

ABSTRACT
The iron-oxidizing Gallionellaceae drive a wide variety of biogeochemical cycles through their metabolisms and biominerals. To better understand the environmental impacts of Gallionellaceae, we need to improve our knowledge of their diversity and metabolisms, especially any novel iron oxidation mechanisms. Here, we used a pangenomic analysis of 103 genomes to resolve Gallionellaceae phylogeny and explore their genomic potential. Using a concatenated ribosomal protein tree and key gene patterns, we determined Gallionellaceae has four genera, divided into two groups: iron-oxidizing bacteria (FeOB) Gallionella, Sideroxydans, and Ferriphaselus with iron oxidation genes (cyc2, mtoA) and nitrite-oxidizing bacteria (NOB) Candidatus Nitrotoga with the nitrite oxidase gene nxr. The FeOB and NOB have similar electron transport chains, including genes for reverse electron transport and carbon fixation. Auxiliary energy metabolisms, including S oxidation, denitrification, and organotrophy, were scattered throughout the FeOB. Within FeOB, we found genes that may represent adaptations for iron oxidation, including a variety of extracellular electron uptake mechanisms. FeOB genomes encoded more predicted c-type cytochromes than NOB genomes, notably more multiheme c-type cytochromes (MHCs) with >10 CXXCH motifs. These include homologs of several predicted outer membrane porin-MHC complexes, including MtoAB and Uet. MHCs efficiently conduct electrons across longer distances and function across a wide range of redox potentials that overlap with mineral redox potentials, which can expand the range of usable iron substrates. Overall, the results of pangenome analyses suggest that the Gallionellaceae genera Gallionella, Sideroxydans, and Ferriphaselus have acquired a range of adaptations to succeed in various environments but are primarily iron oxidizers.
IMPORTANCE
Neutrophilic iron-oxidizing bacteria (FeOB) produce copious iron (oxyhydr)oxides that can profoundly influence biogeochemical cycles, notably the fate of carbon and many metals. To fully understand environmental microbial iron oxidation, we need a thorough accounting of iron oxidation mechanisms. In this study, we show the Gallionellaceae FeOB genomes encode both characterized iron oxidases as well as uncharacterized multiheme cytochromes (MHCs). MHCs are predicted to transfer electrons from extracellular substrates and likely confer metabolic capabilities that help Gallionellaceae occupy a range of different iron- and mineral-rich niches. Gallionellaceae appear to specialize in iron oxidation, so it would be advantageous for them to have multiple mechanisms to oxidize various forms of iron, given the many iron minerals on Earth, as well as the physiological and kinetic challenges faced by FeOB. The multiple iron/mineral oxidation mechanisms may help drive the widespread ecological success of Gallionellaceae.
KEYWORDS: iron oxidation, iron-oxidizing bacteria, pangenome, extracellular electron transfer, multiheme cytochrome
INTRODUCTION
Gallionella are one of the oldest known and most well-studied iron-oxidizing bacteria (FeOB), yet we are still learning how they oxidize iron and adapt to iron-rich niches. Gallionella is the type genus of the family Gallionellaceae, which also includes Sideroxydans, Ferriphaselus, and Ferrigenium. These Gallionellaceae FeOB are found in a wide range of environments, including freshwater creeks, sediment, root rhizospheres, peat, permafrost, deep subsurface aquifers, and municipal waterworks (1–18). FeOB potentially drive the fate of many metals and nutrients via both metabolic reactions and forming iron oxyhydroxides that adsorb and react with many solutes (19). To better understand the biogeochemical effects of Gallionellaceae, we need to improve our knowledge of their phylogeny and metabolic mechanisms, especially for iron oxidation. Recently, the rapid increase in metagenomes from iron-rich environments has significantly expanded the number of available Gallionellaceae genomes, which makes it possible to investigate diversity and mechanisms using genomic analyses of both cultured and uncultured Gallionellaceae.
The Gallionellaceae are named after Gallionella ferruginea, first described by Ehrenberg in 1838 (20), and recognizable by its distinctive, twisted iron oxyhydroxide stalk (21). While the type strain, G. ferruginea Johan (22), no longer exists, there are seven iron-oxidizing Gallionellaceae isolates and several stable enrichment cultures (7, 11, 23–26). Some isolates, such as Ferriphaselus spp., appear to be obligate iron oxidizers, while others also grow on non-iron substrates. In addition to iron, Sideroxydans lithotrophicus ES-1 grows by thiosulfate oxidation (24, 27), while Sideroxydans sp. CL21 shows mixotrophic growth with either lactate or hydrogen (28). Some Ferrigenium are members of the stable autotrophic, nitrate-reducing, iron-oxidizing enrichment cultures Straub, Bremen Pond, and Altingen (29–32). It is unknown how common it is for Gallionellaceae to use electron donors/acceptors besides Fe(II)/O2, though these alternate metabolisms may help their success across different environments and fluctuating conditions typical of many oxic-anoxic interfaces. Even so, since these seven Gallionellaceae isolates are all neutrophilic aerobic chemolithoautotrophic iron oxidizers, this could be the dominant metabolic niche of Gallionellaceae.
In Gallionellaceae and other neutrophilic chemolithotrophic FeOB, there are two potential iron oxidases: Cyc2, a fused monoheme cytochrome-porin and MtoAB, a porin-decaheme cytochrome complex (33–35). Porin-cytochrome complexes conduct electrons across the outer membrane, allowing cells to oxidize Fe(II) outside the cell to avoid internal Fe(III) mineralization (36, 37). The mtoA (metal oxidation) gene was first identified and characterized in FeOB S. lithotrophicus ES-1 (33). It is a homolog of the well-studied pioA (phototrophic iron oxidation) iron oxidase gene for which the function was verified through genetic knockout in the photoferrotroph Rhodopseudomonas palustris TIE-1 (38). The mtoA gene is also a homolog of mtrA (metal reduction), which encodes the MtrA iron reductase in iron-reducing bacteria (FeRB) Shewanella (39). Though known for electron export, MtrA can also conduct electrons into the cell (40). The cyc2 gene is more common than mtoAB and is found in nearly all well-characterized neutrophilic FeOB like the Gallionellaceae (41–43) and Zetaproteobacteria (43), making it a suitable genetic marker for many FeOB. Cyc2 has been demonstrated to oxidize aqueous Fe2+ (34). In S. lithotrophicus ES-1 cultures grown on aqueous Fe2+, cyc2/Cyc2 is highly expressed, whereas mtoA expression is low, and the Mto proteins are not detected, suggesting Cyc2 plays a larger role in aqueous iron oxidation compared to MtoA (27, 44). In contrast, Mto gene/protein expression is upregulated in ES-1 cultures grown on Fe(II) smectite clay, suggesting MtoAB plays a role in the oxidation of solid iron minerals (44). However, Cyc2 and MtoA may not be the only mechanisms for neutrophilic iron oxidation. There are a number of additional uncharacterized cytochromes and electron transport genes (27, 42) within Gallionellaceae genomes such as isolate S. lithotrophicus ES-1 (27, 42), suggesting the existence of novel iron oxidation genes and mechanisms within the family.
The Gallionellaceae also includes a recently identified genus, Candidatus Nitrotoga, which are chemolithotrophic nitrite-oxidizing bacteria (NOB). Like the iron-oxidizing Gallionellaceae, they are widespread in freshwater and engineered environments, including permafrost (45), coastal sediments (46), freshwater (47), freshwater sediments (48), and the activated sludge of wastewater treatment facilities (49, 50). There are only two isolates, Ca. Nitrotoga fabula (49) and Ca. Nitrotoga sp. AM1P (51), along with four genomes from enrichment cultures (48). Ca. Nitrotoga are adapted to niches with low nitrite and oxidize it using a distinct high-affinity Nxr nitrite oxidoreductase (45, 48, 52). Extensive iron uptake mechanisms have been detected in Ca. Nitrotoga genomes, indicating the importance of iron for growth, likely due to the FeS cluster of Nxr (48). However, neither the isolates nor enrichments are known to oxidize Fe(II). If Ca. Nitrotoga lack the capacity to oxidize iron, then we can investigate the iron-oxidizing mechanisms and adaptations of Gallionellaceae through a comparative genomic analysis of iron- vs nitrite-oxidizing members.
Toward this goal, we took advantage of the growing number of environmental metagenomes and collected 103 Gallionellaceae genomes and metagenome assembled genomes (MAGs) with >80% completeness and <7% contamination. We used those sequences to resolve the Gallionellaceae phylogeny and delineate groups of iron and nitrite oxidizers. We searched for known and novel iron oxidation genes, other energy and nutrient metabolisms, and genes found exclusively in FeOB that may represent adaptations for an iron-oxidizing lifestyle. This work increases our understanding of Gallionellaceae family phylogeny and the metabolic traits of its genera. It also highlights some of the key multiheme cytochromes (MHCs) in Gallionellaceae FeOB, which may facilitate extracellular electron uptake and the oxidation of different iron substrates.
RESULTS
Phylogeny
We collected 103 Gallionellaceae isolate genomes and MAGs with >80% completeness and <7% contamination from various databases and collections (Table S1). Many of these MAGs were only classified at the family level, so genus-level designations were initially unclear. To resolve the phylogeny, verify existing classifications, and classify the unknown Gallionellaceae, we examined 16S rRNA gene identity and constructed a concatenated protein tree (Fig. 1) from 13 ribosomal protein sequences. We also present genome average nucleotide identity (ANI) and amino acid identity (AAI) to further evaluate relatedness.
Fig 1.
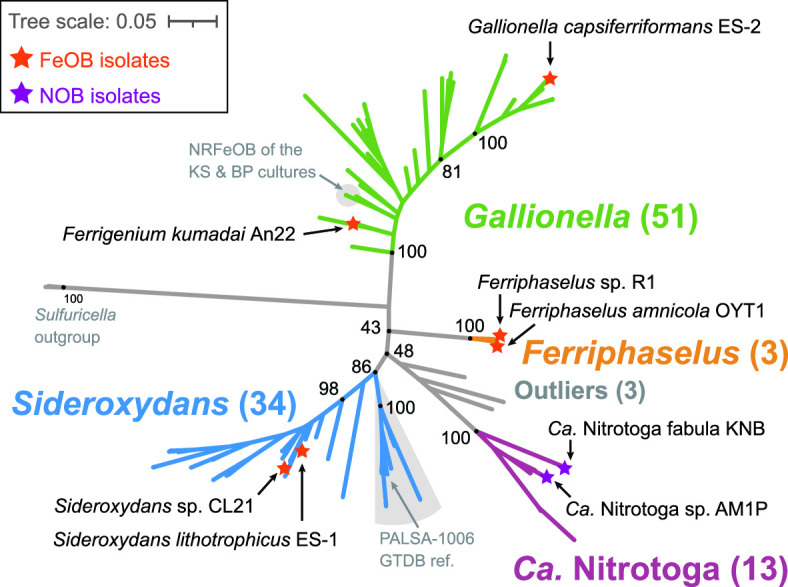
Concatenated ribosomal protein maximum likelihood tree of the Gallionellaceae family showing the four distinct genera: Gallionella, Sideroxydans, Ferriphaselus, and Ca. Nitrotoga. Isolates are labeled and annotated with stars. Numbers in parentheses indicate the number of genomes in each genus or group. Support values from 1,000 bootstraps are shown for major branching nodes (black dots). Detailed tree is shown in Fig. 2.
For genomes that have 16S rRNA sequences, we found the 16S percent identity between organisms ranges from 91.5% to 99.9% (Table S2a). Comparing the 16S percent identities of Gallionellaceae isolates (in bold in Table S2a) at a 94.5% or 95% identity threshold (53, 54), this suggests the existence of three genera (Ferriphaselus, Gallionella-Ferrigenium, and Ca. Nitrotoga-Sideroxydans). For whole genome comparisons, ANI and AAI range from 69.4% to 100% and 65.8% to 99.9%, respectively (Tables S2b and S2c), suggesting the whole family is closely related. These ANI and AAI values do not neatly cluster the genomes into distinct groups, though the values more clearly separate Ca. Nitrotoga and Sideroxydans than 16S (Table S2). However, there are no well-supported ANI or AAI thresholds or discontinuities that can delineate genera (55, 56).
In contrast, a concatenated protein tree (Fig. 1) using 13 ribosomal protein sequences shows distinct, well-supported clades that correspond to the four genera Gallionella, Sideroxydans, Ferriphaselus, and Ca. Nitrotoga (Fig. 1). Most of the MAGs previously classified as Gallionellaceae and Gallionellales were found to be either Gallionella or Sideroxydans, with the exception of one that clustered with the Ca. Nitrotoga (Ca. Nitrotoga SL_21). Although some genomes formed subclades, many were organized along a continuum. Near the base of the Gallionella are Ferrigenium kumadai An22 (25) and the nitrate-reducing FeOB (NRFeOB) of the Straub (KS) (30, 57) and Bremen Pond (BP) (32) enrichments (Fig. 1). There is not a clear boundary between the Gallionella and the relatively new Ferrigenium genus. In addition, the 16S percent identity between F. kumadai An22 and Gallionella species ranges from 94.7% to 95.5%, and the ANI and AAI values also indicate close relationships (Table S2). Therefore, we included the Ferrigenium and NRFeOB with the Gallionella grouping for our analyses. We also constructed a 16S rRNA gene tree containing 22 sequences in our data set along with 941 high-quality, full-length Gallionellaceae sequences from the SILVA database (Fig. S1), but bootstrap support was weaker, and clades were less clearly resolved. Therefore, concatenated ribosomal proteins are a more reliable determinant of Gallionellaceae phylogeny than 16S rRNA genes.
We assessed whether there was a relationship between phylogeny and environment. Each genome and MAG was classified with the Genomes OnLine Database (GOLD) classification schema (58) based on pre-existing GOLD classifications, available metadata, and publications (Fig. 2; Table S3). The majority of aquatic genomes were from freshwater and groundwater environments, while terrestrial genomes were mostly found in soil, peat, and rhizosphere environments. However, some genomes were sequenced from more extreme environments such as thermal hot springs (ENVO:00002181) and acid mine drainage (ENVO:00001997; Table S3). Gallionellaceae are widespread and can inhabit many different environments, but there was no clear pattern between GOLD Ecosystem Type and broad phylogenetic groupings (Fig. 2). Different Gallionellaceae appear to co-exist in some environments, suggesting niches not captured in the ecosystem classification are controlling Gallionellaceae diversity and environmental distribution.
Fig 2.

Maximum likelihood tree of concatenated ribosomal proteins from the Gallionellaceae, annotated with source ecosystem and genes for iron oxidation (cyc2, mtoA), nitrite oxidase (nxrAB), terminal oxidase (ccoN), denitrification (narGH/napAB/nirK/nirS), stalk formation (sfz), and organic utilization (gtsABS, lutABCP). The bar graph to the right shows the number of genes encoding multiheme cytochromes, categorized by number of CXXCH, CX3CH, and CX4CH heme-binding motifs. Phylogeny does not correlate to environments, and key genes, including those for multiheme cytochromes, show distinct distributions between iron and nitrite oxidizer clades. Isolates are shown in bold. % completeness = genome completeness calculated with CheckM. Outgroup omitted for space.
Metabolic potential and diversity
The Gallionellaceae family has few isolates, so to uncover the shared metabolic traits of its FeOB members, we compared and contrasted Gallionella, Sideroxydans, and Ferriphaselus genomes to those of the nitrite-oxidizing Ca. Nitrotoga. We identified key genes within the pangenome for iron oxidation (including predicted c-type cytochromes), carbon fixation, and respiration using a combination of Distilled and Refined Annotation of Metabolism (DRAM) (59), FeGenie (60), MagicLamp (61), a heme motif counter script (62), and BLAST (63, 64). To further uncover genes and pathways specifically enriched in the iron oxidizers, we used Anvi’o (65–67) to analyze a filtered data set of only Gallionella, Sideroxydans, and Ca. Nitrotoga genomes that were >97% complete. This approach enabled us to create a comprehensive picture of Gallionellaceae metabolic diversity and pinpoint promising gene clusters that may be adaptations for an iron-oxidizing lifestyle.
Primary energy metabolisms—iron and nitrite oxidation
Known metabolisms for the few Gallionellaceae isolates suggest Ca. Nitrotoga are nitrite oxidizers, while Sideroxydans, Ferriphaselus, and Gallionella are iron oxidizers. We examined the pangenome for the presence of cyc2 and mtoA iron oxidation genes and nxrAB nitrite oxidase genes to determine if that pattern also holds throughout the uncultured Gallionellaceae. As with the isolates, there is a clear delineation between organisms with marker genes for iron vs nitrite oxidation, which corresponds to the phylogenetic groups (Fig. 2 and 3).
Fig 3.

Plot showing the percent of genomes in each genus/group with genes for key metabolic pathways. The plot indicates the Gallionellaceae are aerobic lithoautotrophs with two main energy metabolisms, iron or nitrite oxidation. Some members also have metabolic potential for S oxidation and/or denitrification. Numbers in parentheses indicate the total number of genomes in each group. Color is used to distinguish groups, while dot size and opacity indicate % presence in the genome groups. Ox. = oxidation, red. = reduction, fix. = fixation, and resp. = respiration.
The cyc2 gene is widespread among clades of iron oxidizers, with at least one copy detected in 83% of the FeOB genomes (Fig. 3; Table S4). The mtoA gene is found in 41% of the FeOB genomes, and 37% of genomes have both mtoA and cyc2. In total, 89% have at least one iron oxidation gene, either cyc2 or mtoA (Table S4). Since the data set includes multiple MAGs with a mean completeness score of 95%, it appears that almost all Gallionellaceae FeOB contain one of these two mechanisms for iron oxidation. Overall, cyc2 homologs are more common than mtoA (Fig. 2 and 3), and some genomes encode multiple copies of cyc2 (Table S4). All of the FeOB Gallionellaceae with cyc2 encode at least one copy of a Cluster 1 Cyc2 [classified as in reference (35)]. Cluster 1 Cyc2 function has been verified through multiple lines of evidence [e.g., biochemistry (34), transcriptomics (27, 35), and proteomics (44)]. The Cyc2 sequences of the Gallionellaceae FeOB are closely related to each other and to a biochemically verified Cluster 1 Cyc2 iron oxidase (34). Therefore, we are confident in inferring that the Cluster 1 Cyc2 of Gallionellaceae is an iron oxidase.
The Ca. Nitrotoga SL_21 MAG contains only a predicted Cluster 2 Cyc2 homolog. Confidently assigning iron oxidation function to Cluster 2 Cyc2s depends on supporting context, which is lacking in this case. Ca. Nitrotoga SL_21 is not from a typical iron-oxidizing environment (permanently stratified, non-marine, saline lake), and although it is within Cluster 2, it is distant from the functionally verified Cyc2 representative from Acidithiobacillus. Currently, there is no clear evidence that this sole Ca. Nitrotoga Cyc2 is an iron oxidase.
In contrast, nxrAB genes are exclusive to the Ca. Nitrotoga. Copies of nxrAB are the most common genes for energy conservation among the Ca. Nitrotoga, present in 85% of the genomes (Fig. 2 and 3). Given that many of the genomes are MAGs with a mean completeness of 94%, the distribution of nxrAB appears to indicate that nitrite oxidation is the main energy metabolism of Ca. Nitrotoga. Thus, our pangenome analysis confirms Gallionellaceae can be divided into two main groups based on primary energy metabolism—FeOB and NOB.
c-type cytochromes
Both Cyc2 and MtoA are c-type cytochromes that transport electrons across the outer membrane. FeOB uses additional c-type cytochromes to transport electrons through the periplasm to the rest of the electron transport chain. We reasoned that novel iron oxidation mechanisms may also utilize c-type cytochromes, so we searched the Gallionellaceae genomes for proteins containing the CXXCH, CX3CH, and CX4CH heme-binding motifs (abbreviated hereafter as CXXCH). There is a stark difference between FeOB and NOB in the distribution of predicted c-type cytochromes. FeOB genomes have an average of 1.5× more CXXCH-containing proteins than NOB, and only the FeOB genomes encode proteins with 10 or more CXXCH motifs (Fig. 2). The abundance of genes for potential c-type cytochromes, in particular MHCs, suggests the presence of additional iron oxidation mechanisms in the Gallionellaceae FeOB.
To find c-type cytochromes of interest, all CXXCH-containing proteins were clustered using MMSeqs2 with bidirectional coverage and an 80% alignment cutoff. Clusters of sequences were then classified with representative sequences from isolates using BLAST to query the Uniprot database. If the cluster did not contain a sequence from an isolate, a consensus classification was used. A cluster of monoheme proteins (Cluster 313) was classified as Cyc2, and three clusters of decaheme proteins were classified as MtoA (Clusters 335 and 451) and MtrC (Cluster 50; Fig. 2 and 4; Table 1). These Cyc2, MtoA, and MtrC clusters largely agree with FeGenie’s HMM-based predicted distributions. Since MMSeqs2 generated two clusters of MtoA sequences, we sought to further verify the classifications. We constructed a tree of all Gallionellaceae MtoA sequences along with reference sequences of MtrA from FeRB (Fig. 5) (68). Although there is some separation of Cluster 335 and Cluster 451 MtoA sequences, many clades are not well defined or supported. In fact, backbone support throughout the tree is poor, and the tree does not indicate a clear separation of the MtoA and MtrA sequences (Fig. 5). There is some evidence that the direction of electron flow through Mto/Mtr can be reversible (33, 69, 70). So, it may be that the functions of MtoA and MtrA are interchangeable, and in fact, they may be indistinguishable proteins that can conduct electrons across the outer membrane in either direction.
Fig 4.

Maximum likelihood tree of concatenated ribosomal proteins from the Gallionellaceae that shows the distribution of MMSeqs2 clusters that represent predicted cytochromes Cyc2, MtoA, MtoC, PCC3, Uet, and Slit_1324. Asterisk (*) for Gallionella_BEO_15 indicates a partial MtoA sequence was detected using HMMs and verified with BLAST but was too short to bin into the MMseqs2 MtoA clusters. Isolates are shown in bold. % completeness = genome completeness calculated with CheckM. Outgroup omitted for space.
TABLE 1.
Clusters of predicted c-type cytochromes and other heme-containing proteins of interest from MMSeqs2a
| Cluster | Functional prediction | # CXXCH, CX3CH, or CX4CH motifs per protein | # FeOB (of 87) |
|---|---|---|---|
| Iron oxidation/reduction proteins | |||
| 313 | Iron oxidase Cyc2‡ | 1 | 70 |
| 451 | Decaheme c-type cytochrome, DmsE family, MtoA‡ | 10 | 19 |
| 335 | Decaheme c-type cytochrome, DmsE family, MtoA‡ | 10 | 17 |
| 50 | Decaheme c-type cytochrome, OmcA/MtrC family† | 10 | 7 |
| Potential extracellular electron transport pathway proteins | |||
| 20 | Cytochrome C family protein; potential periplasmic PCC3 subunit‡ | 21, 24, and 27 | 42 |
| 241 | Cytochrome C family protein; potential extracellular PCC3 subunit‡ | 10, 11, 12, 13, 14, 15, 16, and 18 | 34 |
| 242 | Cytochrome C family protein; potential extracellular PCC3 subunit‡ | 26, 28, 29, 33, and 35 | 7 |
| 331 | Cytochrome C family protein; potential extracellular PCC3 subunit‡ | 15 and 17 | 5 |
| 65 | Doubled CXXCH motif-containing protein; Cytochrome c3 family protein†, potential UetJ subunit | 11 and 12 | 6 |
| 479 | Tetraheme cytochrome—potential UetA subunit | 4 | 6 |
| 330 | Cytochrome C7 domain-containing protein; Triheme cytochrome—potential UetDEG subunit | 3 | 5 |
| 94 | Cytochrome C7 domain-containing protein; Triheme cytochrome—potential UetDEG subunit | 3 | 5 |
| 446 | Diheme cytochrome c‡—potential Slit_1324 | 2 | 51 |
| Sensory proteins | |||
| 152 | Methyl-accepting chemotaxis sensory transducer; YoaH ‡ | 1 | 54 |
| 40 | Methyl-accepting chemotaxis sensory transducer with Pas/Pac sensor; Aerotaxis receptor ‡ | 1 | 43 |
| 400 | Diguanylate cyclase with PAS/PAC sensor; Cyclic di-GMP phosphodiesterase Gmr ‡ | 1 | 36 |
| Other | |||
| 433 | 2Fe-2S ferredoxin‡ | 1 and 2 | 72 |
| 360 | 4Fe-4S ferredoxin iron-sulfur binding domain protein | 1 | 41 |
| 403 | Forkhead-associated protein‡ | 10 | 27 |
| 146 | Cytochrome c; Octaheme tetrathionate reductase† | 8 | 25 |
| 253 | Sulfite reductase, dissimilatory-type, subunit DsrJ† | 3 | 17 |
Functional predictions are based on ‡ isolate annotations and NCBI BLAST or † BLAST of sequences from metagenomes in Uniprot.
Fig 5.

Maximum likelihood tree of the predicted MtoA sequences identified in MMSeqs2 Cluster 335 and Cluster 451 along with MtoA reference sequences from National Center for Biotechnology Information and MtrA reference sequences from Baker et al. 2022. Numbers (1a, 1b, 2, 3, 4, 5, 6, and 7) appended after Mtr denote reference sequences from the seven MtrA groups defined by Baker et al., 2022. MtrA-4 indicates the Group 4 Betaproteobacteria. Tree is rooted using MtrC. Support is the result of 500 bootstrap replicates.
The decaheme cytochrome MtrC is the extracellular partner of the iron-reducing MtrAB complex of Shewanella (71). The MtrAB complex is a homolog of the MtoAB complex of FeOB and can function in reverse to take up electrons (40). MtrC was thought to be exclusive to the Mtr complex of iron-reducers since MtrC homologs were not detected in FeOB isolates with MtoAB. However, we found seven MAGs within both Gallionella and Sideroxydans that encode MtrC (Table S4), leading us to question whether its function is restricted to iron reduction. Another possibility is that MtrC can also work in reverse and be part of an iron oxidation pathway. Differentiating these possibilities would require physiological testing of an FeOB isolate with MtrC, which does not currently exist.
The Gallionellaceae FeOB have other porin-MHC complexes that could potentially catalyze iron oxidation, such as the PCC3 complex, identified through bioinformatic analyses of genomes of several FeOB including S. lithotrophicus ES-1. This predicted complex includes a periplasmic MHC, an extracellular MHC, an outer membrane porin, and a conserved inner membrane protein (72). We identified 26 Gallionellaceae FeOB genomes with a complete predicted PCC3 complex, an additional 11 genomes with a partial complex, and four instances where the PCC3 complex encodes two predicted periplasmic cytochromes instead of one (Fig. 4; Table S4). The predicted periplasmic MHCs grouped in MMSeqs2 Cluster 20, while predicted extracellular MHCs grouped in Clusters 241, 242, and 331. The extracellular MHCs exhibited variability in the number of CXXCH heme motifs (10–35; Table 1), which suggests a range of functions for the extracellular PCC3 MHCs. Based on in silico protein structure models, PCC3 MHCs appear long and mostly linear (Fig. 6; Fig. S2), suggesting an extended conduction range both intra- and extracellularly.
Fig 6.

Models of potential Gallionellaceae extracellular electron transfer mechanisms. All sizes are approximated. Dimensions of Cyc2 with its fused cytochrome-porin and the porin-cytochrome complexes MtoAB, MtoAB+MtrC, MtoD, and Uet drawn from models and measurements in previous literature (34, 72–75). The illustration of PCC3 is based on AlphaFold2 predictions (Fig. S2). The number of hemes and size of PCC3 can vary. The 21/18 heme complex of S. lithotrophicus ES-1 is depicted along with the estimated length of the 10 and 35 heme variants of the extracellular cytochrome.
Another recently described porin-MHC complex is the undecaheme electron transfer (Uet) complex, found in the cathode-oxidizing Tenderiales (73) (Fig. 6). We used a combination of MMSeqs2 and BLAST to identify Uet genes in the Gallionellaceae. While PCC3 is more common to Sideroxydans (59%) than Gallionella (12%), the Uet pathway appears exclusive to Gallionella and two unclassified outliers (Fig. 4). Six Gallionella have predicted undecaheme cytochrome (UetJ), extracellular tetraheme cytochrome (UetA), three predicted periplasmic triheme cytochromes (UetDEG), peptidylprolyl isomerase (UetB), and NHL repeat units (UetHI; Fig. 4; Table S4). We checked for genes encoding the β-barrel porin UetC and found BLAST hits in four of the six genomes (Table S4).
S. lithotrophicus ES-1 has a set of periplasmic cytochrome genes without a predicted porin that was highly upregulated during growth on iron and, therefore, thought to be involved in iron oxidation (27). The genes encode a cytochrome b (Slit_1321), a hypothetical extracellular protein (Slit_1322), a monoheme cytochrome class I (Slit_1323), a periplasmic diheme cytochrome (Slit_1324; Cluster 446 in Table 1), and a molecular chaperone Hsp33 (Slit_1325). We found homologs of the Slit_1321–1324 genes are common, co-located, and well-conserved among Gallionellaceae FeOB, present in 50 genomes (Fig. 4; Table S4). These genes may represent a mechanism of periplasmic electron transport, perhaps as part of an iron oxidation/extracellular electron uptake pathway.
Electron transport chains
We compared electron transport chain component genes of the iron and nitrite oxidizer groups and found them to be largely similar (Fig. 7). High-affinity cbb3-type oxidases are common (Fig. 3), with most genomes containing either the proximal or distal form of ccoN (Fig. 2) (76). Even the four NRFeOB genomes contain ccoNO genes, indicating a potential for both oxygen and nitrate respiration. In contrast, few Gallionellaceae genomes contain narGH or napAB (6 and 10 genomes, respectively, with no overlap). Those that contain narGH include the known NRFeOB of the two separately maintained Straub cultures (Ferrigenium straubiae KS 1 and KS 2) (29, 57), three Gallionella MAGs, and one outlier (Table S4). This indicates the genetic potential for nitrate respiration is relatively rare overall (Fig. 3; Table S4).
Fig 7.

Diagram showing the similarities and differences between the electron transport chains of (A) iron- vs (B) nitrite-oxidizing Gallionellaceae. Pink numbers indicate the percent of FeOB (A) or NOB (B) genomes that encoded each part of the electron transport chain or RuBisCo. Less common components such as FeOB denitrification genes are not shown.
In addition to the cbb3-type oxidase genes, 34.5% of iron oxidizers and 15.4% of nitrite oxidizers possess genes for cytochrome bd-type oxidases (cydAB) (Fig. 7). The presence of bd-type oxidase genes often overlaps with cbb3-type oxidase genes (Table S4). Like cbb3-type oxidases, cytochrome bd-type oxidases have a high affinity for oxygen, and recent studies show they can be more highly expressed than cbb3-type oxidases under low-oxygen, organic-rich conditions (77). Both FeOB and NOB have genes for cytochrome bc1 and Alternative complex III (ACIII) quinol oxidase complexes, which route electrons to the quinone pool where they may be used to form NADH for biosynthetic reactions. Genes for bc1 are more common in FeOB (85.1%) compared to NOB (7.7%), while ACIII is more common in NOB (100%) than FeOB (55.2%; Fig. 7; Table S4). Like the bd- and cbb3-type oxidases, the presence of bc1 and ACIII often overlaps in a single organism, especially in FeOB (Table S4). Possessing both bd- and cbb3-type oxidases and/or having both bc1 and ACIII contributes to flexibility within the electron transport chains of Gallionellaceae. The presence of various terminal oxidases implies adaptation to niches where oxygen and organic carbon availability differ or fluctuate.
Carbon fixation
Gallionellaceae isolates grow autotrophically. To determine if the capacity for autotrophic growth is widespread, we analyzed the pangenome for RuBisCo genes (cbbLS, cbbMQ). Most genomes in the data set (>91%; 94 of 103 genomes) contain genes for either Form I or Form II RuBisCo (Fig. 3; Table S4). FeOB more commonly have Form II, while NOB only have Form I. Form II enzymes are adapted for medium to high CO2 and low O2 concentrations (78), and their predominance in FeOB may correspond to different oxygen niches of FeOB and NOB. The prevalence of RuBisCo genes indicates both iron- and nitrite-oxidizing Gallionellaceae have the capacity to grow autotrophically.
Denitrification
Denitrification has been demonstrated in mixed cultures dominated by Gallionellaceae whose genomes encode various parts of the denitrification pathway (30–32). To better understand the denitrification potential across the Gallionellaceae and its role in FeOB metabolism, we analyzed patterns of denitrification genes (Fig. 2 and 3; Table S4). If autotrophic FeOB are to conserve energy from denitrification, they would in principle require the Nar nitrate reductase, as it is the only denitrification complex proven to generate proton motive force (79). Across our data set, genomes encoding NarGH are uncommon, found only in a cluster of five Gallionella (including the two Ferrigenium KS culture MAGs) plus the outlier Ca. Houarnoksidobacter (Table S4). These six genomes with narGH have at least one dissimilatory nitrite reductase (nirK or nirS), and four of the genomes encoded the eNOR nitric oxide reductase, giving the genetic capability to reduce nitrate to either NO or N2O, respectively. The eNOR nitric oxide reductase has a proposed proton channel, which may also contribute to energy conservation (80). Altogether, the results show a few genomes encode the ability to couple nitrate reduction to iron oxidation for energy, but this is rare amongst Gallionellaceae.
Gallionellaceae may denitrify for reasons other than energy generation, including N assimilation, redox balance, and removal of intermediates nitrite and nitric oxide, which are toxic and may abiotically oxidize Fe(II) (chemodenitrification). We found the denitrification genes napAB, nasA, nirBD, and norBC throughout the Gallionellaceae (Fig. 3; Table S4). The assimilatory nitrate reductase gene, nasA, was found exclusively in FeOB (Fig. 3; Table S3) co-located with nitrite reductase genes nirBD. A similar conserved gene cluster of nasA and nirBD has been observed in iron-oxidizing Zetaproteobacteria (81), allowing for the reduction of nitrate to ammonia for assimilation. Genomes with dissimilatory nitrate reductase genes napAB clustered in two small clades of Gallionella and Sideroxydans (Fig. 2; Table S4). NapAB may be used in aerobic denitrification when oxygen is limiting (82). The nitrite reductase genes nirK/nirS were more common than napAB and spread throughout the Gallionellaceae with little overlap (Fig. 2; Table S4). Both nitrite and NO present major challenges to FeOB metabolism because of their reactivity with iron: they bind to hemes, inhibiting the activity of cytochromes, and also directly oxidize Fe(II), thus competing with enzymatic iron oxidation (83). Therefore, it makes sense that many genomes have both nir genes and nitric oxide reductase (nor) genes (most commonly cNOR). Nitrous oxide reductase (nosZ) was not detected in any genomes, indicating Gallionellaceae are not able to fully denitrify to N2 gas; since N2O is non-toxic and relatively inert, nosZ is unnecessary.
Denitrification is a complex pathway that requires many enzymes and, therefore, a substantial investment of cellular resources. Given the high potential of Fe(II)/Fe(III) couples and the low-energy yield of iron oxidation, it may be difficult for autotrophic FeOB to compete with organoheterotrophic denitrifiers, which may explain the rarity of nar amongst strictly autotrophic FeOB. The ability to assimilate nitrate and detoxify intermediates is more common, suggesting the importance of these functions to FeOB.
Hydrogenases
Gallionellaceae genomes encode a variety of [NiFe]-hydrogenases (Fig. 3; Table S4). The most common type is the reversible, oxygen-tolerant hydrogenase, HoxFUHY, with genes present in 71% of FeOB and 30% of NOB. Other [NiFe]-hydrogenase genes were detected exclusively in FeOB, including genes for the oxygen-tolerant uptake hydrogenase, HyaAB, and the hydrogen-sensing hydrogenase, HupUV. [NiFe]-hydrogenases are capable of different physiological roles (84–87); thus, they may benefit in iron-oxidizing Gallionellaceae in multiple ways. Hydrogenases can enable the use of H2 as an electron donor, as in Sideroxydans sp. CL21 (28). Alternatively, the Hox hydrogenase can directly couple H2 oxidation to the reduction of NAD to NADH (84, 88) and generate reducing power for N2 fixation, CO2 fixation, and/or biosynthetic reactions. Hox can also function in reverse to transfer electrons from NADH to produce H2 as a mechanism of redox balance (89, 90), which can help FeOB cope with dynamic fluxes of Fe(II) and oxygen in redox transition zones.
Auxiliary energy metabolisms
Previous studies showed some Gallionellaceae FeOB possess alternate energy metabolisms such as thiosulfate and lactate oxidation (27, 28). We searched the pangenome for key genes of sulfur, manganese, and organic substrate oxidation pathways to determine how common alternate metabolisms are among Gallionellaceae FeOB. Sulfide:quinone reductase (sqr) is common to both FeOB and NOB (Fig. 3; Table S4). Sqr can oxidize sulfide, transporting electrons to the quinone pool, although it may be a means of detoxification rather than energy conservation (91, 92). In contrast, both soxABXYZ and dsrAB are detected exclusively in the iron-oxidizing Gallionellaceae genomes (Fig. 3; Table S4). To predict the oxidative vs reductive function of dsrAB, we constructed a tree using reference sequences from Loy et al. (93, 94). Gallionellaceae sequences form a discrete clade within the sulfur-oxidizing group (Fig. S3), indicating the DsrAB of Gallionellaceae is likely a reverse dissimilatory sulfite reductase. In contrast, the Ca. Nitrotoga genomes do not contain dsr or sox genes. Instead, Ca. Nitrotoga have sorAB, which may enable oxidation of sulfite to sulfate (Fig. 3). Gallionellaceae are not typically abundant in sulfur-rich environments. These results indicate sulfur oxidation is an auxiliary metabolism in Gallionellaceae with only certain FeOB capable of oxidizing S(0) or thiosulfate.
We analyzed the pangenome for signs of organic carbon utilization. Although not widely distributed, the most common genes were for lactate utilization (lutABCP) and sugar transport (msmX, gtsABC). Only eight Gallionella and five Sideroxydans genomes, including Sideroxydans sp. CL21, have lutABC along with the lutP lactate permease gene (Fig. 2 and 3; Table S4). Likewise, only six genomes contain gtsABC genes for glucose/mannose uptake. None of the NOB contain the lut or gts genes for organic carbon utilization.
We used BLAST to evaluate the Gallionellaceae genomes for manganese oxidase genes mcoA, moxA, mofA, and mnxG. There are a few hits for mcoA, moxA, and mofA genes but none for mnxG (Table S4). Since manganese oxidation activity has not been shown in any of the Gallionellaceae isolates, additional verification is needed to determine whether the genes identified by BLAST are truly Mn oxidases.
Other genes distinct to FeOB, potentially related to iron oxidation
We searched the pangenome for the candidate genes for stalk formation (sfz/sfb) identified in the stalk-forming Ferriphaselus and Zetaproteobacteria isolates (95, 96). The four sfz/sfb genes were found in 12 genomes, restricted to one crown-group cluster of nine Gallionella and all three Ferriphaselus (Fig. 2; Table S4). Thus far, all cultured Gallionellaceae stalk formers belong to these two genera, suggesting stalk formation may be limited and not a trait of Sideroxydans.
Using the Anvi’o subset of only genomes >97% complete, we identified several gene clusters that were present and abundant only in Gallionella and Sideroxydans but lacked a prior connection to an iron-oxidizing lifestyle. These included distinct gene clusters with Clusters of Orthologous Gene (COG) functional annotations for: Cell Wall/Membrane/Envelope Biogenesis, Cytoskeleton formation, Signal Transduction Mechanisms, and Energy Production and Conversion (see selected clusters in Table 2, and additional ones at https://doi.org/10.6084/m9.figshare.22781342). Clusters for Cell Wall/Membrane/Envelope Biogenesis may indicate FeOB have specific adaptations for housing extracellular electron transport mechanisms in the outer membrane or avoiding encrustation by iron oxides. Clusters for Energy Production and Conversion included ferredoxin (Fdx) and subunits of the RnfABCDEG complex. The Rnf complex was originally discovered for its role in N fixation, in which it oxidizes NADH and generates reduced ferredoxin that donates electrons to nitrogenase (97). More recent studies have shown Rnf complexes can conserve energy under anaerobic conditions (98–100), and as a low potential electron donor, ferredoxin can transfer electrons to many metabolic pathways including some that produce secondary metabolites (101). Not all Gallionellaceae with Rnf complex genes have nifDHK nitrogenase genes, implying Gallionellaceae Rnf and ferredoxin have functions beyond N fixation. Although their specific function in Gallionellaceae FeOB are unknown, their ubiquity implies utility for FeOB and an area for additional research.
TABLE 2.
Gene clusters of interest from the Anvi’o pangenome subset that were present in iron-oxidizing Gallionella and Sideroxydans but absent in nitrite-oxidizing Ca. Nitrotoga (Ferriphaselus not considered)
| COG category | COG function | Gene cluster ID |
|---|---|---|
| Cell wall/membrane/envelope biogenesis | Lipid carrier protein ElyC involved in cell wall biogenesis, DUF218 family (ElyC) | GC_00001120 |
| ABC-type lipoprotein export system, ATPase component (LolD) | GC_00000969 | |
| ADP-heptose synthase, bifunctional sugar kinase/adenylyltransferase (RfaE) | GC_00001059, GC_00001084 | |
| ADP-heptose:LPS heptosyltransferase (RfaF) | GC_00001100 | |
| Glycosyltransferase involved in cell wall biosynthesis (RfaB) | GC_00001179 | |
| Outer membrane protein TolC | GC_00000022, GC_00000920 | |
| Glutamate racemase (MurI) | GC_00001047 | |
| Murein L,D-transpeptidase YafK | GC_00001108 | |
| Cytoskeleton | Cytoskeletal protein CcmA, bactofilin family | GC_00000987 |
| Energy production and conversion | Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfA | GC_00000042 |
| Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfB | GC_00001082 | |
| Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfC | GC_00001069 | |
| Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfD | GC_00001055 | |
| Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfE | GC_00001071 | |
| Na+ translocating ferredoxin: NAD+ oxidoreductase RNF, RnfG | GC_00001096 | |
| Ferredoxin (Fdx) | GC_00001052 | |
| Cytochrome c-type biogenesis protein CcmH/NrfF | GC_00001058 | |
| Cytochrome c-type biogenesis protein CcmH/NrfG | GC_00001078 | |
| Signal transduction mechanisms | PAS domain | GAF domain | HAMP domain | Cyclic di-GMP metabolism protein | GC_00000006 |
| cAMP-binding domain of CRP or a regulatory subunit of cAMP-dependent protein kinases | Small-conductance mechanosensitive channel MscK | GC_00001152 |
DISCUSSION
The Gallionellaceae family is historically known for its iron-oxidizing members, but recently, a new candidate genus of nitrite oxidizers, Ca. Nitrotoga, was identified (45). Comparing their genomes to those of FeOB genera has helped identify genes and pathways related to iron oxidation since Ca. Nitrotoga isolates have no documented capacity for that metabolism (45, 46, 48, 49, 51). We resolved the phylogeny of the Gallionellaceae and verified Ca. Nitrotoga lacked iron oxidation marker genes. Given separate groups of FeOB and NOB, we used a pangenomic approach to identify shared features of the Gallionellaceae, as well as FeOB-specific genes that may represent novel iron oxidation pathways.
Phylogeny
Organizing and naming taxa is an essential step toward determining how microbial diversity is connected to function and niches, but it is a challenge to classify microbial taxa. The National Center for Biotechnology Information (NCBI) Taxonomy database and Genome Taxonomy Database (GTDB) place the Gallionellaceae into different higher-level taxa (Betaproteobacteria class and Nitrosomonadales order in NCBI, Gammaproteobacteria class and Burkholderiales order in GTDB). Each database also divides the family differently, with GTDB creating more genus-level classifications based on representative genomes for clades without cultured members. Here, we take a parsimonious approach to minimize the number of genera while maintaining nomenclature that has a long history of use in the literature. In this way, we can discuss groups with potentially distinct features and take advantage of previous findings without undue confusion. Because additional organisms are continuously discovered, we expect that this taxonomy will continue to evolve.
The Gallionellaceae can be described by four genera, Gallionella, Sideroxydans, Ferriphaselus, and Ca. Nitrotoga, based on the current concatenated ribosomal protein tree. Compared to this tree, 16S rRNA phylogeny did a poorer job of resolving these genera, so 16S-based identification should be considered tentative, pending the availability of genomes. ANI and AAI scores were not definitive, as there are no agreed-upon cut-offs to guide genus delineation (55, 56). Therefore, to facilitate consistent classification using the concatenated ribosomal protein phylogeny, the protein sequences and alignments used here (Fig. 1) are available at https://doi.org/10.6084/m9.figshare.21898938, https://doi.org/10.6084/m9.figshare.21898929.
The resolved phylogeny provides a framework for understanding the diversity and major metabolisms of the Gallionellaceae. They are members of Nitrosomonadales (or Burkholderiales), which contain many chemolithotrophic S and N oxidizers. Like their closest relatives, the Sulfuricellaceae (102), many Gallionellaceae retain the ability to oxidize sulfur (Fig. 3; Fig. S3). The Gallionellaceae tree (Fig. 1) shows a deeply branching split between genera, with each of the two major genera, Gallionella and Sideroxydans, containing a continuum of diversity. Within the Gallionella, the isolates G. capsiferriformans ES-2 and Ferrigenium kumadai An22 bracket the Gallionella, with An22 deeply branching and ES-2 at the crown. F. kumadai An22 was originally classified as Ferrigenium based on 16S rRNA distance (25). However, our analyses do not show any clear phylogenetic clustering or functional distinction, with which we could draw a line between Gallionella and Ferrigenium. Moreover, the tree topology suggests continued diversification within both Gallionella and Sideroxydans largely without the formation of subclades that represent distinct niches. There is one subclade of Sideroxydans that corresponds to the GTDB genus level designation PALSA-1006 (Fig. 1). However, 16S/ANI/AAI results (Table S2) indicate there is not enough diversity within the Gallionellaceae to justify further splitting the four major genera any further. Additionally, we did not detect any obvious functional difference in PALSA-1006. Given our phylogenetic analysis, 16S/ANI/AAI, and similar functional profiles, we recommend keeping them within Sideroxydans. Based on the above classification scheme, most of the genomes (84 of 103) fall into either Gallionella or Sideroxydans.
Phylogenetic diversity corresponds to functional diversity that can drive Gallionellaceae success in a variety of environments. Many Gallionella and Sideroxydans do not appear to be obligate iron oxidizers, and some may not be obligate aerobes. Auxiliary metabolisms for S, N, and C are present to varying degrees throughout the iron-oxidizing genera and are not associated with specific subgroups. Some FeOB from organic-rich environments, such as Sideroxydans sp. CL21, have genes for organoheterotrophy. Other FeOB show metabolic flexibility in additional lithotrophic metabolisms, such as oxidation of S or potentially Mn, elements that often co-occur with Fe in the environment. Some Gallionellaceae may also thrive in oxygen-poor environments by reducing nitrate, although this capability appears rare. Such traits contribute to diversity in the Gallionellaceae FeOB genera, which appear to acquire and/or retain additional energy and nutrient metabolisms to adapt to a range of environments.
Ca. Nitrotoga stands out as an exception within the Gallionellaceae. The pangenome analysis shows that Ca. Nitrotoga has distinctive genomic content (Fig. S4). They do not appear to have the capacity for iron oxidation based on available physiological evidence and the genomic analyses presented here. The similarities in Gallionellaceae FeOB and Ca. Nitrotoga electron transport chains enable them to meet the shared challenge of conserving energy from high-potential electron donors. However, Ca. Nitrotoga are a distinct clade that appears to have evolved from the FeOB to occupy a nitrite oxidation niche.
Iron oxidation and extracellular electron uptake (EEU) mechanisms
The Gallionellaceae FeOB genomes encode a wide variety of predicted c-type cytochromes. Of these cytochromes, many appear to be associated with the outer membrane, implying a role in extracellular electron transport. Cyc2 is present in the majority of Gallionellaceae FeOB genomes, while MHCs Mto/Mtr, Uet, and PCC3 are less common, each with different distribution patterns (Fig. 4), suggesting the different cytochromes play distinct roles.
Cyc2 has been shown to oxidize dissolved Fe(II) (27, 34, 44, 103). The monoheme Cyc2 is a small fused cytochrome-porin, and since aqueous Fe2+ is common to many redox transition zones, it makes sense that most FeOB would retain and use the simplest tool. But in Earth’s various environments, iron is largely available as minerals (clays, oxides, and sulfides) and also bound to organics (e.g., humic substances). The decaheme MtoA has been shown to play roles in the oxidation of mineral-bound Fe(II), specifically Fe(II) smectite clay (44). As an MHC, MtoA may have multiple benefits that help in oxidizing minerals. MtoA has a large redox potential window [−350 to +30 mV (33, 37)], which could help with the oxidation of solids, like smectite (44), that also have a range of redox potentials [e.g., −600 to +0 mV for SWa-1 vs −400 to +400 mV for SWy-2 (104)], which change as mineral-bound iron is oxidized or reduced. Assuming the MtoA structure is similar to MtrA, the 10 hemes span the membrane, making a wire that conducts from extracellular substrates to periplasmic proteins (74, 105). The multiple hemes allow for the transfer of multiple electrons at a time (36). Some MAGs with mtoAB also encode the extracellular decaheme cytochrome MtrC. In Shewanella, the MtrCAB complex requires MtrC to reduce solid minerals (ferrihydrite), while MtrAB alone can only reduce dissolved Fe(III) and electrodes (71, 106, 107). Likewise, Gallionellaceae MtrC may help increase interactions with different minerals. Some Gallionellaceae FeOB may retain genes for both Cyc2 and MtoAB (with or without MtrC) to oxidize different Fe(II) substrates in their environments.
Like MtrCAB, the predicted PCC3 complex includes both periplasmic and extracellular MHCs and a porin. A key difference is that the PCC3 cytochromes often have more hemes than MtoA/MtrA and MtrC. The greater number of hemes may serve to store electrons, as in a capacitor. They may also conduct across a greater distance; the PCC3 periplasmic MHC, with 21–27 hemes, is potentially long enough to span the entire periplasm [as noted by Edwards et al. (108)]. Intraprotein electron transfer between hemes is rapid (109–111); therefore, the periplasm-spanning MHC of PCC3 may allow for faster electron transfer compared to complexes containing smaller periplasmic cytochromes like the monoheme MtoD. The extracellular PCC3 MHC contains between 10 and 35 hemes, which could extend further from the outer membrane compared to MtrC. Not only would this extend the range of electron transfer but may also be faster than a “wire” of smaller cytochromes [e.g., Geobacter hexaheme OmcS (112)]. Increasing oxidation rates via larger MHCs would allow FeOB to oxidize substrates faster. Given that Fe(II) is subject to abiotic oxidation under certain conditions and other organisms may compete for EEU, such kinetic advantages would give FeOB a competitive edge.
Conclusions
Gallionellaceae, specifically Gallionella, are best known for lithoautotrophically oxidizing iron to make mineral stalks that come together to form microbial mats at groundwater seeps (18, 113, 114). Although this may contribute to an impression that the niche is relatively restricted, 16S rRNA sequencing of cultures and environmental samples has revealed both the diversity of Gallionellaceae as well as its prevalence across practically any freshwater and some brackish environments where Fe(II) and O2 meet. The pangenome shows that Gallionellaceae possess metabolic flexibility to use non-iron substrates, notably sulfur, and the MHCs likely also confer further metabolic capabilities that may help them occupy a range of different iron- and mineral-rich niches. Gallionellaceae thrive in aquifers, soil, and wetlands, all of which have substantial mineral content. Thus, the widespread ecological success of Gallionellaceae may well correspond to genomes that encode a range of iron oxidation mechanisms as well as adaptations for varied environments.
It is becoming clear that there are multiple ways to oxidize iron, though we have varying levels of evidence for gene/protein function (37, 115, 116). Validating iron oxidation genes/proteins is painstaking work due to challenging cultures, low yield, few genetic systems, and the fact that iron interferes with many molecular extractions and assays. And yet, there are likely even more iron oxidation mechanisms, so we need to be strategic about choosing genes/proteins for deeper characterization. Our pangenome analysis gives a wider view of the distribution and frequency of potentially novel iron oxidation genes, which will help us to prioritize investigations. Furthermore, the varied outer membrane-associated cytochromes inspire us to investigate relationships between structure and function. Why are there so many different multiheme cytochromes? Is there substrate specificity, kinetic advantages, battery-like functions, or some utility we have yet to consider? Addressing these questions will help us understand how these proteins and pathways shape microbial transformations of varied Earth materials.
MATERIALS AND METHODS
Data collection and curation
Gallionellaceae genomes were collected from the NCBI Entrez database (117), the Joint Genome Institute Integrated Microbial Genomes (IMG) database (118), and the European Nucleotide Archive at EMBL-EBI database [Sideroxydans sp. CL21, Ca. Nitrotoga fabula KNB, and the “IN” MAGs (17, 49, 119)] (Table S5). We also received non-public genomes from the Ménez Lab at the Université de Paris [three genomes reconstructed by Aurélien Lecoeuvre from the Carbfix study in Hengill, Iceland (2); metagenomes available at Sequence Read Archive SRR3731039, SRR3731040, SRR4188484, and SRR4188643], and the Banfield Lab at the University of California, Berkeley [three genomes reconstructed by Alex Probst from Crystal Geyser in Utah, USA (120); (Table S5)]. This initial 230-genome data set included isolate genomes, MAGs, and single-cell amplified genomes that were taxonomically classified as members of the Gallionellales order; Gallionellaceae family; or the Gallionella, Sideroxydans, Ferriphaselus, Ferrigenium, or Ca. Nitrotoga genera in their respective databases. Duplicate genomes were identified and removed if they had identical accession numbers, or their ANI were 100%. CheckM v1.1.2 (121) was used to assess genome quality. Genomes with lower than 80% completeness and greater than 7% contamination were removed from the data set. The final filtered data set, referred to as “the Gallionellaceae” or “the data set,” contained 103 genomes (Table S1; 2, 7, 11, 17, 23–26, 32, 42, 47–49, 51, 57, 95, 119, 120, 122–145), including six of the Gallionellaceae FeOB isolates. The seventh isolate, Sideroxyarcus emersonii (26), was not published at the time of our main analysis, but a supplemental of its key metabolic genes and MHCs (https://doi.org/10.6084/m9.figshare.22781912) shows it has similar patterns to Sideroxydans.
Naming conventions
To assign simple, unique names to the metagenomes, codes were appended to genus-level names based on sample location and bin IDs (Tables S1, S5 and S7). Isolates retained their own unique names. Organisms that were taxonomically classified in their original databases at the family Gallionellaceae or order Gallionellales were, if possible, classified at lower taxonomic levels using a combination of AAI, 16S rRNA (if available), classification through the GTDB Toolkit (146), and placement in the concatenated ribosomal protein tree (Fig. 1 and 2).
Ecosystem classifications
To assess whether metabolic diversity correlated to ecosystem type, each genome was assigned to an ecosystem based on the GOLD (147) schema which leverages Environmental Ontology classifications (148). A genome’s pre-existing classification from IMG was used if available. Genomes without prior classification were categorized based on published descriptions of their sample sites and “habitat” information listed in their database of origin. Based on the GOLD classifications (Table S3), genomes were examined for patterns of correspondence between ecosystems and phylogenetic and/or metabolic diversity.
16S rRNA analyses
Twenty-two of the 103 Gallionellaceae genomes contained 16S rRNA gene sequences. All sequences >1,450 bp (35 total) were aligned in Geneious v.10.2.6 (149) using MUSCLE (150). The percent identity of the 16S rRNA sequences is shown in Table S2a.
Calculation of average amino acid and nucleotide identities
AAI and ANI were computed to assess the similarity of genomes in the curated data set (Table S2). AAI was calculated using CompareM (151). ANI was calculated using ANIcalculator v.1.0 (152). Prior to using ANIcalculator, tRNA and rRNA sequences were removed to prevent overinflation of ANI estimates (55, 152). AAI and ANI results were spot checked using the Kostas Lab AAI/ANI Matrix Tool (http://enve-omics.ce.gatech.edu/g-matrix/index) (153) to assure patterns were consistent. Final AAI and ANI tables were formatted using Microsoft Excel.
Tree construction
Concatenated ribosomal protein tree
A concatenated tree of ribosomal proteins (Fig. 1) was constructed to determine the phylogenetic relationships of genomes in the Gallionellaceae data set. Two Sulfuricella genomes, Sulfuricella sp. T08 and Sulfuricella 3300027815, were included as an outgroup to root the tree. The use of a Sulfuricella outgroup was based on previous literature (154, 155), which identified Sulfuricella and other members of the Sulfuricellaceae family as near neighbors of Gallionellaceae. The concatenated sequences were composed of 13 small and large ribosomal proteins (L19, L20, L28, L17, L9_C, S16, L21p, L27, L35p, S11, S20p, S6, and S9) present in 94 or more of the 105 genomes including the outgroup. Protein sequences were aligned in Geneious v.10.2.6 (149) using MUSCLE (150). Ends of the alignments were manually trimmed, and regions with over 70% gaps were masked, after which sequences were concatenated. The tree was constructed using RAxML-NG v1.0.3 (156) with the maximum likelihood method, LG + G model, and 1,000 bootstraps. The final tree was visualized and annotated with iTOL (157). The ribosomal protein sequences used to construct the tree are available on FigShare (https://doi.org/10.6084/m9.figshare.21898938, https://doi.org/10.6084/m9.figshare.21898929).
16S rRNA gene tree
We constructed a 16S rRNA gene tree (Fig. S1) composed of sequences from our data set combined with a selection of sequences from the SILVA database to determine how well 16S rRNA resolves Gallionellaceae phylogeny compared to the concatenated ribosomal protein tree. Full-length (~1,500 bp) 16S rRNA genes were retrieved from 22 of the Gallionellaceae genomes using Anvio’s “anvi-get-sequences-for-hmm-hits” command for “Ribosomal_RNA_16S.” These genes were aligned in SINA (158) along with Gallionellaceae sequences from the Silva database (159) that had >1,475 bp and >85–90 sequence quality score. The outgroup is composed of Thiobacillus, Ferritrophicum, Sulfuricella, Sulfuriferula, and Nitrosomonas sequences acquired from the Silva database. The final alignment contained 965 non-redundant sequences, and the alignment length was 1,500 positions after trimming and masking all sequence gaps greater than 70%. A maximum likelihood tree was constructed using RAxML-NG v1.0.3 (156) with the GTR+G model and 300 bootstraps. Family- and genus-level classifications from the SILVA database were used to annotate the tree in Iroki (160).
Individual protein trees
Trees for DsrAB (Fig. S3) and Mto/Mtr (Fig. 5) were constructed from Gallionellaceae protein sequences along with reference sequences from NCBI, Loy et al. and Baker et al. (68, 93). Sequences were aligned with MUSCLE (150), ends were manually trimmed, and regions with over 70% sequence gaps were masked in Geneious v.10.2.6 (149). For the Dsr tree, DsrA and DsrB sequences were concatenated. Trees were constructed using RAxML-NG v1.0.3 (156) with the LG+G model. Branch support for Mto/Mtr tree is based on 500 bootstraps, and support for the DsrAB tree is based on 300 bootstraps. The final trees were visualized and annotated with Iroki (160).
Pangenome analysis
Metabolic gene analysis
We used DRAM v0.0.2 (59) within KBase (161), LithoGenie within MagicLamp (61), and FeGenie (60) to identify key metabolic genes indicative of various oxidation, respiration, and carbon utilization pathways. NCBI BLAST+ (64) was used to identify additional genes for eNOR, cNOR, SorAB, Mn oxidases, LutABCP, and stalk formation. We then analyzed the presence/absence of the metabolic genes and looked for patterns across the concatenated protein tree, between genera, and between FeOB and NOB.
MHC analysis
To identify potential c-type cytochromes, we used a modified heme counter script (62) to search for CXXCH, CXXXCH, and CXXXXCH motifs within the protein sequences of each genome. The search identified 5,929 protein sequences with one or more CX2-4CH-motifs. To determine which protein sequences were shared between genomes, sequences were clustered using MMSeqs2 (162) with coverage mode 0 for bidirectional coverage of at least 80% of the query and target sequences. Several clusters of interest were identified based on either the number of CX2-4CH-motifs in each sequence or the relative abundance of FeOB sequences in the cluster. Querying with BLASTp (63) against the Uniprot (163) database was used to classify sequences from clusters of interest, thereby identifying clusters of predicted c-type cytochromes. Isolate sequences were used as representative sequences for cluster classification. If a cluster did not contain an isolate sequence, a consensus classification was used. The subcellular localization of proteins was predicted using a combination of PSORTb v3.0.3 (164) and LocTree3 (165).
Some MHCs were predicted to be part of Mto, PCC3, or Uet porin-cytochrome complexes. Therefore, we wanted to determine if the genes for these MHCs were colocalized in their respective genomes with genes for β-barrel porins, periplasmic proteins, and inner membrane proteins previously identified in the literature (72, 73). We searched for the associated genes using BLASTp and amino acid reference sequences from S. lithotrophicus ES-1 (MtoB, MtoD, and CymA), Gallionella AHS-4737 (MtoC), and Ca. Tenderia electrophaga (UetBCDEFGHI). The locus tags of BLASTp hits were then compared to the locus tags of the MHCs to evaluate synteny and colocalization. The same method was used to determine if diheme c-type cytochromes from MMseqs2 cluster 446 which includes Slit_1324 were colocalized with a cytochrome b (Slit_1321), hypothetical extracellular protein (Slit_1322), monoheme cytochrome class I (Slit_1323), and molecular chaperone Hsp33 (Slit_1325).
PCC3 modeling
To model predicted PCC3 proteins, we used ColabFold: AlphaFold2 using MMseqs2 (166). Setting included using MSA mode “MMseqs2 (UniRef+environmental),” pair mode “unpaired+paired,” protein structure prediction with “AlphaFold2-ptm,” and complex prediction with “AlphaFold-multimer-v2” (167, 168). The best scoring model was rendered in PyMol v2.5.4 (169).
Anvi’o subset analysis
We used the Anvi’o v7 (65, 67) to build a pangenome database of all Gallionella (16) , Sideroxydans (15), and Ca. Nitrotoga (6) genomes that were over 97% complete (Fig. S4) to analyze for additional genes important to FeOB lifestyles. Ferriphaselus had too few representatives to define a meaningful core genome and was therefore omitted. Genes were clustered within the Anvi’o pangenome using a min-bit parameter of 0.5 and an mcl inflation parameter of 2. The Anvi’o pangenome was used to compare gene clusters across the data set and to bin: (i) near-core (found in >85% of genomes), (ii) accessory (found in >1 but <85% of genomes), and (iii) strain specific (found in a single genome) sets of gene clusters. Gene annotations were assigned in Anvi’o using Prodigal (170), and functional annotations for Anvi’o gene clusters were assigned using the NCBI’s Database of COGs (171, 172). Data tables of the binned Anvi’o gene clusters were analyzed to identify gene clusters found in the near-core genomes of Gallionella and Sideroxydans but absent in Ca. Nitrotoga.
ACKNOWLEDGMENTS
We thank Aurélien Lecoeuvre, Emmanuelle Gérard, Bénédicte Ménez, Alex Probst, and Jill Banfield for allowing us to use unpublished genomes from their private collections; and all those who granted us permission to use their publicly available, unpublished genomes from the NCBI and IMG databases (Tables S1 and S5).
This research was funded by the National Science Foundation (EAR-1833525 to C.S.C. and S.W.P., MCB-1817651 to C.S.C.) and the Office of Naval Research (N00014-17-1-2640 C.S.C.). R.L.H. was also supported by fellowships from University of Delaware Graduate College and the Microbiology Program/Unidel Foundation. Support from the University of Delaware Center for Bioinformatics and Computational Biology Core Facility (RRID:SCR_017696), including the use of the BIOMIX computer cluster, was made possible through funding from Delaware INBRE (NIH P20GM103446), the State of Delaware, and the Delaware Biotechnology Institute.
Contributor Information
Clara S. Chan, Email: cschan@udel.edu.
Ryan J. Newton, University of Wisconsin-Milwaukee, Milwaukee, Wisconsin, USA
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/msystems.00038-23.
Figures S1–S4.
Tables S1–S6.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Emerson D, Fleming EJ, McBeth JM. 2010. Iron-oxidizing bacteria: an environmental and genomic perspective. Annu Rev Microbiol 64:561–583. doi: 10.1146/annurev.micro.112408.134208 [DOI] [PubMed] [Google Scholar]
- 2. Trias R, Ménez B, le Campion P, Zivanovic Y, Lecourt L, Lecoeuvre A, Schmitt-Kopplin P, Uhl J, Gislason SR, Alfreðsson HA, Mesfin KG, Snæbjörnsdóttir SÓ, Aradóttir ES, Gunnarsson I, Matter JM, Stute M, Oelkers EH, Gérard E. 2017. High reactivity of deep biota under anthropogenic CO2 injection into basalt. Nat Commun 8:1063. doi: 10.1038/s41467-017-01288-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jewell TNM, Karaoz U, Brodie EL, Williams KH, Beller HR. 2016. Metatranscriptomic evidence of pervasive and diverse chemolithoautotrophy relevant to C, S, N and Fe cycling in a shallow alluvial aquifer. ISME J 10:2106–2117. doi: 10.1038/ismej.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Emerson D, Moyer CL. 2002. Neutrophilic Fe-oxidizing bacteria are abundant at the Loihi Seamount hydrothermal vents and play a major role in Fe oxide deposition. Appl Environ Microbiol 68:3085–3093. doi: 10.1128/AEM.68.6.3085-3093.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Emerson D, De Vet W. 2015. The role of FeOB in engineered water ecosystems: a review. Journal AWWA 107:E47–E57. doi: 10.5942/jawwa.2015.107.0004 [DOI] [Google Scholar]
- 6. Wang J, Sickinger M, Ciobota V, Herrmann M, Rasch H, Rösch P, Popp J, Küsel K. 2014. Revealing the microbial community structure of clogging materials in dewatering wells differing in physico-chemical parameters in an open-cast mining area. Water Res. 63:222–233. doi: 10.1016/j.watres.2014.06.021 [DOI] [PubMed] [Google Scholar]
- 7. Kato S, Krepski S, Chan C, Itoh T, Ohkuma M. 2014. Ferriphaselus amnicola gen. nov., sp. nov., a neutrophilic, stalk-forming, iron-oxidizing bacterium isolated from an iron-rich groundwater seep. Int J Syst Evol Microbiol 64:921–925. doi: 10.1099/ijs.0.058487-0 [DOI] [PubMed] [Google Scholar]
- 8. Fabisch M, Beulig F, Akob DM, Küsel K. 2013. Surprising abundance of Gallionella-related iron oxidizers in creek sediments at pH 4.4 or at high heavy metal concentrations. Front Microbiol 4:390. doi: 10.3389/fmicb.2013.00390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fabisch M, Freyer G, Johnson CA, Büchel G, Akob DM, Neu TR, Küsel K. 2016. Dominance of ‘Gallionella capsiferriformans’ and heavy metal association with Gallionella-like stalks in metal-rich pH 6 mine water discharge. Geobiology 14:68–90. doi: 10.1111/gbi.12162 [DOI] [PubMed] [Google Scholar]
- 10. Weiss JV, Rentz JA, Plaia T, Neubauer SC, Merrill-Floyd M, Lilburn T, Bradburne C, Megonigal JP, Emerson D. 2007. Characterization of neutrophilic Fe(II)-oxidizing bacteria isolated from the rhizosphere of wetland plants and description of Ferritrophicum radicicola gen. nov. sp. nov., and Sideroxydans paludicola sp. nov. Geomicrobiol J 24:559–570. doi: 10.1080/01490450701670152 [DOI] [Google Scholar]
- 11. Lüdecke C, Reiche M, Eusterhues K, Nietzsche S, Küsel K. 2010. Acid-tolerant microaerophilic Fe(II)-oxidizing bacteria promote Fe(III)-accumulation in a fen: acid-tolerant Fe(II)-oxidizers in a fen. Environ Microbiol 12:2814–2825. doi: 10.1111/j.1462-2920.2010.02251.x [DOI] [PubMed] [Google Scholar]
- 12. Buongiorno J, Herbert LC, Wehrmann LM, Michaud AB, Laufer K, Røy H, Jørgensen BB, Szynkiewicz A, Faiia A, Yeager KM, Schindler K, Lloyd KG. 2019. Complex microbial communities drive iron and sulfur cycling in Arctic Fjord sediments. Appl Environ Microbiol 85:e00949-19. doi: 10.1128/AEM.00949-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Patzner MS, Logan M, McKenna AM, Young RB, Zhou Z, Joss H, Mueller CW, Hoeschen C, Scholten T, Straub D, Kleindienst S, Borch T, Kappler A, Bryce C. 2022. Microbial iron cycling during palsa hillslope collapse promotes greenhouse gas emissions before complete permafrost thaw. Commun Earth Environ 3:1–14. doi: 10.1038/s43247-022-00407-8 [DOI] [Google Scholar]
- 14. Neubauer SC, Emerson D, Megonigal JP. 2002. Life at the energetic edge: kinetics of circumneutral iron oxidation by lithotrophic iron-oxidizing bacteria isolated from the wetland-plant rhizosphere. Appl Environ Microbiol 68:3988–3995. doi: 10.1128/AEM.68.8.3988-3995.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vet WWJM de, Dinkla IJT, Abbas BA, Rietveld LC, Loosdrecht MCM van. 2012. Gallionella spp. in trickling filtration of subsurface aerated and natural groundwater. Biotechnol Bioeng 109:904–912. doi: 10.1002/bit.24378 [DOI] [PubMed] [Google Scholar]
- 16. Eichinger S, Boch R, Leis A, Koraimann G, Grengg C, Domberger G, Nachtnebel M, Schwab C, Dietzel M. 2020. Scale deposits in tunnel drainage systems – a study on fabrics and formation mechanisms. Sci Total Environ 718:137140. doi: 10.1016/j.scitotenv.2020.137140 [DOI] [PubMed] [Google Scholar]
- 17. Bethencourt L, Bochet O, Farasin J, Aquilina L, Borgne TL, Quaiser A, Biget M, Michon-Coudouel S, Labasque T, Dufresne A. 2020. Genome reconstruction reveals distinct assemblages of Gallionellaceae in surface and subsurface redox transition zones. FEMS Microbiol Ecol 96:fiaa036. doi: 10.1093/femsec/fiaa036 [DOI] [PubMed] [Google Scholar]
- 18. Emerson D, Revsbech NP. 1994. Investigation of an iron-oxidizing microbial mat community located near Aarhus, Denmark: field studies. Appl Environ Microbiol 60:4032–4038. doi: 10.1128/aem.60.11.4032-4038.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kappler A, Bryce C. 2017. Cryptic biogeochemical cycles: unravelling hidden redox reactions. Environ Microbiol 19:842–846. doi: 10.1111/1462-2920.13687 [DOI] [PubMed] [Google Scholar]
- 20. Ehrenberg C. 1838. Die infusionsthierchen Als vollkommene organismen. Leopold Voss, Leipzig. doi: 10.5962/bhl.title.58475 [DOI] [Google Scholar]
- 21. Ghiorse WC. 1984. Biology of iron- and manganese-depositing bacteria. Annu Rev Microbiol 38:515–550. doi: 10.1146/annurev.mi.38.100184.002503 [DOI] [PubMed] [Google Scholar]
- 22. Hallbeck L, Pedersen K. 1991. Autotrophic and mixotrophic growth of Gallionella ferruginea. Microbiology 137:2657–2661. doi: 10.1099/00221287-137-11-2657 [DOI] [Google Scholar]
- 23. Krepski ST, Hanson TE, Chan CS. 2012. Isolation and characterization of a novel biomineral stalk-forming iron-oxidizing bacterium from a circumneutral groundwater seep: a novel stalk-forming Fe-oxidizing bacterium. Environ Microbiol 14:1671–1680. doi: 10.1111/j.1462-2920.2011.02652.x [DOI] [PubMed] [Google Scholar]
- 24. Emerson D, Moyer C. 1997. Isolation and characterization of novel iron-oxidizing bacteria that grow at circumneutral pH. Appl Environ Microbiol 63:4784–4792. doi: 10.1128/aem.63.12.4784-4792.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khalifa A, Nakasuji Y, Saka N, Honjo H, Asakawa S, Watanabe T. 2018. Ferrigenium kumadai gen. nov., sp. nov., a microaerophilic iron-oxidizing bacterium isolated from a paddy field soil. Int J Syst Evol Microbiol 68:2587–2592. doi: 10.1099/ijsem.0.002882 [DOI] [PubMed] [Google Scholar]
- 26. Kato S, Itoh T, Iino T, Ohkuma M. 2022. Sideroxyarcus emersonii gen. nov. sp. nov., a neutrophilic, microaerobic iron- and thiosulfate-oxidizing bacterium isolated from iron-rich wetland sediment. Int J Syst Evol Microbiol 72. doi: 10.1099/ijsem.0.005347 [DOI] [PubMed] [Google Scholar]
- 27. Zhou N, Keffer JL, Polson SW, Chan CS. 2022. Unraveling Fe(II)-oxidizing mechanisms in a facultative Fe(II) oxidizer, Sideroxydans lithotrophicus strain ES-1, via culturing. Appl Environ Microbiol 88:e0159521. doi: 10.1128/AEM.01595-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cooper RE, Finck J, Chan C, Küsel K. 2023. Mixotrophy broadens the ecological niche range of the iron oxidizer Sideroxydans sp. CL21 isolated from an iron-rich peatland. FEMS Microbiol Ecol 99:fiac156. doi: 10.1093/femsec/fiac156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang Y-M, Jakus N, Straub D, Konstantinidis KT, Blackwell N, Kappler A, Kleindienst S. 2022. 'Candidatus ferrigenium straubiae’ sp. nov., ‘Candidatus ferrigenium bremense’ sp. nov., ‘Candidatus ferrigenium altingense’ sp. nov., are autotrophic Fe(II)-oxidizing bacteria of the family Gallionellaceae. Syst Appl Microbiol 45:126306. doi: 10.1016/j.syapm.2022.126306 [DOI] [PubMed] [Google Scholar]
- 30. Straub KL, Benz M, Schink B, Widdel F. 1996. Anaerobic, nitrate-dependent microbial oxidation of ferrous iron. Appl Environ Microbiol 62:1458–1460. doi: 10.1128/aem.62.4.1458-1460.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jakus N, Blackwell N, Osenbrück K, Straub D, Byrne JM, Wang Z, Glöckler D, Elsner M, Lueders T, Grathwohl P, Kleindienst S, Kappler A. 2021. Nitrate removal by a novel lithoautotrophic nitrate-reducing, iron(II)-oxidizing culture enriched from a pyrite-rich limestone aquifer. Appl Environ Microbiol 87:e0046021. doi: 10.1128/AEM.00460-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang Y-M, Straub D, Kappler A, Smith N, Blackwell N, Kleindienst S. 2021. A novel enrichment culture highlights core features of microbial networks contributing to autotrophic Fe(II) oxidation coupled to nitrate reduction. Microb Physiol 31:280–295. doi: 10.1159/000517083 [DOI] [PubMed] [Google Scholar]
- 33. Liu J, Wang Z, Belchik SM, Edwards MJ, Liu C, Kennedy DW, Merkley ED, Lipton MS, Butt JN, Richardson DJ, Zachara JM, Fredrickson JK, Rosso KM, Shi L. 2012. Identification and characterization of MtoA: a decaheme C-type cytochrome of the neutrophilic Fe(II)-oxidizing bacterium Sideroxydans lithotrophicus ES-1. Front Microbiol 3:37. doi: 10.3389/fmicb.2012.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Keffer JL, McAllister SM, Garber AI, Hallahan BJ, Sutherland MC, Rozovsky S, Chan CS. 2021. Iron oxidation by a fused cytochrome-porin common to diverse iron-oxidizing bacteria. mBio 12:e0107421. doi: 10.1128/mBio.01074-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McAllister SM, Polson SW, Butterfield DA, Glazer BT, Sylvan JB, Chan CS, Lloyd KG. 2020. Validating the Cyc2 neutrophilic iron oxidation pathway using meta-omics of Zetaproteobacteria iron mats at marine hydrothermal vents. mSystems 5:e00553-19. doi: 10.1128/mSystems.00553-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Salgueiro CA, Dantas JM. 2016. Multiheme Cytochromes. 1st ed. Springer, Berlin, Heidelberg. doi: 10.1007/978-3-642-44961-1 [DOI] [Google Scholar]
- 37. Paquete CM, Morgado L, Salgueiro CA, Louro RO. 2022. Molecular mechanisms of microbial extracellular electron transfer: the importance of multiheme cytochromes. Front Biosci (Landmark Ed) 27:174. doi: 10.31083/j.fbl2706174 [DOI] [PubMed] [Google Scholar]
- 38. Jiao Y, Newman DK. 2007. The pio operon is essential for phototrophic Fe(II) oxidation in Rhodopseudomonas palustris TIE-1. J Bacteriol 189:1765–1773. doi: 10.1128/JB.00776-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Beliaev AS, Saffarini DA. 1998. Shewanella putrefaciens mtrB encodes an outer membrane protein required for Fe(III) and Mn(IV) reduction. J Bacteriol 180:6292–6297. doi: 10.1128/JB.180.23.6292-6297.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ross DE, Flynn JM, Baron DB, Gralnick JA, Bond DR, Xu S. 2011. Towards electrosynthesis in Shewanella: energetics of reversing the Mtr pathway for reductive metabolism. PLoS ONE 6:e16649. doi: 10.1371/journal.pone.0016649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keffer JL, McAllister SM, Garber AI, Hallahan BJ, Sutherland MC, Rozovsky S, Chan CS. 2021. Iron oxidation by a fused cytochrome-porin common to diverse iron-oxidizing bacteria. mBio 12:e0107421. doi: 10.1128/mBio.01074-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emerson D, Field EK, Chertkov O, Davenport KW, Goodwin L, Munk C, Nolan M, Woyke T. 2013. Comparative genomics of freshwater Fe-oxidizing bacteria: implications for physiology, ecology, and systematics. Front Microbiol 4:254. doi: 10.3389/fmicb.2013.00254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Barco RA, Emerson D, Sylvan JB, Orcutt BN, Jacobson Meyers ME, Ramírez GA, Zhong JD, Edwards KJ. 2015. New insight into microbial iron oxidation as revealed by the proteomic profile of an obligate iron-oxidizing Chemolithoautotroph. Appl Environ Microbiol 81:5927–5937. doi: 10.1128/AEM.01374-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou N, Kupper RJ, Catalano JG, Thompson A, Chan CS. 2022. Biological oxidation of Fe(II)-bearing smectite by microaerophilic iron oxidizer Sideroxydans lithotrophicus using dual Mto and Cyc2 iron oxidation pathways. Environ Sci Technol 56:17443–17453. doi: 10.1021/acs.est.2c05142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Alawi M, Lipski A, Sanders T, Pfeiffer EM, Spieck E. 2007. Cultivation of a novel cold-adapted nitrite oxidizing Betaproteobacterium from the Siberian Arctic. ISME J 1:256–264. doi: 10.1038/ismej.2007.34 [DOI] [PubMed] [Google Scholar]
- 46. Ishii K, Fujitani H, Soh K, Nakagawa T, Takahashi R, Tsuneda S. 2017. Enrichment and physiological characterization of a cold-adapted nitrite-oxidizing Nitrotoga sp. from an eelgrass sediment. Appl Environ Microbiol 83:e00549-17. doi: 10.1128/AEM.00549-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Podowski JC, Paver SF, Newton RJ, Coleman ML. 2022. Genome streamlining, proteorhodopsin, and organic nitrogen metabolism in freshwater nitrifiers. mBio 13:e0237921. doi: 10.1128/mbio.02379-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Boddicker AM, Mosier AC. 2018. Genomic profiling of four cultivated Candidatus Nitrotoga spp. predicts broad metabolic potential and environmental distribution. ISME J 12:2864–2882. doi: 10.1038/s41396-018-0240-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kitzinger K, Koch H, Lücker S, Sedlacek CJ, Herbold C, Schwarz J, Daebeler A, Mueller AJ, Lukumbuzya M, Romano S, Leisch N, Karst SM, Kirkegaard R, Albertsen M, Nielsen PH, Wagner M, Daims H. 2018. Characterization of the first "Candidatus Nitrotoga" isolate reveals metabolic versatility and separate evolution of widespread nitrite-oxidizing bacteria. mBio 9:e01186-18. doi: 10.1128/mBio.01186-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lücker S, Schwarz J, Gruber-Dorninger C, Spieck E, Wagner M, Daims H. 2015. Nitrotoga -like bacteria are previously unrecognized key nitrite oxidizers in full-scale wastewater treatment plants. 3. ISME J 9:708–720. doi: 10.1038/ismej.2014.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ishii K, Fujitani H, Sekiguchi Y, Tsuneda S. 2020. Physiological and genomic characterization of a new ‘Candidatus Nitrotoga’ isolate”. Environ Microbiol 22:2365–2382. doi: 10.1111/1462-2920.15015 [DOI] [PubMed] [Google Scholar]
- 52. Daims H, Lücker S, Wagner M. 2016. A new perspective on microbes formerly known as nitrite-oxidizing bacteria. Trends Microbiol. 24:699–712. doi: 10.1016/j.tim.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, Whitman WB, Euzéby J, Amann R, Rosselló-Móra R. 2014. Uniting the classification of cultured and uncultured bacteria and Archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12:635–645. doi: 10.1038/nrmicro3330 [DOI] [PubMed] [Google Scholar]
- 54. Tindall BJ, Rosselló-Móra R, Busse H-J, Ludwig W, Kämpfer P. 2010. Notes on the characterization of prokaryote strains for taxonomic purposes. Int J Syst Evol Microbiol 60:249–266. doi: 10.1099/ijs.0.016949-0 [DOI] [PubMed] [Google Scholar]
- 55. Barco RA, Garrity GM, Scott JJ, Amend JP, Nealson KH, Emerson D. 2020. A genus definition for bacteria and Archaea based on a standard genome relatedness index. mBio 11. doi: 10.1128/mBio.02475-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Konstantinidis K, Ruiz-Perez C, Gerhardt K, Rodriguez-R L, Jain C, Tiedje J, Cole J. 2022. FastAAI: efficient estimation of genome average amino acid identity and phylum-level relationships using tetramers of universal proteins. In Review. doi: 10.21203/rs.3.rs-1459378/v1 [DOI]
- 57. He S, Tominski C, Kappler A, Behrens S, Roden EE. 2016. Metagenomic analyses of the autotrophic Fe(II)-oxidizing, nitrate-reducing enrichment culture KS. Appl Environ Microbiol 82:2656–2668. doi: 10.1128/AEM.03493-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ivanova N, Tringe SG, Liolios K, Liu W-T, Morrison N, Hugenholtz P, Kyrpides NC. 2010. A call for standardized classification of metagenome projects. Environ Microbiol. 12:1803–1805. doi: 10.1111/j.1462-2920.2010.02270.x [DOI] [PubMed] [Google Scholar]
- 59. Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, Gazitúa MC, Daly RA, Smith GJ, Vik DR, Pope PB, Sullivan MB, Roux S, Wrighton KC. 2020. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res 48:8883–8900. doi: 10.1093/nar/gkaa621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Garber AI, Nealson KH, Okamoto A, McAllister SM, Chan CS, Barco RA, Merino N. 2020. FeGenie: a comprehensive tool for the identification of iron genes and iron gene neighborhoods in genome and metagenome assemblies. Front Microbiol 11:37. doi: 10.3389/fmicb.2020.00037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Garber AI. 2020. MagicLamp: toolkit for annotation of ’omics datasets using curated HMM sets (2020: MagicLamp). GitHub repository. Available from: https://github.com/Arkadiy-Garber/MagicLamp [Google Scholar]
- 62. McAllister SM. 2016. Heme Counter. GitHub repository. Available from: https://github.com/seanmcallister/heme_counter [Google Scholar]
- 63. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 64. Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009. BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi’o: an advanced analysis and visualization platform for ‘omics data. PeerJ 3:e1319. doi: 10.7717/peerj.1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Shaiber A, Willis AD, Delmont TO, Roux S, Chen L-X, Schmid AC, Yousef M, Watson AR, Lolans K, Esen ÖC, Lee STM, Downey N, Morrison HG, Dewhirst FE, Mark Welch JL, Eren AM. 2020. Functional and genetic markers of niche partitioning among enigmatic members of the human oral microbiome. Genome Biol. 21:292. doi: 10.1186/s13059-020-02195-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eren AM, Kiefl E, Shaiber A, Veseli I, Miller SE, Schechter MS, Fink I, Pan JN, Yousef M, Fogarty EC, Trigodet F, Watson AR, Esen ÖC, Moore RM, Clayssen Q, Lee MD, Kivenson V, Graham ED, Merrill BD, Karkman A, Blankenberg D, Eppley JM, Sjödin A, Scott JJ, Vázquez-Campos X, McKay LJ, McDaniel EA, Stevens SLR, Anderson RE, Fuessel J, Fernandez-Guerra A, Maignien L, Delmont TO, Willis AD. 2021. Community-led, integrated, reproducible multi-omics with Anvi’o. Nat Microbiol 6:3–6. doi: 10.1038/s41564-020-00834-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Baker IR, Conley BE, Gralnick JA, Girguis PR, Harwood CS. 2021. Evidence for horizontal and vertical transmission of Mtr-mediated extracellular electron transfer among the bacteria. mBio 13:e0290421. doi: 10.1128/mbio.02904-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ross DE, Flynn JM, Baron DB, Gralnick JA, Bond DR. 2011. Towards electrosynthesis in Shewanella: energetics of reversing the Mtr pathway for reductive metabolism. PLoS One 6:e16649. doi: 10.1371/journal.pone.0016649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Jain A, Kalb MJ, Gralnick JA. 2022. Reconstructing electron transfer components from an Fe(II) oxidizing bacterium. Microbiology (Reading) 168. doi: 10.1099/mic.0.001240 [DOI] [PubMed] [Google Scholar]
- 71. Beliaev AS, Saffarini DA, McLaughlin JL, Hunnicutt D. 2001. MtrC, an outer membrane decahaem C cytochrome required for metal reduction in Shewanella putrefaciens MR-1. Mol Microbiol 39:722–730. doi: 10.1046/j.1365-2958.2001.02257.x [DOI] [PubMed] [Google Scholar]
- 72. He S, Barco RA, Emerson D, Roden EE. 2017. Comparative genomic analysis of neutrophilic iron(II) oxidizer genomes for candidate genes in extracellular electron transfer. Front Microbiol 8. doi: 10.3389/fmicb.2017.01584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Eddie BJ, Bird LJ, Pelikan C, Mussmann M, Martínez-Pérez C, Pinamang P, Malanoski AP, Glaven SM. 2022. Conservation of energetic pathways for electroautotrophy in the uncultivated candidate order Tenderiales. mSphere 7:e0022322. doi: 10.1128/msphere.00223-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Edwards MJ, White GF, Butt JN, Richardson DJ, Clarke TA. 2020. The crystal structure of a biological insulated transmembrane molecular wire. Cell 181:665–673. doi: 10.1016/j.cell.2020.03.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Beckwith CR, Edwards MJ, Lawes M, Shi L, Butt JN, Richardson DJ, Clarke TA. 2015. Characterization of MtoD from Sideroxydans lithotrophicus: a cytochrome C electron shuttle used in lithoautotrophic growth. Front Microbiol 6:332. doi: 10.3389/fmicb.2015.00332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ducluzeau A-L, Ouchane S, Nitschke W. 2008. The cbb3 oxidases are an ancient innovation of the domain bacteria. Mol Biol Evol 25:1158–1166. doi: 10.1093/molbev/msn062 [DOI] [PubMed] [Google Scholar]
- 77. Trojan D, Garcia-Robledo E, Meier DV, Hausmann B, Revsbech NP, Eichorst SA, Woebken D, Bernstein HC. 2021. Microaerobic lifestyle at nanomolar O2 concentrations mediated by low-affinity terminal oxidases in abundant soil bacteria. mSystems 6:e0025021. doi: 10.1128/mSystems.00250-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Badger MR, Bek EJ. 2008. Multiple RuBisCo forms in Proteobacteria: their functional significance in relation to CO2 acquisition by the CBB cycle. J Exp Bot 59:1525–1541. doi: 10.1093/jxb/erm297 [DOI] [PubMed] [Google Scholar]
- 79. Simon J, Klotz MG. 2013. Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations. Biochim Biophys Acta 1827:114–135. doi: 10.1016/j.bbabio.2012.07.005 [DOI] [PubMed] [Google Scholar]
- 80. Schäfer G, Penefsky HS. 2008. Diversity of the Heme–copper Superfamily in Archaea: Insights from Genomics and structural modeling, p 1–31. In Schäfer G, Penefsky HS (ed), Bioenergetics: Energy conservation and conversion. Springer, Berlin, Heidelberg. [Google Scholar]
- 81. McAllister SM, Vandzura R, Keffer JL, Polson SW, Chan CS. 2021. Aerobic and anaerobic iron oxidizers together drive denitrification and carbon cycling at marine iron-rich hydrothermal vents. ISME J 15:1271–1286. doi: 10.1038/s41396-020-00849-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chen J, Strous M. 2013. Denitrification and aerobic respiration, hybrid electron transport chains and co-evolution. Biochim Biophys Acta 1827:136–144. doi: 10.1016/j.bbabio.2012.10.002 [DOI] [PubMed] [Google Scholar]
- 83. Klueglein N, Zeitvogel F, Stierhof Y-D, Floetenmeyer M, Konhauser KO, Kappler A, Obst M. 2014. Potential role of nitrite for abiotic Fe(II) oxidation and cell encrustation during nitrate reduction by denitrifying bacteria. Appl Environ Microbiol 80:1051–1061. doi: 10.1128/AEM.03277-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem Rev 107:4206–4272. doi: 10.1021/cr050196r [DOI] [PubMed] [Google Scholar]
- 85. Wu L-F, Mandrand MA. 1993. Microbial hydrogenases: primary structure, classification, signatures and phylogeny. FEMS Microbiol Rev 10:243–269. doi: 10.1111/j.1574-6968.1993.tb05870.x [DOI] [PubMed] [Google Scholar]
- 86. Vignais PM, Billoud B, Meyer J. 2001. Classification and phylogeny of hydrogenases1. FEMS Microbiol Rev 25:455–501. doi: 10.1111/j.1574-6976.2001.tb00587.x [DOI] [PubMed] [Google Scholar]
- 87. Greening C, Biswas A, Carere CR, Jackson CJ, Taylor MC, Stott MB, Cook GM, Morales SE. 2016. Genomic and metagenomic surveys of hydrogenase distribution indicate H2 is a widely utilised energy source for microbial growth and survival. ISME J 10:761–777. doi: 10.1038/ismej.2015.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schneider K, Schlegel HG. 1976. Purification and properties of soluble hydrogenase from Alcaligenes eutrophus H 16. Biochim Biophys Acta 452:66–80. doi: 10.1016/0005-2744(76)90058-9 [DOI] [PubMed] [Google Scholar]
- 89. Appel J, Phunpruch S, Steinmüller K, Schulz R. 2000. The bidirectional hydrogenase of Synechocystis sp. PCC 6803 works as an electron valve during photosynthesis. Arch Microbiol 173:333–338. doi: 10.1007/s002030000139 [DOI] [PubMed] [Google Scholar]
- 90. Eckert C, Boehm M, Carrieri D, Yu J, Dubini A, Nixon PJ, Maness P-C. 2012. Genetic analysis of the Hox hydrogenase in the Cyanobacterium synechocystis sp. PCC 6803 reveals subunit roles in association, assembly, maturation, and function*. J Biol Chem 287:43502–43515. doi: 10.1074/jbc.M112.392407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Griesbeck C, Schütz M, Schödl T, Bathe S, Nausch L, Mederer N, Vielreicher M, Hauska G. 2002. Mechanism of sulfide-quinone reductase investigated using site-directed mutagenesis and sulfur analysis. Biochemistry 41:11552–11565. doi: 10.1021/bi026032b [DOI] [PubMed] [Google Scholar]
- 92. Marcia M, Ermler U, Peng G, Michel H. 2009. The structure of Aquifex aeolicus sulfide:quinone oxidoreductase, a basis to understand sulfide detoxification and respiration. Proc Natl Acad Sci U S A 106:9625–9630. doi: 10.1073/pnas.0904165106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Loy A, Duller S, Baranyi C, Mussmann M, Ott J, Sharon I, Béjà O, Le Paslier D, Dahl C, Wagner M. 2009. Reverse dissimilatory sulfite reductase as phylogenetic marker for a subgroup of sulfur-oxidizing prokaryotes. Environ Microbiol 11:289–299. doi: 10.1111/j.1462-2920.2008.01760.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. 2015. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. 5. ISME J 9:1152–1165. doi: 10.1038/ismej.2014.208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kato S, Ohkuma M, Powell DH, Krepski ST, Oshima K, Hattori M, Shapiro N, Woyke T, Chan CS. 2015. Comparative genomic insights into ecophysiology of neutrophilic, microaerophilic iron oxidizing bacteria. Front Microbiol 6:1265. doi: 10.3389/fmicb.2015.01265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Koeksoy E, Bezuidt OM, Bayer T, Chan CS, Emerson D. 2021. Zetaproteobacteria pan-genome reveals candidate gene cluster for twisted stalk biosynthesis and export. Front Microbiol 12:679409. doi: 10.3389/fmicb.2021.679409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Schmehl M, Jahn A, Meyer zu Vilsendorf A, Hennecke S, Masepohl B, Schuppler M, Marxer M, Oelze J, Klipp W. 1993. Identification of a new class of nitrogen fixation genes in Rhodobacter capsalatus: a putative membrane complex involved in electron transport to nitrogenase. Mol Gen Genet 241:602–615. doi: 10.1007/BF00279903 [DOI] [PubMed] [Google Scholar]
- 98. Tremblay P-L, Zhang T, Dar SA, Leang C, Lovley DR. 2012. The Rnf complex of Clostridium ljungdahlii is a proton-translocating ferredoxin:NAD+ oxidoreductase essential for autotrophic growth. mBio 4:e00406-12. doi: 10.1128/mBio.00406-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Downing BE, Gupta D, Nayak DD. 2023. The dual role of a multi-heme cytochrome in methanogenesis: MmcA is important for energy conservation and carbon metabolism in Methanosarcina acetivorans. Mol Microbiol 119:350–363. doi: 10.1111/mmi.15029 [DOI] [PubMed] [Google Scholar]
- 100. Liu Y, Chen H, Van Treuren W, Hou B-H, Higginbottom SK, Dodd D. 2022. Clostridium sporogenes uses reductive stickland metabolism in the gut to generate ATP and produce circulating metabolites. Nat Microbiol 7:695–706. doi: 10.1038/s41564-022-01109-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Child SA, Bradley JM, Pukala TL, Svistunenko DA, Le Brun NE, Bell SG. 2018. Electron transfer ferredoxins with unusual cluster binding motifs support secondary metabolism in many bacteria. Chem Sci 9:7948–7957. doi: 10.1039/c8sc01286e [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Watanabe T, Kojima H. 2019. Sulfuricella, p 1–6. In Whitman WB, Rainey F, Kämpfer P, Trujillo M, Chun J, DeVos P, Hedlund B, Dedysh S (ed), Bergey’s Manual of SYSTEMATICS of Archaea and bacteria, 1st ed. Wiley. [Google Scholar]
- 103. Castelle C, Guiral M, Malarte G, Ledgham F, Leroy G, Brugna M, Giudici-Orticoni M-T. 2008. A new iron-oxidizing/O2-reducing supercomplex spanning both inner and outer membranes, isolated from the extreme acidophile Acidithiobacillus ferrooxidans. J Biol Chem 283:25803–25811. doi: 10.1074/jbc.M802496200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gorski CA, Klüpfel LE, Voegelin A, Sander M, Hofstetter TB. 2013. Redox properties of structural Fe in clay minerals: 3 relationships between smectite redox and structural properties. Environ Sci Technol 47:13477–13485. doi: 10.1021/es403824x [DOI] [PubMed] [Google Scholar]
- 105. Gupta D, Sutherland MC, Rengasamy K, Meacham JM, Kranz RG, Bose A, Komeili A. 2019. Photoferrotrophs produce a PioAB electron conduit for extracellular electron uptake. mBio 10:e02668–19. doi: 10.1128/mBio.02668-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Coursolle D, Baron DB, Bond DR, Gralnick JA. 2010. The Mtr respiratory pathway is essential for reducing flavins and electrodes in Shewanella oneidensis. J Bacteriol 192:467–474. doi: 10.1128/JB.00925-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Gorby YA, Yanina S, McLean JS, Rosso KM, Moyles D, Dohnalkova A, Beveridge TJ, Chang IS, Kim BH, Kim KS, Culley DE, Reed SB, Romine MF, Saffarini DA, Hill EA, Shi L, Elias DA, Kennedy DW, Pinchuk G, Watanabe K, Ishii S, Logan B, Nealson KH, Fredrickson JK. 2006. Electrically conductive bacterial nanowires produced by Shewanella oneidensis strain MR-1 and other microorganisms. Proc Natl Acad Sci U S A 103:11358–11363. doi: 10.1073/pnas.0604517103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Edwards MJ, Richardson DJ, Paquete CM, Clarke TA. 2020. Role of multiheme cytochromes involved in extracellular anaerobic respiration in bacteria. Protein Sci 29:830–842. doi: 10.1002/pro.3787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Richardson DJ, Butt JN, Fredrickson JK, Zachara JM, Shi L, Edwards MJ, White G, Baiden N, Gates AJ, Marritt SJ, Clarke TA. 2012. The ‘porin–cytochrome’ model for microbe-to-mineral electron transfer. Mol Microbiol 85:201–212. doi: 10.1111/j.1365-2958.2012.08088.x [DOI] [PubMed] [Google Scholar]
- 110. White GF, Shi Z, Shi L, Wang Z, Dohnalkova AC, Marshall MJ, Fredrickson JK, Zachara JM, Butt JN, Richardson DJ, Clarke TA. 2013. Rapid electron exchange between surface-exposed bacterial cytochromes and Fe(III) minerals. Proc Natl Acad Sci U S A 110:6346–6351. doi: 10.1073/pnas.1220074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. van Wonderen JH, Adamczyk K, Wu X, Jiang X, Piper SEH, Hall CR, Edwards MJ, Clarke TA, Zhang H, Jeuken LJC, Sazanovich IV, Towrie M, Blumberger J, Meech SR, Butt JN. 2021. Nanosecond heme-to-heme electron transfer rates in a multiheme cytochrome nanowire reported by a spectrally unique His/Met-Ligated heme. Proc Natl Acad Sci U S A 118:e2107939118. doi: 10.1073/pnas.2107939118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Wang F, Gu Y, O’Brien JP, Yi SM, Yalcin SE, Srikanth V, Shen C, Vu D, Ing NL, Hochbaum AI, Egelman EH, Malvankar NS. 2019. Structure of microbial nanowires reveals stacked hemes that transport electrons over micrometers. Cell 177:361–369. doi: 10.1016/j.cell.2019.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Chan CS, McAllister SM, Leavitt AH, Glazer BT, Krepski ST, Emerson D. 2016. The architecture of iron microbial mats reflects the adaptation of chemolithotrophic iron oxidation in freshwater and marine environments. Front Microbiol 7. doi: 10.3389/fmicb.2016.00796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hallbeck L, Pedersen K. 2014. The family Gallionellaceae, p 853–858. In Rosenberg E, DeLong EF, Lory S, Stackebrandt E, Thompson F (ed), The Prokaryotes: Alphaproteobacteria and Betaproteobacteria. Springer, Berlin, Heidelberg. [Google Scholar]
- 115. Hedrich S, Schlömann M, Johnson DB. 2011. The iron-oxidizing Proteobacteria. Microbiology (Reading) 157:1551–1564. doi: 10.1099/mic.0.045344-0 [DOI] [PubMed] [Google Scholar]
- 116. Ilbert M, Bonnefoy V. 2013. Insight into the evolution of the iron oxidation pathways. Biochim Biophys Acta 1827:161–175. doi: 10.1016/j.bbabio.2012.10.001 [DOI] [PubMed] [Google Scholar]
- 117. Geer LY, Marchler-Bauer A, Geer RC, Han L, He J, He S, Liu C, Shi W, Bryant SH. 2010. The NCBI BioSystems database. Nucleic Acids Res 38:D492–6. doi: 10.1093/nar/gkp858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Chen I-M, Chu K, Palaniappan K, Ratner A, Huang J, Huntemann M, Hajek P, Ritter S, Varghese N, Seshadri R, Roux S, Woyke T, Eloe-Fadrosh EA, Ivanova NN, Kyrpides NC. 2021. The IMG/M data management and analysis system v.6.0: new tools and advanced capabilities. Nucleic Acids Res. 49:D751–D763. doi: 10.1093/nar/gkaa939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cooper RE, Wegner C-E, McAllister SM, Shevchenko O, Chan CS, Küsel K. 2020. Draft genome sequence of Sideroxydans sp. strain CL21, an Fe(II)-oxidizing bacterium. Microbiol Resour Announc 9:e01444-19. doi: 10.1128/MRA.01444-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Probst AJ, Ladd B, Jarett JK, Geller-McGrath DE, Sieber CMK, Emerson JB, Anantharaman K, Thomas BC, Malmstrom RR, Stieglmeier M, Klingl A, Woyke T, Ryan MC, Banfield JF. 2018. Differential depth distribution of microbial function and putative symbionts through sediment-hosted aquifers in the deep terrestrial subsurface. Nat Microbiol 3:328–336. doi: 10.1038/s41564-017-0098-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. doi: 10.1101/gr.186072.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ahmed M, Saup CM, Wilkins MJ, Lin L-S. 2020. Continuous Ferric iron-dosed anaerobic wastewater treatment: Treatment performance, sludge characteristics, and microbial composition. Journal of Environmental Chemical Engineering 8:103537. doi: 10.1016/j.jece.2019.103537 [DOI] [Google Scholar]
- 123. Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF. 2016. Thousands of microbial Genomes shed light on interconnected Biogeochemical processes in an Aquifer system. Nature communications 7:13219. doi: 10.1038/ncomms13219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Bell E, Lamminmäki T, Alneberg J, Andersson AF, Qian C, Xiong W, Hettich RL, Frutschi M, Bernier-Latmani R. 2020. Active sulfur Cycling in the terrestrial deep subsurface. The ISME journal 14:1260–1272. doi: 10.1038/s41396-020-0602-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Buck M, Garcia SL, Fernandez L, Martin G, Martinez-Rodriguez GA, Saarenheimo J, Zopfi J, Bertilsson S, Peura S. 2021. Comprehensive Dataset of shotgun Metagenomes from oxygen stratified freshwater lakes and ponds. Scientific data 8:131. doi: 10.1038/s41597-021-00910-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Burstein D, Sun CL, Brown CT, Sharon I, Anantharaman K, Probst AJ, Thomas BC, Banfield JF. 2016. Major bacterial lineages are essentially devoid of CRISPR-CAS viral defence systems. Nature communications 7:10613. doi: 10.1038/ncomms10613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Chauhan A, Layton AC, Vishnivetskaya TA, Williams D, Pfiffner SM, Rekepalli B, Stackhouse B, Lau MCY, Phelps TJ, Mykytczuk N, Ronholm J, Whyte L, Onstott TC, Sayler GS. 2014. Metagenomes from thawing low-soil-organic-carbon mineral Cryosols and Permafrost of the Canadian high Arctic. Genome announcements 2:e01217-14. doi: 10.1128/genomeA.01217-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Eze MO, Lütgert SA, Neubauer H, Balouri A, Kraft AA, Sieven A, Daniel R, Wemheuer B. 2020. Metagenome assembly and Metagenome-assembled genome sequences from a historical oil field located in Wietze, Germany. Microbiology resource announcements 9:e00333-20. doi: 10.1128/MRA.00333-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Fortney NW, He S, Converse BJ, Boyd ES, Roden EE. 2018. Investigating the composition and metabolic potential of microbial communities in chocolate pots hot springs. Frontiers in microbiology 9:2075. doi: 10.3389/fmicb.2018.02075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Grettenberger CL, Hamilton TL. n.d. Metagenome assembled Genomes of novel Taxa from an acid mine drainage environment. Microbiology. doi: 10.1101/2020.07.02.185728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. He S, Lau MP, Linz AM, Roden EE, McMahon KD. 2019. Extracellular electron transfer may be an overlooked contribution to Pelagic respiration in Humic-rich freshwater lakes. mSphere 4:e00436-18. doi: 10.1128/mSphere.00436-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Bendall ML, Stevens SL, Chan L-K, Malfatti S, Schwientek P, Tremblay J, Schackwitz W, Martin J, Pati A, Bushnell B, Froula J, Kang D, Tringe SG, Bertilsson S, Moran MA, Shade A, Newton RJ, McMahon KD, Malmstrom RR. 2016. Genome-wide selective sweeps and gene-specific sweeps in natural bacterial populations. The ISME journal 10:1589–1601. doi: 10.1038/ismej.2015.241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Linz AM, He S, Stevens SLR, Anantharaman K, Rohwer RR, Malmstrom RR, Bertilsson S, McMahon KD. 2018. Freshwater carbon and nutrient cycles revealed through reconstructed population Genomes. PeerJ 6:e6075. doi: 10.7717/peerj.6075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kadnikov VV, Ivasenko DA, Beletskii AV, Mardanov AV, Danilova EV, Pimenov NV, Karnachuk OV, Ravin NV. 2016. A novel Uncultured bacterium of the family Gallionellaceae: Description and genome reconstruction based on Metagenomic analysis of microbial community in acid mine drainage. Microbiology 85:449–461. doi: 10.1134/S002626171604010X [DOI] [PubMed] [Google Scholar]
- 135. Lau MCY, Stackhouse BT, Layton AC, Chauhan A, Vishnivetskaya TA, Chourey K, Ronholm J, Mykytczuk NCS, Bennett PC, Lamarche-Gagnon G, Burton N, Pollard WH, Omelon CR, Medvigy DM, Hettich RL, Pfiffner SM, Whyte LG, Onstott TC. 2015. An active atmospheric methane sink in high Arctic mineral Cryosols. The ISME journal 9:1880–1891. doi: 10.1038/ismej.2015.13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. 2017. Recovery of nearly 8,000 Metagenome-assembled Genomes substantially expands the tree of life. Nature microbiology 2:1533–1542. doi: 10.1038/s41564-017-0012-7 [DOI] [PubMed] [Google Scholar]
- 137. Nalven SG, Ward CP, Payet JP, Cory RM, Kling GW, Sharpton TJ, Sullivan CM, Crump BC. 2020. Experimental Metatranscriptomics reveals the costs and benefits of dissolved organic matter photo-alteration for freshwater Microbes. Environmental microbiology 22:3505–3521. doi: 10.1111/1462-2920.15121 [DOI] [PubMed] [Google Scholar]
- 138. Poghosyan L, Koch H, Frank J, van Kessel MAHJ, Cremers G, van Alen T, Jetten MSM, Op den Camp HJM, Lücker S. n.d. Metagenomic profiling of Ammonia- and methane-Oxidizing microorganisms in a Dutch drinking water treatment plant. Microbiology. doi: 10.1101/2020.05.19.103440 [DOI] [PubMed] [Google Scholar]
- 139. Tian R, Ning D, He Z, Zhang P, Spencer SJ, Gao S, Shi W, Wu L, Zhang Y, Yang Y, Adams BG, Rocha AM, Detienne BL, Lowe KA, Joyner DC, Klingeman DM, Arkin AP, Fields MW, Hazen TC, Stahl DA, Alm EJ, Zhou J. 2020. Small and mighty: Adaptation of Superphylum Patescibacteria to Groundwater environment drives their genome simplicity. Microbiome 8:51. doi: 10.1186/s40168-020-00825-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Stewart LC, Stucker VK, Stott MB, de Ronde CEJ. 2018. Marine-influenced microbial communities inhabit terrestrial hot springs on a remote Island volcano. Extremophiles 22:687–698. doi: 10.1007/s00792-018-1029-4 [DOI] [PubMed] [Google Scholar]
- 141. Woodcroft BJ, Singleton CM, Boyd JA, Evans PN, Emerson JB, Zayed AAF, Hoelzle RD, Lamberton TO, McCalley CK, Hodgkins SB, Wilson RM, Purvine SO, Nicora CD, Li C, Frolking S, Chanton JP, Crill PM, Saleska SR, Rich VI, Tyson GW. 2018. Genome-centric view of carbon processing in thawing Permafrost. Nature 560:49–54. doi: 10.1038/s41586-018-0338-1 [DOI] [PubMed] [Google Scholar]
- 142. Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF. 2012. Fermentation, hydrogen, and sulfur metabolism in multiple Uncultivated bacterial phyla. Science 337:1661–1665. doi: 10.1126/science.1224041 [DOI] [PubMed] [Google Scholar]
- 143. Brown CT, Hug LA, Thomas BC, Sharon I, Castelle CJ, Singh A, Wilkins MJ, Wrighton KC, Williams KH, Banfield JF. 2015. Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523:208–211. doi: 10.1038/nature14486 [DOI] [PubMed] [Google Scholar]
- 144. Zhang Y, Kitajima M, Whittle AJ, Liu W-T. 2017. Benefits of Genomic insights and CRISPR-CAS signatures to monitor potential pathogens across drinking water production and distribution systems. Frontiers in microbiology 8:2036. doi: 10.3389/fmicb.2017.02036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Chan CS, Dykes GE, Hoover RL, Limmer MA, Seyfferth AL. n.d. Gallionellaceae in rice root plaque: Metabolic roles in iron oxidation, nutrient Cycling, and plant interactions. Applied and Environmental Microbiology. doi: 10.1128/aem.00570-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the genome taxonomy database. Bioinformatics 36:1925–1927. doi: 10.1093/bioinformatics/btz848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Sundaramurthi JC, Lee J, Kandimalla M, Chen I-M, Kyrpides NC, Reddy TBK. 2021. Genomes OnLine database (GOLD) V.8: overview and updates. Nucleic Acids Res. 49:D723–D733. doi: 10.1093/nar/gkaa983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Buttigieg PL, Morrison N, Smith B, Mungall CJ, Lewis SE, ENVO Consortium . 2013. The environment ontology: contextualising biological and biomedical entities. J Biomed Semantics 4:43. doi: 10.1186/2041-1480-4-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Geneious v.10.2.6. https://www.geneious.com.
- 150. Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32:1792–1797. doi: 10.1093/nar/gkh340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Parks D. 2014. CompareM. Github repository. Available from: https://github.com/dparks1134/CompareM [Google Scholar]
- 152. Varghese NJ, Mukherjee S, Ivanova N, Konstantinidis KT, Mavrommatis K, Kyrpides NC, Pati A. 2015. Microbial species delineation using whole genome sequences. Nucleic Acids Res. 43:6761–6771. doi: 10.1093/nar/gkv657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rodriguez-R LM, Konstantinidis KT. 2016. The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. Peerj Preprints. doi: 10.7287/peerj.preprints.1900v1 [DOI]
- 154. Watanabe T, Kojima H, Fukui M. 2015. Sulfuriferula multivorans gen. nov., sp. nov., isolated from a freshwater lake, reclassification of 'Thiobacillus plumbophilus' as Sulfuriferula plumbophilus sp. nov., and description of Sulfuricellaceae fam. nov. and Sulfuricellales ord. nov. Int J Syst Evol Microbiol 65:1504–1508. doi: 10.1099/ijs.0.000129 [DOI] [PubMed] [Google Scholar]
- 155. Kojima H, Fukui M. 2010. Sulfuricella denitrificans gen. nov., sp. nov., a sulfur-oxidizing autotroph isolated from a freshwater Lake. Int J Syst Evol Microbiol 60:2862–2866. doi: 10.1099/ijs.0.016980-0 [DOI] [PubMed] [Google Scholar]
- 156. Kozlov AM, Darriba D, Flouri T, Morel B, Stamatakis A. 2019. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 35:4453–4455. doi: 10.1093/bioinformatics/btz305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Letunic I, Bork P. 2021. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49:W293–W296. doi: 10.1093/nar/gkab301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Pruesse E, Peplies J, Glöckner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. doi: 10.1093/bioinformatics/bts252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–6. doi: 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Moore RM, Harrison AO, McAllister SM, Polson SW, Wommack KE. 2020. Iroki: automatic customization and visualization of phylogenetic trees. PeerJ 8:e8584. doi: 10.7717/peerj.8584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, Sneddon MW, Henderson ML, Riehl WJ, Murphy-Olson D, Chan SY, Kamimura RT, Kumari S, Drake MM, Brettin TS, Glass EM, Chivian D, Gunter D, Weston DJ, Allen BH, Baumohl J, Best AA, Bowen B, Brenner SE, Bun CC, Chandonia J-M, Chia J-M, Colasanti R, Conrad N, Davis JJ, Davison BH, DeJongh M, Devoid S, Dietrich E, Dubchak I, Edirisinghe JN, Fang G, Faria JP, Frybarger PM, Gerlach W, Gerstein M, Greiner A, Gurtowski J, Haun HL, He F, Jain R, Joachimiak MP, Keegan KP, Kondo S, Kumar V, Land ML, Meyer F, Mills M, Novichkov PS, Oh T, Olsen GJ, Olson R, Parrello B, Pasternak S, Pearson E, Poon SS, Price GA, Ramakrishnan S, Ranjan P, Ronald PC, Schatz MC, Seaver SMD, Shukla M, Sutormin RA, Syed MH, Thomason J, Tintle NL, Wang D, Xia F, Yoo H, Yoo S, Yu D. 2018. KBase: the United States Department of energy systems biology knowledgebase. Nat Biotechnol 36:566–569. doi: 10.1038/nbt.4163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Steinegger M, Söding J. 2017. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol 35:1026–1028. doi: 10.1038/nbt.3988 [DOI] [PubMed] [Google Scholar]
- 163. UniProt Consortium . 2021. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res 49:D480–D489. doi: 10.1093/nar/gkaa1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, Brinkman FSL. 2010. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26:1608–1615. doi: 10.1093/bioinformatics/btq249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Goldberg T, Hecht M, Hamp T, Karl T, Yachdav G, Ahmed N, Altermann U, Angerer P, Ansorge S, Balasz K, Bernhofer M, Betz A, Cizmadija L, Do KT, Gerke J, Greil R, Joerdens V, Hastreiter M, Hembach K, Herzog M, Kalemanov M, Kluge M, Meier A, Nasir H, Neumaier U, Prade V, Reeb J, Sorokoumov A, Troshani I, Vorberg S, Waldraff S, Zierer J, Nielsen H, Rost B. 2014. LocTree3 prediction of localization. Nucleic Acids Res 42:W350–5. doi: 10.1093/nar/gku396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Mirdita M, Schütze K, Moriwaki Y, Heo L, Ovchinnikov S, Steinegger M. 2022. ColabFold: making protein folding accessible to all. Nat Methods 19:679–682. doi: 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D. 2021. Highly accurate protein structure prediction with AlphaFold. Nature 596:583–589. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D. 2022. Protein complex prediction with AlphaFold-Multimer. bioRxiv. doi: 10.1101/2021.10.04.463034 [DOI]
- 169. Schrodinger, LLC. 2010. The PyMOL Molecular Graphics System (2.5.4).
- 170. Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Galperin MY, Kristensen DM, Makarova KS, Wolf YI, Koonin EV. 2019. Microbial genome analysis: the COG approach. Brief Bioinform 20:1063–1070. doi: 10.1093/bib/bbx117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Galperin MY, Wolf YI, Makarova KS, Vera Alvarez R, Landsman D, Koonin EV. 2021. COG database update: focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res 49:D274–D281. doi: 10.1093/nar/gkaa1018 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S4.
Tables S1–S6.