

Abstract
CRISPR gene editing and control systems continue to emerge and inspire novel research and clinical applications. Advances in CRISPR performance such as optimizing the duration of activity in cells, tissues, and organisms, as well as limiting off-target activities, have been extremely important for expanding the utility of CRISPR-based systems. By investigating the effects of various chemical modifications in guide RNAs (gRNAs) at defined positions and combinations, we find that 2′-O-methyl-3′-phosphonoacetate (MP) modifications can be substantially more effective than 2′-O-methyl-3′-phosphorothioate (MS) modifications at the 3′ ends of single-guide RNAs (sgRNAs) to promote high editing yields, in some instances showing an order of magnitude higher editing yield in human cells. MP-modified 3′ ends are especially effective at promoting the activity of guide RNAs cotransfected with Cas messenger RNA (mRNA), as the gRNA must persist in cells until the Cas protein is expressed. We demonstrate such an MP enhancement for sgRNAs cotransfected with a BE4 mRNA for cytidine base editing and also demonstrate that MP at the 3′ ends of prime editing guide RNAs (pegRNAs) cotransfected with PE2 mRNA can promote maximal prime editing yields. In the presence of serum, sgRNAs with MP-modified 3′ ends showed marked improvements in editing efficiency over sgRNAs with MS-modified 3′ ends codelivered with Cas9 mRNA and showed more modest improvements at enhancing the activity of transfected ribonucleoprotein (RNP) complexes. Our results suggest that MP should be considered as a performance-enhancing modification for the 3′ ends of synthetic gRNAs, especially in situations where the guide RNAs may be susceptible to exonuclease-mediated degradation.
One of the significant advantages of producing guide RNAs (gRNAs) by chemical synthesis is that a variety of chemical modifications can be incorporated at defined positions in the sequence to enhance the activity and/or specificity of the gRNA.1−4 Chemical modifications can play various roles in enhancing the activity of gRNAs for maximal editing, one of which is to extend the lifetime of the gRNAs in biological systems by impeding nucleases. The effect of stabilizing the RNA can be important even outside of transfected cells and tissues by preventing degradation during exposure to bodily fluids like serum or routine handling in the laboratory. Initial studies of various modifications including 2′-O-methyl (M), 2′-O-methyl-3′-phosphorothioate (MS), or 2′-O-methyl-3′-thiophosphonoacetate (2′-O-methyl-3′-thioPACE, or MSP) at positions determined to be compatible with guide RNA activity showed that modifications with increasing resistance to exonucleases such as MS and MSP placed at consecutive internucleotide linkages at the 5′ and 3′ ends of single-guide RNAs (sgRNAs) can increasingly enhance CRISPR activity in cells.2 Indeed, since this demonstration, gRNAs with multiple MS modifications at the 5′ and 3′ ends have become commonly used in the field.
Phosphonate modifications can be stably incorporated in DNA and RNA oligonucleotides and have been demonstrated to increase their resistance to nucleases relative to phosphorothioates.5,6 To further explore the potential utility of phosphonate modifications in guide RNAs, we evaluated the performance of gRNAs containing different numbers of consecutive 2′-O-methyl-3′-phosphonoacetate (2′-O-methyl-3′-PACE, or MP) modifications at the 3′ end compared to guide RNAs with MS modifications at that end. We chose to focus on MP rather than MSP because we have found that MP can provide equivalent stability enhancement relative to MSP in cell-based assays (unpublished results) and because it is known that the sulfur atom in phosphorothioates can be lost from the 3′ end during subsequent cycles of nucleotide addition when iodine oxidation is utilized for oligonucleotide synthesis.7 In a previous report exploring the use of MP to enhance the specificity of sgRNAs by incorporating it in the 20-nt guide sequence portion, we found that MP at specific sequence positions such as position 5 or 11 (counted from the 5′ end of the 20 nucleotides) can significantly reduce off-target editing while maintaining high on-target editing.1 However, we also reported that incorporating MP modifications within the first one, two, or three nucleotides at the 5′ ends of sgRNAs can, in some guide sequences, decrease their on-target cleavage activity and/or increase their off-target activities and thus lower specificity.1 Given this observation and the knowledge that 3′ exonucleases account for the majority of exonucleolytic activity in eukaryotic systems,8−10 we focused on evaluating the impact of MP modifications as an alternative to MS modifications at the 3′ ends of sgRNAs for Cas9-based cleavage, base editing, and prime editing.
We present performance advantages for using MP compared to MS to modify the 3′ ends of sgRNAs for creating indels using Cas9 and for editing targeted nucleotides using a cytidine base editor. Our data also show that MP at the 3′ ends of prime editing guide RNAs (pegRNAs) can promote maximal yields for prime editing. To simulate a more challenging cell and tissue environment, we transfected HepG2 hepatocytes that had been cultured with serum but were not rinsed with phosphate-buffered saline (PBS) to remove traces of serum before cotransfecting the cells with sgRNA and Cas9 messenger RNA (mRNA). The advantages of MP over MS modifications for promoting editing yields under these challenging conditions is especially striking, implying that our designs with MP modifications at the 3′ end may give superior performance in other challenging situations, such as in vivo delivery by nanocarriers11−16 or other cell-penetrating formulations.17,18
Materials and Methods
sgRNAs and mRNAs
RNA oligomers were synthesized on Dr. Oligo 48 and 96 synthesizers (Biolytic Lab Performance Inc.) using 2′-O-thionocarbamate-protected nucleoside phosphoramidites (Sigma-Aldrich and Hongene) on controlled pore glass (LGC) according to previously described procedures.19 The 2′-O-methyl-3′-O-(diisopropylamino)-phosphinoacetic acid-1,1-dimethylcyanoethyl ester-5′-O-dimethoxytrityl nucleosides used for synthesis of MP-modified RNAs were purchased from Glen Research and Hongene. For phosphorothioate-containing oligomers, the iodine oxidation step after the coupling reaction was replaced by a sulfurization step using a 0.05 M solution of 3-((N,N-dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-5-thione in a pyridine–acetonitrile (3:2) mixture for 6 min. Unless otherwise noted, reagents for solid-phase RNA synthesis were purchased from Glen Research and Honeywell. The phosphonoacetate (PACE) modifications incorporated in the MP-modified sgRNAs were synthesized using protocols adapted from previous publications5,6 by using the commercially available protected nucleoside phosphinoamidite monomers above. All oligonucleotides were purified using reversed-phase high-performance liquid chromatography (RP-HPLC) and analyzed by liquid chromatography–mass spectrometry (LC–MS) using an Agilent 1290 Infinity series LC system coupled to an Agilent 6545 Q-TOF (time-of-flight) mass spectrometer. In all cases, the mass determined by deconvolution of the series of peaks comprising multiple charge states in a mass spectrum of purified sgRNA matched the expected mass within error of the calibrated instrument (our specification for quality assurance is that the observed mass of purified sgRNA is within 0.01% of the calculated mass), thus confirming the composition of each synthetic sgRNA. Table S1 shows the sequences and confirmed molecular masses of all sgRNAs used in our studies.
CleanCap Cas9 mRNA fully substituted with 5-methoxyuridine was purchased from TriLink (L-7206). BE4-Gam mRNA and PE2 mRNA were purchased from TriLink as custom orders by providing the coding sequences20,21 to which TriLink added their own proprietary 5′ and 3′ UTRs. The custom mRNAs were fully substituted with 5-methylcytidine and pseudouridine, capped with CleanCap AG, and polyA-tailed.
Cell Culture and Nucleofections
Human K562 cells were obtained from ATCC and cultured in RPMI 1640 plus GlutaMax media (Gibco) supplemented with 10% fetal bovine serum (Gibco). K562 cells (within passage nos. 4–14) were nucleofected using a Lonza 4D-Nucleofector (96-well shuttle device, program FF-120) per the manufacturer’s instructions utilizing a Lonza SF cell line kit (V4SC-2960) with 0.2 million cells per transfection in 20 μL of SF buffer combined with 6 μL of 125 pmoles of sgRNA and 1.87 pmoles of BE4-Gam mRNA in PBS buffer for cytidine base editing or combined with 8 μL of 125 pmoles of pegRNA with 100 pmoles of nicking gRNA and 1.35 pmoles of PE2 mRNA in PBS buffer for prime editing. Cells were cultured at 37 °C in ambient oxygen and 5% carbon dioxide and were harvested at 48 h post-transfection.
Human Jurkat clone E6-1 cells were obtained from ATCC and were cultured in RPMI 1640 plus GlutaMax media supplemented with 10% fetal bovine serum. Jurkat cells (within passage nos. 7–20) were nucleofected (program CL-120) utilizing a Lonza SE cell line kit (V4SC-1960) with 0.2 million cells in 20 μL of SE buffer combined with 8 μL of 125 pmoles of pegRNA, 100 pmoles of nicking gRNA, and 1.35 pmoles of PE2 mRNA in PBS buffer. Cultured cells were harvested at 72 h post-transfection.
Human HepG2 cells were obtained from ATCC and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) plus l-glutamine plus 4.5 g/L d-glucose media (Gibco) supplemented with 10% fetal bovine serum. HepG2 cells (within passage nos. 4–13) were spun down from culture media and were either rinsed or not with PBS and spun down again. Cells were nucleofected (program EH-100) utilizing a Lonza SF cell line kit (V4SC-2960) with 0.2 million cells in 20 μL of SF buffer combined with 3 μL of 10 pmoles of sgRNA and 0.0625 pmoles of Cas9 mRNA in PBS buffer or were nucleofected in the presence of residual serum by combining 0.2 million cells in 20 μL of SF buffer with 5 μL of 30 pmoles of sgRNA and 0.5 pmoles of Cas9 mRNA or 12.5 pmoles of SpCas9 protein (Aldevron) in PBS buffer. For 163mer sgRNAs, 0.2 million cells were likewise nucleofected in the presence of residual serum and SF buffer by combining these with 5 μL of 125 pmoles of 163mer sgRNA and 50 pmoles of SpCas9 protein in PBS buffer. For all transfected ribonucleoprotein (RNP) transfections, sgRNA was precomplexed with SpCas9 protein (Aldevron) in PBS buffer by combining and incubating at room temperature for about 20 min before combining with cells in SF buffer for nucleofection. For mRNA transfections, sgRNA was likewise combined with Cas9 mRNA (TriLink) in PBS buffer and kept at room temperature for about 20 min until combined with cells in SF buffer for nucleofection. Cultured HepG2 cells were harvested at about 72 h post-transfection.
Human primary T cells (LP, CR, CD3+, NS) were obtained from AllCells (Alameda, CA) and were cultured in RPMI 1640 plus GlutaMax media supplemented with 10% fetal bovine serum, 5 ng/mL human IL-7, and 5 ng/mL human IL-15 (Gibco). Primary T cells were activated for 48 h with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher) at a beads-to-cells concentration of 3:1. Debeaded primary T cells were nucleofected (program EO-115) utilizing a Lonza P3 primary cell kit (V4SP-3960) with 0.2 million cells in 20 μL of P3 buffer combined with 2.7 μL of 5 pmoles of sgRNA and 0.0625 pmoles of Cas9 mRNA in PBS buffer. Cultured cells were harvested at 7 days post-transfection. Throughout the culture period, T cells were maintained at an approximate density of 1 million cells per mL of media. Following electroporation, additional media was added every 2 days.
Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) Assays
Human K562 cells were cultured as above, and 0.2 million cells per replicate were nucleofected with 125 pmoles of sgRNA (without Cas9 mRNA or protein) as described. For each time point, cells were collected in 1.7 mL Eppendorf tubes, rinsed with PBS, and then resuspended in 750 μL of Qiazol and kept at room temperature for 5 min before being transferred to a −20 °C freezer. Total RNA in PBS was isolated from Qiazol plus chloroform extracts using an miRNeasy kit (Qiagen) on a QiaCube HT, and then immediately reverse-transcribed using a Protoscript II first-strand cDNA synthesis kit (NEB). qRT-PCR was performed on an Applied Biosystems QuantStudio 6 Flex instrument using TaqPath ProAmp master mix with two TaqMan MGB probes, one for sgRNA labeled with FAM and the other for U6 snRNA labeled with VIC (Thermo Fisher) for normalization to the amount of total RNA isolated, calculated as ΔCt. The ΔCt values for triplicate samples were averaged and normalized to the smallest observed mean ΔCt value to calculate ΔΔCt values. Relative sgRNA levels were calculated as 2–ΔΔCt. Guide RNA sequences are listed in Table S1, and primer and probe sequences are listed in Table S2; results are listed in Table S3.
PCR-Targeted Deep Sequencing and Quantification of Targeted Genomic Modifications
Genomic DNA purification and construction of PCR-targeted deep sequencing libraries were performed as previously described.1 Library concentration was determined using a Qubit dsDNA BR assay kit (Thermo Fisher). Paired-end 2 × 220-bp reads were sequenced on a MiSeq (Illumina) at 0.8 ng/μL of PCR-amplified library along with 20.5% PhiX. The primer, index, and target sequences used are listed in Table S4.
Paired-end reads were merged using FLASH ver. 1.2.11 software,22 and then mapped to the human genome using BWA-MEM software (bwa-0.7.10) set to default parameters. Reads were scored as having an indel or not according to whether an insertion or a deletion was found within 10 bp’s of the Cas9 cleavage site. For prime editing analysis, reads were scored as having an edit if the desired edit was identified in the read. For cytidine base editing analysis, reads were scored as base-edited if cytidines were edited within a window of 10–20 bp upstream of the protospacer adjacent motif (PAM) site. For each replicate in each experiment, mapped reads were segregated according to mapped amplicon locus and were binned by the presence or absence of an indel or edit. The tally of reads per bin was used to calculate %indels or %edits produced at each locus. Indel or edit yields and standard deviations for plots were calculated by logit transformation of %indels or %edits, transformed as ln[r/(1 – r)] where r is the %indels or %edits per specific locus, to closely approximate a normal distribution. Triplicate mock transfections provided a mean mock control (or negative control), and triplicate samples showing a mean indel yield or mean edit yield significantly higher (t test p < 0.05) than the corresponding negative control were considered above background.
Results and Discussion
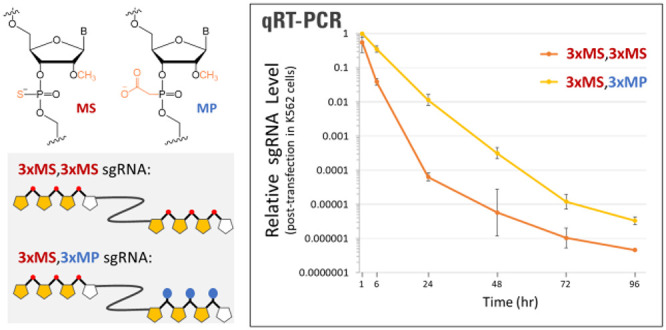
To evaluate the relative lifetimes of sgRNAs with MS or MP modifications at the 3′ end in transfected cells (see Figure 1A for chemical structures of the modifications), we chemically synthesized sgRNAs that had MS modifications at the first three internucleotide linkages at the 5′ end and either MS modifications at the last three internucleotide linkages at the 3′ end (denoted as 3xMS,3xMS) or two, three, or four consecutive MP modifications at the terminal internucleotide linkages at the 3′ end (denoted as 3xMS,2xMP, 3xMS,3xMP, and 3xMS,4xMP, respectively). Sequences of all sgRNAs tested in this publication including chemical modifications at specific nucleotides are listed in Table S1. Each sgRNA was transfected individually into human K562 cells in the absence of Cas9, and qRT-PCR was used to measure the relative amount of sgRNA remaining in cells collected at a series of time points from 1 to 96 h post-transfection.
Figure 1.

Incorporation of 2′-O-methyl-3′-PACE (MP) modifications at the 3′ end of chemically synthesized sgRNAs promotes stability and editing yields. (A) Structures of chemical modifications used in synthetic gRNAs and schematic structures of two examples of sgRNAs with MS or MP 3′ end modifications. (B) sgRNAs modified by 3xMS at the 5′ end and various modification schemes at the 3′ end (as indicated) were transfected into K562 cells in the absence of Cas protein. The amounts of the various gRNAs present at different time points following transfection were measured by qRT-PCR. (C and D) sgRNAs modified by 3xMS at the 5′ end and various modification schemes at the 3′ end, as indicated, without or with an additional MP modification at position 5 to improve specificity, were cotransfected with Cas9 mRNA in HepG2 cells (C) as well as in primary human T cells (D). (E) K562 cells were cotransfected with BE4-Gam mRNA and synthetic sgRNA modified by 3xMS or 3xMP at the 3′ end. Indel and editing yields were measured by deep sequencing of PCR amplicons of the HBB target locus (HBB_ON) and a known highly reactive off-target site on chromosome 9 (HBB_OFF1). Bars on bar graphs or dots and error bars on line graphs represent means with standard deviation (n = 3).
We observed a much steeper decline in the relative level of the 3xMS,3xMS sgRNA detected across 1, 6, and 24 h post-transfection than we observed for any of the sgRNAs modified with MPs at the 3′ end (either two, three, or four consecutive MPs) (Figure 1B, Table S3). Specifically, at 1 h post-transfection, the relative amounts of transfected sgRNA differed by only 2.6-fold with largely overlapping error bars among all four variations of 3′ end protection, whereas much larger differences were observed at 6 h post-transfection, when the remaining amount of 3xMS,3xMS-protected sgRNA had dropped to a relative level of about 1/10 (0.039) that of the 3xMS,3xMP- and 3xMS,4xMP-protected sgRNAs (0.341–0.351). The differences became even larger at the 24 h time point where they varied according to the level of 3′ end protection in a logical progression from having 3xMS to 2xMP to 3xMP to 4xMP at the 3′ end, resulting in residual sgRNA levels that spanned ca. 250-fold, consistent with the level of 3′ end protection. Thus, we found that incorporating MP modifications at the 3′ end of uncomplexed sgRNAs can significantly enhance their stability in transfected cells relative to MS modifications, specifically by 1–2 orders of magnitude for three different MP-modified sgRNAs tested in parallel with an MS-only modified sgRNA. The designs with three or four consecutive MPs at the 3′ end can prolong the lifetimes of the free sgRNAs across even longer time points (72 and 96 h post-transfection).
Although RNP has become the preferred mode for delivering Cas9 into cells ex vivo, delivery of Cas enzymes using mRNA can be preferred in some situations; for example, several newer CRISPR technologies such as base editing and prime editing are not easily served by transfecting a purified protein which is large, prone to precipitation, and not commercially available as a catalog product. These editing techniques often rely on transient transfection of the mRNA to express the protein.23,24 To evaluate the potential impact of MP versus MS modifications at the 3′ ends of sgRNAs on CRISPR editing in cells, modified sgRNAs were cotransfected with Cas9 mRNA using amounts of the RNAs determined to yield about 40–60% editing efficiency with 3xMS,3xMS-modified sgRNA (data not shown), thus enabling detection of potential differences in performance associated with systematic variations in chemically modified sgRNA designs. The guide RNA that targets the HBB sickle cell allele in our experiments is also known to cleave an off-target site on chromosome 9 (referred to as the OFF1 site) with high efficiency, which provides another measure of sgRNA activity. We observed that MP modification of the 3′ end significantly enhanced editing yields in HepG2 cells (Figure 1C, Table S3). For instance, our designs with two, three, or four consecutive MP modifications at the 3′ end gave at least 2-fold more Cas9-mediated indels than a comparable design with 3xMS at the 3′ end (81–83% at the ON-target site for the 2xMP, 3xMP, and 4xMP modifications versus 38% for 3xMS at the 3′ end). A similar trend was observed for the same sgRNAs transfected into primary human T cells although the increase was more modest in that the 2xMP, 3xMP, and 4xMP modifications gave a 1.3-fold higher level of ON-target indels than when using 3xMS at the 3′ end (Figure 1D, Table S3). Incorporation of an additional MP at position 5 in the 20-nt guide sequence portion of the sgRNA significantly lowered editing at the OFF1 site in both cell types while maintaining high on-target editing efficiency, as we reported previously as a means for enhancing specificity.1 Indeed, indels at the OFF1 site were reduced by 7–10-fold in HepG2 cells by incorporating MP at position 5 and similarly by 6–7-fold in primary T cells.
Base editors are a class of alternative genome editing systems originally built around Cas9 nickase (nCas9) or dead Cas9 (dCas9) fused to deaminases that enable editing of genomic DNA in cells without creating double-stranded breaks. Both cytidine base editors (CBE)23−28 and adenosine base editors (ABE)29−34 have been reported, and these have inspired a number of variations for base editing.35 We tested the potential benefits of using MP modifications in contrast to MS modifications at the 3′ ends of such sgRNAs in the context of a CBE, namely, BE4-Gam mRNA.20 We observed a 1.4-fold higher level of cytidine editing using CBE mRNA in K562 cells cotransfected with sgRNA modified with MP at the 3′ end versus an alternative design with MS at the 3′ end (Figure 1E, Table S3).
In prime editing systems,21,36,37 pegRNAs have additional sequences on their 3′ ends: a template sequence that encodes the desired edit and a downstream primer binding sequence where one could incorporate terminal modifications such as MS and/or MP at the 3′ terminus to impede exonucleases, potentially without inhibiting prime editing activity. As illustrated in Figure 2A, we explored two approaches for prime editing adopted from the literature that knock out the PAM in EMX1 or introduce a three-base insertion in RUNX1, both of which utilize pegRNAs with a primer binding sequence comprising 15 nucleotides.21 We compared pegRNAs having 3xMS at the 3′ end for both targets with alternative designs having one, two, or three consecutive MPs at the 3′ end, each cotransfected with PE2 mRNA in K562 or Jurkat cells. Our results show that pegRNAs with MP modifications at the 3′ end performed well and can achieve comparable, or in some cases somewhat higher, editing yields than 3xMS (Figure 2B–E, Table S3). For the two pegRNA sequences tested here, designs with 2xMP and/or 3xMP at the 3′ end performed consistently better than designs with 1xMP at the 3′ end (specifically 1.2–1.4-fold better in Figure 2B–E, Table S3). Given that the 3′ end of a pegRNA functions to capture the nicked end of the target site and then bind the reverse transcriptase portion of the prime editor to initiate reverse transcription, it is important to note that different primer binding sequences, including those of different lengths, may benefit from different end protection designs, and some pegRNAs may benefit from extension of their 3′ ends to accommodate chemical modifications such as by adding a short polyuridine tail to the 3′ end.38
Figure 2.

Incorporation of MP or MS modifications at the 3′ end of chemically synthesized pegRNAs can improve prime editing yields. (A) Prime editing approaches were adopted to knockout the PAM in EMX1 or to introduce a three-base insertion in RUNX1. (B and D) K562 cells were cotransfected with PE2 mRNA and synthetic pegRNA modified by 3xMS at the 5′ end and various modification schemes at the 3′ end (as indicated) for editing EMX1 (B) or RUNX1 (D). (C and E) Jurkat cells were likewise transfected using the same pegRNAs for editing EMX1 (C) or RUNX1 (E). Editing yields were measured by deep sequencing of PCR amplicons of the target loci for both the desired edit (%Edit) and any contaminating indel byproducts (%By-indels). Bars represent means with standard deviation (n = 3).
To simulate harsher cellular environments that CRISPR-Cas components may encounter when delivered in vivo (as by nanocarriers or other cell-penetrating formulations), we cotransfected Cas9 mRNA with sgRNA into cells that were isolated from culture media but not rinsed with PBS to remove residual serum which is known to contain nucleases. Under these conditions, higher amounts of sgRNA and Cas9 mRNA were needed to achieve substantial levels of editing; specifically, we used 3-fold more sgRNA and 8-fold more Cas9 mRNA per transfection for the experiment in Figure 3A than the experiment in Figure 1C where the same numbers of cells per transfection were washed before introducing the CRISPR-Cas components. It seems likely that extracellular exonucleases in serum not rinsed from cells are degrading the transfected RNA. We found that sgRNAs with MP modifications at the 3′ end gave substantially higher editing yields (by an order of magnitude or more) than sgRNAs with MS modifications at the 3′ end when cotransfected with Cas9 mRNA into unrinsed HepG2 cells (Figure 3A, Table S3). Specifically, we observed 15–44% editing for sgRNAs with one or more MPs at the 3′ end versus less than 2% with 3xMS at the 3′ end. In a parallel experiment, an RNP version of each sgRNA was prepared by precomplexation in PBS buffer and transfected into aliquots of unrinsed HepG2 cells. As expected, the unmodified and 3xMS-modified sgRNAs gave higher indel yields as RNP formulations than when these were cotransfected with Cas9 mRNA, as precomplexation of sgRNA with Cas9 protein in RNP is thought to help shield the sgRNA from nucleolytic degradation (Figure 3, part B vs part A). Even though the improvement in editing efficiency between RNPs incorporating sgRNA with MP versus MS modifications at the 3′ end was not as dramatic as when using these modifications in cotransfections with Cas9 mRNA, designs with MPs at the 3′ end gave significantly higher Cas9-mediated indels than a comparable design with 3xMS at the 3′ end (70–73% indels at the ON-target site for the 2xMP, 3xMP, and 4xMP modifications at the 3′ end vs 52% for 3xMS at the 3′ end, a difference of about 1.3-fold) (Figure 3B, Table S3). A similar outcome was observed for a different set of synthetic 163mer sgRNAs designed for CRISPRa SAM systems but used with SpCas9 protein in RNP formulations to produce indels instead of using them for gene activation by CRISPRa (Figure 3C, Table S3).
Figure 3.

Use of MP modifications at the 3′ end of chemically synthesized sgRNAs helps to maximize editing yields in the presence of serum. (A and B) To simulate a harsher biological environment for delivering CRISPR-Cas9 to cells in vivo (where bodily fluids such as serum are present), HepG2 cells were spun down from culture media without rinsing off residual bovine serum, and the cells were transfected with sgRNAs targeting the HBB locus containing different types of 3′ end modifications (as indicated) together with Cas9 mRNA (A) or were transfected with Cas9 RNP prepared by precomplexation of the sgRNA with SpCas9 protein (B). (C) Extended-length 163mer sgRNAs targeting the same HBB locus but designed for CRISPRa SAM systems were precomplexed with SpCas9 protein for targeted double-strand cleavage and indel formation by transfecting the Cas9 RNP into HepG2 cells, again without rinsing off residual serum. Indel yields were measured by deep sequencing of PCR amplicons of the target HBB locus (HBB_ON) and a known highly reactive off-target site on chromosome 9 (HBB_OFF1). Bars represent means with standard deviation (n = 3).
Overall, our results make a compelling case that MP modifications incorporated at the 3′ ends of gRNAs for Cas9-based editing systems can confer significant improvements in editing efficiencies compared to gRNAs with MS modifications at their 3′ ends. Such MP modifications can be especially helpful under conditions where the gRNAs need to persist in cells prior to binding to Cas protein, as when cotransfected with Cas-encoded mRNA or under conditions where the gRNA encounters relatively high levels of 3′ exonuclease activity. Chemical modifications other than PACE have been carefully placed in internal regions of sgRNAs, and several of these designs showed enhanced CRISPR activity beyond that of gRNAs containing only MS modifications at the 5′ and 3′ ends,3,4,11 for example, when chemically synthesized sgRNAs were codelivered in lipid nanoparticles with mRNAs encoding Cas enzymes.3,11 Our results suggest that employing MP modifications at the 3′ end of such guide RNAs may provide further improvements in therapeutic efficacy for CRISPR applications, whether transfecting cells ex vivo or in vivo.
Acknowledgments
We thank R. Brennen, K. Schleifer, and J. Irribarren for engineering solutions, C. Carstens, T. Paredes-Santos, J. Myerson, and A. Apffel for intellectual contributions, and J. Sampson, S. Laderman, and P. Hwung for critical reading of the manuscript. A. Krishnan, P. Chaffey, D. Fisher, and members of the Agilent Genomics division provided helpful input and comments. Agilent Technologies provided funding.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00768.
Sequences of the guide RNAs tested, Table S1(XLSX)
PCR primers and probes for qRT-PCR assays, Table S2(XLSX)
All data in figures including standard deviations for biological triplicates, Table S3(XLSX)
PCR primers and amplicons for deep sequencing of on- and off-target sites in the human genome, Table S4(XLSX)
Author Present Address
§ CellMax Life, Sunnyvale, CA 94085, United States
Author Present Address
∥ Novartis Institutes for Biomedical Research, San Diego, CA 92121, United States
Author Present Address
⊥ UT Southwestern Medical Center, Dallas, TX 75390, United States
Author Present Address
# Cepheid, Bothell, WA 98021, United States
Author Present Address
∇ Quantum-Si, Guilford, CT 06437, United States
Author Present Address
▼ Agilent Technologies, Santa Clara, California, 95051, United States
The authors declare the following competing financial interest(s): D.E.R., I.S., S.T., B.D.L., R.J.K., R.M., W.R.W.W., B.C., D.J.D., and L.B. are employees of Agilent Technologies. T.D.-L., D.T., S.V.-S., M.O., J.T., and M.S. were compensated by Agilent for their work on this research. At the time that some of the experiments were performed for the results presented here, D.J.D. individually owned patent rights for the PACE modification used in this work.
Supplementary Material
References
- Ryan D. E.; Taussig D.; Steinfeld I.; Phadnis S. M.; Lunstad B. D.; Singh M.; Vuong X.; Okochi K. D.; McCaffrey R.; Olesiak M.; et al. Improving CRISPR-Cas specificity with chemical modifications in single-guide RNAs. Nucleic Acids Res. 2018, 46, 792–803. 10.1093/nar/gkx1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendel A.; Bak R. O.; Clark J. T.; Kennedy A. B.; Ryan D. E.; Roy S.; Steinfeld I.; Lunstad B. D.; Kaiser R. J.; Wilkens A. B.; Bacchetta R.; Tsalenko A.; Dellinger D.; Bruhn L.; Porteus M. H. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015, 33, 985–989. 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H.; Song C.-Q.; Suresh S.; Wu Q.; Walsh S.; Rhym L. H.; Mintzer E.; Bolukbasi M. F.; Zhu L. J.; Kauffman K.; Mou H.; Oberholzer A.; Ding J.; Kwan S.-Y.; Bogorad R. L.; Zatsepin T.; Koteliansky V.; Wolfe S. A.; Xue W.; Langer R.; Anderson D. G. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017, 35, 1179–1187. 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir A.; Alterman J. F.; Hassler M. R.; Debacker A. J.; Hudgens E.; Echeverria D.; Brodsky M. H.; Khvorova A.; Watts J. K.; Sontheimer E. J. Heavily and fully modified RNAs guide efficient SpyCas9-mediated genome editing. Nat. Commun. 2018, 9, 2641. 10.1038/s41467-018-05073-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellinger D. J.; Sheehan D. M.; Christensen N. K.; Lindberg J. G.; Caruthers M. H. Solid-phase chemical synthesis of phosphonoacetate and thiophosphonoacetate oligodeoxynucleotides. J. Am. Chem. Soc. 2003, 125, 940–950. 10.1021/ja027983f. [DOI] [PubMed] [Google Scholar]
- Threlfall R. N.; Torres A. G.; Krivenko A.; Gait M. J.; Caruthers M. H. Synthesis and biological activity of phosphonoacetate– and thiophosphonoacetate–modified 2′-O-methyl oligoribonucleotides. Org. Biomol. Chem. 2012, 10, 746–754. 10.1039/C1OB06614E. [DOI] [PubMed] [Google Scholar]
- Wyrzykiewicz T. K.; Cole D. L. Sequencing of oligonucleotide phosphorothioates based on solid-supported desulfurization. Nucleic Acids Res. 1994, 22, 2667–2669. 10.1093/nar/22.13.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseley J.; LaCava J.; Tollervey D. RNA-quality control by the exosome. Nat. Rev. Mol. Cell Biol. 2006, 7, 529–539. 10.1038/nrm1964. [DOI] [PubMed] [Google Scholar]
- Shaw J.-P.; Kent K.; Bird J.; Fishback J.; Froehler B. Modified deoxyoligonucleotides stable to exonuclease degradation in serum. Nucleic Acids Res. 1991, 19, 747–750. 10.1093/nar/19.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham M. J.; Crooke S. T.; Monteith D. K.; Cooper S. R.; Lemonidis K. M.; Stecker K. K.; Martin M. J.; Crooke R. M. In vivo distribution and metabolism of a phosphorothioate oligonucleotide within rat liver after intravenous administration. J. Pharmacol. Exp. Ther. 1998, 286, 447–458. [PubMed] [Google Scholar]
- Finn J. D.; Smith A. R.; Patel M. C.; Shaw L.; Youniss M. R.; van Heteren J.; Dirstine T.; Ciullo C.; Lescarbeau R.; Seitzer J.; Shah R. R.; Shah A.; Ling D.; Growe J.; Pink M.; Rohde E.; Wood K. M.; Salomon W. E.; Harrington W. F.; Dombrowski C.; Strapps W. R.; Chang Y.; Morrissey D. V. A single administration of CRISPR/Cas9 lipid nanoparticles achieves robust and persistent in vivo genome editing. Cell Rep. 2018, 22, 2227–2235. 10.1016/j.celrep.2018.02.014. [DOI] [PubMed] [Google Scholar]
- Sago C. D.; Lokugamage M. P.; Paunovska K.; Vanover D. A.; Monaco C. M.; Shah N. N.; Gamboa Castro M.; Anderson S. E.; Rudoltz T. G.; Lando G. N.; et al. High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, E9944–E9952. 10.1073/pnas.1811276115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q.; Wei T.; Farbiak L.; Johnson L. T.; Dilliard S. A.; Siegwart D. J. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR–Cas gene editing. Nat. Nanotechnol. 2020, 15, 313–320. 10.1038/s41565-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley M. M.; Singh N.; Ravikumar P.; Zhang R.; June C. H.; Mitchell M. J. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020, 20, 1578–1589. 10.1021/acs.nanolett.9b04246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D. N.; Roth T. L.; Li P. J.; Chen P. A.; Apathy R.; Mamedov M. R.; Vo L. T.; Tobin V. R.; Goodman D.; Shifrut E.; Bluestone J. A.; Puck J. M.; Szoka F. C.; Marson A. Polymer-stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat. Biotechnol. 2020, 38, 44–49. 10.1038/s41587-019-0325-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlatkovic I. Non-immunotherapy application of LNP-mRNA: maximizing efficacy and safety. Biomedicines 2021, 9, 530. 10.3390/biomedicines9050530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurthy S.; Wohlford-Lenane C.; Kandimalla S.; Sartre G.; Meyerholz D. K.; Théberge V.; Hallée S.; Duperré A.-M.; Del’Guidice T.; Lepetit-Stoffaes J.-P.; et al. Engineered amphiphilic peptides enable delivery of proteins and CRISPR-associated nucleases to airway epithelia. Nat. Commun. 2019, 10, 4906. 10.1038/s41467-019-12922-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Røise J. J.; He M.; Das R.; Murthy N. Non-viral strategies for delivering genome editing enzymes. Adv. Drug Delivery Rev. 2021, 168, 99–117. 10.1016/j.addr.2020.09.004. [DOI] [PubMed] [Google Scholar]
- Dellinger D. J.; Timar Z.; Myerson J.; Sierzchala A. B.; Turner J.; Ferreira F.; Kupihár Z.; Dellinger G.; Hill K. W.; Powell J. A.; Sampson J. R.; Caruthers M. H. Streamlined process for the chemical synthesis of RNA using 2′-O-thionocarbamate-protected nucleoside phosphoramidites in the solid phase. J. Am. Chem. Soc. 2011, 133, 11540–11556. 10.1021/ja201561z. [DOI] [PubMed] [Google Scholar]
- Komor A. C.; Zhao K. T.; Packer M. S.; Gaudelli N. M.; Waterbury A. L.; Koblan L. W.; Kim Y. B.; Badran A. H.; Liu D. R. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv. 2017, 3, eaao4774. 10.1126/sciadv.aao4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A. V.; Randolph P. B.; Davis J. R.; Sousa A. A.; Koblan L. W.; Levy J. M.; Chen P. J.; Wilson C.; Newby G. A.; Raguram A.; Liu D. R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157. 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoč T.; Salzberg S. L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor A. C.; Kim Y. B.; Packer M. S.; Zuris J. A.; Liu D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016, 533, 420–424. 10.1038/nature17946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblan L. W.; Doman J. L.; Wilson C.; Levy J. M.; Tay T.; Newby G. A.; Maianti J. P.; Raguram A.; Liu D. R. Improving cytidine and adenine base editors by expression optimization and ancenstral reconstruction. Nat. Biotechnol. 2018, 36, 843–846. 10.1038/nbt.4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doman J. L.; Raguram A.; Newby G. A.; Liu D. R. Evaluation and minimization of Cas9-independent off-target DNA editing by cytosine base editors. Nat. Biotechnol. 2020, 38, 620–628. 10.1038/s41587-020-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Y.; Leete T. C.; Born D. A.; Young L.; Barrera L. A.; Lee S.-J.; Rees H. A.; Ciaramella G.; Gaudelli N. M. Cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity. Nat. Commun. 2020, 11, 2052. 10.1038/s41467-020-15887-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koblan L. W.; Erdos M. R.; Wilson C.; Cabral W. A.; Levy J. M.; Xiong Z.-M.; Tavarez U. L.; Davison L. M.; Gete Y. G.; Mao X.; Newby G. A.; Doherty S. P.; Narisu N.; Sheng Q.; Krilow C.; Lin C. Y.; Gordon L. B.; Cao K.; Collins F. S.; Brown J. D.; Liu D. R. In vivo base editing rescues Hutchinson-Gilford progeria syndrome in mice. Nature 2021, 589, 608–614. 10.1038/s41586-020-03086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villiger L.; Rothgangl T.; Witzigmann D.; Oka R.; Lin P. J. C.; Qi W.; Janjuha S.; Berk C.; Ringnalda F.; Beattie M. B.; Stoffel M.; Thöny B.; Hall J.; Rehrauer H.; van Boxtel R.; Tam Y. K.; Schwank G. In vivo cytidine base editing of hepatocytes without detectable off-target mutations in RNA and DNA. Nat. Biomed. Eng. 2021, 5, 179–189. 10.1038/s41551-020-00671-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N. M.; Komor A. C.; Rees H. A.; Packer M. S.; Badran A. H.; Bryson D. I.; Liu D. R. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 2017, 551, 464–471. 10.1038/nature24644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grünewald J.; Zhou R.; Iyer S.; Lareau C. A.; Garcia S. P.; Aryee M. J.; Joung J. K. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 2019, 37, 1041–1048. 10.1038/s41587-019-0236-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M. F.; Zhao K. T.; Eton E.; Lapinaite A.; Newby G. A.; Thuronyi B. W.; Wilson C.; Koblan L. W.; Zeng J.; Bauer D. E.; Doudna J. A.; Liu D. R. Phage-assisted evolution of an adenine base editor with improved Cas domain compatibility and activity. Nat. Biotechnol. 2020, 38, 883–891. 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N. M.; Lam D. K.; Rees H. A.; Solá-Esteves N. M.; Barrera L. A.; Born D. A.; Edwards A.; Gehrke J. M.; Lee S.-J.; Liquori A. J.; Murray R.; Packer M. S.; Rinaldi C.; Slaymaker I. M.; Yen J.; Young L. E.; Ciaramella G. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. 10.1038/s41587-020-0491-6. [DOI] [PubMed] [Google Scholar]
- Musunuru K.; Chadwick A. C.; Mizoguchi T.; Garcia S. P.; DeNizio J. E.; Reiss C. W.; Wang K.; Iyer S.; Dutta C.; Clendaniel V.; Amaonye M.; Beach A.; Berth K.; Biswas S.; Braun M. C.; Chen H.-M.; Colace T. V.; Ganey J. D.; Gangopadhyay S. A.; Garrity R.; Kasiewicz L. N.; Lavoie J.; Madsen J. A.; Matsumoto Y.; Mazzola A. M.; Nasrullah Y. S.; Nneji J.; Ren H.; Sanjeev A.; Shay M.; Stahley M. R.; Fan S. H. Y.; Tam Y. K.; Gaudelli N. M.; Ciaramella G.; Stolz L. E.; Malyala P.; Cheng C. J.; Rajeev K. G.; Rohde E.; Bellinger A. M.; Kathiresan S. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. 10.1038/s41586-021-03534-y. [DOI] [PubMed] [Google Scholar]
- Rothgangl T.; Dennis M. K.; Lin P. J. C.; Oka R.; Witzigmann D.; Villiger L.; Qi W.; Hruzova M.; Kissling L.; Lenggenhager D.; et al. In vivo adenine base editing of PCSK9 in macques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. 10.1038/s41587-021-00933-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzalone A. V.; Koblan L. W.; Liu D. R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. 10.1038/s41587-020-0561-9. [DOI] [PubMed] [Google Scholar]
- Nelson J. W.; Randolph P. B.; Shen S. P.; Everette K. A.; Chen P. J.; Anzalone A. V.; An M.; Newby G. A.; Chen J. C.; Hsu A.; et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 2022, 40, 402. 10.1038/s41587-021-01039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P. J.; Hussmann J. A.; Yan J.; Knipping F.; Ravisankar P.; Chen P.-F.; Chen C.; Nelson J. W.; Newby G. A.; Sahin M.; et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 2021, 184, 5635–5652. 10.1016/j.cell.2021.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petri K.; Zhang W.; Ma J.; Schmidts A.; Lee H.; Horng J. E.; Kim D. Y.; Kurt I. C.; Clement K.; Hsu J. Y.; et al. CRISPR prime editing with ribonucleoprotein complexes in zebrafish and primary human cells. Nat. Biotechnol. 2022, 40, 189. 10.1038/s41587-021-00901-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.