

Abstract
With the emergence of spatial transcriptome sequencing (ST-seq), research now heavily relies on the joint analysis of ST-seq and single-cell RNA sequencing (scRNA-seq) data to precisely identify cell spatial composition in tissues. However, common methods for combining these datasets often merge data from multiple cells to generate pseudo-ST data, overlooking topological relationships and failing to represent spatial arrangements accurately. We introduce GTAD, a method utilizing the Graph Attention Network for deconvolution of integrated scRNA-seq and ST-seq data. GTAD effectively captures cell spatial relationships and topological structures within tissues using a graph-based approach, enhancing cell-type identification and our understanding of complex tissue cellular landscapes. By integrating scRNA-seq and ST data into a unified graph structure, GTAD outperforms traditional ‘pseudo-ST’ methods, providing robust and information-rich results. GTAD performs exceptionally well with synthesized spatial data and accurately identifies cell spatial composition in tissues like the mouse cerebral cortex, cerebellum, developing human heart and pancreatic ductal carcinoma. GTAD holds the potential to enhance our understanding of tissue microenvironments and cellular diversity in complex bio-logical systems. The source code is available at https://github.com/zzhjs/GTAD.
Keywords: cell-type identification, graph attention networks, single-cell RNA sequencing, spatial transcriptomics
INTRODUCTION
In tissues, different types of cells execute their functions through their spatial organization and structure. Revealing the intricate spatial architecture of heterogeneous tissues is of paramount importance for understanding cellular mechanisms and functions in diseases. The rapid development of high-throughput single-cell sequencing technology [1–3] has enabled the study of cellular heterogeneity and gene expression specificity at unprecedented resolutions. Advances in spatial transcriptomics (ST) [4–7] have made it possible to measure gene expression while retaining spatial information, presenting significant opportunities for studying cell heterogeneity [8], intercellular communication [9] and interactions [10] within a spatial context. Breakthrough techniques can capture spatial gene expression of the whole genome at subcellular to single-cell levels [11, 12]. These methods have been applied in various disease models to decipher spatial maps of genes and culprits of interest [13–15].
However, inherent limitations exist in ST analysis, mainly that in most cases, each spot or tile covers multiple cells. Even with high-resolution techniques, a small fraction of several cells can be encompassed within the same spatial barcode region. Additionally, highly heterogeneous tissues, such as cancers, consist of various cell types in every small region of the tissue [16]. Therefore, identifying different cell types at each spot is a crucial task for understanding the pathological and physiological spatial context using spatially resolved transcriptomics.
To comprehend cell-type distributions from ST, the most common strategy is to combine it with scRNA-seq. Currently, mainstream methods primarily rely on deconvolution approaches that aim to estimate the exact cell-type proportions at each spatial position through regression models [17, 18], deep learning models [19, 20] or fitting probability distributions [21, 22]. Deep learning models often combine multiple cells to simulate the composition of real ST data. In this step, the gene expression values of multiple cells are often summed as the gene expression of a single spot to create pseudo-ST data. However, due to different experimental and technical conditions, such as sequencing depth and batch effects [23], pseudo-ST data composed of scRNA-seq are influenced differently compared with real ST data, leading to inconsistency between the datasets, impacting data comparison and overall interpretation. Furthermore, the relationship between pseudo-ST data and real ST data is vital. In research, pseudo-ST data are generated by mixing scRNA-seq data from the same tissue to simulate ST data. The generation of pseudo-ST data aims to provide a theoretically controllable dataset for evaluating and optimizing deconvolution method performance. In this study, the relationship between pseudo-ST data and real ST data is crucial. The objective of deconvolution is to reconstruct the cell-type composition at different locations (spots) from real ST data to gain an in-depth understanding of cell-type distributions and spatial structures in tissues. However, traditional simple graph methods have limitations when dealing with the relationship between pseudo-ST data and real ST data. These methods typically employ a simplified data representation that may not fully capture richer information, such as cell-to-cell topological relationships and spatial correlations. Given the significance of intercellular interactions and relative positions in complex biological tissues, these factors play pivotal roles in the recognition of cell types and the analysis of spatial distribution. Conventional simple graph methods may not adequately account for these critical factors.
To overcome these limitations, we propose a new method called GTAD: A Graph-based Approach for Cell Spatial Composition Inference from Integrated scRNA-seq and ST-seq Data, which utilizes a Graph Attention Network (GAT) model for deconvolution [24]. We use Seurat’s IntegrateData method [25] to integrate the generated pseudo-ST data and real ST data to eliminate batch effects between them, enhancing data consistency and comparability. Subsequently, we employ a random projection forest [26] to construct a weighted adjacency matrix to accurately represent the topological relationship between pseudo-ST and real ST. Importantly, by integrating features and the weighted adjacency matrix into the GAT model, we can infer the cell-type composition at each position in ST. We validated GTAD’s accuracy and sensitivity in predicting cell-type proportions through simulated ST data. Furthermore, to demonstrate the broad applicability of GTAD, we applied it to ST datasets from four different tissues, including mouse cerebral cortex, cerebellum, developing human heart and pancreatic ductal adenocarcinoma (PDAC). Through the joint analysis of spatial and single-cell transcriptomic data, GTAD revealed cell spatial composition and heterogeneity.
Compared with existing methods, GTAD employs a GAT model for deconvolution, accurately revealing the topological relationship between pseudo-ST and real ST and enhancing the resolution of cell-type distribution in tissues. Moreover, GTAD’s versatility has been experimentally validated in four different tissue conditions, showcasing its robustness and reliability across diverse scenarios. Thus, GTAD provides a novel, more accurate and comprehensive approach for the analysis of ST data.
MATERIAL AND METHODS
Implementation of GTAD
First, we apply the enhanced GAT model to predict the cell-type proportions for each spot. The neural network training is implemented using the TensorFlow package [27] (version 2.12) (Figure 1):
Figure 1.

Framework of GTAD for deconvoluting ST data.
1. Feature Selection and Synthetic Pseudo-ST Data Generation: Based on the gene expression matrix from scRNA-seq and the cell types obtained from the metadata, we conducted total count normalization of the gene expression data. Differential gene expression analysis was carried out for each cell subgroup using the Wilcoxon Rank-Sum test. This analysis allowed us to identify and select the most differentially expressed genes with biological significance as the feature genes. These extracted genes were utilized as features to filter the gene expression matrices of both scRNA-seq and ST data. Subsequently, the filtered scRNA-seq data was employed to generate synthetic pseudo-ST data with known cellular components.
2. Integration of Pseudo-ST and Real ST Data. To enhance the consistency and comparability of the generated pseudo-ST data with real ST data, a series of steps were undertaken. Initially, we standardized and reduced the dimensionality of data from each dataset using techniques like principal component analysis. This step facilitated computational efficiency and captured crucial cellular features. Similarity scores between spots within each dataset were computed, often utilizing metrics like cosine similarity. Between datasets, we identified anchor points—pairs of spots with high similarity scores—indicating similar expression patterns. Aligning these anchor points maximized similarity scores, allowing the integration of spot pairs from different datasets. The anchor point information guided the creation of cross-dataset connections, and the feature representations were mapped into an integrated space. The combined dataset, comprising both pseudo-ST and real ST data, served as the feature representation matrix X, where each row represented a spot, and each column represented a feature dimension.
3. Graph Construction Using Random Projection Forest. Pseudo-ST data are derived from the manipulation of genes that exhibit significant expression variations among distinct cell types in scRNA-seq data. These genes in scRNA-seq data hold valuable insights into different cell types. To better analyze the pseudo-ST data alongside real ST data, a graph structure is utilized to depict the topological relationships between pseudo-spots and real spots. This graphical representation aids in unraveling the intricate interplay between these complex datasets, enabling more profound biological insights.
Using a graph structure to depict the relationships between pseudo-spots and real spots is motivated by its capacity to model intricate spatial connections and interactions within ST datasets. This is crucial because neighboring spots in the spatial context often exhibit similarities in gene expression patterns or functional associations, and a graphical representation effectively captures these local dependencies. Furthermore, the utilization of graph-based representations is well founded, as similar methods have been successfully employed in the past within the domain of ST [20].
Using the integrated data as input features, a random projection forest (rpForest) was employed to generate an adjacency matrix by constructing multiple random projection trees (rpTrees). Each rpTree projected and partitioned the data based on random directions, resulting in different leaf nodes. By connecting data points that fell into the same leaf node, an adjacency matrix was constructed. A random projection forest is chosen for graph construction due to its effectiveness in addressing key challenges. Unlike traditional k-nearest neighbors (k-NN) methods that require specifying a neighborhood size, random projection forests adaptively partition data, connecting points in the same leaf node. This dynamic approach adjusts edge weights based on common presence in leaf nodes, resulting in a weighted adjacency matrix reflecting spot similarity. Considering data point distribution during construction, random projection forests overcome equi-weighted edges issues in traditional k-NN graphs, providing a more accurate representation of similarity. The weighted adjacency matrix is represented as A, where each element 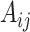 signifies the connection weight between node i and node j.
signifies the connection weight between node i and node j.
4. Model Construction: Traditional graph convolutional networks (such as GCN) [28] employ fixed weights for each node when aggregating node information, meaning that every node attributes the same importance to neighboring nodes’ information. In certain cases, this approach may not be flexible enough, as relationships between different nodes can be diverse and complex, and a fixed weight cannot accurately capture this intricacy. The GAT model addresses this issue by introducing a graph attention mechanism. It adaptively learns the importance and degree of association between nodes by computing attention weights.
Utilizing the relationship between the graph structure and node features, the GAT model adaptively learns the importance and degree of association between nodes through attention weight computation. Graph data typically involve intricate relationships between nodes, where certain connections between nodes hold more significance than others. By introducing graph weights, the model can dynamically allocate distinct weights to each node and its neighboring nodes. This dynamic allocation enhances the model’s capability to effectively capture relationships between nodes. Consequently, the model can more accurately depict information propagation within the graph. The GAT model comprises multiple graph attention heads, each capable of learning different attention weight distributions between nodes, thus capturing contextual information for nodes. Each head generates an aggregated feature representation of nodes by weighting and summing input features and attention weights.
Attention coefficients
For each attention layer, considering N node features, 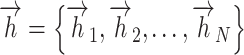 ,
, 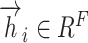 , where the dimension of each node feature is F, the computation of attention coefficients can be represented as follows:
, where the dimension of each node feature is F, the computation of attention coefficients can be represented as follows:
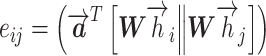 |
(1) |
where 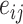 is the attention coefficient of node i relative to node j, and
is the attention coefficient of node i relative to node j, and 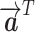 and W are shared learnable parameters.
and W are shared learnable parameters.
Weighted adjacency matrix
Subsequently, the previously calculated weights of the adjacency matrix are incorporated into the model, emphasizing the topological relationships of the graph:
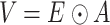 |
(2) |
where V are the attention coefficients, which take into consideration the weights of the adjacency matrix; E are the attention coefficients between all nodes; A is the overall weight of the adjacency matrix.
Normalization
To achieve better weight distribution, it is necessary to uniformly normalize the calculated relevance with respect to all neighbors, typically accomplished through softmax normalization:
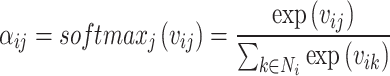 |
(3) |
where 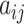 is the normalized attention coefficient of node i with respect to node j.
is the normalized attention coefficient of node i with respect to node j. 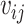 is the attention coefficient of node i relative to node j.
is the attention coefficient of node i relative to node j.
Multi-head attention
By parallelly computing multiple attention heads, the multi-head attention mechanism can capture different patterns of correlations and information interactions, thus providing a more comprehensive understanding of the input sequence:
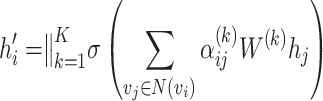 |
(4) |
where 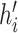 is the new feature of node i after incorporating neighborhood information;
is the new feature of node i after incorporating neighborhood information; 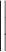 is operations such as vector concatenation or averaging; and
is operations such as vector concatenation or averaging; and 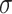 is the activation function.
is the activation function.
ST cell-type deconvolution
In this work, cross-entropy serves as the loss function, quantifying the variance between predicted and actual class distributions in multi-class classification tasks. The softmax activation function is employed to transform model outputs into a probability distribution across classes, simplifying precise class selection based on the highest probability. These choices collectively enhance classification accuracy and render the model’s output readily interpretable. After the completion of training,
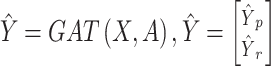 |
(5) |
where 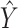 is the ultimately predicted cellular type proportions for all spots,
is the ultimately predicted cellular type proportions for all spots, 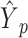 is the cellular type proportions for pseudo-spots and
is the cellular type proportions for pseudo-spots and 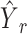 is the cellular type proportions for genuine spots.
is the cellular type proportions for genuine spots.
Hence, the predicted cellular composition for real-ST spots is 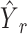 .
.
Evaluating GTAD’s performance using simulated ST data
Simulating ST data from scRNA-seq
We created pseudo-spatial transcriptomics (pseudo-ST) datasets using two distinct single-cell RNA sequencing (scRNA-seq) datasets: the mouse trachea dataset (Montoro_10x and Plasschaert) from the Cell BLAST database and the colorectal cancer dataset from scRNA-seq data of Korean and Belgian colorectal cancer patients (GSE132465 and GSE144735) [29]. Synthetic ST datasets were generated for cell-type deconvolution accuracy evaluation. These synthetic ST datasets comprised spots that represented a mix of two to eight cells randomly selected from the scRNA-seq data. They closely mimic real-world ST data, serving as ground-truth references to assess GTAD’s performance in estimating cell-type proportions within each synthetic spot.
Performance evaluation
We used the Jensen–Shannon distance (JSD) [30], a metric that measures the similarity between two probability distributions. JSD values range from 0 to 1, where 0 indicates identical distributions, and 1 indicates completely dissimilar distributions. Smaller JSD values represent higher accuracy in estimating cell-type compositions.
Benchmarking different parameters
We extracted genes with significant expression differences among different cell types to select appropriate features for model training. To investigate the impact of varying gene numbers, we conducted benchmark tests. Similarly, we performed benchmark tests to explore the influence of different numbers of pseudo-spots on model training. We used spot-level JSD as an evaluation metric to assess the model performance across different attributes.
Benchmarking different deconvolution methods
We employed simulated ST data to compare the performance of GTAD against other ST deconvolution tools, including CellDART [19], STRIDE [22],DSTG [20], RCTD [21] and SPOTlight [17]. When running each of these published methods, we kept all parameters at their default settings as specified in their respective documentation. We evaluated these methods using the JSD metric described in the ‘Performance Evaluation’ section, comparing the average JSD values across all spots.
Sources of scRNA-seq and ST data
We conducted an analysis using ST datasets from various sources. For the mouse brain, we used the ‘Mouse Brain Sequencing Slices (Sagittal - Anterior)’ dataset from the 10X Genomics Data Repository and combined it with scRNA-seq data from the mouse primary visual cortex and anterior lateral motor cortex (GSE115746). This allowed us to gain insights into specific layer-specific excitatory neuron types. In the case of the mouse cerebellum, we utilized the Slide-seq V2 dataset from prior RCTD research along with accompanying scRNA-seq data covering 19 distinct cell types from the DropViz database. Our analysis also included human heart data from a previous study [31], and we performed preprocessing steps to convert ENSEMBL IDs to gene names for seamless dataset integration. Finally, for pancreatic cancer tissue analysis, we utilized data from the study GSE111672. These datasets were selected for their relevance and potential insights into various tissue types and disease conditions.
RESULTS
Assessing the impact of feature gene selection and pseudo-ST data quantity on GTAD deconvolution performance
In GTAD, selecting feature genes and determining the number of pseudo-ST data are critical steps influencing the model’s performance and resource consumption. We conducted benchmark tests using healthy mouse trachea data to explore their impact.
Feature Genes: We examined the influence of the number of differentially expressed genes (feature genes) between cell types on GTAD’s performance (Supplemental Figure S1A). Five quantities of feature genes (20, 30, 40, 50 and 60) were tested. Performance improved with more feature genes up to 30, likely because <30 genes couldn’t represent cell-type differences. However, exceeding 30 genes led to performance decline due to noise, disrupting the model and causing overfitting. Thus, the right number of feature genes is crucial.
Pseudo-ST Data: We investigated the effect of the number of pseudo-ST data (ranging from 2500 to 12 500) on GTAD’s performance (Supplemental Figure S1B). Performance improved up to 10 000 as more data provided better learning. Beyond 10 000, performance declined due to noise and redundancy, resulting in overfitting. Too much data introduced irrelevant information and complexity, affecting generalization to real ST data. Therefore, balancing pseudo-ST data quantity is essential for optimal performance.
In summary, selecting the right number of feature genes and pseudo-ST data is crucial for GTAD’s deconvolution performance. Excessive quantities can introduce noise and overfitting. Achieving the best results requires a careful balance in practical applications.
Evaluate algorithm performance in comparison with state-of-the-art methods
Our goal was to assess the performance of various methods under both normal and pathological conditions. We compared GTAD with other established cell-type deconvolution tools, including CellDART [19], STRIDE [19], DSTG [20], RCTD [21] and SPOTlight [17]. Across simulated datasets from healthy mouse trachea and human colorectal cancer, GTAD consistently outperformed other methods, demonstrating superior performance across various scenarios and pathological tissues (Figure 2A, B).
Figure 2.

Performance assessment of GTAD on benchmark datasets. (A) Assessment and comparative analysis of the performance of various methods on simulated spatial data generated from scRNA-seq data of human colorectal cancer. (B) Evaluation and comparison of the performance of different methods on simulated spatial data generated from scRNA-seq data of healthy mouse trachea. (C) Violin plots illustrating the comparative performance of various methods on the simulated dataset of human colorectal cancer. (D) Violin plots illustrating the comparative performance of different methods on the simulated dataset of healthy mouse trachea. In (A)–(D), the y-axis represents JSD (Jensen-Shannon Divergence) values. Wilcoxon Signed-Rank tests were conducted to compare JSD values between GTAD and other methods. Statistical significance (****P-value < 10–4) is indicated at the top of the violin plots in (C) – (D).
Significant performance variations were observed among methods, with better performance in the healthy mouse trachea dataset compared with the complex human colorectal cancer dataset [29]. However, GTAD consistently excelled in both datasets, achieving JSD values at least 2% lower than CellDART and STRIDE. Statistical analysis, using the Wilcoxon Rank-Sum test [32], confirmed GTAD’s superiority (Supplemental Table S1). The Wilcoxon rank-sum test, selected for its non-parametric nature and robustness, is employed to rigorously compare GTAD with other methods, ensuring that observed differences in performance are statistically significant. This choice allows for a valid assessment of GTAD’s superiority, particularly when dealing with complex and potentially non-normally distributed data, making it a suitable statistical tool for this comparative analysis. Violin plots illustrated GTAD’s accurate deconvolution of cell-type compositions, closely aligning with the ground truth (Figure 2C, D). These findings underscore GTAD’s robust performance in diverse datasets, reaffirming its ability to reconstruct cell-type compositions accurately.
In summary, our analysis of synthetic cell mixtures and comparative assessments validate GTAD as a superior method for deconvolving cell-type compositions across various biological systems and pathological tissues. These results offer valuable insights for spatial transcriptomic data analysis.
Decomposition of spatial cell distribution with GTAD in mouse brain data
To validate the capability of the GTAD method in elucidating the microanatomical structure of complex tissues, we conducted experiments using 10X Visium ST data from the mouse cerebral cortex. This cortex exhibits a well-defined cellular architecture, making it highly suitable for evaluating the performance of the GTAD method. According to the metadata of the scRNA-seq data, this tissue encompasses 28 distinct cell subtypes, each characterized by unique gene expression patterns.
Leveraging this scRNA-seq data, the GTAD method performed spatial deconvolution on the ST data, successfully reconstructing the structure of the cerebral cortex (Figure 3B). We represented the proportions of identified heterogeneous cells at each local spot using pie charts. The presence of these proportions in cortical regions confirmed the high predictive accuracy and sensitivity of the GTAD method. Additionally, the GTAD method predicted and spatially mapped layer-specific excitatory neuron scores for each spot. In mouse brain tissue, we observed seven excitatory neurons displaying spatially restricted patterns in specific cortical layers, aligning with the hierarchical structure of excitatory neurons reported in previous research [33] (Figure 3C). These results demonstrate the GTAD method’s ability to accurately reveal cell types and hierarchical structures in the cerebral cortex.
Figure 3.

Cell analysis of mouse cerebral cortex tissue using GTAD. (A) Slice of mouse brain tissue. (B) Spatial representation in pie chart format displays the predicted cell-type composition for each spot within the cortical layers. (C) Layer-specific arrangement of neurons as reported in previous studies. (D) Spatial mapping of 7 layer-specific excitatory neurons in the mouse cortex. Color mapping indicates the minimum and maximum values of the corresponding cell scores.
Furthermore, the cell compositions predicted by the GTAD method for each region provided more detailed information about their heterogeneity (Figure 3D). Specific investigations revealed that the enrichment of each cell type in an area correlated with its determined proportion. For instance, L2/3 subgroups displayed higher representation in the outer regions of the cortex, while spots with a higher proportion of L6b cells were predominantly located in the inner cortex layers. These findings align with the laminar cellular architecture of cortical tissue. The GTAD method’s capability to identify different spatial cell compositions within each cortical neuron layer further underscores its accuracy and sensitivity.
In summary, through the analysis of mouse cerebral cortex data, the GTAD method demonstrates feasibility and advantages in revealing the microanatomical structure of complex tissues. Our results emphasize the potential of GTAD when applied to brain tissue and other biological systems, providing valuable guidance and methodologies for ST research.
Application of GTAD on Slide-seq V2 mouse cerebellum data
To validate the applicability of the GTAD method to Slide-seq2 sequencing technology [11], we utilized mouse cerebellar slice data obtained through Slide-seq2 technology [34]. The mouse cerebellum exhibits a well-defined hierarchy of cell types, making it an ideal sample for assessing cell-type resolution performance. To further validate the reliability and applicability of the GTAD method, we collected publicly available snRNA-seq datasets as a reference and then used GTAD to map reference cell types to the spatial locations in the Slide-seq2 images (Figure 4A). This approach enabled us to gain a more accurate understanding of the distribution and spatial relationships of different cell types in the mouse cerebellum.
Figure 4.

Cellular analysis of mouse cerebellar tissue using GTAD. (A) Spatial distribution of all predicted cell types by GTAD. Each spot represents pixels captured by Slide-seq V2, with colors indicating different cell types. (B) Spatial distribution of cell types, including Granule cells, Oligodendrocytes, Purkinje cells and MLIs. The color mapping represents the minimum and maximum values of corresponding cell scores.
According to previous literature, the mouse cerebellar cortex is divided into three layers (Table 1). At the top is the molecular layer, in the middle is the Purkinje layer, and at the bottom is the Granular layer, which contains granule cells [35]. Beneath the cortex is the white matter region, rich in oligodendrocytes and astrocytes. The GTAD deconvolution results align with the literature description (Figure 4B), where the MLI1 and MLI2 molecular layer interneurons (MLIs) are mapped to the topmost and outermost layers of the cerebellar cortex. Bergmann and Purkinje cells co-locate in the same middle layer, the Purkinje layer, while granule cells are found in the sub-bottom Granule layer. Oligodendrocytes and astrocytes are distributed below the Granular layer, notably enriched in the innermost regions of the cerebellar slice. In summary, GTAD can accurately deconvolve cell types from ST data generated by Slide-seq and reconstruct the layered structure of the mouse cerebellum.
Table 1.
Anatomy and cellular organization of the cerebellar cortex
| Cerebellar cortex layers | Cell types |
|---|---|
| Molecular layer | MLI1 |
| MLI2 | |
| Purkinje cell dendritic arbors | |
| Purkinje layer | Purkinje cell bodies |
| Bergmann cells | |
| Granular layer | Granule cells |
| White matter | Oligodendrocytes |
| Astrocytes |
It is worth emphasizing that the GTAD method is not only applicable to common single-cell sequencing technologies like 10X Genomics but can also be successfully applied to other sequencing methods such as Slide-seq2. This feature expands the potential applications of GTAD in studying the spatial organization of various biological samples and cell types, further broadening the research scope and application of the GTAD method. By combining Slide-seq2 with GTAD, we have achieved satisfactory results in our study of the mouse cerebellum and provided powerful tools and methods for future spatial organization research in other biological systems.
Application of GTAD on developmental human heart dataset revealed spatial localization patterns of cell types
To validate the applicability of the GTAD method in different biological tissues, especially human organs, we further applied it to the study of organ development and spatial organization. A recent study [31] provided a comprehensive spatiotemporal map of human heart development using an integrated approach combining scRNA-seq, ST and in situ sequencing (ISS). We chose the developmental heart data from the latest 2022 dataset for deconvolution experiments to explore the effectiveness of the GTAD method in cardiac development research (Figure 5A).
Figure 5.
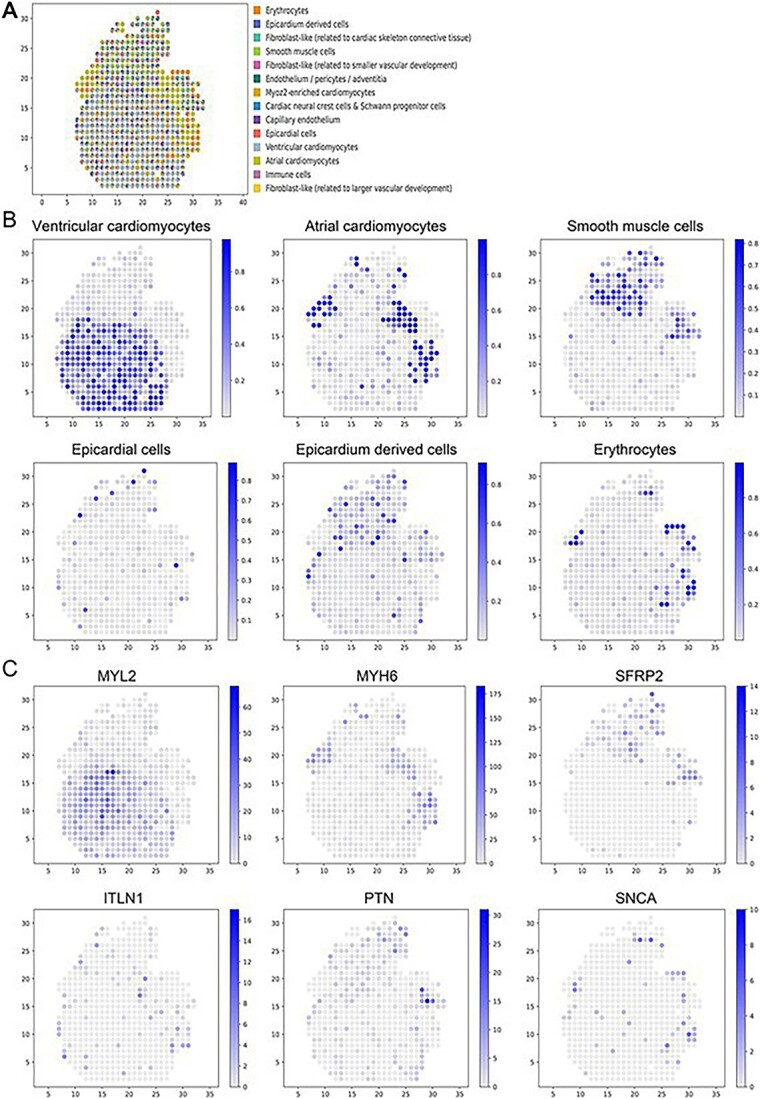
Application of GTAD in the developing human heart. (A) Spatial distribution of all predicted cell types by GTAD. Each spot represents an ST spot, and colors represent different cell types. (B) Spatial distribution of selected cell types, including ventricular cardiomyocytes, atrial cardiomyocytes, smooth muscle cells, epicardial cells, epicardium-derived cells and erythrocytes. (C) Spatial mapping of marker genes for each cell-type depicted in (B). Color mapping indicates the range of corresponding cell scores.
As anticipated, GTAD accurately predicted ventricular and atrial cardiomyocytes’ highest proportions within the ventricles and atria, respectively (Figure 5A). Epicardial cells were precisely localized to the outer heart layer, known as the epicardium. Remarkably, cardiac neural crest cells and Schwann progenitor cells, a rare cell type identified via scRNA-seq, occupied the interstitial region. Smooth muscle cells were predominantly mapped to the outflow tract, consistent with expectations [36] (Figure 5B).
Epicardial cells, forming the thin outer heart layer (epicardium), displayed significantly high distribution proportions covering the heart’s periphery, aligning with GTAD predictions. Cells derived from the epicardium formed a slightly thicker layer, also present within the outflow tract during epicardium development [37]. Our research findings successfully recapitulated these patterns, shedding light on critical processes in cardiac structural development. GTAD’s cell-type map closely aligned with the original study’s ISS cell map.
We examined the distribution of known cell type-specific marker genes [38] (Figure 5C). Top marker gene MYL2 for ventricular cardiomyocytes exhibited high expression in ventricular-dominant spots in the ST data, consistent with predicted proportions. Similarly, top markers MYH6 and Secreted Frizzled Related Protein 2 (SFRP2) for atrial cardiomyocytes and smooth muscle cells showed high expression at their respective locations. These genes had either low expression or went undetected at other spatial spots, reflecting the heterogeneous composition of cell types at specific ST spots.
In summary, through GTAD, we accurately reconstructed cell-type distributions and interactions in the spatial organization of heart development, confirming GTAD’s applicability in human organ research.
GTAD characterized the spatial heterogeneity of tumor cells in human PDAC microenvironment
To validate GTAD’s performance on cancer data, we applied it to human PDAC samples and utilized matched scRNA-seq data (inDrop) as reference data to determine the spatial location of different cell types within the tissue (Figure 6B). We further verified the accuracy of the prediction results through consistency with annotated regions in the original study [39] (Figure 6A), demonstrating the reliability of the GTAD method in revealing the spatial distribution of cell types.
Figure 6.
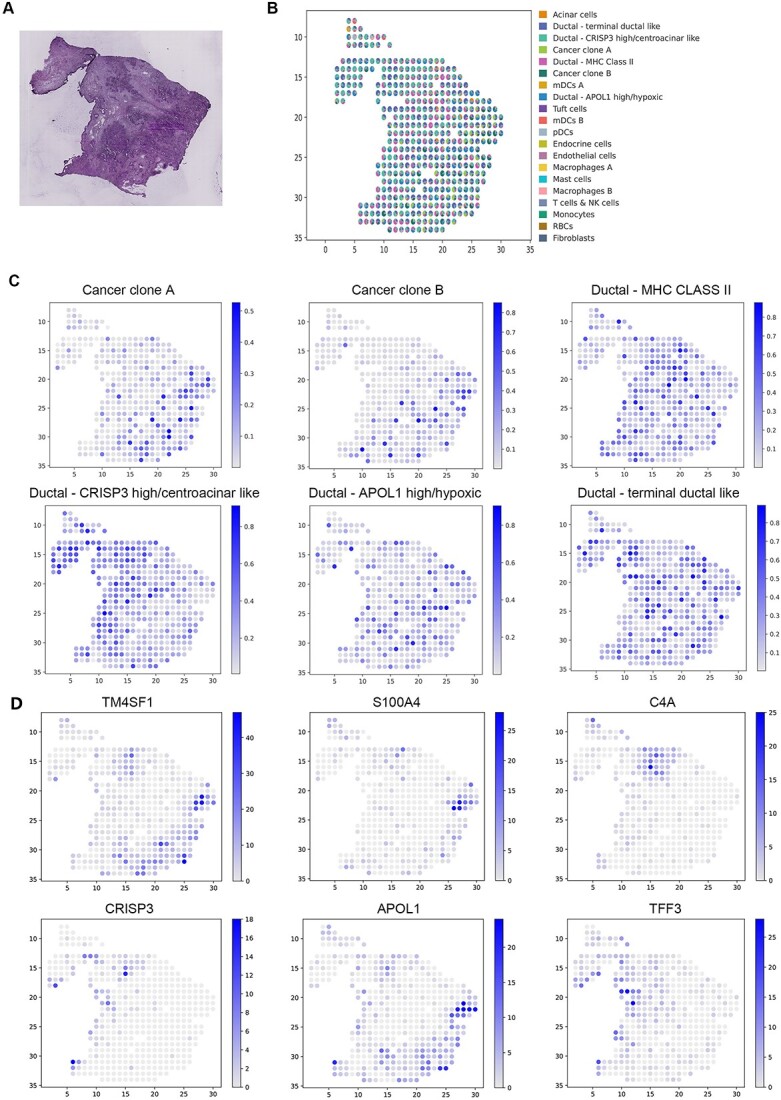
Analysis of the PDAC SRT data. (A) H&E staining image of PDAC. (B) Visualization of the deconvolution results. Spatial scatter plots depict the predicted cell-type composition by GTAD, with each spot representing a spot in the SRT data. (C) Visualization of the abundance of selected cell types at each spatial location. (D) Expression levels of marker genes for the corresponding cell types in (D).
In particular, we emphasized GTAD’s advantage in identifying the enrichment or depletion of cell type-specific genes at their respective spatial locations by examining the distribution of known marker genes for different cell types [38] (Figure 6C, D). We observed that in PDAC, cancer clone A and B cells were located in the cancerous regions, with cancer clone B cells primarily distributed in the top subregion, consistent with the spatial expression patterns of marker genes TM4SF1 and S100A4. Central ductal acinar cells were enriched in the ductal epithelium region, consistent with the expression pattern of the marker gene Cysteine-rich secretory protein 3 (CRISP3). Ductal centroacinar cells were mainly present in the ductal epithelium region and stromal region, in accordance with the expression pattern of the marker gene Apolipoprotein L1 (APOL1). Terminal ductal cells were predominantly found in the ductal epithelium region and stromal region, mirroring the expression pattern of the marker gene Trefoil Factor 3 (TFF3). Meanwhile, the expression levels of these genes in other spots were lower or undetectable.
Furthermore, when examining cell-type enrichment/depletion within annotated tissue regions, we observed distinct enrichment patterns between normal pancreatic and tumor cells in different areas [40]. Notably, our application of the GTAD method in endometrial carcinoma tissues yielded promising results, emphasizing its efficacy in diverse pathological contexts (Figure 7, [41, 42]). This finding further underscores the effectiveness and potential of GTAD in deconvoluting cancer or other pathological tissues. These results highlight the significant utility of GTAD in determining the localization and distribution of cell types in cancer or other pathological tissues, providing valuable insights and understanding for research into related diseases.
Figure 7.

Analysis of the human endometrial carcinoma data. (A) Visualization of the deconvolution results. Spatial scatter plots depict the predicted cell-type composition by GTAD, with each spot representing a spot in the SRT data. (B) Visualization of the abundance of selected cell types at each spatial location.
DISCUSSION
In contemporary biological research, the integration of multi-omics technologies provides a powerful tool for gaining comprehensive and in-depth biological insights. With advancements in technology, we can now simultaneously acquire diverse biological information, enabling the integration and analysis of multi-omics data [43, 44]. This comprehensive analysis not only aids in deciphering the spatial distribution map of cells within tissues but also reveals spatial interactions and regulatory mechanisms among different cell types, thus enhancing our understanding of the complexity and physiological functions of biological systems [45].
In this study, we present GTAD, an innovative method for integrating scRNA-seq and ST data to enhance our understanding of tissue cell composition. By capturing spatial relationships, GTAD improves cell-type identification, revealing intricate tissue microstructures. Benchmarking on two reference datasets demonstrates GTAD’s superior accuracy in inferring cell spatial composition compared with alternative methods. Further experiments showcase GTAD’s efficacy in uncovering spatially located cell states and subpopulations across diverse biological tissues, offering a valuable tool for exploring tissue microenvironments and cellular heterogeneity in complex systems.
However, we acknowledge some limitations in this study. Firstly, the complexity of tissues is often driven by factors such as cellular diversity, intricate microenvironments, and gene expression variability. This complexity can pose challenges to the performance of our model. Specifically, it can lead to increased noise and uncertainty in data analysis. For instance, even within the same cell types, gene expression can vary due to local environments or other factors, particularly in cancer tissues. These differences might be mistakenly interpreted as genuine biological variations, making it difficult to accurately identify cell types or predict their spatial distribution. To address this challenge, approaches like denoising autoencoders for data denoising and the application of pretrained models for preliminary categorization of cancer cells can be employed. These strategies are capable of handling larger and more diverse datasets effectively. Secondly, our method relies on feature gene selection from scRNA-seq data, which may require more precise filtering methods for rare or unknown cell types. Additionally, our method currently does not consider the potential correlations between the spatial positions of cells, which is an area for future improvement.
In summary, the GTAD method provides a novel and effective tool for deciphering the composition of cell types and spatial structures in complex tissues. By integrating scRNA-seq data with ST data and employing GATs for analysis, GTAD demonstrates significant advantages in studying cell heterogeneity and microstructures within tissues. Future efforts will focus on further refining the GTAD method to adapt to more complex and heterogeneous tissues, as well as enhancing the precision and efficiency of data analysis. We believe that the application of GTAD will contribute to the advancement of the field of ST and provide deeper insights into the complexity and functionality of biological tissues.
Key Points
We have developed an innovative model based on graph attention neural networks, known as GTAD. This model extensively leverages the topological relationships among the data with the precise goal of accurately deciphering the gene expressions at each ST data point and reconstructing their cellular components.
GTAD demonstrates remarkable accuracy and robustness, making it applicable across various experimental designs, ST platforms, and tissues originating from diverse organs.
GTAD is provided in an open-source format and can be directly applied for the precise analysis of tissue spatial structures and functions.
Supplementary Material
Acknowledgements
The authors thank the anonymous reviewers for their constructive suggestions.
Author Biographies
Tianjiao Zhang is an associate professor of College of Computer and Control Engineering at Northeast Forestry University of China. His research interests include bioinformatics.
Ziheng Zhang is a master candidate of College of Computer and Control Engineering at Northeast Forestry University of China. His research interests include bioinformatics.
Liangyu Li is a master candidate of College of Computer and Control Engineering at Northeast Forestry University of China. His research interests include bioinformatics.
Benzhi Dong is a professor of College of Computer and Control Engineering at Northeast Forestry University of China. His research interests include bioinformatics.
Guohua Wang is a professor of College of Computer and Control Engineering at Northeast Forestry University of China. He is also a principal investigator at Key Laboratory of Tree Genetics and Breeding, Northeast Forestry University. His research interests are bioinformatics, machine learning and algorithm.
Danan Zhang is an associated professor of Department of Gynaecology and Obstetrics in the First Affiliated Hospital of Harbin Medical University. Her research is bioinformatics of cancers.
Contributor Information
Tianjiao Zhang, College of Computer and Control Engineering, Northeast Forestry University, Harbin 150040, China.
Ziheng Zhang, College of Computer and Control Engineering, Northeast Forestry University, Harbin 150040, China.
Liangyu Li, College of Computer and Control Engineering, Northeast Forestry University, Harbin 150040, China.
Benzhi Dong, College of Computer and Control Engineering, Northeast Forestry University, Harbin 150040, China.
Guohua Wang, College of Computer and Control Engineering, Northeast Forestry University, Harbin 150040, China.
Dandan Zhang, Department of Obstetrics and Gynecology, the First Affiliated Hospital of Harbin Medical University, Harbin 150001, China.
Data availability
This study utilized eight publicly available datasets. The mouse trachea dataset was obtained from the Cell BLAST database (https://cblast.gao-lab.org/) with dataset IDs Montoro_10x and Plasschaert. Colorectal cancer datasets were sourced from GSE132465 and GSE144735, respectively. For the mouse brain dataset, the ST data can be downloaded from the 10X Genomics data repository (https://www.10xgenomics.com/cn/resources/datasets) with the sample_id ‘V1_Mouse_Brain_Sagittal_Anterior,’ while the scRNA-seq data were sourced from GSE115746. Mouse cerebellum ST data were obtained from a previous study, RCTD (https://singlecell.broadinstitute.org/single_cell/study/SCP948). Human developing heart data all originated from a prior study and can be accessed at (https://data.mendeley.com/datasets/mbvhhf8m62/2). The scRNA-seq and ST data for pancreatic ductal adenocarcinoma (PDAC) were sourced from the published dataset GSE111672, while the endometrial cancer data were derived from GSE203612.
The Python source code and R source code for GTAD have been uploaded to https://github.com/zzhjs/GTAD.
Funding
This work was supported by the National Natural Science Foundation of China [62172087, 62102121, 62272095, 62072095] and Heilongjiang Postdoctoral Fund [LBH-Q20138].
References
- 1. Tang FC, Barbacioru C, Wang YZ, et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat Methods 2009;6:377–U386. [DOI] [PubMed] [Google Scholar]
- 2. Macosko EZ, Basu A, Satija R, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 2015;161:1202–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klein AM, Mazutis L, Akartuna I, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 2015;161:1187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science 2016;353:78–82. [DOI] [PubMed] [Google Scholar]
- 5. Wang X, Allen WE, Wright MA, et al. Three-dimensional intact-tissue sequencing of single-cell transcriptional states. Science 2018;361:eaat5691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Eng C-HL, Lawson M, Zhu Q, et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019;568:235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rodriques SG, Stickels RR, Goeva A, et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 2019;363:1463–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Halpern KB, Shenhav R, Matcovitch-Natan O, et al. Single-cell spatial reconstruction reveals global division of labour in the mammalian liver. Nature 2017;542:352–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fawkner-Corbett D, Antanaviciute A, Parikh K, et al. Spatiotemporal analysis of human intestinal development at single-cell resolution. Cell 2021;184:810–26, e823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baccin C, Al-Sabah J, Velten L, et al. Combined single-cell and spatial transcriptomics reveal the molecular, cellular and spatial bone marrow niche organization. Nat Cell Biol 2020;22:38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stickels RR, Murray E, Kumar P, et al. Highly sensitive spatial transcriptomics at near-cellular resolution with Slide-seqV2. Nat Biotechnol 2021;39:313–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cho C-S, Xi J, Si Y, et al. Microscopic examination of spatial transcriptome using Seq-Scope. Cell 2021;184:3559–72, e3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berglund E, Maaskola J, Schultz N, et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018;9:2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thrane K, Eriksson H, Maaskola J, et al. Spatially resolved transcriptomics enables dissection of genetic heterogeneity in stage III cutaneous malignant melanoma. Cancer Res 2018;78:5970–9. [DOI] [PubMed] [Google Scholar]
- 15. Chen W-T, Lu A, Craessaerts K, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell 2020;182:976–91, e919. [DOI] [PubMed] [Google Scholar]
- 16. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol 2018;15:81–94. [DOI] [PubMed] [Google Scholar]
- 17. Elosua-Bayes M, Nieto P, Mereu E, et al. SPOTlight: seeded NMF regression to deconvolute spatial transcriptomics spots with single-cell transcriptomes. Nucleic Acids Res 2021;49:e50–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong R, Yuan G-C. SpatialDWLS: accurate deconvolution of spatial transcriptomic data. Genome Biol 2021;22:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bae S, Na KJ, Koh J, et al. CellDART: cell type inference by domain adaptation of single-cell and spatial transcriptomic data. Nucleic Acids Res 2022;50:e57–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Song Q, Su J. DSTG: deconvoluting spatial transcriptomics data through graph-based artificial intelligence. Brief Bioinform 2021;22:bbaa414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cable DM, Murray E, Zou LS, et al. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat Biotechnol 2022;40:517–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun D, Liu Z, Li T, et al. STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing. Nucleic Acids Res 2022;50:e42–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Goh WWB, Wang W, Wong L. Why batch effects matter in omics data, and how to avoid them. Trends Biotechnol 2017;35:498–507. [DOI] [PubMed] [Google Scholar]
- 24. Velickovic P, Cucurull G, Casanova A, et al. Graph attention networks. Stat 2017;1050:10–48550. [Google Scholar]
- 25. Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902, e1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alshammari M, Stavrakakis J, Ahmed AF, et al. Random projection forest initialization for graph convolutional networks. MethodsX 2023;11:102315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abadi M. TensorFlow: Learning Functions at Scale. Acm Sigplan Notices 2016;51:1–1. [Google Scholar]
- 28. Kipf TN, Welling M. Semi-Supervised Classification with Graph Convolutional Networks. In: International Conference on Learning Representations. 2016.
- 29. Lee H-O, Hong Y, Etlioglu HE, et al. Lineage-dependent gene expression programs influence the immune landscape of colorectal cancer. Nat Genet 2020;52:594–603. [DOI] [PubMed] [Google Scholar]
- 30. Fuglede B, Topsoe F. Jensen-Shannon divergence and Hilbert space embedding. In: 2004 IEEE International Symposium on Information Theory (proceedings). 2004.
- 31. Asp M, Giacomello S, Larsson L, et al. A spatiotemporal organ-wide gene expression and cell atlas of the developing human heart. Cell 2019;179:1647–60, e1619. [DOI] [PubMed] [Google Scholar]
- 32. Wilcoxon F, Katti S, Wilcox RA. Critical values and probability levels for the Wilcoxon rank sum test and the Wilcoxon signed rank test. Sel Tables Math Stat 1970;1:171–259. [Google Scholar]
- 33. Tasic B, Yao Z, Graybuck LT, et al. Shared and distinct transcriptomic cell types across neocortical areas. Nature 2018;563:72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown AM, Arancillo M, Lin T, et al. Molecular layer interneurons shape the spike activity of cerebellar Purkinje cells. Sci Rep 2019;9:1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kozareva V, Martin C, Osorno T, et al. A transcriptomic atlas of mouse cerebellar cortex comprehensively defines cell types. Nature 2021;598:214–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sawada H, Rateri DL, Moorleghen JJ, et al. Smooth muscle cells derived from second heart field and cardiac neural crest reside in spatially distinct domains in the media of the ascending aorta—brief report. Arterioscler Thromb Vasc Biol 2017;37:1722–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eralp I, Lie-Venema H, Bax NA, et al. Epicardium-derived cells are important for correct development of the Purkinje fibers in the avian heart. Anat Rec A Discov in Mol Cell Evol Biol 2006;288:1272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Codeluppi S, Borm LE, Zeisel A, et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat Methods 2018;15:932–5. [DOI] [PubMed] [Google Scholar]
- 39. Moncada R, Barkley D, Wagner F, et al. Integrating microarray-based spatial transcriptomics and single-cell RNA-seq reveals tissue architecture in pancreatic ductal adenocarcinomas. Nat Biotechnol 2020;38:333–42. [DOI] [PubMed] [Google Scholar]
- 40. Fan X, Lu P, Wang H, et al. Integrated single-cell multiomics analysis reveals novel candidate markers for prognosis in human pancreatic ductal adenocarcinoma. Cell Discov 2022;8:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yu X, Xie L, Ge J, et al. Integrating single-cell RNA-seq and spatial transcriptomics reveals MDK-NCL dependent immunosuppressive environment in endometrial carcinoma. Front Immunol 2023;14:1145300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barkley D, Moncada R, Pour M, et al. Cancer cell states recur across tumor types and form specific interactions with the tumor microenvironment. Nat Genet 2022;54:1192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang T, Li L, Sun H, et al. DeepITEH: a deep learning framework for identifying tissue-specific eRNAs from the human genome. Bioinformatics 2023;39:btad375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang T, Li L, Sun H, et al. DeepICSH: a complex deep learning framework for identifying cell-specific silencers and their strength from the human genome. Brief Bioinform 2023;24:bbad316. [DOI] [PubMed] [Google Scholar]
- 45. Longo SK, Guo MG, Ji AL, et al. Integrating single-cell and spatial transcriptomics to elucidate intercellular tissue dynamics. Nat Rev Genet 2021;22:627–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study utilized eight publicly available datasets. The mouse trachea dataset was obtained from the Cell BLAST database (https://cblast.gao-lab.org/) with dataset IDs Montoro_10x and Plasschaert. Colorectal cancer datasets were sourced from GSE132465 and GSE144735, respectively. For the mouse brain dataset, the ST data can be downloaded from the 10X Genomics data repository (https://www.10xgenomics.com/cn/resources/datasets) with the sample_id ‘V1_Mouse_Brain_Sagittal_Anterior,’ while the scRNA-seq data were sourced from GSE115746. Mouse cerebellum ST data were obtained from a previous study, RCTD (https://singlecell.broadinstitute.org/single_cell/study/SCP948). Human developing heart data all originated from a prior study and can be accessed at (https://data.mendeley.com/datasets/mbvhhf8m62/2). The scRNA-seq and ST data for pancreatic ductal adenocarcinoma (PDAC) were sourced from the published dataset GSE111672, while the endometrial cancer data were derived from GSE203612.
The Python source code and R source code for GTAD have been uploaded to https://github.com/zzhjs/GTAD.