

Abstract
The pseudoglycosyltransferase (PsGT) enzyme VldE is a homologue of the retaining glycosyltransferase (GT) trehalose 6-phosphate synthase (OtsA) that catalyzes a coupling reaction between two pseudo-sugar units, GDP-valienol and validamine 7-phosphate, to give a product with α,α-N-pseudo-glycosidic linkage. Despite its biological importance and unique catalytic function, the molecular bases for its substrate specificity and reaction mechanism are still obscure. Here, we report a comparative mechanistic study of VldE and OtsA using various engineered chimeric proteins and point mutants of the enzymes, X-ray crystallography, docking studies, and kinetic isotope effects. We found that the distinct substrate specificities between VldE and OtsA are most likely due to topological differences within the hot spot amino acid regions of their N-terminal domains. We also found that the Asp158 and His182 residues, which are in the active site, play a significant role in the PsGT function of VldE. They do not seem to be directly involved in the catalysis but may be important for substrate recognition or contribute to the overall architecture of the active site pocket. Moreover, results of the kinetic isotope effect experiments suggest that VldE catalyzes a C–N bond formation between GDP-valienol and validamine 7-phosphate via an SNi-like mechanism. The study provides new insights into the substrate specificity and catalytic mechanism of a member of the growing family of PsGT enzymes, which may be used as a basis for developing new PsGTs from GTs.
Keywords: pseudoglycosyltransferase, SNi mechanism, C-N bond formation, validamycin, chimeric protein, kinetic isotope effect
Graphical Abstract

Introduction
Glycosyltransferases are enzymes that catalyze the transfer of sugar (glycon) from a sugar donor to a sugar acceptor (aglycon).1, 2 They play a central role in the biosynthesis of sugar-containing molecules, such as glycosylated natural products, glycolipids, and glycoproteins, many of which are involved in pathogenesis, cellular recognition, signaling, and bacterial cell wall biosynthesis.3–6 Biochemically, glycosyltransferases catalyze a coupling reaction between an activated sugar donor, in the form of nucleoside diphosphates (e.g., UDP-glucose, GDP-mannose), nucleoside monophosphates (e.g., CMP-NeuAc), lipid phosphates (e.g., dolichol phosphate oligosaccharides) or unsubstituted phosphates, and a sugar acceptor to give a glycosylated product.7–10 The products may have either retained or inverted configuration at the anomeric center in respect to the anomeric stereochemistry of the donor substrates.2 Supported by X-ray crystal structures, point mutations, and kinetic isotope effect studies, the catalytic mechanism of inverting glycosyltransferases have been accepted to occur via an SN2 mechanism (Figure 1A).2, 11 On the other hand, retaining glycosyltransferase has been proposed to adopt a double-displacement mechanism, which involves the formation of a covalent enzyme-intermediate binding with an inverted anomeric stereochemistry, followed by a second inversion by the acceptor molecule to give a product with a retained configuration (Figure 1B).12–15 However, amino acid sequence alignments and structural data for many retaining glycosyltransferases did not show conserved nucleophilic amino acid residues within the reasonable distance to the anomeric center to act as a nucleophile.16, 17 Alternatively, an SNi-like mechanism, which involves a highly dissociative oxocarbenium ion-like transition state and a front-face nucleophilic attack, has been proposed in a retaining glycosyltransferase, trehalose 6-phosphate synthase (OtsA) (Figure 1C).18, 19
Figure 1. Proposed catalytic mechanisms of glycosyltransferases and VldE.

(A) glycosyltransferase reaction via an SN2 mechanism that yields an inverted stereoconfiguration at the anomeric center in respect to the stereoconfiguration of the donor substrate; (B) glycosyltransferase reaction via a double SN2 mechanism that yields a retained stereoconfiguration; (C) OtsA glycosyltransferase reaction via an SNi-like mechanism that yields a retained stereoconfiguration; (D) proposed mechanism of VldE that resembles the SNi-like mechanism seen in OtsA.
OtsA is a member of family 20 glycosyltransferase (GT20) that catalyzes a coupling reaction between nucleotidyl diphosphate (NDP)-glucose and glucose 6-phosphate to give α,α−1,1-trehalose-6 phosphate as a product.16, 20, 21 X-ray crystal structures of OtsA in complex with uridine diphosphate (UDP) and a transition state mimicry, validoxylamine A 7´-phosphate (VDO), as well as results from a kinetic isotope effect study suggest the involvement of an oxocarbenium ion-like transition state in OtsA catalysis.18, 22 Moreover, this mechanism involves a hydrogen bonding between the leaving NDP group and the acceptor nucleophile, which allows a front-face nucleophilic attack to form a glycosidic bond with a retained stereochemistry. The leaving group phosphate also acts as a general base that deprotonates the acceptor nucleophile.18
VldE, a pseudoglycosyltransferase (PsGT) that is involved in the biosynthesis of the antifungal agent validamycin A,23–25 is highly homologous to OtsA.26–28 However, VldE catalyzes a coupling reaction between GDP-valienol and validamine 7-phosphate, both of which are pseudo-sugars, to give a product, validoxylamine A 7´-phosphate (VDO), with an α,α-amino linkage – a net retention of stereochemistry.26 Therefore, it is tempting to speculate that VldE adopts the same SNi-like mechanism suggested for OtsA (Figure 1D).18 However, as GDP-valienol is a pseudo-sugar, which lacks the oxygen atom in the ring, the formation of an oxocarbenium ion in the VldE-catalyzed coupling reaction is not possible, raising a question as to how the C–N bond formation actually occurs.
In addition, our studies have shown that OtsA can only catalyze coupling reactions between NDP-glucose and glucose 6-phosphate (or to some extent validol 7-phosphate), both of which contain C-1 hydroxy group.29 On the other hand, VldE can only catalyze coupling reactions between GDP-valienol (or to some extent GDP-glucose) and validamine 7-phosphate or 1-amino-1-deoxyglucose 6-phosphate (both of which contain C-1 amino group).29 Therefore, VldE may be considered as an N-(pseudo)glycosyltransferase, as opposed to an O-glycosyltransferase as in OtsA. However, it is unclear if these differences were due to particular catalytic amino acid residues or certain topologies in the pockets that might be different between OtsA and VldE. Understanding the molecular bases for substrate specificity and catalytic mechanism of VldE and other PsGTs may provide a basis for developing new PsGTs from GTs.
Here, we report comparative bioinformatics and mechanistic studies of VldE and OtsA using various engineered chimeric and point mutant proteins, X-ray crystallography, docking studies, and kinetic isotope effects. The results provide new insights into the molecular bases for the substrate specificity and catalytic activity of the PsGT VldE.
Results
Comparative Bioinformatic Analysis of GT and PsGT Family 20
In addition to VldE, we have identified many putative PsGT family 20 homologues, including SalC (from the salbostatin pathway in Streptomyces albus ATCC 21838), Amir_1997 (from the validoxylamine pathway in Actinosynnema mirum DSM 43827), and Staur_3137 (from the myxobacterium Stigmatella aurantiaca DW4/3–1).30, 31 Using a multiple amino acid sequence alignment, we compared the amino acid sequences of the PsGT-20 proteins with OtsA from E. coli, Mycobacterium tuberculosis and Streptomyces coelicolor, and revealed several conserved amino acid residues that are different between these two families of proteins (Figures 2A and S1). The conserved amino acid residues Leu8, Lys11, Asn109, Thr135, and Leu178 in the N-terminal domains of most PsGT-20 proteins (VldE, Amir_1997, Staur_3137) are replaced with Val, Asn, His, Asn, and Gly, respectively, in OtsA. Interestingly, SalC, whose acceptor molecule is an aminosugar, is more similar to OtsA than VldE in the N-terminal domain (Figures 2A and S1).
Figure 2. Comparisons of VldE and OtsA.

(A) Partial multiple amino acid alignment of VldE and other putative PsGT20 and OtsA enzymes. Conserved residues in PsGT20s are in red and those in OtsAs are in blue. Potential recognition residues for sugar vs nonsugar acceptors are highlighted in yellow. Amino acid numbering is based on the VldE protein; (B) The VldE cyclitol binding sites in the presence of validoxylamine A 7´-phosphate (VDO); (C) The OtsA binding sites in the presence of VDO.
In the C-terminal domains, there are at least 10 amino acid residues that are conserved in PsGT-20 proteins but are consistently different from their corresponding residues in OtsA. Interestingly, many of these residues are located in the active site or adjacent to the amino acids directly interact with the substrates or the products (Figure 2B). Of particular interest were Asn325 and Gln385, which are among the amino acids shown to directly interact with VDO in the X-ray crystal structures but are replaced by Ser299 and Met363 in OtsA (Figure 2C).18, 27
Examination of the significance of the Asn325 and Gln385 residues for substrate specificity and/or catalytic function of VldE
X-ray crystal structures of VldE showed that Asn325 interacts with the phosphate group of the acceptor cyclitol of VDO, suggesting that this residue may help navigate and position the substrate validamine 7-phosphate for catalysis.27 On the other hand, Gln385 interacts with the hydroxy groups of the donor cyclitol.27 To investigate if the Asn325 and Gln385 residues are important for the substrate specificity and/or catalytic function of VldE, we generated N325S and Q385M variants of VldE and tested them against GDP-glucose and glucose 6-phosphate (OtsA substrates) and GDP-valienol and validamine 7-phosphate (VldE substrates). Point mutations were performed following the site-directed mutagenesis protocol using pRSET-B-vldE (Table S1) as a template.32 The sense and antisense PCR primers were designed, at the nucleotide sites that code for N325 and Q385, by changing the nucleotide sequence to code for serine and glutamine, respectively (Table S2). The resulting mutants VldE-N325S and VldE-Q385M were transferred and expressed in E. coli BL21(DE3) pLysS (Figure S2) and the recombinant proteins were tested. We found that these mutations did not change the VldE capability to catalyze the coupling between GDP-valienol and validamine 7-phosphate and its inability to recognize GDP-glucose and glucose 6-phosphate as substrates (Table 1, Figure S3), suggesting that N325 and Q385 are not directly involved in governing the substrate specificity of the proteins.
Table 1.
Characterization of VldE and OtsA chimeras and point mutants.
| Enzyme | Sugar or pseudo-sugar donor | Sugar or pseudo-sugar acceptor | Expected product | Product observed |
|---|---|---|---|---|
| VldE | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| VldE-N325S | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-Q385M | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-N109H | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-D158N | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | – |
| GDP-glucose | validamine 7-P | pseudo-aminotrehalose 7´-P | + | |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-H182A | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | – |
| GDP-glucose | validamine 7-P | pseudo-aminotrehalose 7´-P | + | |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-D383N | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-K11N/L178G | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| VldE-L8V/N109H/T135N | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| GDP-valienol | glucosamine 6-P | val-GlcN 6´-P | – | |
| GDP-glucose | glucosamine 6-P | 2-amino-trehalose 6-P | – | |
| Chimera-1-N109H | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| Chimera-1-D158N | GDP-glucose | validamine 7-P | pseudo-aminotrehalose 7´-P | + |
| Chimera-1-H182A | GDP-glucose | validamine 7-P | pseudo-aminotrehalose 7´-P | + |
| Chimera-1-K11N/L178G | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| Chimera-1-L8V/N109H/T135N | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | + |
| GDP-glucose | glucose 6-P | trehalose 6-P | – | |
| GDP-valienol | glucosamine 6-P | val-GlcN 6´-P | – | |
| GDP-glucose | glucosamine 6-P | 2-amino-trehalose 6-P | – | |
| Chimera-3 | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | – |
| GDP-glucose | glucose 6-P | trehalose 6-P | + | |
| GDP-glucose | validol 7-P | pseudo-trehalose 7´-P | + | |
| OtsA* | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | – |
| GDP-glucose | glucose 6-P | trehalose 6-P | + | |
| OtsA-H154A | GDP-valienol | validamine 7-P | validoxylamine A 7´-P | – |
| GDP-glucose | glucose 6-P | trehalose 6-P | + |
see Abuelizz and Mahmud29
Interrogating the N-terminal domain of VldE
Our earlier studies using recombinant OtsA, VldE, and their chimeric proteins (Chimera-1 and Chimera-2) with a variety of sugar and pseudo-sugar substrates revealed a significant role of the N-terminal domain of VldE in its PsGT activity (Figures 3A–D).29 The N-terminal domain of VldE is highly specific to validamine 7-phosphate (with an NH2-group) and that of OtsA is specific to glucose 6-phosphate and to some extent accepting validol 7-phosphate (a cyclitol similar to validamine 7-phosphate but the amino group has been replaced with a hydroxy group) as acceptor substrates.29 Chimera-1, which contains the N-terminal domain of VldE, is able to accept validamine 7-phosphate, whereas Chimera-2, which contains the N-terminal domain of OtsA, can utilize glucose 6-phosphate and validol 7-phosphate as pseudo-sugar acceptors (Figures 3C and 3D).29
Figure 3. Substrate preferences of VldE, OtsA, and their chimeric proteins.

(A) VldE catalyzes the coupling between GDP-valienol and validamine 7-phosphate; (B) OtsA catalyzes the coupling between GDP-glucose and glucose 6-phosphate; (C) Chimera-1 prefers GDP-glucose and validamine 7-phosphate as substrates; (D) Chimera-2 prefers GDP-glucose and glucose 6-phosphate as substrates, however, it can also recognizes validol 7-phosphate as a sugar acceptor; (E) Chimera-3, which contains the second half of the N-terminal domain of OtsA, can recognize glucose 6-phosphate or validol 7-phosphate as substrate.
To locate the amino acid residues within the N-terminal domain of VldE that might be important for its unique catalytic activity, we engineered an additional chimeric protein, in which the first part of the N-terminal domain of OtsA was replaced with the equivalent portion from VldE. Studies on the crystal structures of VldE revealed several differences in the tertiary structures of the N-terminal domains of VldE and OtsA, particularly within amino acid residues 1–120.27 This includes the presence of a β–hairpin motif between amino acid residues 11–50, which is missing in OtsA. Moreover, this region includes β5 and β6 strands between amino acids 88–110, which together with the β-hairpin motif (β2 and β3) showed a significant movement (10.9 Å) during the opening and closing of the catalytic site.27 Replacing this part of the protein with that of VldE may alter the substrate specificity or catalytic activity of OtsA.
Using splice overlap extension PCR,33 we constructed a chimera-3 gene, which encodes a protein in which one-half of the N-terminal domain is from VldE (residues 1–128) and the rest is from OtsA (EA-A) (Figure 3E). The gene was cloned in the expression vector pRSET B,34 which is then transferred and expressed in E. coli BL21(DE3) pLysS. The recombinant protein was tested against GDP-valienol and validamine 7-phosphate as well as GDP-glucose and glucose 6-phosphate as substrates. The result showed that Chimera-3 could only catalyze a coupling between GDP-glucose and glucose 6-phosphate, but not between GDP-valienol and validamine 7-phosphate (Table 1, Figure S4), suggesting that Chimera-3 is more similar to OtsA than VldE and the first half of the N-terminal domain is not a critical determinant for the distinct functionality of VldE and OtsA.
As OtsA and Chimera-2, which contains the N-terminal domain of OtsA and the C-terminal domain of VldE, were able to utilize validol 7-phosphate, whereas VldE and Chimera-1, which contains the N-terminal domain of VldE and the C-terminal domain of OtsA, do not accept validol 7-phosphate as substrate,29 we predict that Chimera-3 with a half of the N-terminal domain is OtsA-origin may recognize validol 7-phosphate as a substrate. As expected, incubation of Chimera-3 with GDP-glucose and validol 7-phosphate gave a product, pseudo-trehalose 7´-phosphate (Figures 3E and S4), suggesting that amino acid residues within the second half of the N-terminal domain play a role in the substrate specificity of these enzymes.
Determining the importance of the conserved amino acid residues in the N-terminal domains for substrate specificity
To explore the importance of the conserved amino acid residues in the N-terminal domain for substrate selectivity, three sets of site-directed mutants of VldE and Chimera-1 were generated. First, N109H mutants of VldE and Chimera-1 were generated to explore the involvement of His89 in the activation/deprotonation of the acceptor sugar glucose 6-phosphate in OtsA. Although such function may be performed by the diphosphate group of the nucleotidyl diphosphate,18 its relatively close proximity (6 Å) to the C-1 hydroxy group of the acceptor molecule merits closer examinations. Moreover, a careful study of the bifunctional N/O glycosyltransferase UGT72B1 revealed that the H19 residue plays a significant role in the O-glycosyltransferase activity of the protein.35 Point mutation of H19 to Q19 only reduced the N-glycosylation activity by two-fold but reduced the O-glycosyltransferase activity by 300-fold. It is proposed that H19 acts as a general base, abstracting the proton from the C-1 hydroxy group of the acceptor, whereas Q19 may be able to orient the substrate for catalysis but lacks the ability to abstract the proton.35
Second, we generated K11N and L178G double-mutants of VldE and Chimera-1. These residues are the only two amino acids that are conserved in the N-terminal domains of all putative PsGT-20, including SalC. Therefore, K11 and L178 may play a role in the acceptor substrate selectivity.
Third, we generated L8V, N109H, and T135N triple-mutants of VldE and Chimera-1. These amino acid residues are conserved in VldE, Amir_1997, and Staur_3137, but are different in SalC and the OtsAs. The high similarity of SalC and OtsA in this domain is consistent with the fact that SalC utilizes 1-deoxy-2-aminoglucose 6-phosphate (a sugar substrate) as acceptor molecule.36 However, whether or not the conserved residues are responsible for the substrate selectivity is unknown.
All mutant proteins were tested against GDP-valienol and validamine 7-phosphate as well as GDP-glucose and glucose 6-phosphate. Interestingly, despite the above alterations, the mutants did not show any change in their substrate selectivity, as they still utilize GDP-valienol and validamine 7-phosphate to produce validoxylamine A 7´-phosphate (Table 1, Figure S5). Moreover, as the putative acceptor substrate for SalC is a 2-amino-sugar, we tested the L8V, N109H, and T135N triple-mutant proteins using GDP-valienol and glucosamine 6-phosphate as substrates (Figure S6). No products could be observed in the ESI-MS analysis of these reactions either. Combined together, the results suggest that the unique substrate specificity of VldE is not directly controlled by certain conserved amino acid residues in the N-terminal domain but may be due to a partial or an overall topology change of the domain or the protein, most likely due to structural differences in the second half of the N-terminal domain.
Examination of the significance of Asp158, His182, and Asp383 in VldE catalysis
The formation of trehalose 6-phosphate by the retaining GT OtsA has been proposed to occur via an SNi-like mechanism involving an oxocarbenium ion transition state.18, 19 However, oxocarbenium ion formation is not possible in the VldE-catalyzed coupling reaction. Therefore, it is unclear if VldE can catalyze the C-N bond formation in the same way as OtsA forms the glycosidic bond. We hypothesize that the olefinic moiety of GDP-valienol plays a critical role in facilitating the coupling reaction. Moreover, X-ray crystal structures of VldE in complex with validoxylamine A 7´-phosphate showed three amino acid residues, Asp158, His182 and Asp383, are located near the ligand and may be involved in the catalysis. However, these amino acids are conserved in both VldE and OtsA. In the VldE•VDO crystal structure, His182 interacts with the C-7 hydroxy group and Asp383 interacts with the C-2 hydroxy group of the donor cyclitol (Figure 2B). Additionally, Asp158 swings 62–83° toward the catalytic center to interact with the hydroxyls of the acceptor cyclitol in the VldE•VDO complex compared to the VldE•TRE complex.27
To examine the significance of Asp158, His182, and Asp383 in VldE catalysis, we generated three single-point mutants of VldE: D158N, H182A and D383N. The mutated proteins were tested against GDP-valienol and validamine 7-phosphate and the products were analyzed by ESI-MS. The results showed that mutation of Asp383 to Asn did not directly affect the catalytic activity of VldE, whereas replacing Asp158 with Asn or His182 with Ala completely abolished the catalytic activity of VldE, suggesting that Asp158 and His182 are critical for the VldE activity (Table 1, Figure S7). Interestingly, when GDP-valienol was substituted with GDP-glucose, the VldE D158N and H182A variants were able to catalyze the glycosylation reaction to give the expected hybrid product pseudo-aminodisaccharide 7´-phosphate (a.k.a. pseudo-aminotrehalose 7´-P) (Table 1, Figure S8),29 suggesting that Asp158 and His182 are not essential for the coupling reaction involving GDP-glucose. Moreover, we generated two single point mutants of chimera-1, D158N and H182A, and found that these variants were able to catalyze the coupling between GDP-glucose and validamine 7-phosphate to give the expected hybrid product pseudo-aminotrehalose 7´-P (Figure S8).29 This was further confirmed by replacing the corresponding His residue (His154) in OtsA to Ala, leading to an OtsA variant that is still able to catalyze a coupling reaction between GDP-glucose and glucose 6-phosphate (Table 1, Figure S7).
In OtsA, His154 has been shown to interact with the C-6 hydroxy group of GDP-glucose, and the main-chain carbonyl oxygen of His154 has been proposed to participate in catalysis by stabilizing the oxocarbenium ion transition state.16 The fact that the H154A mutant is catalytically active indicates that replacement of His154 to Ala still provides similar main-chain carbonyl oxygen necessary for transition state stabilization. A similar result has been observed when His154 was replaced with Asn, where the mutant protein is still active, albeit in reduced catalytic activity.18 While the imidazole side chain of the His residue is not essential in the OtsA catalysis, it appears to be necessary for the VldE reaction when GDP-valienol is used as a donor molecule (Table 1, Figure 2B).
X-ray crystal structures of the VldE-D158N and VldE-H182A variants
While VldE D158N and VldE H182A can no longer catalyze the coupling between GDP-valienol and validamine 7-phosphate, they still retained some activity for the coupling between GDP-glucose and validamine 7-phosphate. To gain insights into how mutations of His-182 to Ala or Asp158 to Asn affect the pseudoglycosyltransferase activity but not the N-glycosyltransferase activity of VldE, we obtained the X-ray crystal structures of the VldE D158N and H182A variants. The apo crystals were then soaked with GDP-glucose. However, only the crystal structures of the two variants in complex with GDP were obtained (Table S3, Figure S9). Therefore, the ligand VDO was modeled and docked into the active site pocket of the VldE D158N and H182A structures and its possible interactions with the active site amino acid residues were evaluated based on comparisons with structures previously reported for OtsA (in complex with UDP and VDO) and VldE (in complex with GDP and VDO) (Figure 4A–D).18, 27 The results showed that there is no significant difference between the wild-type VldE and the D158N and H182A variants in terms of their interactions with the ligand (Figure 4). Interactions between the main chain amino acid residues of the enzymes with the modeled VDO were also the same as those in OtsA and VldE (Figure 4E–H), leaving the role of His-182 and Asp158 on the pseudoglycosyltransferase activity of VldE remains unclear. In the SNi-like mechanism, the proximity between C-1, the phosphate group of GDP-valienol, and C-4 amino group of validamine 7-phosphate is important for the transition state to form. However, no crystal structures of the H182A and D158N variants with GDP-valienol and validamine 7-phosphate are available to demonstrate that scenario, as the former compound is unstable. Therefore, it is difficult to postulate from the structures the underlying catalytic mechanism, whether it is through an SNi-like mechanism or through another mechanism.
Figure 4. Partial structures of OtsA, VldE, VldE-D158N, and VldE-H182A.

(A) partial active site pocket of OtsA showing interactions between the active site amino acid residues with validoxylamine A 7´-phosphate (VDO); (B) partial active site pocket of VldE showing interactions between the active site amino acid residues with VDO; (C) partial active site pocket of VldE D1158N showing interactions between the active site amino acid residues with modeled VDO; (D) partial active site pocket of VldE-H182A showing interactions between the active site amino acid residues with modeled VDO; (E) partial active site pocket of OtsA showing interactions between the main chain amino acid residues with the valienol moiety of VDO; (F) partial active site pocket of VldE showing interactions between the main chain amino acid residues with the valienamine moiety of VDO; (G) partial active site pocket of VldE D1158N showing interactions between main chain amino acid residues with modeled VDO; (H) partial active site pocket of VldE-H182A showing interactions between main chain amino acid residues with modeled VDO. The OtsA structure was adapted from Lee et al.18 and the VldE structure was adapted from Cavalier et al.27
The possibility of a 1,4-elimination-1,4-addition mechanism
The direct involvement of the Asp158 and His182 residues in the coupling reaction between GDP-valienol and validamine 7-phosphate may also implicate an acid-base reaction in the C–N bond formation with retention of configuration. However, the lack of direct involvement of Asp383 in the process ruled out the possibility of a double SN2 mechanism. Alternatively, it may be speculated that the electron shift occurs through deprotonation at C-7, leading to a 1,4-elimination reaction (Figure 5). This hypothesis is attractive, as the crystal structures of VldE showed that His182 is located in the vicinity of the C-7 of GDP-valienol (Figure 2B).27 To have this to happen, however, all elements would have to line up appropriately to coordinate the deprotonation and the subsequent loss of the nucleotidyl diphosphate. Subsequently, the C–N bond formation may occur via a 1,4-addition mechanism (Figure 5), assuming this reaction takes place faster than the possible tautomerization of the dienol to an α,β unsaturated aldehyde.
Figure 5.

Proposed VldE catalysis via a 1,4-elimination-1,4-addition mechanism.
A preliminary study to probe this possible mechanism was carried out by incubating VldE with its substrates in D2O and analyzed the product by mass spectrometry. Although initially we observed some deuterium replacement in the product, the results were inconsistent, and the exact location of the deuterium could not be clearly identified (data not shown). On the other hand, incubation of VldE with GDP-[7-2H2]valienol and validamine 7-phosphate gave [7-2H2]validoxylamine A 7´-phosphate, indicating that there was no hydrogen replacement at C-7. However, this result does not rule out the possibility of the 1,4-elimination-1,4-addition mechanism, as the hydrogen or deuterium that is subtracted from C-7 during the elimination step may be returned to the same carbon during the addition step (Figure 5).
Kinetic isotope effect studies
To determine whether the coupling reaction by VldE takes place via an SNi-like mechanism or a 1,4-elimination-1,4-addition mechanism, we performed kinetic isotope effect (KIE) studies using GDP-valienol, GDP-[1-2H]valienol, GDP-[2-2H]valienol, and GDP-[7-2H2]valienol. KIE has been used to determine the catalytic mechanism of several glycosyltransferases,37–39 including OtsA, which catalyzes the formation of trehalose 6-phosphate through an SNi-like mechanism.18 Similar to OtsA, the VldE-catalyzed reaction is practically irreversible under the assay conditions, eliminating the possibility of a reverse reaction as a commitment factor. If VldE adopts an SNi-like mechanism, a normal α-secondary KIE for the H-1 isotope substituted GDP-[1-2H]valienol may be expected.18 Similarly, a normal β-secondary KIE may also be observed when H-2 is substituted with deuterium.18 On the other hand, if the VldE reaction takes place via a 1,4-elimination-1,4-addition mechanism, a primary KIE may be observed when GDP-[7-2H2]valienol is used as substrate.
First, we synthesized valienol 1-phosphate, [1-2H]valienol 1-phosphate, [2-2H]valienol 1-phosphate, and [7-2H2]valienol 1-phosphate from glucose or deuterated glucose (Scheme 1). Valienol 1-phosphate, [7-2H2]valienol 1-phosphate, and [2-2H]valienol 1-phosphate were synthesized from D-glucose (1a), [7-2H2]-D-glucose (1b), and [2-2H]-D-glucose (1c), respectively, following a reported procedure (Scheme 1A, Schemes S1–S4).40 [1-2H]valienol 1-phosphate was synthesized from compound 6a by enantioselective reduction of the ketone using BD3 and a chiral oxazaborolidine (Corey-Bakshi-Shibata reagent) as catalyst (Scheme 1B).41
Scheme 1. Chemoenzymatic synthesis of isotopically labeled GDP-valienola.
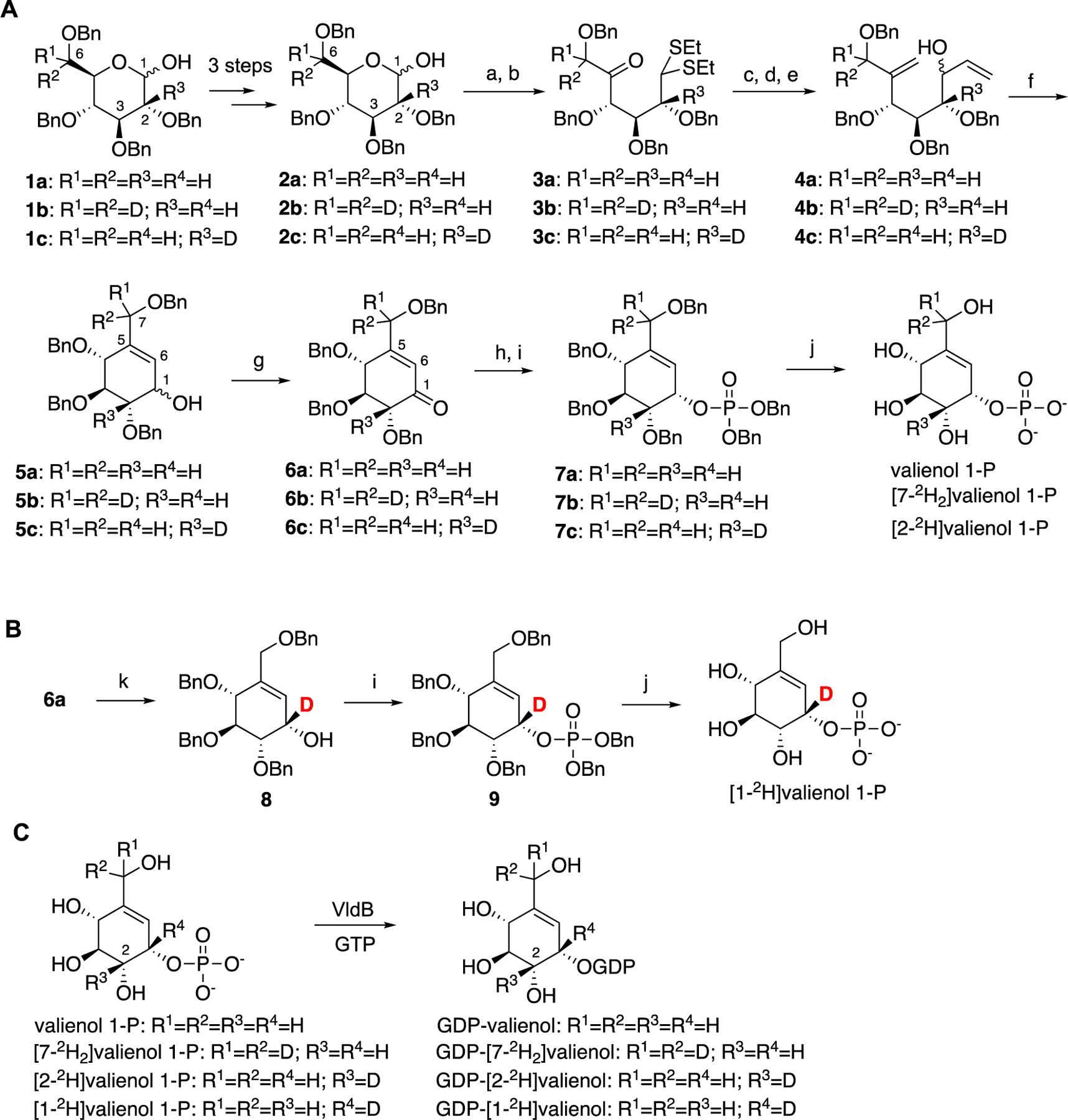
aReaction conditions: a) EtSH, TFA; b) DMSO, Ac2O; c) Ph3PCH3Br, t-BuOK, benzene; d) HgCl2, HgO, aq. CH3CN; e) vinylmagnesium bromide, THF, 0 °C; f) Grubbs’ 2nd gen catalyst, CH2Cl2, reflux; g) DMP; h) L-selectride; i) iPr2NP(OBn)2, 1H-tetrazole, CH2Cl2, m-CPBA; j) BBr3, CH2Cl2; k) BD3, (R)-(+)-2-methyl-CBS-oxazaborolidine (Corey-Bakshi-Shibata reagent).
The phosphorylated valienols were enzymatically converted to their corresponding GDP derivatives using the cyclitol nucleotidyltransferase VldB (Scheme 1C).26, 40 The products GDP-valienols were quantified with a malachite green assay kit (ScienCell) measured at 630 nm.42, 43 Due to their unstable nature, the products were used directly as substrates for the PsGT reactions without further purification. The kinetic values for each of the substrates (Figure S10) were obtained using a procedure reported in our previous publication, employing the pyruvate kinase and lactate dehydrogenase coupled enzyme assay.26, 44, 45 The Lineweaver-Burk plots were used to obtain the Km and Vmax values (Figure S11), whereas the experimental KIEs were obtained by comparing the kcat/Km value of the non-deuterated GDP-valienol with those of the deuterated GDP-valienols (Table 2) according to the procedure reported for OtsA.18
Table 2.
The Km, Vmax, kcat and experimental KIE values for deuterated and non-deuterated GDP-valienols.
| substrate | Km (μM) | Vmax (μM•min−1) | kcat (min−1) | kcat/Km (μM−1 •min−1) | Observed KIE |
|---|---|---|---|---|---|
| GDP-valienol | 147 ± 49 | 2.7 ± 0.4 | 5.4 ± 0.8 | 0.038 ± 0.007 | – |
| GDP-[1-2H]valienol | 103 ± 18 | 1.6 ± 0.2 | 3.1 ± 0.4 | 0.034 ± 0.005 | 1.12 ± 0.04 |
| GDP-[2-2H]valienol | 141 ± 53 | 2.3 ± 0.4 | 4.7 ± 0.8 | 0.035 ± 0.008 | 1.09 ± 0.05 |
| GDP-[7-2H2]valienol | 141 ± 33 | 2.8 ± 0.5 | 5.6 ± 1.0 | 0.039 ± 0.002 | 0.97 ± 0.10 |
The experimental α-secondary KIE value for GDP-[1-2H]valienol was 1.12 ± 0.04 (Table 2), suggesting a shift of hybridization of the “pseudo” anomeric carbon C-1 from sp3 to sp2, similar to that in the oxocarbenium ion-like transition state proposed for OtsA. However, this KIE value is somewhat smaller than that reported for UDP-[1-2H]glucose in the OtsA reaction (1.196 ± 0.003),18 most likely due to the differences in the C–C versus C–O bond order and the geometry around the atom that is subjected to isotope substitution between GDP-valienol and UDP-glucose. While the conversion of UDP-glucose to its oxocarbenium ion-like transition state involves a significant conformational change from a chair to a planer half chair conformation, the conversion of GDP-valienol to its transition state is expected to involve a less dramatic conformational change. The β-secondary KIE value for GDP-[2-2H]valienol (1.09 ± 0.05) is on-par with that observed for UDP-[2-2H]glucose in the OtsA reaction (1.164 ± 0.003),18 suggesting a weakened C-2–H-2 bond due to the hyperconjugation from the bond to the electron deficient C-1. Of note, the standard deviations of the Km and kcat are relatively high. On the other hand, GDP-[7-2H2]valienol gave a small inverse KIE value of 0.97 ± 0.1, which may be interpreted as a more constricted environment or stronger bonding between C-7 and H-7 atoms. However, with the standard deviation value of 0.1, the small inverse KIE may not be real or significant. In addition, the small KIE value does not seem to result from a primary KIE at C-7 by VldE. Overall, the KIE values for the three deuterated substrates are more consistent with the notion that VldE catalyzes the coupling between GDP-valienol and validamine 7-phosphate via an SNi-like mechanism rather than a 1,4-elimination-1,4-addition mechanism.
Discussion
Although VldE is structurally similar to trehalose 6-phosphate synthase (OtsA), the reactions they catalyze are fundamentally different. OtsA catalyzes a coupling between NDP-glucose and glucose 6-phosphate involving a highly dissociative oxocarbenium ion transition state to form a glycosidic bond,18 whereas VldE catalyzes a coupling between GDP-valienol and validamine 7-phosphate to form a non-glycosidic C–N bond.26 There is no oxocarbenium ion transition state that can be formed in the VldE reaction, as the pseudo-sugar substrate lacks the ring oxygen.
Comparative bioinformatics studies between VldE and its homologues and OtsA (GT-20) from several bacteria revealed several conserved amino acid residues shared between VldE and OtsA, as well as those that are unique to VldE. Out of ten amino acid residues unique to the C-terminal domain of VldE, only two (Asn325 and Gln385) are shown in the crystal structures to directly interact with the ligands in the active site pocket. In OtsA, these residues are replaced with Ser and Met.27 However, point mutation of Asn325 and Gln385 in VldE with Ser and Met, respectively, neither change the substrate specificity nor the catalytic activity of the enzyme. This result is consistent with our earlier finding that the C-terminal domain of VldE does not seem to play a major role in governing its PsGT catalysis.29 Moreover, in addition to catalyzing the coupling between GDP-valienol and validamine 7-phosphate, VldE can also couple GDP-glucose and validamine 7-phosphate, albeit in much lower efficiency, confirming that the C-terminal domain of VldE is less specific. On the other hand, the N-terminal domain appears to be the one that is significantly affecting the substrate specificity, particularly of the acceptor molecules, which in the case of VldE need to have a strong nucleophilic group, such as validamine 7-phosphate and 1-amino-1-deoxyglucose 6-phosphate, both contain a C-1 amino group.29
Construction and characterization of Chimera-3, in which the first half of the N-terminal domain is derived from VldE (residues 1–128) and the second half from OtsA, shed more light on the nature and role of the N-terminal domain in the enzyme substrate selectivity and catalysis. We found that the first half of the N-terminal domain of VldE including the unusual β–hairpin motif between amino acid residues 11–33 and β5 and β6 between amino acids 88–110 is not, or at least not by its own, a determinant factor for the VldE unique PsGT activity. We also learned that the conserved amino acid residues Leu8, Lys11, Asn109, Thr135, and Leu178 are not directly involved in determining VldE substrate specificity and/or catalysis. Therefore, the most likely region within the protein that is directly related to its PsGT function is the second half of the N-terminal domain. In this region, in addition to Thr135 and Leu178, which are unique to VldE and related PsGTs, there are other conserved amino acid residues, such as Asp158, Tyr159, and His182. But the latter amino acid residues are also conserved in OtsA. X-ray crystal structures of VldE and OtsA revealed that both Asp158 and His182 are located in the vicinity of the sugar donors. However, they appear to be less important for OtsA catalysis, as replacing the Asp residue with Asn or the His residue with Ala did not significantly affect the activity of the enzyme. Interestingly, similar point mutations in VldE completely abolished the ability of the enzyme to couple GDP-valienol and validamine 7-phosphate, without affecting the enzyme ability to couple GDP-glucose and validamine 7-phosphate (a GT reaction). This finding suggests that both Asp158 and His182 are critical for VldE PsGT activity but are not directly involved in the catalysis. They may be important for substrate recognition or contribute to the overall architecture of the active site pocket (Figure 4). Alternatively, but less likely, VldE catalyzes a PsGT reaction (between GDP-valienol and validamine 7-phosphate) and a GT reaction (between GDP-glucose and validamine 7-phosphate) via two different mechanisms.
The involvement of Asp158 and His182 in the PsGT reaction may suggest an acid-base catalysis, and their locations relative to C-7 of the ligand (GDP-valienol) (Figure 2B) somewhat justifies the possibility of a 1,4-elimination-1,4-addition mechanism (Figure 5). Deprotonation at C-7 followed by a shift of the ring olefin would lead to a 1,4-elimination of GDP. Subsequently, the C-N bond formation may occur via a 1,4-addition mechanism involving a front-face attack of the nucleophilic amino group of the acceptor molecule. It may be postulated that an oxocarbenium-like transition state geometry is an important feature for the GT/PsGT coupling reactions with retained stereochemistry. Intimate hydrogen bonding between the departing GDP and the acceptor nucleophile may direct the nucleophilic attack to occur from the same face as the leaving group. However, the KIE studies suggest that VldE does not catalyze the coupling between GDP-valienol and validamine 7-phosphate via a 1,4-elimination-1,4-addition mechanism. Instead, the data strongly suggest that VldE uses an SNi-like mechanism in its PsGT catalysis. Although oxocarbenium ion formation is not possible in the VldE-catalyzed reaction, the olefinic moiety of GDP-valienol may function similarly to facilitate the C–N bond formation. On the other hand, a double displacement mechanism is highly unlikely because there are no conserved amino acid residues within the reasonable distance to the anomeric center to act as nucleophiles.
Furthermore, the Asp158 and His182 residues, which are required for the VldE coupling reaction, are also present in OtsA. Therefore, it may be concluded that the main determinant for the VldE unique PsGT reaction does not reside in its catalytic mechanism but in its substrate specificity. We have shown in a previous study that the VldE N-terminal domain is specific for the acceptor substrate validamine 7-phosphate, but to some extent also uses 1-amino-1-deoxyglucose 6-phosphate, which contains a C-1 amino group.29 On the other hand, the OtsA N-terminal domain is specific for the acceptor substrate glucose 6-phosphate (to some extent validol 7-phosphate), which contain a C-1 hydroxy group. Davis and coworkers have shown in UGT72B1, a broad spectrum plant bifunctional N- and O-glucosyltransferase, that a deprotonating amino acid residue is necessary for activating the nucleophilic hydroxy group of the acceptor in the O-glycosylation but not for activating the amino group in the N-glycosylation.35 However, such amino acid residue appears to be inessential in the OtsA catalysis, as the departing diphosphate group may function as a base to activate the acceptor molecule.18 In fact, single point mutations of the conserved amino acid residues within the N-terminal domain of VldE and OtsA did not change their substrate selectivity. On the other hand, swapping the N-terminal domains of the enzymes did alter the selection of the sugar acceptors. Therefore, we propose that the distinct substrate specificity of OtsA and VldE is most likely due to the difference in topology of the proteins particularly of the second half of the N-terminal domain.
Conclusions
We have performed a comparative mechanistic study of the pseudoglycosyltransferase VldE and the glycosyltransferase OtsA using engineered chimeric proteins and point mutants of the enzymes, X-ray crystallography, docking studies, and kinetic isotope effects and found that the distinct substrate specificities between VldE and OtsA are most likely due to topological differences within the hot spot amino acid regions of their N-terminal domains. We also found that the Asp158 and His182 residues, which are in the active site, play a significant role in the pseudoglycosyltransferase function of VldE, but they do not seem to be directly involved in the catalysis. We propose that the Asp158 and His182 residues are important for substrate recognition or contribute to the overall architecture of the active site pocket. The proximity of His182 to the C-7 of GDP-valienol has led to the speculation that the C-N bond formation between GDP-valienol and validamine 7-phosphate occurs via a 1,4-elimination-1,4-addition mechanism. However, results of the kinetic isotope effect experiments suggest that the coupling reaction takes place via an SNi-like mechanism. Overall, results of this study may lead to better understanding of the catalytic mechanisms of other retaining PsGTs, including those from other families of PsGTs such as the acarbose PsGTs AcbS/GacS which are similar to the glycogen synthases (GT5 family).46 The knowledge may also be used as a basis for developing new PsGTs from GTs.
MATERIALS AND METHODS
General.
All chemicals were obtained either from Sigma Aldrich, EMD, TCI, or Pharmacia. All reactions were carried out under an inert, Argon atmosphere in oven-dried glassware at 170 °C unless indicated otherwise. Thin-layer chromatography (TLC) was performed using silica gel plates (60 Å), which were visualized using a UV lamp, ceric ammonium molybdate (CAM), potassium permanganate, iodine, and/or vanillin stains. Chromatographic purification of products was performed on silica gel (60 Å, 72–230 mesh). NMR spectra were recorded on Bruker 500 or 700 MHz spectrometers. Proton and carbon chemical shifts are reported in ppm (δ) relative to the residual solvent signals as the internal standard. NMR data were processed using Bruker Topspin (version 3.5). Low-resolution electrospray ionization (ESI) mass spectra were recorded on a ThermoFinnigan Liquid Chromatograph-Ion Trap Mass Spectrometer. High-resolution ESI mass spectra were obtained using an Agilent 1260 HPLC upstream of an Agilent 6545 Q-ToF. The data were processed on Agilent MassHunter workstation (version 10.1). Enzyme assays were measured on a plate reader Spectramax iD3 (Molecular devices). Restriction endonucleases were purchased from Invitrogen or Promega. Preparation of plasmid DNA was done by using a QIAprep Spin Miniprep Kit (QIAGEN). PfuTurbo DNA polymerase was used for amplification of the mutants. GTP, GDP-glucose and glucose 6-phosphate were purchased from Sigma. GDP-valienol, validamine 7-phosphate and validol 7-phosphate according to the published protocols.26, 29, 40 The nucleotide sequences of the generated mutants were determined at the Center for Genome Research and Biocomputing (CGRB) Core Laboratories, Oregon State University. Multiple amino acid sequence alignment was performed using Clustal Omega (EMBL-EBI)47 and visualized with Jalview or MView programs.48, 49
Construction of Chimera-3.
DNA sequences for amino acids 1–128 (384 bp) of VldE and amino acids 102–456 (1062 bp) of OtsA were amplified by PCR using splice overlap extension PCR technique.33 Fragment E was amplified from the plasmid pRSET-B-vldE (Table S1) using primers VldE-F and Chimera-3-R (Table S2) and pfu turbo polymerase, and fragment A was amplified from the plasmid OtsA-pRSET-B using primers Chimera-3-F and OtsA-R. Primers Chimera-3-F and Chimera-3-R were designed with 15 complementary base pair for overlapping (Table S2). Fragments E and A were used as templates for a second PCR using primers VldE-F and OtsA-R. The newly synthesized PCR fragment was digested with XhoI and EcoRI and ligated into pRSET-B (Life technologies) to give pRSET-B-chimera-3. The chimera-3 gene was confirmed by DNA sequencing at the Center for Genome Research and Biocomputing (CGRB) at Oregon State University. The obtained plasmids were then transferred to E. coli BL21(DE3) pLysS.
Site-directed mutagenesis.
Site-directed mutagenesis of VldE, chimera-1 and OtsA was performed using the listed pair primers (N325S, Q385M, N109H, L8V, K11N, T135N, L178G, D158N, H182A and D383N) and amplified by polymerase chain reaction (PCR) using Pfu Turbo DNA polymerase. VldE-pRSET-B, OtsA-pRSETB and chimera-1-pRSET-B were used as templates. PCR amplification reactions were performed using the following conditions: denaturation at 95 °C for 5 min, followed by 18 cycles of 95 °C for 60 s, with primer annealing and extension at 68 °C for 6 min. Extension for 18 min at 68 °C. The PCR product was cooled to 4 °C and then digested with 1 μL of DpnI at 37 °C for 1 h. The DNA was then transferred into E. coli DH10B and the mutants were confirmed by sequencing. The confirmed mutant DNAs were then transferred into E. coli BL21(DE3) pLysS for overexpression and biochemical experiments.
Production and purification of side-directed mutant and chimera-3 proteins.
Site-directed mutant and chimera-3 proteins were produced as N-His6 fusion proteins using pRSET-B in E. coli BL21(DE3) pLysS. Transformants were grown overnight at 37 °C on LB agar plate containing ampicillin (100 μg/mL) and chloramphenicol (25 μg/mL). A single colony was inoculated into LB medium (3 mL) containing ampicillin (100 μg/mL) and chloramphenicol (25 μg/mL) and cultured at 37 °C for 6 h. The seed culture (1 mL) was then transferred into a 500 mL flask containing LB medium (100 mL) with ampicillin (100 μg/mL) and chloramphenicol (25 μg/mL) and grown at 28 °C until OD600 reached 0.6. The temperature was reduced to 18 °C and after 2 h of adaptation IPTG was added to a final concentration of 0.1 mM to induce the N-terminal His6-tagged proteins. After further growth for 14 h, the cells were harvested by centrifugation (5000 rpm, 10 min, 4 °C) and stored at –80 °C until used. Cell pellets from 50 mL of culture were washed with binding (B) buffer (40 mM NaH2PO4 (pH 8), 300 mM NaCl and 10 mM imidazole) (1 mL) and centrifuged (4000 rpm, 10 min, 4 °C). Then, B buffer (1 mL) was added, and the mixtures were sonicated (8 W, 15 s, 4 times). After centrifugation (14,500 rpm, 20 min, 4 °C), the supernatant was purified using Ni-NTA spin column (Qiagen) at 4 °C using washing (W) buffer (40 mM NaH2PO4 pH 8, 300 mM NaCl, and 20 mM imidazole) (2 × 600 μL) and elution (E) buffer (40 mM NaH2PO4 pH 8, containing 300 mM NaCl and imidazole (500 mM, 500 μL). Eluted proteins were dialyzed against dialysis buffer (Tris-HCl (10 mM, pH 7.5), DTT (0.1 mM), MgCl2 (1 mM)) (1 L) 3 times for 3 h each. Purified proteins were analyzed by SDS_PAGE and concentrated by ultrafiltration using Amicon YM-10 (Millipore). Protein concentration was determined using the Bradford assay (Bio-Rad) using BSA as a standard.
Synthesis of substrates.
All substrates, valienol 1-phosphate, [1-2H]Valienol 1-phosphate, [7-2H2]valienol 1-phosphate, and [2-2H]valienol 1-phosphate were synthesized from D-glucose, [7-2H2]-D-glucose, and [2-2H]-D-glucose according to a published procedure and those described in the Supporting Information (Schemes S1–S4).40 Validamine 7-phosphate was synthesized from validamycin A according to a reported procedure (Scheme S5).26
Enzyme assays.
The catalytic activity of the mutants and chimera-3 was examined using GDP-glucose or GDP-valienol as sugar donors and glucose 6-phosphate, validamine 7-phosphate or validol 7-phosphate as acceptors. Each reaction contained (100 μL) donor substrate (5 mM), acceptor substrate (5 mM), protein (10 μM), MgCl2 (10 mM) and Tris-HCl (50 mM, pH 7.5). The reactions mixtures were incubated at 30 °C and aliquots (10 μL) of the reactions were collected at 1, 2, 3, 5, and 1 h and quenched with an equal volume of MeOH and centrifuged (14,500 rpm, 5 min, 4 °C). The supernatants were analyzed by ESI-MS.
Preparation of recombinant VldB and VldE.
The recombinant enzymes of VldB and VldE were prepared according to the procedures described in our previous reports.26, 40 The plasmids pET20-vldB and pRSETB-vldE were used for the production of the recombinant proteins in E. coli BL21(DE3) pLysS. Briefly, E. coli BL21(DE3) pLysS containing each plasmid was cultured at 37 °C, 200 rpm, until OD600 reached 0.4. Then, the cultures were kept on ice for 20 min and IPTG (0.1 mM) was added. The cultures were then incubated at 16 °C, 200 rpm, for 16–20 h. The cells were harvested by centrifugation (4200 rpm, 10 min, 4 °C) and washed with buffer A containing HEPES (40 mM, pH 8.0) and NaCl (300 mM), then stored at 80 °C until used. The cell pellets were re-suspended with buffer A and sonicated (2 W, 5 s, 12 times). After centrifugation (13000 rpm, 40 min, 4 °C), the supernatants were each mixed with His60 Ni Superflow resin (Takara Bio) and incubated for 10 min on ice. The mixtures were then transferred into a column and the resin was washed with buffer A (10 mL) followed by buffer A (20 mL) containing imidazole (50 mM, for VldB) or (20 mM, for VldE). The His-tagged recombinant enzymes were eluted with buffer A (5 – 10 mL) containing imidazole (200 mM) and concentrated with Amicon Ultra (Millipore) until the volume reduced to 2.5 mL. The solutions were passed through PD-10 column pre-equivalated with Tris-HCl buffer (20 mM, pH 7.5). The desalted solutions were concentrated with Amicon Ultra and protein concentration was determined using the Bradford assay (BIO-RAD) with BSA as standard.
Preparation of standard curve using inorganic phosphate with a malachite green assay.
The malachite green assay was conducted with a commercially available kit (ScineCell). Reagent A (ScineCell, 5 µL) was added to mixtures (40 µL each) of Tris-HCl (20 mM, pH 7.5) and inorganic phosphate (10, 5, 2.5, 1 or 0.5 µM, final concentrations in 50 µL) and then incubated at room temperature for 10 min. Subsequently, Reagent B (ScineCell, 5 µL) was added to the mixtures and kept at room temperature for 10 min. The mixtures were then transferred to a 96 wells plate and the absorptions at 630 nm were measured on a plate reader Spectramax iD3 (Molecular devices). Each mixture was prepared triplicate. The values were recorded with SoftMax Pro (version 7). The standard curve was generated and illustrated with Microsoft Excel (ver. 16.54).
X-ray crystallography.
The DNA fragment encoding the H182A and D158N mutant of VldE were ligated into the EcoR1/XhoI-digested pET-22b(+) vector using In-Fusion® HD Cloning Kit (TaKaRa Clontech) to generate His-tagged recombinant H182A and D158N mutant of VldE. The sequences of the inserted DNA fragments were confirmed by DNA sequencing. The vectors were then transformed into E. coli BL21 (DE3). The resulting mutants were cultured in LB medium supplemented with 50 mg/L ampicillin, with shaking at 37 °C. When the OD600 reached 0.6, the cell cultures were cooled down on ice for 30 min, and then IPTG (0.1 mM) was added to induce the expression of target protein at 18°C. After 18 h of post-induction incubation, cell pellets were harvested by centrifugation at 3300 × g for 15 min and suspended in lysis buffer, containing HEPES (40 mM, pH 7.5), NaCl (300 mM), imidazole (5 mM), 2-mercaptoethanol (5 mM), and glycerol (10%). The cell suspension was sonicated for 5 min on ice. After removal of the cell debris by centrifugation at 20,000 × g for 30 min, the supernatant was loaded onto Ni-NTA agarose resin in gravity flow column. Buffer B containing imidazole (20 mM) was used to wash unbound protein from the column. Finally, the His6-tagged H182A and D158N proteins were eluted with buffer B containing imidazole (250 mM). The eluate was concentrated to 10 mg/mL after the removal of imidazole. The eluate was further purified by gel-filtration chromatography on a HiLoad 16/60 Superdex 200 prepacked gel filtration column (4 °C, GE Healthcare), and eluted with a solution containing Tris-HCl (100 mM, pH 7.5), glycerol (5%), DTT (0.1 mM), and MgCl2 (1 mM). The purity of the purified proteins was checked by SDS-PAGE, and the protein concentrations were determined with a Biochem SimpliNano spectrophotometer. The eluate was concentrated to 10 mg/mL, using an Amicon Ultra-4 (MWCO: 30 kDa) filter at 4 °C and used for crystallization.
Crystals were prepared using sitting-drop vapor-diffusion method in aerobic condition. The apo crystals of VldE D158N were obtained after 3–5 days at 20 °C in reservoir containing Tris-HCl (100 mM, pH 8.5) and PEG3350 (26%), while apo crystals of VldE H182A were obtained after a week at 20 °C in reservoir containing Tris-HCl (100 mM, pH 8.0) and PEG3350 (29%). To obtain the crystals of VldE D158N and H182A in complex with GDP-glucose, the apo crystals of VldE D158N and H182A were transferred to a fresh reservoir solution containing GDP-glucose (10 mM), and soaked for 6 h. Prior to analysis, the apo and complexed-VldE D158N and H182A crystals were transferred to a cryoprotectant solution of their respective reservoir solution with ethylene glycol (7.5%).
X-ray diffraction data sets were collected at BL-1A at the Photon Factory, Tsukuba, Japan. The structure was modified using Coot and refined using PHENIX. The final crystal data and intensity statistics are summarized in Table S3. All crystallographic figures were analyzed and visualized with Pymol (ver. 2.5.4).50 The coordinates and the structure factor amplitudes for the VldE D158N and H182A were deposited under accession codes 8IYE and 8IYF, respectively.
Docking studies.
Docking studies were performed using Auto Dock (ver. 4.2.6)51 with the default settings and scoring function. The 3D structure of the ligand validoxylamine A 7´-phosphate (VOD) was obtained using Chem3D Ultra (ver. 19.0.0.22) and MM2 calculations. The structures of VldE D158N and H182A variants used for the docking studies were prepared with Pymol (ver. 2.5.4)50 by removing all ligands and water molecules as well as by calculating the protonation states of the proteins. Polar hydrogen addition and grid box parameter set up were performed using MGLTools (ver. 1.5.4.). D(N)158, H(A)182, D383, G384, Q385, N386, and C387 were selected as flexible residues. The resultant binding free energy was ΔG = −6.4 or −6.6 kcal/mol for D158N or H182A, respectively. The output data were analyzed and visualized with Pymol (ver. 2.5.4).50
GDP-valienol preparation for kinetic isotope effect (KIE) experiments using VldB.
To prepare non-deuterated or deuterated GDP-valienol in situ, reaction mixtures (50 µL, 4 tubes) containing Tris-HCl (20 mM, pH 7.5), GTP (6 mM), MgCl2 (1 mM), VldB (10 µM)26 and valienol 1-phosphate (V1P), [1-2H]valienol 1-phosphate, [2-2H]valienol 1-phosphate, or [7-2H2]valienol 1-phosphate (5 mM each) were individually incubated at 30 °C for 6 h. The reaction mixtures were centrifuged (13,000 x g, at 4 °C for 20 min) with microcons (10K, Thermo Scientific) to remove VldB. The reaction mixtures were quantified with a malachite green assay kit (ScineCell). Briefly, the sample (1 µL) was diluted 100X with Tris Buffer (20 mM, pH 7.5). Subsequently, inorganic phosphatase (IPP, 5 µL, 5 U/mL, Sigma Aldrich) was added to the diluted sample (5 µL) and incubated at 30 °C for 20 min. Tris Buffer (20 mM, pH 7.5, 30 µL) was added to the IPP reaction mixture (10 µL) and the solution was transferred to a 96 well plate followed by addition of Reagent A (ScineCell, 5 µL). After incubation at room temperature for 10 min, Reagent B (ScineCell, 5 µL) was added to the reaction mixture and incubated at room temperature for 10 min. The absorptions at 630 nm were measured on a plate reader Spectramax iD3 (Molecular devices). Each reaction mixture was prepared triplicate. The values were recorded with SoftMax Pro (version 7). The dilution, IPP reaction and quantification with the malachite green assay were repeated three times. In total, nine samples from each substrate were prepared and measured to get the conversion ratios.
KIE experiments of VldE.
The kinetic values for each of the substrates were obtained using a procedure reported in our previous publication.26 The reaction mixtures (50 µL) containing Tris-HCl (20 mM, pH 7.5), phosphoenolpyruvate (1.5 mM), MgCl2 (5 mM), NADH (1 mM), pyruvate kinase and lactate dehydrogenase (37 U/mL and 46 U/mL, respectively, Sigma-Aldrich), validamine 7-phosphate (1 mM) and GDP-valienol, GDP-[1-2H]valienol, GDP-[2-2H]valienol, or GDP-[7-2H2]valienol (500, 250, 125, 100, 75, 50, 25, 12.5 or 0 µM each) were each pre-incubated for several min until no change in absorption at 340 nm was observed. Subsequently, VldE (0.5 µL) was added to the mixtures and the changes in absorption at 340 nm were recorded on a plate reader at 30 °C for 10 min. The assay was repeated three times. The obtained values were analyzed with Microsoft Excel (ver. 16.54) and the Km, Vmax, and kcat values were calculated. Lineweaver-Burk plots were used to obtain the Km and Vmax of the enzymes. The KIE values were calculated as the ratios of the kcat/Km of the non-deuterated substrate and those of the deuterated substrates.
Supplementary Material
ACKNOWLEDGMENT
The authors thanks Y. L. D. Cheung for technical assistance.
Funding Sources
This work was in part supported by Grants GM112068 and AI129957 (to T.M.) from the National Institute of General Medical Sciences (NIGMS) and the National Institute of Allergy and Infectious Diseases (NIAID), respectively. H.A.A. was supported by a scholarship from King Saud University. A.S. was supported by Grant T32 AT010131 from National Center for Complementary and Integrative Health (NCCIH). The content is solely the responsibility of the authors and does not represent the official views of NIGMS, NIAID, NCCIH, or the National Institutes of Health (NIH).
Footnotes
ASSOCIATED CONTENT
Accession Codes
The coordinates and structure factors have been deposited as Protein Data Bank entry 8IYE and 8IYF for VldE D158N and H182A, respectively.
Supporting Information
Additional information of the synthesis of substrates, strains, primers, and plasmids used in the study, X-ray crystallography data, multiple amino acid alignment, SDS-PAGE, ESI-MS data, kinetic data, NMR spectra, and HR-MS data (PDF). The Supporting Information is available free of charge on the ACS Publications website.
The authors declare no competing financial interest.
REFERENCES
- (1).Rini JM; Moremen KW; Davis BG; Esko JD Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology, 4th ed.; Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Mohnen D, Kinoshita T, Packer NH, Prestegard JH, et al. Eds.; 2022; pp 67–78. [Google Scholar]
- (2).Lairson LL; Henrissat B; Davies GJ; Withers SG Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem 2008, 77, 521–555. [DOI] [PubMed] [Google Scholar]
- (3).Hansen SF; Bettler E; Rinnan A; Engelsen SB; Breton C Exploring genomes for glycosyltransferases. Mol. Biosyst 2010, 6, 1773–1781. [DOI] [PubMed] [Google Scholar]
- (4).Xu H; Minagawa K; Bai L; Deng Z; Mahmud T Catalytic analysis of the validamycin glycosyltransferase (ValG) and enzymatic production of 4’’-epi-validamycin A. J. Nat. Prod 2008, 71, 1233–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Perez E; Constant P; Lemassu A; Laval F; Daffe M; Guilhot C Characterization of three glycosyltransferases involved in the biosynthesis of the phenolic glycolipid antigens from the Mycobacterium tuberculosis complex. J. Biol. Chem 2004, 279, 42574–42583. [DOI] [PubMed] [Google Scholar]
- (6).Julien S; Grimshaw MJ; Sutton-Smith M; Coleman J; Morris HR; Dell A; Taylor-Papadimitriou J; Burchell JM Sialyl-Lewis(x) on P-selectin glycoprotein ligand-1 is regulated during differentiation and maturation of dendritic cells: a mechanism involving the glycosyltransferases C2GnT1 and ST3Gal I. J. Immunol 2007, 179, 5701–5710. [DOI] [PubMed] [Google Scholar]
- (7).Elshahawi SI; Shaaban KA; Kharel MK; Thorson JS A comprehensive review of glycosylated bacterial natural products. Chem. Soc. Rev 2015, 44, 7591–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Eida AA; Abugrain ME; Brumsted CJ; Mahmud T Glycosylation of acyl carrier protein-bound polyketides during pactamycin biosynthesis. Nat. Chem. Biol 2019, 15, 795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Raska M; Moldoveanu Z; Suzuki H; Brown R; Kulhavy R; Andrasi J; Hall S; Vu HL; Carlsson F; Lindahl G; Tomana M; Julian BA; Wyatt RJ; Mestecky J; Novak J Identification and characterization of CMP-NeuAc:GalNAc-IgA1 alpha2,6-sialyltransferase in IgA1-producing cells. J. Mol. Biol 2007, 369, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Banerjee DK; Zhang Z; Baksi K; Serrano-Negron JE Dolichol phosphate mannose synthase: a Glycosyltransferase with Unity in molecular diversities. Glycoconj. J 2017, 34, 467–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Bruner M; Horenstein BA Isotope trapping and kinetic isotope effect studies of rat liver alpha-(2-->6)-sialyltransferase. Biochemistry 1998, 37, 289–297. [DOI] [PubMed] [Google Scholar]
- (12).Lairson LL; Chiu CP; Ly HD; He S; Wakarchuk WW; Strynadka NC; Withers SG Intermediate trapping on a mutant retaining alpha-galactosyltransferase identifies an unexpected aspartate residue. J. Biol. Chem 2004, 279, 28339–28344. [DOI] [PubMed] [Google Scholar]
- (13).Forrester TJB; Ovchinnikova OG; Li Z; Kitova EN; Nothof JT; Koizumi A; Klassen JS; Lowary TL; Whitfield C; Kimber MS The retaining beta-Kdo glycosyltransferase WbbB uses a double-displacement mechanism with an intermediate adduct rearrangement step. Nat. Commun 2022, 13, 6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Guerin ME Trapping and retaining intermediates in glycosyltransferases. J. Biol. Chem 2023, 299, 105006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Doyle L; Ovchinnikova OG; Huang BS; Forrester TJB; Lowary TL; Kimber MS; Whitfield C Mechanism and linkage specificities of the dual retaining beta-Kdo glycosyltransferase modules of KpsC from bacterial capsule biosynthesis. J. Biol. Chem 2023, 299, 104609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Gibson RP; Turkenburg JP; Charnock SJ; Lloyd R; Davies GJ Insights into trehalose synthesis provided by the structure of the retaining glucosyltransferase OtsA. Chem. Biol 2002, 9, 1337–1346. [DOI] [PubMed] [Google Scholar]
- (17).Martinez-Fleites C; Proctor M; Roberts S; Bolam DN; Gilbert HJ; Davies GJ Insights into the synthesis of lipopolysaccharide and antibiotics through the structures of two retaining glycosyltransferases from family GT4. Chem. Biol 2006, 13, 1143–1152. [DOI] [PubMed] [Google Scholar]
- (18).Lee SS; Hong SY; Errey JC; Izumi A; Davies GJ; Davis BG Mechanistic evidence for a front-side, SNi-type reaction in a retaining glycosyltransferase. Nat. Chem. Biol 2011, 7, 631–638. [DOI] [PubMed] [Google Scholar]
- (19).Miao Y; Tenor JL; Toffaletti DL; Maskarinec SA; Liu J; Lee RE; Perfect JR; Brennan RG Structural and In Vivo Studies on Trehalose-6-Phosphate Synthase from Pathogenic Fungi Provide Insights into Its Catalytic Mechanism, Biological Necessity, and Potential for Novel Antifungal Drug Design. mBio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).McDougall J; Kaasen I; Strom AR A yeast gene for trehalose-6-phosphate synthase and its complementation of an Escherichia coli otsA mutant. FEMS Microbiol. Lett 1993, 107, 25–30. [DOI] [PubMed] [Google Scholar]
- (21).Mendes V; Acebron-Garcia-de-Eulate M; Verma N; Blaszczyk M; Dias MVB; Blundell TL Mycobacterial OtsA Structures Unveil Substrate Preference Mechanism and Allosteric Regulation by 2-Oxoglutarate and 2-Phosphoglycerate. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Errey JC; Lee SS; Gibson RP; Martinez Fleites C; Barry CS; Jung PM; O’Sullivan AC; Davis BG; Davies GJ Mechanistic insight into enzymatic glycosyl transfer with retention of configuration through analysis of glycomimetic inhibitors. Angew. Chem. Int. Ed. Engl 2010, 49, 1234–1237. [DOI] [PubMed] [Google Scholar]
- (23).Bai L; Li L; Xu H; Minagawa K; Yu Y; Zhang Y; Zhou X; Floss HG; Mahmud T; Deng Z Functional analysis of the validamycin biosynthetic gene cluster and engineered production of validoxylamine A. Chem. Biol 2006, 13, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Dong H; Mahmud T; Tornus I; Lee S; Floss HG Biosynthesis of the validamycins: identification of intermediates in the biosynthesis of validamycin A by Streptomyces hygroscopicus var. limoneus. J. Am. Chem. Soc 2001, 123, 2733–2742. [DOI] [PubMed] [Google Scholar]
- (25).Singh D; Seo MJ; Kwon HJ; Rajkarnikar A; Kim KR; Kim SO; Suh JW Genetic localization and heterologous expression of validamycin biosynthetic gene cluster isolated from Streptomyces hygroscopicus var. limoneus KCCM 11405 (IFO 12704). Gene 2006, 376, 13–23. [DOI] [PubMed] [Google Scholar]
- (26).Asamizu S; Yang J; Almabruk KH; Mahmud T Pseudoglycosyltransferase catalyzes nonglycosidic C-N coupling in validamycin a biosynthesis. J. Am. Chem. Soc 2011, 133, 12124–12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Cavalier MC; Yim YS; Asamizu S; Neau D; Almabruk KH; Mahmud T; Lee YH Mechanistic insights into validoxylamine A 7’-phosphate synthesis by VldE using the structure of the entire product complex. PLoS One 2012, 7, e44934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zheng L; Zhou X; Zhang H; Ji X; Li L; Huang L; Bai L; Zhang H Structural and functional analysis of validoxylamine A 7’-phosphate synthase ValL involved in validamycin A biosynthesis. PLoS One 2012, 7, e32033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Abuelizz HA; Mahmud T Distinct Substrate Specificity and Catalytic Activity of the Pseudoglycosyltransferase VldE. Chem. Biol 2015, 22, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Choi WS; Wu X; Choeng YH; Mahmud T; Jeong BC; Lee SH; Chang YK; Kim CJ; Hong SK Genetic organization of the putative salbostatin biosynthetic gene cluster including the 2-epi-5-epi-valiolone synthase gene in Streptomyces albus ATCC 21838. Appl. Microbiol. Biotechnol 2008, 80, 637–645. [DOI] [PubMed] [Google Scholar]
- (31).Asamizu S; Abugreen M; Mahmud T Comparative Metabolomic Analysis of an Alternative Biosynthetic Pathway to Pseudosugars in Actinosynnema mirum DSM 43827. ChemBioChem 2013, 14, 1548–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Liu H; Naismith JH An efficient one-step site-directed deletion, insertion, single and multiple-site plasmid mutagenesis protocol. BMC Biotechnol 2008, 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Jones ML; Barnard RT Chimerization of multiple antibody classes using splice overlap extension PCR. Biotechniques 2005, 38, 181–182. [DOI] [PubMed] [Google Scholar]
- (34).Schoepfer R The pRSET family of T7 promoter expression vectors for Escherichia coli. Gene 1993, 124, 83–85. [DOI] [PubMed] [Google Scholar]
- (35).Brazier-Hicks M; Offen WA; Gershater MC; Revett TJ; Lim EK; Bowles DJ; Davies GJ; Edwards R Characterization and engineering of the bifunctional N- and O-glucosyltransferase involved in xenobiotic metabolism in plants. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 20238–20243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Vértesy L; Fehlhaber H-W; Schulz A The Trehalase Inhibitor Salbostatin, a Novel Metabolite from Streptomyces albus, ATCC21838. Angew. Chem. Int. Ed. Engl 1994, 33, 1844–1846. [Google Scholar]
- (37).Berti PJ; McCann JA Toward a detailed understanding of base excision repair enzymes: transition state and mechanistic analyses of N-glycoside hydrolysis and N-glycoside transfer. Chem. Rev 2006, 106, 506–555. [DOI] [PubMed] [Google Scholar]
- (38).Paparella AS; Cahill SM; Aboulache BL; Schramm VL Clostridioides difficile TcdB Toxin Glucosylates Rho GTPase by an S(N)i Mechanism and Ion Pair Transition State. ACS Chem. Biol 2022, 17, 2507–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Cleland WW The use of isotope effects to determine enzyme mechanisms. Arch. Biochem. Biophys 2005, 433, 2–12. [DOI] [PubMed] [Google Scholar]
- (40).Yang J; Xu H; Zhang Y; Bai L; Deng Z; Mahmud T Nucleotidylation of unsaturated carbasugar in validamycin biosynthesis. Org. Biomol. Chem 2011, 9, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Corey EJ; Bakshi RK; Shibata S Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc 1987, 109, 5551–5553. [Google Scholar]
- (42).Carter SG; Karl DW Inorganic phosphate assay with malachite green: an improvement and evaluation. J. Biochem. Biophys. Methods 1982, 7, 7–13. [DOI] [PubMed] [Google Scholar]
- (43).Sherwood AR; Paasch BC; Worby CA; Gentry MS A malachite green-based assay to assess glucan phosphatase activity. Anal. Biochem 2013, 435, 54–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Belda FF; Carmona FG; Canovas FG; Gomez JC; Lozano JA Enzymatic assays: optimization of systems by using pyruvate kinase and lactate dehydrogenase as auxiliary enzymes. Rev. Esp. Fisiol 1982, 38, 327–332. [PubMed] [Google Scholar]
- (45).Pistotnik E; Sakamoto H; Pochet S; Namane A; Barzu O Assay of nucleoside 2-deoxyribosyltransferase activity with pyruvate kinase/lactate dehydrogenase coupling system. Anal. Biochem 1999, 271, 192–193. [DOI] [PubMed] [Google Scholar]
- (46).Tsunoda T; Samadi A; Burade S; Mahmud T Complete biosynthetic pathway to the antidiabetic drug acarbose. Nat. Commun 2022, 13, 3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Sievers F; Wilm A; Dineen D; Gibson TJ; Karplus K; Li W; Lopez R; McWilliam H; Remmert M; Soding J; Thompson JD; Higgins DG Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol 2011, 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Waterhouse AM; Procter JB; Martin DM; Clamp M; Barton GJ Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Brown NP; Leroy C; Sander C MView: a web-compatible database search or multiple alignment viewer. Bioinformatics 1998, 14, 380–381. [DOI] [PubMed] [Google Scholar]
- (50).Janson G; Zhang C; Prado MG; Paiardini A PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL. Bioinformatics 2017, 33, 444–446. [DOI] [PubMed] [Google Scholar]
- (51).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.