

Abstract
Glucokinase (GK) catalyzes the phosphorylation of glucose to form glucose‐6‐phosphate as the substrate of glycolysis for energy production. Acetylation of lysine residues in Escherichia coli GK has been identified at multiple sites by a series of proteomic studies, but the impact of acetylation on GK functions remains largely unknown. In this study, we applied the genetic code expansion strategy to produce site‐specifically acetylated GK variants which naturally exist in cells. Enzyme assays and kinetic analyses showed that lysine acetylation decreases the GK activity, mostly resulting from acetylation of K214 and K216 at the entrance of the active site, which impairs the binding of substrates. We also compared results obtained from the glutamine substitution method and the genetic acetyllysine incorporation approach, showing that glutamine substitution is not always effective for mimicking acetylated lysine. Further genetic studies as well as in vitro acetylation and deacetylation assays were performed to determine acetylation and deacetylation mechanisms, which showed that E. coli GK could be acetylated by acetyl‐phosphate without enzymes and deacetylated by CobB deacetylase.
Keywords: deacetylase, genetic code expansion, glucokinase, glycolysis, lysine acetylation
1. INTRODUCTION
Among sugar kinases, hexokinase (HK) and ribokinase (RK) are two distinct families which can phosphorylate glucose (Bork et al., 1993). The HK family uses ATP or polyphosphate as the phosphoryl donor and exists in eukaryotes, bacteria, and archaea, while the RK family uses ADP as the phosphoryl donor and can be found in mammals and Euryarchaeota (Kawai et al., 2005). The HK family in eukaryotes usually has isoenzymes. Humans have four HKs (HK I to HK IV). Only HK IV, named as glucokinase (GK), is specific for glucose and has a much lower affinity of glucose than the other three HKs (Wilson, 1995). GK abnormalities in human can cause type‐II diabetes and neurodegenerative diseases (Ashcroft et al., 2023; Han et al., 2021). GK in microbes can be further categorized into three classes. Class I consists of ATP‐ and ADP‐dependent GKs from archaea (Sakuraba et al., 2004). Class II consists of ATP‐dependent GKs from bacteria, which do not have the repressor open reading frame kinase (ROK) sequence motif CXCGXXGCXE (Titgemeyer et al., 1994). Escherichia coli GK belongs to this group (Fukuda et al., 1983). Class III consists of ATP‐dependent GKs from both archaea and bacteria, which have the ROK sequence motif (Mesak et al., 2004).
Historically, the interest in E. coli GK has been relatively low, mostly because it is not essential for glucose utilization (Curtis & Epstein, 1975). In E. coli and most other bacteria, glucose is phosphorylated when transported by the phosphoenolpyruvate:carbohydrate phosphotransferase system (PTS), thus bypassing the GK function (Postma et al., 1993). However, GK is still important for phosphorylating free intracellular glucose which can arise from disaccharide hydrolysis or PTS‐independent glucose uptake (Hernandez‐Montalvo et al., 2003; Meyer et al., 1997). E. coli GK can also regulate maltose and maltodextrin utilization (Lengsfeld et al., 2009). Recently, GK has obtained industrial interests to be utilized in engineered E. coli strains for increasing production of a wide range of chemicals such as aromatic compounds (Minliang et al., 2021), ethanol (Hernandez‐Montalvo et al., 2003), succinate (Zhu et al., 2020), NADPH (Yuan et al., 2022), hydroxybutyrate (Li et al., 2020), and dihydroxyphenylalanine (Wei et al., 2016). Thus, it is important to investigate the regulation of GK in E. coli which has been less studied before.
During the last decade, a series of proteomic studies have found that lysine acetylation also occurs extensively in bacteria, preferably existing in metabolic enzymes (Carabetta & Cristea, 2017; Christensen, Baumgartner, et al., 2019). Further studies have shown the important role of lysine acetylation in regulating bacterial metabolism (Christensen, Xie, et al., 2019; Liu et al., 2021; VanDrisse & Escalante‐Semerena, 2019). When searching the acetylome database, E. coli GK has multiple lysine acetylation sites identified by proteomic studies (Xu et al., 2017). However, the role of lysine acetylation in regulating GK functions remains largely unknown.
The traditional method to study protein acetylation is to use glutamine as the mimic of acetylated lysine residues. However, glutamine and acetyllysine (AcK) have different lengths of side chains, which could have different impacts on protein functions. Indeed, our previous study on isocitrate dehydrogenase showed that replacement of lysine with glutamine or AcK had different effects on enzyme activities (Venkat et al., 2018). Thus, the most accurate approach to study protein acetylation is to generate site‐specifically acetylated proteins. For this purpose, the genetic code expansion strategy has been applied, which introduces an orthogonal pair of engineered pyrrolysyl‐tRNA synthetase and tRNAPyl to decode an assigned codon (usually a stop codon) as AcK in the target protein (Neumann et al., 2008). As the assigned codon can be easily introduced into a specific position of the target gene by site‐directed mutagenesis, it is feasible to produce the target protein with lysine acetylation at a specific site. In this study, we used the genetic code expansion approach to characterize the effects of lysine acetylation on GK functions as well as the processes of GK acetylation and deacetylation.
2. RESULTS
2.1. Generation of site‐specifically acetylated E. coli GK variants
To date, a series of acetylome studies have identified multiple lysine acetylation sites in E. coli GK (Baeza et al., 2014; Castano‐Cerezo et al., 2014; Colak et al., 2013; Kuhn et al., 2014; Meyer et al., 2016; Schilling et al., 2015; Weinert et al., 2013, 2017; Zhang et al., 2013). However, because of the differences in E. coli strains tested, enrichment methods, detection methods, and identification resolutions, the pools of acetylation sites identified in those studies were not completely overlapped. To understand the role of lysine acetylation, we aimed to study the effects of acetylation on GK functions site‐by‐site. For this purpose, we selected seven lysine residues identified by at least two independent acetylome studies to avoid experimental biases, which are K53, K128, K149, K214, K216, K273, and K288.
We first generated site‐specifically acetylated E. coli GK variants to biochemically characterize the impact of lysine acetylation on GK functions. To achieve this goal, we applied the genetic code expansion approach and used the optimized system for AcK incorporation (Venkat, Gregory, Meng, et al., 2017). E. coli cells usually have background levels of protein acetylation globally, which may interfere our study. To solve this problem, the BL21(DE3) E. coli strain was used to minimize background protein acetylation. E. coli B strains have an activated glyoxylate shunt and a higher acetate consumption rate, so they have lower intracellular acetate concentrations which are associated with global acetylation levels (Weinert et al., 2013). The lysine codon at the corresponding position in the glk gene (the gene of E. coli GK) was mutated to a TAG stop codon as the sign for AcK incorporation. For easy purification by affinity chromatography, we fused a His6‐tag to the C‐terminus of GK. There is another reason for this design. Only when the inserted stop codon is decoded as AcK as planned, full‐length GK with the His6‐tag can be generated and purified with affinity chromatography. If normal termination at the introduced stop codon happens, truncated GK will be generated without the C‐terminal His6‐tag, which can be easily removed during washing steps in purification for high homogeneity of target proteins. Western blotting with the AcK antibody showed that all the seven GK variants produced by the genetic code expansion strategy were acetylated, while WT GK had non‐detectable acetylation in the same condition (Figures 1 and S1). Liquid chromatography–mass spectrometry (LC–MS/MS) analyses further confirmed the correct acetylation site in individual GK variant (Figures S2–S8).
FIGURE 1.
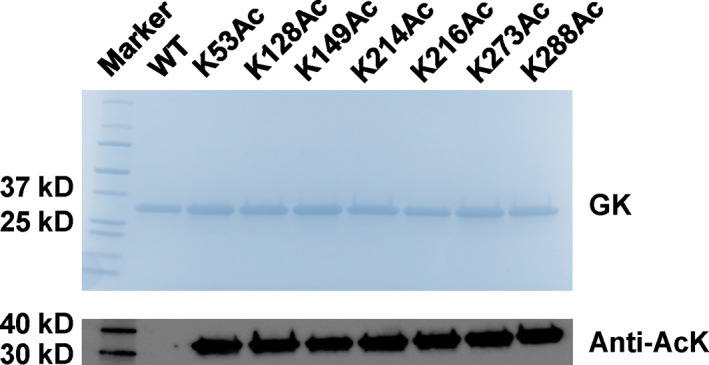
The incorporation of acetyllysine (AcK) at individual lysine acetylation site of Escherichia coli glucokinase (GK). Sodium dodecyl‐sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting analyses of purified GK and its variants from BL21(DE3) cells. Lane 1, marker; Lane 2, wild‐type GK; Lanes 3–9, acetylated GK variant at individual lysine site. The same amounts of proteins were loaded for each lane. The full picture of western blotting is in Figure S1.
2.2. The site‐specific effects of lysine acetylation on GK functions
To illustrate the effects of lysine acetylation on GK functions site‐by‐site, we measured enzyme activities of WT GK and its acetylated variants, individually (Figure 2a). Enzyme assay results showed that most of the acetylated GK variants had similar activities to WT GK. Two acetylated variants had significantly decreased activities, K214 (p < 0.0001) and K216 (p < 0.0001), which retained 43% and 34% of the WT activity, respectively. Proteomics studies have shown that K214 and K216 are acetylated simultaneously in cells (Schilling et al., 2015; Weinert et al., 2013; Zhang et al., 2013), so we generated a GK variant with acetylation at both K214 and K216 by mutating both lysine codons of K214 and K216 in the gene of GK to stop codons for AcK incorporation by genetic code expansion. The resulting doubly acetylated GK variant had an even lower activity, retaining only 22% of the WT activity.
FIGURE 2.
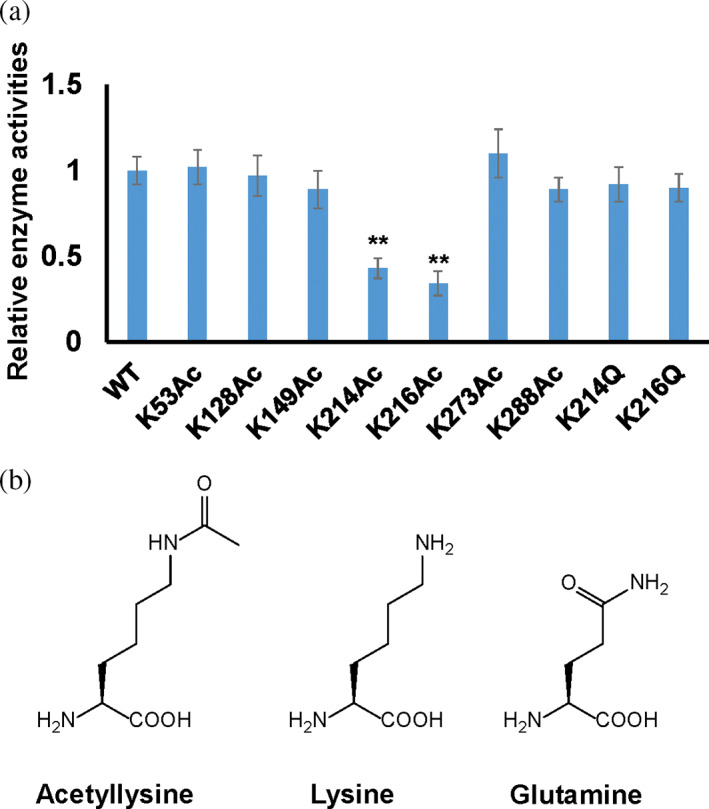
Relative enzyme activities of Escherichia coli GK and its variants. (a) The activity of WT E. coli GK was set as 1. Mean values and standard deviations were calculated based on three replicates. Two‐tailed p values were determined by the t‐test, and the significance level is 0.05. **p Value is less than 0.001. (b) The structures of acetyllysine, lysine, and glutamine.
To better understand how acetylation at K214 and K216 decreases the GK activity, we performed steady‐state kinetic analyses of WT GK and two acetylated variants at K214 or K216, individually (Table 1). Results showed that acetylation increased K m values for both substrates (ATP and glucose) but did not affect the turnover number k cat significantly. The catalytic efficiencies (k cat/K m) of acetylated variants at K214 and K216 decreased nearly 10‐fold compared to that of WT. It indicates that acetylation of those two lysine residues mostly affects substrate binding rather than impairing catalysis. Glutamine is commonly used as the mimic of acetylated lysine residue in protein acetylation studies. Thus, we also generated two K‐to‐Q variants (GK‐K214Q and GK‐K216Q) and performed enzyme activity assays and kinetic analyses (Figure 2a; Table 1). Interestingly, both K‐to‐Q variants had similar activities and kinetic parameters to WT GK but not acetylated GK variants. Although both the glutamine substitution and AcK neutralize the positive charge of the lysine residue, their sizes and hydrophobicity are different. The side chain of glutamine is 1.5 Å shorter than that of lysine, while AcK is 3 Å longer than lysine. The side chain of glutamine is polar, while the side chain of AcK is hydrophobic (Figure 2b). Kinetic results indicated that major factors affecting the GK activity at these two lysine sites are the size and hydrophobicity. It also demonstrated that glutamine is not always a sufficient substitution for an acetylated lysine residue.
TABLE 1.
Steady‐state kinetic parameters of Escherichia coli GK and its acetylated variants.
| k cat (s−1) | K m,glucose (mM) | k cat/K m,glucose (S−1 mM−1) | K m,ATP (mM) | k cat/K m,ATP (S−1 mM−1) | |
|---|---|---|---|---|---|
| WT GK | 135 ± 9 | 0.33 ± 0.08 | 409 | 0.43 ± 0.04 | 312 |
| GK‐K214Ac | 124 ± 11 | 2.73 ± 0.35 | 45.4 | 2.99 ± 0.26 | 41.5 |
| GK‐K216Ac | 132 ± 8 | 3.02 ± 0.21 | 43.7 | 3.91 ± 0.30 | 33.8 |
| GK‐K214Q | 137 ± 7 | 0.35 ± 0.10 | 391 | 0.50 ± 0.02 | 274 |
| GK‐K216Q | 129 ± 7 | 0.32 ± 0.15 | 403 | 0.45 ± 0.06 | 287 |
2.3. Acetylation of E. coli GK
There are two known mechanisms for lysine acetylation in E. coli, acetylation by protein lysine acetyltransferases (KAT) with acetyl‐CoA (AcCoA) as the acetyl‐donor and chemical acetylation using acetyl‐phosphate (AcP) as the acetyl‐donor (Carabetta & Cristea, 2017; Hentchel & Escalante‐Semerena, 2015). To date, YfiQ (originally named as Pat) is the only well‐studied KAT in E. coli (Starai & Escalante‐Semerena, 2004), so we first tested the enzymatic acetylation of E. coli GK by YfiQ. The acetylation level of native GK purified from the WT or ∆yfiQ strain of E. coli BW25113 cells grown in media with 0.2% glucose was determined by western blotting (Figures 3a and S9). The acetylation level of GK in ∆yfiQ cells was not significantly lower than that in WT cells, suggesting that YfiQ is not the major acetyltransferase to acetylate GK. We further implemented an in vitro experiment for YifQ acetylation. We showed that purified WT GK recombinantly expressed from BL21(DE3) cells had non‐detectable acetylation (Figure 1, Lane 1). Then we incubated purified WT GK with purified YfiQ and 0.2 mM AcCoA and determined the acetylation level of GK after YfiQ treatment by western blotting. Consistent with the above in vivo experiment, in vitro incubation with YfiQ could not acetylate purified GK (Figures 3b and S10). Besides YfiQ, one recent study identified four additional KATs (RimI, YiaC, YjaB, and PhnO) in E. coli (Christensen et al., 2018). When searching the proteomic data from this study, we found that overexpression of these four KATs could not increase GK acetylation. Thus, enzymatic acetylation is not the major mechanism of GK acetylation in E. coli.
FIGURE 3.
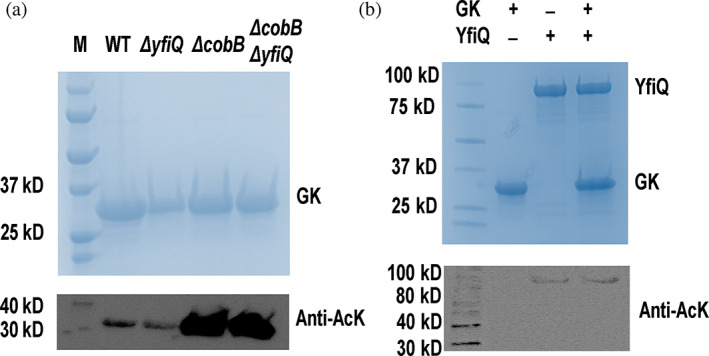
Tests of YfiQ‐mediated acetylation of Escherichia coli glucokinase (GK). SDS–PAGE and western blotting of (a) purified native GK from the WT, ΔyfiQ, ΔcobB, or ΔyfiQ ΔcobB of BW25113 cells growth in 0.2% glucose media. The full picture of western blotting is in Figure S9. (b) Purified WT GK from BL21(DE3) cells incubated with purified YfiQ and 0.2 mM AcCoA for 1 h. YfiQ itself was slightly acetylated when purified from the A Complete Set of Escherichia coli K‐12 ORF Archive (ASKA) collection. The full picture of western blotting is in Figure S10. AcK, acetyllysine.
Next, we tested chemical acetylation of GK by AcP. Again, we used purified WT GK recombinantly expressed from BL21(DE3) cells with non‐detectable acetylation as the substrate and incubated with 1 mM AcP, which corresponds to the intracellular AcP concentration in E. coli cells (Schastnaya et al., 2023). Then we stopped the reaction at 30 or 60 min and used western blotting to determine the acetylation levels of AcP‐treated GK (Figures 4a and S11). Clearly, in vitro incubation with AcP alone was able to acetylate GK, and acetylation levels were increased in a time‐dependent manner. As another acetyl‐group donor in cells, AcCoA can also acetylate proteins chemically (Venkat, Gregory, Sturges, et al., 2017). Thus, we compared the activities of AcP and AcCoA to acetylate GK by performing the similar in vitro acetylation assay. Purified WT GK was incubated with 0.6 mM AcCoA which corresponds to the intracellular concentration of AcCoA in E. coli cells (Takamura & Nomura, 1988). After 1‐h incubation, western blotting was implemented to determine GK acetylation levels (Figure S12). Although AcCoA could also acetylate GK chemically in vitro, AcP had a much higher activity, indicating AcP is the major acetyl‐group donor for GK acetylation in E. coli.
FIGURE 4.
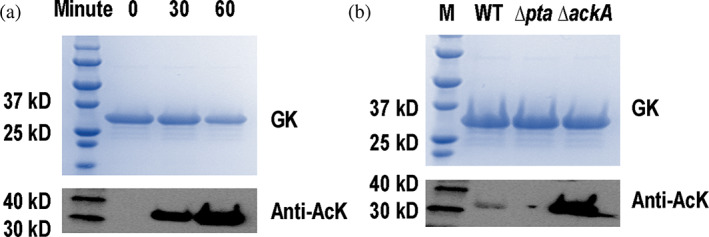
Acetylation of Escherichia coli glucokinase (GK) by acetyl‐phosphate (AcP). SDS–PAGE and western blotting of (a) purified GK from BL21(DE3) cells treated with 1 mM AcP at the start point, 30 and 60 min in vitro. The full picture of western blotting is in Figure S11. (b) Purified native GK from the WT, Δpta, or ΔackA of BW25113 cells growth in 0.2% glucose media. The full picture of western blotting is in Figure S18. AcK, acetyllysine.
Then, we performed mass spectrometry to determine the acetylation sites in AcP‐treated GK. Besides the seven lysine residues we tested in enzyme assays above, we also found other five lysine acetylation sites, which were K3, K57, K113, K204, and K284 (Figures S13–S17). Among them, K57, K113, and K284 have not been identified by proteomic studies before, which indicated the differences between in vitro tests and native acetylation in living cells.
Next, we further investigated AcP acetylation in cells. There are two major enzymes in the AcP metabolic pathway in E. coli. Acetate kinase AckA catalyzes the interconversion of AcP and acetate, while phosphate acetyltransferase Pta catalyzes the interconversion of AcP and AcCoA. Thus, in non‐acetate growth media the ∆ackA strain will accumulate AcP, while the Δpta strain has a much lower AcP level in cells. We purified native GK from the ∆ackA or Δpta strain of BW25113 E. coli cells grown in medium with 0.2% glucose and determined the acetylation levels of GK by western blotting (Figures 4b and S18). There was no detectable acetylation of GK in the Δpta strain, while the acetylation level of GK increased in the ∆ackA strain, showing that AcP‐dependent chemical acetylation is the major mechanism of GK acetylation in E. coli.
2.4. Deacetylation of E. coli GK
Lysine acetylation is a reversible and dynamic process. Deacetylases remove the acetyl group from acetylated lysine residues to control protein acetylation states in cells. To date, CobB is still the only known deacetylase in E. coli (Starai et al., 2002; Zhao et al., 2004), so we tested CobB‐mediated deacetylation for GK. The acetylation level of native GK purified from the ∆cobB strain of E. coli BW25113 cells grown in media with 0.2% glucose was determined by western blotting (Figure 3a). The acetylation level of GK in ∆cobB cells was much higher than that in WT cells, suggesting that CobB can deacetylate E. coli GK. In YfiQ‐mediated acetylation studies above, we showed that the acetylation level of GK in ∆yfiQ cells was not significantly lower than that in WT cells, which could be caused by the existence of CobB in cells, so we also tested the acetylation level of native GK purified from the ∆yfiQ ∆cobB double mutant strain (Figure 3a). The acetylation levels of GK in ∆cobB cells with and without yfiQ were similar, confirming that YfiQ is not the major enzyme for GK acetylation in E. coli cells.
By the genetic code expansion approach, we have generated site‐specifically acetylated GK variants. Those variants were not only useful to determine site‐specific effects of acetylation on GK functions, but also ideal substrates to identify the site specificities of CobB deacetylase. To achieve this goal, individual purified acetylated GK variant was incubated with purified CobB in vitro. The acetylation level of each GK variant after CobB‐treatment was determined by western blotting (Figures 5a and S19). Acetylation of K53, K214, K216, K277, and K288 was not detectable after CobB‐treatment, showing the sensitivities of these acetylation sites to CobB in vitro. On the other hand, acetylation of K128 and K149 remained after CobB‐treatment, indicating that acetylation at these two sites was resistant to CobB in vitro. We mapped these two CobB‐resistant sites onto the GK crystal structure (Figure 5b). K128 and K149 are located at the interface of the GK dimer, which could block CobB for deacetylation. Compared to CobB, AcP is much smaller and could easily reach both lysine residues at GK dimer interface. The other five acetylation sites sensitive to CobB are all located at the surface of the GK dimer, which is a general feature of CobB‐sensitive lysine residues (Figure 5c) (AbouElfetouh et al., 2015). It should be noted that CobB specificity could be different in vivo due to the dimerization state and interactions with other molecules.
FIGURE 5.
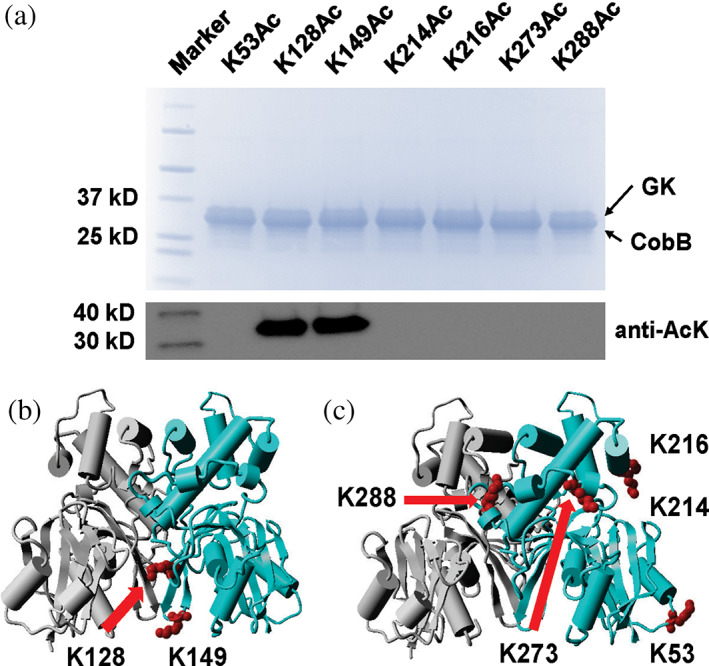
In vitro deacetylation of Escherichia coli glucokinase (GK) by CobB. (a) SDS–PAGE and western blotting of site‐specifically acetylated GK variants incubated with purified CobB in vitro. Numbers above each lane are the positions of acetylated lysine residues. GK and CobB have similar molecular weights and merged in the SDS–PAGE gel. The full picture of western blotting is in Figure S19. (b) Mapping of lysine residues resistant to CobB in vitro on the GK crystal structure protein data bank (PDB ID: 1SZ2). (c) Mapping of lysine residues sensitive to CobB in vitro on the GK crystal structure (PDB ID: 1SZ2). Lysine residues are labeled in red and from the same monomer of the GK dimer. AcK, acetyllysine.
2.5. Regulation of the GK activity by lysine acetylation
To determine the regulatory effect of lysine acetylation on GK functions, we purified native GK from WT, ∆ackA, Δpta, or ∆cobB strains of E. coli BW25113 cells grown in 0.2% glucose media and measured their activities, respectively (Figure 6). GK purified from ∆ackA or ∆cobB cells had significantly decreased activities. We have shown that the acetylation levels of GK in both strains were much higher than that in the WT strain (Figures 3 and 4), indicating that lysine acetylation could impair the GK activity. As we showed that most of the acetylation sites were sensitive to CobB, we further incubated purified GK from above strains with purified CobB in vitro and measured their activities after CobB treatment. Results showed that CobB‐treatment could restore GK activities from ∆ackA or ∆cobB cells. GK enzyme activity assays demonstrated that acetylation of K214 and K216 affected the GK activity most significantly, and CobB specificity assays showed that both K214 and K216 were sensitive to CobB deacetylation. Thus, we conclude that the major regulatory acetylation sites of GK acetylation are K214 and K216. Acetylation of both sites by intracellular AcP impairs the GK activity, while CobB‐mediated deacetylation removes the acetyl group at both sites to restore the GK activity.
FIGURE 6.
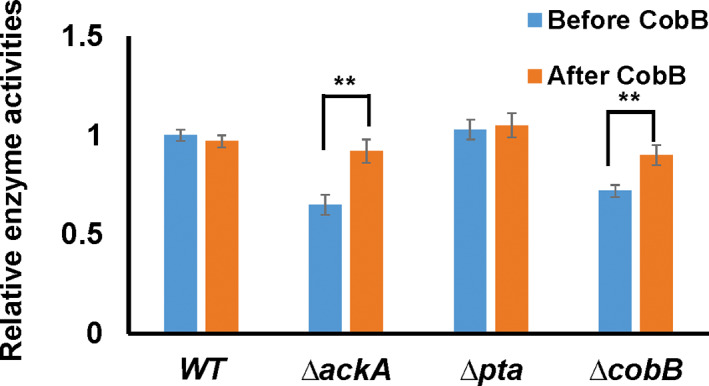
The enzyme activities of purified glucokinase (GK) from WT, ∆ackA, Δpta, or ∆cobB cells grown in 0.2% glucose media before and after 1‐h CobB treatment. The activity of GK purified from the WT strain was set as 1. Mean values and standard deviations were calculated based on three replicates. Two‐tailed p values were determined by the t‐test between the activities of GK from each strain before and after CobB treatment, and the significance level is 0.05. **p Value is less than 0.001.
3. DISCUSSION
Combing all the experiments performed in this study, we conclude that K214 and K216 are the major regulatory acetylation sites, of which acetylation impairs the GK activity. Kinetic analyses showed that acetylation of K214 and K216 increases the K m values of glucose and ATP by seven to nine‐folds, while acetylation of both sites does not affect turnover numbers significantly (Table 1). These results indicate that acetylation of K214 and K216 only influences substrate binding but not catalysis. Kinetic results also showed that glutamine substitutions do not affect both the turnover number and K m values significantly. We mapped both lysine residues onto GK crystal structures (Figure 7). Both acetylation sites are located at the entrance of the active site. The side chain of AcK is about 4 Å longer than glutamine. Besides the size, the hydrophobicity of AcK and glutamine is also different. The side chain of glutamine is polar, while the side chain of AcK is hydrophobic. Both substrates, glucose and ATP, are hydrophilic molecules. Thus, we propose that acetylation of K214 and K216 extends their side chains and increases their hydrophobicity to block the entrance of the active site and weaken binding of both hydrophilic substrates. Both K214 and K216 are at protein surface, so AcP‐dependent acetylation and CobB‐mediated deacetylation can happen quickly to regulate the GK activity, responding to environmental changes or growth demands.
FIGURE 7.
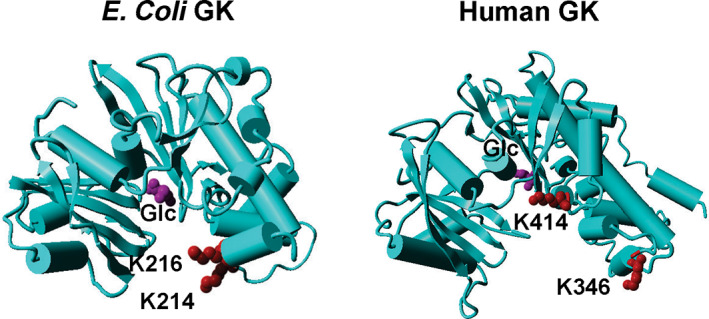
Crystal structures of Escherichia coli glucokinase (GK) and human GK. The PDB ID for E. coli GK is 1SZ2, and the PDB ID for human GK is 4DCH. The acetylation sites are marked as red, and the substrate glucose (Glc) is marked as purple. Both GKs are shown as monomers.
Different from E. coli GK which is not involved in glycolysis directly, human GK performs the first and rate‐limiting step of glycolysis, playing important roles in triggering insulin release, promoting glucose uptake, as well as mediating insulin‐independent glucose utilization in brain (Agius, 2016; De Backer et al., 2016; Matschinsky & Wilson, 2019). Although the sequence identity between E. coli GK and human GK is low (Figure S20), their overall structures are similar (Figure 7). There are no lysine residues in human GK homologous to K214 and K216 of E. coli GK. K346 of human GK has been identified to be acetylated by proteomic studies (Xu et al., 2017), which is also located at the entrance of the active site. However, the opening of the human GK active site is much wider than that of E. coli GK. Even with an extended side chain of the acetyl group, there is still enough space for substrates to enter the active site. Thus, the acetylation of K346 could not affect human GK activity and substrate binding. K414 of human GK is another identified acetylation site and located close to the active site, so acetylation of K414 could affect human GK functions but may not be deacetylated by deacetylases due to limited access. There is no lysine residue in E. coli GK homologous to K414 of human GK. Thus, the regulation of GK functions by acetylation could be distinct between E. coli and humans. Inspired by the impairing effect of K214 and K216 acetylation on E. coli GK, small molecules could be designed or screened from existing libraries to block the entrance of the E. coli GK active site without affecting human GK, as the opening of the human GK active site is much wider than that of E. coli GK, making GK a potential target for antibacterial agent development.
4. MATERIALS AND METHODS
4.1. General molecular biology
Chemical compounds in this study were purchased from Sigma‐Aldrich (St. Louis, MO, USA), VWR International (Radnor, PA, USA), or Chem‐Impex International (Wood Dale, IL, USA). DH5α cells were used for general cloning. Polymerase chain reaction reactions and plasmid constructions were performed by using the NEBuilder HiFi DNA Assembly Kit (New England Biolabs, Ipswich, MA, USA). Point mutations of the glk gene in the plasmid were made by the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs). The strains and plasmids used in this study is listed in Table S1.
Protein concentrations were measured by the Bradford Protein Assay (Bio‐Rad, Hercules, CA, USA). The purified GK and its variants were fractionated on a 4%–20% SDS–PAGE gel and visualized by the Bio‐Safe Coomassie Stain (Bio‐Rad). For western blotting, the fractionated SDS–PAGE gel was transferred onto a polyvinylidene fluoride (PVDF) membrane by using the Trans‐Blot Turbo Transfer System (Bio‐Rad). The PVDF membrane was incubated at room temperature with gentle shacking in the blocking buffer (5% bovine serum albumin, 0.1% Tween 20 in Tris‐buffered saline) for 2 h. The horseradish peroxidase‐conjugated AcK antibody (Cell Signaling Technology, Danvers, MA, USA) was diluted 1:1000 with the blocking buffer and soaked the blocked PVDF membrane overnight at 4°C. The membrane was prepared for chemiluminescence detection by using Pierce ECL Western Blotting substrates (Thermo Scientific, Waltham, MA, USA).
4.2. Expression and purification of acetylated GK variants
The genes of GK and its variants were cloned into the pCDF‐1b plasmid (EMD Millipore, Burlington, MA, USA) with a C‐terminal His6‐tag and transformed into BL21 (DE3) cells (New England Biolabs) together with the pTech plasmids harboring the AcK incorporation system for expression. The AcK‐incorporation system is based on the previous protocol (Venkat, Gregory, Meng, et al., 2017). The gene of optimized acetyllysyl‐tRNA synthetase is under the constitutive lpp promoter (Bryson et al., 2017), while the optimized tRNAPyl is under the constitutive proK promoter (Fan et al., 2015). The expression strain was grown on 500 mL of Luria‐Bertani (LB) medium supplemented with 100 μg mL−1 streptomycin and 50 μg mL−1 chloramphenicol at 37°C to an absorbance of 0.6–0.8 at 600 nm. Then protein expression was induced by the addition of 0.1 mM isopropyl β‐d‐1‐thiogalactopyranoside. For AcK‐containing protein expression, media were additionally supplemented with 10 mM AcK and 20 mM nicotinamine (NAM). Cells were incubated at 30°C for an additional 4 h and harvested by centrifugation at 3200g for 15 min at 4°C. The cell paste was suspended in 12 mL of lysis buffer (50 mM Tris [pH 7.8], 300 mM NaCl, 20 mM imidazole, 20 mM NAM, and 5 mM β‐mercaptoethanol) with cocktail protease inhibitors (Roche, Basel, Switzerland), and broken by sonication. The crude extract was centrifuged at 18,000g for 20 min at 4°C. The soluble fraction was filtered through a 0.45‐μm membrane and loaded onto a column containing 2 mL of Nickel‐Nitriloacetic acid (Ni‐NTA) resin (Qiagen, Hilden, Germany) previously equilibrated with 20 mL lysis buffer. The column was then washed with 20 mL of washing buffer [50 mM Tris (pH 7.8), 300 mM NaCl, and 40 mM imidazole]. The protein bound to the column was finally eluted with 2 mL of elution buffer [50 mM Tris (pH 7.8), 300 mM NaCl, and 150 mM imidazole]. The purified protein was dialyzed with 2 L of storage buffer [25 mM Tris (pH 7.8), 10 mM NaCl, 1 mM dithiothreitol (DTT), and 50% glycerol], and stored at −80°C. Purification of native GK from cells followed previous protocols (Meyer et al., 1997).
4.3. The GK enzyme assay and kinetic analyses
The enzyme activity was measured by using the commercially available kit (ab273303 from Abcam). In this assay, GK converts glucose into glucose‐6‐phosphate, which in turn is converted into a series of intermediates that reduce the probe generating an intense fluorescence product (Ex/Em = 535/587 nm). For K m,ATP determination in kinetic analyses, glucose concentration was fixed at 100 mM, while ATP concentration was varied from 0.01 to 100 mM. For K m,glucose determination, ATP concentration was fixed at 100 mM, while glucose concentration was varied from 0.01 to 100 mM. The kinetic parameters were calculated from nonlinear regression by software Grafit (Erithacus Software, UK).
4.4. Mass spectrometry analyses
The LC–MS/MS analyses were performed by Yale University Keck Proteomics Facility, following previous protocols (Gan et al., 2016). Briefly, proteins were digested in gel by trypsin and analyzed by LC–MS/MS on an LTQ Orbitrap XL equipped with a nanoACQUITY UPLC system. The Mascot search algorithm was used to search for the appropriate AcK incorporation.
4.5. The in vitro acetylation assay
The acetylation assay was modified from previous experiments (Venkat, Gregory, Gan, & Fan, 2017). The reaction was performed in the buffer containing 50 mM 2‐[4‐(2‐hydroxyethyl)piperazin‐1‐yl]ethanesulfonic acid (HEPES) (pH 7.0), 0.1 mM thylenediaminetetraacetic acid (EDTA), 10% glycerol, 1 mM DTT and 10 mM sodium butyrate. The acetylation was carried out by mixing 10 μg GK, 10 μg of YfiQ, 0.2 mM AcCoA in a total volume of 100 μL and incubated at 37°C for 1 h. For nonenzymatic acetylation, 10 μg GK was incubated with 1 mM of AcP at 37°C.
4.6. The in vitro deacetylation assay
The deacetylation reaction was modified from the previous study (Venkat, Nannapaneni, Gregory, et al., 2017). The reaction was performed in buffer containing 50 mM HEPES (pH 7.0), 5 mM MgCl2, 1.0 mM NAD+, 1 mM DTT, and 10% glycerol. The deacetylation was carried out by mixing 10 μg acetylated GK variants, 10 μg CobB proteins in a total volume of 100 μL and incubated at 37°C for 1 h.
AUTHOR CONTRIBUTIONS
Chenguang Fan: Conceptualization; methodology; formal analysis; supervision; funding acquisition; visualization; project administration; writing – review and editing; writing – original draft. Nour Fatema: Data curation; investigation; validation; formal analysis. Xinyu Li: Data curation; investigation. Qinglei Gan: Data curation; investigation; validation; formal analysis; visualization.
Supporting information
Data S1. Supporting information.
ACKNOWLEDGMENTS
This research was funded by the National Institutes of Health (R15GM140433 and P20GM139768) and Arkansas Biosciences Institute (the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000).
Fatema N, Li X, Gan Q, Fan C. Characterizing lysine acetylation of glucokinase. Protein Science. 2024;33(1):e4845. 10.1002/pro.4845
Review Editor: Aitziber L. Cortajarena
REFERENCES
- AbouElfetouh A, Kuhn ML, Hu LI, Scholle MD, Sorensen DJ, Sahu AK, et al. The E. coli sirtuin CobB shows no preference for enzymatic and nonenzymatic lysine acetylation substrate sites. Microbiology. 2015;4:66–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agius L. Hormonal and metabolite regulation of hepatic glucokinase. Annu Rev Nutr. 2016;36:389–415. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Lloyd M, Haythorne EA. Glucokinase activity in diabetes: too much of a good thing? Trends Endocrinol Metab. 2023;34:119–130. [DOI] [PubMed] [Google Scholar]
- Baeza J, Dowell JA, Smallegan MJ, Fan J, Amador‐Noguez D, Khan Z, et al. Stoichiometry of site‐specific lysine acetylation in an entire proteome. J Biol Chem. 2014;289:21326–21338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork P, Sander C, Valencia A. Convergent evolution of similar enzymatic function on different protein folds: the hexokinase, ribokinase, and galactokinase families of sugar kinases. Protein Sci. 1993;2:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryson DI, Fan C, Guo LT, Miller C, Soll D, Liu DR. Continuous directed evolution of aminoacyl‐tRNA synthetases. Nat Chem Biol. 2017;13:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabetta VJ, Cristea IM. Regulation, function, and detection of protein acetylation in bacteria. J Bacteriol. 2017;199:e00107–e00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castano‐Cerezo S, Bernal V, Post H, Fuhrer T, Cappadona S, Sanchez‐Diaz NC, et al. Protein acetylation affects acetate metabolism, motility and acid stress response in Escherichia coli . Mol Syst Biol. 2014;10:762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DG, Baumgartner JT, Xie X, Jew KM, Basisty N, Schilling B, et al. Mechanisms, detection, and relevance of protein acetylation in prokaryotes. mBio. 2019;10:e02708–e02718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DG, Meyer JG, Baumgartner JT, D'Souza AK, Nelson WC, Payne SH, et al. Identification of novel protein lysine acetyltransferases in Escherichia coli . MBio. 2018;9:e01905–e01918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen DG, Xie X, Basisty N, Byrnes J, McSweeney S, Schilling B, et al. Post‐translational protein acetylation: an elegant mechanism for bacteria to dynamically regulate metabolic functions. Front Microbiol. 2019;10:1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colak G, Xie Z, Zhu AY, Dai L, Lu Z, Zhang Y, et al. Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli . Mol Cell Proteomics. 2013;12:3509–3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis SJ, Epstein W. Phosphorylation of D‐glucose in Escherichia coli mutants defective in glucosephosphotransferase, mannosephosphotransferase, and glucokinase. J Bacteriol. 1975;122:1189–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Backer I, Hussain SS, Bloom SR, Gardiner JV. Insights into the role of neuronal glucokinase. Am J Physiol Endocrinol Metab. 2016;311:E42–E55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Xiong H, Reynolds NM, Soll D. Rationally evolving tRNAPyl for efficient incorporation of noncanonical amino acids. Nucleic Acids Res. 2015;43:e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda Y, Yamaguchi S, Shimosaka M, Murata K, Kimura A. Cloning of the glucokinase gene in Escherichia coli B. J Bacteriol. 1983;156:922–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan Q, Lehman BP, Bobik TA, Fan C. Expanding the genetic code of Salmonella with non‐canonical amino acids. Sci Rep. 2016;6:39920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han R, Liang J, Zhou B. Glucose metabolic dysfunction in neurodegenerative diseases—new mechanistic insights and the potential of hypoxia as a prospective therapy targeting metabolic reprogramming. Int J Mol Sci. 2021;22:5887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentchel KL, Escalante‐Semerena JC. Acylation of biomolecules in prokaryotes: a widespread strategy for the control of biological function and metabolic stress. Microbiol Mol Biol Rev. 2015;79:321–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Montalvo V, Martinez A, Hernandez‐Chavez G, Bolivar F, Valle F, Gosset G. Expression of galP and glk in a Escherichia coli PTS mutant restores glucose transport and increases glycolytic flux to fermentation products. Biotechnol Bioeng. 2003;83:687–694. [DOI] [PubMed] [Google Scholar]
- Kawai S, Mukai T, Mori S, Mikami B, Murata K. Hypothesis: structures, evolution, and ancestor of glucose kinases in the hexokinase family. J Biosci Bioeng. 2005;99:320–330. [DOI] [PubMed] [Google Scholar]
- Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, Minasov G, et al. Structural, kinetic and proteomic characterization of acetyl phosphate‐dependent bacterial protein acetylation. PloS One. 2014;9:e94816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengsfeld C, Schonert S, Dippel R, Boos W. Glucose‐ and glucokinase‐controlled mal gene expression in Escherichia coli . J Bacteriol. 2009;191:701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Sun Z, Xu Y, Luan Y, Xu J, Liang Q, et al. Enhancing the glucose flux of an engineered EP‐bifido pathway for high poly(hydroxybutyrate) yield production. Front Bioeng Biotechnol. 2020;8:517336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Guo L, Fu Y, Huo M, Qi Q, Zhao G. Bacterial protein acetylation and its role in cellular physiology and metabolic regulation. Biotechnol Adv. 2021;53:107842. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front Physiol. 2019;10:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesak LR, Mesak FM, Dahl MK. Bacillus subtilis GlcK activity requires cysteines within a motif that discriminates microbial glucokinases into two lineages. BMC Microbiol. 2004;4:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer JG, D'Souza AK, Sorensen DJ, Rardin MJ, Wolfe AJ, Gibson BW, et al. Quantification of lysine acetylation and succinylation stoichiometry in proteins using mass spectrometric data‐independent acquisitions (SWATH). J Am Soc Mass Spectrom. 2016;27:1758–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer D, Schneider‐Fresenius C, Horlacher R, Peist R, Boos W. Molecular characterization of glucokinase from Escherichia coli K‐12. J Bacteriol. 1997;179:1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minliang C, Chengwei M, Lin C, Zeng AP. Integrated laboratory evolution and rational engineering of GalP/Glk‐dependent Escherichia coli for higher yield and productivity of L‐tryptophan biosynthesis. Metab Eng Commun. 2021;12:e00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Peak‐Chew SY, Chin JW. Genetically encoding Nε‐acetyllysine in recombinant proteins. Nat Chem Biol. 2008;4:232–234. [DOI] [PubMed] [Google Scholar]
- Postma PW, Lengeler JW, Jacobson GR. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev. 1993;57:543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuraba H, Goda S, Ohshima T. Unique sugar metabolism and novel enzymes of hyperthermophilic archaea. Chem Rec. 2004;3:281–287. [DOI] [PubMed] [Google Scholar]
- Schastnaya E, Doubleday PF, Maurer L, Sauer U. Non‐enzymatic acetylation inhibits glycolytic enzymes in Escherichia coli . Cell Rep. 2023;42:111950. [DOI] [PubMed] [Google Scholar]
- Schilling B, Christensen D, Davis R, Sahu AK, Hu LI, Walker‐Peddakotla A, et al. Protein acetylation dynamics in response to carbon overflow in Escherichia coli . Mol Microbiol. 2015;98:847–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starai VJ, Celic I, Cole RN, Boeke JD, Escalante‐Semerena JC. Sir2‐dependent activation of acetyl‐CoA synthetase by deacetylation of active lysine. Science. 2002;298:2390–2392. [DOI] [PubMed] [Google Scholar]
- Starai VJ, Escalante‐Semerena JC. Identification of the protein acetyltransferase (pat) enzyme that acetylates acetyl‐CoA synthetase in Salmonella enterica . J Mol Biol. 2004;340:1005–1012. [DOI] [PubMed] [Google Scholar]
- Takamura Y, Nomura G. Changes in the intracellular concentration of acetyl‐CoA and malonyl‐CoA in relation to the carbon and energy metabolism of Escherichia coli K12. J Gen Microbiol. 1988;134:2249–2253. [DOI] [PubMed] [Google Scholar]
- Titgemeyer F, Reizer J, Reizer A, Saier MH Jr. Evolutionary relationships between sugar kinases and transcriptional repressors in bacteria. Microbiology. 1994;140(Pt 9):2349–2354. [DOI] [PubMed] [Google Scholar]
- VanDrisse CM, Escalante‐Semerena JC. Protein acetylation in bacteria. Annu Rev Microbiol. 2019;73:111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S, Chen H, Stahman A, Hudson D, McGuire P, Gan Q, et al. Characterizing lysine acetylation of isocitrate dehydrogenase in Escherichia coli . J Mol Biol. 2018;430:1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S, Gregory C, Gan Q, Fan C. Biochemical characterization of the lysine acetylation of tyrosyl‐tRNA synthetase in Escherichia coli . Chembiochem. 2017;18:1928–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S, Gregory C, Meng K, Gan Q, Fan C. A facile protocol to generate site‐specifically acetylated proteins in Escherichia coli . J Vis Exp. 2017;130:e57061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S, Gregory C, Sturges J, Gan Q, Fan C. Studying the lysine acetylation of malate dehydrogenase. J Mol Biol. 2017;429:1396–1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkat S, Nannapaneni DT, Gregory C, Gan Q, McIntosh M, Fan C. Genetically encoding thioacetyl‐lysine as a non‐deacetylatable analog of lysine acetylation in Escherichia coli . FEBS Open Bio. 2017;7:1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei T, Cheng BY, Liu JZ. Genome engineering Escherichia coli for L‐DOPA overproduction from glucose. Sci Rep. 2016;6:30080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Wagner SA, Scholz C, Gummesson B, Beli P, et al. Acetyl‐phosphate is a critical determinant of lysine acetylation in E. coli . Mol Cell. 2013;51:265–272. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Satpathy S, Hansen BK, Lyon D, Jensen LJ, Choudhary C. Accurate quantification of site‐specific acetylation stoichiometry reveals the impact of sirtuin deacetylase CobB on the E. coli acetylome. Mol Cell Proteomics. 2017;16:759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JE. Hexokinases. Rev Physiol Biochem Pharmacol. 1995;126:65–198. [DOI] [PubMed] [Google Scholar]
- Xu H, Zhou J, Lin S, Deng W, Zhang Y, Xue Y. PLMD: an updated data resource of protein lysine modifications. J Genet Genomics. 2017;44:243–250. [DOI] [PubMed] [Google Scholar]
- Yuan L, Qin YL, Zou ZC, Appiah B, Huang H, Yang ZH, et al. Enhancing intracellular NADPH bioavailability through improving pentose phosphate pathway flux and its application in biocatalysis asymmetric reduction reaction. J Biosci Bioeng. 2022;134:528–533. [DOI] [PubMed] [Google Scholar]
- Zhang K, Zheng S, Yang JS, Chen Y, Cheng Z. Comprehensive profiling of protein lysine acetylation in Escherichia coli . J Proteome Res. 2013;12:844–851. [DOI] [PubMed] [Google Scholar]
- Zhao K, Chai X, Marmorstein R. Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli . J Mol Biol. 2004;337:731–741. [DOI] [PubMed] [Google Scholar]
- Zhu F, San KY, Bennett GN. Improved succinate production from galactose‐rich feedstocks by engineered Escherichia coli under anaerobic conditions. Biotechnol Bioeng. 2020;117:1082–1091. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.