

Conspectus
Nickel excels at facilitating selective radical chemistry, playing a pivotal role in metalloenzyme catalysis and modern cross-coupling reactions. Radicals, being nonpolar and neutral, exhibit orthogonal reactivity to nucleophilic and basic functional groups commonly present in biomolecules. Harnessing this compatibility, we delve into the application of nickel-catalyzed radical pathways in the synthesis of noncanonical peptides and carbohydrates, critical for chemical biology studies and drug discovery.
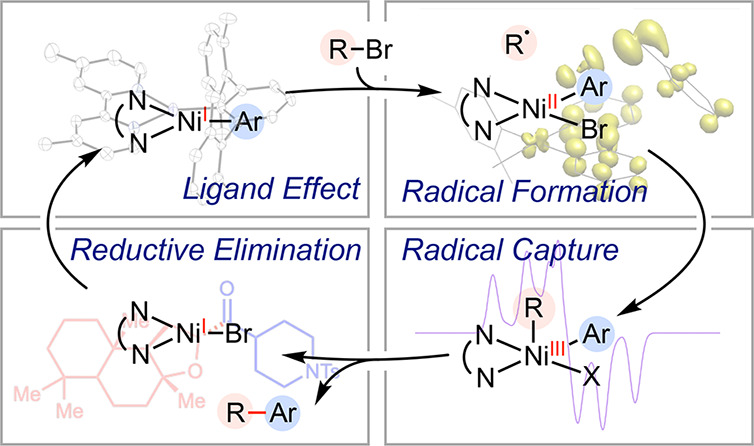
We previously characterized a sequential reduction mechanism that accounts for chemoselectivity in cross-electrophile coupling reactions. This catalytic cycle begins with nickel(I)-mediated radical generation from alkyl halides, followed by carbon radical capture by nickel(II) complexes, and concludes with reductive elimination. These steps resonate with mechanistic proposals in nickel-catalyzed cross-coupling, photoredox, and electrocatalytic reactions. Herein, we present our insights into each step involving radicals, including initiation, propagation, termination, and the nuances of kinetics, origins of selectivity, and ligand effects.
Radical generation from C(sp3) electrophiles via one-electron oxidative addition with low-valent nickel radical intermediates provides the basis for stereoconvergent and cross-electrophile couplings. Our electroanalytical studies elucidate a concerted halogen atom abstraction mechanism, where electron transfer is coupled with halide dissociation. Using this pathway, we have developed a nickel-catalyzed stereoselective radical addition to dehydroalanine, facilitating the synthesis of noncanonical peptides. In this application, chiral ligands modulate the stereochemical outcome through the asymmetric protonation of a nickel-enolate intermediate.
The capture of the alkyl radical by nickel(II) expands the scope of cross-coupling, promotes reductive elimination through the formation of high-valent nickel(III) species, and governs chemo- and stereoselectivity. We discovered that nickel(II)-aryl efficiently traps radicals with a barrier ranging from 7 to 9 kcal/mol, followed by fast reductive elimination. In contrast, nickel(II)-alkyl captures radicals to form a nickel(III) species, which was characterized by EPR spectroscopy. However, the subsequent slow reductive elimination resulted in minimal product formation. The observed high diastereoselectivity of radical capture inspired investigations into C-aryl and C-acyl glycosylation reactions. We developed a redox auxiliary that readily couples with natural carbohydrates and produces glycosyl radicals upon photoredox activation. Nickel-catalyzed cross-coupling of the glycosyl radical with bromoarenes and carboxylic acids leads to diverse non-natural glycosides that can facilitate drug discovery.
Stoichiometric studies on well-defined d8-nickel complexes have showcased means to promote reductive elimination, including ligand association, oxidation, and oxidative addition.
In the final section, we address the influence of auxiliary ligands on the electronic structure and redox activity of organonickel intermediates. Synthesis of a series of low-valent nickel radical complexes and characterization of their electronic structures led us to a postulate that ligand redox activity correlates with coordination geometry. Our data reveal that a change in ligand redox activity can shift the redox potentials of reaction intermediates, potentially altering the mechanism of catalytic reactions. Moreover, coordinating additives and solvents may stabilize nickel radicals during catalysis by adjusting ligand redox activity, which is consistent with known catalytic conditions.
Key References
Lin, Q.; Diao, T. Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc.2019, 141, 17937–17948.1 A mechanistic study that establishes the sequential reduction mechanism for a nickel-catalyzed cross-electrophile coupling reaction and elucidates the mechanistic origin of cross-electrophile selectivity.
Qi, X.; Jambu, S.; Ji, Y.; Belyk, K. M.; Panigrahi, N. R.; Arora, P. S.; Strotman, N. A.; Diao, T. Late-Stage Modification of Oligopeptides by Nickel-Catalyzed Stereoselective Radical Addition to Dehydroalanine. Angew. Chem. Int. Ed.2022, 61, e202213315.2 A method for synthesizing noncanonical peptides via diastereoselective radical addition to dehydroalanine using chiral nickel catalysts.
Lin, Q.; Spielvogel, E. H.; Diao, T. Carbon-Centered Radical Capture at Nickel(II) Complexes: Spectroscopic Evidence, Rates, and Selectivity. Chem.2023, 9, 1295–1308.3 A study that provides spectroscopic evidence for radical capture by nickel, measures the rate of radical capture at nickel(II) complexes and reveals the difference in reactivity between C(sp2)–C(sp3) and C(sp3)–C(sp3) bond formation processes.
Dawson, G. A.; Lin, Q.; Neary, M. C.; Diao, T. Ligand Redox Activity of Organonickel Radical Complexes Governed by the Geometry. J. Am. Chem. Soc.2023, 145, 20551–20561.4 A collection of well-defined nickel radical complexes that reveals an empirical correlation between coordination geometry and ligand redox activity.
1. Introduction
Radical chemistry plays a vital role in biological processes, such as metabolism, replication, and biosynthesis.5 Many of these processes are catalyzed by cofactors featuring first-row base metals, including nickel.6 The relatively high pairing energy combined with weak ligand field stabilization leads to stable open-shell electronic configurations for first-row transition metal catalysts. These paramagnetic complexes are adept at initiating and propagating radical reactions through single-electron transfer (SET) and radical capture mechanisms. For instance, methyl-CoM reductase (MCR), the rate-limiting enzyme in methanogenesis and anaerobic methane oxidation, produces over a billion tons of methane annually. This massive output is achieved through SET from nickel(I) hydrocorphinate F430 cofactor 1 to methyl-CoM, resulting in the generation of a methyl radical (Scheme 1A).7 Other nickel-containing enzymes, including carbon monoxide dehydrogenase8 and nickel superoxide dismutase,9 respectively catalyze the reversible oxidation of CO to CO2 and the disproportionation of superoxide anion radical to molecular oxygen and hydrogen peroxide.
Scheme 1. Nickel Radicals in Enzymes and Catalysis.

Metalloenzyme-catalyzed radical reactions highlight the orthogonal reactivity of radicals and the abundant polar functional groups found in biomolecules.10 While the reactivity of organic radicals are often perceived as overly reactive, their nonpolar and neutral nature make them compatible with the dominant nucleophilic and basic functional groups in biomolecules (Scheme 1B).11 This compatibility can confer chemo- and site-selectivity, evident in recent advancements in late-stage modification of proteins and DNA, achieved through transition metal catalysis,12 photoredox catalysis,13,14 and electrocatalysis.15
Over the past two decades, the rapid growth in synthetic methodology has been propelled by the application of nickel catalysts, due to their ability to mediate radical reactions.16,17 Modern advancements in cross-coupling reactions have broadened the scope, unlocking new routes to quickly access complex molecular architectures found in agrochemicals, pharmaceuticals, and materials. Yet, despite seminal contributions both historically and in recent times, a comprehensive mechanistic understanding of nickel-catalyzed transformations remains elusive.18 This article summarizes our efforts, alongside many others in this field, to unravel the fundamental reactivity of nickel-mediated radical processes, elucidate ligand effects, and develop radical reactions suitable for modifying biomolecules, including peptides and carbohydrates.
A notable synthetic application of nickel catalysis is the cross-electrophile coupling reaction.19 By utilizing two electrophiles, these methods circumvent the necessity for air-sensitive nucleophiles, paving the way for an extensive scope of coupling partners with good functional group compatibility. Our recent study delineated that a cross-electrophile coupling reaction proceeds through a sequential reduction mechanism (Scheme 1C).1 The preference for cross-electrophile coupling over homocoupling stems from the distinct activation of C(sp2) and C(sp3) electrophiles by two separate nickel(I) species, nickel-halide 2 and nickel-aryl 4, via oxidative addition20 and a radical mechanism, respectively. Our program centers around dissecting each radical-involved step: (I) radical generation from the interaction of Ni(I)-aryl 4 with C(sp3)-halides; (II) radical capture by Ni(II) 5; and (III) reductive elimination from nickel(III) 6. Following each mechanistic revelation, we showcase synthetic applications that leverage the insights garnered from these fundamental steps. In the later part of the article (IV), we summarize the redox activity of N-ligands and their significance in nickel-catalyzed reactions proceeding through radical mechanisms.
1.1. Nickel(I)-Mediated Radical Formation
1.1.1. Ni(I)-Mediated Radical Formation via Concerted Halogen-Atom Dissociation
The activation of alkyl electrophiles by a low-valent nickel species can generate an alkyl radical. This mechanism paves the way for the cross-coupling of alkyl electrophiles and attaining stereoconvergence.21,22 Generally, three pathways are postulated: a stepwise SET followed by halide dissociation,23 an SN2 oxidative addition followed by radical ejection,24 or a concerted halogen atom transfer (XAT) (Scheme 2A).25 Our prior studies differentiated these pathways by investigating a series of (Xantphos)Ni(I)-aryl model complexes, supporting a concerted halogen atom abstraction pathway.26 However, characterizing this crucial step with catalytically relevant Ni(I) intermediates remains challenging due to their instability.
Scheme 2. Electroanalytical Determination of a Concerted Halogen-Atom Dissociation Pathway.

To address the challenge of studying catalytically active intermediates that are inherently unstable and thus difficult to isolate, we employed an electroanalytical method. This approach enabled the in situ generation of [(bpy)Ni(Mes)(Br)]•–13 (bpy = 2,2′-bipyridine, Mes = 2,4,6-mesityl) through the electroreduction of (bpy)Ni(Mes)(Br) 12, which allowed for the investigation of the reactivity of 13 with alkyl halides to form radicals (Scheme 2B).27 Cyclic voltammetry (CV) revealed a reversible 12/13 redox couple, evident by an ipa/ipc ratio of 0.92 (ipa = peak anodic current, ipc = peak cathodic current), suggesting the stability of 13 on the CV timescale. In the presence of an electrophile, the redox event shifted from reversible to quasi-reversible or irreversible, as evidenced by a decrease of the ipa/ipc ratio. This change was attributed to the consumption of 13 through its reaction with the alkyl halide. By varying scan rates, we determined the time course and the reaction rates across different alkyl halides. A linear free-energy regression analysis revealed a positive correlation between the bond dissociation free energy (BDFE) of the C–X bond in the electrophile and the activation barrier. Furthermore, when varying the para-substituents on benzyl bromides, a Hammett analysis uncovered the buildup of partial negative charge and radical character at the benzylic position in the transition state.
Benzylic and tertiary halides reacted with 13 faster than secondary and primary variants. Increasing the steric hindrance of the aryl group on nickel substantially reduced the rates of electrophile activation. Collectively, these data support a concerted inner-sphere electron transfer (ISET)/halide dissociation mechanism in the generation of radicals from alkyl halides mediated by [(bpy)Ni(Mes)(Br)]•–13. Additionally, this study rules out the outer-sphere electron transfer and SN2 oxidative addition pathways.
1.1.2. Nickel-Catalyzed Late-Stage Modification of Oligopeptides
The ability of low-valent nickel to initiate radical formation by halogen-atom abstraction prompted us to investigate the application of this fundamental reactivity. Radical addition to dehydroalanine (Dha) provides a versatile and modular approach for synthesizing noncanonical peptide analogues, which are crucial for drug discovery.12,28 Previous studies on radical addition to the Dha residue of peptides and proteins have demonstrated excellent functional group compatibility.12 However, the lack of stereocontrol diminished the broader application of this reaction in preparing peptide analogues.12 To address this challenge, we postulated that chiral nickel catalysts might offer a solution for the diastereoselective late-stage modification of Dha in oligopeptides (Scheme 3).2 We proposed that the generation of a radical under reductive nickel-catalytic conditions could be followed by its efficient addition to Dha, resulting in intermediate 14. A subsequent capture by a nickel(I) metalloradical species would produce enolate 15. Given the fast rate of radical combination, this step would ensure chemo-selectivity. The enolate 15 could then undergo asymmetric protonation to establish the desired chiral center under the stereocontrol of a chiral ligand bound to nickel.
Scheme 3. Late-Stage Functionalization of Dha via Nickel-Catalyzed Radical Addition.
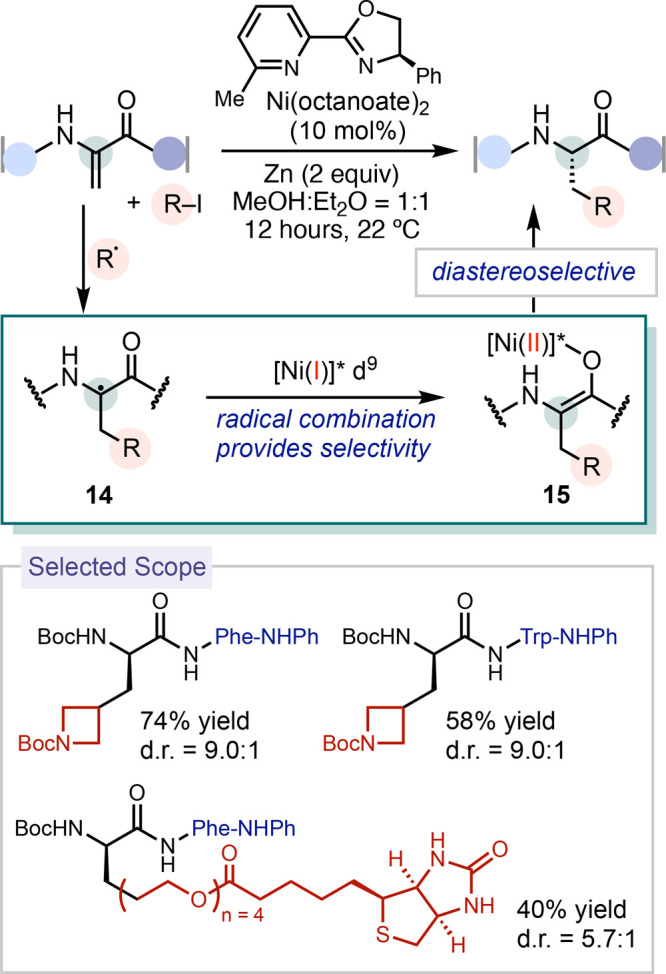
We identified a catalytic condition composed of Ni(octanoate)2 and the chiral ligand, (S)-6-Me-Phpyrox (6-Me-Phpyrox = 4-phenyl-2-(6-methyl-2-pyridyl)oxazoline).2 This system proved effective in promoting diastereoselective radical addition to Dha in dipeptides. We selected alkyl iodides as electrophiles, given their facile reactivity in forming radicals through reduction by zinc or by a low-valent nickel catalyst. This reaction framework has accommodated a variety of primary and secondary electrophiles, enabling the integration of polyethylene glycol, biotin, halo-tag, and both hydrophobic and protected hydrophilic side chains onto dipeptides. Notably, catalyst control predominantly dictates the stereochemical outcome, overcoming substrate control. Despite the versatility of this reaction, limitations remain with hydrophilic side chains and longer peptides.
1.2. Ni(II)-Mediated Radical Capture
1.2.1. Carbon-Centered Radical Capture at Ni(II) Complexes
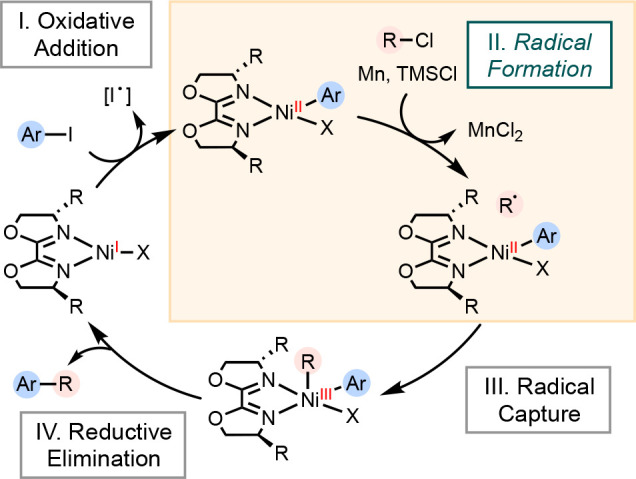
The capture of carbon-centered radicals at the nickel(II) center has been widely applied in recent cross-coupling,29 metallaphotoredox,30 and electrocatalytic reactions (Scheme 4A).31 This step has expanded the scope of potential coupling partners to novel radical precursors, such as C–H bonds, carboxylic acids, boronates, dihydropyridine (DHP), and other redox auxiliaries.32,33 The formation of a high valent Ni(III) intermediate facilitates reductive elimination both kinetically and thermodynamically.34,35 Furthermore, the trapping of radicals by nickel complexes determines chemoselectivity in cross-electrophile coupling reactions1 and, with chiral ligands, controls stereoselectivity in diastereoselective and enantioselective coupling reactions.21,22
Scheme 4. Experimental Evidence and Energy Barriers for Radical Capture at Ni(II) Complexes.

Previously, the capture of radicals by catalytically relevant nickel complexes has been studied computationally. These DFT calculations provided a range of kinetic barriers for this process, which vary based on the type of carbon radicals involved.36 The capture of primary and secondary radicals was calculated to be an inner-sphere and fast process, while tertiary radicals favor a slower outer-sphere pathway to form the C–C bond. In these computational studies, the estimated fast radical capture made it unlikely for this step to be considered rate-limiting when compared to the subsequent reductive elimination. However, there has been limited experimental characterization of radical capture at catalytically competent nickel(II) complexes that would allow for a comparison with computational results.37 We conducted experimental investigations into radical capture at various catalytically relevant nickel(II) complexes, determining the kinetic barriers. Our findings suggest that this step might indeed be rate-determining when compared to reductive elimination.
We investigated the stoichiometric capture of carbon radicals by a variety of nickel complexes,3 applying a previously reported radical precursor, benzyl-DHP (DHP = dihydropyridine), to generate benzyl radical upon photoexcitation (Scheme 4B).38 While bpy, phen, and pybox-ligated nickel-aryl complexes 16 (phen = 1,10-phenanthroline, pybox = pyridine-2,6-bis(oxazoline)) underwent efficient radical capture and reductive elimination, the corresponding nickel-alkyl complex 17 produced only trace amounts of product. Monitoring the addition of benzyl radical to (phen)Ni(p-CF3-o-Tol)(Br) (p-CF3-o-Tol = 2-methyl-4-(trifluoromethyl)phenyl) 16 using EPR spectroscopy revealed a signal that was consistent with benzyl radical. In contrast, performing the same experiment with (dtbpy)Ni(CH2TMS)(Cl), dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine) 17 resulted in a rhombic EPR signal with a giso value of 2.1148, which was assigned to a nickel(III) species. The difference in EPR signals for reactions with (phen)Ni(p-CF3-o-Tol)(Br) 16 and (dtbpy)Ni(CH2TMS)(Cl) 17 highlights the stark contrast in stoichiometric reactivity between nickel-aryl and nickel-alkyl complexes undergoing radical capture.
These results reveal different rate limiting steps for nickel-aryl and nickel-alkyl complexes. In the case of nickel-aryl complexes, radical trapping is the rate-determining step leading to the accumulation of benzyl radical, while the subsequent reductive elimination occurred rapidly. For nickel-alkyl complexes, efficient radical capture leads to the formation of an observed nickel(III) species, but the subsequent C(sp3)–C(sp3) bond-forming reductive elimination is slow (Scheme 4B).
Employing a series of radical clock experiments, we investigated the kinetics of radical capture at nickel-aryl complexes. The rates were determined to be on the scale of 107 M–1 s–1 and 106 M–1 s–1 for primary and secondary radicals, respectively. The corresponding activation energies were 7.0–7.5 kcal/mol for primary radicals and 8.7–9.2 kcal/mol for secondary radicals. Changing the ligands from phen to dtbpy resulted in no significant difference in the rates. These data serve as a benchmark for evaluating the kinetic competence of radical capture as an intermediate step in designing catalytic reactions.
Furthermore, we evaluated the ability of the radical capture step to impart both diastereo- and enantioselectivity (Scheme 4C). Upon irradiating 18 in the presence of 1 equiv of 19, the glycosyl arene product 20 was obtained in 24% yield with a d.r. (d.r. = diastereomeric ratio) of 46:1. This result underscores the potential for achieving diastereoselectivity through the trapping of a glycosyl radical by nickel(II) intermediates. Subsequently, we investigated the enantioselectivity during the capture of α-amino radical 21 by nickel(II) complexes bearing chiral pyrox (pyrox = pyridine-oxazoline) and bi(imidazoline) ligands. The α-amino radical 21 proceeded to generate cross-coupling product (S)-22 and (R)-22, controlled by the respective ligands. While the reversibility of radical trapping by these nickel complexes remains a question, our findings emphasize the pronounced influence of ligands in determining enantioselectivity.
1.2.2. Synthesis of C-Glycosides via Diastereoselective Radical Capture by Nickel
Our observation of high diastereoselectivity in nickel-mediated radical capture motivated us to leverage this observation and develop a method for synthesizing C-glycosides from nickel-catalyzed cross-coupling of glycosyl radicals (Scheme 5).39,40C-aryl glycosyl compounds are prototypical drug candidates due to the in vivo stability of the glycosyl C–C bonds, which are resistant to hydrolysis and enzymatic degradation.41 Furthermore, synthetic C-aryl nucleoside analogues play a crucial role in examining mutagenicity origins and understanding replication and evolution mechanisms.42
Scheme 5. Nickel-Catalyzed C–Aryl and C–Acyl Glycosylation.

To facilitate the formation of glycosyl radicals, we designed a redox auxiliary in the form of a DHP carboxylic acid to activate natural sugars. This auxiliary easily condenses with carbohydrate molecules, resulting in glycosyl ester 23. This promotes glycosyl C–O bond homolysis, leading to the formation of glycosyl radicals through a sequence of photoredox oxidation (23 → 24), Hantzsch pyridine extrusion (24 → 26), and decarboxylation from the alkoxycarbonyl radical intermediate 26 (26 → 27). Once the glycosyl radical 27 is generated, it can promptly engage in the nickel-catalyzed cross-coupling cycle, producing aryl and acyl radical precursors as indicated by our mechanistic analyses.
We achieved efficient cross-coupling of glycosyl esters with bromoarenes and carboxylic acids applying metallaphotoredox conditions, with 4CzIPN (4CzIPN = 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene) as the photocatalyst, NiBr2·DME (DME = 1,2-dimethoxyethane) as the catalyst, and bpy as the ligand.39,40 This reaction proved effective in transforming a diverse scope of furanoses and pyranoses into C-aryl and C-acyl glycosides 28−33. Notably, ribose derivatives 28 and 29 synthesized via this method serve as precursors to tiazofurin, an anticancer and antiviral drug, and dNaM, a non-natural nucleoside, respectively. Additionally, a (+)-sclareolide derivative was conveniently modified with carboxylic acids to afford 33.
Besides the products illustrated in Scheme 5, the reaction scope encompasses common furanose and pyranoses, including d-xylofuranose, d-glucofuranose, d-galactofuranose, d-arabinofuranose, d-mannopyranose, 2-deoxy-d-glucopyranose, l-rhamnopyranose, and 2-deoxy-d-ribopyranose, with various protecting groups, such as acetonide, benzyl, silyl, and benzoyl groups. For furanoses, the d.r. is dependent on C-2 substituents: incoming groups tend to approach from the face opposite to the substituent to reduce steric hindrance. In pyranosides, the d.r. is dictated by steric hindrance at C2 and the kinetic anomeric effect.43 However, challenges remain with 2-deoxyribose, d-glucopyranoses and d-galactopyranose, all resulting in low d.r., due to contradictory preferences by the steric and the stereoelectronic effect.
The electrophile range includes a variety of electron-rich and electron-deficient bromoarenes and heteroarenes. Regarding the carboxylic acid scope, the method proved versatile, showing compatibility with natural acids, such as fenbufen, ursodeoxycholic acid, l-glutamic acid (Glu) 30, α-d-galactopyranuronic acid, d-biotin, and mycophenolic acid. The conjugation of Glu with carbohydrates presents an exciting avenue for glycopeptide synthesis. Given the convenient synthesis of glycosyl esters, their bench-stability, and the wide array of accessible structures, this method holds significant potential in medicinal chemistry.
1.3. Reductive Elimination
Various strategies have been practiced in promoting reductive elimination from a metal center.44 Ligand association or dissociation can lead to odd-numbered coordinate geometries, such as trigonal planar or square pyramidal. This change in geometry lowers the barrier for transferring two electrons to the vacant d-orbital. While this is a kinetic effect, reductive elimination may also be promoted by methods that exploit thermodynamic effects. Incorporating electron-deficient or bulky ligands, or oxidizing the metal center can increase the driving force by destabilizing the starting material and stabilizing the resulting low-valent metal species.45 Seminal reports on reductive elimination from bpy supported dialkyl nickel complexes have displayed means to trigger reductive elimination by both these kinetic and thermodynamic driving forces through oxidation, thermolysis, and ligand association.46,47
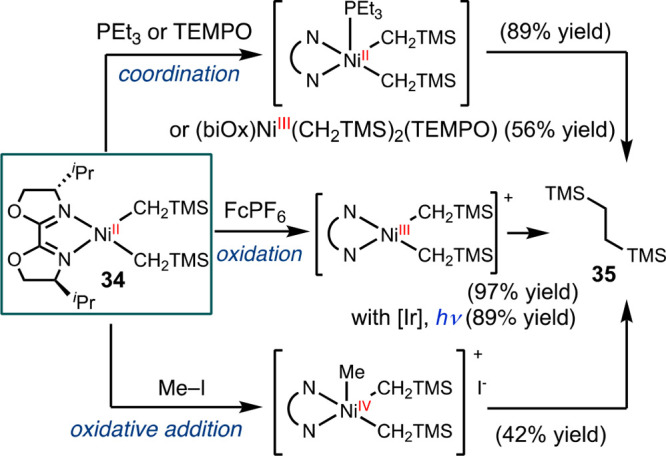
We further contributed to these aspects by investigating strategies to induce reductive elimination from a (biOx)Ni(CH2TMS)2 (biOx = bi(oxazoline)) model complex 34 (Scheme 6).48 The coordination with neutral donor ligands, such as triethylphosphine, and radical ligands, such as TEMPO, triggered reductive elimination to afford 35 in high yields. The oxidation of 34 using a mild oxidant FcPF6 (FcPF6 = ferrocenium hexafluorophosphate), resulted in nearly complete conversion to 35. This oxidation can also be achieved using a photosensitizer under light irradiation. Additionally, we discerned that oxidative addition with methyl iodide generated a high-valent nickel intermediate that rapidly proceeded to reductive elimination. A concurrent study demonstrated that increasing the steric effect of the ligand can accelerate reductive elimination from a bis-alkyl nickel complex.49
Scheme 6. Strategies for Promoting Reductive Elimination from (iPrbiOx)Ni(CH2TMS)2.
1.4. Effect of N-Ligands
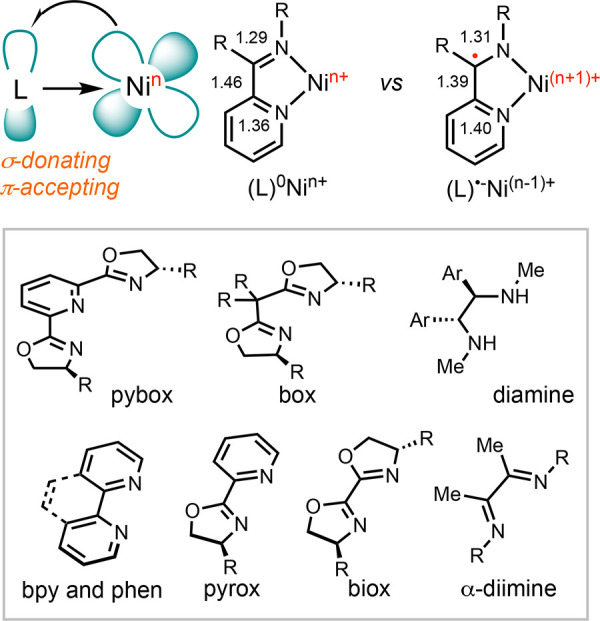
The evolution of ligands has facilitated advancements in transition metal catalysis. Phosphine and N-heterocyclic carbene ligands are frequently applied in cross-coupling reactions proceeding through a two-electron Ni(0)/Ni(II) catalytic cycle,50 whereas N-chelating ligands are most effective in cross-coupling reactions involving radicals.18 Tridentate N-ligands, including terpyridine (terpy) and pybox, along with diamine ligands, are effectively utilized in cross-coupling of C(sp3) partners.51 Bpy and phen ligands, meanwhile, are commonly applied in cross-electrophile coupling reactions.19 Chiral oxazoline ligands, such as pyrox, biOx, and bis(oxazoline) (box), impart enantioselectivity.52 α-Diimine ligands are proficient in promoting polymerization and hydrogenation reactions.53
Polypyridine ligands and related variants are strong σ-donors and π-acceptors, leading to strong field-splitting and stabilization (Scheme 7). Many of these ligands exhibit redox activity, resulting in the transfer of electrons from the metal to the π* orbital of the ligand.54 This effect is assessed and characterized using spectroscopic and computational methods.55 When a radical occupies the ligand’s π* orbital, it leads to a change in bond order. This change manifests in variations in bond lengths, which can be measured using X-ray diffraction (XRD) or predicted through DFT calculations. EPR spectroscopy can reveal the nature of the radical, distinguishing between ligand-centered and nickel-centered radicals. Since the valence orbitals determine the redox potentials of a complex, the redox activity of the ligand can have profound effects on reaction pathways. Consequently, discerning the electronic structures of reaction intermediates becomes crucial to understanding the ligand effect, guiding ligand design for optimizing reactions.
Scheme 7. N-Ligands in Nickel Catalysis with Potential Redox Activity.
1.4.1. Redox Activity of Pyrox in Organonickel Radical Complexes
Pyrox, with its extensive applications in asymmetric catalysis,56 often functions similarly to bpy or phen in catalyzing coupling reactions. Prior to our study, the redox activity of pyrox and its comparison to other N,N-ligands remained ambiguous.
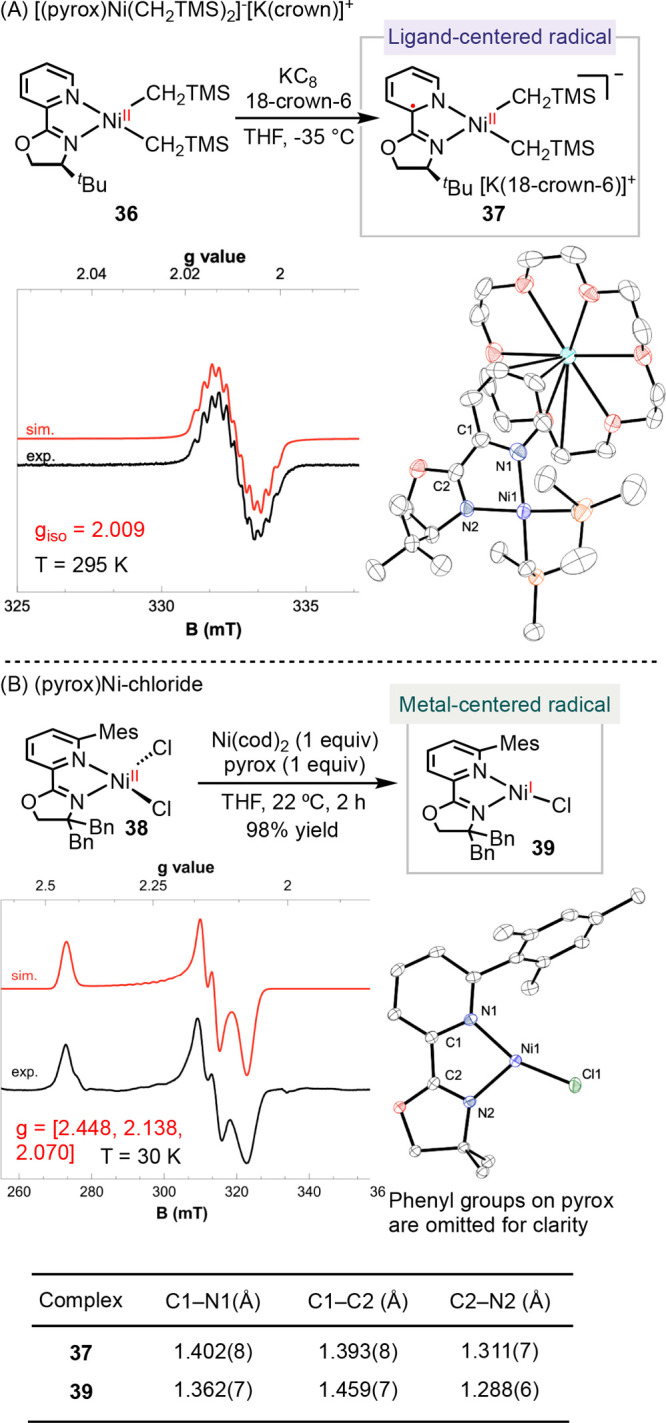
We investigated the electronic structures of a series of pyrox-ligated nickel radical complexes, using two synthetic strategies. In the first approach, the reduction of (tBupyrox)Ni(CH2TMS)236 (tBupyrox = (S)-4-(tert-butyl)-2-(2-pyridyl)oxazoline) by potassium graphite (KC8) in the presence of 18-crown-6 led to the formation of [K(18-crown-6)]+[(tBupyrox)Ni(CH2TMS)2]•–37 (Scheme 8A).57 The second approach involved the comproportionation of (pyrox)NiCl238 and Ni(cod)2, giving (pyrox)NiCl 39 in 98% yield (Scheme 8B).4 X-ray crystallography established unsymmetrical Ni–C bond lengths in 37 and a T-shape geometry in 39, highlighting a pronounced trans-influence of pyridine compared to oxazoline. The elongation of Cox–Nox and Cpy–Npy bond lengths, combined with the contraction of the Cox–Cpy bond, suggests a radical in the π* orbital of 37. This assignment is corroborated with EPR spectra, which reveals a ligand-centered radical in 37 and a nickel-centered radical in 39. Collectively, these data imply that that the redox activity of pyrox varies depending on the coordination environment of each complex.
Scheme 8. Redox Activity of (Pyrox)Ni Complexes.
1.4.2. Bi-Oxazoline Organonickel Radical Complexes
BiOx ligands exhibit intriguing reactivity in asymmetric catalysis, particularly with nickel catalysts.51 For instance, we achieved high yields and enantioselectivity with biOx in the development of an asymmetric diarylation of vinylarenes (Scheme 9A).58 In contrast, using the structurally similar box ligand resulted in no product formation.
Scheme 9. Application and Redox Activity of (biOx)Ni Complexes.
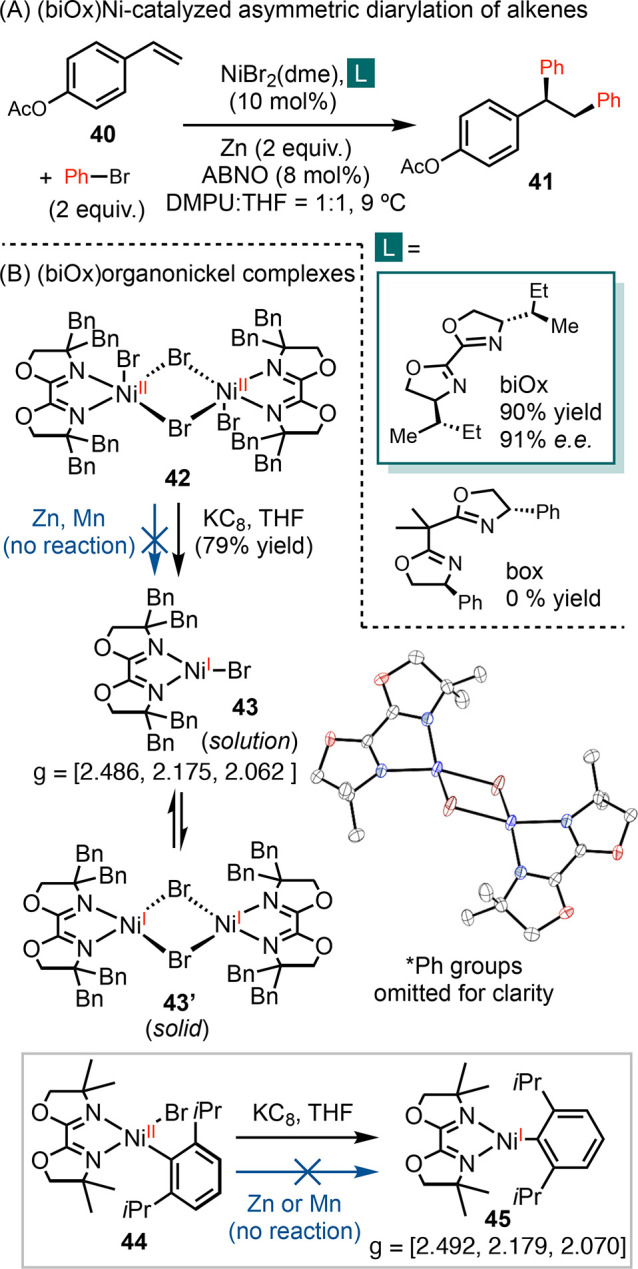
To elucidate the ligand effect of biOx, we synthesized organonickel complexes and characterized their electronic structures (Scheme 9B).59 The reduction of (dBnbiOx)NiBr242 yielded (dBnbiOx)NiBr 43, which exists in equilibrium with the dimer [(dBnbiOx)Ni(μ-Br)]2, 43′. The EPR spectrum of 43 reveals an S = 1/2 nickel complex as the predominant species in solution, whereas the X-ray crystal structure displays a dimer 43′, with a tetrahedral geometry at the nickel center. Reduction of (dMebiOx)Ni(Dipp)Br (Dipp = 2,6-diisopropylphenyl) 44 by KC8 afforded 45, characterized as a nickel-centered radical by its EPR signal. Both the EPR data and bond lengths derived from XRD suggest an absence of redox activity in biOx.
The lack of redox activity in these biOx complexes could be attributed to the π-donating effect of the O-atom of oxazoline. The elevated energy level of the ligand π*-orbital prevented electron transfer from nickel to the ligand. As a result, (biOx)nickel complexes exhibit significantly lower reduction potentials compared to their phen, bpy, and pyrox analogues (Table 1).60 When we tried to synthesize 43 and 45 through reduction, neither zinc nor manganese powder—common reductants in cross-electrophile coupling—initiated any reaction (Scheme 9B). This observation led us to re-evaluate the mechanism of cross-electrophile coupling reactions involving (biOx)Ni catalysts and proposed that a catalyst reduction step was not operational in a reaction catalyzed by (biOx)nickel catalysts (Scheme 10). Instead, our revised proposal suggests a direct interaction between substrates and reductants, facilitated by a Lewis acid, to generate radicals.
Table 1. Comparison of the Redox Activity and Redox Potentials of (N,N-Bidentate)Ni(Aryl) Complexes.
Scheme 10. Revised Mechanism of Cross-Electrophile Coupling Involving (biOx)Ni Catalysts.
1.4.3. Geometry-Dependent Redox Activity in Nickel Radical Complexes
Our study on (pyrox)Ni complexes indicates that the redox activity of a ligand can vary among different complexes (Scheme 8). This observation remains consistent across a range of reported organonickel radical complexes ligated with bidentate N-ligands.4 By analyzing data from the literature combined with our results, we established a general framework for understanding the ligand redox activity of nickel radical complexes.4 These complexes feature ligands that are commonly employed in cross-coupling reactions: such as bpy, phen, and pyrox. This empirical rule entails a correlation between the ligand redox activity and the coordination geometry of the complexes.
We synthesized and characterized a series of (bpy)Ni radical complexes. The reduction of (dtbpy)Ni(Dipp*)Cl 47 (Dipp* = 2,6-di-Dipp-phenyl) led to the formation of complex 48, which was characterized as a ligand-centered radical by EPR (Scheme 11).4 Over time, in solution, chloride dissociation led to the formation of the three-coordinate complex 49, which was identified as a metal-centered radical based on its EPR spectrum. Furthermore, when we reduced [(dtbpy)Ni(Dipp)]+[BArF24]− (BArF24 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate) 50 in THF, we observed an isotropic, organic radical. Intriguingly, performing this reduction in a less coordinating solvent, such as Et2O, yielded a three-coordinated nickel radical 56, showing a rhombic EPR signal with g values of [2.263, 2.086, 2.050].
Scheme 11. Effect of Coordination Geometry on the Redox Activity of (Bpy)Ni-Aryl Complexes.

By increasing the steric hindrance of the bpy ligands and the X-ligand, we synthesized a series of (bpy)Ni radical complexes (Scheme 12).4 The (π-allyl)Ni 52 and the square-planar [(dtbpy)Ni(CH2TMS)2]−[K(18-crown-6)]+53 are characterized as ligand-centered radicals. In contrast, the trigonal planar (Cybpy)NiCl 54 and (Mesdtbpy)Ni(CH2TMS) 55 display metal-centered radicals, evidenced by the lack of significant changes in bond lengths and the rhombic EPR signals corresponding to a nickel-centered radical. Nickel radical complexes with phen ligands follow a similar trend. The four-coordinate [(phen)NiBr2]−[K(DB18C6)]+57 (DB18C6 = dibenzo-18-crown-6) and [(phen)Ni(CH2TMS)2]−[K(18-crown-6)]+58 are determined to be ligand-centered radicals based on EPR and XRD analyses, whereas the three-coordinate (sBuphen)NiBr 59 is a nickel-centered radical.1
Scheme 12. Redox Activity of bpy, phen, and pyrox Ligands in Nickel Radical Complexes and the Correlation with Coordination Geometry.

Overall, both the literature data and our results point to a correlation between ligand redox activity and coordination geometry. Four-coordinate, square-planar nickel radical complexes are characterized as low-spin nickel(II) complexes coordinated with ligand radical anions.63 In contrast, tetrahedral nickel radical complexes, such as 43′, as well as three-coordinate, trigonal planar nickel-halide, alkyl, and aryl complexes typically exhibit nickel(I) centers, supported by non-redox-active ligands. This trend persists irrespective of the nature of the X-ligands or the charge of the molecule across a range of nickel complexes with bpy, phen, pyrox, α-diimine, and biox ligands, characterized by our group and documented in the literature. Notably, (terpy)Ni complexes are exceptions to this trend.64
This relationship between ligand redox activity and coordination geometry can be understood through molecular orbital analysis (Scheme 13). The redox activity of a ligand depends on the relative energy levels of the vacant d-orbital and the π* orbital of the ligand. Molecules with trigonal planar and tetrahedral geometries have low-energy antibonding d-orbitals, often lying beneath the ligand’s π* orbital. As a result, it is energetically favorable for the unpaired electron to remain in the d-orbital, resulting in a d9 electronic configuration. Conversely, for square planar complexes, the high energy level of the dx2-y2 orbital implies a preference for the unpaired electron to transfer to the ligand π* orbital, leading to a d8 configuration.
Scheme 13. Geometry-Governed Ligand Redox Activity.

Our findings provide insights to help optimize catalytic conditions. Nickel-catalyzed cross-coupling reactions often apply additives such as MgCl2 and KI. Besides their role in modulating the speciation of active nickel catalysts,65 the coordinating anions may be pivotal for the stabilization of nickel(I) intermediates and the modulation of redox potentials through the alteration of ligand redox activity. Furthermore, our results imply that coordinating solvents may promote reactions through a similar stabilization effect.
2. Conclusions
Nickel catalysts excel at initiating carbon radical formation and engaging radical intermediates in cross-coupling reactions. We have investigated the mechanisms, kinetics, and ligand effects on fundamental steps that involve radical intermediates. The nonpolar and neutral reactivity of these radical intermediates, orthogonal to polar functional groups in biomolecules, has enabled the diastereoselective modification of dehydroalanine for preparing noncanonical peptides. Furthermore, our study on radical capture by nickel has led to the discovery of a series of C-glycosylation reactions, useful for preparing C-aryl and C-acyl glycosides valuable in drug discovery.
Our continued focus revolves around elucidating the mechanisms of nickel-mediated and catalyzed reactions that involve radical intermediates. We are particularly interested in understanding these pathways at the interface of photoredox catalysis and electrocatalysis. Central to catalyst development is ligand design. Compared to the vast library of phosphine ligands available for palladium, the variety of N-π-acid ligands for nickel are disproportionally insufficient. We are actively exploring and developing new ligands to broaden the scope of nickel catalysis. While the application of the orthogonal reactivity of radicals with polar functional groups in biomolecules remains in its early stages, our methodology projects aim to bridge this knowledge gap and implement methods for accessing noncanonical peptide and nucleoside analogues.
Acknowledgments
This work is supported by the National Institute of General Medical Sciences (R01GM-127778). T.D. acknowledges the Camille-Dreyfus Teacher Scholar Award (TC-19-019) and the Arthur C. Cope Scholar Award.
Biographies
Gregory Dawson received his B.S. in chemistry with an emphasis in biochemistry from San Diego State University in 2018. He is currently a graduate student in Professor Diao’s group. His research seeks to elucidate the electronic structures of nickel intermediates relevant to cross-coupling reactions.
Ethan Spielvogel received his B.S. in chemistry from Binghamton University in 2021. He is currently a graduate student in Professor Diao’s group. His research focuses on the mechanism of nickel-mediated radical capture and the implications of this step in cross-coupling reactions.
Tianning Diao received her Bachelor’s degree in Chemistry from Fudan University in 2007 and her Ph.D. in organic chemistry from the University of Wisconsin–Madison in 2012. Following her postdoctoral research at Princeton University, she joined New York University (NYU) as a faculty member in 2014. Her research interests span fields including organometallic chemistry, organic synthesis, inorganic chemistry, chemical biology, and sustainable energy conversion.
The authors declare no competing financial interest.
This paper originally published ASAP on November 30, 2023. Due to a production error, Scheme 13 was updated and a new version reposted on December 4, 2023.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Cross-Coupling with First-Row Transition Metals”.
References
- Lin Q.; Diao T. Mechanism of Ni-Catalyzed Reductive 1,2-Dicarbofunctionalization of Alkenes. J. Am. Chem. Soc. 2019, 141, 17937–17948. 10.1021/jacs.9b10026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi X.; Jambu S.; Ji Y.; Belyk K. M.; Panigrahi N. R.; Arora P. S.; Strotman N. A.; Diao T. Late-Stage Modification of Oligopeptides by Nickel-Catalyzed Stereoselective Radical Addition to Dehydroalanine. Angew. Chem., Int. Ed. 2022, 61, e202213315 10.1002/anie.202213315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q.; Spielvogel E. H.; Diao T. Carbon-centered radical capture at nickel(II) complexes: Spectroscopic evidence, rates, and selectivity. Chem. 2023, 9, 1295–1308. 10.1016/j.chempr.2023.02.010. [DOI] [Google Scholar]
- Dawson G. A.; Lin Q.; Neary M. C.; Diao T. Ligand Redox Activity of Organonickel Radical Complexes Governed by the Geometry. J. Am. Chem. Soc. 2023, 145, 20551–20561. 10.1021/jacs.3c07031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stubbe J.; Nocera D. G. Radicals in Biology: Your Life Is in Their Hands. J. Am. Chem. Soc. 2021, 143, 13463–13472. 10.1021/jacs.1c05952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale S. W. Nickel-based Enzyme Systems. J. Biol. Chem. 2009, 284, 18571–18575. 10.1074/jbc.R900020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki Y.; Oohora K.; Hayashi T. Focusing on a nickel hydrocorphinoid in a protein matrix: methane generation by methyl-coenzyme M reductase with F430 cofactor and its models. Chem. Soc. Rev. 2022, 51, 1629–1639. 10.1039/D1CS00840D. [DOI] [PubMed] [Google Scholar]
- Can M.; Armstrong F. A.; Ragsdale S. W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase and Acetyl Co-A Synthase. Chem. Rev. 2014, 114, 4149–4174. 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelmenschikov V.; Siegbahn P. E. M. Nickel Superoxide Dismutase Reaction Mechanism Studied by Hybrid Density Functional Methods. J. Am. Chem. Soc. 2006, 128, 7466–7475. 10.1021/ja053665f. [DOI] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenegger P. G.; Davis B. G. Concepts of Catalysis in Site-Selective Protein Modifications. J. Am. Chem. Soc. 2019, 141, 8005–8013. 10.1021/jacs.8b13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom S.; Liu C.; Kölmel D. K.; Qiao J. X.; Zhang Y.; Poss M. A.; Ewing W. R.; MacMillan D. W. C. Decarboxylative alkylation for site-selective bioconjugation of native proteins via oxidation potentials. Nat. Chem. 2018, 10, 205–211. 10.1038/nchem.2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan J. P.; Lang S. B.; Sim J.; Berritt S.; Peat A. J.; Billings K.; Fan L.; Molander G. A. Open-Air Alkylation Reactions in Photoredox-Catalyzed DNA-Encoded Library Synthesis. J. Am. Chem. Soc. 2019, 141, 3723–3732. 10.1021/jacs.9b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flood D. T.; Asai S.; Zhang X.; Wang J.; Yoon L.; Adams Z. C.; Dillingham B. C.; Sanchez B. B.; Vantourout J. C.; Flanagan M. E.; Piotrowski D. W.; Richardson P.; Green S. A.; Shenvi R. A.; Chen J. S.; Baran P. S.; Dawson P. E. Expanding Reactivity in DNA-Encoded Library Synthesis via Reversible Binding of DNA to an Inert Quaternary Ammonium Support. J. Am. Chem. Soc. 2019, 141, 9998–10006. 10.1021/jacs.9b03774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasker S. Z.; Standley E. A.; Jamison T. F. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. 10.1038/nature13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu G. C. Transition-Metal Catalysis of Nucleophilic Substitution Reactions: A Radical Alternative to SN1 and SN2 Processes. ACS Cent. Sci. 2017, 3, 692–700. 10.1021/acscentsci.7b00212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diccianni J. B.; Diao T. Mechanisms of Nickel-Catalyzed Cross-Coupling Reactions. Trends Chem. 2019, 1, 830–844. 10.1016/j.trechm.2019.08.004. [DOI] [Google Scholar]
- Everson D. A.; Weix D. J. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem. 2014, 79, 4793–4798. 10.1021/jo500507s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting S. I.; Williams W. L.; Doyle A. G. Oxidative Addition of Aryl Halides to a Ni(I)-Bipyridine Complex. J. Am. Chem. Soc. 2022, 144, 5575–5582. 10.1021/jacs.2c00462. [DOI] [PubMed] [Google Scholar]
- Cherney A. H.; Kadunce N. T.; Reisman S. E. Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross-Coupling Reactions of Organometallic Reagents to Construct C–C Bonds. Chem. Rev. 2015, 115, 9587–9652. 10.1021/acs.chemrev.5b00162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas E. L.; Jarvo E. R. Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem. 2017, 1, 1–7. 10.1038/s41570-017-0065. [DOI] [Google Scholar]
- Tsou T. T.; Kochi J. K. Mechanism of Oxidative Addition. Reaction of Nickel(0) Complexes with Aromatic Halides. J. Am. Chem. Soc. 1979, 101, 6319–6332. 10.1021/ja00515a028. [DOI] [Google Scholar]
- Funes-Ardoiz I.; Nelson D. J.; Maseras F. Halide Abstraction Competes with Oxidative Addition in the Reactions of Aryl Halides with [Ni(PMenPh(3-n))4. Chem.-Eur. J. 2017, 23, 16728–16733. 10.1002/chem.201702331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X.; Phillips D. L. Density Functional Theory Studies of Negishi Alkyl-Alkyl Cross-Coupling Reactions Catalyzed by a Methylterpyridyl-Ni(I) Complex. J. Org. Chem. 2008, 73, 3680–3688. 10.1021/jo702497p. [DOI] [PubMed] [Google Scholar]
- Diccianni J. B.; Katigbak J.; Hu C.; Diao T. Mechanistic Characterization of (Xantphos)Ni(I)-Mediated Alkyl Bromide Activation: Oxidative Addition, Electron Transfer, or Halogen-Atom Abstraction. J. Am. Chem. Soc. 2019, 141, 1788–1796. 10.1021/jacs.8b13499. [DOI] [PubMed] [Google Scholar]
- Lin Q.; Fu Y.; Liu P.; Diao T. Monovalent Nickel-Mediated Radical Formation: A Concerted Halogen-Atom Dissociation Pathway Determined by Electroanalytical Studies. J. Am. Chem. Soc. 2021, 143, 14196–14206. 10.1021/jacs.1c05255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Davies J. W. Free-radical addition to di- and tripeptides containing dehydroalanine residues. Tetrahedron 1989, 45, 5641–5654. 10.1016/S0040-4020(01)89507-7. [DOI] [Google Scholar]
- Zhu S.; Zhao X.; Li H.; Chu L. Catalytic three-component dicarbofunctionalization reactions involving radical capture by nickel. Chem. Soc. Rev. 2021, 50, 10836–10856. 10.1039/D1CS00399B. [DOI] [PubMed] [Google Scholar]
- Chan A. Y.; Perry I. B.; Bissonnette N. B.; Buksh B. F.; Edwards G. A.; Frye L. I.; Garry O. L.; Lavagnino M. N.; Li B. X.; Liang Y.; Mao E.; Millet A.; Oakley J. V.; Reed N. L.; Sakai H. A.; Seath C. P.; MacMillan D. W. C. Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis. Chem. Rev. 2022, 122, 1485–1542. 10.1021/acs.chemrev.1c00383. [DOI] [PubMed] [Google Scholar]
- Yan M.; Kawamata Y.; Baran P. S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespi S.; Fagnoni M. Generation of Alkyl Radicals: From the Tyranny of Tin to the Photon Democracy. Chem. Rev. 2020, 120, 9790–9833. 10.1021/acs.chemrev.0c00278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvasovs N.; Gevorgyan V. Contemporary methods for generation of aryl radicals. Chem. Soc. Rev. 2021, 50, 2244–2259. 10.1039/D0CS00589D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng B.; Tang F.; Luo J.; Schultz J. W.; Rath N. P.; Mirica L. M. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon-Heteroatom Bond Formation Reactions. J. Am. Chem. Soc. 2014, 136, 6499–6504. 10.1021/ja5024749. [DOI] [PubMed] [Google Scholar]
- Bour J. R.; Camasso N. M.; Meucci E. A.; Kampf J. W.; Canty A. J.; Sanford M. S. Carbon-Carbon Bond-Forming Reductive Elimination from Isolated Nickel(III) Complexes. J. Am. Chem. Soc. 2016, 138, 16105–16111. 10.1021/jacs.6b10350. [DOI] [PubMed] [Google Scholar]
- Yuan M.; Gutierrez O. Mechanisms, challenges, and opportunities of dual Ni/photoredox-catalyzed C(sp2)-C(sp3) cross-couplings. WIREs Comput. Mol. Sci. 2022, 12, e1573 10.1002/wcms.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez O.; Tellis J. C.; Primer D. N.; Molander G. A.; Kozlowski M. C. Nickel-Catalyzed Cross-Coupling of Photoredox-Generated Radicals: Uncovering a General Manifold for Stereoconvergence in Nickel-Catalyzed Cross-Couplings. J. Am. Chem. Soc. 2015, 137, 4896–4899. 10.1021/ja513079r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzzetti L.; Prieto A.; Roy S. R.; Melchiorre P. Radical-Based C-C Bond-Forming Processes Enabled by the Photoexcitation of 4-Alkyl-1,4-dihydropyridines. Angew. Chem., Int. Ed. 2017, 56, 15039–15043. 10.1002/anie.201709571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Ben-zvi B.; Diao T. Diastereoselective Synthesis of Aryl C-Glycosides from Glycosyl Esters via C-O Bond Homolysis. Angew. Chem., Int. Ed. 2021, 60, 9433–9438. 10.1002/anie.202014991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y.; Lam J.; Diao T. Synthesis of C-acyl furanosides via the cross-coupling of glycosyl esters with carboxylic acids. Chem. Sci. 2021, 12, 11414–11419. 10.1039/D1SC03596G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bililign T.; Griffith B. R.; Thorson J. S. Structure, activity, synthesis and biosynthesis of aryl-C-glycosides. Nat. Prod. Rep. 2005, 22, 742–760. 10.1039/b407364a. [DOI] [PubMed] [Google Scholar]
- Moran S.; Ren R. X.-F.; Kool E. T. A thymidine triphosphate shape analog lacking Watson-Crick pairing ability is replicated with high sequence selectivity. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10506–10511. 10.1073/pnas.94.20.10506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H.; Shuto S.; Matsuda A. Highly α- and β-Selective Radical C-glycosylation Reactions Using a Controlling Anomeric Effect Based on the Conformational Restriction Strategy. A Study on the Conformation-Anomeric Effect- Stereoselectivity Relationship in Anomeric Radical Reactions. J. Am. Chem. Soc. 2001, 123, 11870–11882. 10.1021/ja011321t. [DOI] [PubMed] [Google Scholar]
- Hartwig J., Reductive Elimination. In Organotransition Metal Chemistry: From Bonding to Catalysis; Hartwig J., Ed.; University Science Books: Sausalito, 2010; pp 323–324. [Google Scholar]
- Heberer N.; Hu C.-H.; Mirica L. M. High-Valent Ni Coordination Compounds. Comprehensive Coordination Chemistry III 2021, 348–374. 10.1016/B978-0-08-102688-5.00104-5. [DOI] [Google Scholar]
- Saito T.; Uchida Y.; Misono A.; Yamamoto A.; Morifuji K.; Ikeda S. Diethyldipyridylnickel. Preparation, Characterization, and Reactions. J. Am. Chem. Soc. 1966, 88, 5198–5201. 10.1021/ja00974a030. [DOI] [Google Scholar]
- Kohara T.; Yamamoto T.; Yamamoto A. Ligand exchange reaction between NiMe2L2 (L = 12 bpy, PEt3) and ditertiary phosphines Ph2P(CH2)nPPh2 (n = 1–4) and effect of ligand on ease of reductive elimination of C2H6 from NiMe2(Ph2P(CH2)nPPh2). J. Organomet. Chem. 1980, 192, 265–274. 10.1016/S0022-328X(00)94431-0. [DOI] [Google Scholar]
- Ju L.; Hu C. T.; Diao T. Strategies for Promoting Reductive Elimination of Bi- and Bis-Oxazoline Ligated Organonickel Complexes. Organometallics 2022, 41, 1748–1753. 10.1021/acs.organomet.2c00091. [DOI] [Google Scholar]
- Day C. S.; Ton S. J.; McGuire R. T.; Foroutan-Nejad C.; Martin R. Reductive Elimination from Sterically Encumbered Ni-Polypyridine Complexes. Organometallics 2022, 41, 2662–2667. 10.1021/acs.organomet.2c00362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevenger A. L.; Stolley R. M.; Aderibigbe J.; Louie J. Trends in the Usage of Bidentate Phosphines as Ligands in Nickel Catalysis. Chem. Rev. 2020, 120, 6124–6196. 10.1021/acs.chemrev.9b00682. [DOI] [PubMed] [Google Scholar]
- Choi J.; Fu G. C. Transition metal-catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 2017, 356, eaaf7230 10.1126/science.aaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y.; Wang C. Nickel-Catalyzed Asymmetric Cross-Electrophile Coupling Reactions. Synlett 2020, 31, 1843–1850. 10.1055/s-0040-1707216. [DOI] [Google Scholar]
- Ittel S. D.; Johnson L. K.; Brookhart M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100, 1169–1204. 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]
- Chirik P. J.; Wieghardt K. Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. 10.1126/science.1183281. [DOI] [PubMed] [Google Scholar]
- Ganguly S.; Ghosh A. Seven Clues to Ligand Noninnocence: The Metallocorrole Paradigm. Acc. Chem. Res. 2019, 52, 2003–2014. 10.1021/acs.accounts.9b00115. [DOI] [PubMed] [Google Scholar]
- Yang G.; Zhang W. Renaissance of pyridine-oxazolines as chiral ligands for asymmetric catalysis. Chem. Soc. Rev. 2018, 47, 1783–1810. 10.1039/C7CS00615B. [DOI] [PubMed] [Google Scholar]
- Wagner C. L.; Herrera G.; Lin Q.; Hu C. T.; Diao T. Redox Activity of Pyridine-Oxazoline Ligands in the Stabilization of Low-Valent Organonickel Radical Complexes. J. Am. Chem. Soc. 2021, 143, 5295–5300. 10.1021/jacs.1c00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony D.; Lin Q.; Baudet J.; Diao T. Nickel-Catalyzed Asymmetric Reductive Diarylation of Vinylarenes. Angew. Chem., Int. Ed. 2019, 58, 3198–3202. 10.1002/anie.201900228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju L.; Lin Q.; LiBretto N. J.; Wagner C. L.; Hu C. T.; Miller J. T.; Diao T. Reactivity of (bi-Oxazoline)organonickel Complexes and Revision of a Catalytic Mechanism. J. Am. Chem. Soc. 2021, 143, 14458–14463. 10.1021/jacs.1c07139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Q.; Dawson G.; Diao T. Experimental Electrochemical Potentials of Nickel Complexes. Synlett 2021, 32, 1606–1620. 10.1055/s-0040-1719829. [DOI] [Google Scholar]
- Yakhvarov D. G.; Petr A.; Kataev V.; Buchner B.; Gomez-Ruiz S.; Hey-Hawkins E.; Kvashennikova S. V.; Ganushevich Y. S.; Morozov V. I.; Sinyashin O. G. Synthesis, structure and electrochemical properties of the organonickel complex [NiBr(Mes)phen] (Mes = 2,4,6-trimethylphenyl, phen = 1,10-phenanthroline). J. Organomet. Chem. 2014, 750, 59–64. 10.1016/j.jorganchem.2013.11.003. [DOI] [Google Scholar]
- Klein A.; Kaiser A.; Sarkar B.; Wanner M.; Fiedler J. The Electrochemical Behaviour of Organonickel Complexes: Mono-, Di-, and Trivalent Nickel. Eur. J. Inorg. Chem. 2007, 2007, 965–976. 10.1002/ejic.200600865. [DOI] [Google Scholar]
- Mohadjer Beromi M.; Brudvig G. W.; Hazari N.; Lant H. M. C.; Mercado B. Q. Synthesis and Reactivity of Paramagnetic Nickel Polypyridyl Complexes Relevant to C(sp2)–C(sp3)Coupling Reactions.. Angew. Chem. Int. Ed. 2019, 58 (58), 6094–6098. 10.1002/anie.201901866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciszewski J. T.; Mikhaylov D. Y.; Holin K. V.; Kadirov M. K.; Budnikova Y. H.; Sinyashin O.; Vicic D. A. Redox Trends in Terpyridine Nickel Complexes.. Inorg. Chem. 2011, 50, 8630–8635. 10.1021/ic201184x. [DOI] [PubMed] [Google Scholar]
- Day C. S.; Renteria-Gomez A.; Ton S. J.; Gogoi A. R.; Gutierrez O.; Martin R. Elucidating electron-transfer events in polypyridine nickel complexes for reductive coupling reactions.. Nat. Catal. 2023, 6, 244–253. 10.1038/s41929-023-00925-4. [DOI] [Google Scholar]