

ABSTRACT
Virus infection of mammalian cells causes induction of interferon (IFN) synthesis, which, in turn, inhibits virus replication through the actions of the proteins encoded by the IFN-stimulated genes (ISG). Although, in general, ISGs inhibit virus replication in all cell types, there are reports that a few ISGs may function in a cell-specific manner. To address this issue systemically, we used an ISG shRNA library to screen two mouse cell lines, AML-12 and LA-4, for cell-specific actions of ISGs; Ddx3, an RNA helicase, was identified as such an ISG. It was required for both IFN action and IFN induction by the RIG-like receptor (RLR) pathway in AML-12 cells but not LA4 cells. In AML-12 cells, Ddx3 was required for the efficient translation of STAT1 and PACT mRNAs; ectopic expression of STAT1 and PACT in Ddx3-deficient cells restored IFN action and IFN induction, respectively. PACT was required for the activation of RLR signaling in AML-12 cells infected with vesicular stomatitis virus, Sendai virus (SeV), encephalomyocarditis virus (EMCV), or mouse hepatitis virus. Moreover, like PKR activation, RLR activation by PACT required S246 and S287 in its domain 3. This study demonstrated not only cell-specific action of an ISG, Ddx3, but also cell-specific need of PACT to trigger RLR signaling in RNA virus-infected cells.
IMPORTANCE
Interferon-stimulated genes (ISGs) are induced in response to interferon expression due to viral infections. Role of these ISGs can be variable in different cells or organs. Our study highlights such cell-specific role of an ISG, Ddx3, which regulates the translation of mRNAs essential for interferon induction (PACT) and interferon signaling (STAT1) in a cell-specific manner. Our study also highlights the role of PACT in RNA virus-induced RLR signaling. Our study depicts how Ddx3 regulates innate immune signaling pathways in an indirect manner. Such cell-specific behavior of ISGs helps us to better understand viral pathogenesis and highlights the complexities of viral tropism and innate immune responses.
KEYWORDS: Ddx3, RNA virus, RLR signaling, PACT
INTRODUCTION
Viral infections activate different pattern recognition receptors (PRR), which induce interferon (IFN) synthesis (1). RNA virus infections lead to IFN induction by activating RLR signaling (RIG-I, MDA-5) or TLR signaling (TLR3, TLR7) (2). Newly synthesized IFN binds to cell surface receptors and triggers the JAK-STAT signaling pathway leading to induced expression of a plethora of IFN-stimulated genes (ISGs) which mount cellular antiviral response (3). ISGs, in general, are functional in all cell types, but exceptions have been noted in a few studies. We observed that in mice, Ifit2, an ISG, protects the CNS from a variety of neurotropic viruses, such as vesicular stomatitis virus (VSV) (4), rhabdovirus (5), mouse hepatitis virus (MHV) (6), and West Nile virus (WNV) (7). In contrast, Ifit2 expression in other tissues, such as liver, lung, and spleen, is not needed for protection from the same viruses; other ISGs are more relevant there (4). Similarly, Zhang et al. reported the antiviral effect of IFIT1 in human fetal astrocytes which was absent in human embryonic lung fibroblasts (8). Ifit2 deficiency enhanced West Nile virus replication in cerebellar granule and dendritic cells but not in macrophages, cortical neurons, and mouse embryonic fibroblasts (MEFs) (7). To develop a comprehensive understanding of such differential roles of ISGs, we designed a screen to identify ISGs with cell-specific antiviral effects and found Ddx3 to be an ISG with differential roles in two cell lines.
Ddx3 is an ISG, but many cells have a basal level of expression. It belongs to the DEAD-box family of ATP-dependent RNA helicases which participate in multiple aspects of RNA metabolism, translation regulation, and other cellular processes and regulate innate immune responses, cancer, and antiviral responses (9). Focusing on its role in regulating innate immune responses, Ddx3 has been reported to induce IFN expression by binding to MAVS (10), and it has also been demonstrated to bind IKKε/TBK-1 complex triggering its activity (11). Reports also suggest that Ddx3 acts as a checkpoint between inflammasome activation and stress granules formation (12). Ddx3 is required for the replication of certain viruses; it modulates host innate responses by interacting with viral proteins (13). Recent reports proposed Ddx3 as a pharmacological target to suppress latent HIV+ CD4+ T cell (14). Moreover, silencing of Ddx3 inhibits the translation of mRNAs involved in cancer progression (Rac1, TAK1) and antiviral responses (PACT, STAT1) (9, 15, 16).
Here, we report a cell-specific role of Ddx3 in IFN induction and IFN signaling. Both IFN induction by the cytoplasmic RLR signaling pathway and ISG induction by the Jak-STAT pathway required Ddx3 in AML-12 cells. Ddx3 modulated IFN induction by promoting translation of PACT, a dsRNA-binding protein that can directly activate the protein kinase, PKR. PACT can also enhance RLR signaling (17–19) and is a component of the Dicer complex (20). Ddx3 also regulated translation of STAT1 mRNA in AML-12 cells, thus affecting IFN signaling. In contrast, LA4 cells did not need Ddx3 or PACT for IFN induction, and the expression of STAT1 was unaffected in LA4 cells upon silencing of Ddx3. Hence, this study demonstrates cell-specific differences in the role of an ISG, Ddx3, in modulating antiviral responses in an indirect manner.
RESULTS
Identification of Ddx3 as an ISG which regulates antiviral action of interferon differently in two cell lines
Because we were interested in looking for cell-specific role of ISGs, we designed a screen using a lentiviral shRNA library of ISGs transduced in mouse lung LA4 cells and mouse liver AML-12 cell line. Individual ISG shRNA was expressed in cells in each well of 96-well plates; IFN-β pre-treatment was given overnight to the cells to induce IFN signaling followed by VSV virus infection. Virus replication was assessed using flow cytometry of cells stained with antibody to a viral protein, and candidate ISGs were selected as those which inhibited virus replication in one cell line but not the other (Fig. S1 and S2); Ddx3 was one such ISG chosen from the screening due to its differential effect on virus replication in the two cell lines. Silencing of Ddx3 had a different impact on IFN-β-induced antiviral effects in the two cell lines; Ddx3 knockdown (KD) disrupted IFN signaling in AML-12 cells leading to uninhibited VSV replication (Fig. 1A), but similar phenomenon was not observed in LA-4 cells (Fig. 1B). The observation was confirmed using another RNA virus, mouse hepatitis virus, a murine coronavirus, which produced similar results (Fig. 1C and D). Like AML-12 cells, a human liver cell line, HepG2, also displayed a need of Ddx3 for IFN’s antiviral effects (Fig. 1E). Hence, we decided to choose Ddx3 from the ISG screen and dissect further its cell-specific role in antiviral effects of IFN.
Fig 1.

Differential role of Ddx3 in regulating antiviral effects of IFN in LA4 and AML-12 cells. (A) Mouse liver AML-12 cells (WT and Ddx3 KD) were treated with various concentrations of IFN-β overnight followed by VSV infection (MOI 5). The supernatant was harvested after 16 hours, and viral titer was determined by using plaque assay in Vero cells. (B) Mouse lung LA-4 cells (WT and Ddx3 KD) were treated with various concentrations of IFN-β overnight followed by VSV infection (MOI 5). The supernatant was harvested after 16 hours, and viral titer was determined by using plaque assay in Vero cells. (C) Mouse liver AML-12 cells (WT and Ddx3 KD) were treated with IFN-β overnight followed by mouse hepatitis virus infection (MOI 5). The supernatant was harvested after 16 hours, and viral titer was determined by using plaque assay in DBT cells. (D) Mouse lung LA-4 cells (WT and Ddx3 KD) were treated with IFN-β overnight followed by MHV infection (MOI 5). The supernatant was harvested after 16 hours, and viral titer was determined by using plaque assay in DBT cells. (E) Human HepG2 cells (WT and Ddx3 KD) were treated with 100 U/mL IFN-β overnight followed by VSV infection (MOI 5). The supernatant was harvested after 16 hours, and viral titer was determined by using plaque assay in Vero cells. (A, B, C, D, E: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Ddx3 regulates RLR-mediated IFN induction in AML-12 cells
Next, we examined the impact of silencing Ddx3 on virus-triggered IFN induction in these cells. In the absence of Ddx3, MHV could not induce IFN mRNA in AML-12 cells, but in LA-4 cells, IFN mRNA was strongly induced by MHV even in the absence of Ddx3 (Fig. 2A). Similar results were obtained when IFN was induced by VSV (Fig. 2B). In the human liver cell line HepG2, like mouse liver AML-12 cells, IFN induction by VSV required Ddx3 (Fig. 2C). Activation of IRF3 by its phosphorylation is required for IFN induction; this process happened in MHV-infected AML-12 cells but only if Ddx3 was not knocked down (Fig. 2D). Similarly, VSV infection triggered IRF3 phosphorylation in AML-12 cells only if Ddx3 was present, but in LA-4 cells, IRF3 was phosphorylated upon VSV infection even in the absence of Ddx3 (Fig. 2E). These results indicated that the triggering of RLR signaling by VSV and MHV required Ddx3 in AML-12 and HepG2 cells but not in LA-4 cells.
Fig 2.

Differential need of Ddx3 for RLR-mediated IFN induction by VSV and MHV. (A) LA-4 cells and AML-12 cells (WT and Ddx3 KD) were infected with MHV (MOI 5), and cells were harvested 8 hours post infection. IFN-β mRNA expression was determined by RT-PCR. (B) LA-4 cells and AML-12 cells (WT and Ddx3 KD) were infected with VSV (MOI 5), and cells were harvested 8 hours post infection. IFN-β mRNA expression was determined by RT-PCR. (C) WT and Ddx3 KD HepG2 cells were infected with VSV, and cells were harvested 8 hours post infection. IFN-β mRNA levels were determined by RT-PCR. (D) WT and Ddx3 KD AML-12 cells were infected with MHV (MOI 5), and cells were harvested 2 hours post infection. IRF-3 phosphorylation was determined by western blotting. (E) WT and Ddx3 KD LA-4/AML-12 cells were infected with VSV (MOI 5), and cells were harvested 2 hours post infection. IRF-3 phosphorylation was determined by western blotting. (A, B, C: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
In infected cells, the presence of viral RNA is sensed by either cytosolic RLRs (RIG-I, MDA-5) or endosomal TLRs (TLR3, TLR7), which trigger IRF-3 phosphorylation and induction of interferons (21). To further delineate the role of Ddx3 in IFN induction, we looked for the pathway specifically interrupted upon Ddx3 silencing. The synthetic dsRNA, Poly I.C, was either transfected into cells, activating cytosolic RLRs or added to the cell culture media, to be endocytosed and activate TLR3. As expected, IFN mRNA was induced by both RLR and TLR3 in AML-12 cells, but Ddx3 was needed for only the RLR pathway, and IFN was induced strongly by TLR3 even in the absence of Ddx3 (Fig. 3A). In contrast, in LA-4 cells, neither pathway required Ddx3 (Fig. 3B). Because VSV uses RIG-I as the signaling receptor (22) and MHV uses primarily MDA-5 (23), our data suggested that signaling by both RLR (RIG-I and MDA-5) pathways requires Ddx3 in AML-12 cells. To test that experimentally, we knocked down MDA-5 (Fig. 3C) or RIG-I (Fig. 3D) expression in AML-12 cells and measured MHV replication (Fig. 3E) and IFN induction (Fig. 3F) in those cells. Surprisingly, our results showed that MHV used primarily RIG-I, not MDA-5, to induce IFN in AML-12 cells. To probe this issue further, we tested the need of Ddx3 for IFN induction by SeV and EMCV, two prototype viruses that use RIG-I or MDA-5 pathway (24), respectively. Neither virus could induce IFN mRNA in Ddx3 KD AML-12 cells (Fig. 3G and H), demonstrating the need of Ddx3 for triggering signaling by both RIG-I and MDA-5 pathways in these cells.
Fig 3.

In AML-12 cells, Ddx3 is required for activation of both RIG-I and MDA-5 signaling pathways. (A) Mouse AML-12 cells (WT and Ddx3 KD) were either treated (50 µg/ mL) or transfected (10 µg/ mL) with poly I.C, and cells were harvested after 8 hours. IFN-β mRNA expression was determined using RT-PCR. (B) Mouse LA-4 cells (WT and Ddx3 KD) were either treated (50 µg/ mL) or transfected (10 µg/ mL) with Poly I.C, and cells were harvested after 8 hours. IFN-β mRNA expression was determined using RT-PCR. (C) AML-12 cells were transduced with MDA-5 targeting lentivirus, and cells were selected for puromycin resistance. The surviving colonies were cloned for single-cell colony selection, and cells from two colonies were confirmed for MDA-5 knockdown using MDA-5-specific antibody. (D) AML-12 cells were transduced with RIG-I targeting lentivirus, and cells were selected for puromycin resistance. The surviving colonies were cloned for single-cell colony selection, and cells from two colonies were confirmed for RIG-I knockdown using RIG-I-specific antibody. (E) WT, RIG-I KD, and MDA-5 KD AML-12 cells were infected with MHV. And 24 hours later, supernatant was collected, and MHV viral titer was determined by plaque assay. (F) WT, RIG-I KD, and MDA-5 KD AML-12 cells were infected with MHV. And 8 hours later, cells were harvested, and IFN-β mRNA levels were determined by RT-PCR. (G) WT and Ddx3 KD cells were infected with Sendai virus (MOI 5). And 8 hours later, cells were harvested, and IFN-β mRNA levels were determined by RT-PCR. (H) WT and Ddx3 KD cells were infected with EMCV virus (MOI 5). And 8 hours later, cells were harvested, and IFN-β mRNA levels were determined by RT-PCR. (A, B, E, F, G, H: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
In addition to inducing IRF-3-dependent genes, RIG-I signaling also induces NF-κB-dependent genes (25). Two such genes, CCL20 and IL-6, were not induced by poly I.C transfection in Ddx3 KD AML-12 cells, but the induction was rescued by ectopic expression of PACT (Fig. S3A and B). These results demonstrated the need for PACT for activating both IRF-3 and NF-κB pathways by RLR signaling in AML-12 cells.
Ddx3 is needed for translation of PACT and STAT1 mRNAs in AML-12 cells
We next inquired why Ddx3 was needed for IFN induction and action in AML-12 cells. We did not observe any interaction of Ddx3 with IKKε/TBK-1 complex or MAVS in virus-infected AML-12 cells, unlike what was previously observed in other cell lines (10, 11). We did not observe any difference in basal or induced expression of Ddx3 in IFN-β treatment or VSV-infected AML-12 and LA-4 cells (Fig. S3C and D). Other studies highlighted the role of Ddx3 in regulating translation of mRNAs involved in innate immune responses, including PACT and STAT1 mRNAs (15, 16). We observed reduced expression of PACT in AML-12 cells, after Ddx3 knockdown, whereas even WT LA-4 cells did not express any detectable PACT (Fig. 4A). Similarly, little STAT1 was expressed in Ddx3 KD AML-12 cells, whereas STAT1 expression was not reduced in Ddx3 KD LA-4 cells (Fig. 4B). These differential expression patterns of PACT and STAT1 in the two cell lines provided possible clues for their different antiviral responses observed upon Ddx3 silencing. To confirm that low expression of STAT1 in Ddx3 KD AML-12 cells was responsible for their poor response to IFN, we ectopically expressed STAT1 in these cells using a STAT1 mRNA expression vector that was devoid of the 5′ UTR containing the target of Ddx3 regulation (Fig. 4C). The STAT1-reconstituted AML-12 Ddx3 KD cells behaved like Wt AML-12 cells, as measured by the strength of induction by IFN of two ISGs, Ifit-1 (Fig. 4D) and Ifit2 (Fig. 4E); the same was true for MHV replication efficiencies in these cells (Fig. 4F). These results demonstrated that Ddx3 regulates IFN signaling in AML-12 cells by modulating the expression of STAT1.
Fig 4.

Ddx3 is required for PACT and STAT1 expression in AML-12 cells. (A) Expression of PACT in LA-4 and AML-12 cells (WT and Ddx3 KD) was measured by western blotting. (B) Expression of STAT1 in LA-4 and AML-12 cells (WT and Ddx3 KD) was measured by western blotting. (C) STAT1 expression was analyzed in WT, Ddx3 KD, and STAT1-reconstituted cells by western blotting. (D) WT, Ddx3 KD, and STAT1-reconstituted AML-12 cells were treated with IFN-β overnight. Cells were harvested, and Ifit-1 mRNA levels were measured using RT-PCR. (E) WT, Ddx3 KD, and STAT1-reconstituted AML-12 cells were treated with IFN-β overnight. Cells were harvested, and Ifit-2 mRNA levels were measured using RT-PCR. (F) WT, Ddx3 KD, and STAT1-reconstituted cells were treated with IFN-β overnight, and cells were infected by MHV for 16 hours. The supernatant was harvested, and viral titer was determined by plaque assay on DBT cells (D, E, F: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Similarly, reconstitution of PACT in Ddx3 KD AML-12 cells (Fig. 5A) restored poly I.C-induced (Fig. 5B) and MHV-induced (Fig. 5C) IFN expression. However, the antiviral response in KD cells was not reinstated by PACT reconstitution alone. MHV replication in Ddx3 KD and PACT-reconstituted cells was still higher compared to WT cells (Fig. 5D), indicating the need of both PACT and STAT1 in restoring complete antiviral effects. As expected, ectopic expression of both PACT and STAT1 in Ddx3 KD AML-12 cells restricted viral replication to a degree similar to WT cells (Fig. 5E). These experiments confirmed that the defect in the antiviral response of Ddx3 KD AML-12 cells was entirely due to a lack of expression of PACT and STAT1.
Fig 5.

RLR signaling is restored upon ectopic PACT expression, but both PACT and STAT1 are needed for restoring the antiviral effect. (A) PACT expression was analyzed in WT, Ddx3 KD, and PACT-reconstituted cells by western blotting. (B) WT, Ddx3 KD, and PACT-reconstituted AML-12 cells were either mock transfected or Poly I.C transfected (10 µg/mL). Cells were harvested after 8 hours, and IFN-β mRNA expression was analyzed by RT-PCR. (C) Mouse AML-12 cells (WT, Ddx3 KD, and PACT reconstituted) were infected by MHV (MOI-5), and cells were harvested 8 hours post infection. IFN-β mRNA expression was determined using RT-PCR. (D) Mouse AML-12 cells (WT, Ddx3 KD, and PACT reconstituted) were treated with IFN-β overnight, and cells were infected by MHVs for 16 hours. The supernatant was harvested, and viral titer was determined by plaque assay on DBT cells. (E) WT, Ddx3 KD, and PACT + STAT1 double reconstituted cells were treated with IFN-β overnight, and cells were infected by MHV for 16 hours. The supernatant was harvested, and viral titer was determined by plaque assay on DBT cells (B, C, D, E: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Absence of PACT-RIG-I interaction in Ddx3 KD cells prevents RIG-I activation
PACT-RIG-I interaction is crucial for virus-induced IFN expression (17), and many viral proteins tend to disrupt this interaction to evade cellular IFN response (26–29). PACT has also been demonstrated to interact with other RLRs (MDA-5 and LGP2) and trigger antiviral response (18, 19). We looked for interaction between endogenous RIG-I and PACT in the presence and the absence of RNA viruses activating RIG-I, VSV, and MHV in AML-12 cells and concluded that PACT interacts with RIG-I only in the presence of viral infection (Fig. 6A). Also, exogenous FLAG-PACT expressed in Ddx3 KD cells (PACT minus background) interacted with RIG-I only after virus infection (Fig. S4A). RIG-I activation is a multi-step process as binding of viral dsRNA to RIG-I leads to its dephosphorylation and K-63-linked polyubiquitination by E-3 ligases (TRIM25, RIPLET), followed by its oligomerization (2). This oligomerized complex binds to MAVS protein leading to IRF3 phosphorylation and interferon induction. PACT has been reported to bind RIG-I and stimulate its ATPase activity, in vitro (17). In AML-12 Ddx3 KD cells, absence of PACT inhibited K-63 polyubiquitylation of RIG-I upon MHV (Fig. 6B) and VSV and SeV (Fig. S4B) infection which was restored after reconstitution of PACT (Fig. 6C; Fig. S4C). Therefore, we concluded that absence of RIG-I PACT binding in Ddx3 knockdown cells inhibited K-63 polyubiquitination of RIG-I, preventing RIG-I activation.
Fig 6.

PACT is required for RIG-I activation upon virus infection. (A) VSV- and MHV-infected WT AML-12 cells along with mock-infected cells were lysed (1 hour post infection), and immunoprecipitation was done using anti-RIG-I antibody or IgG control antibody conjugated to magnetic dynabeads. The elute was run on SDS-PAGE gel, and western blot detection was done by using PACT antibody. (B) WT and Ddx3 KD cells were mock infected or MHV infected. Cells were harvested 1 hour post infection and lysed for immunoprecipitation using anti-RIG-I antibody conjugated to dynabeads. The elute was run on SDS-PAGE, and K-63 polyubiquitination of RIG-I was analyzed by using specific antibody. (C) WT- and PACT-reconstituted AML-12 cells were mock infected or infected by MHV. Cells were harvested 1 hour post infection and lysed for immunoprecipitation using anti-RIG-I antibody conjugated to dynabeads. The elute was run on SDS-PAGE, and K-63 polyubiquitination of RIG-I was analyzed by western blotting. (D) WT and Ddx3 KD cells were mock infected or MHV infected. Cells were harvested 1 hour post infection and lysed for immunoprecipitation using anti-RIG-I antibody conjugated to dynabeads. The elute was run on SDS-PAGE, and TRIM25 interaction with RIG-I was analyzed by using TRIM25-specific antibody. (E) WT and PACT-reconstituted AML-12 cells were mock infected or MHV infected. Cells were harvested 1 hour post infection and lysed for immunoprecipitation using anti-RIG-I antibody conjugated to dynabeads. The elute was run on SDS-PAGE, and TRIM25 interaction with RIG-I was analyzed by using TRIM25-specific antibody.
PACT-RIG-I interaction is required for TRIM25 binding to RIG-I in AML-12 cells
Binding of dsRNA to RIG-I leads to conformational changes in RIG-I and facilitates binding of E3 ligases to RIG-I causing K-63 polyubiquitination. We looked for binding of the E3 ligase TRIM25 to RIG-I in Ddx3 KD cells devoid of PACT. Our observations revealed that TRIM25 was unable to bind to RIG-I in the absence of PACT-RIG-I interaction in MHV- (Fig. 6D) and VSV- (Fig. S4D) infected AML-12 cells. Restoring expression of PACT in Ddx3 KD cells also restored TRIM25 binding to RIG-I in virus-infected cells (Fig. 6E; Fig. S4E). We concluded that PACT-RIG-I interaction is essential for TRIM-25 binding and K-63 polyubiquitination of RIG-I, activating RIG-I and inducing interferons upon virus infection. Ddx3 KD cells devoid of PACT are thus deficient in virus-induced RIG-I activation and IFN expression.
Domain 3 of PACT containing S246 and S287 phosphorylation sites is essential for binding to RIG-I
We investigated the relevant domains of PACT which are responsible for its binding to RIG-I following leads from our previous studies on PACT-PKR interactions. PACT contains two dsRNA-binding domains, domain 1 and domain 2, and a PKR-activation domain, domain 3 (30). Domain 3 of PACT has two phosphorylation sites S246 and S287 crucial for PACT activation and its binding to PKR (31). We examined the needs of these domains of PACT for RIG-I binding and activation using deletion mutants of each domain and expressing them in Ddx3 KD AML-12 cells. Our immunoprecipitation experiments using the PACT deletion mutants (Δ1, Δ2, and Δ3 deletion mutant) suggested domain 3 of PACT was essential for binding to RIG-I (Fig. 7A). The Δ3 domain mutant was unable to induce IFN-β mRNA upon MHV infection (Fig. 7C), suggesting that domain 3 of PACT was essential for RIG-I activation. We also examined the relevance of the two phosphorylation sites (S246 and S287) in domain 3 of PACT for binding and activation of RIG-I. Our results revealed both phosphodeficient mutants (S246A and S287A) were unable to bind RIG-I (Fig. 7B), even after virus infection, causing impaired IFN-β induction (Fig. 7C). Expression of phosphomimetic mutants (S246D and S287D) did not confer virus-independent IFN induction but exhibited a stronger IFN-β expression in response to viral infection (Fig. 7D). Hence, both phosphorylation sites, S246 and S287, are required for activation of PACT upon virus infection and its binding to RIG-I, leading to IFN induction.
Fig 7.

S246 and S287 in domain 3 of PACT are required for RIG-I activation. (A) Ddx3 KD AML-12 cells were transfected with WT FLAG-PACT vector, and mutant FLAG-PACT vector containing deletion of various domains of PACT (Δ1, Δ2, Δ3 domains). And 24 hours later, MHV infection was given, and cells were harvested after 1 hour. The FLAG-tagged PACT protein was immunoprecipitated by using anti-FLAG antibody-conjugated agarose beads. The elute was run on SDS-PAGE gel, and RIG-I interaction with PACT was analyzed using RIG-I-specific primary antibody. (B) Ddx3 KD AML-12 cells were transfected with WT FLAG-PACT vector, and mutant FLAG-PACT vector containing S246A or S287A substitution mutation. And 24 hours later, MHV infection was given, and cells were harvested after 1 hour. The FLAG-tagged PACT protein was immunoprecipitated by using anti-FLAG antibody-conjugated agarose beads. The elute was run on SDS-PAGE gel, and RIG-I interaction with PACT was analyzed using RIG-I-specific primary antibody. (C) Ddx3 KD AML-12 cells were transfected with WT FLAG-PACT vector, and mutant FLAG-PACT vectors containing various deletions and substitutions domain (Δ1, Δ2, Δ3 deletions, S246A and S287A substitution mutation). And 24 hours later, MHV infection was given, and cells were harvested after 8 hours. WT AML-12 cells were also infected as control. IFN-β mRNA expression was determined using RT-PCR. (D) Ddx3 KD AML-12 cells were transfected with WT PACT and phosphomimetic mutants (S246D and S287D). And 24 hours later, MHV infection was given, and cells were harvested after 8 hours (C, D: mean ± SEM, N = 3). *P < 0.05, **P < 0.01, ***P < 0.001. ns = P > 0.05 (not significant).
DISCUSSION
The DDX family of proteins has been well studied for its role in regulating immune responses, both as positive and negative regulators of immune signaling pathways. Ddx60, an ISG, promotes RLR-induced IFN expression upon virus infection (32). Ddx6 interacts with RIG-I and aids in potentiating IFN response against RNA viruses (33). Ddx41 is an early sensor of viral DNA mediating the activation of STING signaling in dendritic cells (34). Ddx5 and Ddx1 have been reported to promote p65 activation and assist in NF-κB-mediated immune responses (35, 36). DDX24 enhances viral replication by competing with RIG-I to bind viral RNA in VSV-infected cells, acting as a negative regulator (37). Ddx46 inhibits virus-induced immune response by entrapping transcripts that encode antiviral proteins inside nucleus (38). Ddx19 disrupts virus-activated TBK1-IRF3 interaction which inhibits TBK1-mediated phosphorylation of IRF3 (39).
Ddx3 has been reported to be a transcriptional regulator of IFNB promoter, possibly through its interaction with TBK1/IKKε (10) or enhancing IRF3 binding to IFNB promoter (40). Here, we report a new role of Ddx3 in the IFN system, which was indirect and relied on its need for translation of PACT and STAT1 mRNAs. Because ectopic expression of PACT and STAT1 in Ddx3 KD AML-12 cells restored full IFN response, we believe that Ddx3 did not have any additional role, relevant to the antiviral action, in these cells. Surprisingly, this role of Ddx3 was manifested in a cell type-specific way; the mouse liver cell line AML-12 and the human hepatoma line HepG2 exhibited the need of Ddx3, but the mouse lung cell line LA-4 did not. It is not clear why Ddx3 is needed for the efficient translation of STAT1 mRNA in AML-12 cells but not in LA-4 cells. It is speculated that DDX3 facilitates the translation of mRNAs by resolving secondary structures within their complex 5′ UTRs (16, 41). Because the mRNA structure is the same in both cell lines, one can speculate that a different RNA helicase unwinds its 5′ UTR to facilitate translation initiation in LA-4 cells, thereby making Ddx3 redundant. In contrast, even in the presence of Ddx3, there was no detectable expression of PACT in LA-4 cells. It remains to be explored whether this was due to an inhibition of PACT mRNA expression at the transcriptional level.
Our results indicated Ddx3 was required for the expression of PACT in AML-12 cells. Infection of the PACT-expressing cells with RNA viruses activated the RLR pathways, both RIG-I and MDA-5. This was preceded by PACT binding to the receptor, recruitment of TRIM25, and ubiquitination of RIG-I (Fig. 8). In the past, PACT has been shown to enhance RIG-I signaling in virus-infected cells by binding to it, and in vitro, PACT can trigger the helicase activity of RIG-I (17). But in AML-12 and HepG2 cells, the need of PACT for RIG-I signaling was absolute; no IFN mRNA was induced even in robustly infected cells, in the absence of PACT. Ddx3 was also required for the expression of NF-κB-dependent genes upon RIG-I pathway stimulation by poly I.C. It is puzzling why viral RNAs, the widely studied activators of RIG-I, failed to trigger RIG-I signaling. Similarly, transfected synthetic dsRNA, poly I:C, required PACT to activate RIG-I signaling in these cells, although PACT was not needed for TLR3 signaling. Could it be possible that PACT, a dsRNA-binding protein, facilitates viral RNA transport to RIG-I? If this paradigm is true, then AML-12 and LA-4 cell lines represent the two opposite ends; in AML-12 cells, PACT is absolutely needed, whereas in LA-4 cells, it is not needed at all. Most cell lines probably use both PACT and viral RNA to activate RLR signaling efficiently.
Fig 8.
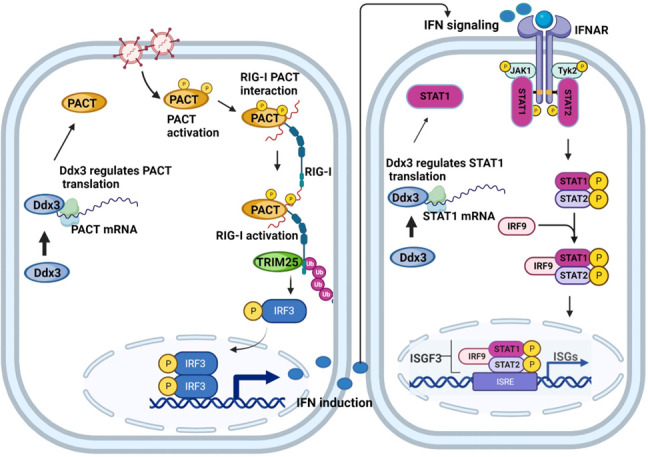
Model depicting regulation of IFN induction and IFN signaling by Ddx3 in AML-12 cells through PACT and STAT1. RNA virus infection activates PACT leading to PACT-RIG-I interaction which causes RIG-I activation and IFN induction. Ddx3 modulates this pathway by regulating the translation of PACT. On the other hand, IFN expression activates the JAK-STAT pathway leading to induction of ISGs. Ddx3 controls this pathway by regulating STAT1 translation. The model depicts how Ddx3 regulates both pathways in an indirect manner. The figure was created using Biorender.com.
PKR activation by PACT has been studied extensively. Like RIG-I, PKR is also a dsRNA-binding protein, and it can be activated by either dsRNA or PACT (42). Domains 1 and 2 of PACT mediate strong binding to both dsRNA and PKR, but the two properties are independent, and there are many mutants of PACT which can bind either PKR or dsRNA but not both (30, 31). Such mutants need to be tested for RIG-I activation in light of our observation that, like PKR activation, it also does not need domains 1 and 2 of PACT. It, however, needs domain 3 and the two serine residues within it that are needed for PKR activation. Therefore, RIG-I and PKR activation by PACT require the same properties of the protein. Further studies are needed to determine the mechanism of RIG-I activation by PACT.
We previously observed organ-specific functions of an ISG, Ifit2, in mice (4), leading us to initiate this study to test whether ISGs can have cell-type-specific functions. We discovered an example of such differential actions of Ddx3, and investigation of its mode of action has revealed new and exciting aspects of the mechanism of activation of RIG-I signaling.
MATERIALS AND METHODS
Reagents and antibodies
Anti-Ddx3 antibody used for western blot was purchased from LS Biosciences (LS-C64576). Anti-PACT (ab-31967) was purchased from Abcam and nti-RIG-I (AG 20B-0009-C100) from Adipogen Life Sciences. K-63 linkage specific polyubiquitin antibody (5621S), phospho IRF3 (4947S), total IRF3 (4302S), actin (3700), STAT1 (9172S), and MDA-5 (5321S) were purchased from Cell Signaling Technology and anti-TRIM-25 antibody (sc-166926) from Santa Cruz Biotechnology. Anti-FLAG antibody (SAB4200071) was purchased from Millipore Sigma. Mouse IgG (sc-2025) and rabbit IgG (sc-2027) control antibodies for immunoprecipitation were purchased from Santa Cruz Biotechnology. Anti-FLAG-conjugated agarose beads (A4596, Millipore Sigma) and Dynabeads protein A (10001D, Thermo Fisher Scientific) were used for immunoprecipitation. Poly I.C (tlrl-pic) was purchased from Invivogen. For reconstitution of PACT (MG52537-NF) and STAT1 (MG53362-UT), cDNA clone expression plasmids were purchased from Sino Biological. Lipofectamine 2000 (Thermo Fisher Scientific, 11668030) was used for all transfection experiments. The clones were transfected into AML-12 cells, and antibiotic selection was used to generate stable cell lines. Recombinant human interferon-β (8499-IF-010) and mouse interferon-β (8234 MB-010) were obtained from R and D Systems.
Cell culture and viruses
Mouse AML-12 cell lines were maintained in DMEM-F12K 1:1 media (obtained from Cleveland Clinic Cell Culture Services) supplemented with 10% FBS, 100 units/mL of penicillin, 100 mg/mL of streptomycin, 1× dexamethasone (10008980, Cayman Chemicals), 1× insulin-selenium-transferrin (41400045, Thermo Fisher Scientific). Mouse LA-4 cells were maintained in F-12 K media (obtained from Cleveland Clinic Cell Culture Services) supplemented with 15% FBS, 100 units/mL of penicillin, and 100 mg/mL of streptomycin. AML-12 cells (CRL-2254), LA-4 cells (CCL-196), Vero cells, and DBT cells were obtained from ATCC (Manassas, VA, USA).
For generation of RIG-I and MDA-5 knockdown cell lines, lentiviral particles were generated in HEK cells using shRNA lentiviral vectors (TRCN0000378444, Millipore Sigma for RIG-I and TRCN0000103646, Millipore Sigma for MDA-5) transfected along with packaging plasmids (psPAX2, #12260 Addgene and pMD2.G, #12259, Addgene). AML-12 cells were infected with lentiviral particles and selected in the presence of puromycin for generating stable cell lines.
Vesicular stomatitis virus Indiana strain, mouse hepatitis virus, and Sendai virus used in the experiments have been described before (4, 6, 43).
Virus infection and quantification
Virus infection was given to cells at MOI 5 for 1 hour in serum-free media. After 1 hour, serum-free media were replaced by normal media. For VSV quantification, plaque assay was performed on Vero cells monolayer as described previously (4). The hepatotropic and neurotropic strain of MHV-A59 known as RSA59 was used in the study. The MHV virus propagation and quantification have been described before (6).
For virus infection experiments, cells were either pre-treated with IFN-β overnight and infected with MHV/VSV virus (MOI-5), or cells were directly infected with virus and harvested post infection. Culture supernatant was collected for viral plaque assays.
mRNA quantification and western blotting
MiRNeasy Mini Kit (Qiagen) was used for RNA isolation, according to manufacturer’s protocol. cDNA synthesis was done using Reverse Transcriptase Kit (#4368813, Thermo Fisher scientific). For real-time PCR, 384 well-format real-time PCR in a Roche Light Cycler 480 II using SYBR Green PCR reagents (#4309155, Thermo Fisher scientific) was used. The primer sequences used for RT-PCR are as follows:
Mouse IFN-β forward—5′ CTTCTCCGTCATCTCCATAGGG 3′.
Mouse IFN-β reverse—5′ CACAGCCCTCTCCATCAACT 3′.
Mouse Ifit1 forward—5′ CAGAAGCACACATTGAAGAA 3′.
Mouse Ifit1 reverse—5′ TGTAAGTAGCCAGAGGAAGG 3′.
Mouse Ifit2 forward—5′ CGGAAAGCAGAGGAAATCAA 3′.
Mouse Ifit2 reverse—5′ TGAAAGTTGCCATACCGAAG 3′.
Mouse CCL 20 forward—5′ GTGGGTTTCACAAGACAGATGGC 3′.
Mouse CCL20 reverse—5′ CCAGTTCTGCTTTGGATCAGCG 3′.
Mouse IL-6 forward—5′ ACACATGTTCTCTGGGAAATCGT 3′.
Mouse IL-6 reverse—5′ AAGTGCATCATCGTTGTTCATACA 3′.
For western blotting, cell pellets were lysed in RIPA lysis buffer containing 20 mM HEPES (pH 7.4), 50 mM NaCl, 2 mM dithiothreitol, 2 mM EGTA, 0.1% SDS, 10 mM Tris-HCl pH 8.0, 10 mM NaF, 12.5 mM β-glycerophosphate, 1 mM Na3VO4, 5 mM Na-pyrophosphate, 0.2% (vol/vol) Triton X-100, and protease inhibitors (Roche Applied Science). The samples were boiled with 4× laemmli buffer and run on SDS-PAGE gel. The protein was transferred to PVDF membrane and blocked in 5% skim milk in TBST buffer (150 mM NaCl; Tris, pH 7.4; and 0.1% Tween-20) for 60 min at room temperature and then incubated with the primary antibody overnight at 4°C. Pierce ECL2 Western Blotting Substrate (Thermo Scientific) was used to visualize the blots.
Immunoprecipitation
Cells were lysed using Pierce IP lysis buffer (#87787, Thermo Fisher scientific). The cell lysates were subjected to preclearing step for 1 hour with anti-mouse or anti-rabbit depending on the source of primary antibody used for IP. After preclearing, the protein supernatant was incubated with IP antibodies overnight at 4°C with rotation followed by incubation with protein A/G magnetic dynabeads for 2 hours with rotation. The beads were washed thrice with IP lysis buffer and boiled in 2× laemmli buffer for 10 min before loading the sample on SDS-PAGE gel. For FLAG-PACT I.P experiments, FLAG-conjugated agarose beads were incubated with protein supernatant overnight at 4°C with rotation followed by 3× washing of beads and boiling with 2× laemmli buffer for 10 min. Western blot experiments were performed via the indicated antibodies and visualized by Pierce ECL2 Western Blotting Substrate (Thermo Fisher Scientific).
For I.P experiments, WT or PACT mutants were transfected into Ddx3 KD AML-12 cells (with PACT minus background). And 24 hours later, virus infection was given, and cells were harvested 1 hour post infection. For K-63 linkage ubiquitination of RIG-I experiments, WT, Ddx3 knockdown, or PACT-reconstituted knockdown AML-12 was infected with VSV or MHV virus (MOI-5), and cells were harvested 1 hour post infection.
Construction of PACT mutants
The deletion mutants for PACT with missing domains Δ1, Δ2, or Δ3 were constructed previously in our lab (30). The phosphodeficient mutants (S246A and S287A) and the phosphomimetic mutants (S246D and S287D) of PACT were previously constructed in our lab (31).
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9.5.1 software. The mean ± SEM of all biological replicates was used to make graphs. Statistical significance was calculated by Student’s t-test or by one way ANOVA followed by Tukey’s post hoc test (P value ≤0.05 was considered significant, *P < 0.05; **P < 0.01, ***P < 0.001).
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health grants CA062220 and CA068782. The funder had no role in study design, data collection and analysis, and decision to publish or preparation of the manuscript.
N.S. performed the experiments, analyzed the results, and wrote parts of the paper; P.K. helped in manuscript writing; G.C.S. designed experiments, interpreted the results, acquired the funding and wrote parts of the paper.
Contributor Information
Ganes C. Sen, Email: seng@ccf.org.
Tom Gallagher, Loyola University Chicago - Health Sciences Campus, Maywood, Illinois, USA.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.01304-23.
ISG shRNA library showing VSV-infected AML-12 cells after IFN treatment.
ISG shRNA library showing VSV-infected LA-4 cells after IFN treatment.
Ddx3 is required for the induction of NF-κB-dependent genes by RIG-I signaling in AML-12 cells.
PACT is required for RNA virus-induced RIG-I activation in AML-12 cells.
Legends for Fig. S1 to S4.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Katze MG, He Y, Gale M. 2002. Viruses and interferon: a fight for supremacy. Nat Rev Immunol 2:675–687. doi: 10.1038/nri888 [DOI] [PubMed] [Google Scholar]
- 2. Rehwinkel J, Gack MU. 2020. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol 20:537–551. doi: 10.1038/s41577-020-0288-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hu X, Li J, Fu M, Zhao X, Wang W. 2021. The JAK/STAT signaling pathway: from bench to clinic. Signal Transduct Target Ther 6:402. doi: 10.1038/s41392-021-00791-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fensterl V, Wetzel JL, Ramachandran S, Ogino T, Stohlman SA, Bergmann CC, Diamond MS, Virgin HW, Sen GC, Hiscott J. 2012. Interferon-induced Ifit2/ISG54 protects mice from lethal VSV neuropathogenesis. PLoS Pathog 8:e1002712. doi: 10.1371/journal.ppat.1002712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davis BM, Fensterl V, Lawrence TM, Hudacek AW, Sen GC, Schnell MJ. 2017. Ifit2 is a restriction factor in rabies virus pathogenicity. J Virol 91:e00889-17. doi: 10.1128/JVI.00889-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das Sarma J, Burrows A, Rayman P, Hwang M-H, Kundu S, Sharma N, Bergmann C, Sen GC. 2020. Ifit2 deficiency restricts microglial activation and leukocyte migration following murine coronavirus (m-CoV) CNS infection. PLoS Pathog 16:e1009034. doi: 10.1371/journal.ppat.1009034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cho H, Shrestha B, Sen GC, Diamond MS. 2013. A role for Ifit2 in restricting West Nile virus infection in the brain. J Virol 87:8363–8371. doi: 10.1128/JVI.01097-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang L, Wang B, Li L, Qian D-M, Yu H, Xue M-L, Hu M, Song X-X. 2017. Antiviral effects of IFIT1 in human cytomegalovirus-infected fetal astrocytes. J Med Virol 89:672–684. doi: 10.1002/jmv.24674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hernández-Díaz T, Valiente-Echeverría F, Soto-Rifo R. 2021. RNA helicase Ddx3: a double-edged sword for viral replication and immune signaling. Microorganisms 9:1206. doi: 10.3390/microorganisms9061206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Oshiumi H, Sakai K, Matsumoto M, Seya T. 2010. DEAD/H BOX 3 (Ddx3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-β-inducing potential. Eur J Immunol 40:940–948. doi: 10.1002/eji.200940203 [DOI] [PubMed] [Google Scholar]
- 11. Gu L, Fullam A, Brennan R, Schröder M. 2013. Human DEAD box helicase 3 couples IκB kinase ε to interferon regulatory factor 3 activation. Mol Cell Biol 33:2004–2015. doi: 10.1128/MCB.01603-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, Guy CS, Briard B, Place DE, Bhattacharya A, Sharma BR, Nourse A, King SV, Pitre A, Burton AR, Pelletier S, Gilbertson RJ, Kanneganti T-D. 2019. Ddx3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 573:590–594. doi: 10.1038/s41586-019-1551-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valiente-Echeverría F, Hermoso MA, Soto-Rifo R. 2015. RNA helicase Ddx3: at the crossroad of viral replication and antiviral immunity. Rev Med Virol 25:286–299. doi: 10.1002/rmv.1845 [DOI] [PubMed] [Google Scholar]
- 14. Rao S, Lungu C, Crespo R, Steijaert TH, Gorska A, Palstra R-J, Prins HAB, van Ijcken W, Mueller YM, van Kampen JJA, Verbon A, Katsikis PD, Boucher CAB, Rokx C, Gruters RA, Mahmoudi T. 2021. Selective cell death in HIV-1-infected cells by Ddx3 inhibitors leads to depletion of the inducible reservoir. Nat Commun 12:2475. doi: 10.1038/s41467-021-22608-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lai M-C, Sun HS, Wang S-W, Tarn W-Y. 2016. Ddx3 functions in antiviral innate immunity through translational control of PACT. FEBS J 283:88–101. doi: 10.1111/febs.13553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ku Y-C, Lai M-H, Lo C-C, Cheng Y-C, Qiu J-T, Tarn W-Y, Lai M-C. 2019. Ddx3 participates in translational control of inflammation induced by infections and injuries. Mol Cell Biol 39:e00285-18. doi: 10.1128/MCB.00285-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kok K-H, Lui P-Y, Ng M-H, Siu K-L, Au SWN, Jin D-Y. 2011. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 9:299–309. doi: 10.1016/j.chom.2011.03.007 [DOI] [PubMed] [Google Scholar]
- 18. Lui P-Y, Wong L-Y, Ho T-H, Au SWN, Chan C-P, Kok K-H, Jin D-Y. 2017. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J Immunol 199:1846–1855. doi: 10.4049/jimmunol.1601493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanchez David RY, Combredet C, Najburg V, Millot GA, Beauclair G, Schwikowski B, Léger T, Camadro J-M, Jacob Y, Bellalou J, Jouvenet N, Tangy F, Komarova AV. 2019. LGP2 binds to PACT to regulate RIG-I- and MDA5-mediated antiviral responses. Sci Signal 12:eaar3993. doi: 10.1126/scisignal.aar3993 [DOI] [PubMed] [Google Scholar]
- 20. Lee Y, Hur I, Park S-Y, Kim Y-K, Suh MR, Kim VN. 2006. The role of PACT in the RNA silencing pathway. EMBO J 25:522–532. doi: 10.1038/sj.emboj.7600942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chattopadhyay S, Sen GC. 2014. dsRNA-activation of TLR3 and RLR signaling: gene induction-dependent and independent effects. J Interferon Cytokine Res 34:427–436. doi: 10.1089/jir.2014.0034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Furr SR, Moerdyk-Schauwecker M, Grdzelishvili VZ, Marriott I. 2010. RIG-I mediates nonsegmented negative-sense RNA virus-induced inflammatory immune responses of primary human astrocytes. Glia 58:1620–1629. doi: 10.1002/glia.21034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zalinger ZB, Elliott R, Rose KM, Weiss SR. 2015. MDA5 is critical to host defense during infection with murine coronavirus. J Virol 89:12330–12340. doi: 10.1128/JVI.01470-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S. 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. doi: 10.1038/nature04734 [DOI] [PubMed] [Google Scholar]
- 25. Zhang H-X, Liu Z-X, Sun Y-P, Zhu J, Lu S-Y, Liu X-S, Huang Q-H, Xie Y-Y, Zhu H-B, Dang S-Y, Chen H-F, Zheng G-Y, Li Y-X, Kuang Y, Fei J, Chen S-J, Chen Z, Wang Z-G. 2013. Rig-I regulates NF-κB activity through binding to Nf-κb1 3'-UTR mRNA. Proc Natl Acad Sci U S A 110:6459–6464. doi: 10.1073/pnas.1304432110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Siu K-L, Yeung ML, Kok K-H, Yuen K-S, Kew C, Lui P-Y, Chan C-P, Tse H, Woo PCY, Yuen K-Y, Jin D-Y. 2014. Middle east respiratory syndrome coronavirus 4a protein is a double-stranded RNA-binding protein that suppresses PACT-induced activation of RIG-I and MDA5 in the innate antiviral response. J Virol 88:4866–4876. doi: 10.1128/JVI.03649-13 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27. Tawaratsumida K, Phan V, Hrincius ER, High AA, Webby R, Redecke V, Häcker H. 2014. Quantitative proteomic analysis of the influenza A virus nonstructural proteins NS1 and NS2 during natural cell infection identifies PACT as an NS1 target protein and antiviral host factor. J Virol 88:9038–9048. doi: 10.1128/JVI.00830-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF. 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14:74–84. doi: 10.1016/j.chom.2013.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kew C, Lui P-Y, Chan C-P, Liu X, Au SWN, Mohr I, Jin D-Y, Kok K-H. 2013. Suppression of PACT-induced type I interferon production by herpes simplex virus 1 Us11 protein. J Virol 87:13141–13149. doi: 10.1128/JVI.02564-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Peters GA, Hartmann R, Qin J, Sen GC. 2001. Modular structure of PACT: distinct domains for binding and activating PKR. Mol Cell Biol 21:1908–1920. doi: 10.1128/MCB.21.6.1908-1920.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peters GA, Dickerman B, Sen GC. 2009. Biochemical analysis of PKR activation by PACT. Biochemistry 48:7441–7447. doi: 10.1021/bi900433y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyashita M, Oshiumi H, Matsumoto M, Seya T. 2011. Ddx60, a DEXD/H box helicase, is a novel antiviral factor promoting RIG-I-like receptor-mediated signaling. Mol Cell Biol 31:3802–3819. doi: 10.1128/MCB.01368-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Núñez RD, Budt M, Saenger S, Paki K, Arnold U, Sadewasser A, Wolff T. 2018. The RNA helicase Ddx6 associates with RIG-I to augment induction of antiviral signaling. Int J Mol Sci 19:1877. doi: 10.3390/ijms19071877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J. 2011. The helicase Ddx41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12:959–965. doi: 10.1038/ni.2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tanaka K, Tanaka T, Nakano T, Hozumi Y, Yanagida M, Araki Y, Iwazaki K, Takagi M, Goto K. 2020. Knockdown of DEAD-box RNA helicase Ddx5 selectively attenuates serine 311 phosphorylation of NF-κB p65 subunit and expression level of anti-apoptotic factor Bcl-2. Cell Signal 65:109428. doi: 10.1016/j.cellsig.2019.109428 [DOI] [PubMed] [Google Scholar]
- 36. Ishaq M, Ma L, Wu X, Mu Y, Pan J, Hu J, Hu T, Fu Q, Guo D. 2009. The DEAD-box RNA helicase Ddx1 interacts with RelA and enhances nuclear factor kappaB-mediated transcription. J Cell Biochem 106:296–305. doi: 10.1002/jcb.22004 [DOI] [PubMed] [Google Scholar]
- 37. Ma Z, Moore R, Xu X, Barber GN, Heise MT. 2013. Ddx24 negatively regulates cytosolic RNA-mediated innate immune signaling. PLoS Pathog 9:e1003721. doi: 10.1371/journal.ppat.1003721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng Q, Hou J, Zhou Y, Li Z, Cao X. 2017. The RNA helicase DDX46 inhibits innate immunity by entrapping m6A-demethylated antiviral transcripts in the nucleus. Nat Immunol 18:1361. doi: 10.1038/ni1217-1361a [DOI] [PubMed] [Google Scholar]
- 39. Zhang K, Zhang Y, Xue J, Meng Q, Liu H, Bi C, Li C, Hu L, Yu H, Xiong T, Yang Y, Cui S, Bu Z, He X, Li J, Huang L, Weng C. 2019. Ddx19 inhibits type I interferon production by disrupting TBK1-IKKε-IRF3 interactions and promoting TBK1 and IKKε degradation. Cell Rep 26:1258–1272. doi: 10.1016/j.celrep.2019.01.029 [DOI] [PubMed] [Google Scholar]
- 40. Saikruang W, Ang Yan Ping L, Abe H, Kasumba DM, Kato H, Fujita T. 2022. The RNA helicase Ddx3 promotes IFNB transcription via enhancing IRF-3/p300 holocomplex binding to the IFNB promoter. Sci Rep 12:3967. doi: 10.1038/s41598-022-07876-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Calviello L, Venkataramanan S, Rogowski KJ, Wyler E, Wilkins K, Tejura M, Thai B, Krol J, Filipowicz W, Landthaler M, Floor SN. 2021. Ddx3 depletion represses translation of mRNAs with complex 5' UTRs. Nucleic Acids Res 49:5336–5350. doi: 10.1093/nar/gkab287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patel RC, Sen GC. 1998. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J 17:4379–4390. doi: 10.1093/emboj/17.15.4379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Raja R, Sen GC. 2022. The antiviral action of the RIG-I induced pathway of apoptosis (RIPA) is enhanced by its ability to degrade Otulin, which deubiquitinates IRF3. Cell Death Differ 29:504–513. doi: 10.1038/s41418-021-00870-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
ISG shRNA library showing VSV-infected AML-12 cells after IFN treatment.
ISG shRNA library showing VSV-infected LA-4 cells after IFN treatment.
Ddx3 is required for the induction of NF-κB-dependent genes by RIG-I signaling in AML-12 cells.
PACT is required for RNA virus-induced RIG-I activation in AML-12 cells.
Legends for Fig. S1 to S4.