

Abstract
The cyanobacterial radiation consists of several lineages of phyletically (morphologically and genetically) related organisms. Several of these organisms show a striking resemblance to fossil counterparts. To investigate the molecular mechanisms responsible for stabilizing or homogenizing cyanobacterial characters, we compared the evolutionary rates and phylogenetic origins of the small-subunit rRNA-encoding DNA (16S rDNA), the conserved gene rbcL (encoding d-ribulose 1,5-bisphosphate carboxylase-oxygenase large subunit), and the less conserved gene rbcX. This survey includes four categories of phyletically related organisms: 16 strains of Microcystis, 6 strains of Tychonema, 10 strains of Planktothrix, and 12 strains of Nostoc. Both rbcL and rbcX can be regarded as neutrally evolving genes, with 95 to 100% and 50 to 80% synonymous nucleotide substitutions, respectively. There is generally low sequence divergence within the Microcystis, Tychonema, and Planktothrix categories both for rbcLX and 16S rDNA. The Nostoc category, on the other hand, consists of three genetically clustered lineages for these loci. The 16S rDNA and rbcLX phylogenies are not congruent for strains within the clustered groups. Furthermore, analysis of the phyletic structure for rbcLX indicates recombinational events between the informative sites within this locus. Thus, our results are best explained by a model involving both intergenic and intragenic recombinations. This evolutionary model explains the DNA sequence clustering for the modern species as a result of sequence homogenization (concerted evolution) caused by exchange of genetic material for neutrally evolving genes. The morphological clustering, on the other hand, is explained by structural and functional stability of these characters. We also suggest that exchange of genetic material for neutrally evolving genes may explain the apparent stability of cyanobacterial morphological characters, perhaps over billions of years.
The current species diversity of the cyanobacterial radiation comprises several lineages of phyletically (morphologically and genetically) related organisms (26). An intriguing question is whether this reflects stability of cyanobacterial characters or whether the phyletic similarities originate from relatively recent common ancestors. Analyses of precambrian microfossils (superficially, hardly distinguishable from recent cyanobacteria) support the view of retention of cyanobacterial properties (1, 11, 28). However, on the basis of molecular data, a 2-billion-year-old mutual ancestor for prokaryotes has been suggested (5), implying that the similarities between the earliest records of cyanobacteria and present-day species do not reflect homologies but rather indicate analogies. In this context, the phyletically clustered groups may reflect a relatively recent divergence of the modern species.
In this work we have addressed, by molecular evolutionary studies, the mechanisms responsible for conserving or homogenizing phyletical characters within groups of cyanobacteria. We investigated the evolutionary rates and origins for two genomic regions, by analyzing strains both within and among groups of phyletically related organisms. This was done by comparative analysis of the small-subunit rRNA-encoding DNA (16S rDNA), which is conserved by the RNA function (37), and the rbcLX region with both conserved and less conserved elements. The rbcLX region contains an intergenic spacer (with no identified functional units), the gene rbcX with a possible chaperonin-like function (18), and the 3′ end of rbcL (encoding the highly conserved d-ribulose 1,5-bisphosphate carboxylase-oxygenase large subunit [LSU]) (23). We analyzed a data set consisting of four phyletically clustered cyanobacterial strain categories, as inferred from microscopic observations and 16S rDNA analysis (26, 31). The data set includes the Microcystis category (16 strains), consisting of unicellular organisms, the Tychonema (6 strains) and Planktothrix (10 strains) categories, which contain multicellular, filamentous organisms, and the Nostoc category (12 strains), which includes both morphologically and genetically slightly divergent organisms (26, 34). The strains in this last category share among other features the ability of cellular differentiation to produce heterocysts with nitrogenase activity.
Our sequence data suggest an evolutionary model involving several events of gene transfer between phyletically closely related organisms but not between less related organisms. We propose that this gene transfer has led to the observed sequence homogeneity for the groups of related organisms and that exchange of genetic material stabilizes the function and structure of proteins encoded by neutrally evolving genes. Our gene transfer model may explain the similarity between the fossil and the recent species.
MATERIALS AND METHODS
Sample preparation.
The organisms investigated are listed in Table 1. The majority of the strains were isolated at the Norwegian Institute for Water Research (33). The strains were grown in laboratory cultures as previously described by Rudi et al. (26).
TABLE 1.
Strains of cyanobacteria used in this study and data bank accession numbers for 16S rDNA and rbcLX sequences
| Taxon and strain designationa | Geographic origin | EMBL accession no.
|
|
|---|---|---|---|
| 16S rDNA | rbcLX | ||
| Microcystis category | |||
| Microcystis aeruginosa N-C 31 | Little Rideau Lake, Canada [strain NRC-1(ss-17)] | y12604 | z94894 |
| M. aeruginosa N-C 43 | Wisconsin (strain ATCC 22663) | z82784 | z94906 |
| M. aeruginosa N-C 57 | Lake Frøylandsvatnet, Norway | z82785 | z94907 |
| M. aeruginosa N-C 143 | Lake Akersvatnet, Norway | z82786 | z94909 |
| M. aeruginosa N-C 228/1 | Lake Akersvatnet, Norway | z82783 | z94908 |
| M. aeruginosa N-C 123/1 | Lake Mälaren, Sweden | y12605 | z94895 |
| M. aeruginosa N-C 166 | Lake Hellesjøvatnet, Norway | y12606 | z94896 |
| M. cf. aeruginosa N-C 118/2 | Lake Gjersjøen, Norway | y12607 | z94897 |
| M. botrys N-C 161/1 | Lake Mosvatnet, Norway | y12608 | z94898 |
| M. botrys N-C 264 | Lake Frøylandsvatnet, Norway | y12609 | z94899 |
| M. cf. flos-aquae N-C 144 | Lake Borrevatnet, Norway | y12610 | z94900 |
| M. cf. ichthyoblabe N-C 279 | Lake Østensjøvatnet, Norway | y12611 | z94901 |
| M. viridis N-C 122/2 | Lake Finjasjön, Sweden | y12612 | z94902 |
| M. viridis N-C 169/7 | Lake Arresø, Denmark | y12613 | z94903 |
| M. cf. wesenbergii N-C 172/5 | Lake Arresø, Denmark | y12614 | z94904 |
| Microcystis sp. strain N-C 324/1 | Lake Tøråssjøen, Norway | z82808 | z94905 |
| Planktothrix category | |||
| Planktothrix agardhii N-C 29 | Lake Gjersjøen, Norway | z82796 | z94866 |
| P. agardhii N-C 299 | Lake Kalvsjøtjern, Norway | z82799 | z94873 |
| P. agardhii N-C 116 | Lake Årungen, Norway | y12676 | z94867 |
| P. mougeotii N-C 56/1 | Lake Steinsfjorden, Norway | y12677 | z94868 |
| P. mougeotii N-C 11 | Lake Akersvatnet, Norway | z82795 | z94874 |
| P. mougeotii N-C 65 | Lake Vansjø, Norway | y12678 | z94869 |
| P. prolifica N-C 18 | Lake Gjersjøen, Norway | y12679 | z94870 |
| P. prolifica N-C 320 | Lake Kolbotnvatnet, Norway | z82798 | z94875 |
| P. rubescens N-C 1 | Lake Zürich, Switzerland | y12680 | z94871 |
| P. cf. rubescens N-C 55 | Lake Steinsfjorden, Norway | y12681 | z94872 |
| Tychonema category | |||
| Tychonema bourrellyi N-C 33/1 | Lake Mjøsa, Norway | y12682 | z94876 |
| T. bourrellyi N-C 33/6 | Lake Mjøsa, Norway | y12683 | z94877 |
| T. bourrellyi N-C 58 | Lake Mjøsa, Norway | y12684 | z94878 |
| T. bourrellyi N-C 70 | River Glåma, Norway | y12685 | z94879 |
| T. bourrellyi N-C 261/1 | River Glåma, Norway | z82791 | z94881 |
| T. bornetii N-C 60 | Lake Mjøsa, Norway | y12686 | z94880 |
| Nostoc category | |||
| Anabaena sp. strain N-C 267/4 | Lake Fammestadtjønni, Norway | z82802 | z94882 |
| A. lemmermannii N-C 281/1 | Lake Storavatnet, Norway | z82797 | z94883 |
| A. lemmermannii N-C 83/1 | Lake Edlandsvatnet, Norway | z82801 | z94884 |
| A. lemmermannii N-C 266/1 | Lake Bergevatnet, Norway | z82800 | z94885 |
| Aphanizomenon gracile N-C 103 | Pond, Vingrom, Norway | z82806 | z94886 |
| A. flos-aquae N-C 142 | Buffalo Pond Lake, Canada (strain NRC-566) | z82809 | z94887 |
| Nostoc sp. strain N-C 246 | United States (strain PCC 7120) | z82803 | z94888 |
| Nostoc sp. strain N-C 124 | Lake Steinsfjorden, Norway | z82776 | z94889 |
| Nostoc sp. strain N-C 194 | Dronning Maud Land, Antarctica | z82805 | z94890 |
| Nostoc sp. strain N-C 308 | Spitsbergen, Svalbard | z82804 | z94891 |
| Nostoc commune | China | y12687 | z94892 |
| N. flagelliforme | China | y12688 | z94893 |
N-C, NIVA-CYA. The strains of N. commune and N. flagelliforme originating from China (Zebo Huang, Institute of Hydrobiology Chinese Academy of Science, Wuhan) are being entered in the NIVA Culture Collection of Algae (32).
The investigated 16S rDNA and rbcLX regions are shown in Fig. 1. To generate sequence data, genomic DNA was isolated, and the rDNA region was amplified and sequenced as described by Rudi et al. (25, 26). The rbcLX region was amplified with the primers CW (5′CGTAGCTTCCGGTGGTATCCACGT3′) and CX (5′GGGGCAGGTAAGAAAGGGTTTCGTA3′). For the Planktothrix category, CW and DF (5′GGGCARYTTCCACAKNGTCCA3′) were used. The PCR program used consisted of an initial denaturation step at 94°C for 4 min and then cycling with the following parameters: 94°C for 30 s, 40°C for 30 s, and 72°C for 2 min for 2 cycles, then 94°C for 30 s, 55°C for 30 s, and 72°C for 2 min for 38 cycles, ending with an extension step for 7 min. In addition to the PCR primers, the internal sequencing primer DN (5′TTGAAGCAATGGATACCGTCTGA3′) was used for the Nostoc category, and primer DL (5′TTGGATTGTGGGTCAGACTTG3′) was used for the Planktothrix category.
FIG. 1.

Structures of the 16S rDNA (A) and rbcLX (B) regions. The primers used for PCR amplification and sequencing are indicated with arrows. Two of the strains (Nostoc sp. strain NIVA-CYA 308 and N. commune) contain an approximately 155-nt insertion between rbcX and rbcS.
Phylogenetic reconstruction.
The sequences were aligned either manually or by using the multisequence alignment algorithm PILEUP (14) in the Wisconsin package version 8.1 for UNIX (Genetic Computer Group, Madison, Wis.). Phylogenetic trees were constructed with the neighbor-joining method (27) or based on parsimony analysis (9), using the Phylogeny Inference Package (PHYLIP; version 3.5) developed by J. Felsenstein (Department of Genetics, University of Washington, Seattle), and the package Phylogenetic Analysis Using Parsimony (PAUP; version 3.1.1) developed by D. L. Swofford (Illinois Natural History Survey, Champaign). The Kimura two-parameter model with a transversion:transition weight of 2:1 (16) was applied to compute the nucleotide distance matrix, and Dayhoff’s PAM 001 matrix (4) was used for calculating the amino acid distance matrix for the neighbor-joining analysis. Bootstrap analysis (8) constituted the basis for inferring confidence of the branch points in the phylogenetic trees. Consensus trees were constructed from 500 bootstrap replicates.
Finally, we tested whether two or more tree topologies generated from different data sets for the same taxa were significantly different. This analysis was done with the Templeton-Felsenstein test (36) for user-defined trees, implemented in the DNAPARS program of the PHYLIP package.
Phyletic structure of the nucleotide alignments.
The tree length distribution based on the informative sites for the phylogenetic trees was analyzed by use of either the exhaustive (for 11 taxa or less) or the random tree (for more than 11 taxa) option in the PAUP package. We used the third moment statistics (g1) implemented in this package to determine the randomization value of the informative sites. This value was used to evaluate the phyletic information, according to the criteria given by Hillis and Huelsenbeck (15).
Models for estimating nucleotide substitution distributions.
For nucleotide alignments with randomized phyletic structures, the observed frequency distributions of nucleotide substitutions in the alignments were compared to the expected distribution from alignments randomized by independent substitutions and to the distribution given that each of the informative sites in the alignment is due to single substitutions (phyletically informative substitutions).
The frequency of nucleotide substitutions in the model based on mutationally randomized data was calculated assuming a star-like phylogenetic structure: p = m/(n × s), where p is mutational frequency, m is nucleotide substitutions (i.e., sites differing from the 50% majority rule consensus sequence for each subgroup of taxa) for each of the individual sequences in the alignment, n is the number of taxa, and s is neutral positions in the alignment (i.e., third codon position and intergenic spacers). The distribution of sites in the alignment for which a given number of taxa have nucleotide substitutions was then calculated by using the binomial probability distribution P(x) = {n!/[(n − x)! × x!]} × [px × (1 − p)n − x] for 1 ≤ x ≤ n/2, where n is the number of taxa, x is the number of taxa with nucleotide substitutions, and P is the probability for this number of taxa with nucleotide substitutions.
For the scenario where each polymorphic site has arisen once from a single (phyletically informative) substitution, we also assume that the current genotypes have arisen from a common ancestor in an expanding population. In this model, the substitution frequencies are given by p = u/s, where u is the number of polymorphic positions in the alignment and s is the number of neutral positions in the alignment. We assume that each of the periods which have resulted in x = 1 up to x ≤ n/2 nucleotide differences per position in the alignment has occupied the same time span during evolution (n is the total number of taxa and x is number of taxa with nucleotide substitutions). Furthermore, we assume that there were (n/x) equally distributed taxa for each of these periods. The frequency distribution per position in the alignment for a given number of taxa with nucleotide is thus inversely proportional to the number of substitutions. Since the integral of the probability density function over the sample space must be 1, a parameter was used to correct the formula. The following probability density function was used in the calculations:
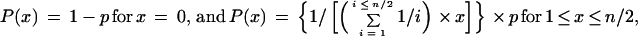 |
where P is the probability for this number of taxa with nucleotide substitutions.
RESULTS
We have sequenced 16S rDNA and rbcLX regions from a total of 44 strains belonging to the cyanobacterial categories Microcystis, Planktothrix, Tychonema, and Nostoc (Fig. 1). This sequence information was used to construct phylogenetic trees and for the comparison of the evolutionary patterns of the regions analyzed.
16S rDNA.
In the 16S rDNA region (Fig. 1A), the sequence divergence between species belonging to different categories varies from 0.1 to 0.15 nucleotide substitutions per position. For the most divergent strains in the Nostoc category, the sequence divergence is about 0.05 nucleotide substitutions per position. This category, however, consists of three genetically distinct lineages—Nostoc lineage I, II, and III. Strains NIVA-CYA 142, 266/1, 83/1, 103, 267/4, 281/1 belong to Nostoc lineage I, strains Nostoc commune, Nostoc flagelliforme, and NIVA-CYA 124, 194, and 308 belong to Nostoc lineage II, and strain NIVA-CYA 246 belongs to Nostoc lineage III. There are only 0.005 nucleotide substitutions per position among the most divergent strains within both the Microcystis and the Planktothrix category, while the strains in the Tychonema category are all monomorphic.
rbcL and rbcX genes.
The investigated strains show an overall homogeneous structure of the rbcLX region (Fig. 1B). The rbcL gene is relatively conserved (≤0.2 nucleotide substitutions per position), while the intergenic region as well as the gene rbcX are variable, with ≥0.3 nucleotide substitutions per position when species belonging to different categories are compared. There are no significant differences in codon usage for the strains within each clustered group. The sequence divergence for the rbcL and the rbcX gene is only 0.002 nucleotide substitutions per position within the Tychonema and Planktothrix categories. There are about 0.01 nucleotide substitutions per position between the most divergent isolates in the Microcystis category. The Nostoc category shows the same three genetically clustered lineages as for 16S rDNA, with approximately 0.05 nucleotide substitutions per position for the most divergent strains within each lineage. On the other hand, for species belonging to different lineages, the sequence divergence is on average 0.1 and 0.2 nucleotide substitutions per position for rbcL and rbcX, respectively.
Notably, a strain isolated from Svalbard (Nostoc sp. strain NIVA-CYA 308) contains a 154-nucleotide (nt) insertion in the rbcLX region, which is highly similar to a 157-nt insertion found in a strain from China (N. commune) (Fig. 1B). The self-splicing ability of this insertion was investigated by reverse transcriptase PCR. No amplification products corresponding to a spliced RNA were visible on ethidium bromide-stained agarose gels (results not shown).
Both the rbcL and rbcX genes are subjected to purifying selection, with 95 to 100% and 50 to 80% synonymous nucleotide substitutions for rbcL and rbcX, respectively. There are no significant differences in the percentages of synonymous relative to replacement mutations when strains both within and between the phyletically related groups are compared.
Evolutionary rates for the 16S rDNA and rbcLX regions.
The evolutionary rates for the 16S rDNA and rbcLX regions were calculated within and between Nostoc lineages I and II. Within Nostoc lineages I and II, the sequence divergence in the rbcX region is only 2 to 2.5 times that of the 16S rDNA divergence. However, the sequence divergence between lineages I and II displays a 4.5- to 35-fold-higher difference in the rbcX region compared to 16S rDNA (Fig. 2). The spacer between rbcL and rbcX for Nostoc lineage I and II has a variation in length from 70 to 132 bp. Within the lineages, the sequence divergence is in the same range as for rbcX, both for the spacer and for the rbcL gene, while between the lineages the divergence is on average 1.5 and 0.3 times the variation in rbcX for the spacer and rbcL, respectively (results not shown). For the rbcL gene, only 0 to 5% of the mutations are replacement substitutions. This indicates that most of the replacement substitutions are eliminated by selection. The rbcX gene, with 20 to 50% replacement substitutions, is also constrained by function, but more mutations are tolerated here. The spacer region, however, displaying the highest evolutionary rate, has probably a low functional constraint.
FIG. 2.

rbcX (A) and rDNA (B) distance trees for Nostoc lineages I, II, and III. The trees were built with the neighbor-joining method (27) from the PHYLIP package, using distance matrices derived from the Kimura two-parameter model (16). Numbers at the nodes indicate the percentage of 500 bootstrap trees (8) in which the cluster descending from the node was found. Abbreviations for strains used: AL 1, Anabaena lemmermannii NIVA-CYA 266/1; AL 2, A. lemmermannii NIVA-CYA 83/1; AL 3, A. lemmermannii NIVA-CYA 281/1; AS, Anabaena sp. strain NIVA-CYA 267/4; AF, Aphanizonenon flos-aquae NIVA-CYA 142; AG, Aphanizomenon gracile NIVA-CYA 103; NS 1, Nostoc sp. strain NIVA-CYA 124; NS 2, Nostoc sp. strain NIVA-CYA 194; NS 3, Nostoc sp. strain NIVA-CYA 308; NS 4, Nostoc sp. strain NIVA-CYA 246; NC, N. commune; NF, N. flagelliforme.
Comparison of the 16S rDNA and rbcLX phylogenies for the genetically distinct clusters of strains.
For this comparison, we used the translated rbcL and rbcX sequences, because the amino acid sequence displays suitable polymorphism for phyletic reconstruction (and the nucleotide sequence has too high variation). Comparison of the phylogeny based on 16S rDNA and the translated (amino acid) sequence of rbcX for the six genetically distinct clusters of strains defined in this work shows the same clustering of strains (Fig. 3).
FIG. 3.

Comparison of distance trees for the rbcX amino acid sequence (A) and 16S rDNA nucleotide sequence (B) for the six evolutionary distinct clusters described in this work. The strains used in the phylogenetic analysis were selected to include all of the genotypes. The distance matrix for the amino acid sequences was calculated by using Dayhoff’s PAM 001 matrix (4), while the 16S rDNA distance matrix was calculated with the Kimura two-parameter model (16). The trees were built by using the neighbor-joining algorithm (27) from the PHYLIP package. The numbering at nodes indicates the percentage of that branch in 500 bootstrap trees (8).
There are, however, some discrepancies in the two trees for the branches leading to Nostoc lineages I, II, and III and for the branches leading to Planktothrix and Tychonema. For the branch leading to Nostoc lineage II in the rbcX amino acid tree, this difference can be due to low statistical confidence, as indicated by the low bootstrap support. The branches leading to Planktothrix and Tychonema receive relatively high bootstrap support in both the rbcX amino acid tree and the 16S rDNA tree. However, the branching pattern for these categories is not stable in the rbcX amino acid tree and is dependent on the trains considered in the phylogenetic analysis (results not shown).
The rbcL amino acid tree is generally congruent with the rbcX amino acid tree. The only differences are the branches leading to Nostoc lineages I, II, and III, where the rbcL amino acid tree is congruent with the 16S rDNA tree (results not shown).
Comparison of the 16S rDNA and rbcLX phylogenies for the genetically clustered strains.
There is low accordance, using both the neighbor-joining method (Fig. 2) and the maximum parsimony analysis (Fig. 4), between the 16S rDNA phylogeny and the rbcLX phylogeny for isolates within Nostoc lineages I and II. The only Nostoc strains which are clustered in both the 16S rDNA and rbcLX trees are Anabaena flos-aquae NIVA-CYA 83/1 (AL 2) and 266/1 (AL 1) (Fig. 2 and 4). The clustering of organisms is highly congruent when results from the two phylogenetic methods are compared (compare Fig. 2 and 4). This is exemplified by the strains Aphanizomenon flos-aquae NIVA-CYA 142 (AF) and Aphanizomenon gracile NIVA-CYA 103 (AG), for which 16S rDNAs are grouped with 84% bootstrap support by the neighbor-joining method (Fig. 2B) and with 96% bootstrap support by the parsimony analysis (90% identity for the informative sites [Fig. 2B and 4B]). In contrast to rDNA, the rbcLX locus reveals a phylogeny where the strains are relatively distantly related (43% identity for the informative sites [Fig. 2A and 4A]). This suggests that the rDNA and rbcLX loci do not have the same evolutionary pattern. The only difference between the parsimony and neighbor-joining analyses is the location of Nostoc sp. strain NIVA-CYA 194 (NS 2) for the rbcLX region (Fig. 2A and 4A). However, when only the strains in Nostoc lineage II are considered in the neighbor-joining analysis, the branching pattern is congruent with that of the parsimony analysis (results not shown).
FIG. 4.

Maximum parsimony tree for Nostoc lineages I and II, displaying the rbcLX locus (A) and the 16S rDNA locus (B). Congruent trees were built both with the DNAPARS program from the software package PHYLIP and by the exhaustive tree search in the software package PAUP (9). The evolutionary distances between the strains are not shown. Informative sites used in the reconstruction are shown for each branch. The numbering at the nodes indicate the percentage of 500 bootstrap trees in which the cluster descending from the node was found with the DNAPARS program. The strict consensus are shown for all trees, except the 16S rDNA tree for Nostoc lineage I, where the majority rule tree is shown. Strain abbreviations are as for Fig. 2.
The Templeton-Felsenstein test (36) implemented in the DNAPARS program in the PHYLIP software package was used to test whether the rbcLX and 16S rDNA tree topologies are significantly different. In this analysis, we assumed that the 16S rDNA topologies correlate with the true phylogeny. The shortest trees generated with both the rbcLX and 16S rDNA data were used as user-defined trees on the rbcLX data set. For Nostoc lineage I, the rbcLX and 16S rDNA topologies were significantly different (variance in step differences as determined by the step differences at individual positions [Its SD] = 5.20. However, for Nostoc lineage II the topological differences were not significant (Its SD = 1.73).
Although not strongly supported statistically, strains in the Microcystis category can also have different evolutionary patterns for 16S rDNA and rbcLX. Strains NIVA-CYA 118/2, 169/7, 161/1, 144, 264, and 324/1 have T in position 234 relative to the published 16S rDNA sequence for Microcystis aeruginosa NIVA-CYA 43, while the other strains have C at this position. Based on informative sites in the rbcLX region, two phyletic clusters could be identified. These clusters are composed of strains NIVA-CYA 264, 324/1 and 161/1 and strains NIVA-CYA 57 and 144, respectively (Fig. 5).
FIG. 5.

Strict consensus maximum parsimony tree for the Microcystis group, displaying the rbcLX locus. Congruent trees were built with the DNAPARS program from the software package PHYLIP and by the branch and bound tree search in the software package PAUP (9). The evolutionary distances between the strains are not indicated. Informative sites used in the reconstruction are shown for each branch. The numbering at the nodes indicate the percentage of 500 bootstrap trees in which the cluster descending from the node was found with the DNAPARS program.
There are no informative sites for the Planktothrix category for 16S rDNA. There are, however, two phyletic clusters of sequences for the rbcLX locus (supported by five of six informative sites). Planktothrix strains NIVA-CYA 29, 299, 56/1, 11, and 65 have the informative sites CTTTA, while strains NIVA-CYA 116, 18, 320, 1 and 55 have the informative sites TCGCG. There are no informative sites in either rbcLX or 16S rDNA for the Tychonema category.
Phyletic structure of the 16S rDNA and rbcLX nucleotide alignments.
Using the exhaustive and the random tree search in the program package PAUP, we determined the tree length distribution of the data. The exhaustive tree search was used for data consisting of 11 or fewer taxa, while the random tree search with 100,000 replicates was used for more than 11 taxa.
The third momentum statistics of the three lengths for each set of trees were calculated, and phyletic structure was evaluated according to the criteria given by Hillis and Huelsenbeck (15). Both for Nostoc lineage I and Nostoc lineage II, the 16S rDNA data have a significant phyletic structure (Table 2). The situation, however, is different for the rbcLX locus. The Planktothrix data do not have significant phyletic structure, because only one branch in the phylogenetic tree is supported by the five informative sites (Table 2). There is some phyletic structure for Microcystis, i.e., for informative sites 1, 2, 6, 7, and 15 shown in Fig. 5. However, when strains NIVA-CYA 57, 144, 264, 324/1, and 161/1 are omitted in the phylogenetic analysis (Fig. 5), the phyletic structure is lost (Table 2). Finally, the analysis shows that there are no phyletic structures in the rbcLX locus for Nostoc lineages I and II (Table 2).
TABLE 2.
Phyletic information in the 16S rDNA and rbcLX alignments
| Category | No. of taxa | Locus | No. of informa- tive sites | g1a | Phyletic informa- tionb |
|---|---|---|---|---|---|
| Nostoc lineage I | 6 | 16S rDNA | 10 | −0.914 | Yes |
| Nostoc lineage II | 5 | 16S rDNA | 7 | −1.50 | Yes |
| Nostoc lineage I | 6 | rbcLX | 14 | −0.479 | No |
| Nostoc lineage II | 5 | rbcLX | 12 | −0.592 | No |
| Microcystis | 16 | rbcLX | 15 | −0.761 | Yes |
| Microcystisc | 11 | rbcLX | 9 | −0.242 | No |
| Planktothrix | 10 | rbcLX | 6 | −0.286 | No |
Measurement of the skewness of a distribution; defined as the third central moment divided by the cube of the standard deviation.
Determined from the critical values given by Hillis and Huelsenbeck (15). For informative sites with phylogenetic information, P < 0.01; for randomized informative sites, P > 0.05. Usually ≤10% phylogenetic information is sufficient to skew the distribution beyond the 95% confidence limits of the random distribution.
Strain NIVA-CYA 57, 144, 264, 324/1, and 161/1 were not considered in this analysis (see Fig. 5).
Nucleotide substitution distribution for the rbcLX locus.
The observed distribution of nucleotide substitutions for the rbcLX locus was compared to the distribution expected for random independent substitutions and to the expected distribution for phyletically informative substitutions (see Materials and Methods). As inferred from Fig. 6, the observed distributions resemble the distribution based on the assumption of phyletically informative substitutions. Thus, the inference from this is that each separate informative site is phyletically informative (from the definition of a single substitution), while the combination of these sites in phylogenetic reconstruction resembles randomly generated data (Table 2).
FIG. 6.

Analysis of the frequency distribution of substitutions in the rbcLX alignment for Nostoc lineage I (A), Planktothrix (B), Microcystis (C), and Nostoc lineage II (D). The frequency distributions of positions in the alignments, where the number of taxa with 1 up to ≤n/2 nucleotide differences from the consensus sequence for each subgroup of taxa, were determined for a model based on independent substitutions (black bars), phyletically informative substitutions (gray bars), and observed distribution (white bars). These distributions were calculated as described in Materials and Methods.
In Nostoc lineage II, the distribution of informative sites fits a distribution consisting of blocs of sequences with different evolutionary affiliations (Fig. 7). This is particularly exemplified by the distribution of informative sites in the comparison of strains AL 1 and AL 2. All first 4 informative sites are different, while the next 10 informative sites are identical. Assuming that these two sequences have diverged by four random substitutions, the probability P (d0 ≤ d) = (10 × 10! × 4!)/14! = 0.00999, where d is the observed distance between the substitutions, and d0 is the distance expected for the substitutions from a random data. A mosaic data structure is also suggested from the distribution of the six informative sites for the Planktothrix data. Four of these sites are located within 80 of the 3′ nucleotides in the 990-nt alignment. We could, however, not easily identify such mosaic structures in the Microcystis or the Nostoc lineage II data (Fig. 4A and 5).
FIG. 7.

Mosaic structure of the informative sites in the rbcLX alignment for Nostoc lineage I. Blocks of sequences with similar phyletic affiliations are boxed. Informative sites 1 and 2 are in the rbcL region, while the other informative sites are in the rbcX region. Strain abbreviations are as for Fig. 2.
DISCUSSION
Data structure of the rbcLX locus.
Assuming that the sites in the spacer between the rbcL and rbcX genes and the third codon position in protein coding genes are neutral, we estimated that there are less than 13.8% polymorphic neutral sites in the rbcLX region for the clustered groups. Based on these observations, we find it unlikely that our data are subjected to errors caused by mutational saturation, which would require ≥75% probability of nucleotide change (15). The randomization values for the rbcLX locus (Table 2) are quite intriguing because only ≤10% phylogenetic signal is usually sufficient to skew the tree length distribution beyond the 95% confidence limit of the random distribution (15).
It can be argued that the above estimate of neutral sites is an overestimate and that there are only a few hypervariable sites in our alignments. But since it is not the same positions that are variable and informative in the alignments for the different groups, we exclude this possibility. Another possible explanation for the randomization of the informative sites in the alignments is that the rbcLX gene region has branched at a distinct point during evolution, and the informative sites therefore are caused by two or more independent substitutions (although the sequences are not saturated by mutations). A consequence of such a model is that the clustering of organisms seen for the rbcLX trees in Fig. 2A and 4A does not reflect the true phylogeny. Nevertheless, the conclusion drawn from the Nostoc data, according to this model, is that the evolutionary pattern for rbcLX is different from that for 16S rDNA. However, the multiple independent substitution model would imply a random distribution of substitution for the neutral nucleotide positions in the alignment, which is clearly not observed (Fig. 6). Thus, the low phyletic signal, according to the criteria given by Hillis and Huelsenbeck (15) of the informative sites in the alignments, cannot be explained by multiple independent substitutions.
Our results suggest that each separate informative site in the rbcLX alignment is due to single substitutions (phyletically informative), while the combination of these sites in phylogenetic reconstruction yields a random tree length distribution. This observation can be explained only by recombinational events between the informative sites. Notably, mosaic structures for the informative sites in Nostoc lineage I further support recombinational events within the rbcLX locus (Fig. 7) (6, 21, 35).
The seemingly random tree length distribution for the rbcLX data (Table 2) could be due to the fact that the tree topologies involving all of the informative sites are compositions of different topologies based on subfragments of the data. Although the rbcLX topologies for Nostoc lineages I and II have low phyletic structure, according to tree length distribution criterion, these topologies receive high support from the bootstrap criterion. Furthermore, the rbcLX tree topology is significantly different from the 16S rDNA topology, according to the Templeton-Felsenstein test (36) for Nostoc lineage I. On the other hand, more sequence data and taxa are needed to rigorously resolve the question about the 16S rDNA and rbcLX topologies for Nostoc lineage II. Taken together, however, our results suggest recombinations between the rbcLX and 16S rDNA loci (intergenic recombination) in addition to the previously discussed recombinations within the rbcLX locus (intragenic recombination).
Possible mechanisms for exchange of genetic material.
Transformation is so far the only known biological mechanism for exchange of chromosomal DNA in cyanobacteria (19). For incorporating DNA into the genome, homologous recombination is a ubiquitous mechanism in eubacteria (19). For homologous recombination in Escherichia coli, there is a sharp decline in the number of recombinants with decreasing sequence identity. Compared to the frequencies for a fragment with 100% similarity in a 400-bp region, there were 42- and 300-fold decreases of cointegrate formation for fragments with 90% and 65 to 70% identity, respectively (30). Accordingly, stable exchange of genetic information solitarily within closely related groups of cyanobacteria observed in this work can be explained by homologous recombination frequencies (20).
However, there are also other possible mechanisms for genetic exchange solitarily within genetically clustered groups. These mechanisms involve, for example, conjugation, transducing bacteriophages, simultaneous competence, recognition sequence, restriction modification barriers, or codon usage (19). The exchange of genetic information could also be an intrinsic property of the rbcLX locus and not a general mechanism. The knowledge of the physiology and genetics involved in the process of exchange of genetic material is, however, limited to a few microorganisms mainly of medical interest. Further studies addressing these mechanisms in aquatic organisms and habitats are necessary (19).
Models for explaining the sequence homogeneity within the phyletically clustered groups of organisms.
The most restrictive explanation for the sequence homogeneity within the clustered groups of organisms is that each cluster has evolved relatively recently from their common ancestors. However, recent common ancestors cannot explain the differences in relative evolutionary rates within, as opposed to among, the genetically clustered groups for rbcX compared to 16S rDNA. In Nostoc lineages I and II, the rbcX divergence between, relative to within the clusters, is 3 to 3.5, while for 16S rDNA this ratio is 0.2 to 1.5 (this effect is not as profound for the rbcL locus, because LSU is highly conserved by function). Presupposing a recent common ancestor, one should expect the opposite result of what we observed (i.e., lower ratios for rbcX compared to 16S rDNA). This is because the neutral sites in protein-coding sequences are relatively rapidly saturated by mutations, leading to a slower evolutionary rate for distantly related species than for closely related species (17). rRNA, on the other hand, has a high degree of functional constancy, resulting in a relative clock-like evolutionary rate (i.e., similar rates for closely and distantly related strains) (37).
The high level of neutral mutations compared to replacement mutations both within and among the clustered groups argues against periods of positive selection, e.g., in a speciation process (22). Although periods of positive selection can explain both the resemblance of the fossil and the recent species and the DNA sequence homogeneity within the clusters, these mechanisms cannot be used as an explanation model for the phyletic clustering of the modern species.
Concerted evolution, however, can be used as a possible model for the phyletic clustering of the modern species. Furthermore, concerted evolution can be explained by the genetic exchange within the clustered groups, as inferred from our data. The high degree of sequence similarity for strains within, as opposed to between, the groups for the rbcX gene is according to this model caused by a relatively higher frequency of transfer of genetic material for this locus compared to 16S rDNA. This argument is also supported by the fact that 16S rRNA is encoded by multicopy genes and that it interacts in a large macromolecular assembly. rRNA is probably more resistant to gene exchange than assemblies which are encoded in a single operon, such as the rbcLXS operon.
Macroevolution of cyanobacterial characters.
In the fossil records, stasis—evolutionary lineages that persist for long periods without change of macroevolutionary (morphological) characters—is common. This led to the theory about punctuated equilibrium, stating that macroevolutionary characters are stable for long time periods, interrupted by short periods of their abrupt changes (12). However, for molecular characters (amino and nucleic acid sequences), the comparison between the time of divergence and the molecular divergence for different species gave rise to the neutral theory of molecular evolution (17). This theory, in contrast, states that the evolution of molecular characters is gradual and clock-like. Although not currently widely accepted, a view of macroevolution—phyletic gradualism—is that morphological characters also can evolve in a gradual fashion (29).
Fossil specimens of cyanobacteria are in many cases rather well preserved, providing similar characters used for the microscopical identification of living species. Of particular interest are organisms with distinct morphological features, such as some species in the taxonomic group Entophysalidaceae. Golubic and Hofmann (11) compared a 2.2-billion-year-old specimen with two modern Entophysalis species. They showed that the fossil and the modern species are morphologically comparable (in cell shape and in the form and arrangement of originally mucilaginous cellular envelopes). Both the fossil and the current species form microtexturally similar stromatolitic structures in comparable intertidal to shallow marine environmental settings. Taken together, the data provide relevant evidence that the fossil cyanobacteria were homologs and not analogs to the recent species.
However, this contradicts the presumed 2-billion-year-old common ancestor of prokaryotes and eukaryotes (5). Nevertheless, this estimate has lately been criticized by several authors as an underestimate (10, 13). Based on our data, we propose an alternative explanation model for this discrepancy. We suggest that only the highly conserved genes used in the estimations could share approximately 2-billion-year-old common ancestors (as discussed below).
Preservation of macroevolutionary characters by exchange of genetic material for neutrally evolving genes.
The work of Stulp (34) has shown that organisms belonging to Nostoc lineage I can occupy different distinct ecological niches, which indicates that they may have diverged by selection for some characters while still exchanging genetic material at the rbcLX locus. Although not thoroughly investigated, this is probably also the case for the other phyletically clustered groups of organisms defined in this work.
Based on a theoretical model, Cohan (2, 3) has deduced that a relatively high level (in prokaryotic terms) of genetic exchange allows differentiation of ecologically selective genes while maintaining ongoing exchange of genetic material for neutrally evolving genes. In this regard, the rbcLX locus examined in this work has the evolutionary pattern expected for a neutral gene locus. Our molecular results show that the divergence between the different bacterial groups (e.g., Nostoc lineages I, II, and III) for the rbcLX locus has mainly been caused by neutral mutations, suggesting homogenization within these groups through neutral gene exchange.
In general, mutations in neutrally evolving genes are most likely to be either indifferent or negative (17). Harmful mutations are removed by selection, while slightly negative mutations may become fixed in the population through linkage with positive mutations or through genetic drift. It is interesting that proteins encoded by neutrally evolving genes can diverge with more than 70% amino acid substitutions and still have a conserved function and three-dimensional structure (24). Thus, there has to exist a strong selection pressure to maintain protein structure and function. The mechanisms responsible for conserving protein structure and function are not likely to be fundamentally different from the mechanisms conserving macroevolutionary characters, because macroevolutionary characters reflect protein function and structure at a higher level of complexity. In terms of neutrally evolving genes, the major obstacle is then to protect the structure and function of the gene products from being lost by fixation of slightly negative mutations in the population. The exchange of genetic material could therefore be a means of protecting what has been invented during billions of years of evolution from being lost by random events. This can contribute to explain the apparent stability of macroevolutionary (morphological) characters, perhaps over billions of years (28).
An interesting interpretation of the results of Doolittle et al. (5), in light of our results, is that the neutral evolving, highly conserved genes used in their calculations shared approximately 2-billion-year-old common ancestors because of exchange of genetic material (see discussion above), while these organisms had diverged at their adaptive genes, giving them different phenotypes. Exchange of genetic material for neutrally evolving genes should be a serious concern for dating prokaryotic divergence by use of molecular clocks, especially clocks calibrated based on eukaryotic species.
Morphological and molecular clustering of modern cyanobacteria.
Finally, we suggest that the morphological clustering of the living cyanobacteria reflects stability through structure and function of relevant characters. The DNA sequence clustering is a result of sequence homogenization and concerted evolution by exchange of genetic material for neutrally evolving genes. Those conclusions are in accordance with both the theory of punctuated equilibrium and the neutral theory of molecular evolution.
ACKNOWLEDGMENTS
This work was part of a research project granted to K.S.J. (grant 107622/420) by the Norwegian Research Council.
We thank Randi Skulberg, Norwegian Institute for Water Research, for excellent work in cultivating the cyanobacteria and Frank Larsen, Dynal A.S., for use of sequencing facilities and laboratory equipment. We are grateful to Zebo Huang, Institute of Hydrobiology, Chinese Academy of Science, Wuhan, for providing the strains of N. commune and N. flagelliforme. We thank William Davies and John G. Ormerod for comments on the manuscript, Per Erik Jorde for help with statistical analysis, and Arne Holst Jensen for help with the phylogenetic analyses. Finally, we are grateful to two anonymous reviewers for helpful comments and suggestions on the manuscript.
REFERENCES
- 1.Castenholz R W. Species usage, concept, and evolution in the cyanobacteria (blue-green algae) J Phycol. 1992;28:737–745. [Google Scholar]
- 2.Cohan F M. The effects of rare but promiscuous genetic exchange on evolutionary divergence in procaryotes. Am Nat. 1994;143:965–986. [Google Scholar]
- 3.Cohan F M. Does recombination constrain neutral divergence among bacterial taxa? Evolution. 1995;49:164–175. doi: 10.1111/j.1558-5646.1995.tb05968.x. [DOI] [PubMed] [Google Scholar]
- 4.Dayhoff M O, Orcutt B C. Methods for identifying proteins by using partial sequences. Proc Natl Acad Sci USA. 1979;76:2170–2174. doi: 10.1073/pnas.76.5.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doolittle R F, Feng D F, Tsang S, Cho G, Little E. Determining divergence times of the major kingdoms of living organisms with a protein clock. Science. 1996;271:470–477. doi: 10.1126/science.271.5248.470. [DOI] [PubMed] [Google Scholar]
- 6.DuBose R F, Dykhuizen D E, Hartl D L. Genetic exchange among natural isolates of bacteria: recombination within the phoA gene of Escherichia coli. Proc Natl Acad Sci USA. 1988;85:7036–7040. doi: 10.1073/pnas.85.18.7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eldredge N, Gould S J. Punctuated equilibria: an alternative to phyletic gradualism. In: Schopf T J M, editor. Models in paleobiology. San Francisco, Calif: Freeman, Cooper, & Co.; 1972. pp. 82–115. [Google Scholar]
- 8.Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- 9.Fitch W M. On the problem of discovering the most parsimonious tree. Am Nat. 1977;111:223–257. [Google Scholar]
- 10.Gogarden J P, Olendzenski L, Hilario E, Simon C, Holsinger K E. Dating the cenancester of organisms. Science. 1996;274:1750–1751. [PubMed] [Google Scholar]
- 11.Golubic S, Hofmann H J. Comparison of Holocene and mid-Precambrian Entophysalidaceae (Cyanophyta) in stromatolitic algal mats: cell division and degradation. J Paleontol. 1976;50:1074–1082. [Google Scholar]
- 12.Gould S J, Eldredge N. Punctuated equilibrium comes of age. Nature. 1993;366:223–227. doi: 10.1038/366223a0. [DOI] [PubMed] [Google Scholar]
- 13.Hasegawa M, Fitch W M. Dating the cenancester of organisms. Science. 1996;274:1750. [PubMed] [Google Scholar]
- 14.Higgins D G, Sharp P M. Fast and sensitive multiple sequence alignments on a microcomputer. CABIOS. 1989;5:151–153. doi: 10.1093/bioinformatics/5.2.151. [DOI] [PubMed] [Google Scholar]
- 15.Hillis D M, Huelsenbeck J P. Signal, noise, and reliability in molecular phylogenetic analyses. J Hered. 1992;83:189–195. doi: 10.1093/oxfordjournals.jhered.a111190. [DOI] [PubMed] [Google Scholar]
- 16.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 17.Kimura M. The neutral theory of molecular evolution. In: Nei M, Koehn R K, editors. Evolution of genes and proteins. Sunderland, Mass: Sinauer Associates; 1983. pp. 208–233. [Google Scholar]
- 18.Li L A, Tabita R. Maximum activity of recombinant ribulose 1,5-bisphosphate carboxylase/oxygenase of Anabaena sp. strain CA requires the product of the rbcX gene. J Bacteriol. 1997;179:3793–3796. doi: 10.1128/jb.179.11.3793-3796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lorenz M G, Wackernagel W. Bacterial gene transfer by natural genetic transformation in the environment. Microbiol Rev. 1994;58:563–602. doi: 10.1128/mr.58.3.563-602.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matic I, Rayssiguier C, Radman M. Interspecies gene exchange in bacteria: the role of SOS and mismatch repair systems in evolution of species. Cell. 1995;80:507–515. doi: 10.1016/0092-8674(95)90501-4. [DOI] [PubMed] [Google Scholar]
- 21.McGuire G, Wright F, Prentice M J. A graphical method for detecting recombination in phylogenetic data sets. Mol Biol Evol. 1997;14:1125–1131. doi: 10.1093/oxfordjournals.molbev.a025722. [DOI] [PubMed] [Google Scholar]
- 22.Messier W, Stewart C B. Episodic adaptive evolution of primate lysozymes. Nature. 1997;385:151–154. doi: 10.1038/385151a0. [DOI] [PubMed] [Google Scholar]
- 23.Pasternak J J, Glick B R. Molecular evolutionary analyses of the small and large subunit of ribulose-1,5-bisphosphate carboxylase/oxygenase. Can J Bot. 1991;70:715–723. [Google Scholar]
- 24.Rost B, Sander C. Bridging the protein sequence-structure gap by structure predictions. Annu Rev Biophys Biomol Struct. 1996;25:113–136. doi: 10.1146/annurev.bb.25.060196.000553. [DOI] [PubMed] [Google Scholar]
- 25.Rudi K, Kroken M, Dahlberg O J, Deggerdal A, Jakobsen K S, Larsen F. Rapid, universal method to isolate PCR-ready DNA using magnetic beads. BioTechniques. 1997;22:506–511. doi: 10.2144/97223rr01. [DOI] [PubMed] [Google Scholar]
- 26.Rudi K, Skulberg O M, Larsen F, Jakobsen K S. Strain characterization and classification of oxyphotobacteria in clone cultures on the basis of 16S rRNA sequences from the variable regions V6, V7, and V8. Appl Environ Microbiol. 1997;63:2593–2599. doi: 10.1128/aem.63.7.2593-2599.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 28.Schopf J W. Are the oldest fossils cyanobacteria? In: Roberts D M, Sharp P, Alderson G, Collins M, editors. Evolution of microbial life. Cambridge, England: Cambridge University Press; 1996. pp. 23–61. [Google Scholar]
- 29.Sheldon P R. Parallel gradualistic evolution of Ordovician trilobites. Nature. 1987;330:561–563. doi: 10.1038/330561a0. [DOI] [PubMed] [Google Scholar]
- 30.Shen P, Huang H V. Homologous recombination in Escherichia coli: dependence on substrate length and homology. Genetics. 1986;112:441–457. doi: 10.1093/genetics/112.3.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Skulberg O M, Carmichael W W, Codd G A, Skulberg R. Taxonomy of toxic Cyanophyceae (cyanobacteria) In: Falconer J R, editor. Algal toxins in seafood and drinking water. London, England: Academic Press Ltd.; 1993. pp. 145–164. [Google Scholar]
- 32.Skulberg, O. M., and R. Skulberg. 1997. Personal communication.
- 33.Skulberg R, Skulberg O M. Forskning med algekulturer.-NIVAs kultursamling av alger. Research with algal cultures.-NIVA’s Culture Collection of Algae. Oslo, Norway: Norsk institutt for vannforskning; 1990. [Google Scholar]
- 34.Stulp B K. Morphological and molecular approaches to the taxonomy of the genus Anabaena (Cyanophyceae, Cyanobacteria). Groningen, Germany: Drukkerij van Denderen B.V.; 1983. [Google Scholar]
- 35.Stephens J C. Statistical methods of DNA sequence analysis: detection of intragenic recombination or gene conversion. Mol Biol Evol. 1985;2:539–556. doi: 10.1093/oxfordjournals.molbev.a040371. [DOI] [PubMed] [Google Scholar]
- 36.Templeton A R. Phylogenetic inference from restriction endonuclease cleavage site maps with particular reference to the evolution of humans and the apes. Evolution. 1983;37:221–244. doi: 10.1111/j.1558-5646.1983.tb05533.x. [DOI] [PubMed] [Google Scholar]
- 37.Woese C R. Bacterial evolution. Microbiol Rev. 1987;51:221–271. doi: 10.1128/mr.51.2.221-271.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]