

ABSTRACT
Inflammatory bowel disease (IBD) is a broad term for a range of chronic intestinal disorders, including Crohn’s disease and ulcerative colitis. The global prevalence of IBD is rising, with over one million patients affected in the United States alone. Adherent-invasive Escherichia coli (AIEC) is a pathobiont frequently found in IBD biopsies. AIEC adhere to and invade epithelial cells, and can survive inside phagocytes in vitro. However, how AIEC contribute to IBD in vivo remains unclear. Here, we established a larval zebrafish (Danio rerio) model to study the interplay between pre-existing intestinal inflammation and AIEC colonization of the gut. We used the pro-inflammatory drug dextran sulfate sodium (DSS) to induce intestinal inflammation. This was followed by food-borne infection of larvae with AIEC using the protozoan Paramecium caudatum, a natural prey, as a vehicle. We show that AIEC more robustly colonize the zebrafish gut and are cleared slower than non-pathogenic E. coli. In addition, DSS-induced enterocolitis increases bacterial burden and decreases bacterial clearance in the larval gut. We benchmark our model against existing rodent models using two mutants deficient in the known AIEC virulence factors FimH and IbeA, which have virulence defects in both rodent and the larval zebrafish model. Finally, we show that AIEC colonization exacerbates DSS-induced enterocolitis and prevents recovery from inflammation-induced damage. In conclusion, we established a high-throughput, genetically tractable model to study AIEC-host interactions in the context of pre-existing inflammation.
IMPORTANCE
Although inflammatory bowel diseases are on the rise, what factors influence IBD risk and severity, and the underlying mechanisms remain to be fully understood. Although host genetics, microbiome, and environmental factors have all been shown to correlate with the development of IBD, cause and effect are difficult to disentangle in this context. For example, AIEC is a known pathobiont found in IBD patients, but it remains unclear if gut inflammation during IBD facilitates colonization with AIEC, or if AIEC colonization makes the host more susceptible to pro-inflammatory stimuli. It is critical to understand the mechanisms that contribute to AIEC infections in a susceptible host in order to develop successful therapeutics. Here, we show that the larval zebrafish model recapitulates key features of AIEC infections in other animal models and can be utilized to address these gaps in knowledge.
KEYWORDS: zebrafish, colitis, AIEC, intestinal colonization
INTRODUCTION
Inflammatory bowel disease (IBD) is a broad term for a range of chronic gastrointestinal disorders, including Crohn’s disease (CD) and ulcerative colitis (UC). IBD is prevalent in industrialized nations, and the number of cases in low-incidence areas is expected to keep rising (1, 2). Although the exact cause of IBD is unknown, host genetics, environmental factors, and the gut microbiota are all known disease modifiers (2).
Adherent-invasive Escherichia coli (AIEC) is a bacterial pathobiont that colonizes the gut of both healthy subjects and IBD patients but has a higher incidence in the diseased mucosae of patients with CD (21%–63%) and UC (0%–35.7%), (3–5). AIEC adhere to and invade intestinal epithelial cells, and survive inside macrophages without inducing host cell death in vitro, but how exactly they contribute to IBD is not well understood (6). It is thought that AIEC modify the pro-inflammatory environment, or inflammation facilitates AIEC colonization because they are often isolated from lesions in patients with chronic CD as opposed to those in remission (3, 7).
Current animal models of AIEC include mice that express the human carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6) receptor (CEABAC10 mice), conventional mice treated with broad-spectrum antibiotics, mice treated with colitis-inducing agents [dextran sulfate sodium (DSS) and 2,4,6-trinitro-benzene sulfonic acid], and mice that are genetically susceptible to spontaneous colitis (8, 9). Although mice are powerful model organisms, they have some limitations including expensive care, long development periods, and laborious genetic manipulation. Furthermore, the scope of intravital imaging, particularly across multiple time points, in mice is limited, and consequently, observation of bacterial invasion, bacteria-phagocyte interactions, and pathophysiological changes often require euthanasia. To address the abovementioned gaps in knowledge, an animal model is needed that allows dynamic, high-throughput analyses and imaging of bacteria-host cell interactions in live animals.
The larval zebrafish (Danio rerio) has emerged as a powerful tool to study bacterial gastrointestinal infections because the gastrointestinal tract of larval zebrafish is physiologically and functionally similar to the human intestine (10–12). Other benefits that make zebrafish an effective high-throughput model organism include high fecundity, genetic tractability, and optical transparency throughout early development (10). Recent studies have used larval zebrafish to identify novel anti-inflammatory therapeutics for IBD and have shown that zebrafish harbor several known IBD susceptibility genes (13–15). A recently developed adult zebrafish model demonstrated the ability of a probiotic E. coli strain to decrease AIEC colonization (16).
Here, we set out to establish a model that combines a drug-inducible DSS enterocolitis model (17) and food-borne colonization with AIEC, to investigate the interplay between host inflammation and AIEC colonization. We use the protozoan Paramecium caudatum, a natural prey of larval zebrafish, as a vehicle to deliver AIEC to the larval intestine, as we have previously described for other enteric pathogens (18, 19). We benchmark this model using mutants of two AIEC virulence factors, FimH and IbeA, with known virulence deficiencies in rodent models (20, 21). We show that deletion of a type 1 pilus gene (fimH) and the gene encoding the invasion of the brain endothelium protein A (ibeA) results in decreased AIEC burden, neutrophil recruitment, and epithelial damage. We also show that IbeA contributes to AIEC invasion in vivo. Finally, we demonstrate that colonization with AIEC hampers recovery of the intestinal epithelium from damages sustained through underlying inflammation.
RESULTS
Adherent-invasive E. coli LF82 colonizes the larval zebrafish intestine better than non-pathogenic E. coli MG1655
We have previously established the protozoan P. caudatum, a natural prey of larval zebrafish, as a vehicle for zebrafish infection with enteric pathogens and non-pathogenic E. coli (18, 19, 22, 23). Internalization of bacteria by P. caudatum and subsequent ingestion of bacteria-loaded paramecia by larvae allow for the delivery of a higher bacterial dosage compared to bath immersion, which is commonly used in other zebrafish infection models including the adult zebrafish AIEC model (16, 19, 24). The uptake of bacteria-loaded paramecia by larvae is followed by digestion of the paramecia in the anterior gut and the subsequent release of bacteria into the intestine within 30 minutes of ingestion (19).
Initially, we investigated the degradation and half-life of AIEC strain LF82 following uptake into P. caudatum vacuoles. The uptake of AIEC by paramecia occurred rapidly, with an average burden of 339 colony-forming units (CFUs) per paramecia quantified minutes after the introduction of AIEC (Fig. 1A). This is in accordance with other studies that show paramecia engulf their target within seconds to minutes (19, 25). The half-life τ of AIEC LF82 inside of paramecia was approximately 2.1 hours (Fig. 1A) and was used to determine the bacterial dosage consumed by larvae following a 2-hour incubation with AIEC-loaded paramecia, as done previously (18). The half-life of AIEC in paramecia was similar to that reported for EHEC (19), so bacteria and P. caudatum concentrations were kept as described previously.
Fig 1.
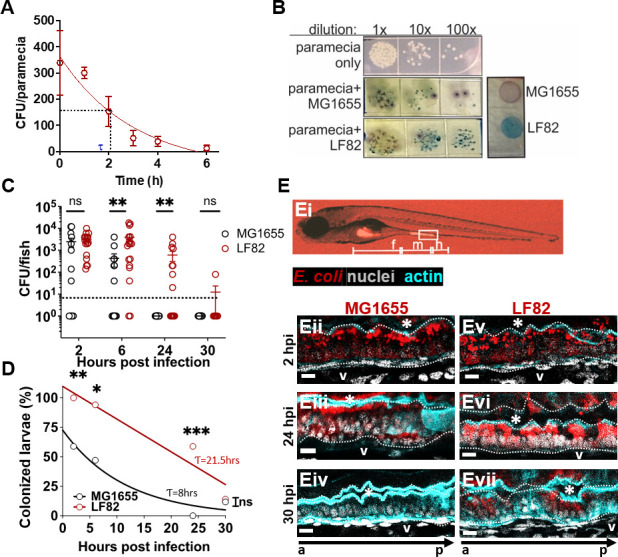
AIEC LF82 colonizes the larval zebrafish intestine better than MG1655. (A) AIEC-loaded paramecia sampled from 0 to 6 hours post incubation, and CFU/paramecia was calculated. AIEC half-life (τ) in paramecia is 2.1 hours. Data are means ± SEM, n = 3. (B) Bacterial colonies from tissue homogenates grown on CHROMagar O157. The zebrafish microbiota (white colonies) can be distinguished from AIEC LF82 (dark blue colonies) and E. coli MG1655 (mauve colonies). (C) Quantification of LF82 and MG1655 CFUs/fish. Fish with CFU below the detection limit (10 CFU/fish, dashed line) were annotated as 1 CFU. Data are from individual fish (n = 14) and means ± SEM. (D) Colonized larvae (%) are the percentage of fish with a burden above the detection limit; n = 14. Non-linear regression, first-order decay, ROUT outlier test with Q = 0.2%, paired t-test and Wilcoxon test. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001, ns, not significant. (E) Images of larvae colonized with E. coli (red), (Ei) whole larva at 10× magnification with intestinal segments (foregut (F), midgut (M), hindgut (H)) marked. (Eii–vii) Sagittal views of the midgut of larvae colonized with MG1655 (Eii–iv) and LF82 (Ev–vii) at 2, 24, and 30 hpi. The dotted white line outlines the intestinal epithelium and separates it from the lumen, indicated by *, and the blood vessel below the basement membrane (V). a to p marks anterior to posterior orientation; Scale bars = 100 um, E. coli (red), phalloidin (cyan, cell outline), nuclei (4′,6-diamidino-2-phenylindole, white), images are representative of n = 3.
Next, we quantified the bacterial burden of AIEC LF82 in zebrafish over 30 hpi, and used the non-pathogenic E. coli strain MG1655 as a control. Tissues from infected fish were homogenized and plated on CHROMagar O157, which allowed us to distinguish AIEC LF82 (steel-blue colonies) from E. coli strain MG1655 (mauve), and the larva’s endogenous microbiota (white, Fig. 1B). Following food-borne delivery, AIEC and MG1655 were taken up by the larvae at similar concentrations (Fig. 1C, 2 hpi). At later time points (6–24 hpi), AIEC formed a significantly higher burden within fish than non-pathogenic E. coli MG1655 (Fig. 1C). The number of fish with a bacterial burden below the detection limit increased after 6 hpi, and by 24 hpi, no MG1655 was detected in any of the fish (Fig. 1C). To get a better representation of the difference in bacterial clearance between LF82 and MG1655, we quantified the percentage of fish that contained a burden of AIEC or MG1655 above the detection limit (≥10 CFU/fish). Although both strains were cleared from the intestine over time, AIEC LF82 (half-life 21.5 hours) was cleared slower than non-pathogenic E. coli MG1655 (half-life of 8 hours), (Fig. 1D). Neither colonization with MG1655 nor LF82 caused any mortality throughout the experimental time course (Fig. S1).
We visualized the site of bacterial colonization within the zebrafish larvae using fluorescent AIEC LF82::mCherry and MG1655::mCherry strains. At 2 hpi, both strains were visible in the foregut lumen and attached to the midgut epithelium (Fig. 1Ei). The localization of E. coli relative to the intestinal epithelium was assessed using a nuclear stain and phalloidin to outline the epithelium (Fig. 1Eii through Evii). High-resolution fluorescence microscopy of the midgut revealed that individual AIEC and MG1655 cells localized both along the epithelial surface and inside the epithelium (Fig. 1Eii and Ev). By 24 hpi, luminal bacteria were no longer observed, and the burden of MG1655 had decreased (Fig. 1Eiii), while the LF82 burden had increased, with more invasion visible (Fig. 1Evi). At 30 hpi, MG1655 was no longer visible (Fig. 1Eiv), while AIEC LF82 was still observed within the epithelium (Fig. 1Evii). Taken together, these experiments showed that AIEC forms a higher burden, and its clearance from the larval gut is slower than for non-pathogenic E. coli, most likely due to invasion of the intestinal epithelium.
Larval immersion in 0.5% DSS recapitulates key morphological and pro-inflammatory features of previously described DSS enterocolitis models
Although AIEC is found in gastrointestinal biopsies from healthy hosts, it is more prevalent in hosts experiencing pre-existing inflammation, such as patients suffering from IBD (26–28).
To address whether pre-existing inflammation affects bacterial colonization and clearance, we expanded the larval model to include drug-induced intestinal inflammation. DSS is a chemical agent that induces enterocolitis in larval zebrafish, with pathological features similar to those of chronic colitis in rodents (17, 29–31).
To replicate previously described DSS models, we tested different DSS concentrations and assessed larval survival, development, and inflammation (Fig. 2 and 3; Fig. S2). The goal was to find a DSS-dosing regimen that would induce a robust pro-inflammatory response without causing excessive mortality. Based on the experimental parameters previously described by Oehlers et al. (17, 29), we immersed larval zebrafish in E3 media containing 0.25%–0.75% DSS from 3 to 6 dpf, replacing the solution daily (Fig. 2A). Over the course of 4–10 dpf (7 days post DSS treatment), the percent survival of larvae administered 0.5% DSS decreased to 48% in comparison to untreated controls (Fig. 2B). The survival of DSS-treated and untreated larvae was similar at 4 and 5 dpf (1 and 2 days post treatment); however, changes in the survival rate were observed at 6 dpf (3 days post treatment) (Fig. 2B). We observed that larval survival stabilized 3 days after the DSS was removed, and no additional mortality was observed from 7 to 10 dpf. In comparison, larvae administered 0.25% DSS had a 100% survival rate, and those administered 0.75% DSS did not survive past 6 dpf (3 days post DSS exposure) (Fig. S2A). Consequently, we further assessed the development and inflammatory responses of larval fish treated with 0.5% DSS.
Fig 2.
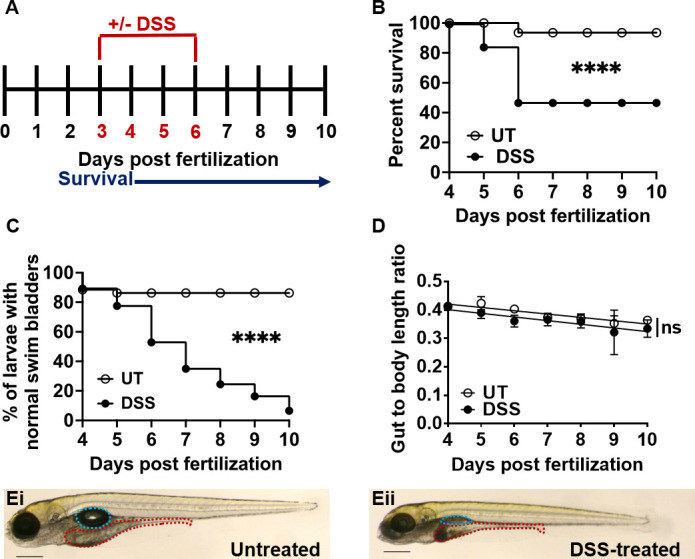
Larval zebrafish treated with 0.5% DSS have decreased survival and intestinal growth rates. (A) Schematic outlining timeline of DSS administration (red, 3–6 dpf) and survival experiments (blue, 1–7 days post exposure). (B) Survival of larvae administered 0.5% DSS (black circles) relative to untreated (UT) controls (empty circles). Data were analyzed using a Kaplan-Meier plot and Mantel-Cox test; ****, P ≤ 0.0001, n = 20. (C) Quantification of swim bladder defects in UT or DSS-treated larvae. Group differences were analyzed using Mantel-Cox test; ****, P ≤ 0.0001, n = 20. (D) Gut-to-body length ratio was analyzed by linear regression; ns = not statistically significant. Data are means ± SEM from n = 20. (E) Representative images of untreated (Ei) and DSS-treated (Eii) larvae at 6 dpf (3 days post DSS exposure), with the swim bladder (teal) and the intestine (red) outlined. Scale bar = 0.3 mm.
Fig 3.
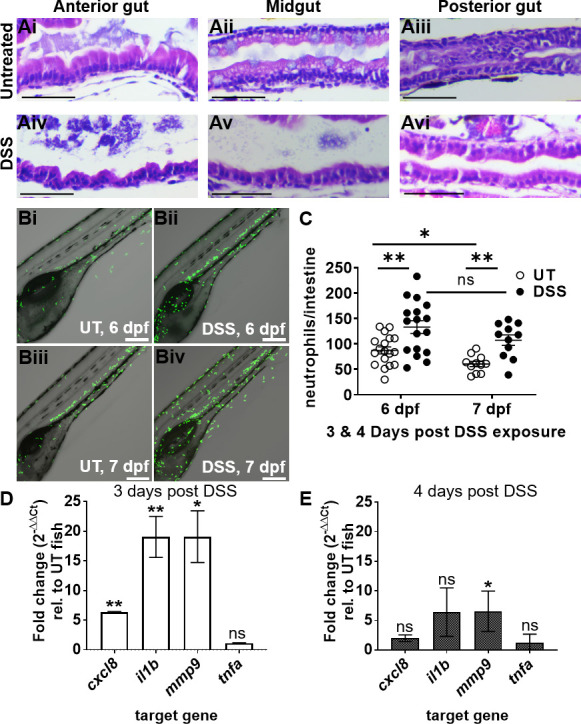
DSS causes intestinal epithelial damage and inflammation consistent with enterocolitis. (A) Representative hematoxylin- and eosin-stained longitudinal sections (n = 4) of the anterior, mid, and posterior intestine from untreated (Ai–iii) and DSS-treated (Aiv–vi) larvae at 6 dpf; scale bars = 50 µm. (B) Representative confocal images of live UT (i, iii) and DSS-treated (ii, iv) Tg(mpo::egfp) larvae at 6 (i-ii) and 7 (iii-iv) dpf; neutrophils (green); larvae were imaged for 18 hours (3–20 hpi). Scale bars = 200 µm. (C) Quantification of neutrophils in the intestine at 6 and 7 dpf (3 and 4 days post DSS treatment); unpaired two-tailed t-test, n ≥ 11. (D) qRT-PCR analyses of cxcl8, il1b, mmp9, and tnfa in DSS-treated larvae relative to untreated controls at 6 dpf and (E) 7 dpf; n = 3. Unpaired two-tailed t-test. Mean ± SEM, *, P ≤ 0.05; **, P ≤ 0.01; ns, not significant.
Prolonged treatment with 0.5% DSS led to abnormal swim bladder development over time (Fig. 2C and E) and slightly stunted the elongation of the larval gut and overall body length (Fig. 2D and E; Fig. S2B and C). Analysis of the gut to whole body ratio of untreated and DSS-treated larvae suggested that DSS did not disproportionally affect gut development and that shorter gut length was a consequence of overall shorter body length since there was no significant difference in slope between untreated and DSS larvae (Fig. 2D). Hematoxylin and eosin (H&E) staining and histology of paraffin embedded, sectioned larvae revealed normal morphology of the anterior, mid, and posterior gut of untreated larvae (Fig. 3A). The intestinal epithelium was intact, with intestinal folds visible in the anterior gut and mucus-producing goblet cells in the midgut epithelium (Fig. 3Ai through iii). In contrast, the epithelium was disrupted in DSS-treated larvae, with visible fraying, corrosion of intestinal folds, and epithelial detachment from the basement membrane in all three gut segments (Fig. 3Aiv through vi).
Next, we studied phagocyte recruitment during DSS-induced intestinal inflammation using transgenic larvae containing fluorescent neutrophils [Tg(mpo::egfp)] and macrophages [Tg(mpeg1::egfp)], respectively. Neutrophils are used as a readout for intestinal inflammation because they are the first responders to injuries and infections (32–35). Macrophages are also involved in the tissue repair and clearance of spent neutrophils but appear at later time points (36). Live imaging of 6–7 dpf larvae allowed us to quantitate the number of neutrophils infiltrating the intestine. We observed that neutrophil recruitment to the intestine was significantly increased in DSS-treated vs untreated larvae at both 6 and 7 dpf (corresponding to 3 and 4 days of DSS treatment, respectively) (Fig. 3B and C). In contrast, there was no change in the number of macrophages infiltrating the gut in untreated vs DSS-treated fish (Fig. S3). To further evaluate pro-inflammatory signaling, we quantified the expression of the key pro-inflammatory markers interleukin 8 (cxcl8-l1), interleukin-1-β (il1b), matrix metallopeptidase 9 (mmp9), and tumor necrosis factor-alpha (tnfa) at 6 and 7 dpf (corresponding to 3 and 4 days of DSS treatment, respectively). At 6 dpf, the relative expression of cxcl8, il1b, and mmp9 was significantly increased in DSS-treated larvae compared to untreated controls, whereas tnfa expression remained constant (Fig. 3D). By 7 dpf, the relative expression of cxcl8, il1b, and tnfa was similar in DSS-treated and untreated fish, whereas mmp9 expression remained elevated (Fig. 3E). Taken together, these data recapitulate key morphological and pro-inflammatory features of previously described DSS enterocolitis models and support our methodology of immersing larvae in 0.5% DSS from 3 to 6 dpf to induce inflammation prior to introducing bacteria.
Pre-existing intestinal inflammation enhances AIEC LF82 colonization and invasion of the gut epithelium
Next, we asked whether DSS-induced intestinal inflammation would affect the outcome of subsequent colonization by AIEC (or the non-pathogenic MG1655 strain as a control). Following the 3-day DSS exposure, we introduced AIEC LF82 to larval zebrafish via food-borne infection (Fig. 4A). Larvae that had become moribund or had a deflated swim bladder following the initial DSS treatment were excluded from subsequent infection experiments. At 2 hpi, the AIEC burden in DSS inflammation fish was similar to the AIEC burden in untreated fish (Fig. 4B). However, the burden of AIEC in DSS-treated larvae was higher than that of the untreated controls at 6 and 12–48 hpi (Fig. 4B). Furthermore, clearance of LF82 from DSS-treated larvae was significantly slower compared to untreated fish (Fig. 4C). Together, these data suggest that pre-existing inflammation enhances the burden and slows the clearance of LF82 in the intestine of larval zebrafish. These results are also in accordance with those of published murine studies that show that AIEC persists longer in mice with IBD compared to healthy controls (37–39).
Fig 4.
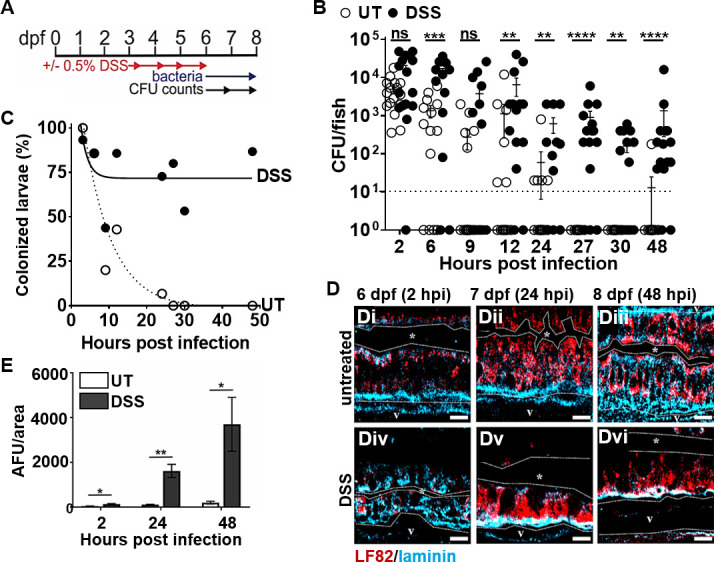
Pre-existing intestinal inflammation enhances the colonization and invasion of AIEC LF82. (A) Timeline of DSS administration, infection of larvae, and sampling for CFU counts. (B) Quantification of LF82 CFUs per larvae with and without DSS treatment, n ≥17; fish with CFU below the detection limit (10 CFU/fish, dashed line) were annotated as 1 CFU. (C) Colonized larvae (%) are percentage of fish with a burden of AIEC above the detection limit; non-linear regression first-order decay, ROUT outlier test with Q = 0.2%. (D) LF82 (red) in the mid-intestine of UT (Di–iii) and DSS-treated (Div–vi) larvae relative to the basement membrane (blue) from 2 to 48 hpi or 6–8 dpf. The dotted white line outlines the intestinal epithelium and separates it from the lumen, indicated by *, and the blood vessel below the basement membrane (V). Scale bars = 10 µm. (E) Quantification of red fluorescence intensity (AFU) (representing AIEC) in the vasculature (V) at 2, 24, and 48 hpi, n = 6; *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001; ns, not statistically significant.
To investigate whether pre-existing inflammation enhances bacterial colonization in general, or specifically for AIEC, the colonization patterns of MG1655 in DSS-treated larvae were also assessed. The burden of LF82 was significantly higher and bacterial clearance slower than that of MG1655 in DSS-treated fish at 2, 6, 24, and 48 hpi (Fig. S4A and B). These results demonstrate that pre-existing intestinal inflammation enhances the burden of both AIEC and non-pathogenic E. coli and that AIEC LF82 still colonized better and was cleared slower compared to non-pathogenic E. coli in fish with enterocolitis.
Intestinal inflammation damages the mucosal barrier and enhances intestinal permeability, allowing for increased bacterial invasion (17, 40, 41). Therefore, we asked whether pre-existing intestinal inflammation would affect AIEC invasion in our model. DSS-treated and untreated larvae were infected with LF82, euthanized at 2, 24, and 48 hpi, and laminin and 4′,6-diamidino-2-phenylindole (DAPI) stained to assess the localization of LF82::mCherry relative to the intestinal lumen, epithelium, and underlying vasculature (Fig. 4D). At 2 hpi, LF82 cells were present within the epithelium of untreated and DSS-treated zebrafish and had begun to invade the underlying vasculature in DSS-treated but not in control fish (Fig. 4Di, Div, and E). At 24 hpi, individual bacterial cells remained visible in untreated larvae, whereas large bacterial aggregates were observed within the epithelium of DSS-treated fish (Fig. 4), and increased bacterial invasion of the underlying vasculature was observed in DSS fish but not untreated controls (Fig. 4E). By 48 hpi, the AIEC burden within the epithelium had lowered (Fig. 4Diii and Dvi), but invasion of the vasculature in DSS-treated fish was further elevated (Fig. 4E). Together, these data suggest that pre-existing enterocolitis facilitates bacterial colonization, slows bacterial clearance, and exacerbates invasion of the bloodstream by AIEC.
AIEC LF82 exacerbates intestinal inflammation in DSS-treated larvae
Murine studies show that colonization with AIEC LF82 exacerbates intestinal inflammation in DSS-treated animals and causes an immunopathology similar to that observed in IBD patients (39, 42, 43). Thus, we investigated whether AIEC could exacerbate inflammation in larvae with pre-existing DSS enterocolitis. Untreated and DSS-treated larvae fed the paramecia vehicle only (uninfected) were used as controls and compared to AIEC-infected fish (Fig. 5). The midgut of untreated fish colonized with LF82 contained more mucus-secreting goblet cells at 2, 24, and 48 hpi compared to control fish (Fig. 5A vs B, cells containing clear/light blue mucous droplets) (44). More goblet cells were also observed in the posterior gut of untreated larvae infected with LF82 from 2 to 48 hpi (Fig. S5F).
Fig 5.
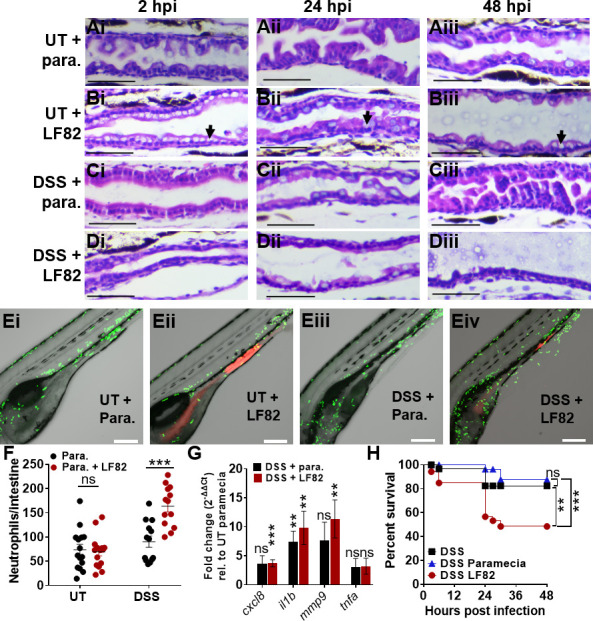
AIEC LF82 exacerbates intestinal inflammation in DSS-treated larvae. (A–D) H&E-stained longitudinal sections of the mid-intestine of larvae without (A and B) and with (C and D) prior DSS treatment, fed paramecia (para.) alone (A, C) or paramecia containing LF82 (B, D) at 2 (i), 24 (ii), and 48 (iii) hpi, n = 3. Black arrows point to goblet cells. Scale bars = 50 µm. (E) Representative confocal images of Tg(mpo::egfp) larvae fed paramecia only (i, iii) or LF82 (ii, iv) at 6 dpf. Larvae were imaged for 18 hours (3–20 hpi), neutrophils (green) and bacteria (red). Scale bars = 200 µm. (F) Quantification of neutrophils per intestine in UT- and DSS-treated fish fed with paramecia only (black) or para. containing LF82 (red); n ≥10. (G) qRT-PCR analyses of cxcl8, il1b, mmp9, and tnfa in DSS-treated (red) larvae infected with LF82- and DSS-treated larvae fed paramecia (black) relative to UT paramecia controls (onefold) at 6 dpf, n = 7. Unpaired two-tailed t-test. Mean ± SEM. (H) Survival of DSS-treated larvae that were uninfected (black), fed paramecia control (blue), or para. containing AIEC (red). N = 17. Kaplan-Meier and Mantel-Cox test, followed by a Bonferroni correction test. **, P ≤ 0.01; ***, P ≤ 0.001; ns, not statistically significant.
Following DSS treatment, we observed corrosion of intestinal folds in the midgut ((Fig. 5Ci) and anterior gut (Fig. S5C) at 6 dpf (corresponding to 3 days post-DSS treatment). In uninfected fish, these folds were partially restored at 7 and 8 dpf (1–2 days after DSS treatment had stopped, Fig. 5Cii through Ciii; Fig. S5C), suggesting that the intestinal epithelium can recover from damage caused by enterocolitis.
In contrast, DSS-treated larvae infected with LF82 were unable to fully recover from enterocolitis by 48 hpi since the anterior and midgut did not recover the original intestinal fold architecture and exhibited a thinner epithelial cell layer compared to DSS-treated larvae that were not infected (Fig. 5D vs C; Fig. S5D vs C). LF82 colonization did not disrupt intestinal folds in the absence of DSS inflammation (Fig. 5B; Fig. S5B). Together, these data suggest that AIEC LF82 alters the architecture of the intestine of larvae; in untreated fish. LF82 increases goblet cell number; and in DSS-exposed fish, it prevents epithelial healing. The increased presence of mucin-producing goblet cells may indicate a host-defense response to fight off bacterial infections, whereas flattening of the intestinal villi may be due to inflammation (45).
To further examine the effect of LF82 on inflammation, neutrophil recruitment was assessed, and induction of inflammatory markers was quantified using qRT-PCR. In untreated fish, AIEC colonization did not affect neutrophil recruitment to the gut (Fig. 5E and F). Similarly, in uninfected fish, neutrophil recruitment to the intestine was unchanged following DSS treatment (Fig. 5E and F). In contrast, DSS treatment and subsequent AIEC colonization had an additive effect and increased neutrophil recruitment (Fig. 5E and F). Macrophage recruitment to the intestine was not significantly affected by either DSS treatment or AIEC infection (Fig. S6). Expression of inflammatory markers cxcl8-l1, il1b, and mmp9 was slightly elevated following DSS treatment alone and significantly increased in DSS-treated fish colonized with AIEC (Fig. 5G). Comparison of inflammatory marker expression following AIEC colonization of untreated or DSS-treated fish further showed that DSS treatment and AIEC infection have an additive effect on pro-inflammatory signaling (Fig. S7).
The observed increase in epithelial damage and pro-inflammatory response following LF82 infection in DSS-treated fish may contribute to the increase in mortality of DSS-treated larvae infected with AIEC LF82, relative to DSS alone or DSS larvae fed paramecia only (Fig. 5H). Together, these data suggest that while AIEC colonization in healthy fish causes little epithelial damage and inflammation, it exacerbates inflammation and tissue damage in hosts with pre-existing enterocolitis.
FimH and IbeA contribute to AIEC virulence in larval zebrafish
Next, we investigated whether the larval zebrafish model is suitable for the characterization and/or identification of virulence factors involved in in vivo infections by characterizing the phenotypes of two known AIEC virulence factors, FimH and IbeA, as a benchmark. FimH is the terminal subunit of type I pili and binds collagen type I and type IV, laminin, fibronectin, and mannosylated glycoproteins (46). FimH of AIEC LF82 adheres to the human CEACAM6 receptor that is abnormally expressed in the ileum of CD patients and expressed in transgenic CEABAC10 mice (8, 21). The presence of CEACAM6 receptors in a host is thought to promote colonization with AIEC and to indirectly contribute to intestinal inflammation since binding of AIEC to CEACAM6 through FimH triggers intestinal inflammation in CEABAC10 mice (47). IbeA is an invasin and outer membrane protein conserved in the E. coli phylogenetic group B2, which includes avian pathogenic E. coli, newborn meningitis-causing E. coli, and AIEC strains NRG857C and LF82 (20). BLAST analyses show that the IbeA protein in these pathogenic E. coli strains is 100% identical (data not shown). IbeA binds to vimentin found in macrophages, fibroblasts, and endothelial cells, and mediates the invasion of Caco-2 and M-like cells by AIEC strain NRG857c (20).
To investigate whether FimH and IbeA play a role in colonization and invasion of AIEC LF82 in zebrafish larvae, these genes were deleted from the parent strain and complemented by inserting fimH or ibeA with their endogenous promoters into the chromosome. Deletion and complementation of either gene did not affect the overall growth of AIEC LF82 (Fig. S8). There were no fortuitous mutations identified in the deletion and complement strains, which were subjected to whole-genome sequencing.
Deletion of fimH but not of ibeA significantly increased larval survival, and the defect was restored in the LF82ΔfimH:fimH complementation strain (Fig. 6A and B). The fimH and ibeA deletion and complementation strains were taken up into the larval gut at similar levels than the wild-type strain (Fig. 6C and D, 0 hpi). Interestingly, deletion of either fimH or ibeA initially increased AIEC colonization but led to a colonization defect at 48 hpi. Complementation of fimH and ibeA restored wild-type colonization levels (Fig. 6C and D). Bacterial clearance was unaffected by fimH deletion (Fig. 6E) but decreased upon deletion of ibeA (Fig. 6F). Next, we asked whether the deletion of fimH or ibeA affected the invasion of the epithelium by AIEC. Infected larvae were euthanized, fixed, and stained with anti-laminin and DAPI to visualize the localization of LF82ΔfimH:mcherry and LF82ΔibeA:mcherry and complementation strains over the course of 48 hpi. Deletion of either fimH or ibeA caused a transient increase in bacterial burden at 2 hpi (Fig. 7A through Ei), followed by significantly decreased colonization at 24–48 hpi (Fig. 7A through Eii and Eiii) consistent with the CFU burden data (Fig. 6C and D). Interestingly, while the fimH mutant was still able to invade the epithelium, the ibeA mutant mainly colonized and formed aggregates at the epithelial surface (Fig. 7D). Complementation of fimH and ibeA restored wild-type adherence and invasion (Fig. 7C and E). These data suggest that FimH and IbeA both contribute to aspects of pathogenesis but play distinct roles in bacterial adherence and invasion.
Fig 6.
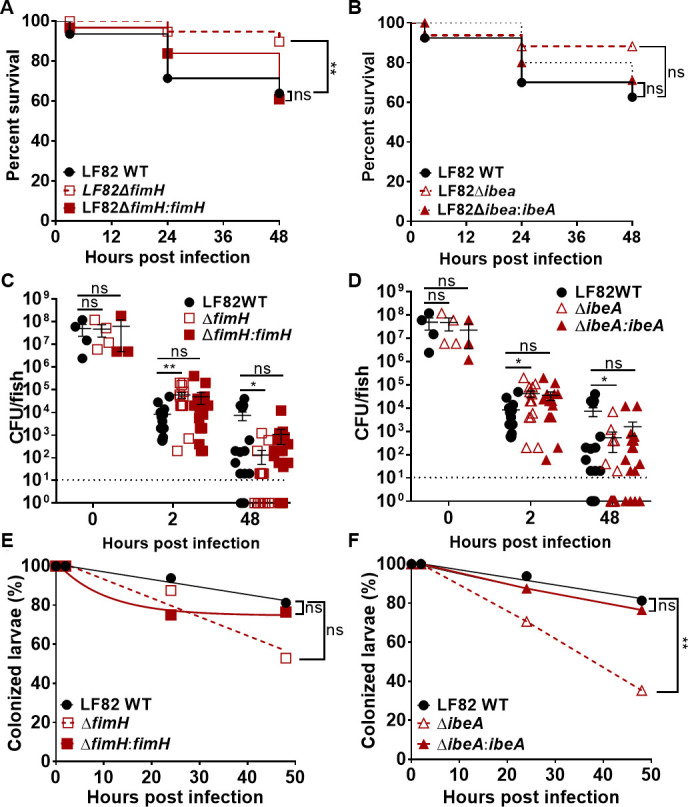
Effects of fimH and ibeA deletion on larval survival, bacterial burden, and bacterial clearance. Survival of larvae infected with (A) LF82 wild-type (WT), LF82ΔfimH, LF82ΔfimH:fimH or (B) LF82, LF82ΔibeA, LF82ΔibeA:ibeA at 2, 24, and 48 hpi. Kaplan-Meier and Mantel-Cox test, followed by a Bonferroni correction test, n = 20. Quantification of bacterial burden and clearance of (C, E) LF82, LF82ΔfimH, LF82ΔfimH:fimH, or (D, F) LF82ΔibeA, and LF82ΔibeA:ibeA in DSS-treated larvae from 2 to 48 hpi. Fish with CFU below the detection limit (10 CFU/fish, dashed line) were annotated as 1 CFU. Significance of difference in burden was analyzed using a Kruskal-Wallis test, n ≥16. Bacterial clearance (percent of fish with a burden of AIEC above the detection limit) was analyzed using a log-rank test. Non-linear regression, first-order decay graph used to model bacterial clearance. *, P ≤ 0.05; **, P ≤ 0.01; ns, not significant; Experiments for the WT and both mutant strains were performed in parallel, and thus, data for the WT strain are duplicated between panels A and B, C and D, and E and F, respectively.
Fig 7.
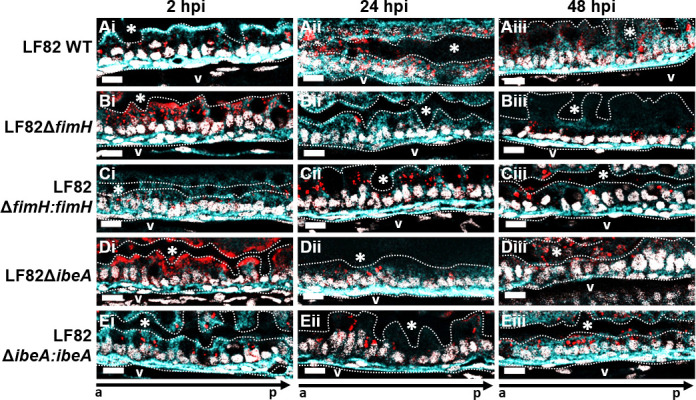
Deletion of ibeA, but not fimH, results in aggregation and retention of AIEC LF82 on the epithelial surface. Representative sections (n = 3) of the mid-intestine of larvae infected with (A) LF82 WT, (B) LF82ΔfimH, (C) LF82ΔfimH:fimH, (D) LF82ΔibeA:ibeA, or (E) LF82ΔibeA:ibeA at (i) 2, (ii) 24, and (iii) 48 hpi. AIEC LF82 (red), laminin (cell surface, cyan), nuclei (DAPI, white). The dotted white line outlines the intestinal epithelium and separates it from the lumen, indicated by *, and the blood vessel below the basement membrane (V). a to p marks anterior to posterior orientation; Scale bars represent 10 µm.
FimH and IbeA elicit a pro-inflammatory response during AIEC colonization and prevent epithelial recovery from enterocolitis
Since both FimH and IbeA are bacterial surface proteins, we next asked if they contribute to the pro-inflammatory response to AIEC colonization in DSS-treated fish. Histology of midgut sections from infected DSS-treated fish showed that colonization with wild-type or complementation strains prevented recovery from DSS intestinal inflammation, and corrosion of intestinal folds persisted even 2 days after DSS treatment had been discontinued (Fig. 8A, 24–48 hpi). In contrast, healthy epithelial morphology was restored following infection with either fimH or ibeA deletion strains (Fig. 8B and D). Lastly, we studied how FimH and IbeA contribute to AIEC immunogenicity, by quantifying neutrophil recruitment to the gut. Fish infected with LF82 WT recruited more neutrophils to the intestine compared to either uninfected, paramecia-fed fish, or fish harboring LF82ΔfimH and LF82ΔibeA (Fig. 8F). Complementation of fimH resulted in increased neutrophil recruitment similar to or in the case of ibeA, more neutrophil recruitment than wild-type infection. Taken together, these data suggest that both FimH and IbeA contribute to pro-inflammatory signaling in response to AIEC infection and also contribute to attenuation of epithelial recovery in DSS enterocolitis fish.
Fig 8.
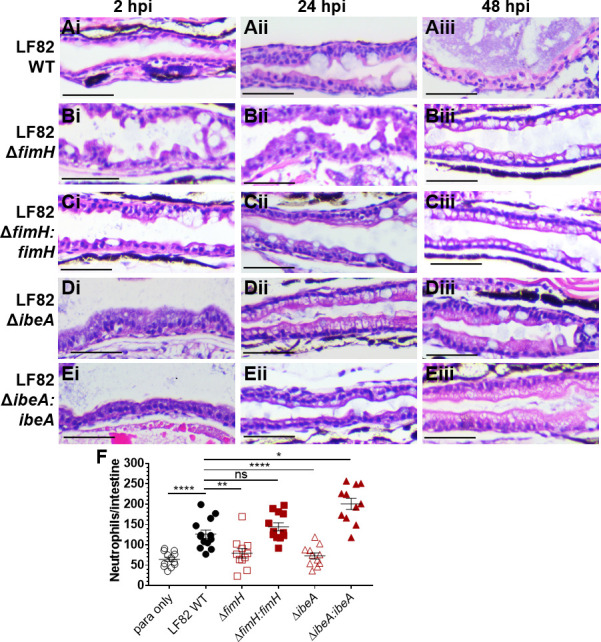
Deletion of fimH and ibeA in AIEC LF82 results in decreased tissue damage and neutrophil recruitment to the intestine compared to LF82. H&E longitudinal sections of the mid-intestine of larvae infected with (A) LF82 WT, (B) LF82ΔfimH, (C) LF82ΔfimH:fimH, (D) LF82ΔibeA, and (E) LF82ΔibeA:ibeA at 2 (i), 24 (ii), and 48 (iii) hpi. Representative images for n = 3. Scale bars = 50 µm. (F) Quantification of neutrophils per intestine for DSS-treated fish infected with abovementioned LF82 strains or paramecia-only control. Kruskal-Wallis test. n ≥11. *, P ≤ 0.05; **, P ≤ 0.001; ****P ≤ 0.0001; ns, not significant.
DISCUSSION
In this study, we establish larval zebrafish as a model to study the interplay between host inflammatory responses and AIEC colonization. During the initial hours post infection, AIEC is observed colonizing the foregut and the midgut; however, over the course of infection, AIEC shows a preference for colonizing the midgut of larvae, similar to EHEC (19). This region of the intestine contains absorptive enterocytes, mucin-secreting goblet cells, and M-like cells, all of which are also found in the mammalian small intestine (48). Accordingly, AIEC predominantly colonizes the small intestine rather than the colon of IBD patients (3, 49–51).
By combining a previously published DSS enterocolitis model (17) with food-borne AIEC infection, we were able to analyze host-microbe interactions in a dynamic fashion, using intravital and high-resolution imaging of live and euthanized larvae. AIEC colonizes the larval gut better than non-pathogenic E. coli, which is in accordance with published murine studies (37). To date, it is unknown whether colonization with AIEC in a susceptible host triggers the onset of intestinal inflammation, or if pre-existing inflammation creates a favorable environment for the AIEC pathotype. Our data suggest that AIEC colonizes and promotes inflammation in healthy hosts but is particularly adapted to colonize hosts with ongoing enterocolitis. While uninfected hosts recover from intestinal inflammation after removal of pro-inflammatory stimuli (here, DSS), tissue repair and healing are impaired in hosts colonized with AIEC.
There are a few differences between rodent and zebrafish models of AIEC. Although both mice and larval zebrafish have an endogenous microbiota, the rodent microbiome renders mice highly colonization resistant, and AIEC models involve antibiotic treatment to remove much of the endogenous microbiome to allow for bacterial colonization. In contrast, larval zebrafish do not need to be treated with antibiotics to remove the endogenous microbiome, and a single dose of 104–105 CFUs of AIEC consumed through food-borne infection is sufficient to promote bacterial colonization. Mouse models are usually challenged with 108–109 CFUs of AIEC through oral gavage daily for 3 or 15 days, making them more labor intensive (8, 38, 52). The existing zebrafish model of AIEC infection only requires bath immersion, but adult zebrafish are needed to achieve robust colonization (16). In larval zebrafish, we found bath immersion ineffective in establishing AIEC colonization. We found that AIEC colonization causes increased mortality in DSS enterocolitis fish, compared to unfed or paramecia-fed DSS-treated fish (Fig. 5H). This is consistent with mouse studies, where AIEC LF82, but not E. coli strain K 12, decreases the survival of CEBAC10 and DSS-treated mice (8).
AIEC LF82 exacerbates intestinal inflammation in hosts with pre-existing inflammation. This is supported by an increase in neutrophil recruitment to the intestine, the inability of the mid-intestine to heal while colonized with AIEC, and the increased expression of pro-inflammatory markers cxcl8-l1, il1b, and mmp9. Cxcl8 is primarily associated with the activation and mobilization of neutrophils, whereas Tnfα and Il-1β are involved in signaling pathways that regulate apoptosis and cell survival (53). Mmp9 degrades the extracellular matrix during inflammation and through this process activates cytokines that mediate tissue and wound healing (54); however, its activation can also contribute to intestinal damage during IBD (55).
To investigate whether fimH and ibeA are important for the colonization of AIEC in the zebrafish intestine, these two genes were deleted from the parent strain. Both fimH and ibeA have been extensively characterized in vivo and in vitro, and thus we used them to benchmark our model against published in vivo and in vitro AIEC models. Deletion of fimH significantly decreased LF82 colonization in two different mouse models that express mammalian CEACAM6 in the intestine (8, 56). Deletion of ibeA did not impact the burden of AIEC strain NRG857c in mice, although it did contribute to invasion and intracellular survival in vitro (20). In the larval zebrafish model, deletion of either fimH or ibeA transiently caused a higher bacterial burden early during infection but decreased bacterial burden at later time points (Fig. 6 and 7). It is possible that LF82 expresses additional virulence factors involved in adhesion, including OmpA, OmpC, long polar fimbriae, and the lipoprotein NlpI (57–61). Alternatively, the transient increase in burden could be due to an altered immune response since both FimH and IbeA are involved in neutrophil recruitment and pro-inflammatory signaling in our model (Fig. 8). It is possible that fimH or ibeA deletion causes a defect in bacterial clearance early during infection, and adhesion and invasion defects during later time points. In addition to their immunogenicity, FimH and IbeA both played a role in sustaining epithelial damage and prevention of healing in DSS-treated fish. It is likely that their role in pro-inflammatory signaling and blocking tissue recovery is linked. Our findings are in-line with other studies showing decreased epithelial corrosion in mice infected with fimH or ibeA mutants (8, 20).
Recently published work established adult zebrafish as a model of AIEC infection and showed that adult zebrafish produce S100A-10b, a calprotectin homolog, in response to intestinal inflammation caused by LF82 (16). This is in-line with our observation that AIEC induce inflammation in zebrafish. The decision to use adult or larval zebrafish to study AIEC depends on the type of readouts required to address a question of interest. In contrast to larvae, adult zebrafish are not transparent, which hinders dynamic imaging of single cells. However, adult fish have a functional adaptive immune system, which allows studies on this aspect of host-microbe interactions.
The reason why AIEC colonizes hosts with pre-existing inflammation more efficiently than healthy fish is not well understood, but there are several potential explanations. First, DSS damages the intestinal barrier and facilitates the adhesion and invasion of AIEC, which results in bacterial localization closer to the epithelial basement membrane (Fig. 4Dvi). As a result, the bacteria are farther away from the lumen and fail to be cleared out by peristaltic contractions (62). Within the basement membrane, fibronectin, collagen types IV, VII, and XVIII, and laminin are abundant, and these host proteins are all known to bind several bacterial adhesins (63). A second reason may be that DSS changes the composition of the intestinal microbiota that may otherwise limit AIEC colonization. Studies show that the administration of the colitis inducing drug 2,4,6-trinitro-benzene sulfonic acid to larval zebrafish changes the proportion of species belonging to the Proteobacteria and Firmicutes phyla (64). Third, intestinal inflammation may cause the overexpression of a receptor important for binding of AIEC. In vitro studies suggest that AIEC can increase the expression of host adhesin receptors. For example, the binding of LF82 through FimH to CEACAM6 induces blebbing of apoptotic cell-derived membranous vesicles, which exposes oligomannosidic glycans that serve as AIEC-binding sites (65). Moreover, the expression of CEACAM6 is increased by TNFα and IFN-γ following AIEC infections (66). CECAM receptors are cell-surface glycoproteins expressed in epithelial, endothelial, and myeloid cells (8, 67). To date, 12 human CEACAM receptors have been identified and fully characterized (67). In contrast, 10 putative CEACAM receptors have been identified in the zebrafish genome, and only 1 CEACAM protein (CEACAMz1) has been characterized. CEACAMz1 is predominantly expressed in gills and, to a lesser extent, in the intestine (68). Mammalian CEACAM6 is also expressed in the alveolar and airway epithelial cells of the lungs under homeostatic conditions and is highly expressed in the gut only during intestinal disease (69). Furthermore, larval zebrafish express a CEACAM6-like protein (encoded by the zgc:198329 gene) in the intestine that is 29% identical to human CEACAM6 (70). Future studies are required to investigate to what extent CEACAM proteins are involved in the binding of AIEC in the zebrafish intestine.
Whether or not AIEC contain specific molecular signatures is not currently known, but it has been proposed that there are undiscovered AIEC-specific genes that are not commonly found in non-pathogenic E. coli strains that are yet to be identified (71). It has been argued that a plausible reason that such molecular markers have not yet been discovered arises from the limitations of currently used infection models and in vitro models to classify E. coli strains as AIEC (72, 73). Currently, classification of strains as AIEC relay on in vitro assays to quantify adhesion, invasion, and replication inside of infected cells since there are no widely conserved genetic features, such as the LEE pathogenicity islands in EHEC/EPEC, or certain toxins, such as in the case of STEC (Shiga toxins) and ETEC (LT/ST enterotoxins). However, it is plausible that there may be genes essential for AIEC in vivo colonization that are not expressed in a simplified in vitro model or are disproportionately important in facilitating colonization only in hosts with pre-existing inflammation. Comparative transcriptomic studies show that the pathogenicity of AIEC changes when AIEC cells are grown in vitro and in the presence of host factors (49, 73, 74). These are hypotheses that may be addressed using transposon mutagenesis and high-throughput assays in larval zebrafish. We also propose that larval zebrafish may facilitate the screening of drugs that target AIEC. Positive results regarding microbial virulence factors, host factors contributing to disease progression, and initial drug candidates in larval zebrafish may then be further evaluated in mammals. We expect this to present a cost-effective way to identify novel genes that link AIEC with the development or progression of IBD.
MATERIALS AND METHODS
Zebrafish maintenance and breeding
The zebrafish lines used in this study were wild-type (AB) and transgenic lines Tg(mpo::egfp) (75) and Tg(mpeg1:egfp) (76), which express EGFP in neutrophils and macrophages, respectively. Adult fish were kept in a recirculating tank system at the University of Texas Health Science Center at Houston Laboratory Animal Medicine and Care on a 14:10-hour light:dark cycle at pH 7.5 and 28°C. Eggs were obtained from natural spawning of adult fish. Fertilized embryos were bleached for 30 s in 0.05% sodium hypochlorite solution (stock 4.00%–4.99%, Sigma-Aldrich) and kept at 30°C on a 14:10-hour light:dark cycle at pH 7.4. Embryos were raised in petri dishes containing E3 buffer (10 mM HEPES, 5 mM NaCl, 0.17 mM KCl, 0.4 mM CaCl2, 0.67 mM MgSO4, pH 7.4). The 1× E3 medium was prepared with 10 mM HEPES to neutralize the acidic (pH 3) solution that arose after dissolving DSS in standard E3 buffer. Larvae maintained past 6 days post fertilization (dpf) were fed GEMMA Micro 75 (Skretting) until euthanized. The larvae were maintained in 150-mm-diameter petri dishes containing 90 mL of E3 medium.
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 1. All strains were grown at 37°C in Luria-Bertani (LB) broth or on LB agar plates, with ampicillin (200 µg/mL), kanamycin (50 µg/mL), chloramphenicol (35 µg/mL), tetracycline (10 µg/mL), or gentamicin (15 µg/mL), when required.
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant characteristic(s) | Source or reference |
|---|---|---|
| Strains | ||
| MG1655 | Non-pathogenic lab E. coli | |
| AIEC LF82 | Adherent-invasive E. coli, parent strain | Torres lab, UTMB |
| LF82∆fimH | LF82 derivative, fimH deletion | This study |
| LF82∆ibeA | LF82 derivative, ibeA deletion | This study |
| LF82∆fimH:fimH | LF82 derivative, fimH complementation | This study |
| LF82∆ibeA:ibeA | LF82 derivative, ibeA complementation | This study |
| E. coli DH5α | Used for cloning experiments | |
| E. coli DH5α λpir | Used for complementation | (77) |
| Plasmids | ||
| pDOC-C | Cloning vector | (78) |
| pDOC-K | Carries the kanamycin cassette | |
| pACBSCE | Recombineering plasmid, encodes the I-SceI and the λ-Red proteins | |
| pME6032:mcherry | Encodes mCherry protein | |
| pSTNSK-Cm | Tn7 transposase expression vector | (77) |
| pGpTn7 | Cloning vector | |
The LF82 deletion strains were generated using recombineering, as previously described (78). Briefly, constructs were generated by amplifying a kanamycin cassette from the plasmid pDOC-K using oligonucleotide pairs that contain at least 45 bp of homology to the DNA immediately upstream and downstream of the target genes (Table 2). The amplified fragment was inserted into the plasmid pDOC-C, and the construct was verified by sequencing (Azenta Life Sciences). The constructed pDOC-C deletion plasmid and the recombineering plasmid pACBSCE were co-transformed into LF82 via electroporation and transformants plated on LB agar containing chloramphenicol, ampicillin, and kanamycin. Selected colonies were grown in LB broth containing 0.5% glucose for 2 hours and then induced with 0.5% arabinose for 4 hours. The cells were then collected by centrifugation and plated on LB agar without NaCl, but containing 5% sucrose and kanamycin. Sucrose-insensitive and kanamycin-resistant recombinant colonies were transferred to LB chloramphenicol plates to confirm loss of the pACBSCE plasmid. Loss of the pDOC-C plasmid was confirmed with pDOC-specific oligonucleotides. Gene deletion was assessed by PCR using primers listed in Table 3.
TABLE 2.
List of primers used to amplify the pDOC-K plasmid with 45 bp homology to the DNA upstream and downstream of ibeA and fimHa
| Gene | Forward primer sequence (5–3) | Reverse primer sequence (5–3) |
|---|---|---|
| ibeA |
CGGAATTCGCGCGGGGGATTGTTTTACTCAATTATTGAAT
ACGGAGATAAAGTATGGAAGACCGGTCAATTGGCTGGAG |
CGGCTAGCGCGCGACATAAAAACTGGGTTTTTCTCTCATAACTTTA
TTCCCTGTTAAAAAATATCCTCCTTAGTTCCTATTCCGAAGTTC |
| fimH |
CGGAATTCTTAGCATCACCTATACCTACAGCTGAACCCGA
AGAGATGATTGTAATGAAAGACCGGTCAATTGGCTGGAG |
CGGCTAGCTCAGGTAATATTGCGTACCTGCATTAGCAATGCCCTG
TGATTTCTTTATTGAATATCCTCCTTAGTTCC |
The restriction site is underlined, and the region homologous to the kanamycin cassette is in italics.
TABLE 3.
List of primers used to verify deletion mutants
| Strain | Forward primer sequence (5–3) | Reverse primer sequence (5–3) |
|---|---|---|
| LF82ΔfimH | CAACCAAAACAGTTCAGG TGG | GCTGATTATTAGCATGGTAGCG |
| LF82ΔibeA | GGCAAAGAGAGATGATCT CCTT | CCCATAACACCGATGCCAATA |
The complementation strains were constructed by insertion of the gene of interest and its endogenous promoter into the respective deletion strains using a Tn7-based vector system (77). Briefly, the genes were cloned in pGp-Tn7-Gm and then introduced into DH5α-λpir by electroporation to construct pGp-Tn7-fimH and pGp-Tn7-ibeA vectors. Positive clones were screened by colony PCR and confirmed by Sanger sequencing. The pGp-Tn7-fimH and pGp-Tn7-ibeA vectors were electroporated into LF82ΔfimH and LF82ΔibeA harboring the Tn7-transposase encoding, temperature-sensitive plasmid pSTNSK-Cm. Transformants were plated on LB agar containing gentamicin and chloramphenicol, and then incubated at 30°C for 20 hours. Selected colonies were further streaked on LB agar plates without antibiotics and incubated at 42°C for 20 hours to promote the loss of plasmid pSTNSK-Cm. The colonies were passaged four to five times on LB agar plates (no antibiotic), incubated at 37°C, and screened for resistance to gentamicin and sensitivity to chloramphenicol.
The deletion of ibeA or fimH and their integration at the attTn7 site was confirmed by PCR (Table 4) and whole-genome sequencing. Genomic DNA was isolated using DNeasy Blood and Tissue kit (QIAGEN, catalog no. 69504) and analyzed by Nanopore sequencing (Plasmidsaurus). Plasmidsaurus also generated a complete genome assembly and annotation. Inspection of those genome assemblies showed that the intended mutations were present in the appropriate strains and that the complementation constructs were correctly integrated at the expected loci. To rule out the possibility that fortuitous mutations were introduced during strain construction, two bioinformatic approaches were used. First, we used Snippy (https://github.com/tseemann/snippy) to compare the nanopore reads to the reference genome (composed of the chromosome https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_021398935.1/ and plasmid https://www.ncbi.nlm.nih.gov/nuccore/NC_011917.1/). Second, we mapped the nanopore reads to the same reference genome using Minimap2 and then used FreeBayes to identify possible single-nucleotide polymorphisms (SNPs). The candidate SNPs identified by either approach were analyzed by inspecting the alignments with IGV (https://software.broadinstitute.org/software/igv/download). This showed that there were no fortuitous mutations that were introduced during strain construction. The E. coli strains were electroporated with the mCherry-expressing pME6032 plasmid to visualize the bacteria inside of the zebrafish intestine.
TABLE 4.
List of primers used to analyze the integration of the Tn7 transposon system at the attTn7 site located downstream of the glmS gene
| Strain | Forward primer sequence (5-3) | Reverse primer sequence (5-3) |
|---|---|---|
| LF82 complementation strain | TGG CTT ACC ACG TTG CGC TG | CAT ACA CCG GCG CAG GGA AG |
Burden of E. coli inside of paramecia and larval zebrafish infections
Paramecia were propagated as described (18) 1 day prior to the infection experiment and every 2 weeks to maintain live cultures. Loading of paramecia with AIEC LF82 and MG1655 was conducted as described previously (18). On the day of the experiment, paramecia were co-cultured with either AIEC LF82 or MG1655, and the amount of E. coli inside of the paramecia was assessed by lysing the paramecia with 1% Triton X-100 followed by dilution plating and colony forming unit counting, as previously described (18).
E. coli-loaded paramecia were counted using an automated cell counter (Life Technologies Countess II), and a final concentration of 2 × 105 paramecia/mL in E3 medium was used to feed the larvae for 2 hours at 30°C in a 6-well sterile plate.
Bacterial colonization and clearance in larvae
The E. coli burden in zebrafish larvae was assessed starting 2 hours post infection (hpi). Briefly, the larvae were anesthetized in the E3 medium with 0.16 mg/mL tricaine and washed six times to remove excess paramecia. Infected zebrafish larvae were euthanized with 1.6 mg/mL of tricaine. The euthanized larvae were then incubated with 100 µL of a 1-mg/mL filter-sterilized pronase solution, vortexed, and placed at 37°C for 6 minutes. The larvae were then homogenized by repeated passage through a 31-gauge needle attached to a 1-mL syringe. In all cases, the samples were serially diluted, and 5 µL of each dilution was plated on CHROMagar O157 plates (DRG International Inc). The plates were incubated at 30°C for 24 hours and then at room temperature for an additional 24 hours to permit full growth of colonies. The number of dark steel-blue (AIEC) and mauve (MG1655) colonies were assessed afterward. Data were analyzed with the GraphPad Prism software, version 9.
DSS administration and survival analysis of DSS-treated larvae
Colitis grade dextran sulfate sodium (36,000–50,000 MW, MP Biomedical) was used to induce enterocolitis as previously described by others (17). At 3 dpf, 120 larvae were anesthetized with 0.16 mg/mL of tricaine and transferred to a 150-mm-diameter petri dish containing 90 mL of freshly prepared 0.5% (wt/vol) DSS dissolved in E3 medium. The DSS treatment was repeated for three consecutive days. Survival was assessed daily by observing the presence or absence of a heartbeat on anesthetized larvae using an Olympus SZX10 stereomicroscope. Dead larvae were removed, and the survivors were transferred to a new petri dish containing DSS in E3 medium every day following assessment.
Measurement of intestinal and body length, and swim bladder assessment
All larvae were imaged on an Olympus SZX10 stereomicroscope at 1.6× magnification. Fish were anesthetized in 0.16 mg/mL tricaine and embedded in 1% low-melting agarose (LMA). ImageJ was used for image analysis to assess whole body and intestinal length. The length of the intestine was measured from the beginning of the bulb to the end of the cloaca, and the total body length was determined from the mouth to the tip of the tail. The presence of a swim bladder was visualized under the stereomicroscope on anesthetized larvae. The data were analyzed using GraphPad Prism.
Histological analysis
Zebrafish larvae were fixed in 4% formaldehyde diluted in PBS and incubated overnight (O/N) at 4°C. Larvae were processed for histological analyses by the UT-Health Core Histopathology Lab. Briefly, larvae were embedded in paraffin, sectioned along the sagittal plane at 2 µm, and stained with H&E. Imaging was performed on an AmScope microscope with an MU1003 camera and the AmScope software version x64, 3.7.11443.20180326.
Neutrophil and macrophage recruitment
Zebrafish larvae were anesthetized, embedded in 1% LMA in a 6-well glass bottom plate, and imaged on an Olympus Fluoview FV3000 confocal microscope for 3–21 hpi. A Z-stack of 190 images of 2 µm slices was analyzed with Fluoview FV31S-SW. The images were then imported into the Imaris software, version 9.7.2, which was used to quantify the number of intestinal GFP-expressing neutrophils or macrophages over the course of 3–21 hpi.
Immunofluorescence
Larvae were euthanized and placed in a 4% formaldehyde solution O/N at 4°C. Then the larvae were washed twice with 1× PBS, permeabilized in acetone for 15 minutes at −20°C, and incubated in PBDT blocking solution (PBS, 1% BSA, 1% DMSO, and 0.5% Triton-X100) O/N. The larvae were then incubated with anti-α-laminin at a 1:25 dilution (Sigma-Aldrich, L9393) O/N at 4°C. The following day, the samples were washed and incubated with goat anti-rabbit IgG Alexa Flour 488 using a 1:250 dilution (Thermo Fisher Scientific, A27034) and 1 µM/mL DAPI O/N at 4°C. The samples were then washed for 30 minutes, three times with a washing solution (1× PBS, 0.1% Tween-20, and 0.1% Triton X-100). Some larvae were stained with phalloidin (300 units/mL) and 1 µM/mL DAPI. Samples were imaged on a confocal microscope (Olympus Fluoview FV3000 confocal microscope at 60× magnification), and images were transferred to cellSENS version 2.3 for deconvolution with five iterations.
Quantification of bacteria inside of epithelium
Bacteria inside of the intestinal epithelium were quantified on deconvoluted images taken after immunofluorescence imaging. ImageJ was used to quantify the fluorescent signal of the mCherry channel (representing bacteria) (79). The data were plotted using Graphpad Prism, and significance was determined using a Mann-Whitney U test.
RNA isolation, reverse transcription, and quantitative PCR
RNA was isolated from 15 zebrafish larvae for each condition. Briefly untreated or DSS-treated larvae, fed or unfed paramecia, were euthanized, homogenized in TRIzol reagent (Thermo Fisher, 15596026) using a disposable pellet pestle (Fisher Scientific, 12-141-364), and RNA was extracted using a standard protocol (80). Isolated RNA was treated with RNase-free DNase (Qiagen) and cleaned and concentrated using a Zymo Research RNA Clean & Concentrator Kit. Removal of DNA contamination was verified by PCR using purified RNA as template.
Reverse transcription was carried out using oligo (dT) primers and the SuperScript IV First-Strand cDNA Synthesis Reaction system. The concentration of the cDNA was measured using a Nanodrop-spectrophotometer, and 45 ng of cDNA was used for each reaction. cDNAs and primers (listed in Table 5) were mixed with Luna Universal qPCR Master mix (New England Biolabs), and amplification was carried out in duplicate in a CFX96 Real-Time System C1000 Touch Thermal Cycler (Bio-Rad, Hercules, CA, USA). The elfα and rpl13 genes were used as internal controls, and the relative fold-change for each gene of interest was expressed in 2−∆∆CT, where ∆∆CT = [(CT gene of interest − CT internal control) one condition − (CT gene of interest − CT internal control)] another condition (81). For DSS experiments, the DSS data were normalized to the untreated group, whereas in the infection experiments, the data were normalized to control-fed paramecia without added bacteria.
TABLE 5.
List of primers used to analyze the transcription of pro-inflammatory genes and housekeeping genes
| Gene | Forward primer sequence (5–3) | Reverse primer sequence (5–3) |
|---|---|---|
| rpl13 | TCTGGAGGACTGTAAGAGGTATGC | AGACGCACAATCTTGAGAGCAG |
| il1b | ATCAAACCCCAATCCACAGAGT | GGCACTGAAGACACCACGTT |
| cxcl8-l1 | TGTTTTCCTGGCATTTCTGACC | TTTACAGTGTGGGCTTGGAGGG |
| mmp9 | CATTAAAGATGCCCTGATGTATCCC | AGTGGTGGTCCGTGGTTGAG |
| tnfa | GTTTATCAGACAACCGTGGCCA | GATGTTCTCTGTTGGGTTTCTGAC |
ACKNOWLEDGMENTS
We thank Alfredo Torres (UTMB) for sharing with us the AIEC LF82 strain and Peter Rady (UTHealth Houston) for microscope use to image the histology slides. We also thank Melissa Stephens and Michelle Nguyen from the UTHealth Histology Core for staining and sectioning the paraffin-embedded larvae.
This work was supported by National Institutes of Health grant R01AI132354 to A.-M.K., and bioinformatic analysis of whole-genome sequencing was supported by National Institutes of Health grant R35GM141710 to A.V.H.
Contributor Information
Anne-Marie Krachler, Email: anne.marie.krachler@uth.tmc.edu.
Sarah E. F. D'Orazio, University of Kentucky College of Medicine, Lexington, Kentucky, USA
ETHICS APPROVAL
Zebrafish care, breeding, and experiments described here are in accordance with the Guide for the Care and Use of Laboratory Animals and have been approved by the Institutional Animal Welfare Committee of the University of Texas Health Science Center, Houston, and protocol number AWC-22-0088.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/msphere.00512-23.
Figures S1-S8.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Kaplan GG. 2015. The global burden of IBD: from 2015 to 2025. Nat Rev Gastroenterol Hepatol 12:720–727. doi: 10.1038/nrgastro.2015.150 [DOI] [PubMed] [Google Scholar]
- 2. Loftus EV Jr. 2004. Clinical epidemiology of inflammatory bowel disease: incidence, prevalence, and environmental influences. Gastroenterology 126:1504–1517. doi: 10.1053/j.gastro.2004.01.063 [DOI] [PubMed] [Google Scholar]
- 3. Darfeuille-Michaud Arlette, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F. 2004. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in crohn's disease. Gastroenterology 127:412–421. doi: 10.1053/j.gastro.2004.04.061 [DOI] [PubMed] [Google Scholar]
- 4. Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. 1998. Presence of adherent Escherichia coli strains in ileal mucosa of patients with crohn's disease. Gastroenterology 115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8 [DOI] [PubMed] [Google Scholar]
- 5. Palmela C, Chevarin C, Xu Z, Torres J, Sevrin G, Hirten R, Barnich N, Ng SC, Colombel J-F. 2018. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut 67:574–587. doi: 10.1136/gutjnl-2017-314903 [DOI] [PubMed] [Google Scholar]
- 6. Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. 2001. Adherent invasive Escherichia coli strains from patients with crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun 69:5529–5537. doi: 10.1128/IAI.69.9.5529-5537.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buisson A, Sokol H, Hammoudi N, Nancey S, Treton X, Nachury M, Fumery M, Hébuterne X, Rodrigues M, Hugot J-P, Boschetti G, Stefanescu C, Wils P, Seksik P, Le Bourhis L, Bezault M, Sauvanet P, Pereira B, Allez M, Barnich N, Remind study group . 2023. Role of adherent and invasive Escherichia coli in crohn's disease: lessons from the postoperative recurrence model. Gut 72:39–48. doi: 10.1136/gutjnl-2021-325971 [DOI] [PubMed] [Google Scholar]
- 8. Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CHF, Stanners CP, Darfeuille-Michaud A. 2009. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 206:2179–2189. doi: 10.1084/jem.20090741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Li J, Dejanovic D, Zangara MT, Chandra J, McDonald C, Rieder F. 2021. Mouse models of intestinal fibrosis. Methods Mol Biol 2299:385–403. doi: 10.1007/978-1-0716-1382-5_26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Flores EM, Nguyen AT, Odem MA, Eisenhoffer GT, Krachler AM. 2020. The zebrafish as a model for gastrointestinal tract-microbe interactions. Cell Microbiol 22:e13152. doi: 10.1111/cmi.13152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ng ANY, de Jong-Curtain TA, Mawdsley DJ, White SJ, Shin J, Appel B, Dong PDS, Stainier DYR, Heath JK. 2005. Formation of the digestive system in zebrafish: iii. Intestinal epithelium morphogenesis. Dev Biol 286:114–135. doi: 10.1016/j.ydbio.2005.07.013 [DOI] [PubMed] [Google Scholar]
- 12. Pack M, Solnica-Krezel L, Malicki J, Neuhauss SC, Schier AF, Stemple DL, Driever W, Fishman MC. 1996. Mutations affecting development of zebrafish digestive organs. Development 123:321–328. doi: 10.1242/dev.123.1.321 [DOI] [PubMed] [Google Scholar]
- 13. Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496:498–503. doi: 10.1038/nature12111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oehlers SH, Flores MV, Hall CJ, Swift S, Crosier KE, Crosier PS. 2011. The inflammatory bowel disease (IBD) susceptibility genes NOD1 and NOD2 have conserved anti-bacterial roles in zebrafish. Dis Model Mech 4:832–841. doi: 10.1242/dmm.006122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hanyang L, Xuanzhe L, Xuyang C, Yujia Q, Jiarong F, Jun S, Zhihua R. 2017. Application of zebrafish models in inflammatory bowel disease. Front Immunol 8:501. doi: 10.3389/fimmu.2017.00501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nag D, Farr D, Raychaudhuri S, Withey JH. 2022. An adult zebrafish model for adherent-invasive Escherichia coli indicates protection from AIEC infection by probiotic E. coli Nissle. iScience 25:104572. doi: 10.1016/j.isci.2022.104572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Oehlers SH, Flores MV, Hall CJ, Okuda KS, Sison JO, Crosier KE, Crosier PS. 2013. Chemically induced intestinal damage models in zebrafish larvae. Zebrafish 10:184–193. doi: 10.1089/zeb.2012.0824 [DOI] [PubMed] [Google Scholar]
- 18. Flores E, Thompson L, Sirisaengtaksin N, Nguyen AT, Ballard A, Krachler A-M. 2019. Using the protozoan paramecium caudatum as a vehicle for food-borne infections in zebrafish larvae. J Vis Exp. doi: 10.3791/58949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stones DH, Fehr AGJ, Thompson L, Rocha J, Perez-Soto N, Madhavan VTP, Voelz K, Krachler AM. 2017. Zebrafish (Danio rerio) as a vertebrate model host to study colonization, pathogenesis, and transmission of foodborne Escherichia coli O157. mSphere 2:e00365-17. doi: 10.1128/mSphereDirect.00365-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cieza RJ, Hu J, Ross BN, Sbrana E, Torres AG. 2015. The IbeA invasin of adherent-invasive Escherichia coli mediates interaction with intestinal epithelia and macrophages. Infect Immun 83:1904–1918. doi: 10.1128/IAI.03003-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dreux N, Denizot J, Martinez-Medina M, Mellmann A, Billig M, Kisiela D, Chattopadhyay S, Sokurenko E, Neut C, Gower-Rousseau C, Colombel J-F, Bonnet R, Darfeuille-Michaud A, Barnich N. 2013. Point mutations in FimH adhesin of crohn's disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 9:e1003141. doi: 10.1371/journal.ppat.1003141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Manneh-Roussel J, Haycocks JRJ, Magán A, Perez-Soto N, Voelz K, Camilli A, Krachler A-M, Grainger DC. 2018. cAMP receptor protein controls vibrio cholerae gene expression in response to host colonization. mBio 9:e00966-18. doi: 10.1128/mBio.00966-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fan Y, Thompson L, Lyu Z, Cameron TA, De Lay NR, Krachler AM, Ling J. 2019. Optimal translational fidelity is critical for salmonella virulence and host interactions. Nucleic Acids Res 47:5356–5367. doi: 10.1093/nar/gkz229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saraceni PR, Romero A, Figueras A, Novoa B. 2016. Establishment of infection models in zebrafish larvae (Danio rerio) to study the pathogenesis of aeromonas hydrophila. Front Microbiol 7:1219. doi: 10.3389/fmicb.2016.01219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Patterson BW, Abraham AO, MacIver MA, McLean DL. 2013. Visually guided gradation of prey capture movements in larval zebrafish. J Exp Biol 216:3071–3083. doi: 10.1242/jeb.087742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Negroni A, Costanzo M, Vitali R, Superti F, Bertuccini L, Tinari A, Minelli F, Di Nardo G, Nuti F, Pierdomenico M, Cucchiara S, Stronati L. 2012. Characterization of adherent-invasive Escherichia coli isolated from pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis 18:913–924. doi: 10.1002/ibd.21899 [DOI] [PubMed] [Google Scholar]
- 27. Meconi S, Vercellone A, Levillain F, Payré B, Al Saati T, Capilla F, Desreumaux P, Darfeuille-Michaud A, Altare F. 2007. Adherent-invasive Escherichia coli isolated from crohn's disease patients induce granulomas in vitro. Cell Microbiol 9:1252–1261. doi: 10.1111/j.1462-5822.2006.00868.x [DOI] [PubMed] [Google Scholar]
- 28. Mazzarella G, Perna A, Marano A, Lucariello A, Rotondi Aufiero V, Sorrentino A, Melina R, Guerra G, Taccone FS, Iaquinto G, De Luca A. 2017. Pathogenic role of associated adherent-invasive Escherichia coli in Crohn's disease. J Cell Physiol 232:2860–2868. doi: 10.1002/jcp.25717 [DOI] [PubMed] [Google Scholar]
- 29. Oehlers SH, Flores MV, Hall CJ, Crosier KE, Crosier PS. 2012. Retinoic acid suppresses intestinal mucus production and exacerbates experimental enterocolitis. Dis Model Mech 5:457–467. doi: 10.1242/dmm.009365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Paola D, Natale S, Iaria C, Cordaro M, Crupi R, Siracusa R, D’Amico R, Fusco R, Impellizzeri D, Cuzzocrea S, Spanò N, Gugliandolo E, Peritore AF. 2022. Intestinal disorder in zebrafish larvae (Danio rerio): the protective action of N-palmitoylethanolamide-oxazoline. Life (Basel) 12:125. doi: 10.3390/life12010125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chuang L-S, Morrison J, Hsu N-Y, Labrias PR, Nayar S, Chen E, Villaverde N, Facey JA, Boschetti G, Giri M, Castillo-Martin M, Thin TH, Sharma Y, Chu J, Cho JH. 2019. Zebrafish modeling of intestinal injury, bacterial exposures and medications defines epithelial in vivo responses relevant to human inflammatory bowel disease. Dis Model Mech 12:dmm037432. doi: 10.1242/dmm.037432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. d’Alençon CA, Peña OA, Wittmann C, Gallardo VE, Jones RA, Loosli F, Liebel U, Grabher C, Allende ML. 2010. A high-throughput chemically induced inflammation assay in zebrafish. BMC Biol 8:151. doi: 10.1186/1741-7007-8-151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Oehlers SH, Flores MV, Okuda KS, Hall CJ, Crosier KE, Crosier PS. 2011. A chemical enterocolitis model in zebrafish larvae that is dependent on microbiota and responsive to pharmacological agents. Dev Dyn 240:288–298. doi: 10.1002/dvdy.22519 [DOI] [PubMed] [Google Scholar]
- 34. Hall C, Flores MV, Crosier K, Crosier P. 2009. Live cell imaging of zebrafish leukocytes. Methods Mol Biol 546:255–271. doi: 10.1007/978-1-60327-977-2_16 [DOI] [PubMed] [Google Scholar]
- 35. Kim ND, Luster AD. 2015. The role of tissue resident cells in neutrophil recruitment. Trends Immunol 36:547–555. doi: 10.1016/j.it.2015.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujiwara N, Kobayashi K. 2005. Macrophages in inflammation. Curr Drug Targets Inflamm Allergy 4:281–286. doi: 10.2174/1568010054022024 [DOI] [PubMed] [Google Scholar]
- 37. Bretin A, Lucas C, Larabi A, Dalmasso G, Billard E, Barnich N, Bonnet R, Nguyen HTT. 2018. AIEC infection triggers modification of gut microbiota composition in genetically predisposed mice, contributing to intestinal inflammation. Sci Rep 8:12301. doi: 10.1038/s41598-018-30055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Carvalho FA, Barnich N, Sauvanet P, Darcha C, Gelot A, Darfeuille-Michaud A. 2008. Crohn's disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis 14:1051–1060. doi: 10.1002/ibd.20423 [DOI] [PubMed] [Google Scholar]
- 39. Imai J, Kitamoto S, Sugihara K, Nagao-Kitamoto H, Hayashi A, Morhardt TL, Kuffa P, Higgins PDR, Barnich N, Kamada N. 2019. Flagellin-mediated activation of IL-33-ST2 signaling by a pathobiont promotes intestinal fibrosis. Mucosal Immunol 12:632–643. doi: 10.1038/s41385-019-0138-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laroui H, Ingersoll SA, Liu HC, Baker MT, Ayyadurai S, Charania MA, Laroui F, Yan Y, Sitaraman SV, Merlin D. 2012. Dextran sodium sulfate (DSS) induces colitis in mice by forming nano-lipocomplexes with medium-chain-length fatty acids in the colon. PLoS One 7:e32084. doi: 10.1371/journal.pone.0032084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Randhawa PK, Singh K, Singh N, Jaggi AS. 2014. A review on chemical-induced inflammatory bowel disease models in rodents. Korean J Physiol Pharmacol 18:279–288. doi: 10.4196/kjpp.2014.18.4.279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hayashi Y, Nakase H. 2022. The molecular mechanisms of intestinal inflammation and fibrosis in crohn’s disease. Front Physiol 13:845078. doi: 10.3389/fphys.2022.845078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Viladomiu M, Metz ML, Lima SF, Jin W-B, Chou L, JRI Live Cell Bank, Guo C-J, Diehl GE, Simpson KW, Scherl EJ, Longman RS. 2021. Adherent-invasive E. coli metabolism of propanediol in crohn's disease regulates phagocytes to drive intestinal inflammation. Cell Host Microbe 29:607–619. doi: 10.1016/j.chom.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Erben U, Loddenkemper C, Doerfel K, Spieckermann S, Haller D, Heimesaat MM, Zeitz M, Siegmund B, Kühl AA. 2014. A guide to histomorphological evaluation of intestinal inflammation in mouse models. Int J Clin Exp Pathol 7:4557–4576. [PMC free article] [PubMed] [Google Scholar]
- 45. Antoni L, Nuding S, Wehkamp J, Stange EF. 2014. Intestinal barrier in inflammatory bowel disease. World J Gastroenterol 20:1165–1179. doi: 10.3748/wjg.v20.i5.1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sokurenko EV, Courtney HS, Maslow J, Siitonen A, Hasty DL. 1995. Quantitative differences in adhesiveness of type 1 fimbriated Escherichia coli due to structural differences in fimH genes. J Bacteriol 177:3680–3686. doi: 10.1128/jb.177.13.3680-3686.1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Martinez-Medina M, Denizot J, Dreux N, Robin F, Billard E, Bonnet R, Darfeuille-Michaud A, Barnich N. 2014. Western diet induces dysbiosis with increased E coli in CEABAC10 mice, alters host barrier function favouring AIEC colonisation. Gut 63:116–124. doi: 10.1136/gutjnl-2012-304119 [DOI] [PubMed] [Google Scholar]
- 48. Wallace KN, Akhter S, Smith EM, Lorent K, Pack M. 2005. Intestinal growth and differentiation in zebrafish. Mech Dev 122:157–173. doi: 10.1016/j.mod.2004.10.009 [DOI] [PubMed] [Google Scholar]
- 49. Zhang Y, Rowehl L, Krumsiek JM, Orner EP, Shaikh N, Tarr PI, Sodergren E, Weinstock GM, Boedeker EC, Xiong X, Parkinson J, Frank DN, Li E, Gathungu G. 2015. Identification of candidate adherent-invasive E. coli signature transcripts by genomic/transcriptomic analysis. PLoS One 10:e0130902. doi: 10.1371/journal.pone.0130902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Conte MP, Longhi C, Marazzato M, Conte AL, Aleandri M, Lepanto MS, Zagaglia C, Nicoletti M, Aloi M, Totino V, Palamara AT, Schippa S. 2014. Adherent-invasive Escherichia coli (AIEC) in pediatric crohn's disease patients: phenotypic and genetic pathogenic features. BMC Res Notes 7:748. doi: 10.1186/1756-0500-7-748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. López-Siles M, Camprubí-Font C, Gómez Del Pulgar EM, Sabat Mir M, Busquets D, Sanz Y, Martinez-Medina M. 2022. Prevalence, abundance, and virulence of adherent-invasive Escherichia coli in ulcerative colitis, colorectal cancer, and coeliac disease. Front Immunol 13:748839. doi: 10.3389/fimmu.2022.748839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Low D, Tran HT, Lee I-A, Dreux N, Kamba A, Reinecker H-C, Darfeuille-Michaud A, Barnich N, Mizoguchi E. 2013. Chitin-binding domains of Escherichia coli ChiA mediate interactions with intestinal epithelial cells in mice with colitis. Gastroenterology 145:602–12. doi: 10.1053/j.gastro.2013.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Osawa Y, Nagaki M, Banno Y, Brenner DA, Asano T, Nozawa Y, Moriwaki H, Nakashima S. 2002. Tumor necrosis factor alpha-induced interleukin-8 production via NF-kappaB and phosphatidylinositol 3-kinase/AKT pathways inhibits cell apoptosis in human hepatocytes. Infect Immun 70:6294–6301. doi: 10.1128/IAI.70.11.6294-6301.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yabluchanskiy A, Ma Y, Iyer RP, Hall ME, Lindsey ML. 2013. Matrix metalloproteinase-9: many shades of function in cardiovascular disease. Physiology (Bethesda) 28:391–403. doi: 10.1152/physiol.00029.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Al-Sadi R, Engers J, Haque M, King S, Al-Omari D, Ma TY. 2021. Matrix Metalloproteinase-9 (MMP-9) induced disruption of intestinal epithelial tight junction barrier is mediated by NF-κB activation. PLoS One 16:e0249544. doi: 10.1371/journal.pone.0249544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sivignon A, Chervy M, Chevarin C, Ragot E, Billard E, Denizot J, Barnich N. 2022. An adherent-invasive Escherichia coli-colonized mouse model to evaluate microbiota-targeting strategies in crohn's disease. Dis Model Mech 15:dmm049707. doi: 10.1242/dmm.049707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rolhion N, Barnich N, Claret L, Darfeuille-Michaud A. 2005. Strong decrease in invasive ability and outer membrane vesicle release in crohn's disease-associated adherent-invasive Escherichia coli strain LF82 with the yfgL gene deleted. J Bacteriol 187:2286–2296. doi: 10.1128/JB.187.7.2286-2296.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chassaing B, Rolhion N, de Vallée A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Söderholm JD, Hugot J-P, Colombel J-F, Darfeuille-Michaud A. 2011. Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human peyer's patches via long polar fimb crohn's diseriae. J Clin Invest 121:966–975. doi: 10.1172/JCI44632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Keita ÅV, Alkaissi LY, Holm EB, Heil SDS, Chassaing B, Darfeuille-Michaud A, McKay DM, Söderholm JD. 2020. Enhanced E. coli LF82 translocation through the follicle-associated epithelium in crohn's disease is dependent on long polar fimbriae and CEACAM6 expression, and increases paracellular permeability. J Crohns Colitis 14:216–229. doi: 10.1093/ecco-jcc/jjz144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rolhion N, Carvalho FA, Darfeuille-Michaud A. 2007. OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the crohn's disease-associated Escherichia coli strain LF82. Mol Microbiol 63:1684–1700. doi: 10.1111/j.1365-2958.2007.05638.x [DOI] [PubMed] [Google Scholar]
- 61. Barnich N, Bringer M-A, Claret L, Darfeuille-Michaud A. 2004. Involvement of lipoprotein NlpI in the virulence of adherent invasive Escherichia coli strain LF82 isolated from a patient with crohn's disease. Infect Immun 72:2484–2493. doi: 10.1128/IAI.72.5.2484-2493.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cremer J, Segota I, Yang C-Y, Arnoldini M, Sauls JT, Zhang Z, Gutierrez E, Groisman A, Hwa T. 2016. Effect of flow and peristaltic mixing on bacterial growth in a gut-like channel. Proc Natl Acad Sci U S A 113:11414–11419. doi: 10.1073/pnas.1601306113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Steukers L, Glorieux S, Vandekerckhove AP, Favoreel HW, Nauwynck HJ. 2012. Diverse microbial interactions with the basement membrane barrier. Trends Microbiol 20:147–155. doi: 10.1016/j.tim.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He Q, Wang L, Wang F, Wang C, Tang C, Li Q, Li J, Zhao Q. 2013. Microbial fingerprinting detects intestinal microbiota dysbiosis in zebrafish models with chemically-induced enterocolitis. BMC Microbiol 13:289. doi: 10.1186/1471-2180-13-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dumych T, Yamakawa N, Sivignon A, Garenaux E, Robakiewicz S, Coddeville B, Bongiovanni A, Bray F, Barnich N, Szunerits S, Slomianny C, Herrmann M, Gouin SG, Lutsyk AD, Munoz LE, Lafont F, Rolando C, Bilyy R, Bouckaert JMJ. 2018. Oligomannose-rich membranes of dying intestinal epithelial cells promote host colonization by adherent-invasive E. coli. Front Microbiol 9:742. doi: 10.3389/fmicb.2018.00742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barnich N, Carvalho FA, Glasser A-L, Darcha C, Jantscheff P, Allez M, Peeters H, Bommelaer G, Desreumaux P, Colombel J-F, Darfeuille-Michaud A. 2007. CEACAM6 acts as a receptor for adherent-invasive E. coli, supporting Ileal mucosa colonization in crohn disease. J Clin Invest 117:1566–1574. doi: 10.1172/JCI30504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hammarström S. 1999. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol 9:67–81. doi: 10.1006/scbi.1998.0119 [DOI] [PubMed] [Google Scholar]
- 68. Kowalewski J, Paris T, Gonzalez C, Lelièvre E, Castaño Valencia L, Boutrois M, Augier C, Lutfalla G, Yatime L. 2021. Characterization of a member of the CEACAM protein family as a novel marker of proton pump-rich ionocytes on the zebrafish epidermis. PLoS One 16:e0254533. doi: 10.1371/journal.pone.0254533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chapin C, Bailey NA, Gonzales LW, Lee J-W, Gonzalez RF, Ballard PL. 2012. Distribution and surfactant association of carcinoembryonic cell adhesion molecule 6 in human lung. Am J Physiol Lung Cell Mol Physiol 302:L216–25. doi: 10.1152/ajplung.00055.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. ZFIN . 1983. zgc:198329 2023. Available from: https://zfin.org/ZDB-GENE-080226-6#summary
- 71. Bustamante P, Vidal R. 2020. Repertoire and diversity of toxin - antitoxin systems of Crohn's disease-associated adherent-invasive Escherichia coli. New insight of T his emergent E. Coli pathotype. Front Microbiol 11:807. doi: 10.3389/fmicb.2020.00807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Camprubí-Font C, Martinez-Medina M. 2020. Why the discovery of adherent-invasive Escherichia coli molecular markers is so challenging? World J Biol Chem 11:1–13. doi: 10.4331/wjbc.v11.i1.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Saitz W, Montero DA, Pardo M, Araya D, De la Fuente M, Hermoso MA, Farfán MJ, Ginard D, Rosselló-Móra R, Rasko DA, Del Canto F, Vidal RM. 2022. Characterization of adherent-invasive Escherichia coli (AIEC) outer membrane proteins provides potential molecular markers to screen putative AIEC strains. Int J Mol Sci 23:9005. doi: 10.3390/ijms23169005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Delmas J, Gibold L, Faïs T, Batista S, Leremboure M, Sinel C, Vazeille E, Cattoir V, Buisson A, Barnich N, Dalmasso G, Bonnet R. 2019. Metabolic adaptation of adherent-invasive Escherichia coli to exposure to bile salts. Sci Rep 9:2175. doi: 10.1038/s41598-019-38628-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Renshaw SA, Loynes CA, Trushell DMI, Elworthy S, Ingham PW, Whyte MKB. 2006. A transgenic zebrafish model of neutrophilic inflammation. Blood 108:3976–3978. doi: 10.1182/blood-2006-05-024075 [DOI] [PubMed] [Google Scholar]
- 76. Ellett F, Pase L, Hayman JW, Andrianopoulos A, Lieschke GJ. 2011. mpeg1 promoter transgenes direct macrophage-lineage expression in zebrafish. Blood 117:e49–56. doi: 10.1182/blood-2010-10-314120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Crépin S, Harel J, Dozois CM. 2012. Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Environ Microbiol 78:6001–6008. doi: 10.1128/AEM.00986-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Lee DJ, Bingle LEH, Heurlier K, Pallen MJ, Penn CW, Busby SJW, Hobman JL. 2009. Gene doctoring: a method for recombineering in laboratory and pathogenic Escherichia coli strains. BMC Microbiol 9:252. doi: 10.1186/1471-2180-9-252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Peterson SM, Freeman JL. 2009. RNA isolation from embryonic zebrafish and cDNA synthesis for gene expression analysis. J Vis Exp:1470. doi: 10.3791/1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative CT method. Nat Protoc 3:1101–1108. doi: 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1-S8.