

Summary
Machine learning is transforming antibody engineering by enabling the generation of drug-like monoclonal antibodies with unprecedented efficiency. Unsupervised algorithms trained on massive and diverse protein sequence datasets facilitate the prediction of panels of antibody variants with native-like intrinsic properties (e.g., high stability), greatly reducing the amount of subsequent experimentation needed to identify specific candidates that also possess desired extrinsic properties (e.g., high affinity). Additionally, supervised algorithms, which are trained on deep sequencing datasets obtained after enrichment of in vitro antibody libraries for one or more specific extrinsic properties, enable the prediction of antibody variants with desired combinations of extrinsic properties without the need for additional screening. Here we review recent advances using both machine learning approaches and how they are impacting the field of antibody engineering as well as key outstanding challenges and opportunities for these paradigm-changing methods.
Keywords: mAb, IgG, variable region, complementarity-determining region, CDR, antigen, affinity, stability, deep learning, protein design, directed evolution
Graphical Abstract
Machine learning is transforming antibody engineering by efficiently generating drug-like antibodies. Unsupervised algorithms trained on large protein datasets enable prediction of antibody variants with native-like intrinsic properties, reducing the amount of experimentation necessary for identifying candidates with desired extrinsic properties. Supervised algorithms trained on deep sequencing datasets enable prediction of antibody variants with favorable extrinsic properties without additional screening. We review how both approaches are transforming the field of antibody engineering as well as key challenges for these paradigm-changing methods.
Introduction
In recent years, monoclonal antibodies have become an increasingly successful class of therapeutics due to their many attractive molecular and pharmacological properties.1-3 Their success has transformed the treatment of many diseases and invigorated efforts to generate candidates against some of the most important and challenging targets.4-9 This has led to intense interest in generating antibody candidates with both high affinity and potency as well as favorable, drug-like biophysical properties. However, current antibody generation methods, while powerful, are inherently limited by the need for high-quality antigens, which in some cases are difficult to generate, and are commonly limited in their ability to generate broad epitope coverage. These and other challenges have motivated the development of advanced computational methods for therapeutic antibody development and are increasingly being integrated into the development pipeline.10 In addition to improving the accuracy of identifying drug-like antibodies, these computational methods offer benefits by reducing the need for costly experimentation, affording greater control over the antibody properties, and facilitating exploration of much larger antibody diversity.
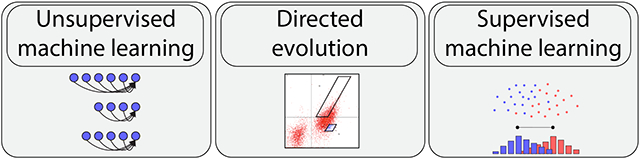
Therefore, there is substantial interest in the continued development of computational methods that guide the design and engineering of therapeutic antibodies. In particular, there is growing emphasis on developing methods that facilitate complete de novo design of therapeutic antibodies given only a target antigen or epitope. However, this is extremely challenging because antibody sequence space is vast and sparsely populated with antibodies that possess native-like properties and are suitable for therapeutic applications. Encouragingly, substantial progress has been made in this area recently, which is primarily due to advances in machine learning. Some of the most powerful advances reported recently are those that combine directed evolution, deep sequencing, and machine learning, which enable efficient exploration of relevant portions of vast antibody sequence space, facilitating the identification and optimization of rare drug-like molecules. In this review, we discuss recent advances that improve the development of therapeutic antibodies through the combination of machine learning and directed evolution (Figure 1). We first focus of unsupervised methods trained on large protein sequence datasets for the identification of antibody sequences with native-like (intrinsic) properties that parallel those of drug-like antibodies, such as high stability, to greatly narrow the search for antibody variants that also have the desired extrinsic, antigen-specific binding activity. We also review supervised machine learning methods trained on deep sequencing data acquired from library sorting analysis for the prediction of individual antibody variants with favorable combinations of drug-like properties, such as high affinity and stability, without the need for additional screening. These methods hold great potential for improving the efficiency and effectiveness of therapeutic antibody development.
Figure 1. Overview of machine learning and directed evolution methods that are being used in concert to simplify complex antibody engineering tasks.

Machine learning-guided antibody design using unsupervised learning from antibody repertoires
A vital component of using directed evolution for therapeutic antibody optimization is the selection of amino acid substitutions predicted to improve antibody properties. This includes discrete mutations that are evaluated individually as well as sets of mutations that are evaluated combinatorially in antibody libraries. There have been substantial advancements in this area recently, with many new machine learning methods adapted from the field of natural language processing to rapidly analyze expanding protein sequence datasets and identify beneficial mutations.11-13 An important concept for many of these methods is that the sequences in large sets of naturally evolved proteins can be parameterized by unsupervised learning algorithms to predict inherent properties of such proteins (i.e., high stability).14,15
One strategy to achieve this goal involves unsupervised or self-supervised machine learning algorithms that are trained to learn the construction convention of protein sequences, not to predict any property-specific information. Recently, a protein language model was used to implement this strategy to affinity mature therapeutic antibodies against several viruses using an ensemble of language models trained on broad sets of protein sequences (UniRef50 and UniRef90), most of which were not antibody sequences.11,16 The resulting models are applicable to a wide variety of protein engineering tasks and broadly useful for applications in diverse protein therapeutics.16 These models employ deep transformer neural networks, trained by masking random amino acid positions in the input sequences and tested by evaluating their ability to predict the held-out amino acids at the masked positions (Figure 2A). The models predict probabilities of mutations, representing the evolutionary likelihoods of the mutations based on the assumption that the databases are comprised of evolutionary fit protein sequences. Higher prediction probabilities represent higher likelihoods that a mutation would be found in an evolutionary fit sequence and therefore a higher likelihood of exhibiting native-like properties. For example, one model was benchmarked against nine sets of deep sequencing data collected for different levels of extrinsic protein properties (e.g., high vs. low levels of β-lactamase activity), which revealed improved or similar ability to predict mutations that improved protein functional properties relative to random predictions (Figure 2B).11
Figure 2. General protein language model identifies mutations that increase antibody affinity.

(A) Self-supervised deep transformer neural networks learn the intrinsic fitness of protein sequences and can predict mutations that increase their fitness. These intrinsic fitness predictions are hypothesized to reflect intrinsic properties of diverse proteins, including antibodies, and can be used in their optimization. (B) Benchmarking of the neural networks on nine high-throughput scanning mutagenesis datasets revealed improved or comparable predictions of single mutations that improved intrinsic properties over background predictions. (C) Mutations with higher predicted intrinsic fitness were identified and introduced to five anti-viral antibodies, resulting in higher affinities in several cases. In Round 1 of optimization, single mutations were evaluated. In Round 2 of optimization, combinations of successful mutations from Round 1 were evaluated. In (B), ADRB2 is adrenoreceptor beta 2, β-la. is β-lactamase, Env is envelope glycoprotein, Ha is hemagglutinin, infA is translation initiation factor 1, MAPK1 is mitogen-activated protein kinase 1, and PafA is phosphate-irrepressible alkaline phosphatase. Moreover, in (B), the p-values are <0.05 (*), <0.01 (**) and <0.001 (***). This figure is adapted from a previous publication.11
One compelling aspect of this model is the ability to identify beneficial mutations that improve extrinsic protein properties by only predicting and testing small sets of protein mutants (e.g., ~10-20 mutants).11 The power of this approach was demonstrated by increasing the affinity of antibodies specific for three viruses, namely influenza, Ebola, and SARS-CoV-2. An initial round of in silico screening of single mutations (~10 mutants per antibody) revealed many successful mutations that increased antibody affinity (Figure 2C). Successful mutations were combined and evaluated in a second round of optimization (~10 mutants per antibody). The final antibody variants displayed up to a 160-fold improvement in binding affinity. Notably, this study demonstrates the impressive capacity of protein language models to distill information relevant for optimizing antibody function, despite not being trained specifically on antibody repertoires.
Despite impressive results for low-N protein engineering, there is little evidence that these predictions of intrinsic fitness levels correlate with experimental measurements of antibody extrinsic properties (e.g., affinity). One possible reason for this is that protein language models trained on large protein databases are not best suited to represent antibody properties such as affinity given the extremely specific paratopes needed to mediate antibody-antigen binding and may require antibody-specific language model representations. Such models have been developed and validated for the prediction of antibody paratope residues, Fv structures, and thermostabilizing mutations,17-20 but they generally do not predict the magnitudes of changes expected for the predicted mutations.
However, there has been recent progress in using unsupervised learning to develop models for predicting metrics that are directly correlated with measurements of certain extrinsic protein properties. One approach for antibody engineering is to train language models directly on antibody sequence sets, instead of broad protein sequence sets. Although this approach reduces the generalizability of the models, the results are potentially more useful for therapeutic antibody development because they enable differentiation of the intraclass variability of biophysical properties rather than broad property classification. This ability is vital for therapeutic development because small differences in biophysical properties can substantially influence the clinical potential of a molecule. In training language models on more specific sequence repertoires, this differentiation can be better achieved.13,21,22 Moreover, in this type of approach, the idea of broad evolutionary fitness from protein language models is adapted for specific property fitness of antibodies, the therapeutic molecule of interest.
One important concept for this type of model is its generative nature, as the model learns the ability to generate antibody sequences that resemble those in the training repertoire. These models enable more efficient exploration of desired sequence space compared to discriminative models and differ from the broad protein language models that cannot be used for de novo generation of antibody sequences. Autoregressive models are one type of generative model adapted from the field of natural language processing that can be readily applied to protein sequences for feed-forward prediction of amino acids in a given sequence. Through this type of training, the model learns sequence construction constraints represented in the training repertoire and enables generation of new sequences with the same properties conferred by those constraints. Notably, autoregressive, feed-forward modeling obviates the need for sequence alignment, which is particularly important for training on antibody repertoires with variable sequence lengths. This approach is particularly important because antibody sequence alignment is challenging, and it is not possible for diverse antibody regions such as HCDR3s, even for such CDRs with same lengths.
Recently, generative autoregressive modeling was applied to predict highly stable nanobody sequences, enabling the production of a library with 1000-fold greater expression than a library designed using conventional methods.22 Specifically, a dilated convolutional neural network was identified as a promising model architecture for feed-forward prediction of amino acids in protein sequences (Figure 3A). The model architecture was first benchmarked against previously validated models by training on various protein libraries, including nanobodies, growth factors, and enzymes, and then evaluated for predicting the effect of mutations on their functional properties, such as protein stability, cellular growth, and enzyme activity (Figure 3B). Differences in the probability predictions between wild-type and mutant sequences were found to be descriptive of the measured functional properties with improved or comparable performance relative to state-of-the-art model architectures.23
Figure 3. Autoregressive neural network accurately predicts stable nanobody variants and libraries.

(A) Autoregressive modeling uses feed-forward amino-acid prediction to parameterize the evolutionary likelihood of sequences. (B) Benchmarking prediction probabilities against 35 high-throughput scanning mutagenesis datasets revealed improved or comparable predictive capacity relative to state-of-the-art (DeepSequence23) and baseline (hidden Markov model) methods. (C) An autoregressive model trained on a naïve nanobody repertoire facilitated accurate prediction of nanobody thermal stability for four benchmark datasets. (D) The model was used to design a library of highly stable nanobody sequences, and the resulting properties of the designed library were superior to those of a conventionally designed library. The conventional library, designed by position-specific scoring matrix analysis of nanobody databases, exhibited lower expression than the library designed using the autoregressive model. This figure is adapted from a previous publication.22
Next, a naïve nanobody repertoire was deep sequenced and used for unsupervised training of a model for predicting novel nanobody sequences with native-like properties.22 A key assumption in this study was that the naïve nanobody repertoire was composed of stably folded nanobodies, and the determinants of such stability were learned by the model. Interestingly, there was a strong correlation between model prediction probabilities and nanobody stability measurements for four independent datasets, demonstrating the impressive potential of such models to generate novel nanobodies with native-like biophysical properties (Figure 3C).
The model was then applied to design a highly stable nanobody library that could be enriched for antigen binding.22 The autoregressive model trained on naïve nanobody sequences was used to predict over 107 binding loop (complementarity-determining region 3 or CDR3) sequences that would result in favorable, native-like properties in the context of a common nanobody framework. This sequence set was filtered to isolate a diverse set of approximately 185,000 CDR3 sequences to be included in the designed library. This library was compared to a conventional library that was designed using a position-specific scoring matrix (PSSM) created from the hundreds of nanobody sequences in the Protein Data Bank. Both libraries were displayed on the surface of yeast for evaluation of expression levels, which revealed that the library designed using machine learning was better expressed with fewer poorly expressing variants (Figure 3D). The designed (better expressing) library was then screened for antigen binding against human serum albumin, identifying a lead variant with modest affinity (KD of ~10 μM). Overall, this study demonstrates the power of methods that learn from nanobody and related repertoires and parameterize intrinsic fitness features that are most relevant for nanobody optimization.
However, this general approach remains limited to native-like antibody properties, such as stability, that are represented in naïve antibody repertoire sequences. To develop models for directly predicting extrinsic antibody properties such as affinity, a typical approach is to train models on large antibody sequence sets that have been experimentally enriched for specific properties of interest. This approach was demonstrated in a recent study where deep sequencing data from an affinity-matured library was used to train an autoregressive neural network to predict binding probabilities.12 A large phage-display library was generated for an anti-kynurenine antibody by mutating sites in the heavy chain CDRs. This library was panned against the antigen and deep sequenced after multiple rounds of enrichment. Next, a generative autoregressive neural network was trained for feed-forward amino acid prediction on a dataset of approximately 1,000 sequences curated from deep sequencing data for the enriched library. Millions of antibody sequences were then generated by the model, and several of the highly scored antibodies were produced and evaluated for antigen binding. Impressively, the predicted probability of binding, which describes the likelihood that a given antibody sequence would be present in the enriched library, correlated well with continuous measurements of antigen binding (R2 of 0.52). Moreover, the sequences generated by the model generally displayed higher affinities than the most enriched clones identified by deep sequencing.
Notably, the theoretical diversity of the library generated in this study was over 1017, far exceeding the achievable transformation efficiencies, panning diversities, and sequencing depths that are possible experimentally.12 This highlights a paradigm shift in antibody engineering enabled by machine learning, as comprehensive experimental observation of library diversity is unnecessary for accurate model training if sufficient mutational diversity is observed in a variety of sequence contexts. Overall, this approach is unique from the studies discussed above because it does not employ naïve sequence repertoires for unsupervised learning but instead requires experimentation to identify a relevant set of sequences enriched for the property of interest. This approach further reduces model generality, but achieves the predictive capacity of specific extrinsic properties necessary for rapid antibody optimization. This study, along with related ones,11,12,22 highlight the many exciting possibilities for further integrating machine learning and directed evolution to develop robust, comprehensive approaches for improving generation of highly potent antibodies.
Machine learning-guided antibody design using supervised learning from antibody libraries
Another valuable use of machine learning for antibody optimization is learning from deep sequencing datasets collected at different stages of enrichment of in vitro antibody libraries for specific extrinsic properties, such as i) before and after selection for high antigen binding or ii) after selection for high and low antigen binding at a terminal stage of library sorting. Machine learning has been increasingly applied to analyze these types of labeled datasets, which have historically been underutilized to identify optimal antibody mutants due to an overreliance on conventional methods such as sequence enrichment ratios and frequencies. One notable study using this approach employed a convolutional neural network to analyze deep sequencing data with binary labels for high and low antigen binding.24 A small sub-library of a therapeutic antibody (trastuzumab) was created by mutating the heavy chain CDR3 and displayed on the surface of mammalian cells (Figure 4A). After two rounds of enrichment for antigen binding, the libraries enriched for high and low antigen binding were deep sequenced. The final curated dataset consisted of over 20,000 sequences of trastuzumab variants with binary labels describing high and low antigen binding. Interestingly, model architectures ranging from simple k-nearest neighbors to complex deep learning classification models performed similarly in their ability to accurately predict antigen binding.
Figure 4. Convolutional neural network predicts levels of antigen binding and identifies highly developable variants of a therapeutic antibody.

(A) A library of trastuzumab variants with mutations in heavy chain CDR3 was displayed on the surface of mammalian cells and sorted for high and low levels of antigen binding. The enriched libraries were deep sequenced, and a neural network was trained to predict sequences with high antigen binding. The predicted antibody sequences were further filtered for favorable developability (viscosity,25 solubility,26 and immunogenicity27) properties to identify drug-like sequences. (B) The resulting neural network was highly accurate with an AUC of 0.91. (C) Several (55) variants predicted with high antigen binding were produced and evaluated, and all of them displayed antigen-specific binding. (D-E) The ten highest affinity trastuzumab variants were further characterized in terms of their (D) expression titers after expansion of hybridoma cells and (E) thermal stabilities. In (E), Tm1 is the first unfolding transition and Tonset is the onset of thermal melting temperature. This figure is adapted from a previous publication.24
The best performing model, a convolutional neural network with high classification accuracy (Figure 4B), was next used to predict millions of antigen-specific trastuzumab variants for additional analysis of their biophysical properties.24 The variants were ranked via a novel developability score that included contributions related to viscosity,25 solubility,26 and immunogenicity.27 In total, 55 variants were predicted to be optimized for both affinity and developability (Figure 4C).24 Notably, all of the predicted variants exhibited antigen binding, with most retaining monovalent affinities (dissociation constants) below 100 nM. Ten of the highest affinity variants were further characterized, revealing that five exhibited comparable or improved expression titers and all ten exhibited comparable or improved thermal stability (Figure 4D). These results illustrate the potential of machine learning to improve the analysis of antibody libraries not only for affinity, but also in concert with analysis of multiple developability criteria at the same time. Nevertheless, a limitation of this study is that most variants (~95%) identified as antigen binders displayed reductions to affinity.
For multi-property optimization of antibodies, it is important not only to predict interclass difference in properties such as high versus low affinity but also intraclass differences such as high versus very high affinity. A recent study addressed this challenge by predicting continuous antibody properties based only on simple deep sequencing datasets using supervised dimensionality reduction.28 In this work, a clinical-stage antibody (emibetuzumab) was optimized to address its high levels of non-specific binding. A large sub-library (107 variants) was generated by mutating residues in the heavy chain CDRs that were predicted to mediated non-specific binding (Figure 5A).28 However, mutating sites in these CDRs was also expected to reduce affinity, highlighting challenges related to co-optimizing affinity and non-affinity (non-specific binding) interactions.29
Figure 5. Linear models trained only on binary deep sequencing data predict continuous properties of a therapeutic antibody.

(A) A library of Fab variants of a therapeutic antibody (emibetuzumab) with mutations in its heavy chain CDRs was displayed on the surface of yeast and sorted for high and low levels of antigen (hepatocyte growth factor receptor, also known as c-MET) and non-specific binding. The enriched libraries were deep sequenced, and simple (linear discriminant analysis or LDA) and more complex (neural network) models were trained to classify antibody sequences for high and low levels of each property. (B) High classification accuracy was observed for the LDA models for both properties. Additionally, the LDA model projections were found to correlate with continuous measurements for both antigen and non-specific binding. (C) Co-optimized antibody mutants were identified along the Pareto frontier. These predictions were confirmed experimentally for soluble IgGs, leading to the identification of several (16) emibetuzumab variants with increased antigen and reduced non-specific binding. This figure is adapted from a previous publication.28
Therefore, the emibetuzumab sub-library displayed on the surface of yeast was sorted for high and low levels of binding to antigen and polyspecificity reagents (soluble membrane proteins30 and ovalbumin31,32). The enriched libraries were deep sequenced and a relatively small dataset of 4,000 sequences were identified and labeled for their high or low levels of antigen and non-specific binding.28 Interestingly, relatively simple models, including those developed using k-nearest neighbors and linear discriminant analysis (LDA), were able to accurately classify both properties. However, classification models are particularly limited for co-optimizing multiple antibody properties because they fail to describe the intraclass variability. Therefore, the investigators evaluated if models developed for classification could also be applied for identifying intraclass differences in antigen and non-specific binding. Surprisingly, the LDA models, which project features into a single dimension to maximize classification accuracy, were also able to describe the intraclass variability, as judged by strong correlations between the model projections and continuous binding measurements (Figure 5B). More complicated models, such as neural networks, only modestly improved the predictions, suggesting that the effects of the CDR mutations on antigen and non-specific binding were largely additive. Finally, the LDA models enabled the co-optimization of emibetuzumab by directly predicting co-optimized variants along the Pareto frontier (Figure 5C). The model predictions were also strongly correlated with antigen and non-specific binding for IgGs, revealing that the datasets collected using Fab fragments enabled generation of models that extend to full-length antibodies.
While these results demonstrated the ability to predict the impact of in-library mutations observed in the training sets on both antigen and non-specific binding, they cannot be extrapolated to novel mutations unseen during training.28 Therefore, the investigators evaluated if models could be developed for predicting emibetuzumab antigen and non-specific binding that generalize to novel mutational space. To do this, it was necessary to change the feature sets used for model training from those that simply encode the observed mutations at the mutated heavy chain CDR sites to those that reflect the entire VH domain. The investigators evaluated conventional feature sets based on physicochemical properties of VH domains (e.g., charge and hydrophobicity) as well as more abstract features, referred to as deep learning (UniRep) features, extracted from protein language models.33 Notably, the antigen and non-specific binding models developed with deep learning features were best at generalizing to novel mutational space and enabled identification of co-optimized emibetuzumab mutants with superior properties relative to those isolated from the initial library. These findings demonstrate the potential of new computational methods for generating better antibody descriptors that lead to more accurate machine learning predictions of antibody properties, which is especially important for overcoming strong tradeoffs between different antibody properties. This study also highlights that the power of relatively simple linear models for predicting protein properties based on analyzing large combinatorial protein spaces up to several millions of sequences. This illustrates another paradigm shift for therapeutic antibody optimization, suggesting that the scale at which library-screening data is frequently acquired does not typically necessitate complex (non-linear) analysis methods.34,35
Conclusions
Much progress has been made in recent years using machine learning to simplify complex antibody engineering. However, there remains a great need for further improvements in the efficiency and accuracy of predictions of optimized antibody variants. The combination of unsupervised and supervised machine learning methods is particularly compelling13,34,36,37 and holds great promise for increasing the efficiency and success of antibody engineering efforts. Hybrid or semi-supervised methods are expected to enable better design of both specific antibody variants and entire libraries that sample the drug-like antibody sequence space. This is particularly important for addressing challenging problems that require co-optimization of several antibody properties, including those related to antigen-binding properties (e.g., affinity, epitope and species cross-reactivity) as well as those related to in vitro (stability, self-association) and in vivo (pharmacokinetics, non-specific binding) properties.
Another important future direction for machine learning-guided antibody engineering is the prediction of antibody epitopes, which is important for targeting pre-selected epitopes of known functional significance and achieving broad epitope coverage when the functional significance of different epitopes is unknown.38 Predicting antibody epitopes requires prediction of both the antibody and antigen structures, as well as antibody/antigen docking.39,40 Alphafold excels at antigen structure prediction,41,42 but it is suboptimal for predicting the structures of antibody CDRs, such as HCDR3, which is being addressed by other methods19,43 and will need to be further improved in the future. Most importantly, future studies need to improve antibody/antigen docking, building on important progress to date,40,44,45 to reliably predict antibody epitopes, which remains one of the greatest needs in the field.
There are two additional future directions that will be particularly important to the field. One is the lack of large datasets relevant to many important antibody engineering problems. Much of the available high-quality data is controlled by biopharmaceutical companies, and increased efforts to generate and share experimental data is critical to further advance the impact of machine learning on antibody engineering. The other key future direction is the need to make it simpler for non-experts, especially for scientists from biological fields, to develop and use emerging machine learning methods. Reducing the barriers of entry to scientists interested in developing and using these exciting methods is expected to usher in a new era of antibody engineering that enables generation of drug-like antibodies with unprecedented efficiency.
Acknowledgments
We thank members of the Tessier lab for their helpful suggestions. This work was supported by the National Institutes of Health (R35GM136300, RF1AG059723 and R21AI171844 to P.M.T., 1T32GM140223-01 to E.K.M.), National Science Foundation (CBET 1813963, 1605266 and 1804313 to P.M.T.), a University of Michigan Rackham Predoctoral Fellowship (to E.K.M.) and the Albert M. Mattocks Chair (to P.M.T).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
Authors declare no competing interests.
References
- 1.Grilo AL & Mantalaris A The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol 37, 9–16, doi: 10.1016/j.tibtech.2018.05.014 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Ecker DM, Jones SD & Levine HL The therapeutic monoclonal antibody market. MAbs 7, 9–14, doi: 10.4161/19420862.2015.989042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu RM et al. Development of therapeutic antibodies for the treatment of diseases. J Biomed Sci 27, 1, doi: 10.1186/s12929-019-0592-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nelson AL, Dhimolea E & Reichert JM Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov 9, 767–774, doi: 10.1038/nrd3229 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Carter PJ & Lazar GA Next generation antibody drugs: pursuit of the 'high-hanging fruit'. Nat Rev Drug Discov 17, 197–223, doi: 10.1038/nrd.2017.227 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Dodd RB, Wilkinson T & Schofield DJ Therapeutic Monoclonal Antibodies to Complex Membrane Protein Targets: Antigen Generation and Antibody Discovery Strategies. BioDrugs 32, 339–355, doi: 10.1007/s40259-018-0289-y (2018). [DOI] [PubMed] [Google Scholar]
- 7.Yu YJ & Watts RJ Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10, 459–472, doi: 10.1007/s13311-013-0187-4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu AM & Senter PD Arming antibodies: prospects and challenges for immunoconjugates. Nat Biotechnol 23, 1137–1146, doi: 10.1038/nbt1141 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Makowski EK, Schardt JS & Tessier PM Improving antibody drug development using bionanotechnology. Curr Opin Biotechnol 74, 137–145, doi: 10.1016/j.copbio.2021.10.027 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makowski EK, Wu L, Gupta P & Tessier PM Discovery-stage identification of drug-like antibodies using emerging experimental and computational methods. MAbs 13, 1895540, doi: 10.1080/19420862.2021.1895540 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hie BL et al. Efficient evolution of human antibodies from general protein language models and sequence information alone. bioRxiv, 2022.2004.2010.487811, doi: 10.1101/2022.04.10.487811 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saka K. et al. Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci Rep 11, 5852, doi: 10.1038/s41598-021-85274-7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Biswas S, Khimulya G, Alley EC, Esvelt KM & Church GM Low-N protein engineering with data-efficient deep learning. Nat Methods 18, 389–396, doi: 10.1038/s41592-021-01100-y (2021). [DOI] [PubMed] [Google Scholar]
- 14.Bloom JD, Wilke CO, Arnold FH & Adami C Stability and the evolvability of function in a model protein. Biophys J 86, 2758–2764, doi: 10.1016/s0006-3495(04)74329-5 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloom JD, Labthavikul ST, Otey CR & Arnold FH Protein stability promotes evolvability. Proc Natl Acad Sci U S A 103, 5869–5874, doi: 10.1073/pnas.0510098103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rives A. et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc Natl Acad Sci U S A 118, doi: 10.1073/pnas.2016239118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruffolo JA, Chu L-S, Mahajan SP & Gray JJ Fast, accurate antibody structure prediction from deep learning on massive set of natural antibodies. bioRxiv, 2022.2004.2020.488972, doi: 10.1101/2022.04.20.488972 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ruffolo JA, Gray JJ & Sulam J Deciphering antibody affinity maturation with language models and weakly supervised learning. arXiv preprint arXiv:2112.07782 (2021). [Google Scholar]
- 19.Ruffolo JA, Sulam J & Gray JJ Antibody structure prediction using interpretable deep learning. Patterns (N Y) 3, 100406, doi: 10.1016/j.patter.2021.100406 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leem J, Mitchell LS, Farmery JHR, Barton J & Galson JD Deciphering the language of antibodies using self-supervised learning. Patterns (N Y) 3, 100513, doi: 10.1016/j.patter.2022.100513 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Amimeur T. et al. Designing Feature-Controlled Humanoid Antibody Discovery Libraries Using Generative Adversarial Networks. bioRxiv, 2020.2004.2012.024844, doi: 10.1101/2020.04.12.024844 (2020). [DOI] [Google Scholar]
- 22.Shin JE et al. Protein design and variant prediction using autoregressive generative models. Nat Commun 12, 2403, doi: 10.1038/s41467-021-22732-w (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Riesselman AJ, Ingraham JB & Marks DS Deep generative models of genetic variation capture the effects of mutations. Nat Methods 15, 816–822, doi: 10.1038/s41592-018-0138-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason DM et al. Optimization of therapeutic antibodies by predicting antigen specificity from antibody sequence via deep learning. Nat Biomed Eng 5, 600–612, doi: 10.1038/s41551-021-00699-9 (2021). [DOI] [PubMed] [Google Scholar]
- 25.Sharma VK et al. In silico selection of therapeutic antibodies for development: viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 111, 18601–18606, doi: 10.1073/pnas.1421779112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sormanni P, Aprile FA & Vendruscolo M The CamSol method of rational design of protein mutants with enhanced solubility. J Mol Biol 427, 478–490, doi: 10.1016/j.jmb.2014.09.026 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Jensen KK et al. Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154, 394–406, doi: 10.1111/imm.12889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Makowski EK et al. Co-optimization of therapeutic antibody affinity and specificity using machine learning models that generalize to novel mutational space. Nat Commun 13, 3788, doi: 10.1038/s41467-022-31457-3 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rabia LA, Desai AA, Jhajj HS & Tessier PM Understanding and overcoming trade-offs between antibody affinity, specificity, stability and solubility. Biochem Eng J 137, 365–374, doi: 10.1016/j.bej.2018.06.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu Y. et al. Addressing polyspecificity of antibodies selected from an in vitro yeast presentation system: a FACS-based, high-throughput selection and analytical tool. Protein Eng Des Sel 26, 663–670, doi: 10.1093/protein/gzt047 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Makowski EK, Wu L, Desai AA & Tessier PM Highly sensitive detection of antibody nonspecific interactions using flow cytometry. MAbs 13, 1951426, doi: 10.1080/19420862.2021.1951426 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y. et al. Physicochemical Rules for Identifying Monoclonal Antibodies with Drug-like Specificity. Mol Pharm 17, 2555–2569, doi: 10.1021/acs.molpharmaceut.0c00257 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alley EC, Khimulya G, Biswas S, AlQuraishi M & Church GM Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16, 1315–1322, doi: 10.1038/s41592-019-0598-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsu C, Nisonoff H, Fannjiang C & Listgarten J Learning protein fitness models from evolutionary and assay-labeled data. Nat Biotechnol, doi: 10.1038/s41587-021-01146-5 (2022). [DOI] [PubMed] [Google Scholar]
- 35.Makowski EK, Schardt JS, Smith MD & Tessier PM Mutational analysis of SARS-CoV-2 variants of concern reveals key tradeoffs between receptor affinity and antibody escape. PLoS Comput Biol 18, e1010160, doi: 10.1371/journal.pcbi.1010160 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shamsi Z, Chan M & Shukla D TLmutation: Predicting the Effects of Mutations Using Transfer Learning. J Phys Chem B 124, 3845–3854, doi: 10.1021/acs.jpcb.0c00197 (2020). [DOI] [PubMed] [Google Scholar]
- 37.Barrat-Charlaix P, Figliuzzi M & Weigt M Improving landscape inference by integrating heterogeneous data in the inverse Ising problem. Sci Rep 6, 37812, doi: 10.1038/srep37812 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krawczyk K, Liu X, Baker T, Shi J & Deane CM Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 30, 2288–2294, doi: 10.1093/bioinformatics/btu190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tsuchiya Y, Yamamori Y & Tomii K Protein-protein interaction prediction methods: from docking-based to AI-based approaches. Biophys Rev 14, 1341–1348, doi: 10.1007/s12551-022-01032-7 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu Z, Davila A, Wilamowski J, Teraguchi S & Standley DM Improved Antibody-Specific Epitope Prediction Using AlphaFold and AbAdapt. Chembiochem 23, e202200303, doi: 10.1002/cbic.202200303 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jumper J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589, doi: 10.1038/s41586-021-03819-2 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Leem J, Dunbar J, Georges G, Shi J & Deane CM ABodyBuilder: Automated antibody structure prediction with data-driven accuracy estimation. MAbs 8, 1259–1268, doi: 10.1080/19420862.2016.1205773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Abanades B, Georges G, Bujotzek A & Deane CM ABlooper: Fast accurate antibody CDR loop structure prediction with accuracy estimation. Bioinformatics 38, 1877–1880, doi: 10.1093/bioinformatics/btac016 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schneider C, Buchanan A, Taddese B & Deane CM DLAB-Deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 38, 377–383, doi: 10.1093/bioinformatics/btab660 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myung Y, Pires DEV & Ascher DB CSM-AB: graph-based antibody-antigen binding affinity prediction and docking scoring function. Bioinformatics 38, 1141–1143, doi: 10.1093/bioinformatics/btab762 (2022). [DOI] [PubMed] [Google Scholar]