

Key Points
-
•
Terminal sialic acids on core-1 O-glycans emanating from GPIIb Ser845 and Ser847 partially mask the human HPA-9b alloantigenic epitope.
-
•
Using iPSC-derived megakaryocytes genetically engineered to lack select sialyltransferases enhances anti-HPA-9b alloantibody detection.
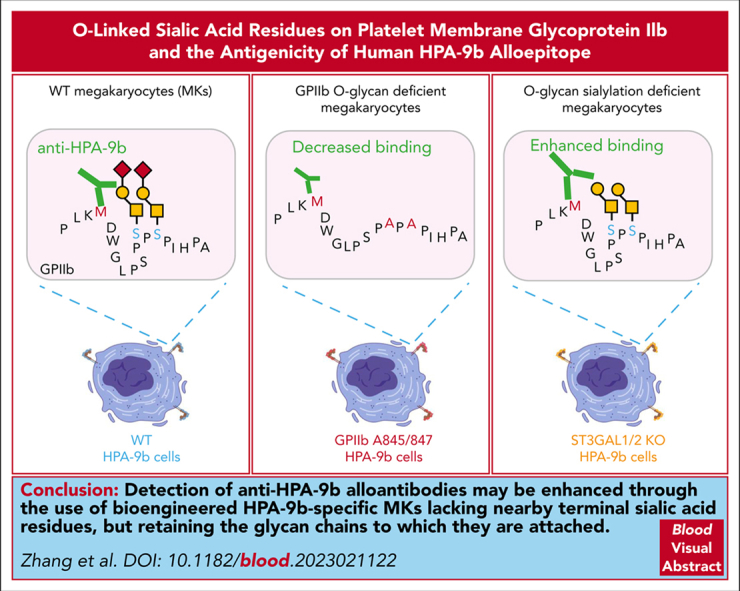
Visual Abstract
Abstract
Sialic acids occupy the terminal position of glycan chains and have the potential to influence the antigenicity of glycoproteins (GP). The polymorphisms of human platelet alloantigens (HPA)-3 and HPA-9, located near the C-terminus of the extracellular domain of platelet membrane GPIIb, are adjacent to sialyl-core 1 O-glycans emanating from serines 845 and 847. Whether the nearby O-glycans affect the antigenicity of HPA-9b or influence the binding of anti-HPA-9b alloantibodies in clinically significant cases of neonatal alloimmune thrombocytopenia is unknown. To address this issue, we generated a series of O-glycan mutant HPA-9 allele-specific induced pluripotent stem cell lines, differentiated them to megakaryocytes (MKs), and examined their ability to bind HPA-9b-specific alloantibodies. We found that both wild-type MKs treated with neuraminidase and those genetically modified to lack the sialidases ST3GAL1 and ST3GAL2 dramatically increased anti-HPA-9b alloantibody binding, indicating that the HPA-9b epitope is partially masked by terminal sialic acids on nearby O-glycans of GPIIb. Interestingly, mutating the serine residues that carry these glycan chains to alanine actually reduced the binding of anti-HPA-9b alloantibodies, indicating that these 2 O-glycan chains contribute to the presentation of the HPA-9b epitope—perhaps by stabilizing the conformation of the GP in this region. Collectively, our data suggest that detection of anti-HPA-9b alloantibodies may be enhanced through the use of HPA-9b-specific MKs that have been genetically altered to lack nearby terminal sialic acid residues but retain the glycan chains to which they are attached.
Accurate diagnosis of fetal/neonatal alloimmune thrombocytopenia is important and requires the presence of both fetomaternal (parental) platelet antigen incompatibility and maternal antibodies specific to the incompatible antigen. Using induced pluripotent stem cells, Zhang and colleagues show that maternal alloantibodies in sera can be either sialylation-dependent or independent. This opens new ways to enhance antibody detection and reveal alloantibodies that would otherwise be missed.
Introduction
Fetal and neonatal alloimmune thrombocytopenia (FNAIT—variously referred to in the literature as NATP, FNIT, and NAIT) is caused by maternal alloantibodies that cross the placenta and clear fetal platelets displaying mismatched human platelet alloantigens (HPAs). FNAIT occurs approximately once in 1000 live births and is the leading cause of severe thrombocytopenia in the fetus and neonate and also the leading cause of intracranial hemorrhage in full-term infants.1, 2, 3 Confirmation of FNAIT requires genotyping to determine platelet antigen incompatibility between mother and baby and the identification of the corresponding maternal alloantibody. Immunization against the HPA-1a and HPA-5b accounts for ∼90% of the serologically proven cases.4,5 However, the majority of apparent FNAIT cases referred for laboratory investigation remain unresolved despite use of the best available diagnostic techniques.4, 5, 6
The HPA-9b alloantigen, historically known as the Maxa alloantigen, was first described in 1995 in a severe case of FNAIT.7 The antigen is located on the extracellular domain of platelet glycoprotein (GP) IIb, and results from a single nucleotide change 2603G>A in the encoding gene leading to a Val837Met substitution. Although HPA-9b is a low-frequency platelet alloantigen with an estimated gene frequency of 0.002 to 0.005, it seems more immunogenic than most of other HPAs and may be the third most important trigger for FNAIT, after HPA-1a and HPA-5b.7, 8, 9, 10 Most FNAIT cases associated with HPA-9b present with severe thrombocytopenia and bleeding, and nearly one-third of such cases present with intracranial hemorrhage.8,10 Unfortunately, detection of anti-HPA-9b alloantibodies can be extremely difficult using standard serologic assays.8,10
Interestingly, the HPA-9 polymorphism (Val837Met) is only 6 amino acids away from the HPA-3 polymorphism (Ile843Ser). Both of them are adjacent to sialyl-core 1 O-glycan chains that emanate from serines 845 and 847 of GPIIb.11 Previous studies have shown that the nearby O-linked glycans are required to support the binding of some HPA-3a alloantibodies.12, 13, 14 Loss of these glycans, especially terminal sialic acid residues, during platelet storage or preparation, can present major difficulties in detecting clinically important anti-HPA-3a alloantibodies in suspected cases of FNAIT. Similarly, detection and identification of anti-HPA-9b alloantibodies from FNAIT patient sera can also be extremely challenging for probably the same reasons, resulting in the inability to resolve clinical cases of this bleeding disorder. Whether the nearby O-glycans on serines 845 and 847 of GPIIb affect the antigenicity of HPA-9b or influence the binding of anti-HPA-9b alloantibodies in clinically significant cases of FNAIT remains unknown.
We recently reported the generation of bioengineered, HLA class I-negative, HPA-3a, HPA-3b, and HPA-9b allele-specific megakaryocytes (MKs) from human induced pluripotent stem cells (iPSCs) that are suitable for whole cell flow cytometry detection of anti-HPA-3a, HPA-3b, and HPA-9b alloantibodies.15 This assay system offers several advantages compared with currently available standard assays in clinical diagnostic laboratories. Employing intact HLA class I-negative MKs provides a distinct advantage when patient samples also contain HLA class I antibodies—the presence of which confounds the traditional diagnostic application of “whole platelet” assays.16 In addition, the identical genetic background of clonally derived, HPA allele-specific, bioengineered MKs provides extremely high specificity for anti-HPA alloantibody detection—a feat that cannot be easily achieved by using donor-derived platelets. In contrast to currently used clinical standard “GP-specific” assays,16 the use of intact cells preserves protein, antigen, and epitope structures on the cell surface found to be essential for the detection of certain anti-HPA alloantibodies against so-called “labile antigens.”17, 18, 19, 20 Finally, appropriately engineered HPA allele-specific iPSC-derived MKs can express the desired GPIIb allele in homozygous form, an advantage particularly important for detection of HPA-9b-specific alloantibodies, since HPA-9b homozygous platelets rarely exist in nature, if at all. HPA allele-specific MK-based whole cell assays, therefore, have the potential to offer high specificity and high sensitivity for identifying anti-HPA alloantibody reactivity in maternal sera derived from previously suspected, but unresolved, cases of FNAIT.15
The use of animal (COS, CHO) and human (K562, HEK293) cell lines expressing GPIIb-IIIa allelic isoforms for alloantibody detection has been largely unsuccessful in clinical diagnostic settings,8,12,21, 22, 23 in part because they likely present different glycan chains than does GPIIb in human MKs and platelets. To overcome this limitation, we have recently shown that human bioengineered iPSC-derived, HPA allele-specific MKs express the same GPIIb-linked sialyl-core 1 O-glycans that are present in human donor-derived platelets.15 In the present study, we have extended the application of these cells to examine the influence of sialylated O-glycan chains on HPA-9b alloantibody detection. Our data suggest that the binding of anti-HPA-9b alloantibodies may be enhanced through the use of iPSC-derived HPA-9b-specific MKs that have been genetically altered to lack nearby terminal sialic acid residues, but retain the glycan chains to which they are attached.
Materials and methods
Patient samples
Anti-HPA-3a P2, anti-HPA-9b P1-P3, and other suspected HPA-9b FNAIT patient sera were provided by the Platelet and Neutrophil Immunology Laboratory, Versiti Blood Center of Wisconsin. Anti-HPA-3a P1 and anti-HPA-9b P4 patient sera were provided by Dr Richard Aster (Versiti Blood Research Institute).
Guide RNA plasmid constructs
Guide ribonucleic acids (gRNA) were designed using the CRISPR Design Tool (https://benchling.com/crispr). gRNA sequences are listed in supplemental Table 1 (available on the Blood website). Oligos were annealed and cloned into the BbsI site of the Cas9 expression plasmids PX459 V2.0 (Addgene, Cambridge, MA).
Design and generation of donor templates
ITGA2B gBlock Gene Fragments (1.6 kb) encoding HPA-9a, HPA-3b, A845/847, or HPA-9b, HPA-3b, A845/847 were synthesized by Integrated DNA Technologies (IDT, Coralville, IA). The sequences of the gBlock Gene Fragment are provided in supplemental Information. Both fragments contain the T→G (HPA-3a to HPA-3b) substitution, mutations to convert Ser845/847 to Ala, as well as silent mutations within the recognition sequence and the protospacer adjacent motif sequence of 2 gRNAs to avoid repetitive digestions by Cas9, and they also introduce an MfeI site into genome for genotyping. The HPA-9b gBlock Gene Fragment contains an additional G→A (HPA-9a to HPA-9b) substitution. The gBlock Gene Fragments were cloned into the pMiniT 2.0 vector using NEB PCR cloning kit (New England Biolabs Inc, Ipswich, MA).
Cell culture and transfection
Human HPA-allele-specific B2MKO OT1-1 iPSCs15 were cultured on Matrigel (Corning, Corning, NY)-coated plates in StemFlex Medium (Thermo Fisher Scientific, Waltham, MA) at 37°C in 4% O2/5% CO2. To generate ST3GAL1/2-deficient iPSC lines, HPA-9a or HPA-9b allele-specific iPSCs (2 × 105) were sequentially transfected with 1 μg of guide plasmid pairs targeting ST3GAL1 and ST3GAL2 using the Amaxa P3 primary cell 4D Nucleofector Kit (Lonza, Allendale, NJ) and Nucleofector Program CB-150. To generate HPA-9a or HPA-9b allele-specific A845/847 mutant iPSCs, 2 × 105 HPA-9a and HPA-3a allele-specific iPSCs were transfected with 1 μg of guide plasmid pairs targeting ITGA2B gene in the presence of 0.5 μg of the HPA-9a, HPA-3b, A845/847 or HPA-9b, HPA-3b, A845/847 plasmid donor using same Nucleofector program. The cells were then cultured on Matrigel-coated plates with 10 μM Y27632. At 24 hours post transfection, puromycin was applied at a concentration of 1 μg/mL for 48 hours. Single clones were harvested at 12 to 14 days post-puromycin selection and replated on Matrigel-coated plates.
Differentiation of iPS cells
CRISPR-edited iPS cell lines were differentiated to hematopoietic progenitor cells (HPCs) as previously described.24,25 Briefly, cells were plated on Matrigel for differentiation. Media and cytokine changes were followed as described except that the GSK-3β inhibitor CHIR99021 (Tocris, Minneapolis, MN) (1 μM) was used instead of Wnt3a. Cells were cultured at 37°C with 4% O2/5% CO2 for 9 days, and loosely adherent HPCs were collected by carefully removing the supernatant. Cells were analyzed by flow cytometry to confirm surface expression of CD41a and CD235a. The HPCs were further differentiated to MKs in serum-free differentiation medium, which is comprised of Iscove's modified Dulbecco's medium (Thermo Fisher Scientific) containing 25% Ham's F-12 (Corning), 0.5% N2 (Thermo Fisher Scientific), 1% B27 without vitamin A (Thermo Fisher Scientific), 0.05% bovine serum albumin (Sigma-Aldrich, St. Louis, MO), 2 mM l-glutamine and penicillin-streptomycin supplemented with 50 ng/mL stem cell factor and 50 ng/mL thrombopoietin (R&D systems, Minneapolis, MN) at 37°C, 5% CO2 for 6 days. MKs were analyzed by flow cytometry to confirm the surface expression of CD41 and CD42b.
Immunoprecipitation and western blot
MKs were lysed in 20 mM Tris (pH 7.4), 150 mM NaCl, 1% Triton X-100, 0.1 mM CaCl2, and protease inhibitor cocktail (Thermo Fisher Scientific). Whole cell lysates were obtained after centrifugation at 17,000g for 15 minutes at 4°C. Whole cell lysates were precleared with Protein G Sepharose (Cytiva, Marlborough, MA) and then incubated with the anti-GPIIIa monoclonal antibody AP3 overnight at 4°C. Immune complexes were collected on Protein G Sepharose beads, then treated with neuraminidase (New England Biolabs Inc) for 1 hour at 37°C and eluted with reducing sodium dodecyl sulfate sample buffer. Following electrophoresis, the samples were electrotransferred onto polyvinylidene difluoride membrane (EMD Millipore, Billerica, MA) and immunoblotted with either biotinylated peanut agglutinin (PNA) (Vector Laboratories Inc, Burlingame, CA) or rabbit anti-GPIIb antibody. Bound lectin and primary antibody were visualized using peroxidase-conjugated streptavidin (BioLegend, San Diego, CA) and peroxidase-conjugated goat anti-rabbit immunoglobulin G (Jackson ImmunoResearch Laboratories, West Grove, PA), respectively.
Flow cytometry analysis
iPSC-derived MKs (5 × 105) were treated with or without 30 mU/mL neuraminidase from Clostridium perfringens (Sigma-Aldrich) at 37°C for 15 minutes. After washing with phosphate-buffered saline, the cells were incubated with 25 to 50 μL of normal human sera or patient sera for 30 minutes at room temperature. After washing, the cells were incubated with fluorescein isothiocyanate-conjugated anti-CD41, allophycocyanin-conjugated anti-CD42b (BioLegend), and phycoerythrin-conjugated donkey anti-human immunoglobulin G (Jackson ImmunoResearch Laboratories) at room temperature for 20 minutes. Flow cytometry was performed using a BD LSRII flow cytometer (BD Biosciences, San Jose, CA). Flow cytometry data were analyzed using FlowJo software (Tree Star Inc, Ashland, OR).
Statistical analysis
P values are calculated from Linear mixed model fit by REML, using R package “lme4,” in which patients are random effects and cell types are fixed effects.
Results
Evaluating the sialic acid dependency of anti-HPA-3a alloantibodies using allele-specific, iPSC-derived MKs
We recently generated bioengineered HLA class I-negative, allele-specific MKs from iPSCs and demonstrated that they contain the same sialyl-core 1 O-glycans that are present on native O-glycosylated GPIIb from human platelets.15 As depicted in Figure 1A, these O-linked chains are adjacent to the amino acid polymorphism responsible for creating the HPA-3a and HPA-3b epitopes. Their influence, however, on the 3-dimensional nature of the alloepitope, as well as its availability for alloantibody binding and detection, is poorly understood. To examine the sialic acid dependency of several representative, patient-derived HPA-3a-specific alloantibodies, iPSC-derived MKs were treated with neuraminidase to remove the terminal sialic acids from cell surface GPs and then probed with PNA—a lectin that binds to nonsialylated core 1 glycans. Flow cytometry analysis (Figure 1B) revealed significantly increased PNA binding to neuraminidase-treated MKs, confirming successful removal of terminal sialic acid from core 1 O-glycan structures on the MK surface. As has been previously seen using human platelets,12, 13, 14 treatment of iPSC-derived MKs with neuraminidase reduced the binding of one such HPA-3a-specific alloantibody (P1) (Figure 1C), demonstrating the contribution of the nearby sialic acid to the complex epitope. In contrast, neuraminidase treatment did not affect the binding of HPA-3a patient antibody P2 (Figure 1C), demonstrating its sialic acid independence. Taken together, these findings validate the use of iPSC-derived MKs to evaluate the epitope requirements for different HPA-3a-specific patient alloantibodies.
Figure 1.

HPA-3 allele-specific MKs can detect sialic acid (SA) dependency of anti-HPA-3a alloantibody binding. (A) Schematic of SA dependency of different anti-HPA-3a alloantibody binding. Neuraminidase treatment, which removes terminal SA from nearby O-glycans on GPIIb, can reduce SA-dependent anti-HPA-3a alloantibody binding but has no effect on SA-independent alloantibody binding. PNA is a lectin that specifically binds to unsialylated Galβ1,3GalNAc structure. (B) Flow cytometry analysis of PNA binding to iPSC-derived MKs confirmed successful removal of terminal SA after neuraminidase treatment. (C) Reactions of anti-HPA-3a patient sera with HPA-3 allele-specific iPSC-derived MKs in the presence or absence of neuraminidase treatment in flow cytometry analysis. Binding to HPA-3b cells serves as background binding for individual patient sample. Color-coded numbers in (B) and (C) indicate median fluorescence intensity of corresponding peaks. NeuAc, N-acetylneuraminic acid; Gal, galactose; GalNAc, N-acetyl-D-galactosamine; IgG, immunoglobulin G; Neu, neuraminidase; PE, phycoerythrin.
Enzymatic removal of terminal sialic acids enhances binding of anti-HPA-9b alloantibodies
As depicted in Figure 2A, the V837M polymorphism responsible for controlling formation of the HPA-9a/9b alloantigenic epitopes is only 6 amino acids away from the I843S polymorphism that defines HPA-3a/3b, with O-linked glycan chains radiating out in proximity to both sites. To examine whether the binding of HPA-9b-specific alloantibodies might be similarly affected by nearby carbohydrate chains, we treated the HPA-9 allele-specific iPSC-derived MKs with neuraminidase to remove the terminal sialic acids. Surprisingly, unlike the decreased anti-HPA-3a alloantibody binding to its target antigen upon antigen desialylation, all 4 of the anti-HPA-9b patient alloantibodies showed enhanced binding to neuraminidase-treated HPA-9b-expressing MKs (Figure 2B). These findings demonstrate that terminal sialic acid residues on O-linked glycans adjacent to the HPA-9b polymorphism partially mask the HPA-9b epitopes and suggest that removal of these terminal sialic acids, either biochemically (Figure 2) or genetically (see later), might facilitate clinical detection of anti-HPA-9b patient alloantibodies.
Figure 2.

Neuraminidase treatment enhanced HPA-9b alloantibody binding. (A) Schematic of HPA-9 alloantigen and nearby O-glycans. (B) Reactions of anti-HPA-9b patient sera with HPA-9 allele-specific iPSC-derived MKs in the presence or absence of neuraminidase treatment in flow cytometry analysis. Color-coded numbers indicate median fluorescence intensity of corresponding peaks. Binding to HPA-9a cells serves as background binding for individual patient sample. Only P1 serum shows increased background binding to HPA-9a cells after neuraminidase treatment, which may be caused by the presence of unknown anti-carbohydrate antibody against certain desialylated glycan in this case. NeuAc, N-acetylneuraminic acid; Gal, galactose; GalNAc, N-acetyl-D-galactosamine; IgG, immunoglobulin G; Neu, neuraminidase; PE, phycoerythrin.
Platelet antibody bead array (PABA),26 an equivalent of the modified antigen capture enzyme-linked immunosorbent assay, is routinely performed in our clinical diagnostic laboratory using HPA-typed donor platelets to detect anti-HPA alloantibodies. Treating HPA-typed donor platelets with neuraminidase prior to a regular PABA procedure enhanced anti-HPA-9b alloantibody binding (supplemental Figure 1), suggesting that using desialylated donor platelets may benefit the anti-HPA-9b alloantibody detection in standard clinical diagnostic assays.
The effect of proximal glycan chains on HPA-9b alloantibody binding
Recent high-resolution mass spectrometry studies have shown that serine residues 845 and 847 on GPIIb bear sialylated core 1 O-glycans.11 To examine the contribution of both these nearby glycans, as well as their terminal sialic acid residues, to the expression of the HPA-9b epitope, we used CRISPR/Cas9 technology to create 2 unique groups of iPSC-derived cell lines. Because the sialyltransferases ST3GAL1 and ST3GAL2 have recently been shown to transfer terminal sialic acid residues to these 2 carbohydrate chains,27 we generated ST3GAL1 and ST3GAL2 double knockout (KO) mutants of HPA-9a and HPA-9b iPSC lines using the strategy illustrated in supplemental Figure 2, so that the glycans attached to serines 845 and 847 would be expressed in unsialylated form (depicted in Figure 3A). The second group of MK cell lines were engineered to express alanine residues in place of serines 845 and 847 so that the entire glycan chain emanating from serine residues would be absent (also depicted in Figure 3A). Supplemental Figure 3 illustrates the detailed CRISPR strategy employed to generate the Ser845Ala, Ser847Ala double mutant cell lines. Mutating serines 845 and 847 to abolish O-glycosylation on GPIIb did not affect MK differentiation or GPIIb surface expression (supplemental Figure 4).
Figure 3.

Gene editing to remove terminal sialic acids or the entire O-glycans on GPIIb differentially affects anti-HPA-9b alloantibody binding. (A) Schematic of local alloantigenic peptide and O-glycan modification of GPIIb from genetically edited iPSC-derived MKs. (B) Flow cytometry analysis of the binding of 4 patient anti-HPA-9b alloantibodies to various HPA-9 allele-specific iPSC-derived MKs. Medium fluorescence intensity of each alloantibody binding to different MKs was normalized to that of WT HPA-9a cells. Green lines represent mean values of 4 patient samples in 3 independent experiments. P values are calculated from Linear mixed model fit by REML, using R package “lme4,” in which patients are random effects and cell types are fixed effects. ∗∗P < .01. (C) Screen of suspected anti-HPA-9b FNAIT maternal sera using WT or ST3GAL1/2 KO HPA-9 allele-specific iPSC-derived MKs. Eight suspected anti-HPA-9b maternal sera that were previously unconfirmable with clinical standard PABA assay were tested with whole cell flow cytometry analysis. All the suspected mothers are HPA-9a homozygous and all the fathers are HPA-9a/9b heterozygous. S1 and S2 contain anti-HPA-9b alloantibodies that are detectable by both WT and ST3GAL1/2 KO HPA-9b cells. Anti-HPA-9b alloantibody in S3 is only detectable by ST3GAL1/KO HPA-9b cells. S2 and S3 also contain anti-HPA-1a alloantibodies that cause high background binding in this assay. S4 is a representative of negative samples for the analysis. Color-coded numbers indicate median fluorescence intensity of corresponding peaks. NeuAc, N-acetylneuraminic acid; Gal, galactose; GalNAc, N-acetyl-D-galactosamine; IgG, immunoglobulin G; PE, phycoerythrin.
To examine the functional effects of these 2 GPIIb glycan modifications, we measured the ability of HPA-9b-specific alloantibodies to bind these cells using a whole cell flow cytometry assay. As shown in Figure 3B, consistent with the increased anti-HPA-9b alloantibody binding after neuraminidase treatment (Figure 2), sialylation-deficient ST3GAL1/2 KO HPA-9b MKs exhibited dramatically increased anti-HPA-9b alloantibody binding, further confirming the notion that HPA-9b epitopes are partially masked by terminal sialic acids on nearby GPIIb O-glycan chains. Surprisingly, however, O-glycan null Ala845/847 mutant MKs carrying the HPA-9b polymorphism exhibited slightly to moderately reduced binding of anti-HPA-9b alloantibodies, indicating that the presence of the core 1 O-glycan chains attached to GPIIb serine residues 845 and 847 contribute to the presentation of the HPA-9b epitope—perhaps by stabilizing the conformation of the GP in this otherwise disordered flexible loop region of GPIIb.28 Taken together, these data suggest that detection of anti-HPA-9b alloantibodies may be enhanced through the use of iPSC-derived HPA-9b-specific MKs that have been genetically altered to lack nearby terminal sialic acid residues, but retain the glycan chains to which they are attached.
To further demonstrate the clinical application potential for our whole cell assay system, we screened 8 suspected anti-HPA-9b FNAIT maternal sera. In these cases, parents are incompatible for HPA-9b; all the mothers are HPA-9a homozygous and all the fathers are HPA-9a/9b heterozygous. HPA-9b alloantibodies were unable to be detected in maternal sera using PABA, a standard clinical diagnostic antigen capture assay. Using our whole cell flow cytometry assay, however, we were able to successfully detect anti-HPA-9b alloantibodies in 3 of 8 suspected cases (S1-S3 in Figure 3C). Anti-HPA-9b alloantibodies in samples S1 and S2 were detected in both wild-type (WT) HPA-9b and ST3GAL1/2 KO HPA-9b cells, with increased alloantibody binding to ST3GAL1/2 KO HPA-9b cells compared with WT HPA-9b cells. Interestingly, the anti-HPA-9b alloantibody present in maternal sample S3 could only be detected using ST3GAL1/2 KO HPA-9b cells, demonstrating their increased sensitivity for anti-HPA-9b alloantibody detection. The unexpected high background binding to HPA-9a cells in S2 and S3 was due to the coexistence of an anti-HPA-1a alloantibody in these 2 samples (unpublished observations, Platelet and Neutrophil Immunology Laboratory, Versiti Blood Center of Wisconsin). These findings highlight the unique properties of bioengineered cells to distinguish anti-HPA-9b alloantibodies from other coexisting anti-platelet antibodies. Such results cannot easily be achieved by standard clinical diagnostic assays because HPA-1b/b and HPA-9a/b donor platelets are unlikely to exist very often in nature. In contrast, the bioengineered cells described herein are easily generated, stored, and distributed.
Finally, it has been known for more than 30 years that the HPA-3b allelic isoform of GPIIb differs from the HPA-3a allele by an I843→S amino acid substitution29 (Figure 4A). It has never been determined, however, whether this serine residue carries what would be a third proximal sialylated glycan chain that might influence HPA-3b antigenicity or alloantibody binding. To address this issue, we subjected HPA-3b, Ala845, Ala847 bioengineered MKs to PNA lectin western blot analysis. As shown in Figures 4A and 4B, GPIIb from these cells showed no PNA binding even after neuraminidase treatment, indicating that Ser843 does not carry an O-glycan chain in the HPA-3b allelic isoform of GPIIb.
Figure 4.

Confirm the complete loss of core 1 O-glycans on GPIIb from A845/847 variant iPSC cell lines. (A) Schematic of local alloantigenic peptide and O-glycan modification of GPIIb from WT or HPA-3b A845/847 mutant cell lines. (B) Coimmunoprecipitation of GPIIb-IIIa complex with anti-GPIIIa monoclonal antibody AP3 from lysates of WT or HPA-3b A845/847-derived MKs, followed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and immunoblot with either PNA or rabbit anti-GPIIb polyclonal antibody. NeuAc, N-acetylneuraminic acid; Gal, galactose; GalNAc, N-acetyl-D-galactosamine; Co-IP, co-immunoprecipitation.
Discussion
Sialic acids often occupy the terminal position of complex carbohydrate chains and have the potential to influence the antigenicity of GPs. Antibody binding sites on a GP can be solely protein in nature or can include or be affected by nearby glycan chains, which may either mask the epitope or, conversely, comprise part of the antibody binding site.
In the current study, we established that maternal anti-HPA-9b alloantibodies show the strongest reaction to the HPA-9b alloantigenic epitope when it is adjacent to unsialylated core 1 glycan chains. These findings suggest that fetal platelets carrying unsialylated exposed core 1 glycans on GPIIb may be the actual immunogen encountered by the maternal immune system during pregnancy. GPIIb-IIIa is expressed on fetal platelets early in gestation30; however the timing of sialylating its O-glycan chains is not known. The developmental expression of ST3GAL1 and ST3GAL2 (the sialyltransferases specifically responsible for O-glycan sialylation) during pregnancy is also not known; however, the fact that fibronectin—a highly O-glycosylated and fully sialylated plasma protein in newborns and adults—has been found to be only partially sialylated in the amniotic fluid of second- and third-trimester pregnancies31 suggests that these 2 sialyltransferases are either not present, or are not fully functional, in early fetal development. It is tempting to speculate, therefore, that GPIIb on fetal platelets starts out adjacent to unsialylated O-glycan chains, thereby directing the maternal immune system to produce an alloantibody specific for the complex protein-carbohydrate epitope shown on the right-hand side of Figure 2A. Alternatively, the O-glycans on fetal platelet GPIIb could start out fully sialylated but become desialylated by neuraminidase under certain pathological conditions, such as following bacterial or viral infection, and present to the maternal immune system after that time. Whether desialylated HPA-9b peptides are more immunogenic than sialylated peptides in mice and whether HPA-9b mouse model can be generated like what has been done for HPA-1a alloantigen32 are interesting topics for future studies.
Mucin-type O-glycosylation is an abundant and highly diverse modification of proteins. It is carried out by a large family of nearly 20 homologous polypeptide N-acetylgalactosamine transferases (GalNAc-Ts) that add GalNAc to serine, threonine, and possibly tyrosine residues.33 Although the mechanism determining O-glycan site occupancy remains unclear, substrate specificities of individual isoform of GalNAc-Ts, normally differentially expressed in different cell types, play important roles in this process. The most abundantly expressed isoforms of GalNAc-Ts in human platelets are GalNAc-T1, GalNAc-T2, and GalNAc-T10,11 consistent with RNA sequencing data from human iPSC-derived MKs (data set GSE119828), suggesting similar O-glycoproteome usage in these cells. Unlike N-linked glycosylation, which takes place at a well-defined consensus sequence, prediction of O-glycosites is challenging due to the lack of a consensus sequence for this posttranslational modification. Recent systematic analysis of native platelet and plasma protein O-glycosylation, however, has revealed that mucin-type O-glycosylation frequently occurs within disordered regions of the extracellular domain of proteins.11 A relevant example is O-glycosylation at serines 845 and 847 of human platelet GPIIb. These sites, adjacent to the HPA-9 (Val837Met) and HPA-3 (Ile843Ser) polymorphisms, are located within a proline-rich unstructured region of the extracellular Calf-2 domain.34,35 Because the O-glycans attached to Ser845/847 are likely spatially very close to Met837 (HPA-9b), they may stabilize the local conformation or participate in HPA-9b alloantigen formation. In contrast, terminal sialylation of the core-1 structure appears to partially mask alloantibody binding sites, perhaps due to the negative charge imparted by the sialic acid residues. Whether site-specific O-glycosylation affects GPIIb-IIIa receptor function deserves further investigation.
We have previously shown that human iPSCs edited to express clinically important HPAs and then differentiated to MKs permit simple, sensitive, and highly specific whole cell flow cytometry detection of anti-platelet alloantibodies from patient sera.15 In this study, we have extended the use of such cells to evaluate the glycan dependency of HPA alloantibody binding. Since iPSC-derived MKs appear to contain the same O-glycoproteome as found in native human platelets, they display intact native epitopes comprised of both peptide and glycan elements, making them particularly suitable for alloantibody detection. The use of this novel platform has enabled determination of how nearby sialylated O-linked glycans on GPIIb regulate HPA-9b epitope formation. In addition to improving our understanding of the etiology of platelet alloimmune disorders, ST3GAL1/2 KO HPA-9 allele-specific MKs can now be added to the growing list of genetically bioengineered human cell lines designed to enhance HPA alloantibody detection.
Conflict-of-interest disclosure: P.J.N. and B.R.C. are consultants for Rallybio in the field of platelet alloimmunity. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Richard Aster for providing valuable human anti-HPA-3a and HPA-9b alloantisera.
This work was supported by grant R35 HL139937 from the National Heart, Lung, and Blood Institute of the National Institutes of Health.
Authorship
Contribution: N.Z. designed and conducted experiments, analyzed and interpreted data, and wrote the manuscript; M.J.S. provided technical support for PABA experiment; B.R.C. provided rare human anti-HPA-3a and anti-HPA-9b alloantisera, designed PABA experiment, and helped interpret data; and P.J.N. designed and interpreted experiments and wrote the manuscript.
Footnotes
For original data, please contact nzhang@versiti.org.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Nanyan Zhang, Email: nzhang@versiti.org.
Peter J. Newman, Email: pjnewman@versiti.org.
Supplementary Material
References
- 1.Dreyfus M, Kaplan C, Verdy E, Schlegel N, Durand-Zaleski I, Tchernia G. Frequency of immune thrombocytopenia in newborns: a prospective study. Immune Thrombocytopenia Working Group. Blood. 1997;89(12):4402–4406. [PubMed] [Google Scholar]
- 2.Sainio S, Jarvenpaa AL, Renlund M, Riikonen S, Teramo K, Kekomaki R. Thrombocytopenia in term infants: a population-based study. Obstet Gynecol. 2000;95(3):441–446. doi: 10.1016/s0029-7844(99)00543-8. [DOI] [PubMed] [Google Scholar]
- 3.Bussel J. Diagnosis and management of the fetus and neonate with alloimmune thrombocytopenia. J Thromb Haemost. 2009;7(Suppl 1):253–257. doi: 10.1111/j.1538-7836.2009.03380.x. [DOI] [PubMed] [Google Scholar]
- 4.Davoren A, Curtis BR, Aster RH, McFarland JG. Human platelet antigen-specific alloantibodies implicated in 1162 cases of neonatal alloimmune thrombocytopenia. Transfusion. 2004;44(8):1220–1225. doi: 10.1111/j.1537-2995.2004.04026.x. [DOI] [PubMed] [Google Scholar]
- 5.Ghevaert C, Campbell K, Walton J, et al. Management and outcome of 200 cases of fetomaternal alloimmune thrombocytopenia. Transfusion. 2007;47(5):901–910. doi: 10.1111/j.1537-2995.2007.01208.x. [DOI] [PubMed] [Google Scholar]
- 6.McQuilten ZK, Wood EM, Savoia H, Cole S. A review of pathophysiology and current treatment for neonatal alloimmune thrombocytopenia (NAIT) and introducing the Australian NAIT registry. Aust N Z J Obstet Gynaecol. 2011;51(3):191–198. doi: 10.1111/j.1479-828X.2010.01270.x. [DOI] [PubMed] [Google Scholar]
- 7.Noris P, Simsek S, de Bruijne-Admiraal LG, et al. Max(a), a new low-frequency platelet-specific antigen localized on glycoprotein IIb, is associated with neonatal alloimmune thrombocytopenia. Blood. 1995;86(3):1019–1026. [PubMed] [Google Scholar]
- 8.Peterson JA, Balthazor SM, Curtis BR, McFarland JG, Aster RH. Maternal alloimmunization against the rare platelet-specific antigen HPA-9b (Max a) is an important cause of neonatal alloimmune thrombocytopenia. Transfusion. 2005;45(9):1487–1495. doi: 10.1111/j.1537-2995.2005.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peterson JA, Gitter M, Bougie DW, et al. Low-frequency human platelet antigens as triggers for neonatal alloimmune thrombocytopenia. Transfusion. 2014;54(5):1286–1293. doi: 10.1111/trf.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan C, Porcelijn L, Vanlieferinghen P, et al. Anti-HPA-9bw (Maxa) fetomaternal alloimmunization, a clinically severe neonatal thrombocytopenia: difficulties in diagnosis and therapy and report on eight families. Transfusion. 2005;45(11):1799–1803. doi: 10.1111/j.1537-2995.2005.00606.x. [DOI] [PubMed] [Google Scholar]
- 11.King SL, Joshi HJ, Schjoldager KT, et al. Characterizing the O-glycosylation landscape of human plasma, platelets, and endothelial cells. Blood Adv. 2017;1(7):429–442. doi: 10.1182/bloodadvances.2016002121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Socher I, Zwingel C, Santoso S, Kroll H. Heterogeneity of HPA-3 alloantibodies: consequences for the diagnosis of alloimmune thrombocytopenic syndromes. Transfusion. 2008;48(3):463–472. doi: 10.1111/j.1537-2995.2007.01550.x. [DOI] [PubMed] [Google Scholar]
- 13.Take H, Tomiyama Y, Shibata Y, et al. Demonstration of the heterogeneity of epitopes of the platelet-specific alloantigen. Br J Haematol. 1990;76(3):395–400. doi: 10.1111/j.1365-2141.1990.tb06374.x. [DOI] [PubMed] [Google Scholar]
- 14.Djaffar I, Vilette D, Pidard D, Wautier JL, Rosa JP. Human platelet antigen 3 (HPA-3): localization of the determinant of the alloantibody Lek(a) (HPA-3a) to the C-terminus of platelet glycoprotein IIb heavy chain and contribution of O-linked carbohydrates. Thromb Haemost. 1993;69(5):485–489. [PubMed] [Google Scholar]
- 15.Zhang N, Santoso S, Aster RH, Curtis BR, Newman PJ. Bioengineered iPSC-derived megakaryocytes for the detection of platelet-specific patient alloantibodies. Blood. 2019;134(22):e1–e8. doi: 10.1182/blood.2019002225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi T, Hirayama F. Advances in alloimmune thrombocytopenia: perspectives on current concepts of human platelet antigens, antibody detection strategies, and genotyping. Blood Transfus. 2015;13(3):380–390. doi: 10.2450/2015.0275-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrison CR, Curtis BR, McFarland JG, Huff RW, Aster RH. Severe neonatal alloimmune thrombocytopenia caused by antibodies to human platelet antigen 3a (Baka) detectable only in whole platelet assays. Transfusion. 2003;43(10):1398–1402. doi: 10.1046/j.1537-2995.2003.00533.x. [DOI] [PubMed] [Google Scholar]
- 18.Barba P, Pallares P, Nogues N, et al. Post-transfusion purpura caused by anti-HPA-3a antibodies that are only detectable using whole platelets in the platelet immunofluorescence test. Transfus Med. 2010;20(3):200–202. doi: 10.1111/j.1365-3148.2009.00978.x. [DOI] [PubMed] [Google Scholar]
- 19.Lin M, Shieh SH, Liang DC, Yang TF, Shibata Y. Neonatal alloimmune thrombocytopenia in Taiwan due to an antibody against a labile component of HPA-3a (Baka) Vox Sang. 1995;69(4):336–340. doi: 10.1111/j.1423-0410.1995.tb00369.x. [DOI] [PubMed] [Google Scholar]
- 20.Kataoka S, Kobayashi H, Chiba K, et al. Neonatal alloimmune thrombocytopenia due to an antibody against a labile component of human platelet antigen-3b (Bak b) Transfus Med. 2004;14(6):419–423. doi: 10.1111/j.1365-3148.2004.00537.x. [DOI] [PubMed] [Google Scholar]
- 21.Goldberger A, Kolodziej M, Poncz M, Bennett JS, Newman PJ. Effect of single amino acid substitutions on the formation of the PlA and Bak alloantigenic epitopes. Blood. 1991;78(3):681–687. [PubMed] [Google Scholar]
- 22.Wihadmadyatami H, Heidinger K, Roder L, et al. Alloantibody against new platelet alloantigen (Lap(a)) on glycoprotein IIb is responsible for a case of fetal and neonatal alloimmune thrombocytopenia. Transfusion. 2015;55(12):2920–2929. doi: 10.1111/trf.13238. [DOI] [PubMed] [Google Scholar]
- 23.Hayashi T, Amakishi E, Matsuyama N, et al. Establishment of a cell line panel as an alternative source of platelet antigens for a screening assay of anti-human platelet antibodies. Transfus Med. 2011;21(3):199–204. doi: 10.1111/j.1365-3148.2010.01064.x. [DOI] [PubMed] [Google Scholar]
- 24.Mills JA, Paluru P, Weiss MJ, Gadue P, French DL. In: Qu KDBaC-K., editor. Vol. 1185. Springer Science+Business Media; 2014. Hematopoietic differentiation of pluripotent stem cells in culture; pp. 181–194. (Methods in Molecular Biology). [DOI] [PubMed] [Google Scholar]
- 25.Paluru P, Hudock KM, Cheng X, et al. The negative impact of Wnt signaling on megakaryocyte and primitive erythroid progenitors derived from human embryonic stem cells. Stem Cell Res. 2014;12(2):441–451. doi: 10.1016/j.scr.2013.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metzner K, Bauer J, Ponzi H, Ujcich A, Curtis BR. Detection and identification of platelet antibodies using a sensitive multiplex assay system-platelet antibody bead array. Transfusion. 2017;57(7):1724–1733. doi: 10.1111/trf.14122. [DOI] [PubMed] [Google Scholar]
- 27.Zhang N, Lin S, Cui W, Newman PJ. Overlapping and unique substrate specificities of ST3GAL1 and 2 during hematopoietic and megakaryocytic differentiation. Blood Adv. 2022;6(13):3945–3955. doi: 10.1182/bloodadvances.2022007001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiong JP, Stehle T, Diefenbach B, et al. Crystal structure of the extracellular segment of integrin aVb3. Science. 2001;294(5541):339–345. doi: 10.1126/science.1064535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lyman S, Aster RH, Visentin GP, Newman PJ. Polymorphism of human platelet membrane glycoprotein IIb associated with the Baka/Bakb alloantigen system. Blood. 1990;75(12):2343–2348. [PubMed] [Google Scholar]
- 30.Gruel Y, Boizard B, Daffos F, Forestier F, Caen J, Wautier JL. Determination of platelet antigens and glycoproteins in the human fetus. Blood. 1986;68(2):488–492. [PubMed] [Google Scholar]
- 31.Kottgen E, Hell B, Muller C, Kainer F, Tauber R. Developmental changes in the glycosylation and binding properties of human fibronectins. Characterization of the glycan structures and ligand binding of human fibronectins from adult plasma, cord blood and amniotic fluid. Biol Chem Hoppe Seyler. 1989;370(12):1285–1294. doi: 10.1515/bchm3.1989.370.2.1285. [DOI] [PubMed] [Google Scholar]
- 32.Zhi H, Ahlen MT, Thinn AMM, et al. High-resolution mapping of the polyclonal immune response to the human platelet alloantigen HPA-1a (Pl(A1)) Blood Adv. 2018;2(21):3001–3011. doi: 10.1182/bloodadvances.2018023341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bennett EP, Mandel U, Clausen H, Gerken TA, Fritz TA, Tabak LA. Control of mucin-type O-glycosylation: a classification of the polypeptide GalNAc-transferase gene family. Glycobiology. 2012;22(6):736–756. doi: 10.1093/glycob/cwr182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell. 2008;32(6):849–861. doi: 10.1016/j.molcel.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anies S, Jallu V, Diharce J, Narwani TJ, de Brevern AG. Analysis of integrin alphaiib subunit dynamics reveals long-range effects of missense mutations on calf domains. Int J Mol Sci. 2022;23(2) doi: 10.3390/ijms23020858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.