

Abstract
Introduction
The high recombinogenic potential of HIV-1 has resulted in the generation of countless unique recombinant forms (URFs) and around 120 reported circulating recombinant forms (CRFs). Here we identify through analyses of near full-length genomes (NFLG) a new HIV-1 CRF derived from subtypes B and F1.
Methods
HIV-1 protease-reverse transcriptase (Pr-RT) sequences were obtained by RT-PCR amplification from plasma RNA. Near full-length genome sequences were obtained after amplification by RT-PCR in 5 overlapping fragments. Phylogenetic sequence analyses were performed via maximum likelihood. Mosaic structures were analyzed by bootscanning and phylogenetic analyses of genome segments. Temporal and geographical estimations of clade emergence were performed with a Bayesian coalescent method.
Results
Through phylogenetic analyses of HIV-1 Pr-RT sequences obtained by us from samples collected in Spain and downloaded from databases, we identified a BF1 recombinant cluster segregating from previously reported CRFs comprising 52 viruses, most from Brazil (n = 26), Spain (n = 11), and Italy (n = 9). The analyses of NFLG genomes of 4 viruses of the cluster, 2 from Spain and 2 from Italy, allowed to identify a new CRF, designated CRF75_BF1, which exhibits a complex mosaic structure with 20 breakpoints. All 4 patients harboring CRF75_BF1 viruses studied by us had CD4+ T-cell lymphocyte counts below 220/mm3 less than one year after diagnosis, a proportion significantly higher (p = 0.0074) than the 29% found in other patients studied in Spain by us during the same period. The origin of the clade comprising CRF75_BF1 and related viruses was estimated around 1984 in Brazil, with subsequent introduction of CRF75_BF1 in Italy around 1992, and migration from Italy to Spain around 1999.
Conclusion
A new HIV-1 CRF, designated CRF75_BF1, has been identified. CRF75_BF1 is the 6th CRF of South American origin initially identified in Western Europe, reflecting the increasing relationship of South American and European HIV-1 epidemics. The finding of low CD4+ T-cell lymphocyte counts early after diagnosis in patients harboring CRF75_BF1 viruses warrants further investigation on the virulence of this variant.
Keywords: HIV-1, circulating recombinant forms, molecular epidemiology, phylogeny, phylodynamics, recombination, genetic diversity
1. Introduction
HIV-1 has a characteristically high recombinogenic potential, which offers some evolutionary advantages to the virus, such as escape from immune responses (Streeck et al., 2008; Ritchie et al., 2014), acquisition of antiretroviral drug resistance (Nora et al., 2007), increased replicative fitness (Gundlach et al., 2000; Moradigaravand et al., 2014; Arenas et al., 2016), and increased viral diversity (Charpentier et al., 2006; Song et al., 2018). The elevated recombination rate of HIV-1 is reflected in the frequent generation of recombinant forms wherever two or more genetic forms circulate in the same population (Nájera et al., 2002). Some HIV-1 recombinant forms have acquired the capacity to spread in the population among epidemiologically-unlinked individuals. These are designated circulating recombinant forms (CRFs) (Robertson et al., 2000), of which around 120 have been reported in the literature (HIV Sequence Database, 2023), with variable epidemiological importance. The number of CRFs is increasing unceasingly, as a consequence of both the generation of new CRFs and the identification of old previously unrecognized CRFs. Their prevalence in the global pandemic has increased, reaching an estimated 17% in a global survey in 2010–2015, where URFs represented around 6% infections (Hemelaar et al., 2020), although a recent systematic literature review estimated the global prevalence of HIV-1 recombinant forms to be 29% (Williams et al., 2023); these percentages could be underestimates, considering that most molecular epidemiological studies are based on the analysis of a small fraction of the viral genome. Among CRFs, those derived from subtypes B and F1 are among the most numerous, of which 19 have been reported in the literature, most of them of South American origin, derived from the F1 strain circulating in Brazil, among which CRF12_BF, widely circulating in Argentina and Uruguay (Thomson et al., 2000, 2002; Carr et al., 2001), is the most prevalent. Five of the CRF_BFs of South American origin have been first identified in Western Europe [CRF47_BF1 (Fernández-García et al., 2010), CRF66_BF1 (Bacqué et al., 2021), CRF89_BF1 (Delgado et al., 2021), and CRF122_BF1 (Cañada-García et al., 2022) in Spain; and CRF42_BF1 in Luxembourg (Struck et al., 2015)], which reflects the increasing immigration from South America to some Western European countries. Here we identify a new CRF_BF of Brazilian origin through near full-length genome analyses of viruses collected in Spain and Italy.
2. Materials and methods
2.1. Samples
Samples from HIV-1-infected individuals were collected in 14 Spanish regions for molecular epidemiological studies or for antiretroviral drug resistance tests. The study was approved by the Committee of Research Ethics of Instituto de Salud Carlos III, Majadahonda, Madrid, Spain. It did not require written informed consent by the study participants, as it used samples and data collected as part of routine clinical practice and patients’ data were anonymized without retaining data allowing individual identification.
2.2. PCR amplification and sequencing
An ~1.4 kb pol fragment in protease-reverse transcriptase (Pr-RT) was amplified from plasma RNA by RT-PCR followed by nested PCR as described previously (Delgado et al., 2015) and sequenced with the Sanger method using a capillary automated sequencer. Near full-length genome (NFLG) sequences were obtained for selected samples by amplification in 5 overlapping segments from plasma RNA and sequenced by the Sanger method, as described (Delgado et al., 2002; Sierra et al., 2005; Cañada et al., 2021). The primers used for PCR and sequencing cover a genome fragment spanning HXB2 nt positions 571–9,509 (~8.9 kb). Newly derived sequences are deposited in GenBank under accessions MK341078, OR466119, OR466120, and OR466121.
2.3. Phylogenetic sequence analyses
Sequences were aligned with MAFFT v7 (Katoh and Standley, 2013). Initial phylogenetic trees with all Pr-RT sequences obtained by us were constructed via approximate maximum likelihood with FastTree2 (Price et al., 2010) using the general time reversible evolutionary model with CAT approximation to account for among-site rate heterogeneity (GTR + CAT), with assessment of node support with Shimodaira-Hasegawa (SH)-like local support values (Guindon et al., 2010). Subsequent maximum likelihood (ML) trees with sequences of interest were constructed with W-IQ-Tree (Trifinopoulos et al., 2016), using the general time reversible with free rate (GTR + FR) evolutionary model, with assessment of node support with the ultrafast bootstrap (UFB) approximation approach (Hoang et al., 2018). Trees were visualized with MEGA v7.0 (Kumar et al., 2016). GenBank accessions of sequences used in phylogenetic analyses are shown in Supplementary Table 1.
Mosaic structures were analyzed by bootscanning (Salminen et al., 1995) with SimPlot v1.3.5 (Lole et al., 1999). In these analyses, trees were constructed using the neighbor-joining method with the Kimura 2-parameter model and a window width of 250 nucleotides moving in 20 nt steps. To further determine subtype assignations, potentially recombinant segments identified with SimPlot were phylogenetically analyzed with relevant subtype references by ML with W-IQ-Tree and PhyML v3 (Guindon and Gascuel, 2003) and by Bayesian inference with MrBayes v3.2 (Ronquist et al., 2012). The analyses with PhyML were done using the best-fit substitution model selected by Smart Model Selection (Lefort et al., 2017), with assessment of node support with the approximate likelihood ratio, SH-like procedure (Guindon et al., 2010). The analyses with MrBayes were performed using the GTR + G + I substitution model, running two simultaneous independent runs and 8 chains 2–5 million generations long, ensuring that both runs reached convergence, as determined by an average standard deviation of split frequencies <0.01. In bootscan analyses and in phylogenetic trees of genome segments constructed to determine mosaic structures, a reconstructed B-F1 ancestral sequence was used as outgroup to avoid artifacts caused by distant outgroups (Travers et al., 2004; Thomson and Fernández-García, 2011; Seager et al., 2014; Hill et al., 2022). For this reconstruction, the ML method implemented in W-IQ-Tree was used, with the GTR + FR evolutionary model, and sequences with the following GenBank accessions and subtypes: AB253421, AB253429 (A1); K03455, U21135, U63632, AY173951, AY331295, AY423387, DQ853463, MG365763 (B); AY772699, U46016 (C); AB485656, AF005494, FJ771006, FJ771007, FJ771008, FJ771009, MG365763, MG365768 (F1); AF061641, AF084936 (G); AF190127, AF190128 (H); J: AF082394, EF614151 (J); and DQ373063 (SIVcpz), used as outgroup.
2.4. Temporal and geographical estimations of clade ancestors
The times and the locations of the most recent common ancestors (MRCA) of the identified CRF and clusters and subclusters within it were estimated using Pr-RT sequences with the Bayesian Markov chain Monte Carlo (MCMC) coalescent method implemented in BEAST v1.10.4 (Suchard et al., 2018). Before the BEAST analysis, the existence of temporal signal in the dataset was analyzed with TempEst v.1.5.3 (Rambaut et al., 2016), which determines the correlation of genetic divergence among sequences (measured as root-to-tip distance) with time. For the BEAST analysis, 7 codon positions with drug resistance mutations in one or more sequences, as determined by CPR v6.0 (Gifford et al., 2009), were removed from the alignment, since we found that their removal increased the temporal signal of the dataset, as reflected in r2 values in the TempEst analysis. The BEAST analysis was performed using the SRD06 codon-based evolutionary model (where the 3rd codon position is in a partition different from the 1st and 2nd positions) (Shapiro et al., 2006). We also specified an uncorrelated lognormal relaxed clock and a Bayesian SkyGrid coalescent tree prior (Gill et al., 2013). The MCMCs were run for 50 million generations. Mixing and convergence were checked with Tracer, ensuring that effective sample size values of all parameters were > 200. We performed runs in duplicate, combining the posterior tree files with LogCombiner v1.10.4. The posterior distribution of trees was summarized in a maximum clade credibility (MCC) tree with TreeAnnotator v1.10.4, after removal a 10% burn-in. MCC trees were visualized with FigTree v1.4.2 (Rambaut1). Parameter uncertainty was summarized in 95% highest posterior density (95% HPD) intervals.
2.5. Coreceptor usage predicition
Coreceptor usage prediction was done with Geno2pheno (Lengauer et al., 2007), based on the analysis of envelope V3 loop sequences, using the recommendations from the European Consensus Group on Clinical Management of HIV-1 Tropism Testing (Vandekerckhove et al., 2011).
2.6. Statistical analyses
Statistical analyses were performed with Fisher’s exact test.
3. Results
3.1. Identification of a HIV-1 BF1 recombinant cluster segregating from reported CRFs
In a HIV-1 molecular epidemiological study in Spain, based on analyses of Pr-RT sequences, we found frequent grouping of HIV-1 infections in clusters (Gil et al., 2022), of which 25 were BF1 recombinant branching apart from reported CRF_BFs. One of these clusters, designated BF13 (numbered according to the order of identification), was further analyzed to determine whether it represents a new CRF. In the initial analyses, with sequences obtained by us, BF13 comprised only four individuals, with Pr-RT sequences showing similar recombinant structures, with a predominance of F1 subsubtype and a short subtype B segment around nt positions 1,000–1,200 (numbered from the 5′ end of protease) (Figure 1). Through inclusion in phylogenetic analyses of all Pr-RT sequences from the Los Alamos HIV Sequence database (HIV Sequence Database, 2023) classified as of F1 subsubtype or BF1 recombinant, we found that the 4 BF13 sequences originally identified by us were included in a much larger cluster (for which we kept the BF13 designation) comprising 52 viruses from 6 countries: 11 from Spain, 9 from Italy, 26 from Brazil, 2 from United Kingdom, 2 from USA, and 1 from Slovakia (Figure 2). All 4 individuals from Spain studied by us were native Spaniards, from 3 cities from two regions (Basque Country and Madrid), and all were men who have sex with men (MSM) (Table 1). In the Pr-RT phylogenetic tree, all viruses from Italy and Spain grouped in a subcluster, that also included a Brazilian virus, in which viruses from Spain grouped in a subsubcluster that also included an Italian virus (Figure 2). The bootscan analyses of Pr-RT sequences from the cluster longer than 1.1 kb showed recombinant structures coincident with that of the 4 viruses of BF13 cluster identified by us (Supplementary Figure S1).
Figure 1.

Bootscan analyses of 4 Pr-RT sequences of the BF13 cluster obtained by us. The horizontal axis represents the position from the 5’ end of protease of the midpoint of a 250 nt window moving in 20 nt increments and the vertical axis represents bootstrap values supporting clustering with subtype reference sequences. As outgroup, a reconstructed B-F1 ancestor sequence was used.
Figure 2.

Maximum likelihood tree of Pr-RT sequences of BF13 cluster. References of subtypes and all CRF_BF1s that are BF1 recombinant in Pr-RT are included in the analysis. Names of sequences obtained by us, all collected in Spain, are in bold type. In database sequences, the country of sample collection is indicated before the virus name with the 2-letter ISO country code (ES, Spain; IT, Italy; BR, Brazil; US, USA; UK, United Kingdom; SK, Slovakia). Only ultrafast bootstrap values ≥90% are shown. For viewing purposes, some clades comprising references of subtypes and CRFs are compressed.
Table 1.
Epidemiological and clinical data of the patients from the BF13 cluster studied by us.
| Sample ID | Gender | Transmission route | Country of origin | Province and region of residence | Year of HIV-1 diagnosis | Year of sample collection | CD4+ T-cell count (cells/mm3) | Plasma viral load (copies/ml) |
|---|---|---|---|---|---|---|---|---|
| P4837 | M | MSM | Spain | Vizcaya, Basque Country | 2017 | 2017 | 219 | 25,300 |
| PV074140 | M | MSM | Spain | Vizcaya, Basque Country | 2017 | 2017 | 96 | 37,300 |
| PV178078 | M | MSM | Spain | Álava, Basque Country | 2022 | 2022 | 92 | 22,900 |
| MD408339 | M | MSM | Spain | Madrid | 2012 | 2013 | 142 | 73,400 |
3.2. Analyses of NFLG sequences
To determine whether the BF13 cluster represents a new CRF, we obtained NFLG sequences of three epidemiologically-unlinked viruses identified by us, from two regions, which were analyzed by bootscanning together with two other viruses from Italy (53,143 and 58,736) from the BF13 cluster whose NFLG sequences were obtained previously by other authors (Saladini et al., 2012). The analyses showed highly similar bootscan plots indicating complex BF1 mosaic structures of two of the Spanish (P4837 and PV074140) and both Italian viruses (Figure 3). ML and Bayesian phylogenetic analyses of the 21 recombinant segments identified in the bootscan analyses in at least 3 of the 4 viruses indicated coincident grouping of all 4 viruses in each segment with B or F1 references (Figure 4). In some apparently recombinant segments seen in the bootscan analyses in only one or two viruses (Figure 3), ML and Bayesian trees failed to support their recombinant origin, since branching with subtype references was not well supported (Supplementary Figure S2). Therefore, these analyses indicated that the 4 viruses exhibited identical mosaic structures.
Figure 3.

Bootscan analyses of 4 NFLG sequences of viruses from the BF13 cluster, 2 from Spain, obtained by us, and 2 from Italy, obtained by Saladini et al. (2012). The horizontal axis represents the position in the HXB2 genome of the midpoint of a 250 nt window moving in 20 nt increments and the vertical axis represents bootstrap values supporting clustering with subtype reference sequences. As outgroup, a reconstructed B-F1 ancestor sequence was used. Breakpoint positions of segments of potential recombinant origin detected in the bootscan analyses in at least 3 viruses are indicated on top, numbered as in the HXB2 genome. These breakpoints were located in the midpoint positions of the transitions between the 75% consensus sequences of subtype B and of the Brazilian F1 subtype variant.
Figure 4.

Phylogenetic trees of interbreakpoint genome segments of the NFLG analyzed by bootscanning. The segment number assigned in the bootscan analysis (Figure 3) and the HXB2 positions delimiting the segments are indicated above the trees. Sequence names of BF1 viruses are in blue or green, according to their branching with B or F1 references, respectively, in the corresponding segment. Names of subtype references are preceded by the corresponding subtype name. As outgroup, a reconstructed B-F1 ancestor sequence was used. Node support values of subtype clades are shown in this order: UFB value (IQ-Tree)/ aLRT SH-like support (PhyML)/ posterior probability (MrBayes). For the other nodes, only UFB values, if ≥70%, are shown.
In a phylogenetic tree of NFLGs, the 4 viruses of the BF13 cluster with coincident mosaic structures grouped in a strongly supported clade segregating from previously identified CRF_BFs (Figure 5).
Figure 5.
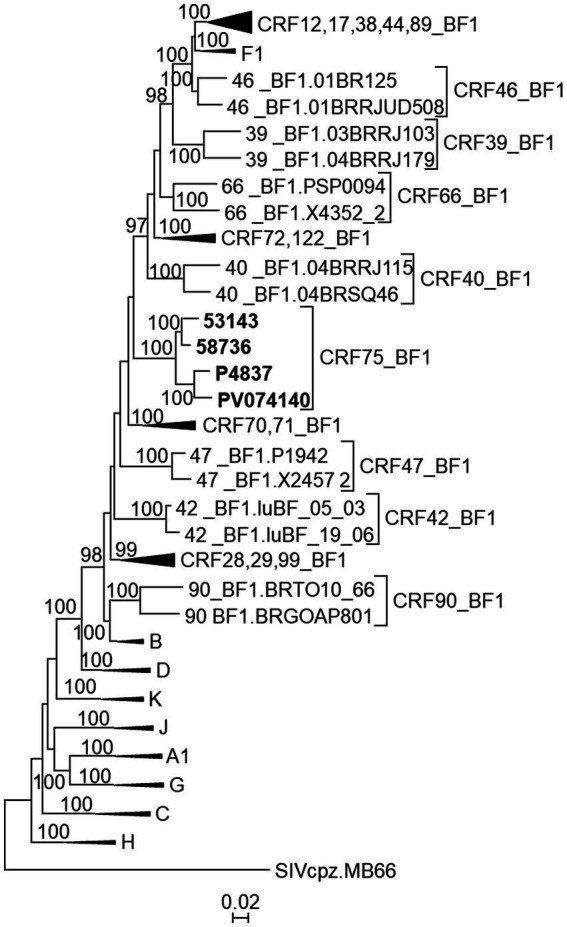
Maximum likelihood tree of NFLG sequences of viruses of the BF13 cluster. References of all published CRF_BF1s and of HIV-1 subtypes are also included in the analysis. The tree is rooted with SIVcpz virus MB66. For viewing purposes, some subtype and CRF reference clades are compressed. Only UFB values ≥90% are shown.
These results allow us to identify a new CRF, designated CRF75_BF1, which exhibits a complex mosaic structure, with 20 breakpoints, delimiting 11 subtype B and 10 subsubtype F1 segments (Figure 6).
Figure 6.

Mosaic structure of CRF75_BF1. Breakpoint positions are numbered as in the HXB2 genome. The drawing was made using the Recombinant HIV-1 Drawing Tool https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html.
The third NFLG sequence from Spain obtained by us (from patient MD408339) showed a mosaic structure slightly different from the other two in the bootscan analysis, with a short additional subtype B segment in HXB2 positions 6,938–7,086 (Supplementary Figure S3). Since this virus branches in Pr-RT in a subcluster with 5 viruses from the Spanish region of Comunidad Valenciana (Figure 2), it would be interesting to know whether viruses from this subcluster show the same mosaic structure as MD408339, in which case they could represent a new CRF closely related to CRF75_BF1.
The bootscan analysis of a third previously sequenced Italian virus for which around 7 kb from two discontinuous segments were obtained by other authors (Saladini et al., 2012) showed a mosaic structure in the sequenced segments coincident with that of the CRF75_BF1 viruses (Supplementary Figure S3).
3.3. Bootscan and phylogenetic analyses of BF1 recombinant sequences from databases related to CRF75_BF1
Three of the viruses whose Pr-RT sequences branched in the BF13 cluster (HI2016-28, from Brazil, and 152,822 and 152,884, from UK) had additional sequences outside of Pr-RT, that were analyzed to determine their mosaic structures. Through BLAST searches of similar sequences in databases and subsequent phylogenetic and bootscan analyses, we found two additional viruses, from Brazil and South Africa, respectively, that were related to CRF75_BF1. Through these analyses (Figure 7 and Supplementary Figures S4, S5), we found that (1) the Brazilian virus 10BR_RJ053, sequenced in the NFLG sequence (Pessoa et al., 2015), exhibited a recombinant structure highly similar, but not identical, to CRF75_BF1, lacking two B subtype segments in env and one F1 subtype segment in vpr found in CRF75_BF1 and having an additional subtype B fragment in pol (Supplementary Figure S6); (2) the Brazilian virus HI2016-28 (Alves et al., 2017), of which a total of 6.1 kb genome sequence in two discontinuos fragments is available, had a mosaic structure highly similar to CRF75_BF1 but with an additional subtype B fragment in pol; (3) the virus from South Africa MSM305 (Middelkoop et al., 2014) had an envelope sequence exhibiting a recombinant structure fully coincident with CRF75_BF1; and (4) the UK viruses 152822 and 152884 had mosaic structures fully coincident with CRF75_BF1 in their available 2.8 kb pol sequences. In phylogenetic trees (Supplementary Figure S5), 10BR_RJ053 branched in a basal position relative to the Spanish-Italian CRF75_BF1 cluster, with HI2016-28 branching in an intermediate position, suggesting that 10BR_RJ053 derives from a BF1 recombinant lineage ancestral to HI2026-28 and the Spanish-Italian CRF75_BF1 strain; the South African virus MSM305 grouped with the Brazilian virus HI2016-28, pointing to its probable Brazilian ancestry; and the UK viruses 152822 and 152884 branched basally to the Spanish-Italian CRF75_BF1, which together to their grouping with Brazilian viruses in Pr-RT (Figure 2), also points to their probable Brazilian ancestry.
Figure 7.

Bootscan analyses of sequences of 5 viruses related to CRF75_BF1. The analyzed viruses are (a) 10BR_RJ053, from Brazil; (b) HI2016-28, from Brazil; (c) MSM305, from South Africa; (d) 152822, from UK; and (e) 152884, from UK. For comparison, the bootscan plot of the NFLG of the CRF75_BF1 virus P4837 is shown on top. The horizontal axis represents the position in the HXB2 genome of the midpoint of a 250 nt window moving in 20 nt increments and the vertical axis represents bootstrap values supporting clustering with subtype reference sequences. As outgroup, a reconstructed B-F1 ancestor sequence was used. Fragments in the analyzed viruses differing in subtype from CRF75_BF1, according to the bootscan analyses and ML phylogenetic trees (Supplementary Figure S4), are indicated as horizontal brackets above the bootscan plots.
3.4. Temporal and geographical estimations
To estimate the times and places of origin of the clade comprising CRF75_BF1 and related viruses and of the clusters within it, Pr-RT sequences from samples with known collection years where analyzed with a Bayesian coalescent method, implemented in BEAST 1.10.4. Two closely related viruses from United Kingdom and two closely related viruses from USA were excluded from this analysis, as they could derive from sporadic acquisitions in another country where CRF75_BF1 or related viruses circulate. Prior to the BEAST analysis, the TempEst analysis indicated the existence of an adequate temporal signal in the dataset (r2 = 0.49). The BEAST analysis estimated a substitution rate of 1.34 × 10−3/sub/site/yr (95% HPD, 8.52 × 10−4-1.86 × 10−3). The time of the MRCA of the clade of CRF75_BF1 and related viruses was estimated around 1984 (95% HPD, 1975–1993), and its most probable location was in Brazil (PP = 0.996) (Figure 8). The BEAST analysis also indicated that in Europe, CRF75_BF1 was first introduced in Italy around 1992 (95% HPD, 1986–1996), with subsequent introduction in Spain from Italy around 1999 (95% HPD, 1994–2003).
Figure 8.

Maximum clade credibility tree of Pr-RT sequences of CRF75_BF1 and CRF75_BF1-like viruses. Branch colors indicate, for terminal branches, country of sample collection, and for internal branches, the most probable location country of the subtending node, according to the legend on the upper left. Nodes supported by PP ≥ 0.95 are marked with filled circles. The most probable countries at the root of the tree and at the nodes corresponding to the Italian-Spanish cluster, the Spanish subcluster, and the largest Brazilian cluster are indicated, together with the PPs supporting each location and the time of the MRCA (mean value, with 95% HPD interval in parentheses).
3.5. Association with low CD4+ T-cell counts
All 4 patients harboring CRF75_BF1 (or a closely related recombinant in one case) viruses studied by us had low CD4+ T-cell counts less than 1 year after HIV-1 diagnosis (3 of them less than 3 months after diagnosis): 219, 96, 92, and 142 cells/mm3, respectively. Compared with 3,548 other patients attended at Spanish clinical centers studied by us in the same period in which infections with CRF75_BF1 had been diagnosed (2012–2022), the difference in the proportion of viruses with CD4+ T-cell counts ≤220 cells/mm3 (100% vs. 29.3%) was statistically significant when analyzed with Fisher’s exact test (p = 0.0074).
3.6. Coreceptor usage predicitions
Genotypic prediction of coreceptor usage in the 3 viruses studied by us whose sequences of the envelope V3 region were obtained indicated that all were R5 viruses.
4. Discussion
The results of this study allow to define an HIV-1 CRF, designated CRF75_BF1, which is the 20th reported CRF derived from subtypes B and F1. Samples harboring CRF75_BF1 or related viruses were collected in 7 countries, but most were from Brazil, Spain, and Italy. Since all 4 individuals from Spain and all 3 from Italy for which data are available were native of the countries of sample collection, it seems that CRF75_BF1 is circulating in both countries among the native population. Transmission mode was men who have sex with men in 4 individuals from Spain and heterosexual in 3 from Italy for which information is available (Saladini et al., 2012).
CRF75_BF1 exhibits a complex mosaic structure, with 20 breakpoints, the largest breakpoint number of any reported CRF (the previous records were for CRF47_BF1 (Fernández-García et al., 2010), CRF18_cpx (Thomson et al., 2005), and CRF56_cpx (Colson et al., 2014), with 16 breakpoints). The highly complex mosaic structures found in CRF75_BF1 and some other CRFs reflect the elevated recombination potential of HIV-1, which can lead to the generation of highly complex recombinant forms through multiple consecutive rounds of recombination (Sierra et al., 2005). In this regard, the identified recombinant strain from Brazil with a mosaic structure slightly less complex than CRF75_BF1 (10BR_RJ053), with 16 breakpoints, 14 of which coincide with those of CRF75_BF1 (Supplementary Figure S6), could derive from an ancestral lineage that led to the generation of CRF75_BF1 through successive recombination events; and the Brazilian virus HI2016-28 (Figure 7B) and Spanish virus MD408339 (Supplementary Figure S3), that show in their available sequences coincidence in mosaic structures with CRF75_BF1, except for the presence of additional subtype B segments, could represent secondary recombinants of CRF75_BF1 with subtype B.
Although no Brazilian CRF75_BF1 virus has been sequenced in the NFLG, we believe that the BF13 cluster as a whole, including Brazilian viruses, most likely corresponds to the CRF75_BF1 clade. We infer this from the following facts and arguments:
The Brazilian virus 10BR_RJ053, for which the NFLG sequence is available, has 14 breakpoints coincident with CRF75_BF1 (Supplementary Figures S4, S6) and branches basally to the CRF75_BF1 Spanish-Italian cluster (Supplementary Figure S5). Therefore it seems reasonable to assume that it derives from a BF1 recombinant lineage ancestral to CRF75_BF1.
The Brazilian virus HI2016-28, for which sequences of two separate genome fragments summing around 6.1 kb are available, has a mosaic structure almost identical to the Spanish-Italian CRF75_BF1 viruses, with 13 coincident breakpoints, except for a subtype B fragment of around 0.5 kb in gag absent from CRF75_BF1 (Figure 7 and Supplementary Figure S4), and in a phylogenetic tree, it branches basally to the Spanish-Italian cluster and closer to it than 10BR_RJ053 (Supplementary Figure S5). It is most parsimonious to assume that the subtype B gag fragment of HI2016-28 that is absent from CRF75_BF1 probably derives from secondary recombination of a CRF75_BF1 virus with a subtype B virus, rather than CRF75_BF1 having being generated by secondary recombination of a HI2016-28-like virus, since the lineage ancestral to the Spanish-Italian CRF75_BF1 viruses and HI2016-28 represented by 10BR_RJ053 is of subtype F1 in the mentioned fragment, and B subtype viruses are much more common in Brazil than F1 subtype viruses, which renders much more probable the acquisition through recombination of a B subtype than of a F1subtype fragment. Since HI2016-28 branches within a Brazilian subcluster comprising 11 viruses (Figure 2), the possibility that it represents a new CRF_BF1 derived from secondary recombination of CRF75_BF1 with a subtype B virus should be considered.
The South African virus MSM305, for which the full-length envelope sequence (~2.6 kb) is available, has a mosaic structure in the envelope identical to CRF75_BF1 (Figure 7 and Supplementary Figure S4) and groups with HI2016-28 in the ~1.6 kb fragment in which they overlap (Supplementary Figure S5). Therefore, it is probably of Brazilian ancestry.
The UK viruses 152822 and 152884, for which pol fragments of ~2.8 kb are available, have mosaic structures identical to CRF75_BF1 (Figure 7 and Supplementary Figure S4) and group with two Brazilian viruses in Pr-RT and branch basally to the Spanish–Italian CRF75_BF1 cluster in the 2.8 kb pol fragment (Supplementary Figure S5). Therefore, they are also probably of Brazilian ancestry.
Consequently, although no Brazilian NFLG sequences of CRF75_BF1 viruses are available, by joining the fragments of two Brazilian viruses and two viruses of probable Brazilian ancestry showing mosaic structures coincident with CRF75_BF1, a Brazilian BF1 recombinant genome fragment of ~8.2 kb exhibiting a mosaic structure fully coincident with the Spanish-Italian CRF75_BF1 viruses can be reconstructed. Therefore, we can reasonably assume that the BF13 cluster probably represents the CRF75_BF1 clade, although the Brazilian subcluster of 11 viruses observed in Pr-RT (Figure 2) could represent a related CRF derived from secondary recombination of CRF75_BF1 with a subtype B virus. However, to prove or disprove these hypotheses, the analysis of NFLG sequences of Brazilian viruses of the BF13 cluster will be needed.
Two NFLG sequences and around 7 kb of a third genome of CRF75_BF1 viruses from Italy were analyzed in a paper published in Saladini et al. (2012). However, the authors failed to recognize that they represented a new CRF, since they observed small differences in recombinant structures, classifying them as URFs. It is difficult to know the reasons for the discrepant interpretations of analyses, since no bootscan plots and no phylogenetic trees of partial segments were shown in the previous paper.
Phylogeographic analyses place the origin of the clade comprising CRF75_BF1 and related viruses most probably in Brazil around 1984, with diffusion to Italy around 1992 and subsequent migration from Italy to Spain around 1999. The Brazilian ancestry of CRF75_BF1 is consistent with the circulation of subtypes B and F1 in the country, which have given rise to at least 12 reported CRF_BF1s that circulate in Brazil and a 13th (CRF47_BF1) circulating in Spain but originated in Brazil (Hill et al., 2022). The estimated year of origin of the CRF75_BF1 clade is congruent with the earlier estimated origin of the Brazilian F1 strain (around 1977) (Bello et al., 2006) and is similar to the emergence dates of most other South American CRF_BFs reported in the literature (Bello et al., 2010; Ristic et al., 2011; Reis et al., 2017; Bacqué et al., 2021; Delgado et al., 2021; Cañada-García et al., 2022).
CRF75_BF1 and related viruses are found at low prevalences in the countries where they circulate. In Brazil, in a large study on HIV-1 genetic diversity (Gräf et al., 2021), they are found in only 4 (0.05%) of 1,791 samples collected in 2017 (the last year of sample collection of the study). In samples collected in a hospital in central Italy in 1996–2004 (Vicenti et al., 2007), out of 169 sequenced, only 4 (2.3%) are of CRF75_BF1. And among 1,804 sequences from samples collected the region of Comunidad Valenciana, east Spain, in 2004–2014 (Patiño-Galindo et al., 2017), only 6 (0.3%) are CRF75_BF1 or related viruses. This implies that there are CRFs that are circulating at low prevalences that are only detected through the analyses of large numbers of samples, and, consequently, that there are probably many more HIV-1 CRFs than currently known, circulating in undersampled areas. Interestingly, in the cited studies in Brazil and eastern Spain, all CRF75_BF1 or related viruses were classified by the authors as F1 subsubtype viruses, a fact that exposes the limitations of the analysis of Pr-RT sequences for the classification of HIV-1 variants in molecular epidemiological studies.
With the identification of CRF75_BF1, a total of 6 CRFs of South American ancestry have been originally identified in Western Europe (5 CRF_BF1 and 1 CRF_BC) (Fernández-García et al., 2010; Simonetti et al., 2014; Struck et al., 2015; Bacqué et al., 2021; Delgado et al., 2021; Cañada-García et al., 2022). This, together with the propagation of HIV-1 variants of South American origin among the European population (De Oliveira et al., 2010; Collaço Verás et al., 2011; Thomson et al., 2012; Lai et al., 2014; Carvalho et al., 2015; Delgado et al., 2015; Fabeni et al., 2015; Vinken et al., 2019), points to a close relationship between the HIV-1 epidemics in both continents. This may reflect migratory phenomena, particularly in Spain, where around 2.7 million people of South American origin live (Instituto Nacional de Estadística, 2023). In Italy, immigration from South America is substantially lower, but still considerable, with nearly 270,000 South Americans living in this country, around 50,000 of whom are from Brazil (Istat, 2023).
An interesting finding of potential biological relevance is that all 4 patients harboring CRF75_BF1 viruses had CD4+ T-cell counts <220/mm3 less than 1 year after HIV-1 diagnosis, a proportion that was significantly higher than that found among 3,548 other patients diagnosed in Spain in the same period (29%). Although the number of CRF75_BF1 infections for which CD4+ T-cell counts are available is low, this finding suggests the possibility of a greater virulence of CRF75_BF1 that should be investigated in a larger number of patients. In this regard, associations with greater pathogenic potentials have been reported for CRF14_BG (Pérez-Álvarez et al., 2014) CRF19_cpx (Kouri et al., 2015), a CRF01_AE cluster (Song et al., 2019; Ge et al., 2021), and a subtype B cluster (Wymant et al., 2022), and CRF55_01B has been associated with lower CD4 T-cell counts than those found in CRF07_BC infections and higher viral loads than those found in CRF01_AE and CRF07_BC infections (Wei et al., 2021).
The identification and genetic characterization of HIV-1 CRFs may be of interest not only for examining their associated biological properties, but also for vaccine immunogen design, since greater intra-CRF susceptibilities to immune responses have been reported for some CRFs (Mascola et al., 1994; Hraber et al., 2014).
In summary, we have identified an HIV-1 CRF, designated CRF75_BF1, with a highly complex mosaic structure, involving at least 20 recombination events, that originated in Brazil and circulates in Brazil, Italy, and Spain, with sporadic cases detected in 4 other countries from 3 continents. The finding of low CD4+ T-cell counts early after diagnosis in the patients infected with CRF75_BF1 studied by us warrants further investigation on its pathogenic associations in a larger number of patients. The results of this and other studies also advocate for the implementation of molecular epidemiological surveillance systems for the rapid detection of the propagation and characterization of HIV-1 variants with potential increased transmission or pathogenic properties.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MK341078; https://www.ncbi.nlm.nih.gov/genbank/, OR466119; https://www.ncbi.nlm.nih.gov/genbank/, OR466120; https://www.ncbi.nlm.nih.gov/genbank/, OR466121.
Ethics statement
The studies involving humans were approved by Committee of Research Ethics, Instituto de Salud Carlos III, Majadahonda, Madrid, Spain. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because the study used samples and data collected as part of routine clinical practice and patients’ data were anonymized without retaining data allowing individual identification.
Author contributions
JB: Formal analysis, Investigation, Methodology, Writing – review & editing. ED: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Writing – review & editing. HG: Data curation, Writing – review & editing. SI: Writing – review & editing. SB: Methodology, Writing – review & editing. IG-A: Writing – review & editing. MM-L: Methodology, Writing – review & editing. ES: Writing – review & editing. CG-G: Writing – review & editing. MS: Methodology, Writing – review & editing. VM: Methodology, Writing – review & editing. MT: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Acknowledgments
We thank the personnel at the Genomic Unit, Instituto de Salud Carlos III, for technical assistance in sequencing.
Funding Statement
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded through Acción Estratégica en Salud Intramural (AESI), Instituto de Salud Carlos III, projects “Estudios sobre vigilancia epidemiológica molecular del VIH- 1 en España,” PI16CIII/00033, “Epidemiología molecular del VIH-1 en España y su utilidad para investigaciones biológicas y en vacunas,” PI19CIII/00042, and “Vigilancia epidemiológica molecular de la infección por VIH-1 en España y su aplicación en salud pública y en investigaciones biológicas,” PI22CIII/00065, and through a scientific agreement with Osakidetza-Servicio Vasco de Salud, Government of the Basque Country.
Footnotes
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1301374/full#supplementary-material
References
- Alves B. M., Siqueira J. D., Garrido M. M., Botelho O. M., Prellwitz I. M., Ribeiro S. R., et al. (2017). Characterization of HIV-1 near full-length proviral genome quasispecies from patients with undetectable viral load undergoing first-line HAART therapy. Viruses 9:392. doi: 10.3390/v9120392, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas M., Lorenzo-Redondo R., López-Galíndez C. (2016). Influence of mutation and recombination on HIV-1 in vitro fitness recovery. Mol. Phylogenet. Evol. 94, 264–270. doi: 10.1016/j.ympev.2015.09.001, PMID: [DOI] [PubMed] [Google Scholar]
- Bacqué J., Delgado E., Benito S., Moreno-Lorenzo M., Montero V., Gil H., et al. (2021). Identification of CRF66_BF, a new HIV-1 circulating recombinant form of south American origin. Front. Microbiol. 12:774386. doi: 10.3389/fmicb.2021.774386, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello G., Guimaraes M. L., Morgado M. G. (2006). Evolutionary history of HIV-1 subtype B and F infections in Brazil. AIDS 20, 763–768. doi: 10.1097/01.aids.0000216377.84313.52, PMID: [DOI] [PubMed] [Google Scholar]
- Bello G., Aulicino P. C., Ruchansky D., Guimaraes M. L., López-Galíndez C., Casado C., et al. (2010). Phylodynamics of HIV-1 circulating recombinant forms 12_BF and 38_BF in Argentina and Uruguay. Retrovirology 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañada J. E., Delgado E., Gil H., Sánchez M., Benito S., García-Bodas E., et al. (2021). Identification of a new HIV-1 BC intersubtype circulating recombinant form (CRF108_BC) in Spain. Viruses 13:93. doi: 10.3390/v13010093, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cañada-García J. E., Delgado E., Gil H., Benito S., Sánchez M., Ocampo A., et al. (2022). Viruses previously identified in Brazil as belonging to HIV-1 CRF72_BF1 represent two closely related circulating recombinant forms, one of which, designated CRF122_BF1, is also circulating in Spain. Front. Microbiol. 13:863084. doi: 10.3389/fmicb.2022.863084, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr J. K., Avila M., Gomez C. M., Salomon H., Hierholzer J., Watanaveeradej V., et al. (2001). Diverse BF recombinants have spread widely since the introduction of HIV-1 into South America. AIDS 15, F41–F47. doi: 10.1097/00002030-200110190-00002, PMID: [DOI] [PubMed] [Google Scholar]
- Carvalho A., Costa P., Triunfante V., Branca F., Rodrigues F., Santos C. L., et al. (2015). Analysis of a local HIV-1 epidemic in Portugal highlights established transmission of non-B and non-G subtypes. J. Clin. Microbiol. 53, 1506–1514. doi: 10.1128/JCM.03611-14, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier C., Nora T., Tenaillon O., Clavel F., Hance A. J. (2006). Extensive recombination among human immunodeficiency virus type 1 quasispecies makes an important contribution to viral diversity in individual patients. J. Virol. 80, 2472–2482. doi: 10.1128/JVI.80.5.2472-2482.2006, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaço Verás N. M., Gray R. R., de Macedo Brígido L. F., Rodrigues R., Salemi M. (2011). High-resolution phylogenetics and phylogeography of human immunodeficiency virus type 1 subtype C epidemic in South America. J. Gen. Virol. 92, 1698–1709. doi: 10.1099/vir.0.028951-0 [DOI] [PubMed] [Google Scholar]
- Colson P., Ravaux I., Tourres C., Stein A., Tamalet C. (2014). Detection of the newly characterized HIV CRF56_cpx in Marseille, southeastern France. Int. J. Infect. Dis. 29, 241–243. doi: 10.1016/j.ijid.2014.10.020 [DOI] [PubMed] [Google Scholar]
- De Oliveira T., Pillay D., Gifford R. J., Resistance, UK Collaborative Group on HIV Drug Resistance (2010). The HIV-1 subtype C epidemic in South America is linked to the United Kingdom. PLoS One 5:e9311. doi: 10.1371/journal.pone.0009311, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado E., Cuevas M. T., Domínguez F., Vega Y., Cabello M., Fernández-García A., et al. (2015). Phylogeny and phylogeography of a recent HIV-1 subtype F outbreak among men who have sex with men in Spain deriving from a cluster with a wide geographic circulation in Western Europe. PLoS One 10:e0143325. doi: 10.1371/journal.pone.0143325, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado E., Fernández-García A., Pérez-Losada M., Moreno-Lorenzo M., Fernández-Miranda I., Benito S., et al. (2021). Identification of CRF89_BF, a new member of an HIV-1 circulating BF intersubtype recombinant form family widely spread in South America. Sci. Rep. 11:11442. doi: 10.1038/s41598-021-90023-x, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado E., Thomson M. M., Villahermosa M. L., Sierra M., Ocampo A., Miralles C., et al. (2002). Identification of a newly characterized HIV-1 BG intersubtype circulating recombinant form in Galicia, Spain, which exhibits a pseudotype-like virion structure. J. Acquir. Immune Defic. Syndr. 29, 536–543. doi: 10.1097/00126334-200204150-00016, PMID: [DOI] [PubMed] [Google Scholar]
- Fabeni L., Alteri C., Orchi N., Gori C., Bertoli A., Forbici F., et al. (2015). Recent transmission clustering of HIV-1 C and CRF17_BF strains characterized by NNRTI-related mutations among newly diagnosed men in Central Italy. PLoS One 10:e0135325. doi: 10.1371/journal.pone.0135325, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-García A., Pérez-Álvarez L., Cuevas M. T., Delgado E., Muñoz-Nieto M., Cilla G., et al. (2010). Identification of a new HIV type 1 circulating BF intersubtype recombinant form (CRF47_BF) in Spain. AIDS Res. Hum. Retrovir. 26, 827–832. doi: 10.1089/aid.2009.0311, PMID: [DOI] [PubMed] [Google Scholar]
- Ge Z., Feng Y., Li K., Lv B., Zaongo S. D., Sun J., et al. (2021). CRF01_AE and CRF01_AE cluster 4 are associated with poor immune recovery in Chinese patients under combination antiretroviral therapy. Clin. Infect. Dis. 72, 1799–1809. doi: 10.1093/cid/ciaa380, PMID: [DOI] [PubMed] [Google Scholar]
- Gifford R. J., Liu T. F., Rhee S. Y., Kiuchi M., Hue S., Pillay D., et al. (2009). The calibrated population resistance tool: standardized genotypic estimation of transmitted HIV-1 drug resistance. Bioinformatics 25, 1197–1198. doi: 10.1093/bioinformatics/btp134, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil H., Delgado E., Benito S., Georgalis L., Montero V., Sánchez M., et al. (2022). Transmission clusters, predominantly associated with men who have sex with men, play a main role in the propagation of HIV-1 in northern Spain (2013-2018). Front. Microbiol. 13:782609. doi: 10.3389/fmicb.2022.782609, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. S., Lemey P., Faria N. R., Rambaut A., Shapiro B., Suchard M. A. (2013). Improving Bayesian population dynamics inference: a coalescent-based model for multiple loci. Mol. Biol. Evol. 30, 713–724. doi: 10.1093/molbev/mss265, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräf T., Bello G., Andrade P., Arantes I., Pereira J. M., da Silva A. B. P., et al. (2021). HIV-1 molecular diversity in Brazil unveiled by 10 years of sampling by the national genotyping network. Sci. Rep. 11:15842. doi: 10.1038/s41598-021-94542-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S., Dufayard J. F., Lefort V., Anisimova M., Hordijk W., Gascuel O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010, PMID: [DOI] [PubMed] [Google Scholar]
- Guindon S., Gascuel O. (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520 [DOI] [PubMed] [Google Scholar]
- Gundlach B. R., Lewis M. G., Sopper S., Schnell T., Sodroski J., Stahl-Hennig C., et al. (2000). Evidence for recombination of live, attenuated immunodeficiency virus vaccine with challenge virus to a more virulent strain. J. Virol. 74, 3537–3542. doi: 10.1128/JVI.74.8.3537-3542.2000, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemelaar J., Elangovan R., Yun J., Dickson-Tetteh L., Kirtley S., Gouws-Williams E., et al. (2020). Global and regional epidemiology of HIV-1 recombinants in 1990–2015: a systematic review and global survey. Lancet HIV 7, e772–e781. doi: 10.1016/S2352-3018(20)30252-6 [DOI] [PubMed] [Google Scholar]
- Hill G., Pérez-Losada M., Delgado E., Benito S., Montero V., Gil H., et al. (2022). The origin, epidemiology, and phylodynamics of human immunodeficiency virus type 1 CRF47_BF. Front. Microbiol. 13:863123. doi: 10.3389/fmicb.2022.863123, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIV Sequence Database . (2023). Available at: https://www.hiv.lanl.gov/content/sequence/HIV/mainpage.html (Accessed, August 9, 2023).
- Hoang D. T., Chernomor O., von Haeseler A., Minh B. Q., Vinh L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hraber P., Korber B. T., Lapedes A. S., Bailer R. T., Seaman M. S., Gao H., et al. (2014). Impact of clade, geography, and age of the epidemic on HIV-1 neutralization by antibodies. J. Virol. 88, 12623–12643. doi: 10.1128/JVI.01705-14, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Instituto Nacional de Estadística (2023). Principales series de población desde 1998. Available at: https://www.ine.es/jaxi/Tabla.htm?path=/t20/e245/p08/l0/&file=01005.px&L=0 (Accessed August 11, 2023).
- Istat . (2023). Istituto Nazionale di Statistica. Available at: https://demo.istat.it/app/?i=P03&a=2020 (Accessed August 11, 2023).
- Katoh K., Standley D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouri V., Khouri R., Alemán Y., Abrahantes Y., Vercauteren J., Pineda-Peña A. C., et al. (2015). CRF19_cpx is an evolutionary fit HIV-1 variant strongly associated with rapid progression to AIDS in Cuba. EBioMedicine 2, 244–254. doi: 10.1016/j.ebiom.2015.01.015, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016). MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai A., Bozzi G., Franzetti M., Binda F., Simonetti F. R., Micheli V., et al. (2014). Phylogenetic analysis provides evidence of interactions between Italian heterosexual and south American homosexual males as the main source of national HIV-1 subtype C epidemics. J. Med. Virol. 86, 729–736. doi: 10.1002/jmv.23891, PMID: [DOI] [PubMed] [Google Scholar]
- Lefort V., Longueville J. E., Gascuel O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer T., Sander O., Sierra S., Thielen A., Kaiser R. (2007). Bioinformatics prediction of HIV coreceptor usage. Nat. Biotechnol. 25, 1407–1410. doi: 10.1038/nbt1371, PMID: [DOI] [PubMed] [Google Scholar]
- Lole K. S., Bollinger R. C., Paranjape R. S., Gadkari D., Kulkarni S. S., Novak N. G., et al. (1999). Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 73, 152–160. doi: 10.1128/JVI.73.1.152-160.1999, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascola J. R., Louwagie J., McCutchan F. E., Fischer C. L., Hegerich P. A., Wagner K. F., et al. (1994). Two antigenically distinct subtypes of human immunodeficiency virus type 1: viral genotype predicts neutralization serotype. J. Infect. Dis. 169, 48–54. doi: 10.1093/infdis/169.1.48, PMID: [DOI] [PubMed] [Google Scholar]
- Middelkoop K., Rademeyer C., Brown B. B., Cashmore T. J., Marais J. C., Scheibe A. P., et al. (2014). Epidemiology of HIV-1 subtypes among men who have sex with men in Cape Town, South Africa. J. Acquir. Immune Defic. Syndr. 65, 473–480. doi: 10.1097/QAI.0000000000000067, PMID: [DOI] [PubMed] [Google Scholar]
- Moradigaravand D., Kouyos R., Hinkley T., Haddad M., Petropoulos C. J., Engelstadter J., et al. (2014). Recombination accelerates adaptation on a large-scale empirical fitness landscape in HIV-1. PLoS Genet. 10:e1004439. doi: 10.1371/journal.pgen.1004439, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nájera R., Delgado E., Pérez-Álvarez L., Thomson M. M. (2002). Genetic recombination and its role in the development of the HIV-1 pandemic. AIDS 16, S3–S16. doi: 10.1097/00002030-200216004-00002, PMID: [DOI] [PubMed] [Google Scholar]
- Nora T., Charpentier C., Tenaillon O., Hoede C., Clavel F., Hance A. J. (2007). Contribution of recombination to the evolution of human immunodeficiency viruses expressing resistance to antiretroviral treatment. J. Virol. 81, 7620–7628. doi: 10.1128/JVI.00083-07, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patiño-Galindo J. Á., Torres-Puente M., Bracho M. A., Alastrué I., Juan A., Navarro D., et al. (2017). The molecular epidemiology of HIV-1 in the Comunidad Valenciana (Spain): analysis of transmission clusters. Sci. Rep. 7:11584. doi: 10.1038/s41598-017-10286-1, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Álvarez L., Delgado E., Vega Y., Montero V., Cuevas T., Fernández-García A., et al. (2014). Predominance of CXCR4 tropism in HIV-1 CRF14_BG strains from newly diagnosed infections. J. Antimicrob. Chemother. 69, 246–253. doi: 10.1093/jac/dkt305, PMID: [DOI] [PubMed] [Google Scholar]
- Pessoa R., Watanabe J. T., Calabria P., Alencar C. S., Loureiro P., Lopes M. E., et al. (2015). Enhanced detection of viral diversity using partial and near full-length genomes of human immunodeficiency virus type 1 provirus deep sequencing data from recently infected donors at four blood centers in Brazil. Transfusion 55, 980–990. doi: 10.1111/trf.12936, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., Arkin A. P. (2010). FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Lam T. T., Max C. L., Pybus O. G. (2016). Exploring the temporal structure of heterochronous sequences using TempEst (formerly path-O-gen). Virus Evol. 2. doi: 10.1093/ve/vew007, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis M., Bello G., Guimaraes M. L., Stefani M. M. A. (2017). Characterization of HIV-1 CRF90_BF1 and putative novel CRFs_BF1 in central west, north and northeast Brazilian regions. PLoS One 12:e0178578. doi: 10.1371/journal.pone.0178578, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ristic N., Zukurov J., Alkmim W., Diaz R. S., Janini L. M., Chin M. P. (2011). Analysis of the origin and evolutionary history of HIV-1 CRF28_BF and CRF29_BF reveals a decreasing prevalence in the AIDS epidemic of Brazil. PLoS One 6:e17485. doi: 10.1371/annotation/25854e06-a961-4fec-8664-38667cc278b8, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie A. J., Cai F., Smith N. M., Chen S., Song H., Brackenridge S., et al. (2014). Recombination-mediated escape from primary CD8+ T cells in acute HIV-1 infection. Retrovirology 11:69. doi: 10.1186/s12977-014-0069-9, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson D. L., Anderson J. P., Bradac J. A., Carr J. K., Foley B., Funkhouser R. K., et al. (2000). HIV-1 nomenclature proposal. Science 288, 55–56. doi: 10.1126/science.288.5463.55d, PMID: [DOI] [PubMed] [Google Scholar]
- Ronquist F., Teslenko M., van der Mark P., Ayres D. L., Darling A., Hohna S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saladini F., Foley B. T., Rosi A., Vicenti I., Nannetti G., Meini G., et al. (2012). Near full-length sequence analysis of HIV type 1 BF recombinants from Italy. AIDS Res. Hum. Retrovir. 28, 299–303. doi: 10.1089/aid.2011.0002, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen M. O., Carr J. K., Burke D. S., McCutchan F. E. (1995). Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 11, 1423–1425. doi: 10.1089/aid.1995.11.1423, PMID: [DOI] [PubMed] [Google Scholar]
- Seager I., Travers S. A., Leeson M. D., Crampin A. C., French N., Glynn J. R., et al. (2014). Coreceptor usage, diversity, and divergence in drug-naive and drug-exposed individuals from Malawi, infected with HIV-1 subtype C for more than 20 years. AIDS Res. Hum. Retrovir. 30, 975–983. doi: 10.1089/aid.2013.0240, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro B., Rambaut A., Drummond A. J. (2006). Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol. Biol. Evol. 23, 7–9. doi: 10.1093/molbev/msj021, PMID: [DOI] [PubMed] [Google Scholar]
- Sierra M., Thomson M. M., Ríos M., Casado G., Castro R. O., Delgado E., et al. (2005). The analysis of near full-length genome sequences of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Chile, Venezuela and Spain reveals their relationship to diverse lineages of recombinant viruses related to CRF12_BF. Infect. Genet. Evol. 5, 209–217. doi: 10.1016/j.meegid.2004.07.010, PMID: [DOI] [PubMed] [Google Scholar]
- Simonetti F. R., Lai A., Monno L., Binda F., Brindicci G., Punzi G., et al. (2014). Identification of a new HIV-1 BC circulating recombinant form (CRF60_BC) in Italian young men having sex with men. Infect. Genet. Evol. 23, 176–181. doi: 10.1016/j.meegid.2014.02.007, PMID: [DOI] [PubMed] [Google Scholar]
- Song H., Giorgi E. E., Ganusov V. V., Cai F., Athreya G., Yoon H., et al. (2018). Tracking HIV-1 recombination to resolve its contribution to HIV-1 evolution in natural infection. Nat. Commun. 9:1928. doi: 10.1038/s41467-018-04217-5, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H., Ou W., Feng Y., Zhang J., Li F., Hu J., et al. (2019). Disparate impact on CD4 T cell count by two distinct HIV-1 phylogenetic clusters from the same clade. Proc. Natl. Acad. Sci. U. S. A. 116, 239–244. doi: 10.1073/pnas.1814714116, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streeck H., Li B., Poon A. F., Schneidewind A., Gladden A. D., Power K. A., et al. (2008). Immune-driven recombination and loss of control after HIV superinfection. J. Exp. Med. 205, 1789–1796. doi: 10.1084/jem.20080281, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struck D., Roman F., De L. S., Servais J. Y., Lambert C., Masquelier C., et al. (2015). Near full-length characterization and population dynamics of the human immunodeficiency virus type I circulating recombinant form 42 (CRF42_BF) in Luxembourg. AIDS Res. Hum. Retrovir. 31, 554–558. doi: 10.1089/aid.2014.0364, PMID: [DOI] [PubMed] [Google Scholar]
- Suchard M. A., Lemey P., Baele G., Ayres D. L., Drummond A. J., Rambaut A. (2018). Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus. Evol. 4. doi: 10.1093/ve/vey016, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson M. M., Casado G., Posada D., Sierra M., Nájera R. (2005). Identification of a novel HIV-1 complex circulating recombinant form (CRF18_cpx) of Central African origin in Cuba. AIDS 19, 1155–1163. doi: 10.1097/01.aids.0000176215.95119.1d [DOI] [PubMed] [Google Scholar]
- Thomson M. M., Delgado E., Herrero I., Villahermosa M. L., Vázquez de Parga E., Cuevas M. T., et al. (2002). Diversity of mosaic structures and common ancestry of human immunodeficiency virus type 1 BF intersubtype recombinant viruses from Argentina revealed by analysis of near full-length genome sequences. J. Gen. Virol. 83, 107–119. doi: 10.1099/0022-1317-83-1-107 [DOI] [PubMed] [Google Scholar]
- Thomson M. M., Fernández-García A. (2011). Phylogenetic structure in African HIV-1 subtype C revealed by selective sequential pruning. Virology 415, 30–38. doi: 10.1016/j.virol.2011.03.021, PMID: [DOI] [PubMed] [Google Scholar]
- Thomson M. M., Fernandez-García A., Delgado E., Vega Y., Díez-Fuertes F., Sánchez-Martínez M., et al. (2012). Rapid expansion of a HIV-1 subtype F cluster of recent origin among men who have sex with men in Galicia, Spain. J. Acquir. Immune Defic. Syndr. 59, e49–e51. doi: 10.1097/QAI.0b013e3182400fc4, PMID: [DOI] [PubMed] [Google Scholar]
- Thomson M. M., Villahermosa M. L., Vázquez-de-Parga E., Cuevas M. T., Delgado E., Manjón N., et al. (2000). Widespread circulation of a B/F intersubtype recombinant form among HIV-1-infected individuals in Buenos Aires, Argentina. AIDS 14, 897–899. doi: 10.1097/00002030-200005050-00020, PMID: [DOI] [PubMed] [Google Scholar]
- Travers S. A., Clewley J. P., Glynn J. R., Fine P. E., Crampin A. C., Sibande F., et al. (2004). Timing and reconstruction of the most recent common ancestor of the subtype C clade of human immunodeficiency virus type 1. J. Virol. 78, 10501–10506. doi: 10.1128/JVI.78.19.10501-10506.2004, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifinopoulos J., Nguyen L. T., von Haeseler A., Minh B. Q. (2016). W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–W235. doi: 10.1093/nar/gkw256, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove L. P., Wensing A. M., Kaiser R., Brun-Vézinet F., Clotet B., De Luca A., et al. (2011). European guidelines on the clinical management of HIV-1 tropism testing. Lancet Infect. Dis. 11, 394–407. doi: 10.1016/S1473-3099(10)70319-4, PMID: [DOI] [PubMed] [Google Scholar]
- Vicenti I., Razzolini F., Saladini F., Romano L., Zazzi M. (2007). Use of peripheral blood DNA for genotype antiretroviral resistance testing in drug-naive HIV- infected subjects. Clin. Infect. Dis. 44, 1657–1661. doi: 10.1086/518287, PMID: [DOI] [PubMed] [Google Scholar]
- Vinken L., Fransen K., Cuypers L., Alexiev I., Balotta C., Debaisieux L., et al. (2019). Earlier initiation of antiretroviral treatment coincides with an initial control of the HIV-1 sub-subtype F1 outbreak among men-having-sex-with-men in Flanders, Belgium. Front. Microbiol. 10:613. doi: 10.3389/fmicb.2019.00613, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei L., Li H., Lv X., Zheng C., Li G., Yang Z., et al. (2021). Impact of HIV-1 CRF55_01B infection on the evolution of CD4 count and plasma HIV RNA load in men who have sex with men prior to antiretroviral therapy. Retrovirology 18:22. doi: 10.1186/s12977-021-00567-z, PMID: [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A., Menon S., Crowe M., Agarwal N., Biccler J., Bbosa N., et al. (2023). Geographic and population distributions of HIV-1 and HIV-2 circulating subtypes: a systematic literature review and meta-analysis (2010-2021). J Infect Dis. (in press). doi: 10.1093/infdis/jiad327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymant C., Bezemer D., Blanquart F., Ferretti L., Gall A., Hall M., et al. (2022). A highly virulent variant of HIV-1 circulating in the Netherlands. Science 375, 540–545. doi: 10.1126/science.abk1688, PMID: [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MK341078; https://www.ncbi.nlm.nih.gov/genbank/, OR466119; https://www.ncbi.nlm.nih.gov/genbank/, OR466120; https://www.ncbi.nlm.nih.gov/genbank/, OR466121.