

Key Points
-
•
In patients with active HLH, B-cell development and maturation are impaired along with a decrease in cTfh cells.
-
•
Control of T-cell activation during an active HLH state improves cTfh cell frequency and B-cell maturation.
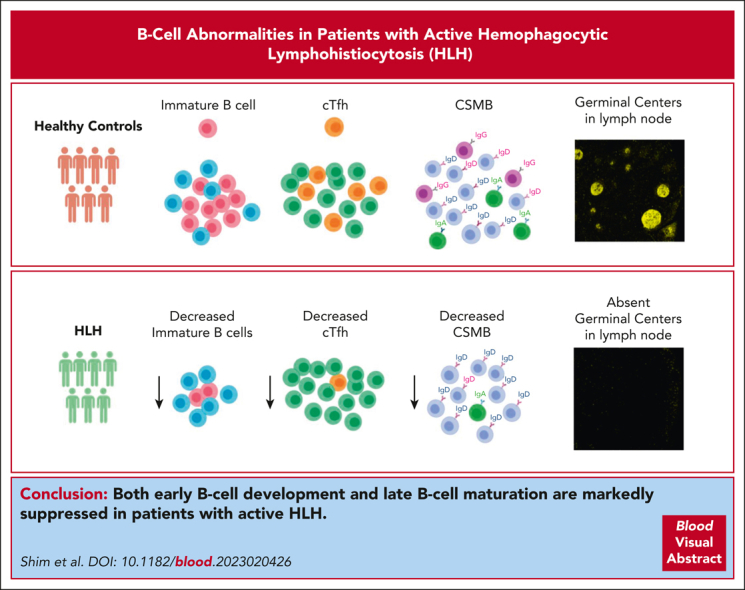
Visual Abstract
Abstract
Hemophagocytic lymphohistiocytosis (HLH) is characterized by hyperinflammation and multiorgan dysfunction. Infections, including the reactivation of viruses, contribute to significant disease mortality in HLH. Although T-cell and natural killer cell–driven immune activation and dysregulation are well described, limited data exist on the status of B-cell compartment and humoral immune function in HLH. We noted marked suppression of early B-cell development in patients with active HLH. In vitro B-cell differentiation studies after exposure to HLH-defining cytokines, such as interferon gamma (IFN-γ) and tumor necrosis factor, recapitulated B-cell development arrest. Messenger RNA sequencing of human CD34+ cells exposed to IFN-γ demonstrated changes in genes and pathways affecting B-cell development and maturation. In addition, patients with active HLH exhibited a marked decrease in class-switched memory B (CSMB) cells and a decrease in bone marrow plasmablast/plasma cell compartments. The decrease in CSMB cells was associated with a decrease in circulating T follicular helper (cTfh) cells. Finally, lymph node and spleen evaluation in a patient with HLH revealed absent germinal center formation and hemophagocytosis with associated lymphopenia. Reassuringly, the frequency of CSMB and cTfh improved with the control of T-cell activation. Taken together, in patients with active HLH, these changes in B cells may affect the humoral immune response; however, further immune studies are needed to determine its clinical significance.
Hemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory syndrome characterized by cytotoxic T-cell and natural killer cell–dysfunction. Shim and colleagues investigated the B-cell compartment in active HLH, demonstrating marked suppression of early B-cell development. The authors show decreased emergence of class-switched memory B cells (CSMB) and decreased plasma cell maturation in active HLH, associated with a decrease in circulating T follicular helper (cTfh) cells. The profile is ameliorated with control of the T-cell activation with increases in CSMB and cTfh. Further study is needed to determine whether the B-cell compartment fully recovers with control of HLH.
Introduction
Hemophagocytic lymphohistiocytosis (HLH) is a life-threatening disorder. Patients present with hyperinflammation, cytopenia, splenomegaly, coagulopathy, and multiorgan dysfunction.1 Primary HLH (P-HLH) pathogenesis is characterized by genetic mutations associated with defects in T-cell and natural killer (NK) cell cytotoxic functions or inflammasome disorders, leading to overwhelming immune activation.2, 3, 4 Secondary HLH occurs in patients with infection, malignancy, or rheumatological illnesses. Several cytokines and chemokines are markedly elevated and drive the hyperinflammatory process in HLH. Of these, interferon gamma (IFN-γ) is a critical driver of HLH: in preclinical animal models of HLH, IFN-γ neutralization or blockade of downstream signaling resulted in significant disease amelioration.5, 6, 7 Correspondingly, improvement in morbidity and mortality was noted with the use of an IFN-γ neutralizing antibody, emapalumab, or JAK-STAT inhibitors that block signaling of INF-γ and several other inflammatory cytokines for the management of patients with P-HLH and HLH associated with rheumatologic illnesses (R-HLH), also known as macrophage activation syndrome.8, 9, 10
T-cell– and NK cell–driven immune activation and dysregulation have been the primary focus of research in HLH. However, the effect of overwhelming immune activation and inflammation in other aspects of the immune system, including the humoral compartment, has not been well characterized in HLH. Interestingly, hypogammaglobulinemia has been reported as the presenting manifestation in patients with STXBP2, UNC13D, and XIAP deficiency.11, 12, 13, 14, 15 Although hypogammaglobulinemia and ineffective humoral response could also increase the risk of infections, increased infection risk has been generally attributed to neutropenia and T-cell suppression during HLH therapy. It is unclear whether hypogammaglobinemia and B-cell lymphopenia noted in patients with HLH are secondary to treatment with chemoimmunotherapy or a direct effect of inflammation. Several inflammatory cytokines and chemokines affect B-cell development, maturation, and function.16 Thus, evaluating the effects of the inflammatory milieu in HLH on B-cell development and maturation is critical.
To gain insight, we analyzed bone marrow B-cell development in patients with HLH. Furthermore, we performed in vitro B-cell differentiation studies with or without exposure to cytokines known to be critical drivers of HLH pathology, namely IFN-γ and tumor necrosis factor (TNF).17 We also performed messenger RNA (mRNA) sequencing of human CD34+ cells with and without exposure to IFN-γ to evaluate genes and pathways affected by IFN-γ–driven inflammation. We further assessed B-cell class switching and circulating T follicular helper (cTfh) cell frequency in a subset of patients with HLH and longitudinally evaluated its changes. Finally, we evaluated a lymph node and spleen from a patient with HLH. Altogether, we show that early B-cell development and maturation are affected in patients with HLH and provide evidence that control of inflammation improves the humoral immune profile.
Methods
Patients
A total of 42 patients with a diagnosis of HLH from April 2009 to August 2021 were included in this study and categorized into groups of P-HLH (n = 6), secondary HLH due to infection (I-HLH; n = 22), and secondary HLH due to rheumatological illnesses (R-HLH; n = 14) (supplemental Table 1, available on the Blood website). Bone marrow aspirates and biopsies were performed as part of the standard clinical evaluation at diagnosis, and flow cytometry data were subsequently obtained and then analyzed retrospectively. Eight bone marrow aspirate samples from age-matched healthy controls were concurrently evaluated. Chart review was performed using EPIC (Epic Systems, Verona, WI) to extract patient data. Peripheral T- and B-cell immunophenotyping was performed in a cohort of 6 patients with HLH at diagnosis before any therapies and while receiving HLH therapy to collect longitudinal data and in 16 healthy controls (supplemental Table 2). Peripheral blood was analyzed for cytokine profiling in an additional 9 patients with HLH (P-HLH, n = 3; I-HLH, n = 6). The study was approved by the institutional review board of Children’s Healthcare of Atlanta and Emory University.
Flow cytometry analysis
The clinical hematopathology laboratory at Children’s Healthcare of Atlanta performed flow cytometry of bone marrow aspirates from patients with HLH. Data from flow panel A was available for all patients; flow panel B with extended B-cell phenotyping data was available in a subset of patients (supplemental Table 1). Lymphoid and myeloid compartments were characterized with the following cell surface markers: B cells (CD19+), T cells (CD3+), myeloid cells (CD33+), NK cells (CD56+CD16+), hematopoietic stem and progenitor cells (CD34+CD38−), hematopoietic progenitor cells (HPCs) (CD34+CD38+), hematogones (CD19+CD45+), pre–pro-B cells (CD34+CD19−CD10+), pro-B cells (CD34+CD19+CD10+), immature B cells (CD19+CD10+), transitional B cells (CD19+CD10−CD20−), mature B cells (CD19+CD10−CD20+), and plasma cells/blasts (CD19+CD20−CD38bright).18,19 Based on CD19 and CD45 staining intensity, hematogones were further categorized as stage 1 (CD45dim), stage 2 (CD45intermediate), and stage 3 (CD45bright) (supplemental Figure 1).20,21 Additional peripheral blood mononuclear cells immunophenotyping was performed to evaluate activated CD4+ T cells (CD4+ effector memory cells, CD38+HLA-DR+), cTfh cells (CD4+CD45RA−CXCR5+), and class-switched memory B (CSMB) cells (CD19+CD27+IgD−) in a research setting.22 Supplemental Table 3 lists the surface antibodies used. Flow cytometry data were acquired on BD FACSymphony A5 and analyzed using FlowJo software version 10.
In vitro B-cell differentiation assay
In vitro differentiation of human CD34+ cells to B cells was carried out following previously established protocols using a mesenchymal stromal cell coculture system.23 Details are provided in the supplemental Methods. B-cell differentiation was assessed by flow cytometry between weeks 3 and 4. The surface antibodies used are listed in supplemental Table 3.
RNA sequencing
Total RNA was extracted from human CD34+ cells with and without IFN-γ (10 ng/mL) exposure for 48 hours (3 paired samples each) using an RNA extraction kit (Zymo) and quantified by nanodrop (Thermo Fisher). RNA integrity numbers were determined for samples on a bioanalyzer (Agilent 2100), and samples were only used if the values of RNA integrity numbers were >7.0. Complementary DNA libraries were prepared and sequenced at 30 million paired-end depth (150 base pairs) using the DNBSEQ platform system by BGI USA. The supplemental Methods detail the methods for differential gene expression analysis and gene ontology and pathway analysis using the R software.
Immunohistochemistry
Plasma cells were quantified in bone marrow core biopsy samples from patients with HLH (P-HLH, n = 4; I-HLH, n = 6; and R-HLH, n = 6) using CD138 (Novacastra Ready to Use; Leica PA0088) immunohistochemistry staining. Whole slides were scanned for digital imaging, and 3 representative unbiased fields were chosen for analysis using the Image J (National Institutes of Health) software. In addition, lymph node and spleen samples from controls (1-month-old and 3-year-old) and a patient with P-HLH (aged 2 months) were stained with hematoxylin and eosin, CD68 (Novacastra Ready to Use; Leica PA0273), and CD163 (Novacastra Ready to Use; Leica PA0090).
Multiplex immunofluorescence
Sections from lymph node and spleen samples from controls and the patient with P-HLH as noted earlier were cut at 5-μM thickness and mounted on positively charged slides for staining. Opal (Akoya Biosciences) multiplex staining using a Ventana DISCOVERY ULTRA system (Roche) autostainer was performed by staining tissue sections with antibodies to detect CD3 (1:150; Sigma SAB5500057), CD19 (1:250; Abcam 134114), BCL6 (1:500; Sigma HPA004899), and spectral DAPI (4′,6-diamidino-2-phenylindole) (Akoya). Details of staining and imaging processes are provided in the supplemental Methods.
Cytokine profiling and statistical analysis are detailed in the supplemental Methods.
Results
Patient characteristics and laboratory findings portray different subtypes of HLH
There was a similar distribution of males (n = 24) and females (n = 18) with a diagnosis of HLH. The age distribution showed that patients with P-HLH were young (80% aged <2 years) (supplemental Table 1). The laboratory data at the time of bone marrow sampling showed a significantly elevated white blood cell count, absolute neutrophil count, and platelet count in the R-HLH group (supplemental Figure 2).24 The P-HLH group had a significantly elevated mean soluble interleukin-2 (IL-2) receptor value compared with the I-HLH and R-HLH groups, suggesting higher T-cell activation.25
Attrition of bone marrow B cells in patients with active HLH
The total B-cell compartment in bone marrow in all forms of HLH was decreased, whereas the T-cell compartment increased compared with that of healthy controls. In addition, the myeloid cell compartments were expanded in the P-HLH and I-HLH groups. The NK cell compartment was similar among all groups (Figure 1A; supplemental Figure 3). There was no significant difference in the percentage of hematopoietic stem and progenitor cell (CD34+CD38−) populations among the groups, but the R-HLH group exhibited a lower percentage of CD34+ cells and HPC (CD34+CD38+) compared with the control group (Figure 1B-C; supplemental Figure 4).
Figure 1.

Early B-cell development is suppressed in the bone marrows of patients with HLH. (A) The individual percentages of CD19+ (B-cell lineage), CD3+ (T-cell lineage), and CD33+ (myeloid lineage) cells in the bone marrow compartment for age-matched healthy control (HC) group (n = 8), P-HLH group (n = 5), I-HLH group (n = 19), and R-HLH group (n = 12). B-cell compartment (HC, 16.59% ± 1.85%; P-HLH, 10.78% ± 4.05%; I-HLH, 6.04% ± 1.19%; and R-HLH, 3.64% ± 0.76%); T-cell compartment (HC, 11.16% ± 1.01%; P-HLH, 28.38% ± 4.36%; I-HLH, 18.21% ± 2.48%; and R-HLH, 14.78% ± 3.36%); myeloid compartment (HC, 4.20% ± 0.62%; P-HLH, 8.93% ± 1.84%; I-HLH, 8.02% ± 1.09%; and R-HLH, 5.95% ± 1.07%). (B) Percentage of hematopoietic stem and progenitor cells (CD34+CD38−) population in the bone marrow compared among HC (n = 8; 0.17% ± 0.06%), P-HLH (n = 3; 0.055% ± 0.04%), I-HLH (n = 4; 0.024% ± 0.005%), and R-HLH (n = 4; 0.096% ± 0.04%). (C) Percentage of HPC (CD34+CD38+) population in the bone marrow compared among HC (n = 8; 2.94% ± 0.19%), P-HLH (n = 3; 1.87% ± 0.63%), I-HLH (n = 4; 1.90% ± 0.48%), and R-HLH (n = 4; 0.90% ± 0.49%) groups. (D) Percentages of pre–pro-B cells (CD34+CD19−CD10+) in HC (n = 8; 0.12% ± 0.03%) and HLH (n = 7; 0.0047% ± 0.0020%) groups and (E) pro-B cells (CD34+CD19+CD10+) in in the bone marrow of HC (n = 8; 1.26% ± 0.13%) and HLH (n = 7; 0.023% ± 0.017%) groups. (F) Representative flow cytometry dot plot of CD19/CD45 showing the depletion of hematogones (CD19+CD45+) in a patient with P-HLH (right) compared with that in an HC (left). (G) Percentages of total hematogones (stages 1-3), (H) stage 1 hematogones (CD19+CD45dim), and (I) stage 2 hematogones (CD19+CD45intermediate) in the bone marrow from HC (n = 8), P-HLH (n = 6), I-HLH (n = 20), and R-HLH (n = 14) groups. Total: HC (n = 8) 12.70% ± 1.74%, P-HLH (n = 6) 6.74% ± 2.67%, I-HLH (n = 20) 4.78% ± 1.04%, and R-HLH (n = 14) 2.68% ± 0.65%; stage 1: HC 1.38% ± 0.13%, P-HLH 0.21% ± 0.12%, I-HLH 0.077% ± 0.035%, and R-HLH 0.019% ± 0.01%; stage 2: HC 8.38% ± 1.28%, P-HLH 0.79% ± 0.46%, I-HLH 0.27% ± 0.11%, and R-HLH 0.16% ± 0.05%. (J) Percentages of immature B cells (CD19+CD10+), (K) transitional B cells (CD19+CD10−CD20−), and (L) mature B cells (CD19+CD10−CD20+) in the bone marrow of patients in each group. Immature: HC (n = 8; 8.36% ± 1.06%), P-HLH (n = 6; 1.32% ± 0.65%), I-HLH (n = 19; 0.54% ± 0.15%), and R-HLH (n = 14; 0.20% ± 0.05%); transitional: HC 2.91% ± 0.38%, P-HLH 8.33% ± 3.12%, I-HLH 5.97% ± 1.09%, and R-HLH 3.02% ± 0.60%; and mature: HC 2.35% ± 0.42%, P-HLH 2.72% ± 1.01%, I-HLH 4.52% ± 1.13%, and R-HLH 1.77% ± 0.42%. The two-tailed Student t test or the Mann-Whitney test was used depending on normality for statistical comparison between 2 groups. The Kruskal-Wallis 1-way analysis of variance (ANOVA) followed by the Dunn multiple comparison test for non-normally distributed samples and an ordinary 1-way ANOVA followed by the Tukey multiple comparison test for normally distributed samples were used for statistical comparison of HC and HLH groups. Data represent mean ± standard error of the mean (SEM); ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001; ns, not significant.
Early B-cell development is suppressed in the bone marrows of patients with HLH
In a subset of patients, we found that the early B cells (CD34+CD19−CD10+ pre–pro-B cells and CD34+CD19+CD10+ pro–B cells) in the bone marrow were significantly decreased compared with healthy controls (Figure 1D-E). In a larger patient panel, we assessed the hematogones, which have been described as early B cells developing in the bone marrow and immunologically recognized as precursor B cells.21,26 We found a significant decrease in the percentage of total hematogones in the bone marrows of the different groups of patients with HLH compared with that in the control group (Figure 1F-G). Stage 1 hematogones were significantly decreased in all patients with different forms of HLH (P-HLH, 0.21% ± 0.12%; I-HLH, 0.077% ± 0.035%; and R-HLH, 0.019% ± 0.01%; P < .0001) compared with healthy controls (1.38% ± 0.13%; Figure 1H). Stage 2 hematogones also showed a similar trend (healthy controls, 8.38% ± 1.28%; P-HLH, 0.79% ± 0.46%; I-HLH, 0.27% ± 0.11%; and R-HLH, 0.16% ± 0.05%; Figure 1I). Of note, the R-HLH group had significantly lower hematogones in both stages than the P-HLH group (Figure 1H-I). Although the P-HLH, I-HLH, and R-HLH groups had significantly lower percentages of immature B cells (CD10+ fraction) than the control group (Figure 1J), there was no significant difference in the percentages of transitional and mature B cells (CD10− fraction) between the control and HLH groups (Figure 1K-L).
Human HPCs exposed to IFN-γ and TNF in vitro have impaired B-cell development
To validate the clinical findings of lower B-cell counts in patients with HLH, we performed in vitro assays to study the role of critical HLH-defining inflammatory cytokines IFN-γ and TNF on B-cell development. Human CD34+ cells cocultured with mesenchymal stromal cells and B-cell differentiation cytokines in the presence of IFN-γ or TNF or both demonstrated suppression of B-cell maturation and development, as shown by the decreased presence of total CD19+ B cells, CD19+CD34− cells, and CD19+CD10+ immature B cells. IFN-γ had a more profound effect on B-cell development than TNF, yet a combination of IFN-γ and TNF led to an exaggerated suppression of B-cell development (>90% decrease compared with the media-only condition in all B-cell populations) (Figure 2A-B).
Figure 2.

Human CD34+ cells cocultured with mesenchymal stromal cells exposed to IFN-γ and TNF exhibit suppressed B-cell development. (A) Representative flow cytometry panel of CD19+CD20+ mature B cells exposed to media, TNF, IFN-γ, or the combination of TNF and IFN-γ. (B) Quantified values of the populations of interest in each treatment group normalized to media control group represented as fold change for total CD19+ B cells (TNF, 0.31 ± 0.043; IFN-γ, 0.17 ± 0.090; and IFN-γ + TNF, 0.030 ± 0.014), CD19+CD34− (TNF, 0.33 ± 0.061; IFN-γ, 0.13 ± 0.059; and IFN-γ + TNF, 0.030 ± 0.017), and CD19+CD10+ immature B cells (TNF, 0.43 ± 0.076; IFN-γ, 0.22 ± 0.094; and IFN-γ + TNF, 0.081 ± 0.052) (n = 5 per treatment group). A Kruskal-Wallis 1-way ANOVA followed by the Dunn multiple comparison test for nonnormally distributed samples and an ordinary 1-way ANOVA followed by the Tukey multiple comparison test for normally distributed samples were used for statistical comparison of media and treated groups. Data represent mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
Genes and pathways involved in B-cell development are affected by IFN-γ–mediated inflammation
To gain insight into the profound effect of IFN-γ on B-cell development, mRNA sequencing of human CD34+ stem cells from healthy bone marrow donors, with or without exposure to IFN-γ was performed. The first 2 principal components from the entire transcriptomic profile showed 2 distinct clusters for the treated and control groups (Figure 3A). Differential expression (DE) analysis identified 704 genes significantly different with a false discovery rate < 0.05. Of these genes, 511 were increased (upregulated) in the treated group, whereas 193 were decreased (downregulated) (Figure 3B; supplemental Tables 4 and 5). A hierarchal clustering heatmap classified 2 independent clusters for the treated and control groups using the 704 differentially expressed genes (Figure 3C). A similar heat map analysis focused on genes involved in B-cell activation (Gene ontology term GO:0042113) and early B-cell development showed 2 separate clusters between the treated and control groups (Figure 3D). Genes known to impact B-cell development, such as LRRK2 and SAMD9L, were significantly upregulated, whereas S1PR1, IGF1, MMP2, and GPR183 were significantly downregulated in the IFN-γ–treated group (Figure 3D). Panther pathway and WikiPathways analysis with overrepresentation analysis revealed type 2 IFN signaling, allograft rejection, host response to ebola virus, and human immune response to tuberculosis pathways among the most significantly affected (Figure 3E; supplemental Figure 5). Similarly, gene set enrichment analysis revealed IFN-γ, allograft rejection, human immune responses to tuberculosis pathways, and the DE genes enriched in selected pathways (Figure 3F; supplemental Figure 6). Both DE and pathway analyses validated the effect of IFN-γ treatment. Genes driven by IFN-γ, IRF1, CXCL10, and GBP1 were among the most upregulated (Figure 3D; supplemental Figure 6).27, 28, 29 Some of the most significantly affected pathways were all disease states, such as viral and mycobacterial infection or allograft rejection, driven by IFN-γ (Figure 3E-F; supplemental Figure 5).30,31
Figure 3.

DE gene analysis among human CD34+ stem cells treated with IFN-γ compared with that of the control group reveals distinct genes and pathways affected by inflammation. (A) The principal component analysis was performed with all the samples, and the first 2 principal components are plotted. Data show clear separation of treated (n = 3) and nontreated samples (n = 3) into 2 distinct groups. (B) The DE analysis results are plotted as a volcano plot. x-axis shows log2FC, and y-axis shows −log10 (P value) for each gene. Each dot represents a gene and the colors indicate significance level. (C) Genes that are significant by log2 fold change (log2FC) > 2 and false discovery rate (FDR) < 0.05 are plotted in the heat map. (D) Genes involved in B-cell activation and early development are differentially expressed between treated and nontreated groups. (E) Overrepresentation analysis shows significant pathway hits (FDR < 0.05) for 704 DE genes. Size of the circle represents the number of genes in each pathway; color represents the significance level. (F) Gene set enrichment analysis identifies 3 significant pathways based on the enrichment score (top). The value of the ranking metric is shown along the list of the ranked genes (bottom).
Active HLH impairs peripheral B-cell development and class switching
To further evaluate peripheral B-cell development during a massive T-cell activation state, we evaluated B-cell maturation to CSMB cells in healthy controls and patients with HLH (supplemental Table 2). We found that the age-normalized CSMB cell values were significantly lower in the HLH group (2.19 ± 0.24 vs 0.41 ± 0.14; P < .0001), suggesting potentially impaired B-cell maturation (Figure 4A). As expected, there was high T-cell activation in CD4 and CD8 effector memory compartments in patients with HLH (Figure 4A). Interestingly, decreased CSMB cells is associated with a significant decrease in cTfh cell percentage in the HLH group (16.47% ± 0.96% vs 6.10% ± 2.45%, P < .0001), suggesting decreased T-cell help for B-cell maturation (Figure 4A). Sequential peripheral blood flow cytometry data from the same patients showed that decreased T-cell activation with the initiation of therapy improved the percentage of cTfh cells and fold change of CSMB cells compared with the active HLH state before any therapies (supplemental Figure 7A; Figure 4B). Soluble IL-2 receptor values, followed as a marker for HLH, decreased significantly on treatment (supplemental Figure 7B). Cytokine profiling performed to evaluate the inflammatory milieu during active HLH showed an expected increase in IFN-γ, CXCL9, and IL-6. However, CXCL13, which has previously been used as a biomarker of germinal center activity, showed a marked increase despite poor B-cell maturation (Figure 4C).32 To assess whether the effects on peripheral B-cell development were specific to HLH, we evaluated peripheral blood CSMB cells in patients with multisystem inflammatory syndrome in children, which is another hyperinflammatory condition associated with T-cell activation,33 and found no significant decrease in proportion of age-normalized CSMB cells compared with that of healthy controls (supplemental Figure 8).
Figure 4.

B-cell activation and class switching are impaired in active HLH. (A) Age-normalized values of CSMB (CD19+CD27+IgD−) (2.19 ± 0.24 vs 0.41 ± 0.14) represented as fold change and percentages of CD4+ effector memory T cells (CD4+TEM, CD4+HLA-DR+CD38+; 0.94% ± 0.14% vs 58.02% ± 7.93%), CD8+TEM cells (CD8+HLA-DR+CD38+; 2.42% ± 0.43% vs 75.12% ± 8.10%), and cTFh (CD45RA−CD4+CXCR5+; 16.47% ± 0.96% vs 6.10% ± 2.45%) were determined in the peripheral blood of HCs (n = 16) vs patients with HLH (P-HLH, n = 5; I-HLH, n = 1; total, n = 6). (B) Percentage of activated CD4+ TEM cells (n = 6) decreased when patients with HLH received therapy (top), whereas percentages of CXCR5+ cTfh cells (n = 6) (middle) and age-normalized fold change values of CSMB cells (n = 5) (bottom) in patients with HLH increased while on therapy. Representative flow cytometry dot plots from HCs and patients with HLH are shown adjacently (left), with a green or black rectangle highlighting the population of interest. (C) Cytokine profiling of IFN-γ (0.74 ± 0.30 pg/mL vs 296.1 ± 130.3 pg/mL), CXCL9 (76.90 ± 14.69 pg/mL vs 2163.0 ± 82.19 pg/mL), IL-6 (0.42 ± 0.14 pg/mL vs 187.2 ± 156.5 pg/mL), and CXCL13 (51.04 ± 9.78 pg/mL vs 355.0 ± 84.02 pg/mL) in HCs (n = 16) vs patients with HLH (P-HLH, n = 3; I-HLH, n = 6; Total, n = 9). The two-tailed Student t test or the Mann-Whitney test was used, depending on the normality for statistical comparison between 2 groups. Data represent mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
Germinal centers and BCL6-expressing Tfh cells and B cells are decreased in the lymph node and spleen of a patient with HLH
To further validate our findings, we assessed the lymph node and spleen of a 2-month-old infant who died of HLH within 12 hours of presentation and before initiating therapy. The diagnosis of P-HLH was made post mortem; genetic testing revealed a biallelic STXBP2 pathogenic mutation. The lymph node and spleen of the infant with HLH were compared with those of controls, a 1-month-old infant and a 3-year-old, who died of other causes and without known immune deficiencies. The lymph node of the 1-month-old infant control featured primary follicles but lacked discrete germinal centers, whereas that of the older control displayed discrete germinal centers (Figure 5A). The spleen of the control infant showed areas of discrete white pulp (Figure 5B). The lymph node and spleen of the infant with HLH demonstrated hemophagocytosis and the presence of macrophage activation (CD68+/CD163+) (supplemental Figure 9). In addition, we found generalized severe lymphopenia with evidence of histiocytic activation. Correspondingly, no appreciable germinal centers or areas of white pulp were noted with a marked diminution of BCL6-expressing germinal center B cells and Tfh cells in the lymph node and spleen of the patient with HLH (Figure 5A-B).
Figure 5.

Germinal centers and BCL6-expressing Tfh and B cells are decreased in the lymph node and spleen of a patient with P-HLH. Panel showing multiplex immunofluorescence (CD3, red; CD19, green; BCL6, yellow; and DAPI, blue) images from (A) lymph node and (B) spleen of controls (a 1-month-old infant and a 3-year-old) and a patient with active P-HLH (aged 2 months). Primary follicles present in lymph nodes of both controls. Germinal centers not prominent in lymph node of younger control but present in older control. Lymph nodes of the patient with HLH lack primary follicles and germinal centers. The spleen of the control demonstrates areas of distinct white pulp, whereas the spleen of the patient with HLH lacks white pulp.
Plasma cell/blast and immunoglobulin levels are low in patients with HLH
In addition to early and peripheral B-cell development, we evaluated later stages of B-cell maturation in the bone marrow. We compared the plasma cell/blast (CD19+CD20−CD38bright) population in the bone marrow of patients with P-HLH, I-HLH, and R-HLH. We found that the plasma cell/blast percentage was lower in the bone marrows of patients with P-HLH/I-HLH (0.014% ± 0.0038%) than that of the control (0.043% ± 0.0064%; P < .01) and R-HLH (0.057% ± 0.0081%; P < .001) group, but the percentage of plasma cell/blast in R-HLH group was comparable with that of the control (Figure 6A-B). To quantify the plasma cell number in the bone marrow between the P-HLH/I-HLH and R-HLH groups, we compared CD138 immunohistochemistry staining on bone marrow biopsy specimen. We found no significant difference in the frequency of CD138+ plasma cells between the P-HLH/I-HLH groups and the R-HLH group (Figure 6C-D). Among 21 patients with HLH who had an immunoglobulin G (IgG) level checked before receiving any therapies including immunoglobulin replacement, 19% had low IgG levels for their age (Figure 6E). In addition to decreases in IgG levels at presentation in a subset of patients with HLH, we noted a decrease in absolute B cell numbers in 50% of patients with HLH (Figure 6F).
Figure 6.

Plasma cell/blast and immunoglobulin production are low in patients with HLH. (A) Representative flow cytometry dot plot of CD38/CD19 gated on CD20− cells showing depletion of the CD19+CD38bright population in the HLH group (right) compared with the control group (left). (B) Percentage of plasma cell/blast (CD19+CD20−CD38bright) population in P-HLH/I-HLH groups (n = 6) and R-HLH group (n = 4) compared with the HC (n = 8) (HC, 0.043% ± 0.006%; P-HLH/I-HLH, 0.014% ± 0.004%; and R-HLH, 0.056% ± 0.008%). (C) Representative immunohistochemistry stains of CD138 in bone marrow biopsy specimen from a patient with P-HLH (left) and 1 with R-HLH (right). (D) Percentage of CD138+ immunohistochemistry staining from bone marrow biopsy samples in patients with P-HLH/I-HLH (n = 10) and R-HLH (n = 6) (P-HLH/I-HLH, 0.61% ± 0.21%; R-HLH, 0.92% ± 0.41%). The two-tailed Student t test or the Mann-Whitney test was used depending on normality for statistical comparison between 2 groups. Data represent mean ± SEM; ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001. (E-F) Peripheral blood IgG values and absolute B cells from patients with HLH before receiving steroid or immunoglobulin treatment. The open shapes represent values lower than normal range for patient age. Of the 21 patients with HLH, 19% have low IgG levels for their age. Of the 12 patients with HLH, 50% have low absolute B-cell levels for their age.
Discussion
Most studies of HLH and hyperinflammatory state have focused on T-cell and innate immune activation and dysregulation. Our data suggest that active HLH is an acquired humoral immune deficiency state. Active HLH impairs several stages of B-cell development and maturation, leading to both quantitative and qualitative defects. Characteristic findings are early B-cell development arrest in the bone marrow and decreased CSMB cells with suppression of germinal center activity. We attribute some of these effects to the inflammatory state driven by IFN-γ.
We report profoundly decreased early B-cell development in all forms of active HLH. Defects in B-cell development are more prominent in R-HLH than in P-HLH.34 Unlike the acute presentation of P-HLH, patients with R-HLH have a prolonged exposure to an inflammatory milieu. The prior use of immune suppressive or modulatory therapies for patients with R-HLH potentially affects early B-cell development even further.35 Moreover, R-HLH generally occurs at an older age, and hematogones are known to decrease with increased age.21
Murine models of toll-like receptor 9 and IFN-γ hyperstimulation,36 along with overexpression of IFN-γ and human interferon regulatory factor 1 (IRF1),37,38 have shown a severe reduction in B-cell development. We show that in humans with disease physiology associated with marked IFN-γ exposure, such as in HLH, there is a profound decrease in early B-cell development in the bone marrow, as recapitulated in the in vitro B-cell differentiation system, following exposure to IFN-γ alone. The addition of TNF results in marked synergistic suppression of B-cell development.39 Our findings are supported by the previous report of low κ-deleting element combination circles in infants with UNC13D gene defects, one of the common causes of P-HLH.40
To further explore the mechanism underlying IFN-γ–mediated B-cell suppression in humans, we performed mRNA sequencing of human CD34+ cells treated with IFN-γ and identified genes and pathways involved in B-cell development and regulation in the setting of inflammation. The treated group had significantly upregulated expression levels in genes such as IRF1, GBP1, and CXCL10 and pathways related to IFN-γ signaling, suggesting the induction of the IFN-γ response.28,29 IRF1, SAMD9L, and LRRK2 were among the top upregulated genes directly related to B-cell development. A transgenic murine model with aberrant expression of IRF1 showed a marked decrease in B-cell development, thereby validating IRF1 as a critical mediator of the IFN-γ effect on B-cell development.41 In addition, gain-of-function mutations in SAMD9L have been recently reported in patients with myelodysplastic syndrome and immune deficiency characterized by B-cell and NK cell lymphopenia.41,42 LRRK2 negatively affects B-cell terminal differentiation and antibody production in diseases such as systemic erythematous lupus.43 Therefore, these upregulated genes are likely to suppress B-cell development. Furthermore, genes involved in B-cell migration and maturation, such as S1PR1, IGF1, MMP2, and GPR183, were downregulated in the setting of inflammation, supporting the findings of suppressed B-cell maturation in the presence of IFN-γ.44, 45, 46, 47, 48 Because IFN-γ is primarily produced by activated T cells, pathological states that result in hyperactivation of T cells, such as viral and mycobacterial infections,49, 50, 51, 52 and those pathways indicative of T-cell activation, such as allograft rejection and graft-versus-host disease,53,54 could suppress early B-cell development and maturation, resulting in an occult or overt secondary humoral immune defect worthy of further investigations.
In addition to early B-cell development arrest, we show a peripheral B-cell class switch defect associated with a decrease in cTfh cells in patients with HLH.55,56 Furthermore, the decrease in CSMB cells occurs despite high CXCL13 levels. CXCL13 binding to CXCR5-expressing Tfh and B cells facilitates the germinal center reaction. Marked decrease in CXCR5-expressing cells could result in high levels of its ligand CXCL13; however, increased production cannot be ruled out.57 Near-complete effacement of germinal centers with a marked decrease in BCL6-expressing B and Tfh cells suggest that there may be an overall shift from BCL6 to an effector transcriptional module in active HLH. Interestingly, we noted improvement in the frequency of cTfh and CSMB cells once T-cell activation was controlled in HLH. These findings suggest that the suppression of B-cell development and cTfh cells is reversible with control of T-cell activation and hyperinflammation. In addition to steroids, targeted therapies, such as JAK-STAT inhibitors and IFN-γ–blocking antibody emapalumab, could, in part, reverse the effects of IFN-γ on B-cell development, thereby potentially improving humoral immune responses.6,8,58,59 Further studies are needed to evaluate whether JAK-STAT inhibitors or IFN-γ–blocking antibody emapalumab may restore humoral function in addition to controlling hyperinflammation.
Finally, we show that the proportions of plasma cells/blasts were significantly decreased in the bone marrow of patients with P-HLH/I-HLH compared with those of the R-HLH group. Despite markedly decreased early B-cell numbers in both groups, patients with R-HLH are clinically known to display normal levels of immunoglobulin or hypergammaglobulinemia.60,61 The discordant high IgG levels in R-HLH could be due to the high proportion of plasmablasts in R-HLH when compared with P-HLH or I-HLH. Elevated IL-6 and IL-18 levels in patients with rheumatologic diseases, such as systemic juvenile idiopathic arthritis and R-HLH, could contribute to the enhanced late-stage B-cell maturation, resulting in higher numbers of plasmablasts and antibody-secreting cells.34,62, 63, 64, 65, 66, 67, 68
Our data demonstrate that despite the suppression of early B-cell development and subsequent reduction in B-cell maturation to CSMB cells, the number of plasma cells remains unaffected at the diagnosis of HLH. Given that IgG levels are primarily maintained by plasma cells, it is unsurprising that IgG levels did not decrease in the majority of patients at the diagnosis of HLH. Further immunobiology studies are needed to assess whether impaired early B-cell development and poor germinal center activity during acute HLH result in clinically significant impairment of the humoral immune response to pathogens (neoantigens). The observation that patients with P-HLH gene defects such as STXBP2 and UNC13D mutations present with the common variable immune deficiency phenotype, characterized by hypogammaglobulinemia and reduced CSMB cells, suggests that our B-cell findings might be clinically relevant.11, 12, 13, 14 Additional studies are needed to evaluate whether acquired B-cell defects in the setting of marked T-cell activation could have relevance in other disorders characterized by T-cell activation with high IFN-γ activity, such as in STAT1 gain of function,69 primary immune dysregulation disorders presenting with a common variable immune deficiency phenotype,70,71 and acute graft-versus-host disease.
To conclude, our study shows that in patients with active HLH, there is marked suppression of B-cell development spanning all stages, that is, early, peripheral, and late stages of B-cell development and maturation. Our observations may have clinical relevance, but further specific immunobiology studies are needed to characterize whether these B-cell changes affect the humoral immune function of patients with HLH.
Conflict-of-interest disclosure: S.C. serves on the ad hoc advisory board of Swedish Orphan Biovitrum. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Gavin Statham and his family for their contribution to HLH research.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grant 1K08HL141635-01A1, and Henagan Foundation (S.C.), and the Atlanta Pediatric Scholars Program K12 Scholar supported by the National Institutes of Health, Eunice Kennedy Shriver National Institute of Child Health and Human Development grant K12HD072245 (J.S.). S. Prahalad is supported in part by the Marcus Foundation, Atlanta. Research reported in this publication was supported, in part, by the Cancer Tissue and Pathology shared resource of Winship Cancer Institute of Emory University and National Institutes of Health, National Cancer Institute under award number P30CA138292.
Authorship
Contribution: J.S. was involved in data collection, data analysis, figure creation, and manuscript writing; S. Park was involved in data collection, data analysis, and manuscript writing; S.V. was involved in data analysis, figure creation, and manuscript writing; D.K. was involved in data collection, data analysis, and figure creation; C.P., V.P., L.M., M.L., and C.J.H. were involved in data collection and analysis; M.B. was involved in clinical care and manuscript writing; E.K.W., S.K., and S. Prahalad were involved in manuscript writing and critical inputs; and S.C. was responsible for the project concept, design, data collection and analysis, manuscript writing, and project oversight.
Footnotes
The gene expression data are available in the Sequence Read Archive database (accession number BioProject PRJNA944951) at https://www.ncbi.nlm.nih.gov/bioproject/PRJNA944951.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Ishii E. Hemophagocytic lymphohistiocytosis in children: pathogenesis and treatment. Front Pediatr. 2016;4:47. doi: 10.3389/fped.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Filipovich AH, Chandrakasan S. Pathogenesis of hemophagocytic lymphohistiocytosis. Hematol Oncol Clin North Am. 2015;29(5):895–902. doi: 10.1016/j.hoc.2015.06.007. [DOI] [PubMed] [Google Scholar]
- 3.Marsh RA, Madden L, Kitchen BJ, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010;116(7):1079–1082. doi: 10.1182/blood-2010-01-256099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romberg N, Vogel TP, Canna SW. NLRC4 inflammasomopathies. Curr Opin Allergy Clin Immunol. 2017;17(6):398–404. doi: 10.1097/ACI.0000000000000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. 2004;104(3):735–743. doi: 10.1182/blood-2003-10-3413. [DOI] [PubMed] [Google Scholar]
- 6.Das R, Guan P, Sprague L, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. 2016;127(13):1666–1675. doi: 10.1182/blood-2015-12-684399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albeituni S, Verbist KC, Tedrick PE, et al. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood. 2019;134(2):147–159. doi: 10.1182/blood.2019000761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Locatelli F, Jordan MB, Allen C, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. 2020;382(19):1811–1822. doi: 10.1056/NEJMoa1911326. [DOI] [PubMed] [Google Scholar]
- 9.De Benedetti F, Grom A, Brogan P, et al. Efficacy and safety of emapalumab in macrophage activation syndrome. Ann Rheum Dis. 2023;82(6):857–865. doi: 10.1136/ard-2022-223739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keenan C, Nichols KE, Albeituni S. Use of the JAK inhibitor ruxolitinib in the treatment of hemophagocytic lymphohistiocytosis. Front Immunol. 2021;12 doi: 10.3389/fimmu.2021.614704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pagel J, Beutel K, Lehmberg K, et al. Distinct mutations in STXBP2 are associated with variable clinical presentations in patients with familial hemophagocytic lymphohistiocytosis type 5 (FHL5) Blood. 2012;119(25):6016–6024. doi: 10.1182/blood-2011-12-398958. [DOI] [PubMed] [Google Scholar]
- 12.Meeths M, Entesarian M, Al-Herz W, et al. Spectrum of clinical presentations in familial hemophagocytic lymphohistiocytosis type 5 patients with mutations in STXBP2. Blood. 2010;116(15):2635–2643. doi: 10.1182/blood-2010-05-282541. [DOI] [PubMed] [Google Scholar]
- 13.Rohr J, Beutel K, Maul-Pavicic A, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. 2010;95(12):2080–2087. doi: 10.3324/haematol.2010.029389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Esmaeilzadeh H, Bemanian MH, Nabavi M, et al. Novel patient with late-onset familial hemophagocytic lymphohistiocytosis with STXBP2 mutations presenting with autoimmune hepatitis, neurological manifestations and infections associated with hypogammaglobulinemia. J Clin Immunol. 2015;35(1):22–25. doi: 10.1007/s10875-014-0119-z. [DOI] [PubMed] [Google Scholar]
- 15.Pachlopnik Schmid J, Canioni D, Moshous D, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency) Blood. 2011;117(5):1522–1529. doi: 10.1182/blood-2010-07-298372. [DOI] [PubMed] [Google Scholar]
- 16.Pieper K, Grimbacher B, Eibel H. B-cell biology and development. J Allergy Clin Immunol. 2013;131(4):959–971. doi: 10.1016/j.jaci.2013.01.046. [DOI] [PubMed] [Google Scholar]
- 17.Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. 2019;3(1):47–50. doi: 10.1182/bloodadvances.2018025858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.LeBien TW, Tedder TF. B lymphocytes: how they develop and function. Blood. 2008;112(5):1570–1580. doi: 10.1182/blood-2008-02-078071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miosge LA, Goodnow CC. Genes, pathways and checkpoints in lymphocyte development and homeostasis. Immunol Cell Biol. 2005;83(4):318–335. doi: 10.1111/j.1440-1711.2005.01353.x. [DOI] [PubMed] [Google Scholar]
- 20.Carulli G, Ottaviano V, Guerri V, et al. Multiparameter flow cytometry to detect hematogones and to assess B-lymphocyte clonality in bone marrow samples from patients with non-Hodgkin lymphomas. Hematol Rep. 2014;6(2):5381. doi: 10.4081/hr.2014.5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna RW, Washington LT, Aquino DB, Picker LJ, Kroft SH. Immunophenotypic analysis of hematogones (B-lymphocyte precursors) in 662 consecutive bone marrow specimens by 4-color flow cytometry. Blood. 2001;98(8):2498–2507. doi: 10.1182/blood.v98.8.2498. [DOI] [PubMed] [Google Scholar]
- 22.Sanz I, Wei C, Jenks SA, et al. Challenges and opportunities for consistent classification of human B cell and plasma cell populations. Front Immunol. 2019;10:2458. doi: 10.3389/fimmu.2019.02458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corcione A, Benvenuto F, Ferretti E, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107(1):367–372. doi: 10.1182/blood-2005-07-2657. [DOI] [PubMed] [Google Scholar]
- 24.Ravelli A, Minoia F, Davi S, et al. 2016 classification criteria for macrophage activation syndrome complicating systemic Juvenile idiopathic arthritis: a European League against rheumatism/American College of Rheumatology/Paediatric Rheumatology International Trials Organisation Collaborative Initiative. Ann Rheum Dis. 2016;75(3):481–489. doi: 10.1136/annrheumdis-2015-208982. [DOI] [PubMed] [Google Scholar]
- 25.Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007;48(2):124–131. doi: 10.1002/pbc.21039. [DOI] [PubMed] [Google Scholar]
- 26.Chantepie SP, Cornet E, Salaun V, Reman O. Hematogones: an overview. Leuk Res. 2013;37(11):1404–1411. doi: 10.1016/j.leukres.2013.07.024. [DOI] [PubMed] [Google Scholar]
- 27.Roy S, Guler R, Parihar SP, et al. Batf2/Irf1 induces inflammatory responses in classically activated macrophages, lipopolysaccharides, and mycobacterial infection. J Immunol. 2015;194(12):6035–6044. doi: 10.4049/jimmunol.1402521. [DOI] [PubMed] [Google Scholar]
- 28.Liu M, Guo S, Hibbert JM, et al. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011;22(3):121–130. doi: 10.1016/j.cytogfr.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Honkala AT, Tailor D, Malhotra SV. Guanylate-binding protein 1: an emerging target in inflammation and cancer. Front Immunol. 2019;10:3139. doi: 10.3389/fimmu.2019.03139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farrar MA, Schreiber RD. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611. doi: 10.1146/annurev.iy.11.040193.003035. [DOI] [PubMed] [Google Scholar]
- 31.Halloran PF, Miller LW, Urmson J, et al. IFN-gamma alters the pathology of graft rejection: protection from early necrosis. J Immunol. 2001;166(12):7072–7081. doi: 10.4049/jimmunol.166.12.7072. [DOI] [PubMed] [Google Scholar]
- 32.Havenar-Daughton C, Lindqvist M, Heit A, et al. CXCL13 is a plasma biomarker of germinal center activity. Proc Natl Acad Sci U S A. 2016;113(10):2702–2707. doi: 10.1073/pnas.1520112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar D, Rostad CA, Jaggi P, et al. Distinguishing immune activation and inflammatory signatures of multisystem inflammatory syndrome in children (MIS-C) versus hemophagocytic lymphohistiocytosis (HLH) J Allergy Clin Immunol. 2022;149(5):1592–1606.e16. doi: 10.1016/j.jaci.2022.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weiss ES, Girard-Guyonvarc'h C, Holzinger D, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. 2018;131(13):1442–1455. doi: 10.1182/blood-2017-12-820852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Papadaki HA, Kritikos HD, Gemetzi C, et al. Bone marrow progenitor cell reserve and function and stromal cell function are defective in rheumatoid arthritis: evidence for a tumor necrosis factor alpha-mediated effect. Blood. 2002;99(5):1610–1619. doi: 10.1182/blood.v99.5.1610. [DOI] [PubMed] [Google Scholar]
- 36.Baratono SR, Chu N, Richman LP, Behrens EM. Toll-like receptor 9 and interferon-gamma receptor signaling suppress the B-cell fate of uncommitted progenitors in mice. Eur J Immunol. 2015;45(5):1313–1325. doi: 10.1002/eji.201445319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamada G, Ogawa M, Akagi K, et al. Specific depletion of the B-cell population induced by aberrant expression of human interferon regulatory factor 1 gene in transgenic mice. Proc Natl Acad Sci U S A. 1991;88(2):532–536. doi: 10.1073/pnas.88.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Young HA, Klinman DM, Reynolds DA, et al. Bone marrow and thymus expression of interferon-gamma results in severe B-cell lineage reduction, T-cell lineage alterations, and hematopoietic progenitor deficiencies. Blood. 1997;89(2):583–595. [PubMed] [Google Scholar]
- 39.Popescu M, Cabrera-Martinez B, Winslow GM. TNF-alpha contributes to lymphoid tissue disorganization and germinal center B cell suppression during intracellular bacterial infection. J Immunol. 2019;203(9):2415–2424. doi: 10.4049/jimmunol.1900484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Borte S, Meeths M, Liebscher I, et al. Combined newborn screening for familial hemophagocytic lymphohistiocytosis and severe T- and B-cell immunodeficiencies. J Allergy Clin Immunol. 2014;134(1):226–228. doi: 10.1016/j.jaci.2014.04.026. [DOI] [PubMed] [Google Scholar]
- 41.Tesi B, Davidsson J, Voss M, et al. Gain-of-function SAMD9L mutations cause a syndrome of cytopenia, immunodeficiency, MDS, and neurological symptoms. Blood. 2017;129(16):2266–2279. doi: 10.1182/blood-2016-10-743302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Allenspach EJ, Soveg F, Finn LS, et al. Germline SAMD9L truncation variants trigger global translational repression. J Exp Med. 2021;218(5) doi: 10.1084/jem.20201195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang M, Yao C, Cai J, et al. LRRK2 is involved in the pathogenesis of system lupus erythematosus through promoting pathogenic antibody production. J Transl Med. 2019;17(1):37. doi: 10.1186/s12967-019-1786-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allende ML, Tuymetova G, Lee BG, Bonifacino E, Wu YP, Proia RL. S1P1 receptor directs the release of immature B cells from bone marrow into blood. J Exp Med. 2010;207(5):1113–1124. doi: 10.1084/jem.20092210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Landreth KS, Narayanan R, Dorshkind K. Insulin-like growth factor-I regulates pro-B cell differentiation. Blood. 1992;80(5):1207–1212. [PubMed] [Google Scholar]
- 46.Clutter SD, Fortney J, Gibson LF. MMP-2 is required for bone marrow stromal cell support of pro-B-cell chemotaxis. Exp Hematol. 2005;33(10):1192–1200. doi: 10.1016/j.exphem.2005.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gatto D, Brink R. B cell localization: regulation by EBI2 and its oxysterol ligand. Trends Immunol. 2013;34(7):336–341. doi: 10.1016/j.it.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 48.Gatto D, Paus D, Basten A, Mackay CR, Brink R. Guidance of B cells by the orphan G protein-coupled receptor EBI2 shapes humoral immune responses. Immunity. 2009;31(2):259–269. doi: 10.1016/j.immuni.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 49.Moir S, Buckner CM, Ho J, et al. B cells in early and chronic HIV infection: evidence for preservation of immune function associated with early initiation of antiretroviral therapy. Blood. 2010;116(25):5571–5579. doi: 10.1182/blood-2010-05-285528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moir S, Fauci AS. Pathogenic mechanisms of B-lymphocyte dysfunction in HIV disease. J Allergy Clin Immunol. 2008;122(1):12–19. doi: 10.1016/j.jaci.2008.04.034. quiz 20-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tipton TRW, Hall Y, Bore JA, et al. Characterisation of the T-cell response to Ebola virus glycoprotein amongst survivors of the 2013-16 West Africa epidemic. Nat Commun. 2021;12(1):1153. doi: 10.1038/s41467-021-21411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Winslow GM, Cooper A, Reiley W, Chatterjee M, Woodland DL. Early T-cell responses in tuberculosis immunity. Immunol Rev. 2008;225:284–299. doi: 10.1111/j.1600-065X.2008.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Turka LA, Linsley PS, Lin H, et al. T-cell activation by the CD28 ligand B7 is required for cardiac allograft rejection in vivo. Proc Natl Acad Sci U S A. 1992;89(22):11102–11105. doi: 10.1073/pnas.89.22.11102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merli P, Caruana I, De Vito R, et al. Role of interferon-gamma in immune-mediated graft failure after allogeneic hematopoietic stem cell transplantation. Haematologica. 2019;104(11):2314–2323. doi: 10.3324/haematol.2019.216101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinuesa CG, Linterman MA, Yu D, MacLennan IC. Follicular helper T cells. Annu Rev Immunol. 2016;34:335–368. doi: 10.1146/annurev-immunol-041015-055605. [DOI] [PubMed] [Google Scholar]
- 56.Breitfeld D, Ohl L, Kremmer E, et al. Follicular B helper T cells express CXC chemokine receptor 5, localize to B cell follicles, and support immunoglobulin production. J Exp Med. 2000;192(11):1545–1552. doi: 10.1084/jem.192.11.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wong CK, Wong PT, Tam LS, Li EK, Chen DP, Lam CW. Elevated production of B cell chemokine CXCL13 is correlated with systemic lupus erythematosus disease activity. J Clin Immunol. 2010;30(1):45–52. doi: 10.1007/s10875-009-9325-5. [DOI] [PubMed] [Google Scholar]
- 58.Moura RA, Fonseca JE. JAK inhibitors and modulation of B cell immune responses in rheumatoid arthritis. Front Med (Lausanne) 2020;7 doi: 10.3389/fmed.2020.607725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gabr JB, Liu E, Mian S, et al. Successful treatment of secondary macrophage activation syndrome with emapalumab in a patient with newly diagnosed adult-onset Still's disease: case report and review of the literature. Ann Transl Med. 2020;8(14):887. doi: 10.21037/atm-20-3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lo MS, Zurakowski D, Son MB, Sundel RP. Hypergammaglobulinemia in the pediatric population as a marker for underlying autoimmune disease: a retrospective cohort study. Pediatr Rheumatol Online J. 2013;11(1):42. doi: 10.1186/1546-0096-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Granata G, Didona D, Stifano G, Feola A, Granata M. Macrophage activation syndrome as onset of systemic lupus erythematosus: a case report and a review of the literature. Case Rep Med. 2015;2015 doi: 10.1155/2015/294041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Calame KL. Plasma cells: finding new light at the end of B cell development. Nat Immunol. 2001;2(12):1103–1108. doi: 10.1038/ni1201-1103. [DOI] [PubMed] [Google Scholar]
- 63.Nguyen DC, Joyner CJ, Sanz I, Lee FE. Factors affecting early antibody secreting cell maturation into long-lived plasma cells. Front Immunol. 2019;10:2138. doi: 10.3389/fimmu.2019.02138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fairfax KA, Kallies A, Nutt SL, Tarlinton DM. Plasma cell development: from B-cell subsets to long-term survival niches. Semin Immunol. 2008;20(1):49–58. doi: 10.1016/j.smim.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 65.de Benedetti F, Massa M, Robbioni P, Ravelli A, Burgio GR, Martini A. Correlation of serum interleukin-6 levels with joint involvement and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis Rheum. 1991;34(9):1158–1163. doi: 10.1002/art.1780340912. [DOI] [PubMed] [Google Scholar]
- 66.Choy EH, De Benedetti F, Takeuchi T, Hashizume M, John MR, Kishimoto T. Translating IL-6 biology into effective treatments. Nat Rev Rheumatol. 2020;16(6):335–345. doi: 10.1038/s41584-020-0419-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Enoksson SL, Grasset EK, Hagglof T, et al. The inflammatory cytokine IL-18 induces self-reactive innate antibody responses regulated by natural killer T cells. Proc Natl Acad Sci U S A. 2011;108(51):E1399–1407. doi: 10.1073/pnas.1107830108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jego G, Bataille R, Pellat-Deceunynck C. Interleukin-6 is a growth factor for nonmalignant human plasmablasts. Blood. 2001;97(6):1817–1822. doi: 10.1182/blood.v97.6.1817. [DOI] [PubMed] [Google Scholar]
- 69.Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127(25):3154–3164. doi: 10.1182/blood-2015-11-679902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chandrakasan S, Chandra S, Davila Saldana BJ, Torgerson TR, Buchbinder D. Primary immune regulatory disorders for the pediatric hematologist and oncologist: a case-based review. Pediatr Blood Cancer. 2019;66(5) doi: 10.1002/pbc.27619. [DOI] [PubMed] [Google Scholar]
- 71.Kumar D, Prince C, Bennett CM, et al. T-follicular helper cell expansion and chronic T-cell activation are characteristic immune anomalies in Evans syndrome. Blood. 2022;139(3):369–383. doi: 10.1182/blood.2021012924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.