

Abstract
The ubiquity of mass spectrometry-based bottom-up proteomic analyses as a component of biological investigation mandates the validation of methodologies that increase acquisition efficiency, improve sample coverage, and enhance profiling depth. Chromatographic separation is often ignored as an area of potential improvement with most analyses relying on traditional reversed-phase liquid chromatography (RPLC); this consistent reliance on a single chromatographic paradigm fundamentally limits our view of the observable proteome. Herein, we build upon early reports and validate porous graphitic carbon chromatography (PGC) as a facile means to substantially enhance proteomic coverage without changes to sample preparation, instrument configuration, or acquisition method. Analysis of offline fractionated cell line digests using both separations revealed increase peptide and protein identifications by 43% and 24%, respectively. Increased identifications provided more comprehensive coverage of cellular components and biological processes independent of protein abundance, highlighting the substantial quantity of proteomic information that may go undetected in standard analyses. We further utilize these data to reveal that label-free quantitative analyses using RPLC separations alone may not be reflective of actual protein constituency. Together, these data highlight the value and comprehension offered through PGC-MS proteomic analyses. RAW proteomic data have been uploaded to the MassIVE repository with primary accession code MSV000091495.
Keywords: Porous Graphitic Carbon, Proteomics, Data Completeness, Liquid Chromatography, Mass Spectrometry, LC-MS
Graphical Abstract
Introduction
The long-standing need for human health- and disease-related biomolecular investigation has promoted the widespread development of numerous analytical disciplines. Among others, proteomic analyses remain a vital component of biological investigations, as these studies provide a more robust representation of functioning cells and living systems. Holistic proteomic investigations require analysis of protein expression1, modification2, structure3, and function4, each presenting unique instrumental, preparatory and bioinformatic requirements. Mass spectrometry (MS) is now the tool-of-choice in contemporary proteomics, as this modality facilitates the breadth of measurements required and remains the only high-throughput strategy for peptide sequencing and high-resolution mass measurements5. The current acceptance, ubiquity, and ever-increasing expertise of MS-based proteomic analyses continues to expand the conduit towards rapid investigation of biomolecular alteration in response to external stress, disease, and treatment. However, this analytical pursuit demands continual method development and optimization. While the improvements desired in MS-based proteomics are diverse and may be discussed separately6–9, the most fundamental need is for methodologies that enhance acquisition efficiency10, increase sample coverage11, and enhance profiling depth12. Efforts seeking to provide such improvements target either the sample preparation or instrumentation levels while chromatographic separation is relatively constant and potential improvements are underexplored12.
By in large, high-throughput bottom-up proteomic investigations utilize reversed-phase liquid chromatography (RPLC) due to its reliability, availability, relatively low cost, and extensive innovation13–15. This modality is preferred in bottom-up experiments as the hydrophobicity-based retention mechanism retains and separates a large portion of the average proteolytic mixture. RPLC does not, however, effectively retain hydrophilic peptides, a shortcoming exacerbated in various analyses such as post-translational modification (PTM) discovery16, 17. Additionally, any hydrophilic peptides that do not elute in the void volume may be poorly resolved and suffer from significant ionization suppression in the presence of more hydrophobic, basic peptides18. Within RPLC-MS analyses, the willful disposal of these peptides and their potential insight into protein structure and function is considered an acceptable loss in favor of high identification rates and simpler experimental setups. For this reason, there is a critical need to implement facile, flexible experimental components that allow for these often-discarded analytes to be effectively retained, separated, and identified.
Traditionally, there have been few options when in search of chromatographic paradigms capable of retaining hydrophilic peptides. Hydrophilic interaction chromatography (HILIC) is the most common and widely reported19–21 modality but may be considered disadvantageous as it requires mixtures to undergo phase change into organic buffers prior to analysis – an obvious limitation for hydrophilic analytes. Electrostatic repulsion-hydrophilic interaction chromatography (ERLIC)22–25 is a recent addition to the chromatographic toolbox, reporting greater retention of hydrophilic peptides26 and polar analytes27–29. A limitation of both HILIC and ERLIC is the requirement of salt-containing buffers to mitigate charge effects23, 30 or to maintain and improve separation capacity31, which can hinder mass spectrometry detection. Porous graphitic carbon (PGC) chromatography is an emerging chromatographic regime that has gained popularity for its ability to retain polar, hydrophilic analytes32–34 with particular favor in the analysis of released glycans35–45. This separation strategy was shown to be suitable for the analysis of tryptic glycopeptides46, 47, suggesting the utility of PGC may extend beyond metabolomic and glycomic analyses. With growing understanding of the retention mechanism, it was recently hypothesized that chromatography of this nature may be a suitable complement to traditional RPLC in untargeted, high-throughput analyses. Early reports validated this hypothesis as PGC revealed a substantial improvement in peptide and protein identification, with additional benefits seen when column temperature is optimized48. Stating broadly the advantages that may be seen when PGC separations are included, these initial studies did not acutely detail to what extent this additional information serves to increase sample coverage, improve profiling depth, and affect our understanding of sample constituency.
Herein, we expand on the benefits of PGC chromatography within discovery proteomics experiments. Utilizing offline fractionation to partition prostate cancer cell line lysate, sequential analyses revealed a 43% increase in peptide identification when PGC separations are included, with almost all fractions revealing competitive identification rates between RPLC and PGC. Confident protein identifications were also increased by 23% when including PGC separations, providing greater coverage of numerous cellular compartments and biological process pathways. Interestingly, there was no significant difference in the known abundances of proteins identified through each separation, suggesting proteomic profiling can be significantly improved without the need to explore deeper into a given mixture. Finally, we compiled these data into spectral libraries that were deployed in data-independent label-free quantitative analyses. These evaluations reveal highly reproducible quantitation between PGC and RPLC separations when using the same collection of peptide precursors for quantitation. However, including the additional, complementary peptide identifications provided through PGC during quantitation produces significantly different protein expression levels than those found through RPLC alone. Overall, our work demonstrates the level of information that may go undiscovered in traditional proteomic analyses and how a narrow view of the observable proteome can impact qualitative and quantitative measurements. Despite the incalculable number of experimental optimizations intended to improve analytical throughput, each will be fundamentally hindered by a limited, chromatography-specific view of the proteome. For this reason, future development of PGC that increases retention capacity and reduces time needed to perform sequential RPLC and PGC will play a pivotal role in comprehensive proteome profiling.
Experimental
Materials
Water (H2O, 223623) acetonitrile (ACN, A955), methanol (MeOH, A456), chloroform (C298), formic acid (FA, A117), tris base (BP152), urea (U15), and hydrochloric acid (HCl, A144SI) were purchased from Thermo Scientific. Acetone (179124), sodium dodecyl sulfate (SDS, 7173C), dithiothreitol (DTT, D9779), and iodoacetamide (IAA, I6125) were purchased from Millipore Sigma. Trypisn (V5113) was purchased from Promega (Madison, WI). RPLC packing materials (4451IP, 4472IP) were purchased from Osaka Soda Co (Osaka, Japan). PGC packing material was harvested from ThermoFisher PGC guard columns (35003-014001). Capillary tubing (1068150019) was purchased from PolyMicro. All other sources are listed.
Cell Growth
Benign prostate hyperplasia to prostate cancer (BCaP) cell lines were generated and described previously49. A tumorigenic cell line (BCaP-T10) and an aggressive, metastatic, tumorigenic cell line (BCaP-MT10) are used throughout these analyses. Growth conditions are listed in the Supporting Information.
Protein Extraction and Digestion
Cell pellets were resuspended in 4 volumes 50mM Tris-HCl, 4% SDS prior to lysis via ultrasonication. Lysates were centrifuged to remove cell debris and protein concentration was estimated via bicinchoninic acid (BCA) assay (ThermoFisher Scientific, 23225). Disulfide bonds were reduced with 450mM DTT for 30 minutes at 55°C followed by alkylation with 10mM IAA at room temperature for 15 minutes. Protein was extracted through repeated additions of cold 80% acetone and incubation at −20°C. Protein was reconstituted in 8M urea with 1x protease inhibitor cocktail (Roche, 05892791001 and 04906837001). Aliquots of crude protein were diluted 1:10 with 50mM Tris-HCl to reduce urea concentration to <1 M followed by two additions of trypsin for digestion. Trypsin was added 1:100 w:w and incubated for four hours at 37°C followed by a second addition of trypsin 1:50 that was left to incubate overnight at 37°C. Proteolytic mixtures were desalted, dried under vacuum, and peptide concentration was estimated via peptide assay (ThermoFisher Scientific, 23275).
HPLC Fractionation
Samples were fractionated using a Waters e2695 separation module equipped with a Waters 2489 UV-Vis detector operating in acquiring at 214 and 280nm. A Phenomenex Kinetex 2.6um PS C18 100Å column (150mm x 4.6mm) was used for separation. Buffers A and B were H2O+0.1% FA and ACN+0.1%FA, respectively. 100ug each of T10 and MT10 lysate digest were combined, dried, and reconstituted in buffer A prior to separation. Samples were separated using a 94 min gradient of the following composition: 1% buffer B from minute 0-5, 40% buffer B at minute 50, 60% buffer B at minute 54, 70% buffer B at minute 58, 100% buffer B from minutes 59-74, 1% buffer B from minutes 74.5-94. Flow rate was set to 0.2 mL/min. Fractions were collected in 1.5-minute intervals between minutes 10-70 and were combined as described below (see Results and Discussion).
LC-MS/MS
Samples were analyzed using a Dionex nanoUltimate 3000 chromatography stack coupled to a ThermoFisher Scientific Orbitrap Fusion Lumos. Separation was performed on 15cm custom-packed capillary columns, which were prepared as described in the Supporting Information. Buffers A and B were H2O+0.1% FA and ACN+0.1%FA, respectively. A flow rate of 350 nL/min and the following 110 gradient were used for all analyses: 3% buffer B from minutes 0-18.3, 35% buffer B at minute 90, 95% buffer B from minutes 90.5-100, 3% buffer B from minutes 101-110. The following MS1 parameters were used for DDA analyses: resolution, 120,000; scan range, 400-1250m/z; AGC target, 2e5; maximum injection time, 50ms; intensity threshold, 2e4; charge state, 2-6; dynamic exclusion, after 1 occurrence for 45s. The MS2 parameters were as follows: resolution, 60,000; isolation width 1 m/z; activation, HCD 30; AGC target, 1e4; fixed first mass, 100m/z. For DIA analyses, MS1 settings were resolution, 120,000; scan range, 400-1250m/z; AGC target, 1e6; maximum injection time, 50ms. DIA MS2 parameters were resolution, 60,000; scan range, 200-2000m/z; isolation window 24m/z; activation, HCD 30; AGC target, 1e5; maximum injection time, 45ms; loop control N=9. All fractions and all samples were analyzed in technical duplicate.
Database Searching
DDA data were processed using FragPipe 18.0 with MSFragger50 3.5. An open-source Python library, easypqp, was used to generate spectral libraries from processed DDA runs; RPLC and PGC libraries were generated separately. These spectral libraries were imported to DIA-NN51 for analysis of data-independent analyses. All parameters used within MSFragger and DIA-NN may be found in the Supporting Information.
Data and Code Availability
RAW proteomic data have been uploaded to the MassIVE repository with primary accession code MSV000091495. All code and files required to reproduce the analyses and figures presented within can be found at https://github.com/lingjunli-research/pgc-rplc-frac-profiling.
Results and Discussion
Profiling Fractionated Prostate Cancer Cell Lysate
Within mass spectrometry-based proteomic analyses, the profiling depth that may be achieved is directly tied to the efficiency with which biological mixtures are simplified during separation. Often, a single chromatographic modality does not provide the requisite simplicity for deep profiling, leading many to employ offline fractionation. As previous analyses have directly compared RPLC and PGC in shotgun analyses48, we chose to employ offline fractionation both to further profile the level of information gained through the addition of PGC separations, as well as mimic common decomplexation techniques within bottom-up proteomics. Pooling tumorigenic and metastatic prostate cancer cell lysate, reversed phase offline fractionation was performed to partition the complex mixture and each fraction was analyzed sequentially via RPLC- and PGC-MS analysis (Figure 1a). After fractionation (see Experimental), the 280nm absorbance trace was integrated across the fractionation window (minutes 10-70). The integrated area was divided into 8 approximately equal segments, all fractions within a segment were pooled for LC-MS analysis (Figure 1b). As previous studies have reiterated the capacity of PGC to separate polar, hydrophilic analytes, we elected to combine fractions sequentially, keeping peptides of similar hydrophobicity together.
Figure 1.
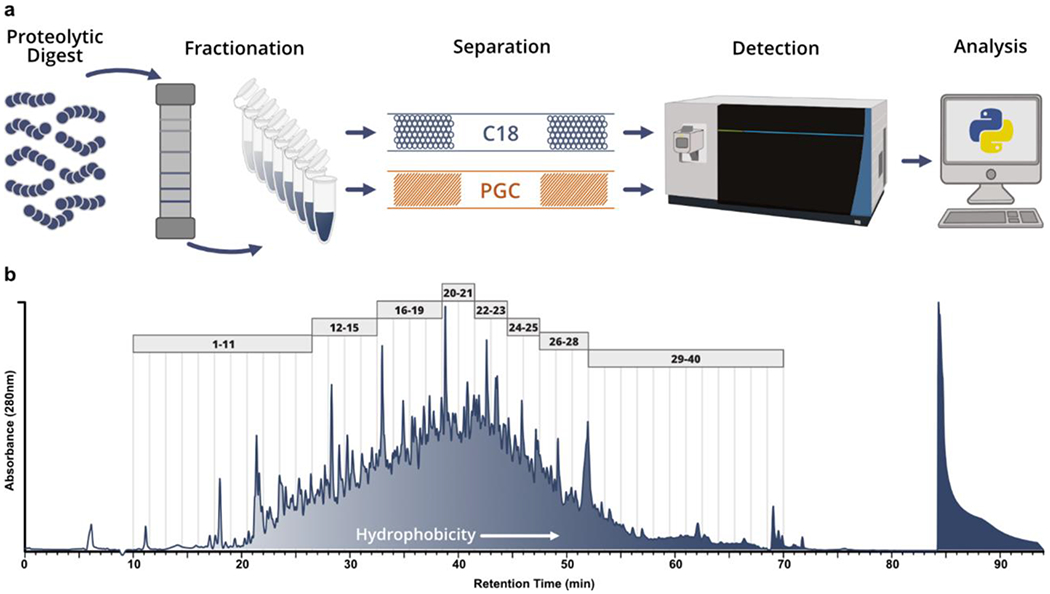
Analytical workflow and offline fractionation. a) Graphical representation of proposed analytical workflow whereby pooled prostate cancer cell line digests are fractionated offline and analyzed through both RPLC- and PGC-MS. b) Absorbance (280nm) trace collected during fractionation of pooled BCaP-T10 and BCaP-MT10; vertical lines represent the 1.5 minute divisions of each fraction collected. The trace was integrated between minutes 10-70 and divided into 8 approximately equal components. All fractions within these 8 components (depicted by the gray boxes) were combined to make 8 final fractions used for analysis.
We hypothesized the sequential combination would result in the greatest contrast between RPLC and PGC analyses. Theoretically, the early fractions containing predominantly hydrophilic analytes should be poorly retained and elute early in subsequent RPLC-MS, whereas PGC should retain these analytes far longer and have elution profiles inversely correlated with fraction number (i.e., peptides in early fractions elute late and vice versa). Examining the time points of all confidently identified peptides, we see this theoretical expectation largely holds true in RPLC analyses but not for PGC separations (Figure 2a, Table S1). Rather, PGC separations demonstrate a progressive trend in peptide retention times, similar to that of RPLC analyses, suggesting that peptide hydrophobicity is not solely responsible for PGC retention. As well, we had anticipated PGC elution profiles to be more broadly distributed than those in RPLC separations, which was only marginally observed. This observation indicates the LC gradient used within our analyses – one modeled from typical RPLC experiments – is not the most appropriate for PGC separations and later optimizations will result in more effective PGC peptide separation (Supplemental Figure S1).
Figure 2.
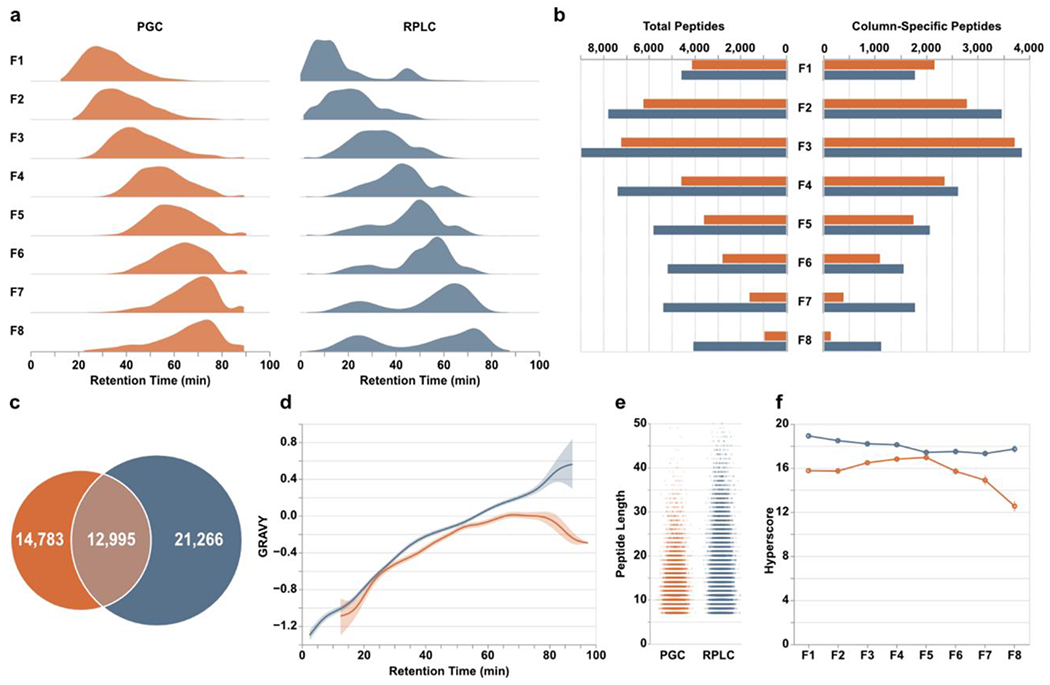
Peptide-level differences between PGC (orange) and RPLC (blue) analyses. a) Density plots displaying the time points during which peptides were identified. b) Total peptides (left) and number of column-specific peptides (right) identified in each fraction. “Column-specific” refers to peptides identified only through that single separation modality. c) Overlap of all peptides identified in all fractions. d) Relative hydrophilicity of all peptides identified within a given separation method. Grand average of hydropathy (GRAVY) calculations are grouped by retention time and averaged across fractions. e) Jitter plots displaying the length of peptides identified through both separations. f) Line plots displaying the average hyperscore, an MSFragger metric of confidence, for all peptides in a single fraction, partitioned according to the separation that retained them.
Examining the overall peptide identifications within each fraction, initial comparisons show RPLC outpaces PGC across all fractions (Figure 2b), mirroring those observations seen elsewhere48. However, given the anticipated redundancy in identifications, removing peptides detected through both separation modalities reveals PGC separations are competitive, especially for those early, predominantly hydrophilic fractions (Figure 2b). Furthermore, the number of peptides specific to a single separation paradigm serves to highlight how much proteomic information may be lost during typical RPLC-MS analyses. Overall, RPLC analysis of offline fractions revealed 34,261 peptides with 21,266 unique to this separation. The inclusion of PGC separations revealed an additional 14,783 peptides, a 43% increase compared to RPLC alone, culminating in 49,044 total peptide sequences (Figure 2c). As anticipated, PGC provided greater access to those hydrophilic peptides across all fractions (Figure 2d) in addition to selectively retaining shorter analytes compared to RPLC (Figure 2e). While these high-level results are encouraging at face value, they should be further contextualized within this experiment. Here, we utilized offline fractionation, which empirically improves profiling capability of RPLC analyses. As we compare the analysis of eight separate fractions between two independent, complementary separation paradigms may be questioned whether the strategy used here is more beneficial than analyzing 16 less complex fractions through RPLC alone. This question should be investigated independently. We hypothesize implementing a more extensive fractionation strategy would improve the number of peptide identifications but would not recover the entirety those peptides found only in PGC analyses; the characteristics of those PGC-compatible peptides is incongruent with RPLC separations. Nevertheless, given that we are still able to extract such an extensive quantity of additional information through PGC analyses even when modest fractionation is performed, it is even more clear the level of information that is lost in single-separation, RPLC shotgun proteomics.
One consideration in PGC analyses, however, is the software and parameters used during peptide identification. Within this study we utilized MSFragger, a well-recognized suite of tools with demonstrated merit50. A highly beneficial component of this software is the ability generate in silico tandem MS spectra and theoretical retention times that may be used as a scoring mechanism for identified peptides. Within our analyses, peptides identified in RPLC separations regularly scored higher and may be considered more confident matches than those in PGC separations (Figure 2f). Certainly, it is possible that all RPLC-retained peptides produced better spectra; however, peptides identified in PGC analyses also fell behind in score of the next-best peptide sequence identification, match to theoretical retention time, and PeptideProphet expectation52 (Supplemental Figure S2). Given the consistency with which PGC peptides score below RPLC peptides, this is most likely a reflection of how database searching tools, statistical models, and predicted expectations are largely trained upon datasets that utilize RPLC separations. We do not argue that the retention and separation capacity of RPLC is superior to that of PGC, as demonstrated here and previously48, but given these observations and further discussion provided below, we posit that the heavy emphasis on RPLC separations in the construction and utilization of bioinformatic tools presents a fundamental limitation in the ability to correctly and confidently identify peptides in PGC-MS experiments.
Finally, we must consider the peak capacity of each separation paradigm. A limitation of this experimental design is inconsistent particle diameter between RPLC and PGC columns (RPLC d.p.=1.7um, PGC d.p.=3.0um). For this reason, the theoretical peak capacity and plate heights is substantially different between the two. If we assume all factors are consistent between the two separations, plate height should be approximately three-fold shorter in the RPLC column. This discrepancy in plate height, and therefore peak capacity, does provide a substantial benefit to RPLC separations and may help explain the higher identification rates in RPLC analyses. Though, if we look at the empirical differences in peak capacity, the two separations are comparable. Peak width and height are not reported directly in our analysis pipeline making analysis of thousands of peptides untenable. Examining, however, a collection of peptides identified in every sample and every replicate of our DIA analyses (read below), we binned peptides according to their RPLC retention time and selected the most abundant peptide within each 10-minute bin. Extracting the signal for each of the 10 representative peptides and determining FWHM allows us to calculate plate number at a given time using the formula . As shown in Supplemental Figure S3 and Table S2, plate number is generally higher in PGC columns for all eligible time points until the later portions of the gradient. Though these peptides are only a subset of the overall data, we can infer that it is not peak capacity or plate height that significantly drives differences in analyte recognition, instead it is the compatibility of peptides with the retention mechanism of each column. These data demonstrate PGC separations do not yield limitations in analyte resolution, peak capacity or plate heights and is a performant separation modality when compatible analytes are presented.
Enhanced Protein Identification, Compartment Coverage and Pathway Completeness
Encouraged by the improved peptide recognition provided when PGC separations are used in addition to RPLC, we anticipated the number of peptides identified would directly correlate to the number of proteins identified through both separations. Considering all proteotypic peptides identified in a given fraction, PGC and RPLC yielded virtually identical numbers of proteins except for those later fractions where RPLC excelled (Figure 3a). However, knowing the redundancy in peptide identifications between the two separations (Figure 2c), removing these redundant identifications reveals notable improvements in protein recognition enabled through PGC separations. Though both separations provided access to different collections of proteins, PGC outpaces RPLC in the number of unique protein identifications, especially within the inner-most fractions (Figure 3a). This observation is particularly valuable when considered alongside the differences in peptide identifications shown in Figure 2. RPLC identified more peptides overall and in all fractions except for one; however, those peptides do not map to a larger collection of proteins. This likely speaks to the known limitations in typical RPLC-MS analyses where data-dependent acquisition experiments bias towards identification of those highly abundant, hydrophobic peptides that ionize well and can cause signal suppression of unique, low-abundance analytes. PGC, which provided a greater number of unique protein species, likely benefits from the smaller number of peptides within each protein that are compatible with the separation modality, reducing overall number of peptides but increasing number of proteins being identified.
Figure 3.
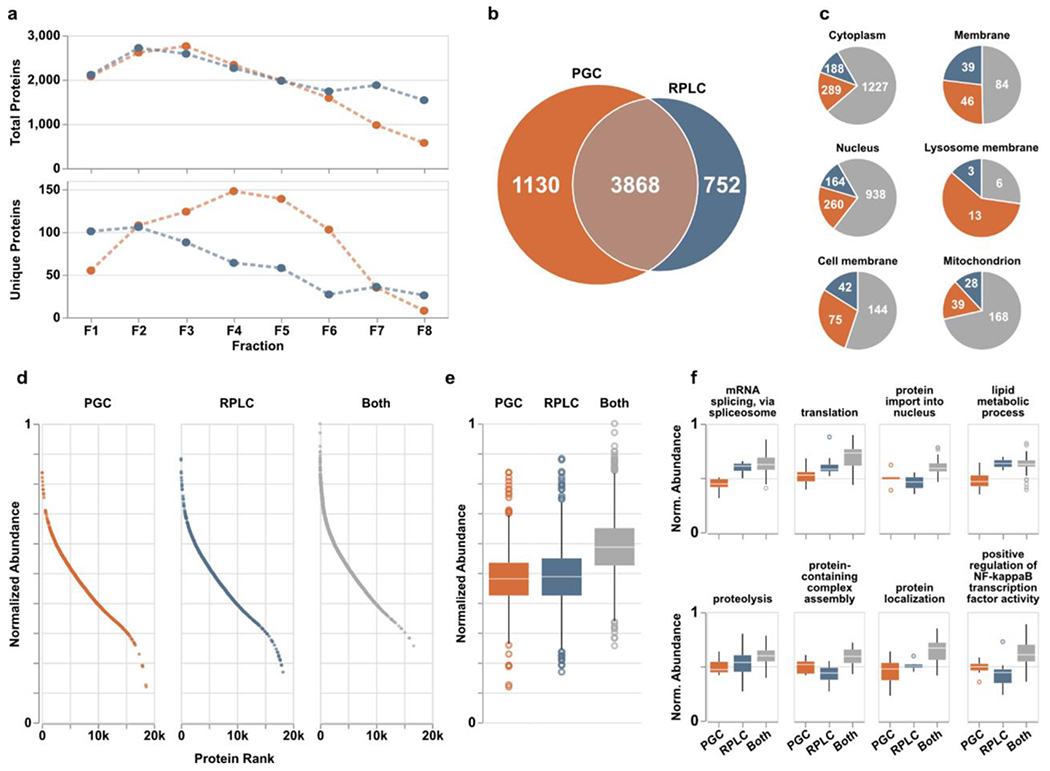
PGC analyses (orange) provide enhanced protein identification compared to RPLC (blue) alone. a) Quantities of total (top) and column-specific (bottom) proteins identified in each fraction. “Column-specific” refers to peptides identified only through that single separation modality. b) Overlap of proteins identified across all fractions. c) Six representative cellular compartments displaying the number of proteins localized within those compartments and through what separations they were identified; PGC (orange), RPLC (blue), both columns (gray). d) Proteins identified across all fractions sorted and ranked according to expected protein abundance within the human proteome of ~20,000 proteins. Expected abundances are normalized according to quantities estimated in the protein abundance database, Pax-DB (see Supporting Information). e) Boxplots displaying the distribution of protein abundances identified in either separation. Statistical differences noted in text or in available. f) Representative biological processes identified across all fractions with boxplots displaying the distribution of protein abundances identified within those pathways. Statistical significance may be generated from supplementary Table S3.
In total, 3,868 proteins were identified through both separations with PGC and RPLC revealing 1,130 and 752 separation-specific proteins, respectively (Figure 3b). Increased identification rates alone are notable, though we hypothesized the significant increase in protein identification rates likely signaled greater profiling depth across the experiment. Organizing all identified proteins into their known subcellular compartments (as listed in the UniProt knowledgebase) reveals the improved compartment coverage when PGC separations are used (Figure 3c). While we anticipated PGC would enable more comprehensive coverage of the cytoplasm, nucleus and other compartments with predominantly cytosolic proteins, our data also revealed PGC was able to improve the detection of membrane and membrane-associated proteins. These observations are encouraging as it demonstrates that PGC broadly provides more effective protein recognition and is not biased towards compartments dominated by hydrophilic species.
As PGC separations noticeably augmented the proteome coverage achieved through traditional RPLC-MS analyses, we allowed ourselves to consider the possibility that PGC was sampling deeper into the biological matrix, identifying species lower in abundance. To evaluate this possibility, we referenced the proteins in our dataset against the anticipated expression of all proteins within the human proteome, as provided by Pax-DB53 (see Supplemental Information, Table S3). As shown in Figure 3d–e, the proteins identified through both separations were those known to be highest in abundance. However, proteins unique to a single separation were not significantly different in abundance (p-value=8.68e-2, Mann-Whitney U test), with PGC separations showing only a slightly greater density of lower abundance species. This observation alone would lead us to believe PGC does not significantly enhance profiling depth, rather it provides greater breadth. However, comparing the global protein populations provides an obtuse conclusion, as there is no information as to protein relatedness or activity. As such, we further categorized proteins according to their biological processes to determine whether PGC separations provide any better coverage of physiological pathways or protein communities. Of those biological processes represented by at least 50 members, many were enhanced through the inclusion of PGC separations, providing detection of lower abundance proteins (Figure 3f, Table S4). For example, PGC provided greater coverage of mRNA splicing, translation, lipid metabolic process, and protein localization by identifying species lower in abundance than those seen in RPLC analyses. Noting there are other pathways where RPLC provides identification of lower abundance species (Figure 3f), PGC does still provide benefit in amplifying pathway coverage and revealing information that may be otherwise lost.
Overall, the peptide- and protein-level results presented here serve to illustrate the breadth and depth of information reclaimed when utilizing PGC analyses in addition to RPLC. Within this experiment, we utilized offline fractionation to reduce matrix complexity and enable greater sample coverage, anticipating PGC separations would benefit analyses of those predominantly hydrophilic fractions but would provide negligible enhancement of others. However, these expectations were largely subverted as PGC separations provided substantial increases to peptide and protein recognition in almost all cases. More interestingly, the additional proteins identified in PGC experiments showed virtually no difference in known abundance as those seen in RPLC analyses. These observations indicate current proteomics should not only focus on exploring deeper into the proteome but should also consider exploring broader coverage. Our data shows within all analyses – not just shotgun experiments – using a single separation will most likely provide a limited, biased view of the proteome. By utilizing and optimizing facile, complementary separation strategies, these limitations may be systematically addressed and overcome.
PGC Analyses Enable More Representative Label-Free Protein Quantitation
Data-independent acquisition (DIA) MS is rapidly gaining favor in analysis of biological mixtures as it provides higher throughput and greater profiling depth54. Critically, the comprehensive and reproducible nature of DIA-MS has promoted its widespread use in label-free protein quantitation54–56. After confident precursor assignment, protein quantitation in DIA analyses is enabled through summating peptide or transition ion abundances or peak area, though variations to this workflow have been described57. As we have established the vast, complementary proteomic information provided when PGC separations are used to augment RPLC-MS analyses, our ability to quantify proteins is similarly enhanced. However, while PGC enables identification, and therefore quantitation, of proteins previously unseen in RPLC-MS, PGC also enabled the retention and identification of additional peptides from protein sequences already identified. Knowing common label-free protein quantitation in DIA analyses utilizes summated precursor abundances, the additional peptides identified through PGC are likely to significantly impact quantitative estimations.
To investigate this claim, we compiled the data-dependent analyses of the offline fractions into two spectral libraries, one for each separation regime. After DIA analyses of tumorigenic (T10) and metastatic (MT10) prostate cancer cell line digests, these libraries were deployed for precursor assignment. Peptide identification rates resembled the trend observed in DDA analyses of fractions, though fewer were identified overall (Figure 4a, Table S5). The number of identified proteins, however, was comparable between the two separations. This observation, which does not coincide with our DDA analyses, is likely a result of the compressed elution profile observed through PGC separations (Figure 4b), rearticulating the need to investigate the optimal gradient composition for this paradigm. During manual interrogation of these identifications, we noted an additional aspect of chromatographic behavior that may present limitation. Focusing on peptides identified in both separations, these peptides were almost unanimously retained longer in PGC separations (Figure 4c), 11.68 minutes longer on average. Expectedly, those with greatest retention time difference were generally those with highest hydrophilicity and polarity (Supplemental Figure S4). These differences in retention time do not impact our DIA analyses as we are using empirical spectral libraries where the experimental MS spectra and retention time are known and used for identification. However, library-free analyses are gaining popularity as they are efficient and can expand profiling capacity while eliminating the need to generate extensive libraries. We posit library-free analysis built into current software is not suitable for PGC-DIA-MS analyses.
Figure 4.
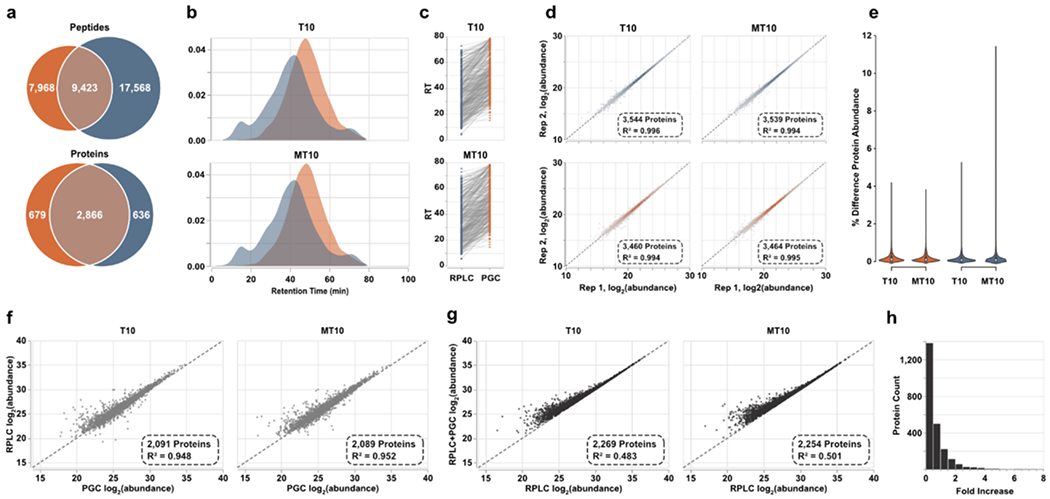
Spectral library-based DIA-MS analysis of prostate cell lines. a) Overlap of peptides and proteins identified through both separations. Results are combined across the two cell lines analyzed. b) Density plots representing the elution profiles of peptides identified in both PGC and RPLC experiments. c) Retention times of representative peptides identified through both separation paradigms displaying the significantly later times associated with PGC analysis. d) Intrasample reproducibility of protein-level abundance calculated after DIA-MS analyses. e) Violin plots displaying the percent difference in protein abundance between technical replicates. f) Scatter plot displaying the agreement of protein abundances when using peptide precursors identified in both separation regimes. g) Protein abundances calculated using all proteotypic peptide precursors plotted against protein abundances estimated using only precursors found in RPLC analyses. h) The count of proteins showing increased abundance estimations after PGC peptides are included. Vertical bars represent count and horizontal axis is the binned fold increase as calculated by .
Library-free analyses, such as that offered through DIA-NN, work by using machine learning approaches to generate theoretical tandem-MS spectra and peptide retention times. These tools are constructed on the extensive body of RPLC-MS proteomics data, making them accurate, reliable tools when RPLC is the separation regime. However, the significant difference in peptide retention time observed in our PGC analyses means that library-free tools such as DIA-NN would struggle to make accurate retention time predictions and would provide limited peptide and protein identifications. We briefly investigated this claim by performing library-free analyses of our DIA datasets (see Supplemental Information). Agreeing with previous literature, library-free data processing resulted in significant improvements in the number of precursors and proteins identified in RPLC analyses (Supplemental Figure S5). These improvements are largely due to the rigorous, well-aligned in silico spectra and retention times predicted for our RPLC separations. Nevertheless, library-free results for our PGC datasets were rather poor, as expected, identifying only marginally more peptides but far fewer proteins compared to our chosen spectral-library approach (Supplemental Figure S5). As we are confident these deficiencies stem from the inability to correctly predict precursor retention time, users must either rely on empirical spectral libraries or develop custom machine learning approaches that provide rigorous, accurate retention time predictions for PGC-DIA-MS analyses.
These limitations notwithstanding, we turned our attention to ensuring technical reproducibility and quantitative accuracy. DIA-NN implements strict requirements for precursor assignment, offers matching between runs, and has a built-in FDR estimation. These features, alongside the implementation of the MaxLFQ algorithm58, allow for highly reproducible protein-level estimations. Within our analyses, both separation paradigms provided excellent intra-sample reproducibility (Pearson R2>0.99, Figure 3d) and low variance (Figure 3e) in protein abundance estimations, indicating both separations are suitable for high-throughput quantitative DIA-MS analyses. To directly compare protein abundance estimations between PGC and RPLC experiments, all proteotypic peptides identified in both separations were compiled, grouped by protein precursor, and peptide MS1 areas were summed and then averaged across technical duplicates. For each of the two prostate cancer samples analyzed, approximately 2,100 proteins could be directly compared between each separation paradigm, demonstrating excellent correlation (R2≈0.95, Figure 4f). This observation indicates that extracted precursor area is conserved regardless of the separation modality employed and that protein-level estimations made through one separation modality will largely hold true in the other. Knowing this, we may reliably combine extracted precursor areas of separation-specific peptides to achieve more representative protein abundances.
To evaluate how protein quantity estimations change when PGC separations are used in tandem with RPLC, we compiled all proteotypic peptides regardless of their identifying separation, summated peptide areas and averaged technical replicates as above. We observed poor correlation (Pearson R2≤0.5, Figure 4g) of these new, adjusted protein abundances to those calculated using peptides from RPLC experiments alone. As well, protein quantities were significantly different between the two calculations, 963 proteins having notably greater calculated abundance (≥0.25-fold increase) with 465 and 133 proteins shown to be at least 1- and 2-fold greater, respectively (Figure 4h). These substantial differences in protein level estimations further evidence the swath of information lost or left undiscovered in routine RPLC analyses. Even if our quantitative approach was altered to utilize averaged peptide abundances or only the N-most abundant peptides from each protein, our data suggest the resulting protein abundance estimations could be significantly impacted.
Within this single experiment, PGC separations enable the retention and identification of topical peptide precursors that not only enhance proteomic coverage but also present the possibility of significantly impacting our perception of protein regulation, pathway activity, and sample constituency. As such, we hold the position that RPLC separations may never be replaced or supplanted but we can, and should, turn our attention to developing facile, high-throughput strategies that enable complementary proteomic investigations. Our data validate PGC not as the single solution to these endeavors but as one suitable strategy that enables more comprehensive, representative analyses. We are confident PGC can gain purchase within the ever-changing analytical landscape and that engineering developments, targeted optimization, and increased utilization will help drive the future proteomic investigations.
Conclusions
Validated methodologies that increase efficiency and enable more comprehensive sample coverage are an ever-present need in mass spectrometry-based proteomics. Whereas high-throughput measurements continue to rely on RPLC as the principal separation strategy, this report details to what extent the incorporation of PGC chromatography may enhance routine analyses. Without changes to sample preparation, gradient composition, or acquisition parameters, the inclusion of porous graphitic carbon provided a significant increase in peptide and protein identifications and resulted in greater coverage of cellular compartments and biological pathways. Our report also demonstrates how these additional peptide identifications significantly impact downstream protein quantitation when compared against RPLC-MS based measurements. These data further highlight the utility of PGC separations within broad proteome profiling and the effort to provide more comprehensive analyses.
Though our data suggests PGC is not biased towards any cellular compartment or towards proteins at higher abundance, the retention mechanism of PGC make it naturally adept for the retention and identification of shorter, more hydrophilic peptides and those with a high proportion of aromatic residues. Furthermore, the analyses presented here do not target or take into consideration protein post-translational modification. Empirical evidence suggests there is benefit in utilizing PGC separations in the analysis of glycopeptides – where the relative hydrophilicity of analytes will shift higher – though other modifications have yet to be explored. It may very well be the case that other classes of modified peptides see a similar augmentation in retention and identification when PGC separations are employed. We are confident that the results presented here, alongside our previous works and others in the field, provide readers with sufficient evidence to make rational decisions over experimental design, and to determine when PGC separations will significantly support analytical objectives. In all, this report should serve to highlight the emerging potential of this separation paradigm in discovery analyses. Upon development of tailored chromatographic and MS acquisition parameters, it is clear PGC separations present a valid, worthwhile avenue towards comprehensive analysis across a range of applications.
Supplementary Material
Supplemental Methods: Cell Growth, Column Preparation, Data Processing, Expected Protein Abundance; Supplemental Figures: Figures S1-S5. Figure S1. Comparison of peptide elution/identification times with respect to gradient composition; Figure S2. Quality metrics of peptides identified through MSFragger; Figure S3. Evaluations of peak width, peak capacity, and plate number across a panel of representative peptides and time points; Figure S4. Peptide characteristics driving retention time differences in PGC separations; Figure S5. Overlap of peptide precursor and protein identifications identified in library free analysis.
The supplemental tables listed below are found within a single Excel file with multiple, titled sheets. As a note, this data is not needed to recreate the analyses used within the manuscript; the database search output from MSFragger and DIA-NN have been uploaded to the github repository. Cloning the repository will let you recreate all figures.
Table S1. Fractionated DDA Results
Table S2. Plate number determinations.
Table S3. DDA data joined to Pax-DB data.
Table S4. DDA data joined to Pax-DB and Uniprot Pathways
Table S5. DIA results.
Acknowledgements
Support for this research is provided in part by the National Institutes of Health (NIH) grants R01 AG052324 (LL), R01 DK071801 (LL), and U54DK104310 (WAR, LL). The Orbitrap instruments were purchased through the support of an NIH shared instrument grant (NIH-NCRR S10RR029531) and Office of the Vice Chancellor for Research and Graduate Education at the University of Wisconsin-Madison. DGD acknowledges a Luminex Summer Fellowship provided by Department of Chemistry at UW-Madison. LL acknowledges the National Science Foundation funding support (CHE-2108223 and IOS-2010789), NIH grant support R01 AG078794, R21AG065728, S10OD028473, and S10OD025084 as well as a Vilas Distinguished Achievement Professorship and Charles Melbourne Johnson Distinguished Chair Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
Footnotes
The Supporting Information is available free of charge.
The authors declare no competing interests.
References
- 1.Ankney JA; Muneer A; Chen X, Relative and Absolute Quantitation in Mass Spectrometry–Based Proteomics. Annual Review of Analytical Chemistry 2016, 11 (1), 49–77. [DOI] [PubMed] [Google Scholar]
- 2.Brandi J; Noberini R; Bonaldi T; Cecconi D, Advances in enrichment methods for mass spectrometry-based proteomics analysis of post-translational modifications. Journal of Chromatography A 2022, 1678, 463352. [DOI] [PubMed] [Google Scholar]
- 3.Timp W; Timp G, Beyond mass spectrometry, the next step in proteomics. Science Advances 2020, 6 (2), eaax8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Parker CG; Pratt MR, Click Chemistry in Proteomic Investigations. Cell 2020, 180 (4), 605–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gillet LC; Leitner A; Aebersold R, Mass Spectrometry Applied to Bottom-Up Proteomics: Entering the High-Throughput Era for Hypothesis Testing. Annual Review of Analytical Chemistry 2016, 9 (1), 449–472. [DOI] [PubMed] [Google Scholar]
- 6.Addie RD; Balluff B; Bovée JVMG; Morreau H; McDonnell LA, Current State and Future Challenges of Mass Spectrometry Imaging for Clinical Research. Analytical Chemistry 2015, 87 (13), 6426–6433. [DOI] [PubMed] [Google Scholar]
- 7.Reinders J; Lewandrowski U; Moebius J; Wagner Y; Sickmann A, Challenges in mass spectrometry-based proteomics. PROTEOMICS 2004, 4 (12), 3686–3703. [DOI] [PubMed] [Google Scholar]
- 8.Smits AH; Vermeulen M, Characterizing Protein–Protein Interactions Using Mass Spectrometry: Challenges and Opportunities. Trends in Biotechnology 2016, 34 (10), 825–834. [DOI] [PubMed] [Google Scholar]
- 9.Karch KR; Snyder DT; Harvey SR; Wysocki VH, Native Mass Spectrometry: Recent Progress and Remaining Challenges. Annual Review of Biophysics 2022, 51 (1), 157–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang F; Ge W; Ruan G; Cai X; Guo T, Data-Independent Acquisition Mass Spectrometry-Based Proteomics and Software Tools: A Glimpse in 2020. PROTEOMICS 2020, 20 (17–18), 1900276. [DOI] [PubMed] [Google Scholar]
- 11.Furtwangler B; Uresin N; Motamedchaboki K; Huguet R; Lopez-Ferrer D; Zabrouskov V; Porse BT; Schoof EM, Real-Time Search-Assisted Acquisition on a Tribrid Mass Spectrometer Improves Coverage in Multiplexed Single-Cell Proteomics. Mol Cell Proteomics 2022, 21 (4), 100219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenz C; Urlaub H, Separation methodology to improve proteome coverage depth. Expert Review of Proteomics 2014, 11 (4), 409–414. [DOI] [PubMed] [Google Scholar]
- 13.Mao Z; Hu C; Li Z; Chen Z, A reversed-phase/hydrophilic bifunctional interaction mixed-mode monolithic column with biphenyl and quaternary ammonium stationary phases for capillary electrochromatography. Analyst 2019, 144 (14), 4386–4394. [DOI] [PubMed] [Google Scholar]
- 14.Liang Y; Zhang L; Zhang Y, Monolithic Materials-Based RPLC-MS for Proteoform Separation and Identification. In Proteoform Identification: Methods and Protocols, Sun L; Liu X, Eds. Springer US: New York, NY, 2022; pp 43–53. [DOI] [PubMed] [Google Scholar]
- 15.Rozing G, Micropillar array columns for advancing nanoflow HPLC. Microchemical Journal 2021, 170, 106629. [Google Scholar]
- 16.Badgett MJ; Boyes B; Orlando R, Predicting the Retention Behavior of Specific O-Linked Glycopeptides. J Biomol Tech 2017, 28 (3), 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu R; Zacharias L; Wooding KM; Peng W; Mechref Y, Chapter Twenty-One - Glycoprotein Enrichment Analytical Techniques: Advantages and Disadvantages. In Methods in Enzymology, Shukla AK, Ed. Academic Press: 2017; Vol. 585, pp 397–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oss M; Kruve A; Herodes K; Leito I, Electrospray Ionization Efficiency Scale of Organic Compounds. Analytical Chemistry 2010, 82 (7), 2865–2872. [DOI] [PubMed] [Google Scholar]
- 19.Boersema PJ; Mohammed S; Heck AJR, Hydrophilic interaction liquid chromatography (HILIC) in proteomics. Analytical and Bioanalytical Chemistry 2008, 391 (1), 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun Z; Ji F; Jiang Z; Li L, Improving deep proteome and PTMome coverage using tandem HILIC-HPRP peptide fractionation strategy. Analytical and Bioanalytical Chemistry 2019, 411 (2), 459–469. [DOI] [PubMed] [Google Scholar]
- 21.Badgett MJ; Boyes B; Orlando R, Peptide retention prediction using hydrophilic interaction liquid chromatography coupled to mass spectrometry. Journal of Chromatography A 2018, 1537, 58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yeh T-T; Ho M-Y; Chen W-Y; Hsu Y-C; Ku W-C; Tseng H-W; Chen S-T; Chen S-F, Comparison of different fractionation strategies for in-depth phosphoproteomics by liquid chromatography tandem mass spectrometry. Analytical and Bioanalytical Chemistry 2019, 411 (15), 3417–3424. [DOI] [PubMed] [Google Scholar]
- 23.Cui Y; Tabang DN; Zhang Z; Ma M; Alpert AJ; Li L, Counterion Optimization Dramatically Improves Selectivity for Phosphopeptides and Glycopeptides in Electrostatic Repulsion-Hydrophilic Interaction Chromatography. Analytical Chemistry 2021, 93 (22), 7908–7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cui Y; Yang K; Tabang DN; Huang J; Tang W; Li L, Finding the Sweet Spot in ERLIC Mobile Phase for Simultaneous Enrichment of N-Glyco and Phosphopeptides. Journal of the American Society for Mass Spectrometry 2019, 30 (12), 2491–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alpert AJ, Electrostatic Repulsion Hydrophilic Interaction Chromatography for Isocratic Separation of Charged Solutes and Selective Isolation of Phosphopeptides. Analytical Chemistry 2008, 80 (1), 62–76. [DOI] [PubMed] [Google Scholar]
- 26.Zhen J; Kim J; Zhou Y; Gaidamauskas E; Subramanian S; Feng P, Antibody characterization using novel ERLIC-MS/MS-based peptide mapping. mAbs 2018, 10 (7), 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yan J; Ding J; Jin G; Duan Z; Yang F; Li D; Zhou H; Li M; Guo Z; Chai W; Liang X, Profiling of Human Milk Oligosaccharides for Lewis Epitopes and Secretor Status by Electrostatic Repulsion Hydrophilic Interaction Chromatography Coupled with Negative-Ion Electrospray Tandem Mass Spectrometry. Analytical Chemistry 2019, 91 (13), 8199–8206. [DOI] [PubMed] [Google Scholar]
- 28.Qing G; Yan J; He X; Li X; Liang X, Recent advances in hydrophilic interaction liquid interaction chromatography materials for glycopeptide enrichment and glycan separation. TrAC Trends in Analytical Chemistry 2020, 124, 115570. [Google Scholar]
- 29.Bermudez A; Pitteri SJ, Enrichment of Intact Glycopeptides Using Strong Anion Exchange and Electrostatic Repulsion Hydrophilic Interaction Chromatography. In Mass Spectrometry of Glycoproteins: Methods and Protocols, Delobel A, Ed. Springer US: New York, NY, 2021; pp 107–120. [DOI] [PubMed] [Google Scholar]
- 30.Alpert AJ, Effect of salts on retention in hydrophilic interaction chromatography. Journal of Chromatography A 2018, 1538, 45–53. [DOI] [PubMed] [Google Scholar]
- 31.West C; Auroux E, Deconvoluting the effects of buffer salt concentration in hydrophilic interaction chromatography on a zwitterionic stationary phase. J Chromatogr A 2016, 1461, 92–7. [DOI] [PubMed] [Google Scholar]
- 32.West C; Elfakir C; Lafosse M, Porous graphitic carbon: A versatile stationary phase for liquid chromatography. Journal of Chromatography A 2010, 1217 (19), 3201–3216. [DOI] [PubMed] [Google Scholar]
- 33.Bapiro TE; Richards FM; Jodrell DI, Understanding the Complexity of Porous Graphitic Carbon (PGC) Chromatography: Modulation of Mobile-Stationary Phase Interactions Overcomes Loss of Retention and Reduces Variability. Analytical Chemistry 2016, 88 (12), 6190–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stavenhagen K; Hinneburg H; Kolarich D; Wuhrer M, Site-Specific N- and O-Glycopeptide Analysis Using an Integrated C18-PGC-LC-ESI-QTOF-MS/MS Approach. In High-Throughput Glycomics and Glycoproteomics: Methods and Protocols, Lauc G; Wuhrer M, Eds. Springer New York: New York, NY, 2017; pp 109–119. [DOI] [PubMed] [Google Scholar]
- 35.Xu J; Liu X; Zhou H, Recent advances in separation methods for post-translational modification proteomics. Sepu 2016, 34 (12), 1199–1205. [Google Scholar]
- 36.Zhou S; Huang Y; Dong X; Peng W; Veillon L; Kitagawa DAS; Aquino AJA; Mechref Y, Isomeric Separation of Permethylated Glycans by Porous Graphitic Carbon (PGC)-LC-MS/MS at High Temperatures. Analytical Chemistry 2017, 89 (12), 6590–6597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou S; Dong X; Veillon L; Huang Y; Mechref Y, LC-MS/MS analysis of permethylated N-glycans facilitating isomeric characterization. Analytical and Bioanalytical Chemistry 2017, 409 (2), 453–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huang Y; Zhou S; Zhu J; Lubman David M; Mechref Y, LC‐MS/MS isomeric profiling of permethylated N‐glycans derived from serum haptoglobin of hepatocellular carcinoma (HCC) and cirrhotic patients. ELECTROPHORESIS 2017, 38 (17), 2160–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ashwood C; Lin C-H; Thaysen-Andersen M; Packer NH, Discrimination of Isomers of Released N- and O-Glycans Using Diagnostic Product Ions in Negative Ion PGC-LC-ESI-MS/MS. Journal of the American Society for Mass Spectrometry 2018, 29 (6), 1194–1209. [DOI] [PubMed] [Google Scholar]
- 40.Hinneburg H; Chatterjee S; Schirmeister F; Nguyen-Khuong T; Packer NH; Rapp E; Thaysen-Andersen M, Post-Column Make-Up Flow (PCMF) Enhances the Performance of Capillary-Flow PGC-LC-MS/MS-Based Glycomics. Analytical Chemistry 2019, 91 (7), 4559–4567. [DOI] [PubMed] [Google Scholar]
- 41.Ashwood C; Pratt B; MacLean BX; Gundry RL; Packer NH, Standardization of PGC-LC-MS-based glycomics for sample specific glycotyping. The Analyst 2019, 144 (11), 3601–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashwood C; Waas M; Weerasekera R; Gundry RL, Reference glycan structure libraries of primary human cardiomyocytes and pluripotent stem cell-derived cardiomyocytes reveal cell-type and culture stage-specific glycan phenotypes. Journal of Molecular and Cellular Cardiology 2020, 139, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei J; Tang Y; Bai Y; Zaia J; Costello CE; Hong P; Lin C, Toward Automatic and Comprehensive Glycan Characterization by Online PGC-LC-EED MS/MS. Analytical Chemistry 2020, 92 (1), 782–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C-H; Lin Y-P; Ren C-T; Shivatare SS; Lin N-H; Wu C-Y; Chen C-H; Lin J-L, Enhancement of fucosylated N-glycan isomer separation with an ultrahigh column temperature in porous graphitic carbon liquid chromatography-mass spectrometry. Journal of Chromatography A 2020, 1632, 461610. [DOI] [PubMed] [Google Scholar]
- 45.Riley NM; Bertozzi CR; Pitteri SJ, A Pragmatic Guide to Enrichment Strategies for Mass Spectrometry-Based Glycoproteomics. Mol Cell Proteomics 2021, 20, 100029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu R; Huang Y; Zhao J; Zhong J; Mechref Y, Isomeric Separation of N-Glycopeptides Derived from Glycoproteins by Porous Graphitic Carbon (PGC) LC-MS/MS. Analytical Chemistry 2020, 92 (14), 9556–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Delafield DG; Miles HN; Ricke WA; Li L, Higher Temperature Porous Graphitic Carbon Separations Differentially Impact Distinct Glycopeptide Classes. J Am Soc Mass Spectrom 2023, 34 (1), 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Delafield DG; Miles HN; Liu Y; Ricke WA; Li L, Complementary proteome and glycoproteome access revealed through comparative analysis of reversed phase and porous graphitic carbon chromatography. Anal Bioanal Chem 2022, 414 (18), 5461–5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu TT; Ewald JA; Ricke EA; Bell R; Collins C; Ricke WA, Modeling human prostate cancer progression in vitro. Carcinogenesis 2018, 40 (7), 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kong AT; Leprevost FV; Avtonomov DM; Mellacheruvu D; Nesvizhskii AI, MSFragger: ultrafast and comprehensive peptide identification in mass spectrometry–based proteomics. Nature Methods 2017, 14 (5), 513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Demichev V; Messner CB; Vernardis SI; Lilley KS; Ralser M, DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nature Methods 2020, 17 (1), 41–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Keller A; Nesvizhskii AI; Kolker E; Aebersold R, Empirical Statistical Model To Estimate the Accuracy of Peptide Identifications Made by MS/MS and Database Search. Analytical Chemistry 2002, 74 (20), 5383–5392. [DOI] [PubMed] [Google Scholar]
- 53.Wang M; Herrmann CJ; Simonovic M; Szklarczyk D; von Mering C, Version 4.0 of PaxDb: Protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 2015, 15 (18), 3163–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li KW; Gonzalez-Lozano MA; Koopmans F; Smit AB, Recent Developments in Data Independent Acquisition (DIA) Mass Spectrometry: Application of Quantitative Analysis of the Brain Proteome. Frontiers in Molecular Neuroscience 2020, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meyer JG; Schilling B, Clinical applications of quantitative proteomics using targeted and untargeted data-independent acquisition techniques. Expert Review of Proteomics 2017, 14 (5), 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyer JG; Niemi NM; Pagliarini DJ; Coon JJ, Quantitative shotgun proteome analysis by direct infusion. Nature Methods 2020, 17 (12), 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ludwig C; Gillet L; Rosenberger G; Amon S; Collins BC; Aebersold R, Data-independent acquisition-based SWATH-MS for quantitative proteomics: a tutorial. Molecular Systems Biology 2018, 14 (8), e8126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cox J; Hein MY; Luber CA; Paron I; Nagaraj N; Mann M, Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 2014, 13 (9), 2513–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods: Cell Growth, Column Preparation, Data Processing, Expected Protein Abundance; Supplemental Figures: Figures S1-S5. Figure S1. Comparison of peptide elution/identification times with respect to gradient composition; Figure S2. Quality metrics of peptides identified through MSFragger; Figure S3. Evaluations of peak width, peak capacity, and plate number across a panel of representative peptides and time points; Figure S4. Peptide characteristics driving retention time differences in PGC separations; Figure S5. Overlap of peptide precursor and protein identifications identified in library free analysis.
The supplemental tables listed below are found within a single Excel file with multiple, titled sheets. As a note, this data is not needed to recreate the analyses used within the manuscript; the database search output from MSFragger and DIA-NN have been uploaded to the github repository. Cloning the repository will let you recreate all figures.
Table S1. Fractionated DDA Results
Table S2. Plate number determinations.
Table S3. DDA data joined to Pax-DB data.
Table S4. DDA data joined to Pax-DB and Uniprot Pathways
Table S5. DIA results.
Data Availability Statement
RAW proteomic data have been uploaded to the MassIVE repository with primary accession code MSV000091495. All code and files required to reproduce the analyses and figures presented within can be found at https://github.com/lingjunli-research/pgc-rplc-frac-profiling.