

Abstract
The integration of multi‐omics information (e.g., epigenetics and transcriptomics) can be useful for interpreting findings from genome‐wide association studies (GWAS). It has been suggested that multi‐omics could circumvent or greatly reduce the need to increase GWAS sample sizes for novel variant discovery. We tested whether incorporating multi‐omics information in earlier and smaller‐sized GWAS boosts true‐positive discovery of genes that were later revealed by larger GWAS of the same/similar traits. We applied 10 different analytic approaches to integrating multi‐omics data from 12 sources (e.g., Genotype‐Tissue Expression project) to test whether earlier and smaller GWAS of 4 brain‐related traits (alcohol use disorder/problematic alcohol use, major depression/depression, schizophrenia, and intracranial volume/brain volume) could detect genes that were revealed by a later and larger GWAS. Multi‐omics data did not reliably identify novel genes in earlier less‐powered GWAS (PPV <0.2; 80% false‐positive associations). Machine learning predictions marginally increased the number of identified novel genes, correctly identifying 1–8 additional genes, but only for well‐powered early GWAS of highly heritable traits (i.e., intracranial volume and schizophrenia). Although multi‐omics, particularly positional mapping (i.e., fastBAT, MAGMA, and H‐MAGMA), can help to prioritize genes within genome‐wide significant loci (PPVs = 0.5–1.0) and translate them into information about disease biology, it does not reliably increase novel gene discovery in brain‐related GWAS. To increase power for discovery of novel genes and loci, increasing sample size is required.
Keywords: genetics, GWAS, human, multi‐omics, sample size, transcriptomics
Multi‐omics cannot reliably identify true‐positive novel genes. Genes that are not proximal to a GWAS‐significant locus but are identified by multi‐omics methods in the smaller GWAS are not likely to contain a GWAS‐significant locus in the larger GWAS. (A) The positive predictive value and sensitivity for each method, and for all methods combined (Any). (B) Performance for increasing agreement between multi‐omics methods. (C) Performance of machine‐learning (ML) in the larger GWAS (ML was trained in the smaller GWAS). Points represent the estimates, while horizonal bars reflect the 95% CI. AUD/PAU = Alcohol use disorder/Problematic alcohol use; MDD/DEP = Major depressive disorder/Depression; SCZ = Schizophrenia; ICV/BV = Intracranial volume/Brain volme.
1. INTRODUCTION
Genome‐wide association studies (GWAS) have proven to be a uniquely effective tool for investigating the genetic architecture of complex traits, providing insights into the polygenic contributions of common variants, and yielding replicable genetic signals, 1 , 2 , 3 , 4 , 5 in contrast to many candidate gene studies. However, for complex genetic traits, particularly for psychopathology, the number of cases needed for discovery is high, and depends on the heritability, polygenicity, heterogeneity, diagnostic accuracy, and prevalence of the trait. 3 , 4 , 5 , 6 Schizophrenia, a highly heritable disorder, has witnessed linear increases in GWAS discoveries once a critical threshold was reached 5 (e.g., from 7 loci for 17,836 cases 7 to 270 loci for 67,390 cases 8 ). Depression required a much larger sample size to reach the inflection point. 5 Discoveries for substance use disorders have lagged, with evidence of greater polygenicity. Despite hundreds of loci for smoking behavior phenotypes, 9 only 5 genome‐wide significant loci have been identified for nicotine dependence. 10 A broad index of problematic alcohol use 2 yielded 29 loci, but findings are still quite limited for other drugs: only 10 loci for opioid use disorder (31,473 cases) 11 and 2 for cannabis use disorder (14,080 cases). 12 In parallel to GWAS, there has been an explosion in multi‐omics data (e.g., gene expression, chromatin structure) which are now available to interrogate GWAS findings to reveal underlying biology and potential therapeutics. 13 , 14 For some traits, expression data can also be drawn from model organisms in experimentally controlled exposure and behavioral paradigms. 15 , 16 As greater evidence arises for genome‐wide significant signals to be enriched in regulatory regions, these multi‐omics data have proved to be valuable in gene prioritization and interpretation. 15 , 16
Some have speculated that multi‐omics could serve as a substitute for additional sample size, and “recover” true signal from smaller GWAS by increasing support for signals that do not meet criteria for genome‐wide significance. 17 , 18 , 19 This is an appealing proposition, because it would reduce the need (and expense) for collection and phenotyping of larger samples. Some early work using expression‐based methods (e.g., transcriptome‐wise association analyses – TWAS) found that they can identify genes that would eventually achieve genome‐wide significance in subsequent, larger GWAS, 20 , 21 although these studies did not evaluate what proportion of all novel genes were first identified by TWAS.
To date, there have not been empirical tests of whether multi‐omics methods can sift through the nominally significant signals of a smaller GWAS to pinpoint future true positives, nor of the ratio of false to true positives from such analyses. Therefore, we tested the hypothesis that the application of existing omics data and methods to a smaller‐sized GWAS will yield additional “true positive” discoveries that would be found in the next, larger GWAS. We selected four brain‐related phenotypes for testing: (a) Alcohol use disorder/Problematic alcohol use (AUD/PAU), representing either diagnostic alcohol use disorder or an amalgam of diagnostic AUD and a non‐diagnostic screener for problem drinking that is highly correlated with AUD; (b) Schizophrenia (SCZ), one of the earliest psychiatric disorders with many genome‐wide significant findings that have increased with increasing sample size, making it an ideal reference trait for this test; (c) Major depression/depression (MDD/DEP), representing either diagnostic major depressive disorder or broad depression/mood phenotypes that are highly correlated with MDD/DEP, which is more common and less heritable than SCZ, clinically heterogeneous and highly polygenic, and in that way similar to AUD/PAU; and (d) Intracranial volume/brain volume (ICV/BV), a highly heritable phenotype, which we selected to assess whether brain‐derived omics data would work in this highly related brain phenotype.
We addressed three primary hypotheses: (a) does incorporation of multiple omics (multi‐omics) sources to annotate the findings of the smaller GWAS recapitulate genes identified in the subsequent GWAS of the same or genetically closely‐related trait (i.e., genetic correlation >0.7), while minimizing false positives? (b) If multi‐omics data can recover novel genes, which omics data and methods produce the most reliable predictions? and (c) does the multivariate consideration of these different data sources improve prediction? To test these, we examined sequential pairs of GWAS for each of the 4 traits, where the latter GWAS that included more subjects increased the number of genome‐wide significant findings. Across analyses, we used genes, rather than variants, as the unit of discovery, because multi‐omics data are gene‐focused. We used a combination of omics data sources (including cross‐species data) and statistical methods to annotate each GWAS. Key criteria for our analyses were the positive predictive value (PPV, the proportion of genes identified in a larger GWAS relative to all genes predicted to be relevant applying omics approaches to the prior, smaller GWAS), and the sensitivity (the proportion of genes identified by both the larger and smaller GWAS relative to all genes identified in the larger GWAS).
2. METHODS
2.1. GWAS summary statistics
For each trait, analyses started with summary statistics from the smaller GWAS of the trait. We applied a series of bioinformatic methods to identify additional genes, and compared the results to a subsequent GWAS of the trait with a larger sample size. Analyses focused on four traits: alcohol use disorder/problematic alcohol use (AUD/PAU), 1 , 2 major depressive disorder/depression (MDD/DEP), 22 , 23 schizophrenia (SCZ), 8 , 24 and intracranial volume/brain volume (ICV/BV). 25 See Table 1 for study details.
TABLE 1.
Data sources. (A) Publicly available summary statistics for each trait used in post‐GWAS multi‐omics analyses. * = Sample size omits data that is not publicly available. The number of significant risk loci (p < 0.05 × 10−8) was calculated in FUMA, using publicly available summary statistics. (B) Publicly available data used in post‐GWAS multi‐omics methods (see Table 2). (C) Published data on genes which are differentially expressed in the brain, comparing participants with each disorder to controls. (D) Published data on genes which are differentially expressed in mouse models of each disorder. (E) Published rodent gene set on genes implicated in rodent models of alcohol use.
| A: GWAS summary statistics | |||
|---|---|---|---|
| Trait | Study | Sample size | # Genome‐wide risk loci |
| Alcohol Use Disorder | Kranzler et al. 2019 1 |
Ncases = 35,105 Ncontrol = 172,697 |
11 |
| Problematic Alcohol Use | Zhou et al. 2020 2 | Neffective = 300,789 | 29 |
| Major Depressive Disorder | Howard et al. 2019 23 |
Ncases = 170,756* Ncontrol = 329,443* |
51 |
| Depression | Levey et al. 2021 22 |
Ncases = 320,212* Ncontrol = 581,929* |
100 |
| Schizophrenia | Ripke et al. 2014 24 |
Ncases = 36,989 Ncontrol = 113,075 |
96 |
| Schizophrenia | Ripke et al. 2020 8 |
Ncases = 67,390 Ncontrol = 161,405 |
242 |
| Intracranial Volume (UK Biobank only) | Jansen et al. 2020 25 | N = 17,062 | 8 |
| Brain Volume | Jansen et al. 2020 25 | N = 47,316 | 18 |
| B: Post‐GWAS multi‐omics data sources | |||
|---|---|---|---|
| Name/type | Study | Sample size | Notes |
| GTEx – brain RNA | GTEx Consortium, 2017 36 | 80–154 | 13 brain regions |
| PsychEncode – brain RNA | Wang et al. 2018 37 | 1387 | 1 region |
| Brain‐eMeta – brain RNA | Qi et al., 2018 38 | 1194 | Meta analysis across regions and samples |
| INTERVAL – plasma protein levels | Sun et al., 2018 39 | 3301 | ‐ |
| C: Human post‐mortem brain gene expression | |||
|---|---|---|---|
| Trait | Study | Case/Control | # Brain regions |
| Alcohol Use Disorder | Rao et al., 2020 40 | 30/30 | 4 |
| Major Depressive Disorder | Wu et al., 2021 41 | 101/96 | 7 |
| Schizophrenia | Collado‐Torres et al., 2019 42 | 286/265 | 2 |
| D: Rodent post‐mortem brain gene expression | |||
|---|---|---|---|
| Trait | Study | # Models | # Brain regions |
| Alcohol Use Disorder (genetic model of binge drinking) | Ferguson et al., 2019 43 | 1 | 7 |
| Major Depressive Disorder (chronic stress) | Scarpa et al., 2020 44 | 3 | 2 |
| Schizophrenia (developmental disruption) | Donegan et al., 2020 45 | 3 | 1 |
| E: Rodent gene set | |||
|---|---|---|---|
| Trait | Study | Method | ‐ |
| Alcohol Use Disorder (multiple alcohol outcomes) | Huggett et al., 2021 15 | GeneWeaver | ‐ |
2.2. Post‐GWAS multi‐omics methods
Eight post‐GWAS multi‐omics methods (Table 2) were used to identify genes associated with each trait, based on the results of each GWAS: the max‐SNP p‐value, 26 MAGMA, 27 H‐MAGMA, 28 fastBAT, 29 DEPICT, 30 FUSION, 21 , 31 S‐MultiXcan, 32 and SMR. 33 These methods were selected because they are among the most widely‐used and are representative of the major method types (i.e., positional: MAGMA, H‐MAGMA, and fastBAT; and expression‐based: FUSION, S‐MultiXcan, SMR, and DEPICT). These methods were applied to both the larger and smaller GWAS of each trait. Analyses focused on gene‐level associations, to incorporate information across methods and species. MAGMA was applied using the FUMA web platform, 34 fastBAT, DEPICT, S‐MultiXcan, and SMR were applied using the Complex‐Traits Genetics Virtual Lab. 35 Among the multi‐omics methods used, FUSION and S‐MultiXcan used RNA expression in the brain from the GTEx Consortium, 36 and SMR used RNA expression in the brain from PsychENCODE 37 and the Brain‐eMeta 38 study, as well as plasma protein expression from INTERVAL 39 (Table 1B).
TABLE 2.
Post‐GWAS multi‐omics methods. Methods used to augment GWAS summary statistics and identify gene‐level associations.
| Method | Description |
|---|---|
| SNP‐based | Gene is assigned the p‐value of the most significant SNP within 10 kb of its boundaries. 26 |
| MAGMA | Set‐based analysis, regression‐based p‐values. 27 |
| H‐MAGMA | Incorporates chromatin interaction profiles for gene boundaries 28 . |
| fastBAT | Fast set‐based analysis, simulation‐based p‐values. 29 |
| DEPICT | Predicts gene function to prioritize the most likely causal genes. 30 |
| FUSION | Transcriptome‐wide association study (TWAS) using GTEx gene expression to impute gene expression. Integrates across multiple tissues using an aggregated Cauchy association test. 21 , 31 |
| S‐MultiXcan | Transcriptome‐wide association study (TWAS) using GTEx gene expression to impute gene expression. Integrates across multiple tissues using multivariate regression. 32 |
| SMR | Summary mendelian randomization. Tests for mediation of genetic effects by gene or protein expression, using Brain‐eMeta, PsychEncode, and INTERVAL. 33 |
2.3. Additional multi‐omics data
Additional multi‐omics data were drawn from published data sets: genes differentially expressed in human brain tissue (Table 1C) 40 , 41 , 42 and genes differentially expressed in brains of mouse models of each disorder 43 , 44 , 45 (Table 1D). For AUD/PAU, data came from alcohol‐naïve mice from a line bred for binge‐drinking‐like behavior, High Drinking in the Dark mice. 43 Results from 7 tissues were integrated using an aggregated Cauchy association test, 46 which is also used by the FUSION TWAS method to integrate across tissues. 31 An additional list of genes from rodent studies enriched for the heritability of alcohol use disorder was included as an additional method. 15 MDD/DEP associated genes came from a study examining differentially expressed genes in two brain tissues across three mouse chronic stress models. 44 Results within each model were similarly combined across tissues using an aggregated Cauchy association test. As results were only reported for nominally significant genes, precluding a meta‐analysis combining models, the minimum p‐value across the three models was assigned as a gene‐level p‐value. SCZ‐associated genes came from a study examining differentially expressed genes in one brain tissue across three mouse models of developmental disruption. 45 Results were combined across models using Fisher's combined probability test. 47
2.4. Predictive value of significant genes
Multi‐omics methods were applied to statistics‐level data from the smaller GWAS. Genes identified by each multi‐omics method were defined as those surviving false discovery rate correction within each method. Genes that were either not measured in a data source (e.g., RNA expression was too low to measure), or unreported (i.e., some data sources only reported genes that were at least nominally‐significant) were marked as showing no association for that source. Analyses for novel gene discovery examined genes that were not proximal to a genome‐wide significant locus in the smaller GWAS but were identified by individual multi‐omics methods or combinations of methods. These analyses tested whether these genes were identified as genome‐wide significant in the larger GWAS. Analyses for prioritization examined genes identified by proximity to a genome‐wide significant locus in the smaller GWAS. These analyses tested if these genes were more likely to be identified as genome‐wide significant in the larger GWAS if they were also identified by individual multi‐omics methods or combinations of methods. For each trait, the target set of genes was defined as genes close (i.e., ± 10 kb) to a genome‐wide significant locus (p < 5 × 10−8) in the larger of the two paired GWAS. Supplemental post‐hoc analyses expanded the target set of genes to those within ±20, ±50, and ±100 kb.
Two primary performance metrics were used, the positive predictive value (PPV) and sensitivity. 48 The PPV, which ranges from 0–1, reflects the probability that a positive prediction reflects a true genetic signal (i.e., the ratio of the true positive rate to the sum of the true and false positive rates). PPV reflects what proportion of target genes identified in the later, larger, GWAS, are genes predicted to be relevant using omics approaches in the prior, smaller GWAS. For example, a PPV of 0.75 would mean that 75% of the genes identified by multi‐omics data in the smaller GWAS are positionally significant in the larger GWAS. Sensitivity, which also ranges from 0–1, reflects the proportion of the positionally significant genes in the larger GWAS captured by the test (the ratio of the true positive rate to the sum of the true positive and false negative rates). That is, of the genes positionally identified in the later, larger, GWAS, what proportion are found using multi‐omics data in the prior, smaller, GWAS? For example, a sensitivity of 0.5 would mean that multi‐omics identified 50% of all the positionally significant genes in the later GWAS. Thus, an ideal test would have both a high PPV and a high sensitivity (i.e., the test captures the majority of the significant genes in the larger GWAS, with very few incorrect predictions). A test with a high PPV and low sensitivity misses the majority of the significant genes in the larger GWAS, but the few that are identified are mostly correct predictions. Conversely, a test with high sensitivity and a low PPV captures the majority of the significant genes in the larger GWAS, but at the cost of a high number of incorrect predictions. All analyses were conducted in R 49 and performance metrics were computed using the epiR package. 48
2.5. Multivariate machine learning gene prioritization
2.5.1. Training procedure
As the predictions of individual methods are not perfectly correlated (Figure S1), and methods may only partially contribute to prediction (i.e., may need to be weighted), a multivariate combination of methods might improve performance. Models were trained, using only the earlier GWAS, to predict which genes were positionally linked to genome‐wide significant SNPs using data from multi‐omics methods. Summary data in the smaller GWAS were split into a training and testing component. We estimated PPV and sensitivity using the hold‐out (testing) set, and then subsequently in the larger future GWAS. For example, in MDD/DEP, ML models were trained using half the chromosomes from Howard et al. 23 These models were then tested in the remaining chromosomes of Howard et al., which yielded a predicted probability of how likely a gene is to be proximal to a genome‐wide significant locus, based on multi‐omics data. A probability threshold was selected by examining PPV and sensitivity distributions in Howard et al. We then took the genes which surpassed the identified threshold and assessed the PPV and sensitivity of those predictions in the larger Levey et al. 22 GWAS.
2.5.2. ML algorithms
GWAS of psychiatric disorders and brain‐related traits are currently only powered to find small proportions of variants associated with these traits. Standard ML classifiers may thus not be appropriate, as these methods perform best when data are evenly balanced (i.e., when 50% of genes are significant). Therefore, we used a combination of up‐sampling, bootstrapping, bagging, and ensemble learning in two algorithms. Missing data across methods was handled with a median impute (bagimpute and K‐means impute were unreliable due to high missingness). The first method was AdaBoosting 50 ; a tree ensemble method that was originally developed to predict rare outcomes. 51 AdaBoosted trees were bagged with up‐sampling minority cases. The second method was model‐average neural network (ANN) 52 ; a series of simple functions which attempt to capture patterns in the data. The model ANN averages across many runs to develop a prediction. An ensemble prediction was then generated by averaging the predictions from the two approaches. All machine learning models were trained with 3‐fold cross‐validation with up‐sampling of genes containing GWAS‐significant SNPs. This cross‐validation and training steps were repeated 10 times (bootstrapping with replacement), increasing the proportion of genes containing GWAS‐significant SNPs in the training data.
Finally, we determined feature importance to identify which multi‐omics methods most contributed to gene identification. For ANN, an ROC curve was generated for each variable using sensitivity and specificity, and the probabilities were cut off at a series of (random) points. Using the trapezoidal rule, the area under the ROC was calculated for each multi‐omics method and used as a measure of feature importance. 53 When using AdaBoost we used tree‐specific feature importance, computed by summing how much the model improved each time it used a multi‐omics method. 54
3. RESULTS
Correlations between gene sets identified by different multi‐omics approaches were quite low (median = 0.12, range = 0–0.7; Figure S1). Related methods identified more similar gene sets, though correlations remained moderate (positional: median = 0.4, range = 0.27 to 0.63; expression‐based: median = 0.38, range = 0.22 to 0.72). Genes identified by methods using GWAS summary statistics showed low overlap with genes identified by post‐mortem gene expression studies in humans and rodents and with rodent‐based gene set analyses (range = −0.01 to 0.02).
For novel gene discovery (i.e., genes that are distal from GWAS significant loci in the smaller GWAS, but proximal to significant loci in the larger GWAS; Figure 1), no method achieved a PPV greater than 0.2 for any trait (i.e., 80% of genes identified by multi‐omics were not proximal to a genome‐wide significant locus in the larger GWAS) with the exception of fastBAT for AUD/PAU, which correctly identified a single gene (ADH1A), yielding a PPV of 1.0 (Figure 1A). Similarly, when examining genes identified by multiple methods, none surpassed a PPV of 0.22 for any trait (Figure 1B). Performance improved only a little in post‐hoc analyses that broadened the definition of ‘proximal’ in the larger GWAS to genes that are within 20 kb, 50 kb, or 100 kb of a significant locus (Figure S2), but the average performance of individual methods did not surpass a PPV of 0.12 and the average performance when methods were combined did not surpass a PPV of 0.25.
FIGURE 1.
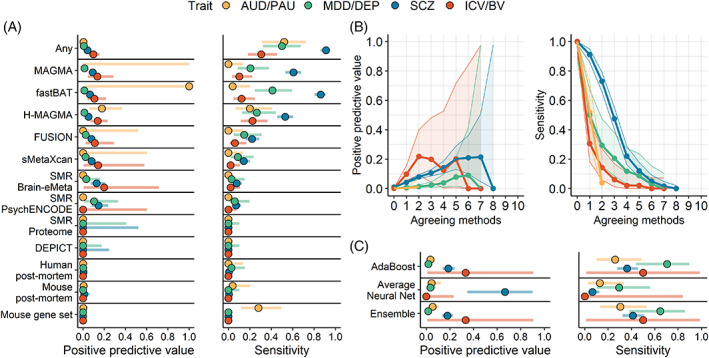
Multi‐omics cannot reliably identify true‐positive novel genes. Genes that are not proximal to a GWAS‐significant locus but are identified by multi‐omics methods in the smaller GWAS are not likely to contain a GWAS‐significant locus in the larger GWAS. (A) The positive predictive value and sensitivity for each method, and for all methods combined (Any). (B) Performance for increasing agreement between multi‐omics methods. (C) Performance of machine‐learning (ML) in the larger GWAS (ML was trained in the smaller GWAS). Points represent the estimates, while horizonal bars reflect the 95% CI. AUD/PAU = Alcohol use disorder/Problematic alcohol use; MDD/DEP = Major depressive disorder/Depression; SCZ = Schizophrenia; ICV/BV = Intracranial volume/Brain volume.
For gene prioritization (i.e., genes that are proximal to GWAS significant loci in the smaller GWAS; Figure 2), genes that were identified both by proximity to a genome‐wide significant locus and by multi‐omics were more likely to be significant in the larger GWAS than genes only identified by a genome‐wide significant locus (Figure 2A). However, no method had both a high PPV and a high sensitivity for any trait. Expression‐based methods had a slightly higher PPV than position‐based methods, but position‐based methods attained a much greater sensitivity for all traits. Analyses examining agreement between methods found that genes identified by a larger number of methods (regardless of the specific method) had a higher PPV across all traits, although with a lower sensitivity (Figure 2B). That is, when more methods agreed, the gene was more likely to be positionally significant in the larger GWAS. However, the low sensitivity indicates that this approach misses the majority of findings in the larger GWAS.
FIGURE 2.
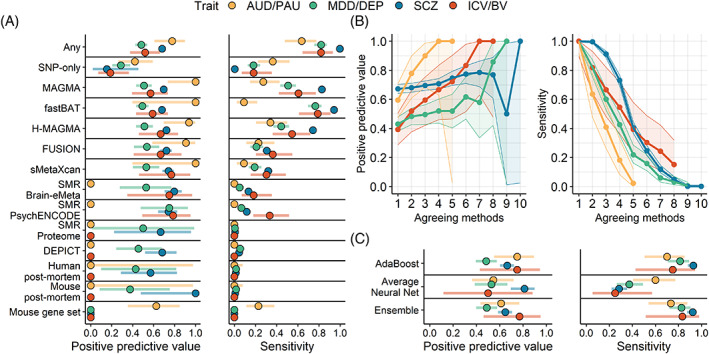
Multi‐omics is useful for prioritization of GWAS‐significant genes. Genes that are proximal to a GWAS‐significant locus and are identified by multi‐omics methods in the smaller GWAS are likely to contain a GWAS‐significant locus in the larger GWAS. (A) The positive predictive value and sensitivity for each method, and for all methods combined (Any). ‘SNP‐only’ = the gene is proximal to a GWAS‐significant locus, but is not identified by multi‐omics. (B) Performance for increasing agreement between multi‐omics methods. (C) Performance of machine‐learning (ML) in the larger GWAS (ML was trained in the smaller GWAS). Points represent the estimates, while horizonal bars reflect the 95% CI. AUD/PAU = Alcohol use disorder/Problematic alcohol use; MDD/DEP = Major depressive disorder/Depression; SCZ = Schizophrenia; ICV/BV = Intracranial volume/Brain volume.
Across both novel gene discovery and gene prioritization analyses, genes identified by more methods in the smaller GWAS tended to have a higher ‐log10(p) value in the larger GWAS and similarly tended to be identified by more methods in the larger GWAS (Supplemental Results; Figures S3 & S4). Supplemental analyses examined performance across all possible combinations of multi‐omics methods, as well as for different definitions of a ‘significant’ gene in the larger GWAS. No combination performed best across all traits, and the pattern of associations for different definitions of ‘significant’ (i.e., p < 5 × 10−6 or identified by multi‐omics in the larger GWAS) remained similar (Supplemental Results; Figures S5 & S6).
3.1. Machine learning
Model predictions are probability scores (i.e., the probability that a gene contains a genome‐wide significant SNP, based on multi‐omics). The performance (PPV and sensitivity) of different cut‐offs was evaluated in the hold‐out data from the smaller GWAS (Figure S7). A probability cut‐off of 75% was selected for AdaBoost, 90% for the model‐average neural net, and 50% for the ensemble of the two. The performance of each gene set was then assessed in the later, larger GWAS (Table S1). ML performance in the held‐out data from the smaller GWAS was moderately reflective of performance in the larger GWAS (Figure S8 A & B), with the exception of AUD/PAU. Novel gene discovery (Figure 1C) was the most successful for Schizophrenia (PPV = 0.67), though only a minority of novel genes were found (sensitivity = 0.066; i.e., 8 genes). Similarly, for ICV/BV, one of the three identified genes was significant. Novel gene discovery in AUD/PAU and MDD/DEP did not surpass a PPV of 0.1. AdaBoost attained moderate performance for gene prioritization across all traits (PPV = 0.48–0.75, sensitivity = 0.42–0.88; Figure 2C). While the average neural net had a higher PPV for SCZ (0.8), this was at the cost of much lower sensitivity (0.28). Examination of variable importance scores revealed that fastBAT, MAGMA, and H‐MAGMA were the primary contributors to prediction across models and methods (Figure 3). DEPICT also attained a comparable level of importance for AUD/PAU in the average neural net and for MDD/DEP in AdaBoost. In general, gene expression methods were the least informative predictors.
FIGURE 3.
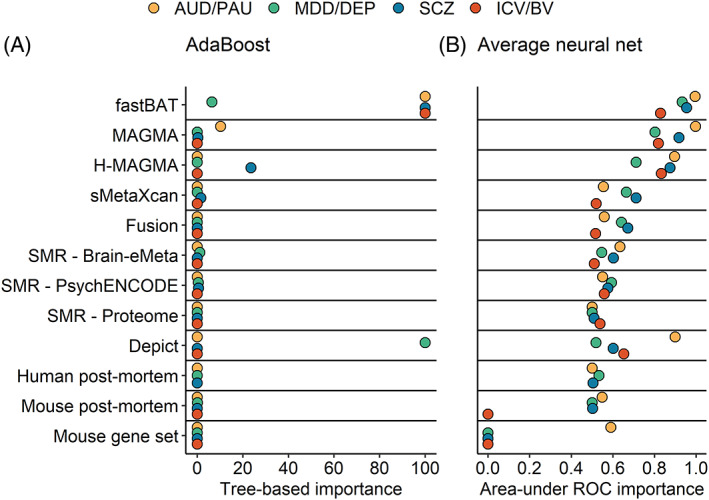
Machine learning variable importance. (A) Average neural net variable importance using the area‐under the ROC method. The maximum possible importance is ‘1.0’. (B) Tree‐based importance in AdaBoost. The maximum possible importance is ‘100’. AUD/PAU = Alcohol use disorder/Problematic alcohol use; MDD/DEP = Major depressive disorder/Depression; SCZ = Schizophrenia; ICV/BV = Intracranial volume/Brain volume.
4. DISCUSSION
GWAS have provided unique insights into the genetic architecture of complex traits. Their informativeness, particularly for complex and heterogeneous traits, is dependent on sample size. Because increasing GWAS sample size is arduous and expensive, there is a natural desire to identify additional sources of data that may increase signal within existing GWAS data. In particular, it has been hypothesized that augmenting GWAS with various kinds of omics data (e.g., transcriptomics and data from non‐human animal models) may boost signals within existing less powered GWAS to circumvent the need for additional sample size. That hypothesis has not previously been rigorously tested. Here we report that for 4 different complex traits, applying omics data by a variety of approaches to an existing smaller GWAS does not reliably detect novel genes that are later discovered by a subsequent GWAS. Increasing GWAS sample size is a requirement to increase the statistical power for discovery of novel genes.
Specifically, we tested whether applying multi‐omics to gene identification in the smaller GWAS could identify genes that will become significant in a later, larger GWAS of the same trait. We applied eight post‐GWAS multi‐omics methods and 2 multivariate machine learning approaches, leveraging both human and model organism omics data, to four different complex traits. No single method or combination of methods achieved both a high PPV and sensitivity when “predicting” novel signals in a larger subsequent GWAS (Figure 1). This is particularly striking for positional methods, where prior expectations were high that “omics” might be helpful: most genes enriched for nominally significant loci did not contain a genome‐wide significant hit within the larger sample. Similarly, while early work evaluating expression‐based methods suggested that they could be used to identify genes that would contain significant loci in a future, larger GWAS, 20 , 21 this was not the case for the four traits evaluated here. While machine learning out‐performed individual methods and even linear combinations of methods, it only correctly identified an additional 1–8 genes, and only for highly heritable traits with an already well‐powered smaller GWAS (SCZ and ICV/BV). This observation converges with related work evaluating methods for SNP and locus annotation, 13 , 14 , 55 which concluded that such methods can only marginally increase the number of true‐positive observations. Similarly, prior work examining positional gene‐based methods in a simulated trait with relatively few causal SNPs (n = 602) observed a tradeoff between sensitivity and specificity, 56 which is also seen here. Overall, multi‐omics data for novel gene discovery incur either a high false‐negative burden (i.e., they miss many novel discoveries) or a high false‐positive burden (i.e., they have many findings which are not found when the sample size increases), and are thus cannot serve as a replacement for larger discovery samples.
The application of multi‐omics does aid in prioritizing already GWAS‐significant genes in the smaller GWAS; those that are also prioritized by multi‐omics were more likely to replicate in the larger GWAS (Figure 2). However, the number of agreeing methods necessary to attain a good PPV (i.e., PPV > = 0.9) differed for each trait (between 4 and 10), and no single method or combination of methods performed the best across all traits (Figure 2, Figure S5). Here, machine learning (ML) performed similarly to the best‐performing individual and combinations of methods. Broadly, these results support current practices in the field, wherein associations that are GWAS‐significant and robust across multi‐omics methods are given the greatest credence, and suggest that further development of ML approaches to post‐GWAS multi‐omics integration may be a fruitful avenue for prioritization within loci. 57
Despite the variation in performance of individual methods, positional methods (i.e., fastBAT, MAGMA, and H‐MAGMA) were frequently among the top performers of multi‐omics methods in both the individual and multivariate ML analyses, for both novel gene discovery and gene prioritization. These achieved a comparable PPV to expression‐based methods, with superior sensitivity. Positional methods were also the top ranked by both machine learning algorithms (with the exception of MDD/DEP analyses with AdaBoost, where fastBAT was second to DEPICT; Figure 3). However, while positional methods are computational, expression‐based methods rely on the power and accuracy of external datasets (e.g., GTEx; Table 1) and will likely improve with larger sample sizes.
Analyses defined the significance of each gene in each GWAS as the p‐value of the most‐significant proximal (within 10 kb) SNP. This definition was selected as an efficient method that permits exploration of different p‐value cut‐offs (i.e., Bonferroni vs. FDR; Figures S3 & S6). However, it is an imperfect approach, as many significant SNPs are not proximal to a protein‐coding region 58 , 59 and significant cis‐acting loci may be further than 10 kb from a gene's boundaries. Indeed, this was a major impetus for the development of the expression‐based multi‐omics methods used here. 21 , 28 Therefore, we conducted two follow‐up analyses: first, post‐hoc analyses expanded the proximal region to 20, 50, and 100 kb (Figure S2); second, we tested whether multi‐omics could identify genes that would be Bonferroni‐significant in a similar multi‐omics analysis of the larger GWAS (Figure S9). While the performance of expression‐based methods improved in both cases, position‐based methods still had better performance, both for novel gene discovery and for gene prioritization. This suggests that the superiority of position‐based methods is not due to our definition of a gene's significance.
Across analyses, the success of multi‐omics approaches appeared largely to depend on trait heritability and GWAS power. Analyses were the most successful in SCZ (the best‐powered GWAS), attaining the largest PPV and sensitivity. However, despite a relatively large sample size, analyses were largely the least successful in MDD/DEP (the trait with the lowest SNP‐based heritability). AUD/PAU was an occasional exception to this general pattern, wherein PPV greatly increased for each additional agreeing method for gene prioritization (Figure 2). Substance use disorders are unique among psychiatric disorders, because there are a few large‐effect loci mapping to substance‐specific receptors and metabolic pathways. 60 , 61 The increase in AUD/PAU PPV likely reflects the relatively large effect size of genes in these pathways, and indeed it largely included genes implicated in alcohol metabolism (i.e., ADH1B, ADH5, and ADH7). 61 However, these results more broadly suggest that multi‐omics will be the least able to successfully prioritize findings and to identify novel associations in traits where it would be the most useful (i.e., where GWAS are particularly underpowered and no loci of large effect are evident).
We note some limitations of the present study. We tested four different traits that are quite different, to cover a range of trait characteristics, but we cannot exclude that some other traits might respond more or less successfully to the integration of omics data. Analyses focused on trait associations at the gene level, rather than individual SNPs. 18 This enabled the additional integration of multi‐omics data that did not use GWAS information, including results from post mortem studies of gene expression in patients and rodent models, and a gene‐set for AUD/PAU that integrates information from a variety of rodent data sources. 15 These methods were uniformly the least informative across all analyses. However, data from post‐mortem studies of MDD/DEP and SCZ, and the AUD/PAU rodent gene set, had a moderate PPV and low sensitivity for gene prioritization. We note that we did not systematically query the literature to derive comprehensive gene‐sets for all traits. Indeed, the AUD/PAU rodent gene set, which was derived from a systematic review of the literature, 15 achieved a higher PPV and sensitivity for AUD/PAU than data from individual studies of rodent models did for their respective traits. The gene‐level focus reflects an additional weakness of the omics‐integration approach, in that it could lead to improved knowledge of biology without narrowing down the identity of causal loci in human populations. Thus, while recent related studies using locus‐level analyses yielded similar findings, 18 we cannot rule out that alternative methods could have led to stronger results. Also, analyses only used GWAS from European samples. Recent work suggests that gene‐level findings from expression‐based methods may be more replicable across ancestries than SNP‐level effects. 62 Thus, expression‐based methods may have superior performance in contexts that could not be evaluated in the present study, owing to the lack of well‐powered GWAS for brain‐related traits in non‐European samples. The smaller GWAS were subsets of the subsequent larger GWAS. This should have favorably biased the tests, but nevertheless they did not perform well. Lastly, larger GWAS achieved a greater sample size in part by broadening the trait definition (i.e., AUD was expanded to PAU, MDD to DEP, and ICV to BV). While this may have contributed to the poor performance of multi‐omics in some of these traits, it does not explain the poor performance in SCZ, the best‐powered of the smaller GWAS, which did not use proxy‐phenotypes in the larger GWAS.
5. CONCLUSIONS
The present results demonstrate that multi‐omics are not a replacement for increasing GWAS sample size. Instead, results support the use of multi‐omics as methods for prioritizing genes within GWAS‐significant loci. Underpowered traits (e.g., cannabis use disorder 12 ) will require much larger sample sizes before even prioritization will be possible. We view the combination of larger GWAS sample sizes and multi‐omics method advancements as likely the most fruitful avenue for identifying multiple plausible causal loci for brain‐related traits.
Supporting information
Data S1. Supporting Information.
ACKNOWLEDGMENTS
This manuscript was posted as a preprint on the bioRxiv server prior to submission (https://doi.org/10.1101/2022.04.13.487655). The authors acknowledge the following funding from the United States National Institutes of Health: R21AA027827 (DAAB, RB), R01DA054750 (DAAB, RB), T32DA007261 (ASH), R33DA047527 (RP), R01DA054869 (AA, JG, HE), U01DA055367 (RB), DA54750 (AA, RB), K02DA32573 (AA). Funders were not involved in the preparation of this manuscript in any way. Dr. Gelernter is named as an inventor on PCT patent. GENOTYPE‐GUIDED DOSING OF OPIOID RECEPTOR AGONISTS (U.S. Patent 10,900,082) (2021). Drs. Gelernter and Polimanti are paid for their editorial work on the journal Complex Psychiatry. All authors have no other potential conflicts of interest.
Baranger DAA, Hatoum AS, Polimanti R, et al. Multi‐omics cannot replace sample size in genome‐wide association studies. Genes, Brain and Behavior. 2023;22(6):e12846. doi: 10.1111/gbb.12846
DATA AVAILABILITY STATEMENT
Code and gene‐level statistics used in these analyses are available at: https://osf.io/g4u9a/?view_only=9613ba48eaa046e3a6614f3d28071cde. The publicly available GWAS summary statistics analyzed here can be found on the PGC website (https://pgc.unc.edu/for-researchers/download-results/), the CTG Lab website (https://ctg.cncr.nl/software/summary_statistics), and dbGap (https://www.ncbi.nlm.nih.gov/projects/gap/cgi‐bin/study.cgi?study_id=phs001672.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001672.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v1.p1). Gene expression data are available in the Supplementary Materials of the respective studies.
REFERENCES
- 1. Kranzler HR, Zhou H, Kember RL, et al. Genome‐wide association study of alcohol consumption and use disorder in 274,424 individuals from multiple populations. Nat Commun. 2019;10:1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhou H, Sealock JM, Sanchez‐Roige S, et al. Genome‐wide meta‐analysis of problematic alcohol use in 435,563 individuals yields insights into biology and relationships with other traits. Nat Neurosci. 2020;23:809‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sullivan PF, Agrawal A, Bulik CM, et al. Psychiatric genomics: An update and an agenda. Am J Psychiatry. 2018;175:15‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Visscher PM, Wray NR, Zhang Q, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101:5‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sullivan PF, Geschwind DH. Defining the genetic, genomic, cellular, and diagnostic architectures of psychiatric disorders. Cell. 2019;177:162‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cai N, Revez JA, Adams MJ, et al. Minimal phenotyping yields genome‐wide association signals of low specificity for major depression [no. 4]. Nat Genet. 2020;52:437‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Consortium TSPG‐WAS (GWAS) . Genome‐wide association study identifies five new schizophrenia loci. Nat Genet. 2011;43:969‐976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Consortium TSWG of the PG , Ripke S, Walters JT, O'Donovan MC. Mapping Genomic Loci Prioritises Genes and Implicates Synaptic Biology in Schizophrenia. 2020. 2020.09.12.20192922.
- 9. Liu M, Jiang Y, Wedow R, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet. 2019;51:237‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Quach BC, Bray MJ, Gaddis NC, et al. Expanding the genetic architecture of nicotine dependence and its shared genetics with multiple traits [no. 1]. Nat Commun. 2020;11:5562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kember RL, Vickers‐Smith R, Xu H, et al. Cross‐ancestry meta‐analysis of opioid use disorder uncovers novel loci with predominant effects in brain regions associated with addiction. Nat Neurosci. 2022;25:1279‐1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Johnson EC, Demontis D, Thorgeirsson TE, et al. A large‐scale genome‐wide association study meta‐analysis of cannabis use disorder. Lancet Psychiatry. 2020;7:1032‐1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Breen G, Li Q, Roth BL, et al. Translating genome‐wide association findings into new therapeutics for psychiatry. Nat Neurosci. 2016;19:1392‐1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Edenberg HJ. Perspective on beyond statistical significance: finding meaningful effects. Complex Psychiatry. 2021;7:1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huggett SB, Johnson EC, Hatoum AS, et al. Genes identified in rodent studies of alcohol intake are enriched for heritability of human substance use. Alcohol Clin Exp Res. 2021;45:2485‐2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Palmer RHC, Johnson EC, Won H, et al. Integration of evidence across human and model organism studies: a meeting report. Genes Brain Behav. 2021;20:e12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pickrell JK. Joint analysis of functional genomic data and genome‐wide association studies of 18 human traits. Am J Hum Genet. 2014;94:559‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moore A, Marks J, Quach BC, et al. Evaluation of methods incorporating biological function and GWAS summary statistics to accelerate discovery. bioRxiv. 2022; 2022.01.10.475153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ayalew M, Le‐Niculescu H, Levey DF, et al. Convergent functional genomics of schizophrenia: from comprehensive understanding to genetic risk prediction [no. 9]. Mol Psychiatry. 2012;17:887‐905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mancuso N, Shi H, Goddard P, Kichaev G, Gusev A, Pasaniuc B. Integrating gene expression with summary association statistics to identify genes associated with 30 complex traits. Am J Hum Genet. 2017;100:473‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gusev A, Ko A, Shi H, et al. Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet. 2016;48:245‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Levey DF, Stein MB, Wendt FR, et al. Bi‐ancestral depression GWAS in the million veteran program and meta‐analysis in >1.2 million individuals highlight new therapeutic directions. Nat Neurosci. 2021;24:954‐963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Howard DM, Adams MJ, Clarke T‐K, et al. Genome‐wide meta‐analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343‐352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ripke S, Neale BM, Corvin A, et al. Biological insights from 108 schizophrenia‐associated genetic loci. Nature. 2014;511:421‐427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jansen PR, Nagel M, Watanabe K, et al. Genome‐wide meta‐analysis of brain volume identifies genomic loci and genes shared with intelligence. Nat Commun. 2020;11:5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Helgeland Ø. oyhel/vautils [R]. 2019. Retrieved December 15, 2021, from https://github.com/oyhel/vautils
- 27. de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene‐set analysis of GWAS data. PLoS Comput Biol. 2015;11:1‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sey NYA, Hu B, Mah W, et al. A computational tool (H‐MAGMA) for improved prediction of brain‐disorder risk genes by incorporating brain chromatin interaction profiles. Nat Neurosci. 2020;23:583‐593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bakshi A, Zhu Z, Vinkhuyzen AAE, et al. Fast set‐based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci Rep. 2016;6:32894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pers TH, Karjalainen JM, Chan Y, et al. Biological interpretation of genome‐wide association studies using predicted gene functions. Nat Commun. 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng H, Mancuso N, Gusev A, et al. Leveraging expression from multiple tissues using sparse canonical correlation analysis and aggregate tests improves the power of transcriptome‐wide association studies. PLoS Genet. 2021;17:e1008973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Barbeira AN, Pividori M, Zheng J, Wheeler HE, Nicolae DL, Im HK. Integrating predicted transcriptome from multiple tissues improves association detection. PLoS Genet. 2019;15:e1007889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhu Z, Zhang F, Hu H, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481‐487. [DOI] [PubMed] [Google Scholar]
- 34. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cuéllar‐Partida G, Lundberg M, Kho PF, et al. Complex‐Traits Genetics Virtual Lab: A Community‐Driven Web Platform for Post‐GWAS Analyses. 2019. 518027.
- 36. Aguet F, Brown AA, Castel SE, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang D, Liu S, Warrell J, et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362:eaat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qi T, Wu Y, Zeng J, et al. Identifying gene targets for brain‐related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9:2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sun BB, Maranville JC, Peters JE, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rao X, Thapa KS, Chen AB, et al. Allele‐specific expression and high‐throughput reporter assay reveal functional genetic variants associated with alcohol use disorders. Mol Psychiatry. 2021;26:1142‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu W, Howard D, Sibille E, French L. Differential and spatial expression meta‐analysis of genes identified in genome‐wide association studies of depression. Transl Psychiatry. 2021;11:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Collado‐Torres L, Burke EE, Peterson A, et al. Regional heterogeneity in gene expression, regulation, and coherence in the frontal cortex and hippocampus across development and schizophrenia. Neuron. 2019;103:203‐216.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ferguson LB, Zhang L, Kircher D, et al. Dissecting brain networks underlying alcohol binge drinking using a systems genomics approach. Mol Neurobiol. 2019;56:2791‐2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scarpa JR, Fatma M, Loh Y‐HE, et al. Shared transcriptional signatures in Major depressive disorder and mouse chronic stress models. Biol Psychiatry. 2020;88:159‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Donegan JJ, Boley AM, Glenn JP, Carless MA, Lodge DJ. Developmental alterations in the transcriptome of three distinct rodent models of schizophrenia. PLOS One. 2020;15:e0232200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu Y, Chen S, Li Z, Morrison AC, Boerwinkle E, Lin X. ACAT: a fast and powerful p value combination method for rare‐variant analysis in sequencing studies. Am J Hum Genet. 2019;104:410‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Moulos P, Hatzis P. Systematic integration of RNA‐Seq statistical algorithms for accurate detection of differential gene expression patterns. Nucleic Acids Res. 2015;43:e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stevenson M, Sergeant E, Nunes T, et al. epiR: Tools for the Analysis of Epidemiological Data, Version 2.0.40. 2021. Retrieved December 16, 2021, from https://CRAN.R-project.org/package=epiR
- 49. R Core Team . R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2014. Retrieved from http://www.r-project.org/ [Google Scholar]
- 50. Alfaro E, Gamez M, García N. Adabag: An R package for classification with boosting and bagging. J Stat Softw. 2013;54:1‐35. [Google Scholar]
- 51. Rojas R. AdaBoost and the Super Bowl of Classifiers a Tutorial Introduction to Adaptive Boosting. Freie Univ Berl Tech Rep; 2009. [Google Scholar]
- 52. Venables WN, Ripley BD. Introduction. In: Venables WN, Ripley BD, eds. Modern Applied Statistics with S. Springer; 2002. doi: 10.1007/978-0-387-21706-2_1 [DOI] [Google Scholar]
- 53. Kuhn M. Building predictive models in R using the caret package. J Stat Softw. 2008;28:1‐26.27774042 [Google Scholar]
- 54. Friedman JH. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29:1189‐1232. [Google Scholar]
- 55. Dey KK, van de Geijn B, Kim SS, Hormozdiari F, Kelley DR, Price AL. Evaluating the informativeness of deep learning annotations for human complex diseases. Nat Commun. 2020;11:4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wojcik GL, Kao WL, Duggal P. Relative performance of gene‐ and pathway‐level methods as secondary analyses for genome‐wide association studies. BMC Genet. 2015;16:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nicholls HL, John CR, Watson DS, Munroe PB, Barnes MR, Cabrera CP. Reaching the end‐game for GWAS: machine learning approaches for the prioritization of complex disease loci. Front Genet. 2020;11: Retrieved February 22, 2022, from 10.3389/fgene.2020.00350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease‐associated variation in regulatory DNA. Science. 2012;337:1190‐1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Nasser J, Bergman DT, Fulco CP, et al. Genome‐wide enhancer maps link risk variants to disease genes [no. 7858]. Nature. 2021;593:238‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hatoum AS, Colbert SMC, Johnson EC, et al. Multivariate Genome‐Wide Association Meta‐Analysis of over 1 Million Subjects Identifies Loci Underlying Multiple Substance Use Disorders. 2022. 2022.01.06.22268753. [DOI] [PMC free article] [PubMed]
- 61. Edenberg HJ, McClintick JN. Alcohol dehydrogenases, aldehyde dehydrogenases, and alcohol use disorders: a critical review. Alcohol Clin Exp Res. 2018;42:2281‐2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu Z, Gopalan S, Yuan D, et al. Multi‐ancestry fine‐mapping improves precision to identify causal genes in transcriptome‐wide association studies. Am J Hum Genet. 2022;109:1388‐1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting Information.
Data Availability Statement
Code and gene‐level statistics used in these analyses are available at: https://osf.io/g4u9a/?view_only=9613ba48eaa046e3a6614f3d28071cde. The publicly available GWAS summary statistics analyzed here can be found on the PGC website (https://pgc.unc.edu/for-researchers/download-results/), the CTG Lab website (https://ctg.cncr.nl/software/summary_statistics), and dbGap (https://www.ncbi.nlm.nih.gov/projects/gap/cgi‐bin/study.cgi?study_id=phs001672.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001672.v3.p1, https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs001672.v1.p1). Gene expression data are available in the Supplementary Materials of the respective studies.