubMed] [Google Scholar]
- 41. Banno K, Omori S, Hirata K. et al. Systematic cellular disease models reveal synergistic interaction of trisomy 21 and GATA1 mutations in hematopoietic abnormalities. Cell Rep 2016;15:1228–41. [DOI] [PubMed] [Google Scholar]
- 42. Liang C-C, Park AY, Guan J-L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007;2:329–33. [DOI] [PubMed] [Google Scholar]
- 43. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc 2010;5:628–35. [DOI] [PubMed] [Google Scholar]
- 44. Tsuru H, Ishida H, Narita J. et al. Cardiac fibroblasts play pathogenic roles in idiopathic restrictive cardiomyopathy. Circ J 2021;85:677–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing raw fastq files, gene by cell transcript counts matrix, and metadata files were available in Gene Expression Omnibus database (GSE203257).