

Abstract
Actinic keratosis (AK) is a common precancerous skin lesion that can develop into cutaneous squamous cell carcinoma (CSCC). AK is characterized by atypical keratinocytes in the skin’s outer layer and is commonly found in sun-exposed areas. Like many precancerous lesions, the development of AK is closely associated with genetic mutations. The molecular biology and transcriptional mechanisms underlying AK development are not well understood. Ultraviolet (UV) light exposure, especially UVA and UVB radiation, is a significant risk factor for AK, causing DNA damage and mutagenic effects. Besides UV exposure, comorbidities like diabetes, rheumatoid arthritis, and psoriasis may also influence AK development. AK patients have shown associations with various internal malignancies, indicating potential vulnerability in cancer-associated genes. Treatment for AK includes cryosurgery, electrodesiccation and curettage, chemotherapeutic creams, photodynamic therapy, or topical immune-modulators. Genomic studies have identified genetic aberrations in AK, with common mutations found in genes like TP53, NOTCH1, and NOTCH2. The progression from AK to CSCC involves chromosomal aberrations and alterations in oncogenes and tumor-suppressor genes. The functional relationships among these genes are not fully understood, but network analysis provides insights into their potential mechanisms. Further research is needed to enhance our understanding of AK’s pathogenesis and develop novel therapeutic approaches.
Keywords: Actinic keratosis, Cutaneous squamous cell carcinoma
INTRODUCTION
Actinic keratosis (AK) is a common precancerous lesion that can progress into cutaneous squamous cell carcinoma (CSCC). AK is histologically defined by atypical keratinocytes arising in the stratum spinosum which are packed and enlarged with a loss of polarity (Fig. 1). Clinically, AKs present as red, skin-colored, or dark patches often with crusts. The most common sites of occurrence include sun-exposed areas, such as the face, ears, and dorsum of the hands and arms. Approximately 65% of CSCC is estimated to arise from AKs1. AK is one of the strongest independent risk factors for CSCC development, and the concept of field cancerization applies to AK with multiple lesions often occurring in the same region (Fig. 2). Although the precise rate of progression is unknown, a study that prospectively followed 169 patients with a total of 7,784 AKs in the United States estimated a 2.6% risk of progression for an individual lesion during a 4-year follow-up period1. Evidence for progression of AK to CSCC is also provided by genetic studies that report AKs have a similar karyotypic profile to CSCC but display a reduced degree of complexity, consistent with an earlier stage of tumor development2.
Fig. 1. Histology of actinic keratosis. Parakeratosis and atypical keratinocytes are shown in the epidermis, and there is a loss of cell polarity. Widening of the intercellular spaces and cytologic atypia with large nuclei are shown in the hematoxylin and eosin findings (×100). The underlying dermis shows solar elastosis and mild inflammatory infiltrates.
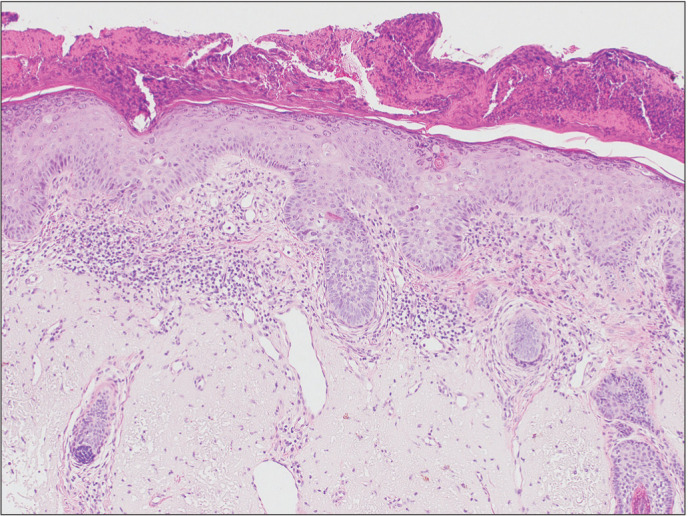
Fig. 2. A schematic diagram of development of damaged keratinocytes and the progression into actinic keratosis and cutaneous squamous cell carcinoma. UV: ultraviolet.

Despite the high prevalence of AK and the possibility of developing cancer, molecular biology and transcriptional studies on the pathogenesis of AK are not well understood. CSCC occurs in sun-exposed areas and is characterized by a high mutation burden. Aged, sun-exposed skin has been reported to be a patchwork of thousands of evolving clones, with >25% of cells carrying carcinogenic mutations while maintaining normal physiological functions of the epidermis. Normal-looking sun-exposed skin had already been found to have driver mutations, such as NOTCH1, NOTCH2, FAT1, and TP53, in 18% to 32% of skin cells at a density of ~140/cm23. Therefore, it is difficult to identify true driver mutations in AKs that arise in sun-exposed areas. No sufficient studies have been conducted on the pathogenesis of AK. In this review, we summarized the results of previous genomic studies on AK with a brief introduction to AK.
EPIDEMIOLOGY OF AK IN KOREA AND WORLDWIDE INCIDENCE
In Korea, according to data obtained from the Korean National Health Insurance System, the incidence rate of AK increased from 17.9 to 54.0 per 100,000 person-years between 2006 and 2015, and it increased consistently with age4. In Korea, which already boasts a super-aged society, the incidence of AK is expected to increase in line with the growth of the elderly population. The incidence of skin cancer and medical expenses associated with treating AK and skin cancer are also expected to increase. In the United States, the cost of treating AK exceeded $920 million in 20065. In Australia, 9% of the population aged ≥60 years developed AK in 1996, and treatment costs for AK were predicted to increase by >30% in 2020 compared to 20126.
ETIOLOGY OF AK
Ultraviolet light exposure
Ultraviolet (UV) A and UVB radiation are indisputable carcinogens to human skin7. UVB causes direct DNA damage, characterized by a typical signature of C→T substitutions, which occur due to the formation of covalent cyclobutane dimers between adjacent pyrimidine rings. UVA exposure, on the other hand, induces mutagenic effects through indirect mechanisms involving the production of reactive oxygen species, resulting in G→T transversions during replication8. Additionally, UVA leads to chromosomal aberrations and widespread alterations in cellular proteins and lipids9,10. Furthermore, apart from promoting cancer cells, UV exposure has similar precancerous effects on the surrounding tissue environment, inducing inflammation and and also cutaneous immune suppression11.
Associated comorbidities of AK
Although UV exposure is widely recognized as the primary risk factor for AK, a comprehensive survey conducted on 2.05 million diabetic patients from the Korean National Health Insurance database revealed that diabetic patients with end-stage kidney disease, a diabetes complication, had a 4.24 times higher risk (95% confidence interval [CI], 3.28~5.47) of developing AK compared to diabetic patients without complications, after adjusting for age, gender, drinking, smoking, exercise, blood pressure, and obesity12. The results suggest that not only UV exposure, but also immune status influence the development of AK.
An association between AK and rheumatoid arthritis (hazard ratio [HR], 1.336; 95% CI, 1.16~1.54) as well as psoriasis (HR, 1.51; 95% CI, 1.44~1.60) has also been reported13, indicating a possible link between AK and systemic inflammatory diseases13.
In another previous study, we compared the incidence of internal malignancies in Korean AK patients (n=61,438) with age- and gender-matched controls using a ratio of 1:5 (n=307,190). After adjusting for economic income, residence, diabetes, hypertension, and hyperlipidemia, the incidence of overall internal malignancies was found to be higher AK patients (HR, 1.43) compared to non-AK controls14. A positive association with AK was observed in skin cancer (HR, 3.43; 95% CI, 2.47~4.75), oral cavity and pharyngeal cancer (HR, 1.99; 95% CI, 1.57~2.52), lymphoma (HR, 1.59; 95% CI, 1.28~1.96), leukemia (HR, 1.35; 95% CI, 1.03~1.77), prostate cancer (HR, 1.35; 95% CI, 1.21~1.51), renal cancer (HR, 1.29; 95% CI, 1.02~1.63), liver cancer (HR, 1.21; 95% CI, 1.09~1.35), thyroid cancer (HR, 1.20; 95% CI, 1.05~1.38), and gastric cancer (HR, 1.13; 95% CI, 1.03~1.23)14. These findings suggest a potential vulnerability in cancer-associated genes in patients with AK (Table 1).
Table 1. The risk of developing actinic keratosis according to the presence of associated comorbidities.
| Comorbidity | Hazard ratio | 95% confidence interval |
|---|---|---|
| Diabetic mellitus with end-stage renal disease12* | 4.24 | 3.28~5.47 |
| Rheumatoid arthritis13 | 1.336 | 1.16~1.54 |
| Psoriasis13 | 1.51 | 1.44~1.60 |
| Skin cancer14 | 3.43 | 2.47~4.75 |
| Oral cavity and pharyngeal cancer14 | 1.99 | 1.57~2.52 |
| Lymphoma14 | 1.59 | 1.28~1.96 |
| Leukemia14 | 1.35 | 1.03~1.77 |
| Prostate cancer14 | 1.35 | 1.21~1.51 |
| Renal cancer14 | 1.29 | 1.02~1.63 |
| Liver cancer14 | 1.21 | 1.09~1.35 |
| Thyroid cancer14 | 1.20 | 1.05~1.38 |
| Gastric cancer14 | 1.13 | 1.03~1.23 |
*The risk was compared to diabetic patients without diabetic complications.
TREATMENT FOR AK
Treatment for AK is necessary due to the potential progression to skin cancer. In many cases, treatment encompasses large areas to address field cancerization. Conservative and effective approaches for managing precancerous AK lesion include cryosurgery, electrodesiccation and curettage, chemotherapeutic creams (5-fluorouracil, tricholoroacetic acid, and ingenol mebutate), photodynamic therapy, or topical immunemodulators (imiquimod)15. Cryotherapy, a commonly and widely used treatment option in Korea, can cause pain and blistering, resulting in patient discomfort and potentially affecting patients’ compliance with the treatment. The efficacy of topical treatments for AK remains limited compared to conservative ablative treatments. Therefore, it is essential to further understand the mechanisms underlying AK development and explore the development of novel therapeutic drugs.
REPORTED GENETIC STUDIES ON AK
Mutational analysis of candidate genes has been recently developed by whole-exome sequencing (WES) of many tumor types with expression profiles. Recent whole-genome analyses have highlighted the presence of mutations in gene-regulatory regions and the post-transcriptional potential of epigenetic alterations and non-coding RNAs.
We conducted a search for studies on genomic mutations of AK using research methods such as polymerase chain reaction (PCR), immunofluorescence staining, expression microarray analysis, and WES published between 1994~2022 found in PubMed (Fig. 3). Herein, we present a chronological summary of the findings from these previous studies, organized according to their respective analytical methods (Table 2).
Fig. 3. A schematic diagram showing common etiologic factors involved in the development of actinic keratosis and squamous cell carcinoma as well as the time-window of possible genetic investigations, such as microarray, sequencing, and single cell seqeuncing.
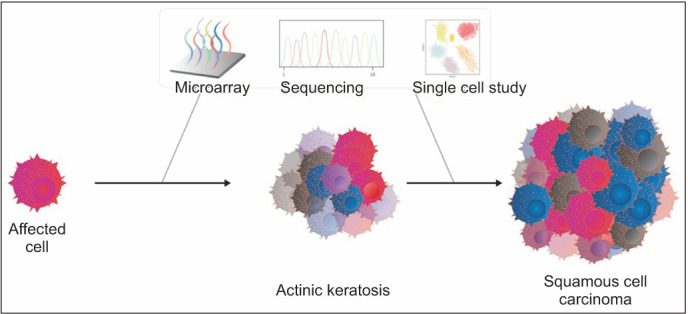
Table 2. Reported studies on genomic mutations of AK in the English literature from 1994 to 2021.
| Study | Methods | Significant results of the study |
|---|---|---|
| Nelson et al. (1994)23 | PCR and IHC analyses in FFPE AK and CSCC | TP53 mutations in both AK and CSCC |
| Rehman et al. (1996)16 | LOH analysis of AK and CSCC | High frequency of LOH on chromosome 3p, 9p, 9q, 13q, 17p, and 17q in AK compared to CSCC |
| Rehman et al. (1997)17 | LOH analysis of AK and CSCC FFPE | LOH rate was significantly lower in AKs from immune-suppressed patients compared to AKs from immune-competent patients |
| Jin et al. (2002)18 | Short-term culture and cytogenetic (karyotype) analysis of AK and CSCC | LOH on chromosome 3p13, 3q10, and 21q10 in both AK and CSCC |
| Mortier et al. (2002)19 | LOH analysis and immunohistochemistry of AK and CSCC FFPE tissue | LOH on 17p13.1 (p53) and 9p21 (p16INK4a/ARFAK) in both AK and CSCC |
| Ashton et al. (2003)2 | Whole-genome comparative genomic hybridization analysis of chromosomal aberrations on non-matched FFPE AK and CSCC | Chromosomal copy number gains on 17q, 4p, 14q, Xq, 5p, 9q, 8q, 17p, and 20q and losses on 3p, 13q, 17p, 11p, 8q, and 18p in both AK and CSCC |
| Nindl et al. (2006)24 | Expression microarray analysis on FF-matched AK and CSCC from organ-transplanted immunosuppressed patients | Significant up-regulation of nine genes (CDH1, MAP4K4, IL1RN, IL4R, NMI, GRN, RAB31, TNC, and MMP1) and down-regulation of four genes (ERCC1, APR-3, CGI39, and NKEFB) in both AK and CSCC |
| Dang et al. (2006)25 | Quantitative real-time RT-PCR | Large splice variant of Tenasin-C might serve as a useful diagnostic marker for AK and CSCC |
| Nindl et al. (2007)36 | PCR by direct sequencing | p53 mutations rather than p16(INK4a) and/or Ha-ras mutations may be an early event in the development of AK to CSCC |
| Kanellou et al. (2008)37 | real-time RT-PCR; mutation analysis and genomic instability analysis | Expression levels of CDKN2A, CDKN2B, and TP53 examined in AKs and CSCC |
| Toll et al. (2009)20 | FISH on FFPE AK and CSCC | MYC numerical aberrations in 35% of AK and 63% of CSCC |
| Padilla et al. (2010)26 | Expression microarray analysis in AK, CSCC, and normal skin sample | 89 unique genes were identified that most likely contribute to the molecular evolution of CSCC |
| Ra et al. (2011)27 | Expression microarray analysis of FFPE non-matched AK and CSCC | Nine differentially expressed genes (HOXC4, HOXC6, ROBO4, COL6A3, RPL13A, CASZ1, RTN4, NFIB, and RPS6) were identified between AK and CSCC |
| Hameetman et al. (2013)38 | Genome-wide SNP microarray of AK and matched CSCC from fresh-frozen tissue and expression microarray analysis from fresh-frozen tissue | Upregulated in CSCCs were hyperproliferation related genes (KRT6, KRT16 and KRT17) |
| Activation of downstream targets of RAS and cMYC in CSCCs and of NFκB and TNF in AKs | ||
| Lambert et al. (2014)39 | Expression microarray analysis from fresh-frozen AK and CSCC tissue from immunocompetent and immunosuppressed patients | 196 genes were differentially expressed between AK and CSCC. Gene set enrichment analysis highlighted a key role for the mitogen activated protein kinase (MAPK) pathway |
| Jacobs et al. (2015)28 | Genetic susceptibility to AK according to GWAS | IRF4, MC1R, and TYR are associated with oncogenic function and increased risk of AK |
| Chitsazzadeh et al. (2016)29 | WES, RNAseq, and microRNAseq on matched fresh AK, CSCC, and normal skin | Key pathways include cell cycle progression, mitotic roles of polo-like kinase and DNA damage checkpoint functions, as well as upstream regulators E2F1, E2F4, CDK4, TP53, RABL6 and ERBB2 |
| Albibas et al. (2018)32 | WES and target-enriched sequencing of FFPE AK and CSCC | TP53 were mutated in both AK and CSCC |
| Rodríguez-Paredes et al. (2018)31 | DNA methylation array on fresh-frozen AK and CSCC | AK methylation patterns already display classical features of cancer methylomes and are highly similar to CSCC profiles |
| García-Díez et al. (2019)21 | Expression microarray, cytogenetic, and copy number array analyses on matched AK, CSCC, and normal skin | Copy number gains on 3q and 8q, as well as a copy number loss on 9p in AK |
| Hervás-Marín et al. (2019)47 | DNA methylation array on fresh-frozen AK and CSCC | Massive non-sequential changes in DNA-methylome were observed from AK to CSCC |
| Srinivas et al. (2020)33 | RT-PCR | TERT promoter mutations in AK |
| Azimi et al. (2020)41 | Data-independent acquisition proteomics workflow; sequential window acquisition of all theoretical mass spectra | VPS45, TIMM8B, DHPS, NKTR, and GCSH were sugguested as AK markers |
| Das Mahapatra et al. (2020)40 | Representative transcripts were validated by NanoString nCounter assays using an extended cohort in AK and CSCC | Pathways related to cell cycle, apoptosis, inflammation and epidermal differentiation were enriched in CSCC |
| Zou et al. (2021)42 | Transcriptome sequencing was performed on AK, CSCC, and normal sun-exposed skin tissues | 78 significant Kyoto Encyclopedia of Genes and Genomes pathways in AK and CSCC |
| Trager et al. (2021)34 | Transcriptomic analyses of 788 genes were performed using the nanoString assay | The reversal of overexpressed oncogenes and decreased tumor- suppressor genes in the imiquimod treated AK lesions |
| Thomson et al. (2021)22 | WES on AK | Copy number losses on chromosome regions 9p, 13q, 3p, and 5q, and gains of chromosome regions 3q, 8q, 5p, and 1q in AK |
| CCDC71L and STRN in AK, while LCLAT1, HERC6, MAP3K9, and TMEM51 were more frequently altered in CSCC | ||
| Alterations in signaling pathways also differ, with immune-related signaling and TGFβ signaling significantly more mutated in CSCC | ||
| Kim et al. (2022)30 | WES on AK, CSCC | CSCC-specific genomic alterations included mutations in TP53, PIK3CA, FBXW7, and CDKN2A mutations, as well as copy number gain in MYC |
AK: actinic keratosis, CSCC: cutaneous squamous cell carcinoma, DNA: deoxyribonucleic acid, FFPE: formalin-fixed paraffin-embedded, FISH: fluorescence in situ hybridization, GWAS: genome-wide association study, LOH: loss of heterozygosity, RNA: ribonucleic acid, RT-PCR: reverse transcription polymeration chaing reaction, SNP: single nucleotide polymorphism, WES: whole-exome sequencing.
Chromosomal aberrations in AK
In the early stages of genomic research on AK, chromosomal aberrations were assessed using loss of heterozygosity (LOH) analysis. Rehman et al.16,17 reported that AK exhibited a higher frequency of LOH on chromosomes 3p, 9p, 9q, 13q, 17p, and 17q compared to CSCC. They also found that the LOH rate in AK showed a statistically significant difference depending on whether it occurred in immune-competent or immunocompromised patients. Specifically, the LOH rate was significantly lower in AKs from immune-suppressed patients compared to AKs from immune-competent patients.
In 2002, Jin et al.18 analyzed karyotypes and identified structural aberrations in both AK and CSCC, affecting 3p13, 3q10, and 21q10. These structural rearrangements are common features observed in other types of malignant tumors, indicating their association with early genetic events in malignant transformation.18 Mortier et al.19 analyzed LOH on 17p13.1 (p53) and 9p21 (p16INK4a/ARFAK) in CSCC and AK. They suggested that the progression of AK to CSCC involves deletion of the 9p21 region, which encodes the p16INK4a/ARFAK tumor-suppressor gene.
In 2003, Ashton et al.2 conducted a whole-genome comparative genomic hybridization analysis of chromosomal aberrations in AK and CSCC. They reported common frequent gains (3q, 17q, 4p, 14q, Xq, 5p, 9q, 8q, 17p, and 20q) and losses (9p, 3p, 13q, 17p, 11p, 8q, and 18p), which suggest a clonal relationship between AK and CSCC. In 2009, Toll et al.20 reported MYC cytogenetic profiling by FISH analysis indicating MYC numerical aberrations in 35% of AK and 63% of CSCC cases.
Most recently, in 2019, García-Díez et al.21 reported cytogenetic and copy number array results related to AK and CSCC using expression microarray analysis. They observed copy number gains on 3q and 8q, as well as a copy number loss on 9p, in the context of AK. Another recent study utilizing WES revealed loss of chromosome regions 9p, 13q, 3p, and 5q, and gains of chromosome regions 3q, 8q, 5p, and 1q in AK22.
Unveiling the molecular landscape of AK and cCSCC: insights from genomic studies and key mutated genes
In 1994, during the early stages of genomic studies, Nelson et al.23 utilized PCR and immunohistochemistry to report a p53 mutation in both AK and CSCC. In 2006, Nindl et al.24 examined the expression levels of 22,283 genes in normal skin versus AK and CSCC samples using microarray technology. They observed significant up-regulation of nine genes (CDH1, MAP4K4, IL1RN, IL4R, NMI, GRN, RAB31, TNC, and MMP1) and down-regulation of four genes (ERCC1, APR-3, CGI39, and NKEFB) in AK and CSCC samples compared to normal skin. Also in 2006, Dang et al.25 reported the dysregulation of Tenasin-C, an extracellular matrix protein involved in cell adhesion, migration, growth, and angiogenesis, in AK and CSCC using PCR methods. They suggested that the large splice variant of Tenasin-C might serve as a useful diagnostic marker for AK and CSCC. However, the these studies did not differentiate between AK and CSCC.
Padilla et al.26 conducted a study where they combined AK ans CSCC samples for comparision with normal samples using expression microarray analysis. They identified 89 genes that contribute to the molecular evolution of CSCC.
In 2011, Ra et al.27 performed an expression microarray analysis to compare AK and CSCC. They found 382 differentially expressed genes between CSCC and normal skin, 423 differentially expressed genes between AK and normal skin, but only nine differentially expressed genes between AK and CSCC. Among the nine genes that were differently expressed in AK compared to CSCC, four were upregulated (HOXC4, HOXC6, ROBO4, and COL6A3) and five were down-regulated (RPL13A, CASZ1, RTN4, NFIB, and RPS6). Jacob et al.28 conducted a genome-wide association study in a northwestern European population to investigate genomic susceptibility to AK. They reported that IRF4, MC1R, and TYR are associated with oncogenic function and increased risk of AK.
In recent years, next-generation sequencing techniques such as WES have been utilized in several studies investigating AKs22,29,30,31,32. These studies have identified similar mutational profiles in AK and CSCC, with common mutations found in genes such as TP53, NOTCH1, and NOTCH222,29,31,32. Thomson et al.22 conducted a WES study and found differences in significantly mutated genes between AK ans CSCC. They observed greater alterations in CCDC71L and STRN in AK, while LCLAT1, HERC6, MAP3K9, and TMEM51 were more frequently altered in CSCC. Kim et al.30 also performed a WES study and identified driver mutations, including NOTCH1 and TP53 mutations, in both AK ans CSCC. CSCC-specific genomic alterations included mutations in TP53, PIK3CA, FBXW7, and CDKN2A mutations, as well as copy number gain in MYC. However, these alterations varies among cases, suggesting that the pregression of AK to CSCC is not solely explained by a single gene or pathway. Srinivas et al.33 investigated TERT promoter mutations in AK, which were not detected after photodynamic therapy for AK. Trager et al.34 analyzed transcriptomes of AK before and after imiquimod treatment and reported the reversal of overexpressed oncogenes and decreased tumor-suppressor genes in the treated AK lesions (Table 2).
Potential mechanisoms in the progression from AK to CSCC
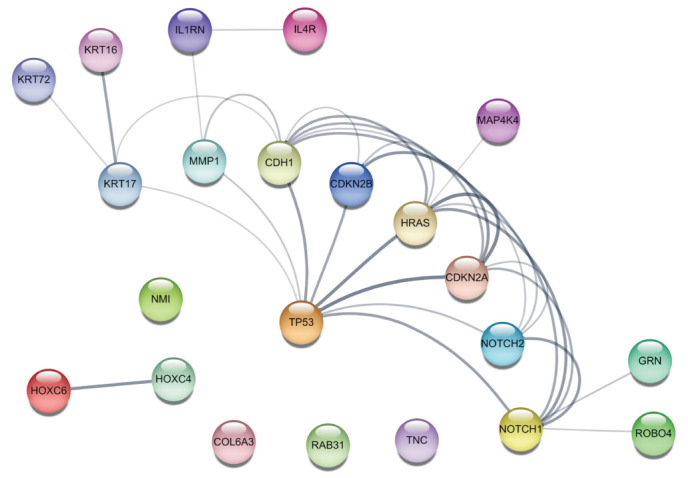
The previous studies in Table 223,24,25,26,27,28,29,30,31,32,33,34 have highlighted the significance of upregulated genes in AK (TP53, HRAS, CDKN2A, CDKN2B, NOTCH1, NOTCH2, HOXC4, HOXC6, ROBO4, COL6A3, KRT6, KRT16, KRT17, CDH1, MAP4K4, IL1RN, IL4R, NMI, GRN, RAB31, TNC, and MMP1) as key players in AK-related mutations. To better understand their functional relationships, a network analysis was performed using the Cytoscape StringApp35, and the results were depicted in Fig. 4. This visualization provides insights into the interconnectedness and potential interactions among these genes, shedding light on their collective involvement in AK development and progression. TP53 occupies a central position and exhibits extensive connections with other proteins in Fig. 4. While the pathways in the progression from normal skin to AK have not been elucidated yet, plausible pathways involved in the development from AK to CSCC can be inferred.
Fig. 4. Functional associations among the upregulated genes in actinic keratosis are visualized using Cytoscape StringApp.
1) Mutations in tumor-suppressor genes are key players in progression from AK to CSCC
Several gene mutations were revealed to play a role in progression from AK to CSCC, including CDKN2A, CDKN2B, and TP5336,37. Cyclin-dependent kinase inhibitor 2A (CDKN2A) locus is the second-most-commonly altered gene locus in human cancer after p53. The locus CDKN2A encodes two tumor-suppressor genes, p16INK4a and p14ARF, both of which are involved in negative control of cell proliferation37.
2) Nuclear factor κB1 and the tumor necrosis factor pathways are activated in both AK and CSCC
Hameetman et al.38 provided evidence that several signaling pathwyas, which are activated in CSCC, are already activated in AKs, thus confirming that AKs serve as precursor lesions of CSCCs. Theses pathways, such as nuclear factor κB1 and the tumor necrosis factor pathways, were identified through comprehensive genome-wide SNP microarray and expression microarray analyses. It is worth noting that both the nuclear factor κB1 and the tumor necrosis factor pathways are prototypical proinflammatory signaling pathways.
3) MAPK signaling pathway plays a key role in AK and CSCC
Lambert et al.39 conducted a comparison of the transcriptome between tumor cells isolated from CSCC and dyskeratotic cells from AK. The analysis confirmed the significance of the mitogen activated protein kinase (MAPK) pathway as a key biological pathway, as evidenced by the enrichment of differentially expressed genes. Notably, genes associated with the MAPK family, including JUN, FOS, MET, MAPK1, MAP2K2, and MAP3K5, were found to be enriched. Additionally, genes associated with apoptosis, such as PAK2, BAX, DEDD, DAXX, and DFFA, were also identified as differentially expressed in this context. This study sheds light on the involvement of the MAPK pathway and apoptosis-related genes in the pathogenesis of CSCCs and AKs39.
4) Epidermal differentiation, migration, and adhesion pathways are important in AK occurrence
In a comparative analysis of gene expression between AK and CSCC conducted by Hameetman et al.38, it was observed that hyperproliferation related genes (KRT6, KRT16, and KRT17) were upregulated in CSCC. Furthermore, Lambert et al.39 reported that genes involved in epidermal differentiation (KRT10, FLG, LOR, and LCE1B), migration (MMP1 and MMP10), and adhesion (FLNA, FLNB, and TIGA5) were found to be differentially expressed in CSCC compared to AK. These findings indicated the involvement of pathways related to cell cycle regulation, apoptosis, inflammation, and epidermal differentiation in the development and progression of CSCC from AK. Similarly, Das Mahapatra et al.40 utilized RNA sequencing analysis and identified alterations in gene expression associated with these pathways in AK and CSCC. These studies provide valuable insights into the molecular mechanisms underlying the pathogenesis of AK and its progression to CSCC, as well as the effects of treatment on gene expression profiles in AK.
5) Other reported pathways in development of AK
Azimi et al.41 utilized data-independent acquisition methods during mass spectrometry–based proteomic analysis to identify molecular markers in AK samples. A comparision to Bowen’s disease and CSCC, it revealed 19 AK markers, with the top five being VPS45, TIMM8B, DHPS, NKTR, and GCSH. Additionally, Reactome pathway annotations showed significant association with AK, including the inhibition of protein kinase R, regulation of the MAPK pathway, and apoptosis and DNA damage repair. In contrast, the most enriched pathways in CSCC were related to Wnt signaling and DNA damage responses41.
In a study by Zou et al.42, investigated differentially expressed genes in normal skin, AK, and CSCC and reported 78 significant Kyoto Encyclopedia of Genes and Genomes pathways in their CSCC vs. AK analysis. The upregulated differentially expressed genes in CSCC, compared to AK, were primarily associated with extracellular matrix receptor interaction, the interleukin-17 signaling pathway, cytokine-cytokine receptor interaction, the PI3K/Akt signaling pathway, focal adhesion, human papillomavirus infection, and the tumor necrosis factor signaling pathway. On the other hand, the down-regulated differentially expressed genes in CSCC, compared to AK, were predominantly linked to immunodeficiency and calcium signaling pathways. These findings provide insights into the molecular mechanisms underlying the progression from AK to CSCC and highlight potential targets for further research and therapeutic interventions42.
DNA methylation in AK
DNA methylation is a covalent epigenetic modification of cytosines within CpG dinucleotides. The modification is controlled by DNA methyltransferases and regulates cellular identity through the modulation of gene expression43. Healthy cells are characterized by the absence of DNA methylation at CpG islands which are short CpG-rich regions present in up to 60% of human promoters. Otherwise healthy cells show extensive methylation of gene bodies and repetitive regions43. Dysregulation of the normal methylome is considered as a hallmark of human cancers44. Altered DNA methylation patterns emerge early in tumorigenesis21 and can be used as biomarkers for tumor detection, diagnosis, and prognosis45,46.
DNA methylation array has also been evaluated for use with AK in some studies31,47,48. During transition form normal skin to AK and CSCC, it has been suggested that E-cadherin promoter hypermethylation might increase48, and a significant malignant potential of AK was demonstrated by observing AK methylomes with typical cancer-related features, such as CpG island promoter hypermethylation and hypomethylation of lamina-associated domains31. Hervás-Marín et al.47 investigated the DNA methylation profile in patients at different stages of disease ranging from premalignant AK to low-risk invasive and high-risk non-metastatic and metastatic CSCC. They identified a minimal methylation signature that discriminates between stages and might predict the overall survival of CSCC patients.
CONCLUSION
As shown in previous studies, AKs have genomic mutations similar to those of CSCC, including those in tumor-suppressors, oncogenes, and epigenome alterations. However, the exact pathologic mechanisms in the transitions from normal skin to AK and from AK to CSCC have not been elucidated. Although there has been enormous development in the genomic era, including next-generation sequencing, studies on AK have not been suitable for distinguishing information from dyskeratotic cells in AKs, normal keratinocytes, or CSCC cells. The WES method does not require separation of normal keratinocytes and atypical keratocytes in AK, which are limited to the basal or spinous layer depending on stage. Because atypical keratinocytes compose a small fraction of the epidermis, their allele frequency is likely to be ignored or treated as not significant in an analysis of WES data. Therefore, it is necessary to analyze the genome and transcriptome of AK in a single-cell unit. Single-cell transcriptome sequencing is a new technology for high-throughput sequencing analysis at the level of the single cell and is promising tool for exploring the heterogeneity and development of skin cancer.
In addition, AK is characterized by many inflammatory cells infiltrating the dermal layer of the skin, and the type and role of these inflammatory cells are not yet known. Due to the nature of AKs, genetic mutations should be identified in a single nuclear unit, and studies on inflammatory cells in the dermis are needed. Based on the results of genomic and transcriptomic studies, development of a new drug for AK should be pursued in the future.
Footnotes
CONFLICTS OF INTEREST: The authors have nothing to disclose.
FUNDING SOURCE: This research was supported by National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIP) (NRF-2022R1A2C1009194) and Korea Basic Science Institute (National research Facilities and Equipment Center) grant funded by the Ministry of Education. (grant No. 2021R1A6C101A445).

Young Bok Lee, currently a Professor at The Catholic University of Korea, College of Medicine, is an esteemed scholar in the field of dermatology. Lee obtained a Ph.D. from the same institution (2008~2012) and honed clinical skills during a dermatology residency at the Catholic Medical Center (2006~2010). Lee’s research interests focus on atopic dermatitis, psoriasis, and skin cancer, pivotal areas in medical dermatology. In addition to teaching and research, Lee served as a Commissioned Researcher at Seoul National University’s Genomic Medicine Institute (2021.9-2022.8), furthering the field of genomic medicine.
References
- 1.Criscione VD, Weinstock MA, Naylor MF, Luque C, Eide MJ, Bingham SF. Actinic keratoses: natural history and risk of malignant transformation in the Veterans Affairs Topical Tretinoin Chemoprevention Trial. Cancer. 2009;115:2523–2530. doi: 10.1002/cncr.24284. [DOI] [PubMed] [Google Scholar]
- 2.Ashton KJ, Weinstein SR, Maguire DJ, Griffiths LR. Chromosomal aberrations in squamous cell carcinoma and solar keratoses revealed by comparative genomic hybridization. Arch Dermatol. 2003;139:876–882. doi: 10.1001/archderm.139.7.876. [DOI] [PubMed] [Google Scholar]
- 3.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Kim YH, Han KD, Park YM, Lee JY, Park YG, et al. Incidence of actinic keratosis and risk of skin cancer in subjects with actinic keratosis: a population-based cohort study. Acta Derm Venereol. 2018;98:382–383. doi: 10.2340/00015555-2854. [DOI] [PubMed] [Google Scholar]
- 5.Warino L, Tusa M, Camacho F, Teuschler H, Fleischer AB, Jr, Feldman SR. Frequency and cost of actinic keratosis treatment. Dermatol Surg. 2006;32:1045–1049. doi: 10.1111/j.1524-4725.2006.32228.x. [DOI] [PubMed] [Google Scholar]
- 6.Perera E, McGuigan S, Sinclair R. Cost for the treatment of actinic keratosis on the rise in Australia. F1000Res. 2014;3:184. doi: 10.12688/f1000research.4671.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y, He YY. Ultraviolet radiation-induced non-melanoma skin cancer: regulation of DNA damage repair and inflammation. Genes Dis. 2014;1:188–198. doi: 10.1016/j.gendis.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schuch AP, Moreno NC, Schuch NJ, Menck CFM, Garcia CCM. Sunlight damage to cellular DNA: focus on oxidatively generated lesions. Free Radic Biol Med. 2017;107:110–124. doi: 10.1016/j.freeradbiomed.2017.01.029. [DOI] [PubMed] [Google Scholar]
- 9.Ridley AJ, Whiteside JR, McMillan TJ, Allinson SL. Cellular and sub-cellular responses to UVA in relation to carcinogenesis. Int J Radiat Biol. 2009;85:177–195. doi: 10.1080/09553000902740150. [DOI] [PubMed] [Google Scholar]
- 10.Wischermann K, Popp S, Moshir S, Scharfetter-Kochanek K, Wlaschek M, de Gruijl F, et al. UVA radiation causes DNA strand breaks, chromosomal aberrations and tumorigenic transformation in HaCaT skin keratinocytes. Oncogene. 2008;27:4269–4280. doi: 10.1038/onc.2008.70. Erratum in: Oncogene 2008;27:6790. [DOI] [PubMed] [Google Scholar]
- 11.Narayanan DL, Saladi RN, Fox JL. Ultraviolet radiation and skin cancer. Int J Dermatol. 2010;49:978–986. doi: 10.1111/j.1365-4632.2010.04474.x. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y, Lee J, Choi J, Yu D, Han K, Park YG. Actinic keratosis and diabetes complications: a nationwide population-based study in South Korea (2009-2015) Diabetes Metab. 2019;45:32–38. doi: 10.1016/j.diabet.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 13.Kim HJ, Kim YH, Yun SY, Yu DS, Lee YB. Association of actinic keratosis with rheumatoid arthritis and psoriasis: a nationwide population-based study in Korea. Acta Derm Venereol. 2021;101:adv00510. doi: 10.2340/00015555-3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee YB, Lee JH, Kim YH, Seo JM, Yu DS, Park YG, Han KD. Positive association between actinic keratosis and internal malignancies: a nationwide population-based cohort study. Sci Rep. 2021;11:19769. doi: 10.1038/s41598-021-99225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Worley B, Harikumar V, Reynolds K, Dirr MA, Christensen RE, Anvery N, et al. Treatment of actinic keratosis: a systematic review. Arch Dermatol Res. 2023;315:1099–1108. doi: 10.1007/s00403-022-02490-5. [DOI] [PubMed] [Google Scholar]
- 16.Rehman I, Takata M, Wu YY, Rees JL. Genetic change in actinic keratoses. Oncogene. 1996;12:2483–2490. [PubMed] [Google Scholar]
- 17.Rehman I, Quinn AG, Takata M, Taylor AE, Rees JL. Low frequency of allelic loss in skin tumours from immunosuppressed individuals. Br J Cancer. 1997;76:757–759. doi: 10.1038/bjc.1997.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin Y, Jin C, Salemark L, Wennerberg J, Persson B, Jonsson N. Clonal chromosome abnormalities in premalignant lesions of the skin. Cancer Genet Cytogenet. 2002;136:48–52. doi: 10.1016/s0165-4608(01)00517-9. [DOI] [PubMed] [Google Scholar]
- 19.Mortier L, Marchetti P, Delaporte E, Martin de Lassalle E, Thomas P, Piette F, et al. Progression of actinic keratosis to squamous cell carcinoma of the skin correlates with deletion of the 9p21 region encoding the p16(INK4a) tumor suppressor. Cancer Lett. 2002;176:205–214. doi: 10.1016/s0304-3835(01)00757-1. [DOI] [PubMed] [Google Scholar]
- 20.Toll A, Salgado R, Yébenes M, Martín-Ezquerra G, Gilaberte M, Baró T, et al. MYC gene numerical aberrations in actinic keratosis and cutaneous squamous cell carcinoma. Br J Dermatol. 2009;161:1112–1118. doi: 10.1111/j.1365-2133.2009.09351.x. [DOI] [PubMed] [Google Scholar]
- 21.García-Díez I, Hernández-Muñoz I, Hernández-Ruiz E, Nonell L, Puigdecanet E, Bódalo-Torruella M, et al. Transcriptome and cytogenetic profiling analysis of matched in situ/invasive cutaneous squamous cell carcinomas from immunocompetent patients. Genes Chromosomes Cancer. 2019;58:164–174. doi: 10.1002/gcc.22712. [DOI] [PubMed] [Google Scholar]
- 22.Thomson J, Bewicke-Copley F, Anene CA, Gulati A, Nagano A, Purdie K, et al. The genomic landscape of actinic keratosis. J Invest Dermatol. 2021;141:1664–1674.e7. doi: 10.1016/j.jid.2020.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nelson MA, Einspahr JG, Alberts DS, Balfour CA, Wymer JA, Welch KL, et al. Analysis of the p53 gene in human precancerous actinic keratosis lesions and squamous cell cancers. Cancer Lett. 1994;85:23–29. doi: 10.1016/0304-3835(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 24.Nindl I, Dang C, Forschner T, Kuban RJ, Meyer T, Sterry W, et al. Identification of differentially expressed genes in cutaneous squamous cell carcinoma by microarray expression profiling. Mol Cancer. 2006;5:30. doi: 10.1186/1476-4598-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dang C, Gottschling M, Roewert J, Forschner T, Stockfleth E, Nindl I. Tenascin-C patterns and splice variants in actinic keratosis and cutaneous squamous cell carcinoma. Br J Dermatol. 2006;155:763–770. doi: 10.1111/j.1365-2133.2006.07401.x. [DOI] [PubMed] [Google Scholar]
- 26.Padilla RS, Sebastian S, Jiang Z, Nindl I, Larson R. Gene expression patterns of normal human skin, actinic keratosis, and squamous cell carcinoma: a spectrum of disease progression. Arch Dermatol. 2010;146:288–293. doi: 10.1001/archdermatol.2009.378. [DOI] [PubMed] [Google Scholar]
- 27.Ra SH, Li X, Binder S. Molecular discrimination of cutaneous squamous cell carcinoma from actinic keratosis and normal skin. Mod Pathol. 2011;24:963–973. doi: 10.1038/modpathol.2011.39. [DOI] [PubMed] [Google Scholar]
- 28.Jacobs LC, Liu F, Pardo LM, Hofman A, Uitterlinden AG, Kayser M, et al. IRF4, MC1R and TYR genes are risk factors for actinic keratosis independent of skin color. Hum Mol Genet. 2015;24:3296–3303. doi: 10.1093/hmg/ddv076. [DOI] [PubMed] [Google Scholar]
- 29.Chitsazzadeh V, Coarfa C, Drummond JA, Nguyen T, Joseph A, Chilukuri S, et al. Cross-species identification of genomic drivers of squamous cell carcinoma development across preneoplastic intermediates. Nat Commun. 2016;7:12601. doi: 10.1038/ncomms12601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim YS, Shin S, Jung SH, Park YM, Park GS, Lee SH, et al. Genomic progression of precancerous actinic keratosis to squamous cell carcinoma. J Invest Dermatol. 2022;142(3 Pt A):528–538.e8. doi: 10.1016/j.jid.2021.07.172. [DOI] [PubMed] [Google Scholar]
- 31.Rodríguez-Paredes M, Bormann F, Raddatz G, Gutekunst J, Lucena-Porcel C, Köhler F, et al. Methylation profiling identifies two subclasses of squamous cell carcinoma related to distinct cells of origin. Nat Commun. 2018;9:577. doi: 10.1038/s41467-018-03025-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Albibas AA, Rose-Zerilli MJJ, Lai C, Pengelly RJ, Lockett GA, Theaker J, et al. Subclonal evolution of cancer-related gene mutations in p53 immunopositive patches in human skin. J Invest Dermatol. 2018;138:189–198. doi: 10.1016/j.jid.2017.07.844. [DOI] [PubMed] [Google Scholar]
- 33.Srinivas N, Neittaanmäki N, Heidenreich B, Rachakonda S, Karppinen TT, Grönroos M, et al. TERT promoter mutations in actinic keratosis before and after treatment. Int J Cancer. 2020;146:2932–2934. doi: 10.1002/ijc.32878. [DOI] [PubMed] [Google Scholar]
- 34.Trager MH, Rizk E, Rose S, Zhu K, Lau B, Fullerton BT, et al. Transcriptomic analysis identifies differences in gene expression in actinic keratoses after treatment with imiquimod and between responders and non responders. Sci Rep. 2021;11:8775. doi: 10.1038/s41598-021-88424-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape stringApp: network analysis and visualization of proteomics data. J Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nindl I, Gottschling M, Krawtchenko N, Lehmann MD, Röwert-Huber J, Eberle J, et al. Low prevalence of p53, p16(INK4a) and Haras tumour-specific mutations in low-graded actinic keratosis. Br J Dermatol. 2007;156 Suppl 3:34–39. doi: 10.1111/j.1365-2133.2007.07857.x. [DOI] [PubMed] [Google Scholar]
- 37.Kanellou P, Zaravinos A, Zioga M, Stratigos A, Baritaki S, Soufla G, et al. Genomic instability, mutations and expression analysis of the tumour suppressor genes p14(ARF), p15(INK4b), p16(INK4a) and p53 in actinic keratosis. Cancer Lett. 2008;264:145–161. doi: 10.1016/j.canlet.2008.01.042. [DOI] [PubMed] [Google Scholar]
- 38.Hameetman L, Commandeur S, Bavinck JN, Wisgerhof HC, de Gruijl FR, Willemze R, et al. Molecular profiling of cutaneous squamous cell carcinomas and actinic keratoses from organ transplant recipients. BMC Cancer. 2013;13:58. doi: 10.1186/1471-2407-13-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lambert SR, Mladkova N, Gulati A, Hamoudi R, Purdie K, Cerio R, et al. Key differences identified between actinic keratosis and cutaneous squamous cell carcinoma by transcriptome profiling. Br J Cancer. 2014;110:520–529. doi: 10.1038/bjc.2013.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Das Mahapatra K, Pasquali L, Søndergaard JN, Lapins J, Nemeth IB, Baltás E, et al. A comprehensive analysis of coding and non-coding transcriptomic changes in cutaneous squamous cell carcinoma. Sci Rep. 2020;10:3637. doi: 10.1038/s41598-020-59660-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Azimi A, Yang P, Ali M, Howard V, Mann GJ, Kaufman KL, et al. Data independent acquisition proteomic analysis can discriminate between actinic keratosis, Bowen's disease, and cutaneous squamous cell carcinoma. J Invest Dermatol. 2020;140:212–222.e11. doi: 10.1016/j.jid.2019.06.128. [DOI] [PubMed] [Google Scholar]
- 42.Zou DD, Xu D, Deng YY, Wu WJ, Zhang J, Huang L, et al. Identification of key genes in cutaneous squamous cell carcinoma: a transcriptome sequencing and bioinformatics profiling study. Ann Transl Med. 2021;9:1497. doi: 10.21037/atm-21-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baubec T, Schübeler D. Genomic patterns and context specific interpretation of DNA methylation. Curr Opin Genet Dev. 2014;25:85–92. doi: 10.1016/j.gde.2013.11.015. [DOI] [PubMed] [Google Scholar]
- 44.Baylin SB, Jones PA. A decade of exploring the cancer epigenome-biological and translational implications. Nat Rev Cancer. 2011;11:726–734. doi: 10.1038/nrc3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stirzaker C, Taberlay PC, Statham AL, Clark SJ. Mining cancer methylomes: prospects and challenges. Trends Genet. 2014;30:75–84. doi: 10.1016/j.tig.2013.11.004. [DOI] [PubMed] [Google Scholar]
- 46.Costa-Pinheiro P, Montezuma D, Henrique R, Jerónimo C. Diagnostic and prognostic epigenetic biomarkers in cancer. Epigenomics. 2015;7:1003–1015. doi: 10.2217/epi.15.56. [DOI] [PubMed] [Google Scholar]
- 47.Hervás-Marín D, Higgins F, Sanmartín O, López-Guerrero JA, Bañó MC, Igual JC, et al. Genome wide DNA methylation profiling identifies specific epigenetic features in high-risk cutaneous squamous cell carcinoma. PLoS One. 2019;14:e0223341. doi: 10.1371/journal.pone.0223341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiles MC, Ai L, Zuo C, Fan CY, Smoller BR. E-cadherin promoter hypermethylation in preneoplastic and neoplastic skin lesions. Mod Pathol. 2003;16:1014–1018. doi: 10.1097/01.MP.0000089779.35435.9D. [DOI] [PubMed] [Google Scholar]