

Abstract
Ever since the second world symposium on pulmonary hypertension (PH) held in Evian, France, in 1998, PH has been classified into five major clinical groups. Group 5 PH includes a variety of distinct conditions with unclear and/or multifactorial underlying pathologies. Management of these patients is challenging as the number of patients within these groups is often small, not all individuals with certain underlying conditions are affected by PH and patients exhibit distinct symptoms due to different underlying diseases. Studies and clinical trials in these groups are largely lacking and mostly restricted to case series and registry reports. Nonetheless, the worldwide burden of group 5 PH is estimated to be significant in terms of the prevalence of some associated diseases. Group 5 PH encompasses six subgroups, including haematological disorders (inherited and acquired chronic haemolytic anaemia and chronic myeloproliferative disorders), systemic disorders (sarcoidosis, pulmonary Langerhans's cell histiocytosis and neurofibromatosis type 1), metabolic disorders (glycogen storage diseases and Gaucher disease), chronic renal failure with or without haemodialysis, pulmonary tumour thrombotic microangiopathy and fibrosing mediastinitis.
Tweetable abstract
A review of clinical, radiological and pathological features of group 5 PH, highlighting the importance of identifying and treating the underlying disease. Focus on PH in sickle cell disease, sarcoidosis and pulmonary tumour thrombotic microangiopathy. https://bit.ly/47F6HP3
Introduction
Following the second world symposium on pulmonary hypertension (PH) held in Evian, France, in 1998, PH has been classified into five major clinical groups [1]. Although the architecture of this classification was kept over subsequent world symposia and publications of guidelines, it was also continuously adapted in order to reflect the growing knowledge on the underlying aetiologies and pathologies [2–5]. Group 5 PH, characterised by unclear and/or multifactorial mechanisms, summarises several conditions that involve a variety of distinct disorders, as outlined in the revised European Society of Cardiology/European Respiratory Society guidelines for the diagnosis and treatment of PH. Each of these definitions is associated with largely unclear and multifactorial underlying pathologies [5]. The management of these patients is challenging as the number of patients in these groups is often small, not all individuals with certain underlying conditions are affected by PH and patients suffer from distinct symptoms due to different underlying diseases. Studies and clinical trials in these groups are largely lacking and mostly restricted to case series and registry reports, even though the worldwide burden of group 5 PH is estimated to be significant in terms of the prevalence of some associated diseases [5]. Group 5 PH encompasses six subgroups, including haematological disorders (inherited and acquired chronic haemolytic anaemia and chronic myeloproliferative disorders), systemic disorders (sarcoidosis, pulmonary Langerhans's cell histiocytosis and neurofibromatosis type 1), metabolic disorders (glycogen storage diseases and Gaucher disease), chronic renal failure with or without haemodialysis, pulmonary tumour thrombotic microangiopathy (PTTM) and fibrosing mediastinitis (table 1). As the exact underlying mechanisms of PH in these groups are not clear, the treatment should be focused on the underlying disease. Additionally, treatment of potential involved pathomechanisms should be evaluated, such as supplemental oxygen in hypoxaemia and tissue hypoxia, anticoagulation in thrombosis, revascularisation therapy in mechanical pulmonary artery obstructions, anti-inflammatory drug therapies in inflammatory diseases, and heart failure therapies in cardiomyopathies according to guidelines [6].
TABLE 1.
Overview of group 5 pulmonary hypertension with unclear and/or multifactorial mechanisms
| 5.1) Haematological disorders |
| Inherited and acquired chronic haemolytic anaemias: |
| Sickle cell disease |
| β-thalassaemia |
| Spherocytosis |
| Stomatocytosis |
| Autoimmune disorders |
| Chronic myeloproliferative disorders: |
| Chronic myelogenous leukaemia |
| Polycythaemia vera |
| Idiopathic myelofibrosis |
| Essential thrombocytopenia |
| Others |
| 5.2) Systemic disorders |
| Sarcoidosis |
| Pulmonary Langerhans's cell histiocytosis |
| Neurofibromatosis type 1 |
| 5.3) Metabolic disorders |
| Glycogen storage diseases and Gaucher disease |
| 5.4) Chronic renal failure with or without haemodialysis |
| 5.5) Pulmonary tumour thrombotic microangiopathy |
| 5.6) Fibrosing mediastinitis |
The scope of this review is to give the reader a practical insight into potential clinical cases from group 5 PH. Due to the complexity of the group, only three examples were chosen for this clinical–pathological–radiological mini-review.
Methods
This paper was written in light of the recent European Respiratory Society webinar series [7]. Three cases within group 5 PH were chosen to represent the various forms within this group.
Key publications related to clinical, radiological and pathological features of the presented cases were identified.
Case 1 – sarcoidosis
Case 1 part I
A 54-year old woman diagnosed sarcoidosis with cutaneous, pulmonary and nodal manifestation 8 years ago presented with increasing dyspnoea over 2 years. Her past medical history included allergic asthma with concomitant allergic rhinoconjunctivitis, arterial hypertension and hypercholesterinaemia, all of them well controlled with medical treatment. The symptoms at initial presentation 8 years ago were dyspnoea and fatigue. After a diagnostic work-up, sarcoidosis was histologically confirmed in biopsies of the mediastinal lymph nodes. As lung function was preserved, no specific treatment at initial diagnosis had been started. In the further course of the disease, several cutaneous manifestations were diagnosed, which were treated locally by a dermatologist, with the last episode 2 years ago. For 2 years, the patient had experienced progressive dyspnoea at exercise (World Health Organization (WHO) functional class (FC) II) without other symptoms such as chest pain, palpitations or peripheral oedema. Because of new echocardiographic signs of PH, she was referred to our PH clinic at the University Hospital of Zurich in 2016 for further diagnostic work-up and treatment. The regular treatment at the time of presentation consisted of aspirin, losartan, hydrochlorthiazide, rosuvastatin and seretide.
Clinical presentation and baseline diagnostics
Clinical examination showed a body mass index (BMI) of 26.7 kg·m−2, heart rate (HR) 94 bpm, blood pressure (BP) 157/108 mmHg and peripheral oxygen saturation (SpO2) 97% on room air. Physical examination revealed no abnormalities except for one little cutaneous lesion at the left forearm. The ECG showed no pathologies with a normal axis and no signs of right heart hypertrophy. Lung function revealed reduced dynamic volumes (forced expiratory volume in 1 s 1.8 L (65%), forced vital capacity 2.6 L (70%) without a manifest restrictive pattern (total lung capacity 5.1 L (95%)) and a reduced diffusion capacity for CO (DLCO 68%). Blood cell count was normal except for a mild lymphopaenia; renal and hepatic function showed normal values, N-terminal pro-brain natriuretic peptide (NT-proBNP) was in the normal range (84 ng·L−1). Blood gas analysis showed normal oxygenation (PaO2 85 mmHg) without a relevant gas exchange disturbance (normal alveolar–arterial gradient). Echocardiography revealed normal right and left heart function. A slightly elevated right ventricular/right atrial (RV/RA) gradient of 45 mmHg was noted without other indirect signs of PH. Six-minute walking distance was preserved (584 m), but showed a severe exercise-induced desaturation from 97 to 83% at end of exercise (Borg dyspnoea scale 3/10). Regarding sarcoidosis activity parameters, only neopterin was slightly elevated with 3.8 ng·L−1 (normal <2.5 ng·L−1), whereas angiotensin-converting enzyme activity and serum soluble interleukin 2 receptor values were normal.
Radiology
The baseline imaging features were consistent with pulmonary sarcoidosis. Chest radiography showed bilateral peri-hilar fibrosis (figure 1), but the overall lung volumes appeared to be preserved. On computed tomograph pulmonary angiography (CTPA), in addition to the fibrosis, there were ill-defined centrilobular nodules and calcified mediastinal and hilar lymphadenopathy (figure 2a). Over the course of the disease, the patient had also undergone dual-energy CTPA, which elegantly demonstrated large perfusion deficits on the iodine map but no thromboembolic disease. These defects are likely a consequence of pulmonary vascular compression by the lymph-nodal enlargement. The presence of subtle centrilobular ground glass changes amongst the background fibrosis raises the possibility of underlying small vessel vasculopathy. In concordance with the echocardiography, there were no computed tomography (CT) features to suggest PH (figure 2b).
FIGURE 1.
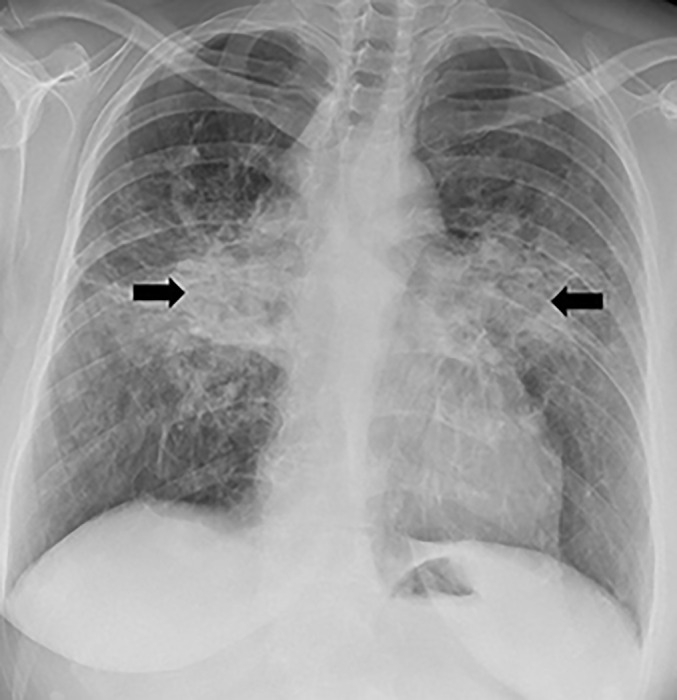
Bilar perihilar fibrosis (arrows).
FIGURE 2.

a) Coronal view computed tomography pulmonary angiography (CTPA) (left panel: lung window; right panel: mediastinal window). There is bilateral upper zone predominant pulmonary fibrosis with ill-defined centrilobular nodules (notched black arrow) and calcified mediastinal and hilar lymph nodes (thin white arrows). The main pulmonary artery (star) was nondilated. b) Axial dual-energy CTPA images demonstrating heterogenous reduction of perfusion in the right lung (white star) on iodine maps (left panel) with markedly calcified bilar hilar lymphadenopathy (thin arrows). There are few centrilobular ground glass opacities (block arrows) in the lower zones (right panel). Of note, the main pulmonary artery (MPA) is not particularly dilated and there is no right-sided cardiac chamber enlargement. RA: right atrium; RV: right ventricle.
Right heart catheterisation
Considering the exertional dyspnoea, the elevated tricuspid regurgitation pressure gradient and the exercise-induced desaturation, right heart catheterisation was performed, which revealed precapillary pulmonary hypertension, as shown in table 2.
TABLE 2.
Case 1 right heart catheterisation
| Right heart catheterisation | At rest | Under exercise (40 Watt) |
| mPAP, mmHg | 32 | 66 |
| Wedge pressure, mmHg | 11 | 8 |
| Cardiac index, L·min−1·m−2 | 2.8 | 5.1 |
| Pulmonary vascular resistance, WU | 3.8 | 5.9 |
| mPAP/CO slope | NA | 5.9 |
mPAP: mean pulmonary arterial pressure; CO: cardiac output; NA: not applicable; WU: Wood unit.
Background – epidemiology and pathophysiology
Sarcoidosis-associated pulmonary hypertension (SAPH) has remained in group 5 of the current clinical classification because of its multifactorial pathogenesis and clinical appearance [5, 8]. It can be caused by different and overlapping mechanisms and its precise origin is still unknown. According to registry data, the incidence of SAPH is 5–20% in sarcoidosis and its prevalence reaches up to 74% in advanced stages listed for lung transplantation [9, 10]. The median time between the initial diagnosis of sarcoidosis and SAPH is 15 years, suggesting that SAPH is a complication of the advanced disease stage [11, 12, 13]. However, interestingly, a correlation between lung function impairment and pulmonary haemodynamics has not yet been shown. There is an important impact on survival with an 8-to-10-fold increase of mortality in SAPH versus sarcoidosis without PH and a 5-year survival rate of 59% [10, 14, 15]. In SAPH, complex interactions may occur between the pulmonary vasculature and the parenchymal, mediastinal and cardiovascular compartments. Therefore, elevation of pulmonary artery pressures can be induced directly by involvement of pulmonary vessels or indirectly by advanced lung parenchymal destruction or compressive mediastinal infiltration. The most common cause of SAPH is pulmonary fibrosis with a related destruction of the vascular bed and alveolar hypoxia with resulting hypoxic pulmonary vasoconstriction contributing to the development of PH. However, up to one fifth of patients with SAPH have no radiographic evidence of interstitial lung disease [16, 17]. In about 20% of SAPH patients, cardiomyopathy caused by left ventricular involvement of sarcoidosis can lead to development of post-capillary PH [10]. Less commonly, isolated pulmonary vasculopathy with granulomatous involvement of the pulmonary arteries and veins can lead to pulmonary arterial hypertension (PAH) or pulmonary veno-occlusive-like disease. In addition, granulomatous mediastinal lymphadenopathy with compression of pulmonary vessels, chronic thromboembolic disease and liver involvement may contribute to PH in sarcoidosis patients.
Pathology
Granulomatous pulmonary vasculopathy may occur in all calibres of pulmonary vessels, but appears particularly compressive/obstructive in smaller calibres with thin vessel walls. Typical sarcoid granulomas are well defined and composed of epithelioid histiocytes with scattered giant cells (Langhans cells) and a peripheral lymphocytic rim. They lack any sign of necrosis, as compared to tuberculosis-type granulomas. Typically, they occur within the collagen-rich adventitia of pulmonary arteries, small airways and veins (figure 3). They are also found independently of the broncho-vascular bundles throughout the interstitial space, though more frequently next to the interlobular septa, where lymphatics and veins are passing through. This might explain why venous involvement is common in SAPH pathology and has been described in the past [15]. As stated above, venular and arteriolar remodelling is also observed independently of granulomas in SAPH and in this case appears more or less similar to the vascular lesions found in group 1 PH (PAH); plexiform lesions, however, are very unusual in SAPH and the possibility of independent concomitant disease should be at least considered. Interstitial lung fibrosis leading to late-stage SAPH may resemble atypical unusual interstitial pneumonia/idiopathic pulmonary fibrosis and is usually distinguished by the distribution pattern of collagen-rich fibrosis. So-called fibroblastic foci, particularly aggressive myofibroblasts at the boundaries of progressive collagen deposition, are more common in the latter and less frequently seen in SAPH with interstitial fibrosis.
FIGURE 3.

Explanted lungs from a patient with pulmonary sarcoidosis and pulmonary hypertension. a) A muscular-type pulmonary artery is compressed by a small (left) and a larger (right) epitheloid granuloma containing some rare giant cells. Note the adjacent bronchiole above the larger granuloma which also appears to be mildly compressed. Magnification: ×40. b) Two pulmonary vein branches displaying granulomatous inflammation within the adventitia; some epithelioid histiocytes appear to reach the intimal layer of the vessel. Magnification: ×100. Case provided by Vincent Thomas de Montpréville and Jean-Francois Bernaudin (Hôpital Marie Lannelongue, Le Plessis Robinson, France).
Management
With regard to the management of SAPH patients, the identification of the predominant cause of PH is crucial and an individual approach is essential. In addition to supportive measures such as oxygen supplementation and diuretic therapy, there are two main principles of treatment, namely anti-inflammatory and PAH-targeted therapy. As data is scarce, treatment should be discussed on an individual case-by-case basis and no PAH-specific treatment can be recommended over another. Careful evaluation is needed and fluorodeoxyglucose (FDG)–positron emission tomography–CT might support decision making by evaluation of mediastinal uptake as a sign of inflammatory disease activity favouring anti-inflammatory therapy [13]. Additionally, mechanical treatment by pulmonary vascular stenting might be an option in patients with pulmonary artery stenosis. In cases of insufficient treatment response or deterioration of lung function, early referral for lung transplantation is recommended.
Case 1 part II
In this patient with preserved lung function, vasculopathy (PAH) was suspected as the predominant cause of SAPH, aggravated by pulmonary fibrosis and pulmonary vascular compression. PAH-targeted therapy with macitentan was started (10 mg once daily) [18]. One month after initiation of macitentan, the patient suffered from progressive dyspnoea (WHO FC III). In line with this, pulmonary gas exchange was found to deteriorate with increasing hypoxaemia at rest (PaO2 71 mmHg) and during exercise (SpO2 76% at end of exercise). On follow-up CTPA, there was no significant change in the distribution of the interstitial fibrosis, but the lymph-nodal activity showed intense FDG avidity, in keeping with active inflammation. Cardiac magnetic resonance imaging showed a nondilated right ventricle with preserved systolic function (figure 4).
FIGURE 4.

Follow-up imaging shows the bilateral hilar lymphadenopathy to be 2-fluoro-2-deoxy-d-glucose-avid, in keeping with active sarcoidosis (middle panel). There was no significant uptake in the pulmonary fibrosis, which is likely burnt-out disease. On cardiac magnetic resonance imaging (right panel), there is no right ventricular (RV) enlargement, but there is mild RV insertion site late gadolinium enhancement (block arrows) suggestive of increased shear stress. Also, note the enhancement of the hilar nodes (thin arrows). LV: ventricle.
After multidisciplinary discussion, additional immunosuppressive therapy with steroids (prednisone 40 mg once daily) was started as a flare up of inflammatory disease activity was suspected. PAH-targeted therapy was maintained, although progressive ventilation–perfusion mismatch in regard of the pulmonary involvement might have contributed to the deterioration. Within several days, the patient recorded less dyspnoea and oxygenation improved. In view of the good clinical and functional response, immunosuppressive maintenance therapy was sought. To avoid the long-term side-effects of steroids, prednisone was tapered with simultaneous up-dosing of azathioprine. Currently, under established dual therapy (macitentan and azathioprine), the patient is in good clinical condition with normal exercise capacity (maximal oxygen uptake 87%) without signs of relevant residual PH on echocardiography and improved dynamic lung volumes (see table 3).
TABLE 3.
Summary of the course of the disease for case 1
|
2016
baseline |
2016
1 month macitentan |
2017
6 months macitentan 3 months prednisone |
2017
14 months macitentan 12 months prednisone 8 months AZA |
2022
7 years macitentan 6 years AZA |
|
| NYHA | II | III | II | II | II |
| 6MWD, end SpO2, Borg scale | 584 m, 83%, 3 | 558 m, 76%, 4 | 570 m, 77%, 4 | 582 m, 76%, 4 | – |
| PaO2, kPa (mmHg) | 11.3 (85) | 9.51 (71) | 9.68 (73) | 9.70 (73) | 10.3 (77) |
| A–a gradient, kPa (mmHg) | 3.0 (23) | 4.2 (32) | 4.2 (32) | 3.5 (26) | 2.2 (17) |
| RV/RA gradient, mmHg | 45 | – | 60 | 51 | 38 |
| FVC, L (%) | 2.6 (70) | – | 2.7 (75) | 3.01 (95) | 3.22 (93) |
| DLCO, % | 58 | – | 63 | 70 | 60 |
–: Data unavailable; 6MWD: 6-min walk distance; A–a: alveolar–arterial; AZA: azathioprine; Borg: Borg dyspnoea scale; DLCO: diffusing capacity of the lung for carbon monoxide; FVC: forced vital capacity; NYHA: New York Heart Association class; PaO2: arterial oxygen tension; RA: right atrial; RV: right ventricular; SpO2: peripheral oxygen saturation.
Conclusion
In summary, classification of SAPH in WHO group 5 still seems reasonable due to the multifactorial aetiology and variable clinical picture. This case should remind the reader that the complex and multifaceted nature of the disease must be taken into account in the diagnostic work-up as well as in treatment decisions. Patients suspected of SAPH should be referred to expert PH centres.
Case 2 – sickle cell disease
Case 2 part I
A woman born 1987 was diagnosed with homozygote sickle cell disease (SCD) in early childhood. Her past medical history included deep vein thrombosis and pulmonary embolism in 2008; oral anticoagulation was stopped by the patient in 2009 and restarted in 2017 due to recurrent venous thromboembolism 2017. She experienced approximately monthly vaso-occlusive crisis needing chronic opioid therapy, was treated with regular blood exchange transfusions, revealed consecutive severe hyperferritinaemia, had sickle cell intrahepatic cholestasis with an enlarged liver, was functionally asplenic, had retinopathy, was a persistent smoker and suffered from chronic depression. She was referred to the PH clinic at the University Hospital of Zurich in 2021 due to increasing shortness of breath. Her treatment at the time in addition to regular erythrocytapheresis consisted of rivaroxaban, torasemide, deferiprone, deferasirox, sustained-release morphine, metamizole and esomeprazole.
Clinical presentation and baseline diagnostics
Her clinical examination revealed a BMI of 19 kg·m−2, BP 113/68 mmHg, HR 93 bpm, SpO2 89%, rales on both lung bases, jugular vein distension, severe leg oedema, suspicion of ascites and a split S2. Her haemoglobin (Hb) was 6.7 g·dL−1, after transfusion of two erythrocyte packs 8.7 g·dL−1 (normal 13–17 g·dL−1), chromatography revealed HbA 86.6%, HbS 8.7%, HbF 1.8%, the NT-proBNP was 2640 ng·L−1 (normal <130) and serum ferritin 7999 µg·dL−1 (normal 30–150 µg·dL−1). Her 6-min walk test distance was 345 m and she desaturated from SpO2 92 to 84%. Echocardiography revealed a tricuspid regurgitation pressure gradient of 42 mmHg, a dilated right ventricle with reduced function (tricuspid annular plane systolic excursion 14 mm, tissue Doppler imaging of RV free wall (S′) 11 cm·s−1), the left ventricular ejection fraction was normal with 61%, the left atrium was seriously enlarged (left atrial volume index 63 mL·m−2) and there were signs of diastolic dysfunction.
Radiology
A dual-energy CTPA demonstrated generic features of PH such as dilatation of the main pulmonary artery, enlargement of the right atrium, a dilated right ventricle with right ventricular free wall hypertrophy and flattening of the interventricular septum. There was good opacification of the pulmonary vasculature with no acute or chronic pulmonary embolism on angiography. The corresponding iodine maps showed homogenous perfusion in both lungs, thereby allowing confident exclusion of thromboembolic disease (figure 5a). There were multifocal subsegmental atelectatic bands, predominantly in both lower lobes, likely sequelae of sickle crisis. On limited views of the upper abdomen, there was hepatomegaly with congestion and evidence of autosplenectomy with small calcified spleen. Review of bone windows revealed characteristic “H”-shaped sickle vertebrae due to end-plate depression (figure 5b). A subsequent cardiac magnetic resonance imaging demonstrated biatrial dilatation, with left atrial predominance and a small pericardial effusion (figure 6).
FIGURE 5.

a) Axial images from a dual-energy computed tomography (CT) showing dilated main pulmonary artery (MPA), mildly dilated right atrium (RA), mildly dilated right ventricle (RV) with mild right ventricular hypertrophy and flattening of interventricular septum (arrow). Iodine maps (right panel) demonstrate homogenous perfusion in both lungs. b) Lung windows from the same CT series show bilateral subsegmental atelectatic bands in both lower lobes (top panel, thin arrows). The liver is enlarged and congested (white star) and spleen is atrophic (back star). Coronal views (bottom right) show the characteristic end-plate depression of sickle cell disease (block arrows).
FIGURE 6.
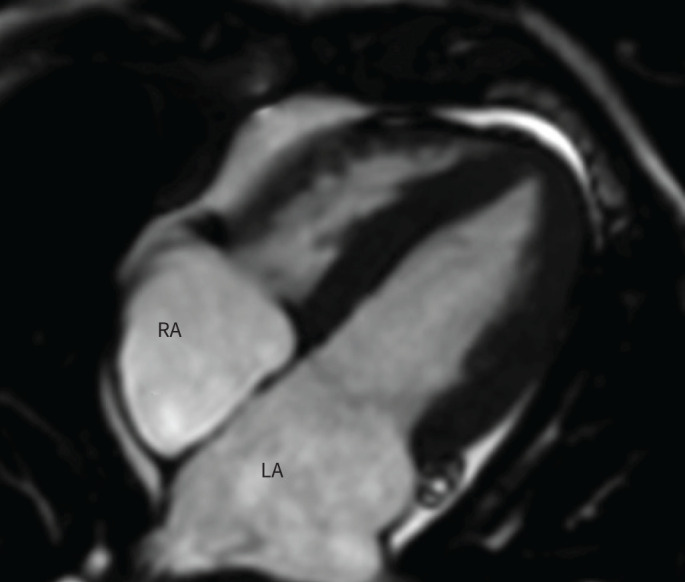
Still image from a steady state free-precession cardiac magnetic resonance imaging examination showing biatrial dilatation (left→right) and a small pericardial effusion (block arrow). LA: left atrium; RA: right atrium.
Right heart catheterisation
Diuretic therapy was increased. Shortly after, the patient needed hospitalisation at the intensive care unit, where right heart catheterisation was performed and, after stabilisation of the acute crisis, pre-and post-capillary PH was diagnosed according to the haemodynamic measures shown in table 4.
TABLE 4.
Case 2 right heart catheterisation
| Right heart catheterisation | FIO2 25% |
| Heart rate, bpm | 54 |
| Mean pulmonary artery pressure, mmHg | 43 |
| Wedge pressure, mmHg | 22 |
| Cardiac output, L·min−1 | 8.5 |
| Pulmonary vascular resistance, WU | 2.4 |
FIO2: inspiratory oxygen fraction; bpm: beats per min; WU: Wood unit.
Background – epidemiology and pathophysiology
SCD-related PH is a multi-aetiological and factorial condition and may include chronic thromboembolic disease and microvascular pulmonary occlusions, haemolytic anaemia associated with low nitric oxide and increased pro-inflammatory factors such as endogenous damage-associated molecular patterns, adenosine deaminase, purine nucleoside phosphorylase, asymmetric dimethylarginine, reactive oxygen species, NADPH, superoxide dismutases and others, left ventricular dysfunction due to anaemia-induced hypercirculation and hypoxic pulmonary vasoconstriction associated with chronic lung disease related parenchymal destruction [19, 20]. According to registry data, half of patients with SCD reveal pulmonary complications and 6–10% of patients with SCD reveal PH with an equal distribution of pre- and post-capillary PH [20, 21]. As known from other groups, PH is also associated with increased mortality in SCD. Screening of SCD patients for PH using echocardiography is questionable, as tricuspid regurgitation velocity is increased in anaemia. However, echocardiography is helpful for diagnosing, treating and following-up symptomatic patients with left heart disease and to guide diagnostic right heart catheterisation in patients with suspected predominant pre-capillary PH [21–23].
Management
According to data from the French PH registry, in addition to SCD-targeted haematological management, about half of the patients with pre-capillary PH were treated with drug therapies approved for PAH; a minority was treated for predominant chronic thromboembolic disease with balloon pulmonary angioplasty or even endarterectomy, but prognosis of SCD-associated PH was poor [21].
Case 2 part II
The patient presented above received treatment for left heart disease and SCD, but no treatment in analogy to patients with PAH due to the relatively low pulmonary vascular resistance, a predominant left heart component and hypercirculation, and continued with blood exchange transfusions. She died in 2022 during a subsequent vaso-occlusive crisis.
Conclusion
This case is intended to remind the reader of the complexity of the pathogenesis of PH in SCD, the poor prognosis and the importance of finding new treatment options for this devastating disease. Specific genetic therapies are currently being developed and we hope that they will be effective and affordable for the millions of affected patients worldwide [24].
Case 3 – PTTM
Case 3 part I
A 37-year-old woman presented to the emergency department after she suffered from her first syncope. She reported exertional dyspnoea WHO FC II–III for several weeks. This case has been published as a single case report with the extended clinical history and follow-up since survival for >12 months in this rare form of PH had not been reported previously [25].
She is a mother of three healthy children and works as a beautician. She had been treated for Crohn's disease with tumour necrosis factor-α inhibitors since 2017. She is a current smoker of two cigarettes per day. She had no other comorbidities and her family history revealed an early death at 44 years from gastrointestinal carcinoma in her grandmother.
Clinical presentation and baseline diagnostics
Clinical examination showed a BMI of 26.8 kg·m−2, HR 109 bpm, BP 100/70 mmHg, SpO2 87% on room air. Physical examination revealed no abnormalities except for a split S2. The ECG revealed new onset of T-wave inversions. Lung function testing was normal but DLCO was reduced with 57%.
Laboratory investigations revealed thrombocytopenia, anaemia, normal renal function, elevated D-dimers and NT-proBNP (3769 ng·L−1). Results of screening for connective tissue disease and HIV were negative. Echocardiography showed a dilated right ventricle, a normal right atrium and an elevated RV/RA gradient of 55 mmHg without signs of an intracardiac shunt nor a left ventricular pathology. 6-min walking distance was reduced (360 m).
Radiology
Ventilation–perfusion scintigraphy showed heterogenous perfusion with a few subsegmental mismatched perfusion defects (figure 7) and hence a diagnosis of thromboembolic disease was considered. Subsequent CTPA showed features of PH such as a dilated central pulmonary artery, right-sided cardiac chamber enlargement and flattening of the interventricular septum (figure 8a). Morphologically, there was no evidence of acute pulmonary embolism or proximal chronic thromboembolic disease. Whilst it is acknowledged that CTPA can miss distal disease, the lack of classic mosaic attenuation on lung windows made chronic thromboembolism less likely. In addition, there were subtle but widespread bilateral ground glass opacities in both central and peripheral distribution with dilatation of a few peripheral subsegmental vessels. A peripheral subpleural wedge-shaped opacity in the left lung was suggestive of pulmonary infarction but overall CT appearances were atypical for thromboembolic disease (figure 8b).
FIGURE 7.
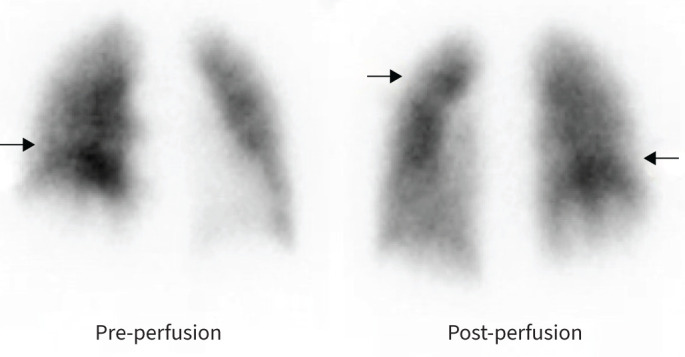
Perfusion scintigraphy showing heterogeneous perfusion with subsegmental perfusion defects (arrows). Reproduced from [25].
FIGURE 8.

a) Axial computed tomography (CT) images showing dilated main pulmonary artery (MPA), mildly dilated right atrium (RA) and mildly dilated right ventricle (RV) with flattening of the interventricular septum (arrow). b) Lung windows from the same CT demonstrating widespread lung parenchymal abnormalities. There are multifocal subtle ground glass opacities (white arrows), a few dilated subsegmental arteries (black arrows) and subpleural consolidation (middle panel). b) Reproduced from [25].
Right heart catheterisation
Right heart catheterisation measurements are shown in table 5.
TABLE 5.
Case 3 right heart catheterisation
| Right heart catheterisation | At rest |
| mPAP, mmHg | 33 |
| Wedge pressure, mmHg | 7 |
| Cardiac index, L·min−1·m−2 | 2.8 |
| Pulmonary vascular resistance, WU | 4.8 |
| mPAP/CO slope | Exercise right heart catheterisation not performed |
mPAP: mean pulmonary arterial pressure; CO: cardiac output; WU: Wood unit.
Case 3 part II
A specific PAH-targeted therapy with macitentan and tadalafil was started. Within a week after treatment initiation, an episode of haematemesis occurred and gastric adenocarcinoma was diagnosed during immediate gastroscopy. Chest radiography showed marked cardiomegaly with dilatation of proximal pulmonary arteries and bilateral pleural effusions (figure 9). Due to clinical worsening with severe hypoxaemia, treated by high flow oxygen treatment, an escape chemotherapy (mFOLFOX regimen – fluorouracil, calcium folinate, oxaliplatin) was initiated before completion of tumour staging. Within days, the hypoxaemia alleviated, thus allowing for diagnostic laparoscopy, which did not reveal a metastatic spread. Accordingly, a neoadjuvant chemotherapy was launched (FLOT regimen – docetaxel, oxaliplatin, fluorouracil, leucovorin) and subsequent gastrectomy was performed. The patient's symptoms resolved completely within 2 months. Follow-up CTPA showed complete resolution of lung parenchymal abnormalities and vascular lesions with normalisation of the right ventricular size and main pulmonary artery calibre (figure 10). Retrospectively, we considered a PTTM the most probable cause of the PH. Follow-up after successful oncologic treatment and 10 cumulative months of PAH-specific medication showed an excellent clinical response without remaining dyspnoea (WHO FC 0–I), normal echocardiography and NT-proBNP levels. Specific PAH therapy was stopped and follow-up right heart catheterisation confirmed complete remission of PH at rest 12 months after the initial diagnosis.
FIGURE 9.
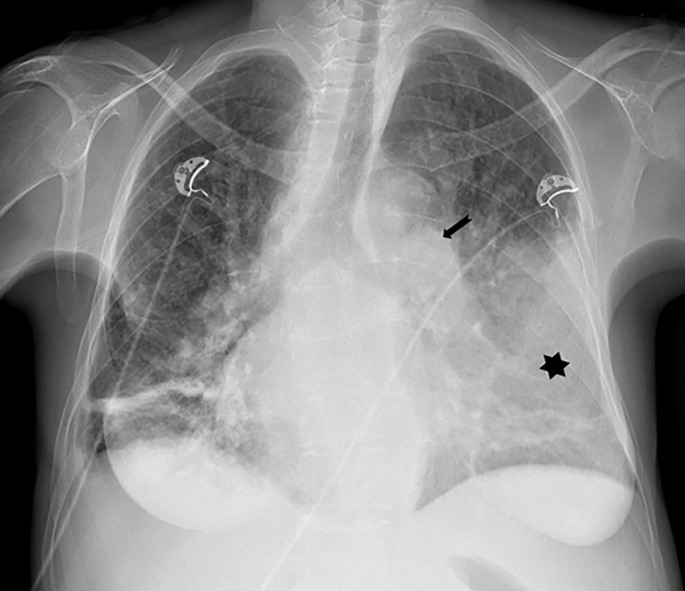
Chest radiograph showing marked cardiomegaly with central pulmonary artery dilatation (arrow) and large left (star) and small right pleural effusions with dependent atelectasis.
FIGURE 10.

Computed tomography pulmonary angiography (CTPA) 2 months post-chemotherapy showing normal calibre main pulmonary artery (MPA) with a nondilated right ventricle (RV). Lung windows show complete resolution of the ground glass opacities.
Background – epidemiology and pathophysiology
PTTM is a devastating disease that is largely underdiagnosed due to its rapid disease progression with a fatal outcome before a full diagnosis can be made [26, 27]. Post mortem studies show a high prevalence of tumour emboli in the pulmonary microcirculation in up to 26% of patients with a solid tumour, markedly associated with adenocarcinoma. PTTM, however, is a rare disease only found in post mortem studies in up to 3.3% of patients with extrathoracic malignancies, with the highest frequency (26%) in gastric carcinomas [28–30].
PTTM with pre- and post-capillary microvascular remodelling (figure 11) is most probably due to the interaction of growth-factor-expressing tumour cells, most commonly carcinoma cells that are derived from epithelial cells. Vasculopathy-like pressure-impacting remodelling of pulmonary arteries and veins beyond mere obstruction through tumour emboli is not a common observation in metastatic tumour disease and has been shown to occur in only 1–3% of carcinoma patients in an autopsy study [31]. In particular, platelet-derived growth factor and vascular endothelial growth factor signalling have been described during the process of tumour cell–endothelial cell interaction [32, 33], both important pathways in the natural history of PAH. In addition, the osteopontin–CD44 axis, which is well-known to promote growth and migration in cancer cells, has been shown to exert similar properties in adventitial fibroblasts of experimental hypoxic PH and to substantially contribute to pulmonary vascular remodelling in this setting [34]. Hence, the rather infrequent development of PH in the context of metastatic cancer might depend on a predisposition for PH with an additive effect of similar molecular pathways.
FIGURE 11.

25-year-old female patient with gastric carcinoma and severe pulmonary hypertension. Autopsy findings. a) Muscular-type pulmonary arteries display irregular/eccentric intimal fibrosis; the arterial lumen contains fresh fibrinous clot material (arrows) intermingled with carcinoma cells (arrowheads) and scattered leucocytes; note the adjacent bronchioles to the upper right. Magnification: ×40. b) Similar clots and tumour cells are found in small pulmonary arterioles, with complete occlusion (arrows). Magnification: ×100. Case provided by Teodora Radonic and Esther Nossent (UMC, Amsterdam, the Netherlands).
Management
A small case series and single case reports form our current understanding and knowledge of disease progression and possible treatment and therefore only low evidence exists regarding therapeutic strategies and follow-up procedures. Those reports, however, suggest that the initiation of cancer therapy is of utmost importance [35, 36]. Furthermore, administration of PAH-targeted therapy and antiproliferative drugs, specifically tyrosine-kinase inhibitors such as imatinib, have been used to treat patients with PTTM. In most cases, patients did not survive >12 months [32, 37–41].
Conclusion
This case shows the complexity of diagnosing and treating patients with PTTM. Physicians should consider the diagnosis especially in patients with unusually rapid disease progression with severe hypoxaemia. A rapid multidisciplinary approach with initial oncological and PAH therapy seems most beneficial.
Points for clinical practice
Group 5 PH encompasses patients with PH and haematological disorders, systemic disorders such as sarcoidosis, pulmonary Langerhans's cell histiocytosis and neurofibromatosis type 1, metabolic disorders such as glycogen storage diseases and Gaucher disease, chronic renal failure with or without haemodialysis, PTTM, and fibrosing mediastinitis.
The underlying pathomechanisms in group 5 PH are complex and manifold.
Treatment of group 5 PH is based on expert opinion; therefore, patients should be treated in PH centres.
Conclusion
Group 5 PH encompasses patients with PH and haematological disorders, systemic disorders such as sarcoidosis, metabolic disorders, chronic renal failure with or without haemodialysis, PTTM and fibrosing mediastinitis.
The underlying pathomechanisms are complex and manifold and a dedicated diagnostic work-up is of importance to identify potentially treatable forms of PH. Treatment is often only based on expert opinion and therefore treatment should be established in an expert centre for PH.
Footnotes
Provenance: Commissioned article, peer reviewed.
Previous articles in this series: No. 1: Condliffe R, Durrington C, Hameed A, et al. Clinical–radiological–pathological correlation in pulmonary arterial hypertension. Eur Respir Rev 2023; 32: 230138.
This article has an editorial commentary: https://doi.org/10.1183/16000617.0237-2023
Number 2 in the Series “Clinical–radiological–pathological correlation in pulmonary hypertension” Edited by Robin Condliffe, Anton Vonk Noordegraaf, Olivier Sitbon, Peter Dorfmüller and Deepa Gopalan
Conflict of interest: M. Lichtblau reports lecture honoraria and advisory board participation from MSD; and travel support from Orpha Swiss, Janssen and MSD, outside the submitted work. S. Ulrich reports grants from the Swiss National Science Foundation, Swiss Lung Foundation, MSD SA and Orpha Swiss; consulting fees, travel support and lecture honoraria from Janssen SA, Actelion SA, MSD SA and Orpha Swiss; and advisory board participation for Janssen SA, Actelion SA and MSD SA, outside the submitted work. All other authors have nothing to disclose.
References
- 1.Rich S. Primary pulmonary hypertension. Executive summary from the World Symposium – Primary Pulmonary Hypertension 1998. Date last accessed: 13 September 2023. Date last updated: 16 September 2019. www.wsphassociation.org/wp-content/uploads/2019/04/Primary-Pulmonary-Hypertension-Evian-1998.pdf
- 2.Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43: Suppl. S12, 5S–12S. doi: 10.1016/j.jacc.2004.02.037 [DOI] [PubMed] [Google Scholar]
- 3.Simonneau G, Gatzoulis MA, Adatia I, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62: Suppl. 25, D34–D41. doi: 10.1016/j.jacc.2013.10.029 [DOI] [PubMed] [Google Scholar]
- 4.Simonneau G, Montani D, Celermajer DS, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 2018; 53: 1801913. doi: 10.1183/13993003.01913-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022; 43: 3618–3731. doi: 10.1093/eurheartj/ehac237 [DOI] [PubMed] [Google Scholar]
- 6.McDonagh TA, Metra M, Adamo M, et al. 2021 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J 2021; 42: 3599–3726. doi: 10.1093/eurheartj/ehab368 [DOI] [PubMed] [Google Scholar]
- 7.European Respiratory Society . Pulmonary hypertension: clinical–radiological–pathological case series. Date last accessed: 13 September 2023. Date last updated: 13 March 2023. https://www.ersnet.org/events/pulmonary-hypertension-clinical-radiological-pathological-case-series/
- 8.Savale L, Huitema M, Shlobin O, et al. WASOG statement on the diagnosis and management of sarcoidosis-associated pulmonary hypertension. Eur Respir Rev 2022; 31: 210165. doi: 10.1183/16000617.0165-2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baughman RP, Field S, Costabel U, et al. Sarcoidosis in America. Analysis based on health care use. Ann Am Thorac Soc 2016; 13: 1244–1252. doi: 10.1513/AnnalsATS.201511-760OC [DOI] [PubMed] [Google Scholar]
- 10.Baughman RP, Engel PJ, Taylor L, et al. Survival in sarcoidosis-associated pulmonary hypertension: the importance of hemodynamic evaluation. Chest 2010; 138: 1078–1085. doi: 10.1378/chest.09-2002 [DOI] [PubMed] [Google Scholar]
- 11.Baughman RP, Valeyre D, Korsten P, et al. ERS clinical practice guidelines on treatment of sarcoidosis. Eur Respir J 2021; 58: 2004079. doi: 10.1183/13993003.04079-2020 [DOI] [PubMed] [Google Scholar]
- 12.Shlobin OA, Kouranos V, Barnett SD, et al. Physiological predictors of survival in patients with sarcoidosis-associated pulmonary hypertension: results from an international registry. Eur Respir J 2020; 55: 1901747. doi: 10.1183/13993003.01747-2019 [DOI] [PubMed] [Google Scholar]
- 13.Boucly A, Cottin V, Nunes H, et al. Management and long-term outcomes of sarcoidosis-associated pulmonary hypertension. Eur Respir J 2017; 50: 1700465. doi: 10.1183/13993003.00465-2017 [DOI] [PubMed] [Google Scholar]
- 14.Kirkil G, Lower EE, Baughman RP. Predictors of mortality in pulmonary sarcoidosis. Chest 2018; 153: 105–113. doi: 10.1016/j.chest.2017.07.008 [DOI] [PubMed] [Google Scholar]
- 15.Nunes H, Humbert M, Capron F, et al. Pulmonary hypertension associated with sarcoidosis: mechanisms, haemodynamics and prognosis. Thorax 2006; 61: 68–74. doi: 10.1136/thx.2005.042838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sulica R, Teirstein AS, Kakarla S, et al. Distinctive clinical, radiographic, and functional characteristics of patients with sarcoidosis-related pulmonary hypertension. Chest 2005; 128: 1483–1489. doi: 10.1378/chest.128.3.1483 [DOI] [PubMed] [Google Scholar]
- 17.Baughman RP, Shlobin OA, Wells AU, et al. Clinical features of sarcoidosis associated pulmonary hypertension: results of a multi-national registry. Respir Med 2018; 139: 72–78. doi: 10.1016/j.rmed.2018.04.015 [DOI] [PubMed] [Google Scholar]
- 18.Mathijssen H, Huitema MP, Bakker ALM, et al. Safety of macitentan in sarcoidosis-associated pulmonary hypertension: a case-series. Sarcoidosis Vasc Diffuse Lung Dis 2020; 37: 74–78. doi: 10.36141/svdld.v37i1.9292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheikh AB, Nasrullah A, Lopez ED, et al. Sickle cell disease-induced pulmonary hypertension: a review of pathophysiology, management, and current literature. Pulse 2021; 9: 57–63. doi: 10.1159/000519101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gordeuk VR, Castro OL, Machado RF. Pathophysiology and treatment of pulmonary hypertension in sickle cell disease. Blood 2016; 127: 820–828. doi: 10.1182/blood-2015-08-618561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Savale L, Habibi A, Lionnet F, et al. Clinical phenotypes and outcomes of precapillary pulmonary hypertension of sickle cell disease. Eur Respir J 2019; 54: 1900585. doi: 10.1183/13993003.00585-2019 [DOI] [PubMed] [Google Scholar]
- 22.Fonseca GH, Souza R, Salemi VM, et al. Pulmonary hypertension diagnosed by right heart catheterisation in sickle cell disease. Eur Respir J 2012; 39: 112–118. doi: 10.1183/09031936.00134410 [DOI] [PubMed] [Google Scholar]
- 23.Parent F, Bachir D, Inamo J, et al. A hemodynamic study of pulmonary hypertension in sickle cell disease. N Engl J Med 2011; 365: 44–53. doi: 10.1056/NEJMoa1005565 [DOI] [PubMed] [Google Scholar]
- 24.Hoban MD, Orkin SH, Bauer DE. Genetic treatment of a molecular disorder: gene therapy approaches to sickle cell disease. Blood 2016; 127: 839–848. doi: 10.1182/blood-2015-09-618587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aeberhard J, Lichtblau M, Arenja N, et al. An unexpected cause of pulmonary hypertension in a young woman – a case report. Eur Heart J Case Rep 2023; 7: ytad474. doi: 10.1182/blood-2015-09-618587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajdev K, Madan U, McMillan S, et al. Pulmonary tumor embolism and pulmonary tumor thrombotic microangiopathy causing rapidly progressive respiratory failure: a case series. J Investig Med High Impact Case Rep 2022; 10: 23247096221086453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kane RD, Hawkins HK, Miller JA, et al. Microscopic pulmonary tumor emboli associated with dyspnea. Cancer 1975; 36: 1473–1482. doi: [DOI] [PubMed] [Google Scholar]
- 28.Shields DJ, Edwards WD. Pulmonary hypertension attributable to neoplastic emboli: an autopsy study of 20 cases and a review of literature. Cardiovasc Pathol 1992; 1: 279–287. doi: 10.1016/1054-8807(92)90038-P [DOI] [PubMed] [Google Scholar]
- 29.Winterbauer RH, Elfenbein IB, Ball WC, Jr. Incidence and clinical significance of tumor embolization to the lungs. Am J Med 1968; 45: 271–290. doi: 10.1016/0002-9343(68)90044-2 [DOI] [PubMed] [Google Scholar]
- 30.Kawakami N, Moriya T, Kato R, et al. Pulmonary tumor thrombotic microangiopathy in occult early gastric cancer that was undetectable on upper endoscopy: a case report and review of similar cases. BMC Gastroenterol 2021; 21: 423. doi: 10.1186/s12876-021-02009-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Uruga H, Fujii T, Kurosaki A, et al. Pulmonary tumor thrombotic microangiopathy: a clinical analysis of 30 autopsy cases. Intern Med 2013; 52: 1317–1323. doi: 10.2169/internalmedicine.52.9472 [DOI] [PubMed] [Google Scholar]
- 32.Minatsuki S, Miura I, Yao A, et al. Platelet-derived growth factor receptor-tyrosine kinase inhibitor, imatinib, is effective for treating pulmonary hypertension induced by pulmonary tumor thrombotic microangiopathy. Int Heart J 2015; 56: 245–248. doi: 10.1536/ihj.14-220 [DOI] [PubMed] [Google Scholar]
- 33.Wakabayashi Y, Iwaya M, Akita M, et al. Pulmonary tumor thrombotic microangiopathy caused by urothelial carcinoma expressing vascular endothelial growth factor, platelet-derived growth factor, and osteopontin. Intern Med 2016; 55: 651–656. doi: 10.2169/internalmedicine.55.5758 [DOI] [PubMed] [Google Scholar]
- 34.Anwar A, Li M, Frid MG, et al. Osteopontin is an endogenous modulator of the constitutively activated phenotype of pulmonary adventitial fibroblasts in hypoxic pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2012; 303: L1–L11. doi: 10.1152/ajplung.00050.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Imakura T, Tezuka T, Inayama M, et al. A long-term survival case of pulmonary tumor thrombotic microangiopathy due to gastric cancer confirmed by the early diagnosis based on a transbronchial lung biopsy. Intern Med 2020; 59: 1621–1627. doi: 10.2169/internalmedicine.3630-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merad M, Alibay A, Ammari S, et al. Pulmonary tumor thrombotic microangiopathy. Rev Mal Respir 2017; 34: 1045–1057. doi: 10.1016/j.rmr.2017.02.008 [DOI] [PubMed] [Google Scholar]
- 37.Gainza E, Fernandez S, Martinez D, et al. Pulmonary tumor thrombotic microangiopathy: report of 3 cases and review of the literature. Medicine 2014; 93: 359–363. doi: 10.1097/MD.0000000000000219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fukada I, Araki K, Minatsuki S, et al. Imatinib alleviated pulmonary hypertension caused by pulmonary tumor thrombotic microangiopathy in a patient with metastatic breast cancer. Clin Breast Cancer 2015; 15: e167–e170. doi: 10.1016/j.clbc.2014.10.008 [DOI] [PubMed] [Google Scholar]
- 39.Ogawa A, Yamadori I, Matsubara O, et al. Pulmonary tumor thrombotic microangiopathy with circulatory failure treated with imatinib. Intern Med 2013; 52: 1927–1930. doi: 10.2169/internalmedicine.52.0718 [DOI] [PubMed] [Google Scholar]
- 40.Higo K, Kubota K, Takeda A, et al. Successful antemortem diagnosis and treatment of pulmonary tumor thrombotic microangiopathy. Intern Med 2014; 53: 2595–2599. doi: 10.2169/internalmedicine.53.2379 [DOI] [PubMed] [Google Scholar]
- 41.Ma G, Wang D, Xu X, et al. Case report: apatinib plus selexipag as a novel therapy for pulmonary tumor thrombotic microangiopathy accompanied by pulmonary hypertension associated with gastric carcinoma. Medicine 2022; 101: e29412. doi: 10.1097/MD.0000000000029412 [DOI] [PMC free article] [PubMed] [Google Scholar]