

Abstract
Uniquely positioned as sentinel cells constantly exposed to the environment, pulmonary macrophages are vital for the maintenance of the lung lining. These cells are responsible for the clearance of xenobiotics, pathogen detection and clearance, and homeostatic functions such as surfactant recycling. Among the spectrum of phenotypes that may be expressed by macrophages in the lung, the pulmonary lipid-laden phenotype is less commonly studied in comparison to its circulatory counterpart, the atherosclerotic lesion-associated foam cell, or the acutely activated inflammatory macrophage. Herein, we propose that lipid-laden macrophage formation in the lung is governed by lipid acquisition, storage, metabolism, and export processes. The cellular balance of these four processes is critical to the maintenance of homeostasis and the prevention of aberrant signaling that may contribute to lung pathologies. This review aims to examine mechanisms and signaling pathways that are involved in lipid-laden macrophage formation and the potential consequences of this phenotype in the lung.
1.0. Introduction
Pulmonary macrophages play a critical role in innate immunity through pathogen detection, foreign substance clearance, and maintenance of surfactant homeostasis1,2. Due to its constant exposure to the environment, the lung is uniquely susceptible to external influence, which can alter macrophage function. The lung lining is inherently lipid rich, however, it can become hyperlipidemic in response to acute injury. As macrophages are constantly taking up lipid through phagocytosis of cells and catabolism of surfactant, this hyperlipidemia can promote the formation of lipid-laden cells. Lipid-laden macrophages, commonly referred to as “foam cells” have been implicated in the early development of atherosclerotic lesions3,4, leading to their extensive study within the vasculature. However, macrophage lipid accumulation has the potential to play a role in other organ systems, such as the lung, where there is a growing appreciation of a role for cholesterol and lipoproteins in homeostasis and signaling5, and lipid-laden macrophages have also been observed in various models of lung injury6,7. The presence of these large, activated cells during injury and their subsequent reduction with treatment indicate that they may play a role in the promotion of lung injury6. Despite these observations, it is still unclear how lipid-laden macrophages in the lung are formed or how their presence affects the inflammatory process. The purpose of this review is to examine potential mechanisms involved in lipid-laden macrophage formation and to postulate the outcomes of this phenotype in the lung.
2.0. Principles governing lipid-laden macrophage formation
Lipid accumulation in circulating macrophages in the vasculature results in foam cell formation4,8–10, which is attributed to the uptake of damaged, oxidized, acetylated or otherwise-modified low-density lipoproteins (LDL)8,11. Lipoprotein uptake is facilitated through a step-wise process: receptor-mediated uptake, lysosomal processing, esterification, and storage in droplets4, all of which if dysregulated or saturated, could contribute to excess lipid accumulation in macrophages. Mechanisms to combat excess accumulation include lipolysis and subsequent cholesterol efflux from the cell, primarily mediated through the ATP-binding cassette (ABC) transporters ABCA1 and ABCG13,12,13. When intracellular cholesterol levels accumulate beyond the cellular capacity for handling and efflux mechanisms become overwhelmed, free cholesterol is esterified to form cholesterol esters (CE) which are stored as lipid droplets in the cytoplasm4,11. Excess CE/lipid accumulation results in the formation of lipid droplets and transforms the macrophage into a foam cell4. These processes can be summarized by consideration of four basic mechanisms that, when aberrant, can lead to lipid accumulation: acquisition, storage, export, and metabolism. Critically, intracellular metabolism can operate as a regulator of these processes14. Major proteins involved in each of these four mechanisms are summarized in Figure 1.
Figure 1.
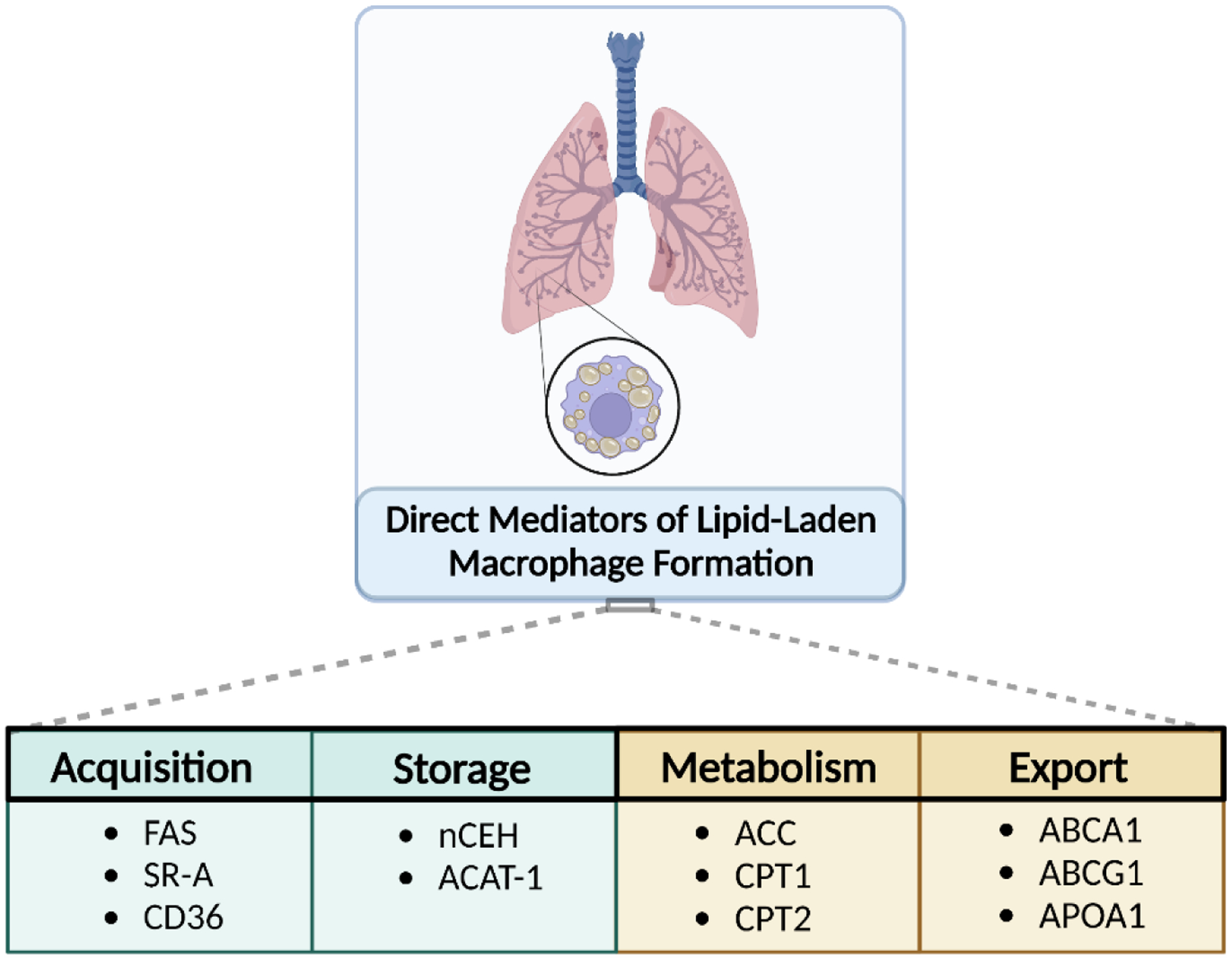
Balance between lipid acquisition, export, metabolism, and storage in pulmonary macrophages are the primary processes that prevent the development of a lipid-laden phenotype. Dysregulation or imbalance of these processes leading to the excess storage of lipid are the most significant when considering the development of this cell phenotype and potential therapeutic intervention in the lung.
Pulmonary macrophages, in particular resident alveolar macrophages, exist in a lipid-rich environment due to the presence of lung surfactants15–17. These cells are critical to maintaining lung lipid homeostasis by constantly removing oxidized and damaged lipids, such that surfactant functions normally. The high partial pressure of oxygen in the lung and high lipid content favors lipid peroxidation and ROS generation; this leads to a disproportionate quantity of modified and damaged LDL. Lung injury has the potential to increase the quantity of lipids, especially damaged lipids, within the lung lining, which increases the load on macrophages to clear these species18–20. Both excessive lipid storage, uptake of oxidized lipids, and free cholesterol can result in apoptosis of macrophages, adding to the burden on the remaining phagocytic cells in the environment. Excessive oxidation and cell death contributes to injury through the release of cytotoxic mediators such as damage associated molecular-patterns (DAMPs), including heat shock protein 7221 and high mobility group box 1 protein (HMGB1)22, which upregulate downstream inflammatory pathways such as NF-κB, stimulate the production of reactive oxygen and nitrogen species (RONS), and recruit inflammatory cells11,23. Both oxidized lipids and free cholesterol can initiate the pro-inflammatory signaling cascade, however, macrophages can also esterify cholesterol, which can transform the cell into a lipid-laden macrophage and potentiate persistence and pro-fibrotic signaling24. In the following section, we will describe the mechanisms by which macrophages handle lipids and how dysregulation of these processes contributes to lipid-laden macrophage formation, lung injury, and inflammation.
3.0. Cellular processes governing lipid accumulation and function of pulmonary macrophages.
3.1. Acquisition of Lipids
The physicochemical properties of lipids, which are determined by their composition and size, determine the ability of receptors to bind and transport them into the cell. Indeed, whether a particular class of lipid is taken up by scavenger receptors or endocytosed is dependent on these properties25–27. Anionic lipids are taken up more efficiently than their neutral counterparts28–30, with larger diameter also increasing uptake by macrophages31. The lipids of the lung lining, including phosphatidylserine, phosphatidylglycerol, and phosphatidylcholine, are negatively charged, making them prone to uptake by alveolar macrophages32–34. Thus, one can see that the environment surrounding the alveolar macrophage will bias these cells towards lipid uptake and accumulation under physiological conditions. However, modified lipid species that may rapidly increase in the context of injury, such as oxidized cholesterol products like 7-ketocholesterol and hydroxycholesterol, common oxysterols,35–37 have been shown to influence macrophage signaling and apoptosis. High concentrations of oxysterols affect cholesterol homeostasis and induce cytotoxicity37,38, contributing to injury and inflammation in the lung through activation of pro-inflammatory signaling pathways, such as NF-κB39. This indicates that while lipid uptake and incorporation of lipid species into comparatively more inert intracellular lipid droplets promote cell survival and prolonged signaling, lack of sufficient lipid droplet formation leads to cell death and inflammatory signaling.
3.1.1. Receptor-Mediated Lipid Uptake
The scavenger receptor family of cell surface proteins are inherently vital to macrophage function recognizing a wide range of motifs that include lipids40. As opposed to the classical and singular uptake pathway of LDL by the LDLR41, modified sterols, such as oxidized LDL (oxLDL) and acetylated LDL, are taken up through a variety of scavenger receptors, which can recognize both native and modified lipids14,40,42. Receptors known to regulate lipid uptake include Scavenger receptor A (SR-A) I/II43,44, cluster of differentiation 36 (CD36)44–46, scavenger receptor B (SR-B) I44,47, cluster of differentiation factor 68 (CD68)47, chemokine (C-X-C motif) ligand 16 (CXCL16)47, lectin-like oxidized low-density-lipoprotein receptor-1 (LOX-1)48,49, and cleavage factor polyribonucleotide kinase subunit 1 (CL-P1)47. All of these receptors have been reported to bind oxidized sterols, with the majority of oxLDL internalization attributed to SR-A and CD3650,51. Uptake through receptors aside from LDLR may circumvent negative feedback signaling via sterol regulatory element binding protein-2 (SREBP-2)52, leading to excess LDL accumulation and increased cholesterol efflux. As such, macrophage scavenger receptor expression significantly contributes to the lipid-laden phenotype.
CD36 is widely expressed on macrophages and binds various modified or native proteins with a particular affinity for oxLDL45,46. It has been found to account for most of the CE accumulation in macrophages exposed to oxLDL46, implicating this receptor as one of particular interest in lipid-laden macrophage formation. Further, CD36 expression is not downregulated by the presence of intracellular cholesterol,53 and thus lack of feedback regulation may point to this mechanism as a significant contributor to lipid-laden macrophage formation. Upregulation of CD36 has been found to be mediated through PPAR-γ activation in the presence of oxLDL, increasing its expression through a positive feedback loop54,55. Increased expression of uptake transporters without a concomitant increase in efflux of lipid-associated material can contribute to the development of a lipid-laden phenotype in macrophages.
3.2. Lipid Synthesis
The endomembrane, comprised of the endoplasmic reticulum (ER) and the Golgi apparatus, constitutes the primary site of lipid synthesis in the cell56 and produces high levels of phospholipids57,58. The ER is directly affected by cellular stress, as can occur during lung injury, and this can impact lipid accumulation. Intracellular accumulation of free cholesterol initiates the ER stress response in macrophages59,60, impacting a host of cellular mechanisms, including direct modification of lipid synthesis. Furthermore, macrophage functional responses are critically tied to lipid homeostasis, as the fundamental phagocytic response of macrophages requires an increase in membrane lipid production to repair cell damage and to envelop foreign matter for phagocytosis61,62. Below we will discuss several factors involved in lipid synthesis and its regulation.
3.2.1. Fatty Acid Synthase and Acetyl CoA Carboxylase
In the cytoplasm, Acetyl CoA Carboxylase 1 (ACC1) converts acetyl-CoA to malonyl-CoA, and in the ER, fatty acid synthase (FAS converts acetyl-CoA and malonate to malonyl-CoA63. In addition, the acyltransferase family of enzymes is involved in the de novo synthesis of membrane lipids. However, more commonly they mediate the modification of acyl chains to generate different classes of lipids56,64,65. Enzymes located in the mitochondria and peroxisomes also play a role in overall lipogenesis in the cell; however, the ER/Golgi and acyltransferases remain the primary drivers.
Cytosolic ACC1 regulates de novo FAS-mediated FA synthesis. However, in contrast to other reports of FAS-mediated lipid signaling, the ACC1-FAS pathway of lipogenesis is not critical to the macrophage mediated innate immune defenses against Mycobacterium tuberculosis infection66. Furthermore, regulation of innate immunity through lipid-mediated mechanisms may be pathogen or toxicant-specific, which is consistent with the idea that lipid content can alter chemokine response14. Within resident liver macrophages, inhibition of ACCs using a phosphorylation mimic results in a switch from pro-inflammatory to anti-inflammatory phenotype67. This suggests that the inhibition of lipogenesis may be associated with pro-inflammatory signaling.
3.2.2. SREBP Signaling
Sterol regulatory element binding proteins (SREBPs) SREBP1 and 2 are critical transcription factors for fatty acid and sterol biosynthesis68–70. SREBP-1a is abundantly expressed in macrophages and has been established as one link between lipid homeostasis and innate immune response62,71,72. SREBP1 accumulates upon acute proand anti-inflammatory activation of macrophages, and the absence of the protein is associated with decreased fatty acid synthesis62,73. For example, SREBP1 is transcriptionally regulated by the pro-inflammatory Toll-like receptor 4 (TLR-4)-nuclear factor kappa B (NF-κB) pathway in macrophages14,74. During lung injury, saturated fatty acid levels increase and act as endogenous TLR-4 ligands to activate SREBP1 expression and subsequent lipogenesis75,76. Increased lipogenesis within macrophages is permissive of inflammatory activation. However, IL-4 stimulation leads to increased AKT phosphorylation, STAT6 signaling, and SREBP1-mediated upregulation of fatty acid synthesis77, which leads to alternative activation14. In this regard, SREBP1 enhancement is required for alternative macrophage activation, whereas SREBP2, is not required. Thus, it is possible that SREBP1 potentiation during inflammation may contribute to eventual lipid-laden macrophage formation.
3.2.3. Mechanistic/mammalian target of rapamycin (mTOR)
mTOR complex 1 (mTORC1) is involved in lipid metabolism, proliferation, and growth, as its activation occurs in times of nutrient abundance and the absence of cellular stress78. The main regulatory element of mTORC1 is mTOR itself. mTOR activates SREBP1, and via mTORC1 controls the expression of binding proteins to promote lipogenesis and storage. Its inhibition has been shown to increase LDL levels in the circulation by diminishing cholesterol clearance68. Furthermore, a loss of mTORC function reduces macrophage mediated scavenging of LDL and increases efflux, which results in a decrease in lipid storage systemically69,79. The importance of mTOR activation within lung macrophages appears to be important in metabolic shifts required for activation and signaling in cancer models73,80,81, both of which are critical to metabolic functions in lipid-laden macrophages. Despite the clear capacity of mTOR to play a role in lipid regulation, its importance in lipid-laden macrophage formation within the lung has not been delineated. This may be critical within resident alveolar macrophages, which exist in a lipid rich environment and thus can be considered to be in a state of nutrient abundance.
4.0. Lipid export in macrophages
4.1. Lysosomal Processing for Export
Prior to export, lipids are processed in lysosome-related organelles known as lamellar bodies. Lipids are broken down in the acidic interior compartment, which contains acid phosphatase, cathepsin C, and cathepsin H82,83. Lipolysis transforms lipids into free cholesterol and fatty acids through the action of lysosomal acid lipase84. Free fatty acids can then feed into the FAO metabolic pathway and subsequent mitochondrial metabolism or be incorporated as triglycerides into lipid droplets for storage within the cell. In contrast, released free cholesterol must be effluxed and chaperoned by lipoproteins, such as high-density lipoprotein or APOA185 or esterified in the cell to avoid pro-inflammatory signaling, cytotoxicity, and cell death85,86 (Figure 2).
Figure 2. Cellular processes governing lipid accumulation in alveolar macrophages.
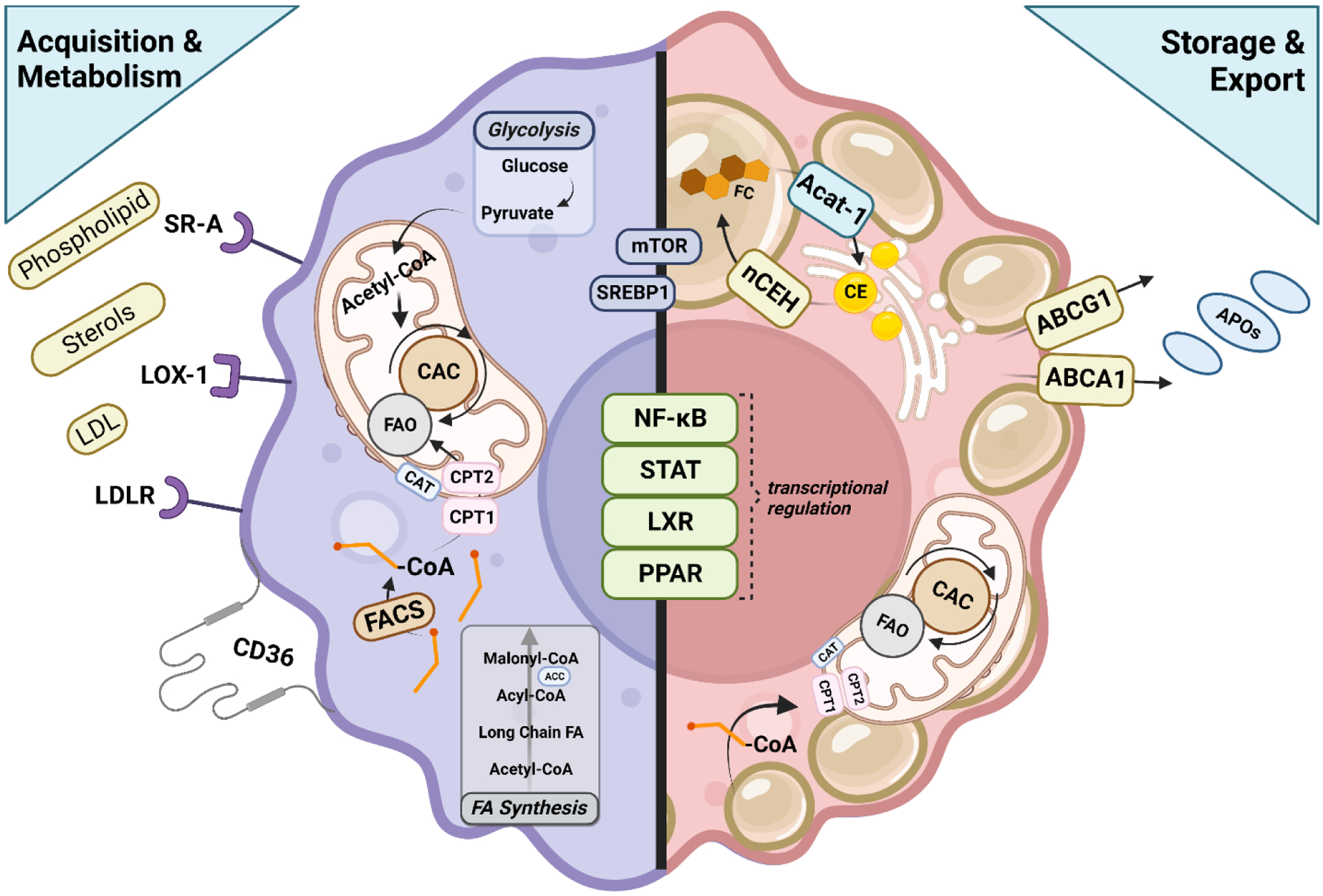
Macrophages acquire lipids and cholesterol through a variety of receptors including SR-A, LOX-1, LDLR and CD36. These cells also acquire lipid through de novo fatty acid synthesis. FACS conjugates free Co-A to the molecule, allowing for the conversion of the fatty acyl CoA to acyl carnitine, which can then be transported across the mitochondria membrane by CAT. CPT2 converts acyl carnitine back to fatty acyl CoA which can then be oxidized and feed into the CAC. NADH, FADH2, and GTP produced from glycolysis/CAC are oxidized and the resulting electrons flow through the complexes of the electron transport chain, creating a protein gradient that drives ATP synthase to produce usable energy for the cell in the form of ATP. Upregulated storage of lipids in the macrophage drive sterol-sensitive signaling pathways, some of which oppose lipid accumulation. LXR signaling induces the transcription of cholesterol efflux transporters like ABCG1 and ABCA1 to promote lipid homeostasis within the cell, similar to PPARα-mediated reduction in triglyceride levels. PPARγ signaling increases glucose metabolism and upregulates the expression of CD36, promoting the uptake of lipids in the macrophage and, along with STAT6, also induces FAO. Contributing to increased substrate availability for FAO, NFκB controls the transcription of pro-inflammatory genes and contributes to macrophage signaling, including activation of SREBP1 which promotes lipogenesis, a process also controlled by mTOR signaling. Importantly, the cell must also have mechanisms to store lipid to be oxidized at a later time for energy and to counteract free cholesterol-induced cytotoxicity, thus storage of these molecules is vitally important. Free cholesterol is esterified by ACAT-1, forming CE, the critical reaction leading to lipid droplet formation in the macrophage. This action is opposed by nCEH which releases free cholesterol for export for loading onto various apolipoprotein carriers.
4.2. Transporter-mediated efflux
ABC Transporters A1 and G1 are the predominant cholesterol efflux transporters in macrophages, known to promote the flow of intracellular stores to extracellular carriers such as ApoA-1 or HDL87. The expression of these transporters is controlled by the PPAR-γ/LXR axis88,89. Activation of PPAR-γ in macrophages has been shown to stimulate ABCA1 and ABCG1-dependent efflux88,90. Antagonists to these transporters promote lipid-laden cell formation, while activators of the PPAR-γ/LXR pathways upregulate efflux, potentially limiting the formation of lipid-laden cells.
These processes can be opposed by other signaling mechanisms that maintain lipid homeostasis in the cell. For example, the nuclear transcription factor LXR is crucial for lipid homeostatic signaling in macrophages91,92. Oxysterols ingested by macrophages during phagocytosis lead to increased cholesterol and oxysterol loading, inducing LXR signaling and increased transcription of ABCA1, ABCG191 and the inducible degrader of the LDL receptor (IDOL)93. Transcription of these genes oppose lipid accumulation by increasing cholesterol efflux mechanisms and degradation of the LDLR through ubiquitin-mediated mechanisms. Although the relevance of the LDLR pathway within the lung lining is unknown.
4.3. Mechanisms contributing to decreased lipid export.
As the major efflux transporters, the ABC proteins function to promote cholesterol export and mitigate lipid accumulation in the cell87. Therefore, dysfunction or reduced expression of these transporters can promote lipid accumulation in macrophages. Other factors contributing to reduced cholesterol efflux include depletion of cholesterol carrier proteins94, such as apolipoprotein A-195, but it is unclear if this mechanism is of consequence in the lung.
Gene silencing by non-coding RNAs has been implicated in lipid homeostasis. miR-33 has been observed to decrease ABCA1 expression while knockdown of miR-33 increases reverse cholesterol transport in macrophage foam cells96. miR-33 targets the 3’- untranslated region of many genes involved in cholesterol homeostasis, including ABCA197–99. The miRNA suppression of ABCA1 and other cholesterol efflux transports could contribute to increases in macrophage lipid accumulation.
5.0. Lipid Metabolism in Macrophages
5.1. Lipid Recycling by Macrophages
Within the lung, macrophages are continuously exposed to a lipid-rich environment, regardless of injury. They play a significant role in surfactant homeostasis by recycling the lipid. Pulmonary surfactant is comprised of primarily phospholipids and neutral lipids, the most common of the latter being cholesterol17,100. Alveolar type II (AT2) and Clara cells are also highly involved in lipid metabolism, producing lipids critical to surface active function101. If not recycled by AT2 cells, surfactant is degraded by alveolar macrophages16,101. Alveolar macrophages are thus needed to clear lipids even under normal physiological conditions.
5.2. Fatty acid oxidation (FAO)
Fatty acids in the cytosol are enzymatically converted to fatty acid acyl-CoA. Further downstream oxidation and energy release occurs within the mitochondria. Carnitine conjugation and transport via carnitine palmitoyl transferase 1 (CPT1) facilitates the movement of the fatty acid acyl-CoA into the mitochondria, where carnitine is removed by carnitine palmitoyl transferase 2 (CPT2)102. Oxidation of these molecules yields the reducing equivalents NADH and FADH2 as well as acetyl-CoA, which is used to generate energy throughout the citric acid cycle and the electron transport chain. ACC2, which is located in the mitochondrial membrane, regulates FAO by controlling fatty acid uptake to the mitochondria via CPT1103.
Reliance on FAO is associated with enhanced cellular lifespan and limits lipid and fatty acid accumulation in macrophages102. In contrast, in vivo work has shown that constitutive activation of the transferase responsible for long-chain fatty acid import to the mitochondria, CPT1, reduces lipid accumulation in the cell105. Furthermore, FAO is induced by STAT6 and PPAR-γ-co-activator 1β84,106 which are anti-inflammatory mediators, and CPT2 deletion was shown to impair FAO105,107, oxidative phosphorylation and anti-inflammatory activation. Collectively, these data indicate a critical role for FAO in regulating lipid accumulation and macrophage phenotype. Products of lipid metabolism, such as free fatty acids, triglycerides, diacylglycerides, and ceramides, as well as oxidized lipids, influence pro-inflammatory signaling23. High concentrations of oxysterols affect cholesterol homeostasis and can induce cytotoxicity37,38, contributing to injury and inflammation in the lung.
6.0. Lipid Storage in Macrophages
6.1. Lipid droplet formation
Triglycerides and cholesterol esters are the primary components of lipid droplets, encapsulated by a phospholipid membrane, and thus the accumulation of all of these components is critical to lipid droplet formation108. Esterification is a critical step in mitigating free cholesterol-induced cytotoxicity. The Acyl-coenzyme A cholesterol acyltransferase/sterol O acyl transferase (ACAT/Soat) subset of enzymes catalyzes cholesterol esterification in macrophages, with two primary isoforms identified as ACAT-1/Soat1 and ACAT-2/Soat2109–111. ACAT-2/Soat2 is localized to hepatocytes and the intestine, whereas ACAT-1/Soat1 is expressed primarily in macrophages and is the principal mechanism in lipid droplet formation in this cell type65,112,113. ACAT-1/Soat1 converts cholesterol and oleoyl coenzyme A to esterified cholesterol with coenzyme A as a by-product, contributing to lipid droplet formation109–111. The cholesterol esterification rate can be modified via extracellular signaling, as ACAT-1/Soat1 expression can be induced through the leptin-JAK/PI3K pathway114 and the insulin-Erk/JNK pathway115,116. Esterification of cholesterol by ACAT-1/Soat1 is opposed by the action of neutral cholesterol ester hydrolase (nCEH), which releases free cholesterol for export from the cell117.
6.1.1. Triglycerides in lipid droplet formation and turnover
In addition to esterified products, triglycerides can form the core of intracellular lipid droplets. ER-associated transferase enzymes acyl-CoA:diacylglycerol acyl transferase-1 (DGAT-1) and acylCoA:diacylglycerol acyl transferase-2 (DGAT-2) participate in triglyceride synthesis from diacylglycerol and acyl-CoA derived from FAs118. Conversely, triglycerides are liberated back to diacylglycerol and acyl-CoA through various lipases found in the cytoplasm119. Triglycerides may also be broken down within macrophages through lipase activity in the autophagosome120. Thus, the dynamic balance between triglyceride formation and breakdown is a key regulator of lipid droplet formation in macrophages.
6.2. Lipid droplet signaling
Lipid droplets, comprised of the neutral lipids triacylglycerol and CE108,121, have enzymes localized on their surface that regulate various pathways such as triacylglycerol synthesis and rates of their own accumulation and degradation122,123. Because lipid droplets store many different lipid types, their release can result in them directly acting as or being converted to signaling molecules121,122. The breakdown of triacylglycerol and CE results in free fatty acid formation, which can bind many cell surface and intracellular receptors that are stimulators of inflammatory signaling122,124–126. Notably, free fatty acids are able to bind and activate intra- or extracellular G-protein coupled receptors (GCPRs), toll-like receptors (TLRs), PPARs, and NF-κB, all of which are highly involved in macrophage regulation, signaling, and phenotype, potentially contributing to the pathophysiological conditions present in the lung during injury122,124–126.
Additionally, lipid droplet formation may also impact cell signaling by removing substrates necessary for bioactive, pro-inflammatory lipid signaling. By acting as a repository for fatty acids, cholesterol, and other species, lipid droplet formation reduces the capacity for the formation of lipid peroxidation, preventing pro-inflammatory signaling122,127. Though this may help to prevent or lessen oxidative damage in the lung, the excessive accumulation of lipids within macrophages will alter lipid droplet metabolism and the function of lipid droplet-associated proteins122,128. As such, it is proposed that accumulation of lipid droplets in macrophages bias the cell towards reliance on FAO and oxidative phosphorylation rather than glycolysis. This has the potential to negatively affect macrophage function, as macrophages will persist, taking on a pro-resolution phenotype rather than undergoing homeostatic turnover and cell death which could lead to long-term signaling effects such as fibrosis. Similar dysfunction has been observed in dendritic cells, where lipid droplets prevent proper antigen presentation and chaperone-mediated autophagy128–131, drastically altering cell function through fundamental metabolic mechanisms.
7.0. Conclusions
As reviewed here, lipid-laden macrophage formation has been observed in a variety of pathophysiologies, such as atherosclerosis, but knowledge is lacking as to their role in lung pathology. Recently, there has been considerable interest in the pulmonary macrophage and its heterogeneous roles in lung injury1. In this review, we have focused on the mechanisms that can lead to the formation of lipid-laden macrophages, or “foam cells”, as these cells seem to have their own unique roles. It appears that these cells may limit acute activation and bias the injury response to repair, as well as significantly altering phagocytosis and surfactant recycling1. When considering lipid-laden macrophage formation, there are four main processes to consider: lipid import, metabolism, storage, and export. The balance between these processes is critical to maintaining normal macrophage function under homeostatic conditions. However, these four processes are influenced and altered by signaling, genetic factors, and – vital in the case of injury – the tissue microenvironment132. These processes and their regulation are summarized in Figure 2. In this way the formation of a lipid-laden macrophage can be considered as being dependent upon the balance between acquisition and synthesis with export and metabolism.
Distinct from other tissue environments in which foam cells have been characterized, the pulmonary environment is heavily lipid laden at normal physiological levels, as approximately 90% of the lung lining fluid is comprised of phospholipid133. In addition to their role in innate immunity, macrophages contribute significantly to the regulation of lung function and work of breathing through the catabolism of surfactant. With this role in the consistent turnover of lipid species, it is significant that pulmonary macrophages do not become inherently lipid laden under homeostatic conditions. It is proposed that the accumulation of lipid-laden macrophages in the lung is significant in lung pathophysiology, especially in the context of lung injury and inflammation. This phenotypic change may reduce the capacity of the lung to sufficiently resolve injury. The accumulation of lipids in macrophages has the potential to prolong the life of the cell134, likely due to the reliance on FAO and anti-inflammatory phenotypic switching. These cells may be persistent and contribute to chronic signaling, promoting the transition to fibrosis rather than injury resolution24. Increased cellular persistence, aberrant signaling, and maintenance of a pro-fibrotic phenotype stand out as the most significant potential consequences of lipid-laden cell formation in the context of lung injury. Thus, limiting the formation of lipid-laden macrophages in lung pathologies may be an advantageous proposition from a translational standpoint. Though any of the four processes may be a feasible target for pharmacological development, we propose that targeting the excess lipid storage may be the most viable and least disruptive to normal macrophage processes. With a limited storage capacity, macrophages exposed to excess lipids would become susceptible to cell death via the cytotoxic effects of sterols84. Limiting excess lipid accumulation and restoring macrophage homeostasis in the context of injury may also have significant effects on the phenotypic differentiation of both resident and recruited cells.
The formation and persistence of lipid-laden macrophages in the lung have the capacity to impact the spectrum of inflammation and resolution in virtually all pulmonary pathologies. For example, the prevalence of lipid-laden macrophages may prime these cells towards the use of FAO due to substrate availability rather than glycolysis, which may ultimately impair macrophage function in various pathologies (Fig 3). More research is needed to assess how inhibition of lipid accumulation in the lung specifically may represent a pharmacological target in lung injury and disease. Additionally, there are many outstanding questions regarding the formation, prevalence, composition, and significance of this phenotype in experimental animal models and in human pulmonary insult and disease. It would be most pertinent to document the occurrence or lack of lipid-laden macrophages in lung pathologies, as well as interrogate the mechanism leading to their formation, as it may be that regulating key proteins in lipid and cholesterol handling are differential within and among lung pathologies.
Figure 3.
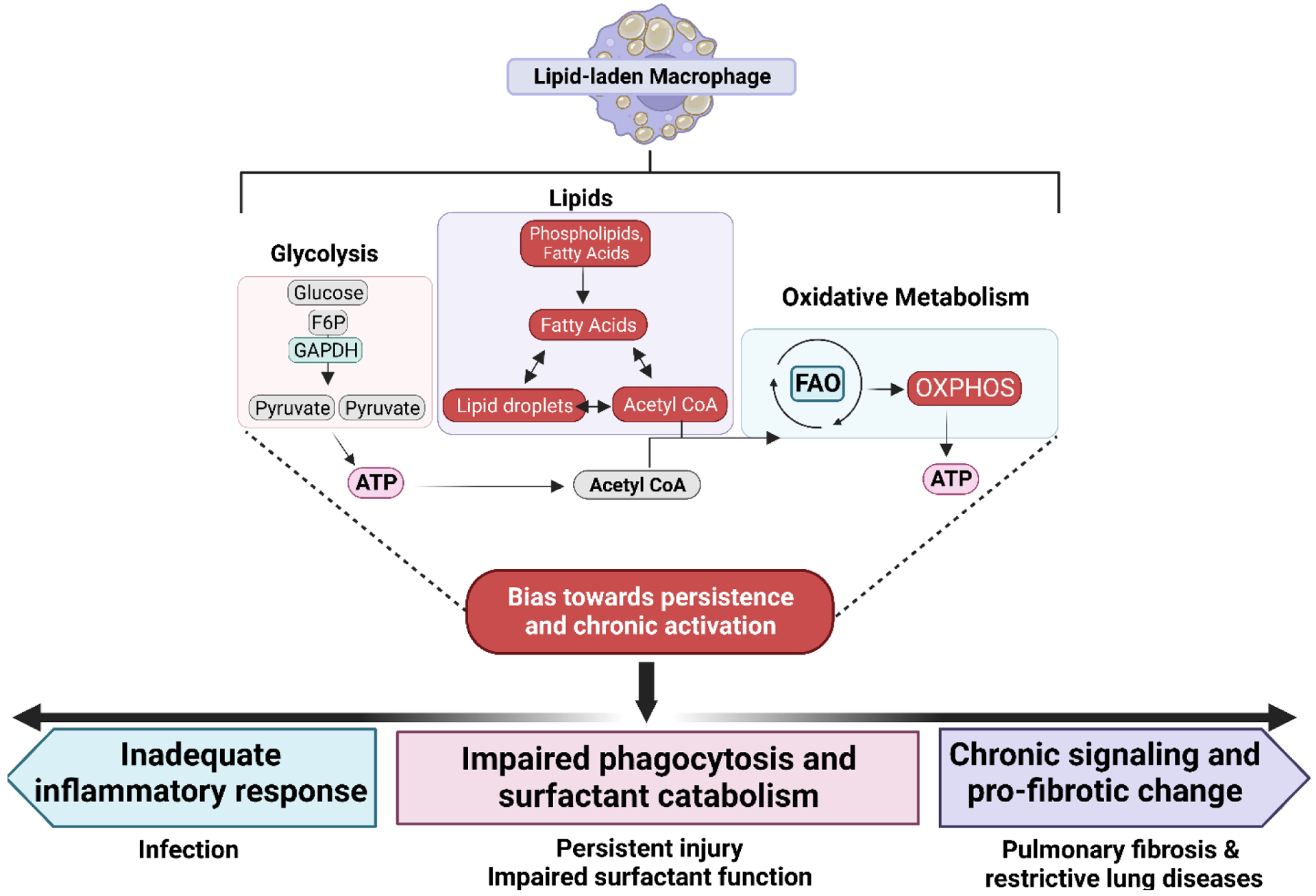
Persistence of lipid laden macrophages in the lung may lead to dampened inflammation and fibrotic change. Due to excess lipid accumulation in macrophages of this phenotype, it is proposed that there is significant reliance on fatty acid oxidation (FAO) rather than glycolysis which is typical of an acute inflammatory response. Increased reliance on these processes is represented by red boxes. The downstream effects of these changes are proposed to bias towards chronic activation, which may ultimately lead to an inadequate inflammatory response upon stimulation, priming the lung for susceptibility to infection and persistent injury. Furthermore, this may impair the phagocytic capacity of the macrophage, leading to both ineffective inflammation and poor surfactant catabolism (pale blue box), further negatively impacting lung function. Persistence of this phenotype may also contribute to persistence of this traditionally “anti-inflammatory phenotype,” leading to pro-fibrotic signaling and collagen deposition in the lung (purple box), leading to fibrosis or other restrictive lung diseases.
From the various gatekeepers of lipid-laden macrophages presented within, there are many potential therapeutic targets to prevent the formation of this cell phenotype in the lung. It is of note that many of these, such as limiting scavenger receptor expression and activity135 have been explored in the context of other disease states like atherosclerosis. However, altering receptor uptake in pulmonary macrophages inherently blunts their phagocytic capacity, pointing to this strategy as ineffectual in this organ-specific context. Activation of the LXR signaling pathway leads to the inhibition of enzymes directly catalyzing lipid droplet storage such as the DGAT or ACAT enzymes. This may present as a logical target to inhibit lipid-laden cell formation, however, macrophage-specific targeting of LXR in the lung remains challenging. The use of mi-RNA knockdown, such as with the investigation of miR27 for the prevention of atherosclerosis136 may eventually useful. Ultimately, there are inherent risks in immunomodulation as well as in the practical application of the strategies, limiting the discussion of viable therapeutic targets at this time. More detailed research into the formation of pulmonary-specific lipid-laden macrophages is necessary to understand their role in lung pathologies.
Highlights.
Lipid-laden macrophages are observed in the lung in vivo following pulmonary injury, and this phenotype may significantly contribute to pulmonary pathologies.
This phenotype is proposed to form due to the imbalance between lipid acquisition, metabolism, storage, and export.
The complex interplay between metabolism and storage of lipids are discussed as they pertain to disease progression.
Herein, we discuss critical regulators and signaling pathways that may play a role in the formation of lipid-laden macrophages in the lung.
The consequences of lipid-laden macrophage formation within the context of pulmonary inflammation are considered.
Acknowledgments
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant 086621]; and the National Institutes of Health National Institute of Environmental Health Sciences [Grant T32ES01984; P30ES005022], ES030984, ES032473.
Footnotes
Figures were created with Biorender.com.
No author has an actual or perceived conflict of interest with the contents of this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bain CC & MacDonald AS The impact of the lung environment on macrophage development, activation and function: diversity in the face of adversity. Mucosal Immunol 15, 223–234 (2022). 10.1038/s41385-021-00480-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laskin DL et al. Macrophages, reactive nitrogen species, and lung injury. Ann N Y Acad Sci 1203, 60–65 (2010). 10.1111/j.1749-6632.2010.05607.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shibata N & Glass CK Macrophages, oxysterols and atherosclerosis. Circ J 74, 2045–2051 (2010). 10.1253/circj.cj-10-0860 [DOI] [PubMed] [Google Scholar]
- 4.Aluganti Narasimhulu C et al. Atherosclerosis--do we know enough already to prevent it? Curr Opin Pharmacol 27, 92–102 (2016). 10.1016/j.coph.2016.02.006 [DOI] [PubMed] [Google Scholar]
- 5.Gowdy KM & Fessler MB Emerging roles for cholesterol and lipoproteins in lung disease. Pulm Pharmacol Ther 26, 430–437 (2013). 10.1016/j.pupt.2012.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Venosa A et al. Regulation of Macrophage Foam Cell Formation During Nitrogen Mustard (NM)-Induced Pulmonary Fibrosis by Lung Lipids. Toxicological sciences : an official journal of the Society of Toxicology 172, 344–358 (2019). 10.1093/toxsci/kfz187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stevenson ER, Wilkinson ML, Abramova E, Guo C & Gow AJ Intratracheal Administration of Acat-1 Inhibitor K-604 Reduces Pulmonary Inflammation Following Bleomycin-induced Lung Injury. Journal of Pharmacology and Experimental Therapeutics (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brown MS & Goldstein JL LIPOPROTEIN METABOLISM IN THE MACROPHAGE: Implications for Cholesterol Deposition in Atherosclerosis. Annual Review of Biochemistry 52, 223–261 (1983). 10.1146/annurev.bi.52.070183.001255 [DOI] [PubMed] [Google Scholar]
- 9.Libby P Inflammation in atherosclerosis. Nature 420, 868–874 (2002). 10.1038/nature01323 [DOI] [PubMed] [Google Scholar]
- 10.Yu XH, Fu YC, Zhang DW, Yin K & Tang CK Foam cells in atherosclerosis. Clin Chim Acta 424, 245–252 (2013). 10.1016/j.cca.2013.06.006 [DOI] [PubMed] [Google Scholar]
- 11.Galkina E & Ley K Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol 27, 165–197 (2009). 10.1146/annurev.immunol.021908.132620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oram JF ATP-binding cassette transporter A1 and cholesterol trafficking. Curr Opin Lipidol 13, 373–381 (2002). 10.1097/00041433-200208000-00004 [DOI] [PubMed] [Google Scholar]
- 13.Wang X et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest 117, 2216–2224 (2007). 10.1172/jci32057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan J & Horng T Lipid Metabolism in Regulation of Macrophage Functions. Trends Cell Biol 30, 979–989 (2020). 10.1016/j.tcb.2020.09.006 [DOI] [PubMed] [Google Scholar]
- 15.Haagsman HP, Hogenkamp A, Van Eijk M & Veldhuizen EJ Surfactant collectins and innate immunity. Neonatology 93, 288–294 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Trapnell BC & Whitsett JA Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol 64, 775–802 (2002). 10.1146/annurev.physiol.64.090601.113847 [DOI] [PubMed] [Google Scholar]
- 17.Wright JR Host defense functions of pulmonary surfactant. Neonatology 85, 326–332 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Sunil VR et al. Protective Role of Surfactant Protein-D Against Lung Injury and Oxidative Stress Induced by Nitrogen Mustard. Toxicol Sci 166, 108–122 (2018). 10.1093/toxsci/kfy188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quintero OA & Wright JR Clearance of surfactant lipids by neutrophils and macrophages isolated from the acutely inflamed lung. Am.J.Physiol Lung Cell Mol.Physiol 282, L330–L339 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Matute-Bello G, Frevert CW & Martin TR Animal models of acute lung injury. Am J Physiol Lung Cell Mol Physiol 295, L379–399 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ganter MT et al. Extracellular heat shock protein 72 is a marker of the stress protein response in acute lung injury. Am J Physiol Lung Cell Mol Physiol 291, L354–361 (2006). 10.1152/ajplung.00405.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell CW, Jiang W, Reich CF & Pisetsky DS The extracellular release of HMGB1 during apoptotic cell death. Am J Physiol Cell Physiol 291, C1318–1325 (2006). 10.1152/ajpcell.00616.2005 [DOI] [PubMed] [Google Scholar]
- 23.S. N SG, Raviraj R, Nagarajan D & Zhao W Radiation-induced lung injury: impact on macrophage dysregulation and lipid alteration – a review. Immunopharmacology and Immunotoxicology 41, 370–379 (2019). 10.1080/08923973.2018.1533025 [DOI] [PubMed] [Google Scholar]
- 24.Romero F et al. A pneumocyte-macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol 53, 74–86 (2015). 10.1165/rcmb.2014-0343OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schwenk RW, Holloway GP, Luiken JJ, Bonen A & Glatz JF Fatty acid transport across the cell membrane: regulation by fatty acid transporters. Prostaglandins Leukot Essent Fatty Acids 82, 149–154 (2010). 10.1016/j.plefa.2010.02.029 [DOI] [PubMed] [Google Scholar]
- 26.Ahsan F, Rivas IP, Khan MA & Torres Suárez AI Targeting to macrophages: role of physicochemical properties of particulate carriers—liposomes and microspheres—on the phagocytosis by macrophages. Journal of Controlled Release 79, 29–40 (2002). 10.1016/S0168-3659(01)00549-1 [DOI] [PubMed] [Google Scholar]
- 27.Abumrad NA, Sfeir Z, Connelly MA & Coburn C Lipid transporters: membrane transport systems for cholesterol and fatty acids. Curr Opin Clin Nutr Metab Care 3, 255–262 (2000). 10.1097/00075197-200007000-00003 [DOI] [PubMed] [Google Scholar]
- 28.Raz A, Bucana C, Fogler WE, Poste G & Fidler IJ Biochemical, morphological, and ultrastructural studies on the uptake of liposomes by murine macrophages. Cancer research 41, 487–494 (1981). [PubMed] [Google Scholar]
- 29.Hsu M & Juliano R Interactions of liposomes with the reticuloendothelial system: II. Nonspecific and receptor-mediated uptake of liposomes by mouse peritoneal macrophages. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research 720, 411–419 (1982). [DOI] [PubMed] [Google Scholar]
- 30.Heath TD, Lopez NG & Papahadjopoulos D The effects of liposome size and surface charge on liposome-mediated delivery of methotrexate-γ-aspartate to cells in vitro. Biochimica et Biophysica Acta (BBA)-Biomembranes 820, 74–84 (1985). [DOI] [PubMed] [Google Scholar]
- 31.Allen T, Austin G, Chonn A, Lin L & Lee K Uptake of liposomes by cultured mouse bone marrow macrophages: influence of liposome composition and size. Biochimica et Biophysica Acta (BBA)-Biomembranes 1061, 56–64 (1991). [DOI] [PubMed] [Google Scholar]
- 32.Fidler IJ, Raz A, Fogler WE, Hoyer LC & Poste G The role of plasma membrane receptors and the kinetics of macrophage activation by lymphokines encapsulated in liposomes. Cancer Res 41, 495–504 (1981). [PubMed] [Google Scholar]
- 33.Schroit AJ & Fidler IJ Effects of liposome structure and lipid composition on the activation of the tumoricidal properties of macrophages by liposomes containing muramyl dipeptide. Cancer Res 42, 161–167 (1982). [PubMed] [Google Scholar]
- 34.Fidler IJ Targeting of immunomodulators to mononuclear phagocytes for therapy of cancer. Advanced Drug Delivery Reviews 2, 69–106 (1988). 10.1016/0169-409X(88)90006-3 [DOI] [Google Scholar]
- 35.Brown AJ, Dean RT & Jessup W Free and esterified oxysterol: formation during copper-oxidation of low density lipoprotein and uptake by macrophages. J Lipid Res 37, 320–335 (1996). [PubMed] [Google Scholar]
- 36.Panini SR et al. Arachidonate metabolism and the signaling pathway of induction of apoptosis by oxidized LDL/oxysterol. J Lipid Res 42, 1678–1686 (2001). [PubMed] [Google Scholar]
- 37.Aupeix K et al. Oxysterol-induced apoptosis in human monocytic cell lines. Immunobiology 194, 415–428 (1995). 10.1016/s0171-2985(11)80108-7 [DOI] [PubMed] [Google Scholar]
- 38.Yang L & Sinensky MS 25-Hydroxycholesterol activates a cytochrome c release-mediated caspase cascade. Biochem Biophys Res Commun 278, 557–563 (2000). 10.1006/bbrc.2000.3855 [DOI] [PubMed] [Google Scholar]
- 39.Young LR et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest 138, 674–681 (2010). 10.1378/chest.10-0573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Canton J, Neculai D & Grinstein S Scavenger receptors in homeostasis and immunity. Nature Reviews Immunology 13, 621–634 (2013). [DOI] [PubMed] [Google Scholar]
- 41.Luchetti F et al. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br J Pharmacol 178, 3104–3114 (2021). 10.1111/bph.15272 [DOI] [PubMed] [Google Scholar]
- 42.Plüddemann A, Mukhopadhyay S & Gordon S The interaction of macrophage receptors with bacterial ligands. Expert Rev Mol Med 8, 1–25 (2006). 10.1017/s1462399406000159 [DOI] [PubMed] [Google Scholar]
- 43.Peiser L, Mukhopadhyay S & Gordon S Scavenger receptors in innate immunity. Curr Opin Immunol 14, 123–128 (2002). 10.1016/s0952-7915(01)00307-7 [DOI] [PubMed] [Google Scholar]
- 44.Moore KJ & Freeman MW Scavenger Receptors in Atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 26, 1702–1711 (2006). https://doi.org:doi: 10.1161/01.ATV.0000229218.97976.43 [DOI] [PubMed] [Google Scholar]
- 45.Kunjathoor VV et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem 277, 49982–49988 (2002). 10.1074/jbc.M209649200 [DOI] [PubMed] [Google Scholar]
- 46.Podrez EA et al. Macrophage scavenger receptor CD36 is the major receptor for LDL modified by monocyte-generated reactive nitrogen species. J Clin Invest 105, 1095–1108 (2000). 10.1172/jci8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greaves DR & Gordon S The macrophage scavenger receptor at 30 years of age: current knowledge and future challenges. Journal of lipid research 50 Suppl, S282–S286 (2009). 10.1194/jlr.R800066-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Martín-Fuentes P et al. Individual Variation of Scavenger Receptor Expression in Human Macrophages with Oxidized Low-Density Lipoprotein Is Associated with a Differential Inflammatory Response. The Journal of Immunology 179, 3242–3248 (2007). 10.4049/jimmunol.179.5.3242 [DOI] [PubMed] [Google Scholar]
- 49.Chen M, Masaki T & Sawamura T LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: implications in endothelial dysfunction and atherosclerosis. Pharmacology & therapeutics 95, 89–100 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Geloen A et al. CD36 inhibitors reduce postprandial hypertriglyceridemia and protect against diabetic dyslipidemia and atherosclerosis. PLoS One 7, e37633 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cai Y et al. Neuropeptide Y regulates cholesterol uptake and efflux in macrophages and promotes foam cell formation. Journal of Cellular and Molecular Medicine (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Ma KL, Ruan XZ & Liu BC Dysregulation of the Low-Density Lipoprotein Receptor Pathway Is Involved in Lipid Disorder-Mediated Organ Injury. Int J Biol Sci 12, 569–579 (2016). 10.7150/ijbs.14027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghodsian N, Yeandle A & Gieseg SP Foam cell formation but not oxLDL cytotoxicity is inhibited by CD36 down regulation by the macrophage antioxidant 7,8-dihydroneopterin. The International Journal of Biochemistry & Cell Biology 133, 105918 (2021). https://doi.org: 10.1016/j.biocel.2021.105918 [DOI] [PubMed] [Google Scholar]
- 54.Nagy L, Tontonoz P, Alvarez JG, Chen H & Evans RM Oxidized LDL regulates macrophage gene expression through ligand activation of PPARγ. Cell 93, 229–240 (1998). [DOI] [PubMed] [Google Scholar]
- 55.Feng J et al. Induction of CD36 expression by oxidized LDL and IL-4 by a common signaling pathway dependent on protein kinase C and PPAR-gamma. J Lipid Res 41, 688–696 (2000). [PubMed] [Google Scholar]
- 56.Fagone P & Jackowski S Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res 50 Suppl, S311–316 (2009). 10.1194/jlr.R800049-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fagone P & Jackowski S Membrane phospholipid synthesis and endoplasmic reticulum function. J Lipid Res 50 Suppl, S311–316 (2009). 10.1194/jlr.R800049-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacquemyn J, Cascalho A & Goodchild RE The ins and outs of endoplasmic reticulum-controlled lipid biosynthesis. EMBO Rep 18, 1905–1921 (2017). 10.15252/embr.201643426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Feng B et al. The endoplasmic reticulum is the site of cholesterol-induced cytotoxicity in macrophages. Nat Cell Biol 5, 781–792 (2003). 10.1038/ncb1035 [DOI] [PubMed] [Google Scholar]
- 60.Devries-Seimon T et al. Cholesterol-induced macrophage apoptosis requires ER stress pathways and engagement of the type A scavenger receptor. J Cell Biol 171, 61–73 (2005). 10.1083/jcb.200502078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Aderem A & Underhill DM Mechanisms of phagocytosis in macrophages. Annu Rev Immunol 17, 593–623 (1999). 10.1146/annurev.immunol.17.1.593 [DOI] [PubMed] [Google Scholar]
- 62.Im SS et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab 13, 540–549 (2011). 10.1016/j.cmet.2011.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Semenkovich CF Regulation of fatty acid synthase (FAS). Prog Lipid Res 36, 43–53 (1997). 10.1016/s0163-7827(97)00003-9 [DOI] [PubMed] [Google Scholar]
- 64.Shindou H & Shimizu T Acyl-CoA:lysophospholipid acyltransferases. J Biol Chem 284, 1–5 (2009). 10.1074/jbc.R800046200 [DOI] [PubMed] [Google Scholar]
- 65.Chang TY, Li BL, Chang CCY & Urano Y Acyl-coenzyme A: cholesterol acyltransferases. American Journal of Physiology-Endocrinology and Metabolism 297, E1–E9 (2009). 10.1152/ajpendo.90926.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stüve P et al. De Novo Fatty Acid Synthesis During Mycobacterial Infection Is a Prerequisite for the Function of Highly Proliferative T Cells, But Not for Dendritic Cells or Macrophages. Front Immunol 9, 495 (2018). 10.3389/fimmu.2018.00495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lally JSV et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab 29, 174–182.e175 (2019). 10.1016/j.cmet.2018.08.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Caron A, Richard D & Laplante M The Roles of mTOR Complexes in Lipid Metabolism. Annu Rev Nutr 35, 321–348 (2015). 10.1146/annurev-nutr-071714-034355 [DOI] [PubMed] [Google Scholar]
- 69.Martinet W, De Loof H & De Meyer GRY mTOR inhibition: a promising strategy for stabilization of atherosclerotic plaques. Atherosclerosis 233, 601–607 (2014). 10.1016/j.atherosclerosis.2014.01.040 [DOI] [PubMed] [Google Scholar]
- 70.Shimano H & Sato R SREBP-regulated lipid metabolism: convergent physiology — divergent pathophysiology. Nature Reviews Endocrinology 13, 710–730 (2017). 10.1038/nrendo.2017.91 [DOI] [PubMed] [Google Scholar]
- 71.Gurcel L, Abrami L, Girardin S, Tschopp J & van der Goot FG Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell 126, 1135–1145 (2006). 10.1016/j.cell.2006.07.033 [DOI] [PubMed] [Google Scholar]
- 72.Castoreno AB et al. Transcriptional regulation of phagocytosis-induced membrane biogenesis by sterol regulatory element binding proteins. Proc Natl Acad Sci U S A 102, 13129–13134 (2005). 10.1073/pnas.0506716102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Covarrubias AJ, Aksoylar HI & Horng T Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol 27, 286–296 (2015). 10.1016/j.smim.2015.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Suganami T et al. Role of the Toll-like Receptor 4/NF-κB Pathway in Saturated Fatty Acid–Induced Inflammatory Changes in the Interaction Between Adipocytes and Macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 27, 84–91 (2007). https://doi.org:doi: 10.1161/01.ATV.0000251608.09329.9a [DOI] [PubMed] [Google Scholar]
- 75.Horton JD, Goldstein JL & Brown MS SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 109, 1125–1131 (2002). 10.1172/jci15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Osborne TF & Espenshade PJ Evolutionary conservation and adaptation in the mechanism that regulates SREBP action: what a long, strange tRIP it’s been. Genes Dev 23, 2578–2591 (2009). 10.1101/gad.1854309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bidault G et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab 3, 1150–1162 (2021). 10.1038/s42255-021-00440-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Laplante M & Sabatini DM Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci 126, 1713–1719 (2013). 10.1242/jcs.125773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kurdi A, De Meyer GR & Martinet W Potential therapeutic effects of mTOR inhibition in atherosclerosis. Br J Clin Pharmacol 82, 1267–1279 (2016). 10.1111/bcp.12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding C et al. Tumor Microenvironment Modulates Immunological Outcomes of Myeloid Cells with mTORC1 Disruption. J Immunol 202, 1623–1634 (2019). 10.4049/jimmunol.1801112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Xiao X et al. Dihydroartemisinin inhibits Lewis Lung carcinoma progression by inducing macrophages M1 polarization via AKT/mTOR pathway. Int Immunopharmacol 103, 108427 (2022). 10.1016/j.intimp.2021.108427 [DOI] [PubMed] [Google Scholar]
- 82.Ridsdale R, Na CL, Xu Y, Greis KD & Weaver T Comparative proteomic analysis of lung lamellar bodies and lysosome-related organelles. PLoS One 6, e16482 (2011). 10.1371/journal.pone.0016482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yayoi Y et al. Specific localization of lysosomal aminopeptidases in type II alveolar epithelial cells of the rat lung. Arch Histol Cytol 64, 89–97 (2001). 10.1679/aohc.64.89 [DOI] [PubMed] [Google Scholar]
- 84.Huang SC et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat Immunol 15, 846–855 (2014). 10.1038/ni.2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chistiakov DA, Bobryshev YV & Orekhov AN Macrophage-mediated cholesterol handling in atherosclerosis. J Cell Mol Med 20, 17–28 (2016). 10.1111/jcmm.12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tabas I Free cholesterol-induced cytotoxicity a possible contributing factor to macrophage foam cell necrosis in advanced atherosclerotic lesions. Trends Cardiovasc Med 7, 256–263 (1997). 10.1016/s1050-1738(97)00086-8 [DOI] [PubMed] [Google Scholar]
- 87.Frambach SJ et al. Brothers in arms: ABCA1-and ABCG1-mediated cholesterol efflux as promising targets in cardiovascular disease treatment. Pharmacological reviews 72, 152–190 (2020). [DOI] [PubMed] [Google Scholar]
- 88.Jiang T et al. Leonurine prevents atherosclerosis via promoting the expression of ABCA1 and ABCG1 in a Pparγ/Lxrα signaling pathway-dependent manner. Cellular Physiology and Biochemistry 43, 1703–1717 (2017). [DOI] [PubMed] [Google Scholar]
- 89.Zhao G-J, Yin K, Fu Y. c. & Tang C-K The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Molecular medicine 18, 149–158 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hu H-J et al. PLK1 promotes cholesterol efflux and alleviates atherosclerosis by up-regulating ABCA1 and ABCG1 expression via the AMPK/PPARγ/LXRα pathway. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids 1867, 159221 (2022). [DOI] [PubMed] [Google Scholar]
- 91.Janowski BA et al. Structural requirements of ligands for the oxysterol liver X receptors LXRalpha and LXRbeta. Proc Natl Acad Sci U S A 96, 266–271 (1999). 10.1073/pnas.96.1.266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leussink S, Aranda-Pardos I & A-Gonzalez N Lipid metabolism as a mechanism of immunomodulation in macrophages: the role of liver X receptors. Curr Opin Pharmacol 53, 18–26 (2020). 10.1016/j.coph.2020.02.003 [DOI] [PubMed] [Google Scholar]
- 93.Zelcer N, Hong C, Boyadjian R & Tontonoz P LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science 325, 100–104 (2009). 10.1126/science.1168974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Tall AR, Costet P & Wang N Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest 110, 899–904 (2002). 10.1172/jci16391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Francis GA, Knopp RH & Oram JF Defective removal of cellular cholesterol and phospholipids by apolipoprotein A-I in Tangier Disease. J Clin Invest 96, 78–87 (1995). 10.1172/jci118082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhao L et al. miR-33–5p knockdown attenuates abdominal aortic aneurysm progression via promoting target adenosine triphosphate-binding cassette transporter A1 expression and activating the PI3K/Akt signaling pathway. Perfusion 35, 57–65 (2020). [DOI] [PubMed] [Google Scholar]
- 97.Wei Y & Schober A MicroRNA regulation of macrophages in human pathologies. Cell Mol Life Sci 73, 3473–3495 (2016). 10.1007/s00018-016-2254-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Najafi-Shoushtari SH et al. MicroRNA-33 and the SREBP host genes cooperate to control cholesterol homeostasis. Science 328, 1566–1569 (2010). 10.1126/science.1189123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rayner KJ et al. MiR-33 contributes to the regulation of cholesterol homeostasis. Science 328, 1570–1573 (2010). 10.1126/science.1189862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chander A & Fisher AB Regulation of lung surfactant secretion. American Journal of Physiology-Lung Cellular and Molecular Physiology 258, L241–L253 (1990). [DOI] [PubMed] [Google Scholar]
- 101.Agudelo CW, Samaha G & Garcia-Arcos I Alveolar lipids in pulmonary disease. A review. Lipids Health Dis 19, 122 (2020). 10.1186/s12944-020-01278-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.O’Neill LA, Kishton RJ & Rathmell J A guide to immunometabolism for immunologists. Nat Rev Immunol 16, 553–565 (2016). 10.1038/nri.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wakil SJ & Abu-Elheiga LA Fatty acid metabolism: target for metabolic syndrome. J Lipid Res 50 Suppl, S138–143 (2009). 10.1194/jlr.R800079-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Palmieri EM et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat Commun 11, 698 (2020). 10.1038/s41467-020-14433-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Malandrino MI et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am J Physiol Endocrinol Metab 308, E756–769 (2015). 10.1152/ajpendo.00362.2014 [DOI] [PubMed] [Google Scholar]
- 106.Vats D et al. Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab 4, 13–24 (2006). 10.1016/j.cmet.2006.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nomura M et al. Fatty acid oxidation in macrophage polarization. Nature Immunology 17, 216–217 (2016). 10.1038/ni.3366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Thiam AR, Farese RV Jr & Walther TC The biophysics and cell biology of lipid droplets. Nature reviews Molecular cell biology 14, 775–786 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chang CC, Huh HY, Cadigan KM & Chang TY Molecular cloning and functional expression of human acyl-coenzyme A:cholesterol acyltransferase cDNA in mutant Chinese hamster ovary cells. J Biol Chem 268, 20747–20755 (1993). [PubMed] [Google Scholar]
- 110.Chang T-Y, Li B-L, Chang CCY & Urano Y Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab 297, E1–E9 (2009). 10.1152/ajpendo.90926.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chang TY et al. Roles of acyl-coenzyme A:cholesterol acyltransferase-1 and −2. Curr Opin Lipidol 12, 289–296 (2001). 10.1097/00041433-200106000-00008 [DOI] [PubMed] [Google Scholar]
- 112.Sakashita N et al. Localization of human acyl-coenzyme A : cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. American Journal of Pathology 156, 227–236 (2000). 10.1016/s0002-9440(10)64723-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Miyazaki A et al. Expression of ACAT-1 protein in human atherosclerotic lesions and cultured human monocytes-macrophages. Arterioscler Thromb Vasc Biol 18, 1568–1574 (1998). 10.1161/01.atv.18.10.1568 [DOI] [PubMed] [Google Scholar]
- 114.Hongo S et al. Leptin modulates ACAT1 expression and cholesterol efflux from human macrophages. Am J Physiol Endocrinol Metab 297, E474–482 (2009). 10.1152/ajpendo.90369.2008 [DOI] [PubMed] [Google Scholar]
- 115.Xin C et al. Study of the insulin signaling pathways in the regulation of ACAT1 expression in cultured macrophages. Cell Biol Int 33, 602–606 (2009). 10.1016/j.cellbi.2009.02.011 [DOI] [PubMed] [Google Scholar]
- 116.Ge J et al. Insulin induces human acyl-coenzyme A: cholesterol acyltransferase1 gene expression via MAP kinases and CCAAT/enhancer-binding protein α. J Cell Biochem 114, 2188–2198 (2013). 10.1002/jcb.24568 [DOI] [PubMed] [Google Scholar]
- 117.Igarashi M et al. The critical role of neutral cholesterol ester hydrolase 1 in cholesterol removal from human macrophages. Circ Res 107, 1387–1395 (2010). 10.1161/circresaha.110.226613 [DOI] [PubMed] [Google Scholar]
- 118.Kassan A et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. Journal of Cell Biology 203, 985–1001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Salah RB et al. Biochemical and molecular characterization of a lipase produced by Rhizopus oryzae. FEMS Microbiol Lett 260, 241–248 (2006). 10.1111/j.1574-6968.2006.00323.x [DOI] [PubMed] [Google Scholar]
- 120.Soto-Avellaneda A & Morrison BE Signaling and other functions of lipids in autophagy: a review. Lipids in Health and Disease 19, 214 (2020). 10.1186/s12944-020-01389-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Olzmann JA & Carvalho P Dynamics and functions of lipid droplets. Nature reviews Molecular cell biology 20, 137–155 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Jarc E & Petan T A twist of FATe: Lipid droplets and inflammatory lipid mediators. Biochimie 169, 69–87 (2020). 10.1016/j.biochi.2019.11.016 [DOI] [PubMed] [Google Scholar]
- 123.Arrese EL, Saudale FZ & Soulages JL Lipid droplets as signaling platforms linking metabolic and cellular functions. Lipid insights 7, LPI. S11128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zechner R et al. FAT SIGNALS--lipases and lipolysis in lipid metabolism and signaling. Cell Metab 15, 279–291 (2012). 10.1016/j.cmet.2011.12.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Papackova Z & Cahova M Fatty acid signaling: the new function of intracellular lipases. Int J Mol Sci 16, 3831–3855 (2015). 10.3390/ijms16023831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Schmitz G & Ecker J The opposing effects of n-3 and n-6 fatty acids. Prog Lipid Res 47, 147–155 (2008). 10.1016/j.plipres.2007.12.004 [DOI] [PubMed] [Google Scholar]
- 127.Coleman RA & Mashek DG Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem Rev 111, 6359–6386 (2011). 10.1021/cr100404w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Herber DL et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med 16, 880–886 (2010). 10.1038/nm.2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kaushik S & Cuervo AM Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol 17, 759–770 (2015). 10.1038/ncb3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Radons J The human HSP70 family of chaperones: where do we stand? Cell Stress Chaperones 21, 379–404 (2016). 10.1007/s12192-016-0676-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ramakrishnan R et al. Oxidized lipids block antigen cross-presentation by dendritic cells in cancer. J Immunol 192, 2920–2931 (2014). 10.4049/jimmunol.1302801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Verdeguer F & Aouadi M Macrophage heterogeneity and energy metabolism. Exp Cell Res 360, 35–40 (2017). 10.1016/j.yexcr.2017.03.043 [DOI] [PubMed] [Google Scholar]
- 133.Wright JR Clearance and recycling of pulmonary surfactant. Am J Physiol 259, L1–12 (1990). 10.1152/ajplung.1990.259.2.L1 [DOI] [PubMed] [Google Scholar]
- 134.Tall AR & Yvan-Charvet L Cholesterol, inflammation and innate immunity. Nat Rev Immunol 15, 104–116 (2015). 10.1038/nri3793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Febbraio M, Hajjar DP & Silverstein RL CD36: a class B scavenger receptor involved in angiogenesis, atherosclerosis, inflammation, and lipid metabolism. J Clin Invest 108, 785–791 (2001). 10.1172/jci14006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen W-J, Yin K, Zhao G-J, Fu Y-C & Tang C-K The magic and mystery of microRNA-27 in atherosclerosis. Atherosclerosis 222, 314–323 (2012). [DOI] [PubMed] [Google Scholar]