

Abstract
Clathrin assembles into a dynamic two-dimensional lattice on the plasma membrane where it plays a critical role in endocytosis. To probe the regulation of this process, we used siRNA against clathrin, in combination with single cell assays for transferrin uptake as well as total internal reflection microscopy, to examine how endocytic rates and membrane dynamics depend upon cellular clathrin concentration ([Clathrin]). We find that endocytosis is tightly controlled by [Clathrin] over a very narrow dynamic range such that small changes in [Clathrin] can lead to large changes in endocytic rates, indicative of a highly cooperative process (apparent Hill coefficient, n > 6). The number of clathrin assemblies at the cell surface was invariant over a wide range of [Clathrin]; however, both the amount of clathrin in each assembly and the subsequent membrane dynamics were steeply dependent on [Clathrin]. Thus clathrin controls the structural dynamics of membrane internalization via a strongly cooperative process. We used this analysis to show that one important regulator of endocytosis, the actin cytoskeleton, acts noncompetitively as a modulator of clathrin function.
INTRODUCTION
Clathrin-mediated endocytosis is one of the most studied internalization routes for membrane proteins and extracellular ligands, yet the details of its regulation are poorly understood (Brodsky et al., 2001). A recent proteomic analysis of clathrin-coated vesicles in neurons indicates that up to 200 different gene products are associated with this pathway (Blondeau et al., 2004). As with the entire genome it is likely that a large fraction of these constituents are devoted to regulation of clathrin function. Thus, appropriate methods to study how endocytosis is regulated will be necessary to decipher the functions of the various components.
Prior studies addressing clathrin's role in membrane traffic primarily focused on the complete intracellular depletion or functional inactivation of clathrin. Such studies utilized environmental perturbations (Larkin et al., 1983; Heuser, 1989), genetic ablation (Payne and Schekman, 1985; Lemmon and Jones, 1987; Ruscetti et al., 1994; Wettey et al., 2002), targeted repression of expression (Hinrichsen et al., 2003; Motley et al., 2003; Huang et al., 2004), and mutational analysis of clathrin-binding proteins (Damke et al., 1994; Benmerah et al., 1998; Nesterov et al., 1999). The majority of the information gleaned from prior studies on clathrin function was obtained with either unperturbed clathrin function or in the complete absence of clathrin. Thus, these studies largely illustrated which cellular pathways and processes involve clathrin, but did not provide much information about their regulation.
To better understand the regulatory control points of endocytosis, we developed a novel approach to vary the intracellular concentration of clathrin within living cells. This method allows the examination of how endocytosis depends on the total cellular concentration of the main component itself, clathrin ([Clathrin]). The clathrin coat that forms on the plasma membrane around sites of internalization is comprised of a repeating two-dimensional array of clathrin triskelia that are in constant exchange with the cytoplasm (Wu et al., 2001). The clathrin triskelion is composed of three clathrin heavy chains joined at their C-termini and three clathrin light chains, each of which is attached to a single heavy chain (Kirchhausen, 2000). The intricate arrangement of the clathrin triskelia is thought to impose important restrictions on self-assembly (Wakeham et al., 2003). In addition, clathrin assembly should be highly sensitive to the free concentration of triskelia as assembly is likely to be very cooperative. Accordingly, we have attempted to determine how variations in intracellular clathrin concentration impact clathrin-mediated endocytosis.
To examine how endocytosis depends on [Clathrin], we used siRNA duplexes targeted against clathrin heavy chain. We utilized transfection conditions that generated a wide-spectrum of clathrin depletion within the population of cells. Our analysis of clathrin function involved a variety of single cell assays, including quantification of endocytic function and budding frequency. Our results indicate that membrane internalization involving clathrin is a highly cooperative process. In addition, this novel method to study clathrin function can be adapted to probe the specific role that other proteins serve in modulating the efficacy of this pathway.
MATERIALS AND METHODS
Design of siRNA Duplex
Using standard siRNA design techniques, a suitable region of the clathrin heavy-chain molecule was chosen to serve as a target for siRNA-mediated reduction of clathrin molecules. The target sequence is as follows: 5′-AACAUUGGCUUCAGUACCCUG-3′. The siRNA duplex was ordered from Dharmacon RNA Technologies (Dallas, TX) using processing option B.
Antibodies
Clathrin heavy chain was detected using the monoclonal antibodies X22 (Affinity BioReagents, Golden, CO) and TD.1 (Santa Cruz Biotechnology, Santa Cruz, CA). AP2 molecules were detected using the monoclonal antibody (mAb) AP6 that detects the α-adaptin subunit (Affinity BioReagents, Golden, CO). Dynamin was detected using the mAb Hudy-1 (Upstate Cell Signaling Solutions, Lake Placid, NY).
Transfection
siRNAs were transfected by the Oligofectamine or Lipofectamine 2000 method (Life Technologies, Rockville, MD). Cells were analyzed 72 h after transfection. Control cells were treated with the same reagents as the CHC siRNA-treated cells throughout the experiments, with the exception that no siRNA was added to the transfection media.
Cell Culture
Chinese hamster ovary (CHO) cells used in this report are derived from TRVb CHO cells that do not express any functional transferrin receptor (McGraw et al., 1987). TRVb-EFL cells stably express human transferrin receptor and EGFP-FKBP-LC and were cultured in McCoy's 5A medium containing 5% fetal bovine serum, penicillin-streptomycin (Life Technologies), 26 mM sodium bicarbonate, 0.2 mg/ml G418 (Life Technologies) and 400–500 U/ml hygromycin (Calbiochem-Novabiochem Corporation, San Diego, CA).
Immunolocalization of Antibodies
Cells were grown on coverslip bottom dishes and fixed with 3.7% formaldehyde for 10 min. Dishes were stained with primary antibody in 250 μg/ml saponin in phosphate-buffered saline (PBS) with 1% bovine serum albumin (permeabilization buffer) for 1 h at room temperature. Cells were washed in permeabilization buffer for 15 min (three 5-min washes). Cells were subsequently stained with a goat anti-mouse secondary antibody conjugated to Alexa-546 (Molecular Probes, Eugene, OR) at a 1:2000 dilution for 1 h, washed for 15 min (three 5-min washes), and examined by fluorescence microscopy.
Western Blot Analysis
Cells treated with or without CHC siRNA were plated in parallel cultures for biochemical extraction or fluorescence microscopy. At 72 h post-transfection, cells were fixed for imaging or harvested by trypsin detachment and extensive washing in PBS. Cell pellets were resuspended in 20 mM Tris-HCl (pH 7.6) and 150 mM NaCl (tris-buffered saline) supplemented with 1% Triton X-100 and protease inhibitors and quantitated with a modified Lowry assay (DC; Bio-Rad, Hercules, CA). Proteins were resolved on an 8% Laemmli gel and transferred to PVDF membrane (Hybond P; Amersham Biosciences, Piscataway, NJ) for immunoblotting using standard procedures. Primary antibody:antigen complexes were bound with goat anti-mouse IgG-conjugated HRP (Pierce, Rockford, IL) and subjected to chemiluminescent detection on autoradiographic film. Relative quantitation of specific bands was performed by scanning the film and analytical processing in Photoshop 6.0 (Adobe Systems, San Jose, CA) and Origin 7.0 (Microcal, Northampton, MA). Pixel densities were in the linear range of the film and relative knockdown was calibrated against dilutions of the control sample. Sum pixels for 50 μgof total protein: CHC control (1940), CHC knockdown (1283), CLC control (2714), and CLC knockdown (1430).
Transferrin Internalization and Measurements of Total Clathrin Concentration in Each Cell
Cells were incubated with 3 μg/ml Cy3 transferrin (Tf) in McCoy's 5A medium supplemented with 26 mM sodium bicarbonate, 20 mM HEPES, pH 7.2 (McBB) for 0, 2, 4, 6, 8, or 10 min. After incubation at 37°C the plates were placed on ice and washed twice with medium 2 (150 mM NaCl, 20 mM HEPES, 1 mM CaCl2, 5 mM KCl, 1 mM MgCl2, pH 7.2) at 4°C. The cells were incubated for 5 min at 4°C in 200 mM NaCl, 50 mM MES, pH 5.0, and washed with medium 2, which effectively strips greater than 95% of the any surface bound transferrin (unpublished data). After this wash, the cells were incubated with 3 μg/ml Cy5-Tf in medium 2 at 4°C for 2 h. After the Cy5-Tf incubation, the cells were washed six times with medium 2 at 4°C. The cells were subsequently fixed, imaged in three channels (GFP, Cy-3, and Cy-5) using wide-field microscopy and subjected to analysis using MetaMorph (Universal Imaging, Downingtown, PA). Briefly, cell outlines were created around the cells, and the fluorescence intensity and area of the cell were recorded. The ratio of fluorescence intensity per total cell area (I/A) was calculated for all cells in all conditions. The average value for I/A from all of the cells in the 0-min time point serves as the marker for background fluorescence and was subtracted from all other I/A values at the other time points. For GFP-clathrin, the average background was determined for identical imaging and fixation conditions from non-GFP–transfected cells to determine the non-GFP–associated background to be used for subtraction. The average value of I/A for the GFP channel after background correction was used as the measure of [Clathrin] for each cell.
Nonspecific Membrane Uptake
Cells were washed with warmed medium 2 and placed at 37°C for 5 min. Cells were incubated with 15 μM FM4-64 (Molecular Probes) in warmed medium 2 for 0, 2, 4, 6, or 8 min at 37°C. Immediately after the addition of FM4-64 to the 0-min time point, the plates were placed on ice and washed twice with medium 2 at 4°C. The cells were incubated with 2 mg/ml β-cyclodextrin sulfobutyl ether, 7 sodium salt (Advasep 7; CyDex, Overland Park, KS) in medium 2 for 2 min to remove noninternalized, membrane-associated FM4-64. This step was repeated three times. The cells were subsequently analyzed using MetaMorph to quantitate the internalized FM4-64 levels as described for Tf internalization (above).
Optical Measurements, Microscopy, and Analysis
Cells were viewed with an inverted microscope (IX-70, Olympus America, Melville, NY) through a 1.45 NA objective (60×, Olympus). Fluorophores were excited using a 100 W high-pressure mercury lamp (Olympus America) and standard filter cubes to specifically obtain the fluorescence signals from each of the fluorophores used during the course of this study. Images were projected onto a CCD camera (Roper Scientific, Trenton, NY). Cells were imaged using MetaMorph (Universal Imaging). Quantitative data were fit to Eq. 1 using a nonlinear least squares fitting procedure in Origin 7.0 (Origin Lab, Northampton, NY).
Total Internal Reflection Fluorescence Microscopy
Cells were plated on glass coverslips and viewed with an inverted microscope (IX-70, Olympus America) through a 1.45 NA objective (60×, Olympus) equipped with a custom-built objective heater used to maintain the specimen at 37°C. An argon ion laser was used to excite EGFP in total internal reflection mode, by steering the beam to the outer-edge of the microscope objective back-aperture using an Olympus TIR illuminator. The angle of illumination was adjusted to provide an evanescent field depth of ∼150 nm. Images were projected onto a CCD camera (Roper Scientific). Cells were imaged using MetaMorph (Universal Imaging).
TRVb-EFL cells were grown on coverslips and were imaged 72 h after transfection. The microscope was focused on a region of 6–20 cells. Images were taken every 10 s for 3 min. Images were subdivided into a 16-region grid (2 × 2 μm/region) and chosen to avoid direct apposition to the Golgi apparatus. These regions were inspected manually for the appearance or disappearance of spots from the field of view. Two types of activity with respect to these fluorescent spots were described: 1) Stationary: visualized throughout the duration of image collection even if they drifted to another region in the field of view; 2) Membrane budding: the sum of events that were initially in the field of view, but disappeared at a later frame and events that were not in the field at the beginning of the image collection, appeared at some point, and then disappeared before the end of the image collection.
Puncta Analysis
The fluorescence intensity of individual puncta was measured over a 16-pixel (0.125 × 0.125 μm) area, which is slightly smaller than the diffraction limited resolution in this system. Thus this measurement reflects the density of GFP per punctum and not the total size of the lattice.
RESULTS
Targeted Reduction in Clathrin Heavy and Light Chains
To examine how endocytosis depends upon [Clathrin], we used stably transfected fibroblasts in which clathrin light-chain expression has been completely replaced with that of an EGFP-labeled variant (Moskowitz et al., 2003). We designed siRNA duplexes targeted against clathrin heavy chain and utilized transfection conditions in which we could not only transfect a large percentage of the cell population, but also generate a wide-spectrum of clathrin concentrations within the population of cells (Figure 1A). Alterations in clathrin light-chain expression should mirror any changes in clathrin heavy-chain abundance, due to the rapid destruction of any clathrin light chain molecules not bound to a clathrin heavy chain (Brodsky, 1985). We confirmed the siRNA-mediated reduction in clathrin heavy chain via retrospective immunofluorescence against clathrin heavy chain (Figure 1B) as well as Western blot analysis (see below). Our experiments revealed that the fluorescence derived from our EGFP-labeled clathrin light chain provides a robust and linear readout of clathrin content (Figure 1C). Clathrin light chain expression shows a one-to-one correspondence with clathrin heavy-chain expression.
Figure 1.

Levels of clathrin heavy and light chains are coordinately decreased due to siRNA-mediated decreases in clathrin heavy chain. (A and B) TRVb-EFL cells transiently transfected with CHC siRNA (n = 341) exhibited an expanded range of clathrin levels compared with cells under control conditions (n = 187). Levels of clathrin light chain (A) and clathrin heavy chain (B) were decreased as a result of CHC siRNA transfection. Clathrin light chain levels were determined via the stably expressed EGFP-clathrin light chain fusion protein and clathrin heavy-chain levels were determined via immunostaining against clathrin heavy chain. (C) The concentration of the fluorescence associated with clathrin heavy chain and clathrin light chain scale directly. The data points represent the averaged fluorescence values over several cells ± SEM. Scale bar, 10 μm.
These data indicate that the use of siRNA can effectively expand the range of clathrin concentrations across a population of cells by extending the range to lower, barely detectable values. Under control conditions, the total EGFP fluorescence derived from clathrin light chain typically varies only ∼3–4-fold over the population of cells with a roughly normal distribution (Figure 2A). The variation in the control condition indicates that cells in this population can vary their intracellular clathrin concentration. After siRNA treatment, the range of EGFP values expands to ∼65-fold, with the appearance of a bimodal distribution. Depletion of cellular clathrin leads to a new population of expression with a mean value ∼400% lower than the mean of the control population (Figure 2A). We confirmed that EGFP fluorescence provides a robust measure of both the total light and heavy chain clathrin as measured by Western blot analysis of whole cell lysates (Figure 2B) prepared from cells treated in parallel to those used to derive the frequency distribution of EGFP-clathrin after siRNA treatment (shown in Figure 2A). The correspondence between the fractional change induced by siRNA treatment in total protein levels for both heavy- and light-chain clathrin, and the mean intensity of EGFP-clathrin over the whole population was excellent. The observed decrease in protein levels of 34% for clathrin heavy chain and 47% for clathrin light chain from control was in agreement with the 39% reduction in mean EGFP-FKBP-LC fluorescence in CHC siRNA-treated cells. The variable reduction in clathrin levels, combined with single-cell assays of endocytic function and the use of EGFP-clathrin light chain to provide a robust measure of [Clathrin], permits the study of the concentration-dependent effects of clathrin in living cells.
Figure 2.

Specific reduction in clathrin expression with siRNA that targets clathrin heavy chain (CHC). (A) Histogram of the expression levels of clathrin per unit area in control cells (□) or cells transfected with CHC siRNA (▪; n = 3916 and 3052 for CHC siRNA and control, respectively). The range of expression levels for CHC siRNA cells overlaps with the control cells. However, there is also a significant expansion in the range of clathrin expression to lower cellular clathrin levels in CHC siRNA cells. (B) CHC siRNA transfection results in reduced levels of clathrin heavy and light chain protein in accordance with parallel fluorescence measurements. Total protein (μg) from TRVb-EFL cells (Control) or same transfected with CHC siRNA (RNAi) was examined for relative clathrin levels by Western blotting. EGFP-FKBP-LC, and CHC migrate at relative molecular masses of ∼70 and ∼170 kDa, respectively (arrows) with molecular-mass markers indicated on the left of the blot in kDa. The observed decrease in protein levels of 34% for CHC and 47% for CLC from control was in agreement with the 39% reduction in mean EGFP-FKBP-LC fluorescence in CHC siRNA-treated cells. (C) Cells transfected with CHC siRNA (n = 316) or control media (n = 223) were immunostained against dynamin. Histogram of dynamin intensities in this population of cells indicates that the average value of dynamin in CHC siRNA cells is only reduced 38% relative to the untransfected cells. (D) Cells transfected with or without CHC siRNA (n = 328 and 185, respectively) were immunostained against the AP-2 complex. The level of clathrin light chain and AP-2 expression per unit area for each cell was determined. The histogram illustrates the increase in the average AP-2 values for cells treated with CHC siRNA compared with control cells. (E) The total number of AP-2 puncta within regions of CHC siRNA or control cells was counted (n = 20 regions from separate cells for each condition). CHC siRNA cells exhibited a slightly increased number of AP-2 puncta compared with control cells. (F) Histogram of the fluorescence intensity of the AP-2 puncta selected for analysis in E.
To verify the targeted destruction of only clathrin without commensurate changes in associated endocytic proteins, we examined the intensity distribution of both dynamin as well as the adaptor protein α-adaptin, part of the AP-2 complex, after clathrin heavy-chain siRNA treatment. These data indicate that siRNA treatment against clathrin led to a modest decrease in the total amount of dynamin per cell (Figure 2C). The lack of increase in cellular dynamin in response to clathrin depletion is in contrast to a recently published report in another cell line using an alternative approach to reduce the cellular abundance of clathrin (Iversen et al., 2003). A similar analysis indicated that the average AP-2 immunostaining per cell, as well as the total number of AP-2 assemblies showed a modest increase. However, we did not detect a change in the average amount of AP-2 per clathrin assembly (Figure 2, D–F), and thus the increase does not appear to be due simply to an unmasking of new binding sites at clathrin assembly sites. Thus, the major impact of clathrin siRNA is the direct reduction of clathrin itself, with only small changes in other major endocytic proteins.
[Clathrin] Regulates Transferrin Internalization
Clathrin-mediated endocytosis is the principal mode of entry for several cell surface proteins. In particular, the internalization of transferrin receptor is prototypically clathrin-dependent. Therefore, we measured the rate constant for endocytosis (kendo) of transferrin receptor in individual cells using a combination of Cy3- and Cy5-labeled transferrin to measure both internalization and total surface concentration of transferrin receptors, respectively. The rate constant kendo is defined as the fraction of surface receptors internalized per unit time, measured over sufficiently short time periods such that little or no recycling of the receptor back to the plasma membrane occurs within the time examined (2–10 min; Mayor et al., 1993). We verified that transferrin uptake varied linearly with time for the entire range of time points that we examined (our unpublished results). In addition to measuring the value of kendo for each cell, we also measured the total cellular EGFP fluorescence per unit area to quantify [Clathrin] and examined the correlation between kendo and [Clathrin] on a cell-by-cell basis (Figure 3A).
Figure 3.

Highly cooperative control of transferrin internalization. (A) Cells transiently transfected with CHC siRNA or control conditions were analyzed for transferrin internalization (Cy3-labeled transferrin) and steady state surface transferrin levels (Cy5-labeled transferrin). The intensity of the total cellular EGFP, Cy3-Tf uptake, and surface Cy5-Tf per total cell area was ascertained for each cell. (B) The ratio of Cy3-Tf to Cy5-Tf was calculated for each cell. The scatterplot shows the distribution of a data subset from several experiments (n = 1700 cells). The data for each experiment was normalized to the maximal Cy3-Tf/Cy5-Tf ratio of the population. (C) Cells from many experiments were grouped and ordered according to clathrin concentration (n = 3916 and 3052 for CHC siRNA and control, respectively). Each graphed point represents the averaged values from 100 cells ± SEM. The data are fit to the simplified Hill equation (Eq. 1) with an apparent Hill coefficient n = 6.25.
When a large sample size of kendo values from individual cells is analyzed via scatterplot there is a general correlation with increasing uptake in cells that express relatively more clathrin (Figure 3B). Notably, the density in the scatter plot indicates two densely populated regions of kendo values that suggests a bistable relationship between kendo and [Clathrin]. When these data sets are binned according to [Clathrin] values and averaged, a clearer view of the aggregate ensemble appears and three main characteristics become evident (Figure 3C). First, over the majority of clathrin concentrations found in control cells, the rate constant of internalization remains constant. Similarly, as evidenced by the siRNA-treated cells, above a critical level of clathrin the rate constant of internalization reaches a plateau. Second, below the threshold of the plateau level of internalization, the concentration range of [Clathrin] that has been expanded as a result of siRNA targeting clathrin heavy chain, the rate constant of internalization decreases rapidly. Finally, extrapolation of the lowest values of clathrin to zero intracellular clathrin indicates that the rate constant for non–clathrin-mediated endocytosis (knc) of transferrin is ∼18% of the maximal rate constant for endocytosis. This figure agrees very well with a previous estimate of the reliance of transferrin uptake on a non-clathrin-dependent pathway (Moskowitz et al., 2003).
The steep dependence of endocytosis over a relatively narrow concentration range of [Clathrin] is indicative of an underlying cooperative process and the data are well fit with a simplified Hill equation:
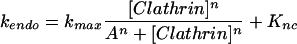 |
(1) |
where kmax is the rate constant at saturating [Clathrin], A is a constant, and n is the apparent Hill coefficient. The data here are best fit with a value of n = 6.25, indicating that the molecular mechanism by which clathrin participates in endocytosis is highly cooperative. The onset of the steep dependence (roughly given by the parameter A) occurs roughly at [Clathrin] = 266 arbitrary fluorescence units (afu). By contrast, we examined the internalization profile of FM4-64, an amphipathic dye whose quantum yield increases greatly upon insertion in a membrane. FM4-64 is not specifically recruited to areas of the membrane, and therefore the majority of its uptake is likely due to general membrane uptake. Accordingly, when the uptake profiles of cells incubated with FM4-64 for a variety of time points are examined, only a very weak dependence on [Clathrin] is observed (see Supplementary Figure).
Reductions in [Clathrin] Alter the Intensity, but Not the Number of Clathrin Puncta
We next performed analyses aimed at determining the possible source of the cooperativity in clathrin function. Because clathrin triskelia assembled at the plasma membrane are in equilibrium with the cytoplasmic pool (Wu et al., 2001; Moskowitz et al., 2003), we expect that lowering the total [Clathrin] would potentially diminish the number of assembled lattices and/or decrease the number of triskelia per lattice. We measured the total number of detectable clathrin puncta per unit area for both control and clathrin siRNA cells over a fixed area size at the dorsal surface of the cell to give an approximate measure of the density of clathrin assemblies and correlated this with the total EGFP-light chain fluorescence in each cell. These data are shown in Figure 4A and indicate that the density of clathrin puncta is largely invariant over the majority of the concentration range of [Clathrin]. In particular, the density of clathrin puncta does not change significantly over values of [Clathrin] for which kendo is changing rapidly. Furthermore, the density of clathrin puncta is not altered in the control cells across the approximately threefold variation in normal [Clathrin]. Accordingly, variations in the number of clathrin puncta are not the source of the high cooperativity described in Figure 3.
Figure 4.

Highly cooperative control of lattice intensity. (A) The total number of puncta within regions of cells transiently transfected with CHC siRNA or control conditions was counted. The number of puncta remained constant over the majority of the concentration range, decreasing only at very low clathrin concentrations in CHC siRNA cells. (B) The intensity of the fluorescence associated with each punctum selected in A was obtained for control and CHC siRNA-treated cells. These data were fit to Eq. 1 yielding the values n = 3.3, A = 313, and an offset of 26 U to show the nonlinear relationship between [clathrin] and the punctum intensity. The inset shows the same data on a log-log plot overlayed with a slope of n = 3.3. The graphed points in A and B represent the averaged values from several cells (n = 112 and 40 for CHC siRNA and control, respectively) ± SEM. (C) The left panel shows an image of clathrin spots taken from a cell whose clathrin was depleted significantly and resulted in very dim clathrin assemblies. The two circled assemblies (pink circle 46 afu, blue circle 24 afu) are each clearly visible above background (scale bar, 1 μm). The right panel shows the relative intensity distribution for all clathrin assemblies (in red) measured in cells whose [Clathrin] was severely depleted (ranging from [Clathrin] = 66 to [Clathrin] = 126 afu) compared with background areas (in green) in the same cells. The arrows indicate the position of the two circled clathrin assemblies shown in the left panel in this histogram.
We next we examined whether the average fluorescence intensity of each punctum varies as a function of the total EGFP-light chain fluorescence per cell ([Clathrin], Figure 4B). The fluorescence intensity should give a measure of the number of triskelia assembled into each punctum. Interestingly, the cells not incubated with the siRNA duplexes maintain a largely invariant range of clathrin intensity per punctum. Thus, over the approximately threefold variation in clathrin expression found normally in the population of cells, the cells maintain a consistent amount of clathrin per punctum. However, cells incubated with the siRNA duplexes reveal a significant nonlinear relationship between the average punctum intensity and the total cellular EGFP-light chain fluorescence. At low [Clathrin], the average punctum intensity appears relatively flat but begins to increase significantly around the same range of [Clathrin] values as that for kendo. A log-log plot indicates the cooperativity in this parameter to have an apparent Hill coefficient n = 3.3 (Figure 4B, inset). We verified that we could still reliably detect clathrin assemblies for cells whose value of [Clathrin] was significantly reduced. An example of a representative image of individual dim clathrin puncta is shown in the left panel of Figure 4C. The key feature for detection of these puncta is the degree to which the mean intensity exceeds the average fluctuation in the background. The right panel of Figure 4C shows a frequency distribution of background pixel intensities as well as the mean intensity of the distribution of clathrin puncta obtained from cells whose [Clathrin] was between 66 and 126 afu (as seen in Figure 4B the average punctum intensity is largely invariant over this range of [Clathrin]). These data reveal that the signal-to-noise ratio is adequate to detect even the dimmest clathrin puncta as even at low values of [Clathrin] the average punctum intensity was clearly distinguishable from background fluctuations.
Membrane Budding Frequency Depends on [Clathrin]
As it is thought that the assembly of the clathrin lattice might be responsible for driving internalization, we examined how the rate of clathrin-mediated plasma membrane budding depends on [Clathrin] using time-lapse analysis of clathrin puncta with total-internal reflection fluorescence (TIRF) microscopy in both control and clathrin siRNA-treated cells. TIRF utilizes a shallow, evanescent field of fluorescence illumination within several hundred nanometers of the interface. The evanescent field permits the excitation of only those fluorescent molecules located near the plasma membrane. TIRF eliminates much of the out-of-focus fluorescence associated with wide-field fluorescence microscopy and permits the characterization of the presence and disappearance of assembled clathrin lattices at the cell surface (Steyer and Almers, 2001).
An example of a putative budding event as viewed in TIRF is shown in Figure 5A, indicated by the arrow. During this episode, a fluorescent punctum corresponding to an assembled clathrin lattice disappears from the field of view while neighboring spots remain. To estimate the number of dynamic events that would likely be related to coated pit budding from the cell surface, we counted the number of puncta that disappeared during a fixed recording period. This quantification provides a direct means to physically determine the rate of internalization on a per punctum basis. These data indicate that clathrin-mediated budding from the plasma membrane is also highly cooperative with respect to [Clathrin]. This relationship is best fit with an apparent Hill coefficient n = 3 (Figure 5B).
Figure 5.

Highly cooperative control of budding frequency. (A) Time-lapse imaging of a region of mock-treated cells using TIRF. The arrow indicates a clathrin punctum that is in the initial frame and disappears in a subsequent frame. Scale bar, 0.4 μm. (B) CHC siRNA and control cells were examined for the frequency of clathrin-mediated membrane budding from the cell surface. The graphed points in B represent the averaged values from several cells (n = 112 and 40 for CHC siRNA and control, respectively) ± SEM. These data were fit to Eq. 1, which gave n = 3, A = 248.
The Actin Cytoskeleton Regulates Clathrin-mediated Endocytosis
Such strong cooperativity in biological processes is often emblematic of tight regulatory control, as it affords a large change in output over a relatively small change in some control parameter such as [Clathrin] in this case. We therefore used this type of analysis to examine how one specific type of regulation of endocytosis, i.e., that imparted by the actin cytoskeleton, relates to the explicit dependence of kendo on [Clathrin]. A role for the actin cytoskeleton in clathrin-mediated endocytosis has been suggested by numerous studies (Munn, 2001; Schafer, 2002). Actin plays a fundamental role in endocytosis in yeast, although actin's role in higher eukaryotic cells has yet to be fully appreciated. However, agents that destabilize actin filaments significantly reduce the rate of endocytosis in certain cell types (Lamaze et al., 1997; Fujimoto et al., 2000; Moskowitz et al., 2003).
The relationship between clathrin dynamics and function and the actin cytoskeleton remains unclear, although it has been suggested that a possible function for actin in endocytosis is to localize components of the endocytic machinery to sites of endocytosis and/or to stabilize these sites. If actin served to regulate the assembly or stability of clathrin itself, then one would expect a shift in the relationship between kendo and [Clathrin] compared with that shown in Figure 3C. Thus, in order to determine how actin disruption might impact the details of clathrin assembly and dynamics, we repeated the measurements of kendo as a function of [Clathrin] using siRNA under conditions in which the actinmonomer sequestering agent Latrunculin-A (Lat-A) has destabilized actin filaments (Figure 6).
Figure 6.

Actin acts noncompetitively in clathrin-mediated endocytosis. kendo as a function of [Clathrin] was measured after disruption of actin with Lat-A (10 μM, 30 min) in CHC siRNA-treated (n = 804) and control cells (n = 677). The data for these cells was processed as described above (Figure 3), although the fluorescence data represents the average of 50 cells per data point ± SEM. The blue curve is the fit to the data from siRNA-treated cells in the absence of Lat-A, taken from Figure 3 for comparison. Inset shows a graph of the siRNA-treated cells in the presence of Lat-A and siRNA-treated cells not treated with Lat-A (n = 3916), but whose average values have been decreased by 50%. The data were fit with the simplified Hill equation (Eq. 1) to give n = 6.49, A = 249, kmax = 0.44, and knc = 0.09. The inset shows the same data along with that from Figure 3C rescaled to the same saturating value of kendo to indicate that the impact of Lat-A is only to change the value of kmax and knc, indicating a noncompetitive impact on clathrin function.
In control cells, Lat-A reduces the rate constant of internalization by ∼50% over the normal range of [Clathrin], corresponding to previous estimates of the effect of perturbing actin function on endocytosis. The data from the siRNA-treated cells were fit as in Figure 3, using the simplified Hill equation described in Eq. 1. This analysis reveals that Lat-A reduces the value of kmax as well as knc by ∼50% in the siRNA-treated cells additionally incubated with Lat-A. However, there was no impact on either the apparent Hill coefficient, n, or the concentration of [Clathrin] at which the steep [Clathrin] dependence occurs (the parameter A).
DISCUSSION
Clathrin utilizes an intricate arrangement of weak interactions to maintain the structure of lattices and the outer coat of vesicles. In addition, polymerized clathrin is in constant equilibrium with the free cytosolic pool of triskelia. Thus, regulation of this free pool of clathrin is a likely source of cellular regulation of clathrin function. Accordingly, we assayed how clathrin-dependent processes vary because of [Clathrin] in living cells using siRNA that targeted clathrin heavy chain. We determined that many of the clathrin-dependent processes including the intensity of clathrin puncta, membrane budding, and internalization of clathrin-dependent ligands are highly cooperative processes that are steeply dependent upon clathrin concentration below a critical value of [Clathrin].
The transfection procedure we utilized here resulted in a wide variation in intracellular clathrin depletion across the cell population. The reductions in clathrin heavy chain were mirrored by similar changes in the expression levels of clathrin light chain. Because the siRNA duplex only targets clathrin, we detected only slight changes in two closely associated endocytic proteins, α-adaptin of the AP-2 complex and dynamin. These changes are likely compensatory changes due to altered clathrin expression and not a direct modification due to the siRNA duplexes. Furthermore, the expression levels of these important endocytic proteins likely indicate that the remainder of the endocytic apparatus exists intact, in spite of clathrin depletion. On the whole the siRNA treatment resulted in a significant expansion of the range of values of [Clathrin] now present in the cell population. The dependence of the endocytic rate constant on [Clathrin] was similar for siRNA treated cells compared with controls where the values of [Clathrin] overlapped; however, some differences at the lowest end of the [Clathrin] values in the control population appear to have higher endocytic rate constants than for the equivalent value in the siRNA-treated population. The origin of this discrepancy is unknown, but perhaps it reflects additional contributions from the modest change in dynamin concentration that accompanies the reduction in [Clathrin].
Our data suggest that the dynamics of assembly and the subsequent impact of the frequency of internalization both display strong cooperativity with regard to [Clathrin]; however, in both cases the apparent Hill coefficient (n ≈ 3) is smaller than that displayed for the rate constant for endocytosis of transferrin receptors (n = 6.25). Thus it appears quite likely that a step before the actual internalization of transferrin receptor also has a nonlinear dependence on [Clathrin], which together with the nonlinear behavior of clathrin assembly and membrane budding would account for the overall apparent Hill coefficient. A hypothesis that remains to be tested is that the number of transferrin receptors that are bound or trapped in the clathrin lattice before internalization also depends nonlinearly on the number of triskelia assembled, and therefore on [Clathrin].
Furthermore, we have shown that we can adapt our technique to determine the specific role other proteins serve in the regulation of clathrin-dependent trafficking. Specifically, we used Lat-A to inhibit actin function and determined that Lat-A acts as a noncompetitive inhibitor in the control of endocytosis by [Clathrin], because it reduced the values of kmax and knc by ∼50% in siRNA-treated cells, but did not alter the apparent Hill coefficient. This result demonstrates that actin does not directly control clathrin function, but rather acts in a step upstream or downstream of clathrin function in endocytosis. Furthermore, little is known about the nature of the clathrin-independent pathway, characterized here by the parameter knc, although given that its sensitivity to Lat-A is identical to that of the clathrin-mediated internalization, it suggests that the ultimate target of actin-based modulation is common to both pathways. As it is generally thought that most internalization processes require dynamin function, one strong possibility is that actin impacts the efficiency of the dynamin-dependent step during endocytosis.
The quantitative approach we outlined here should open a new window in understanding the regulation of endocytosis and is well suited to investigate the role of many clathrin-associated proteins in this process. Although the rate constant for endocytosis is almost constant in the control cell population, the steep cooperativity strongly suggests that the clathrinmediated pathway is poised for tight regulatory control that might be utilized for physiological regulation. As a large part of this cooperativity probably arises from the fact that clathrin assembly is a two-dimensional polymerization process, it seems likely that most membrane-coat–driven steps in membrane trafficking would also behave cooperatively, thus potentially providing opportunities for interesting allosteric regulation in many steps of the secretory pathway.
Supplementary Material
Acknowledgments
We thank members of the Ryan lab for useful discussions. The National Institutes of Health and the Irma T. Hirschl Trust supported this work. We thank members of the T. McGraw and F. Maxfield laboratories for protein reagents and helpful discussions. H. S. M. was supported by a National Institutes of Health MSTP grant GM-07739.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04–08–0739) on February 2, 2005.
The online version of this article contains supplemental material at MBC Online (http://www.molbiolcell.org).
References
- Benmerah, A., Lamaze, C., Begue, B., Schmid, S. L., Dautry-Varsat, A., and Cerf-Bensussan, N. (1998). AP-2/Eps15 interaction is required for receptor-mediated endocytosis. J. Cell Biol. 140, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau, F. et al. (2004). Tandem MS analysis of brain clathrin-coated vesicles reveals their critical involvement in synaptic vesicle recycling. Proc. Natl. Acad. Sci. USA 101, 3833–3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, F. M. (1985). Clathrin structure characterized with monoclonal antibodies. II. Identification of in vivo forms of clathrin. J. Cell Biol. 101, 2055–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky, F. M., Chen, C. Y., Knuehl, C., Towler, M. C., and Wakeham, D. E. (2001). Biological basket weaving: formation and function of clathrin-coated vesicles. Annu. Rev. Cell. Dev. Biol. 17, 517–568. [DOI] [PubMed] [Google Scholar]
- Damke, H., Baba, T., Warnock, D. E., and Schmid, S. L. (1994). Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol. 127, 915–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto, L. M., Roth, R., Heuser, J. E., and Schmid, S. L. (2000). Actin assembly plays a variable, but not obligatory role in receptor-mediated endocytosis in mammalian cells. Traffic 1, 161–171. [DOI] [PubMed] [Google Scholar]
- Heuser, J. (1989). Effects of cytoplasmic acidification on clathrin lattice morphology. J. Cell Biol. 108, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichsen, L., Harborth, J., Andrees, L., Weber, K., and Ungewickell, E. J. (2003). Effect of clathrin heavy chain- and alpha-adaptin-specific small inhibitory RNAs on endocytic accessory proteins and receptor trafficking in HeLa cells. J. Biol. Chem. 278, 45160–45170. [DOI] [PubMed] [Google Scholar]
- Huang, F., Khvorova, A., Marshall, W., and Sorkin, A. (2004). Analysis of clathrin-mediated endocytosis of epidermal growth factor receptor by RNA interference. J. Biol. Chem. 279, 16657–16661. [DOI] [PubMed] [Google Scholar]
- Iversen, T. G., Skretting, G., van Deurs, B., and Sandvig, K. (2003). Clathrin-coated pits with long, dynamin-wrapped necks upon expression of a clathrin antisense RNA. Proc. Natl. Acad. Sci. USA 100, 5175–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen, T. (2000). Clathrin. Annu. Rev. Biochem. 69, 699–727. [DOI] [PubMed] [Google Scholar]
- Lamaze, C., Fujimoto, L. M., Yin, H. L., and Schmid, S. L. (1997). The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. J. Biol. Chem. 272, 20332–20335. [DOI] [PubMed] [Google Scholar]
- Larkin, J. M., Brown, M. S., Goldstein, J. L., and Anderson, R. G. (1983). Depletion of intracellular potassium arrests coated pit formation and receptor-mediated endocytosis in fibroblasts. Cell 33, 273–285. [DOI] [PubMed] [Google Scholar]
- Lemmon, S. K., and Jones, E. W. (1987). Clathrin requirement for normal growth of yeast. Science 238, 504–509. [DOI] [PubMed] [Google Scholar]
- Mayor, S., Presley, J. F., and Maxfield, F. R. (1993). Sorting of membrane components from endosomes and subsequent recycling to the cell surface occurs by a bulk flow process. J. Cell Biol. 121, 1257–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw, T. E., Greenfield, L., and Maxfield, F. R. (1987). Functional expression of the human transferrin receptor cDNA in CHO cells deficient in endogenous transferrin receptor. J. Cell Biol. 105, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz, H. S., Heuser, J., McGraw, T. E., and Ryan, T. A. (2003). Targeted chemical disruption of clathrin function in living cells. Mol. Biol. Cell 14, 4437–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motley, A., Bright, N. A., Seaman, M. N., and Robinson, M. S. (2003). Clathrin-mediated endocytosis in AP-2-depleted cells. J. Cell Biol. 162, 909–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn, A. L. (2001). Molecular requirements for the internalisation step of endocytosis: insights from yeast. Biochim. Biophys. Acta 1535, 236–257. [DOI] [PubMed] [Google Scholar]
- Nesterov, A., Carter, R. E., Sorkina, T., Gill, G. N., and Sorkin, A. (1999). Inhibition of the receptor-binding function of clathrin adaptor protein AP-2 by dominant-negative mutant mu2 subunit and its effects on endocytosis. EMBO J. 18, 2489–2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne, G. S., and Schekman, R. (1985). A test of clathrin function in protein secretion and cell growth. Science 230, 1009–1014. [DOI] [PubMed] [Google Scholar]
- Ruscetti, T., Cardelli, J. A., Niswonger, M. L., and O'Halloran, T. J. (1994). Clathrin heavy chain functions in sorting and secretion of lysosomal enzymes in Dictyostelium discoideum. J. Cell Biol. 126, 343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, D. A. (2002). Coupling actin dynamics and membrane dynamics during endocytosis. Curr. Opin. Cell Biol. 14, 76–81. [DOI] [PubMed] [Google Scholar]
- Steyer, J. A., and Almers, W. (2001). A real-time view of life within 100 nm of the plasma membrane. Nat. Rev. Mol. Cell. Biol. 2, 268–275. [DOI] [PubMed] [Google Scholar]
- Wakeham, D. E., Chen, C. Y., Greene, B., Hwang, P. K., and Brodsky, F. M. (2003). Clathrin self-assembly involves coordinated weak interactions favorable for cellular regulation. EMBO J. 22, 4980–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettey, F. R., Hawkins, S. F., Stewart, A., Luzio, J. P., Howard, J. C., and Jackson, A. P. (2002). Controlled elimination of clathrin heavy-chain expression in DT40 lymphocytes. Science 297, 1521–1525. [DOI] [PubMed] [Google Scholar]
- Wu, X., Zhao, X., Baylor, L., Kaushal, S., Eisenberg, E., and Greene, L. E. (2001). Clathrin exchange during clathrin-mediated endocytosis. J. Cell Biol. 155, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.