

Abstract
To understand cell cycle control mechanisms in early development and how they change during differentiation, we used embryonic stem cells to model embryonic events. Our results demonstrate that as pluripotent cells differentiate, the length of G1 phase increases substantially. At the molecular level, this is associated with a significant change in the size of active cyclin-dependent kinase (Cdk) complexes, the establishment of cell cycle-regulated Cdk2 activity and the activation of a functional Rb–E2F pathway. The switch from constitutive to cell cycle-dependent Cdk2 activity coincides with temporal changes in cyclin A2 and E1 protein levels during the cell cycle. Transcriptional mechanisms underpin the down-regulation of cyclin levels and the establishment of their periodicity during differentiation. As pluripotent cells differentiate and pRb/p107 kinase activities become cell cycle dependent, the E2F–pRb pathway is activated and imposes cell cycle-regulated transcriptional control on E2F target genes, such as cyclin E1. These results suggest the existence of a feedback loop where Cdk2 controls its own activity through regulation of cyclin E1 transcription. Changes in rates of cell division, cell cycle structure and the establishment of cell cycle-regulated Cdk2 activity can therefore be explained by activation of the E2F–pRb pathway.
INTRODUCTION
Cell proliferation is coordinated by the activity of cyclin-dependent kinase (Cdk) activities (Nigg, 1995). This family of kinases functions by regulating the activity of proteins required for progression through the different cell cycle phases and hence must themselves be tightly cell cycle regulated. At one level, this is achieved through the cell cycle-regulated synthesis of cyclin regulatory subunits, which bind and activate their catalytic Cdk partner. Inactivation of Cdk activity from one cell cycle phase is required for transition into the next by a mechanism involving cyclin destruction (Tyers and Jorgensen, 2000; Breeden, 2003). Hence, the ordered synthesis and destruction of phase-specific cyclin regulatory subunits are essential elements of normal cell cycle progression. Cyclin expression levels are controlled, in part at least, through transcriptional regulation. Cell cycle-regulated cyclin E1 and A2 transcription is controlled by E2F transcription factors that are subject to repression through recruitment of “pocket proteins,” such as pRb and p107, to the promoter regions of these genes. Repression of E2F-dependent transcription is lifted through phosphorylation of pocket proteins by Cdk activities that become active during G1 phase (Harbour et al., 1999; Zhang et al., 2000), allowing for the transcriptional activation of genes required for the G1-S transition, such as cyclin E1.
Pluripotent cells of late preimplantation and early postimplantation embryos exhibit two remarkable features. First, their cell division rates are very short, and their division is controlled through unusual mechanisms of Cdk regulation. Second, they have extraordinary developmental plasticity, illustrated by their unlimited differentiation potential. At gastrulation, pluripotent cells commit to become one of the three embryonic germ layers and coinciding with this, cell division rates decrease dramatically (Snow, 1977). These early developmental events can be reproduced in vitro with embryonic stem (ES) cells and their differentiated derivatives, making them an ideal model on which to study pluripotency and early embryonic differentiation. ES cells are equivalent to cells comprising the inner cell mass of blastocyst stage (4.5 days postcoitum [dpc]) embryos and can be converted into a second pluripotent cell type (early primitive ectoderm-like, EPL), that closely resembles the early primitive ectoderm of the embryonic epiblast (5.25 dpc) in terms of their gene expression profile and differentiation potential (Rathjen et al., 1999; Lake et al., 2000; Pelton et al., 2002). ES and EPL cells divide with generation times of <10 h (Savatier et al., 1994; Stead et al., 2002) by a mechanism that does not conform to that typically seen in differentiated cells. Rapid division of these pluripotent cells and their embryonic equivalents is associated with an unusual cell cycle structure, composed largely of S-phase cells and a truncated G1 phase (Solter et al., 1971; Snow, 1977; Stead et al., 2002). At the molecular level, this is underpinned by precocious, cell cycle-independent Cdk activities (Stead et al., 2002; Faast et al., 2004) and an absence of Cdk inhibitory molecules such as p27Kip1, p21Cip1, and p16INK4a (Savatier et al., 1996; Stead et al., 2002). The only Cdk activity that shows any clear sign of periodicity is the mitotic kinase Cdk1-cyclin B1 (Stead et al., 2002). This is reminiscent of Cdk activity in the early cleavage stage divisions in Xenopus (Newport and Kirschner, 1982, 1984; Murray and Kirschner, 1989; Hartley et al., 1996) and Drosophila development (Foe and Alberts, 1983; Edgar and Schubiger, 1986; Edgar et al., 1994; Stiffler et al., 1999), which consist of alternating rounds of DNA replication and mitosis. Although ES cells express the pocket proteins pRb and p107, they are hyperphosphorylated and consequently held in a biochemically inactive state, presumably due to the activity of cell cycle-independent pRb kinase activities (Savatier et al., 1994; Stead et al., 2002). Consequently, E2F transcription factors are not subject to pocket protein repression during the cell cycle in ES cells, and so their target genes are transcribed independently of cell cycle position (Stead et al., 2002).
On withdrawal of maintenance factors that promote self-renewal, such as leukemia inhibitory factor (LIF), cells differentiate and divide at significantly reduced rates; however, the mechanisms underpinning changes in cell cycle dynamics that occur during embryonic differentiation have not been defined. We previously proposed that as a function of ES cell differentiation, changes in rates of cell division could be accounted for by a fundamental change in the cell cycle machinery (Stead et al., 2002; Faast et al., 2004). In this report, we set out to characterize Cdk regulation in differentiating ES cells and asked whether changes in cell cycle structure and division rates corresponded to changes in Cdk activity during the cell cycle and status of the pRb–E2F pathway.
MATERIALS AND METHODS
Cell Culture, Synchronization, and Flow Cytometry
D3 murine ES cells (Doetschman et al., 1985) were cultured as described previously (Stead et al., 2002). mEPL cells and EPL embryoid bodies (EBs) were formed as described previously (Faast et al., 2004). Early passage (passage 3) mouse embryonic fibroblasts (MEFs) were prepared from outcrossed Swiss mice (Hogan et al., 1994) and cultured in DMEM supplemented with 10% fetal calf serum (FCS) and 1 mM l-glutamine, at 37°C under 5% CO2. Cells were statically synchronized in prophase/mitosis by the addition of 45 ng/ml nocodazole (Sigma-Aldrich, St. Louis, MO) to media for 12 h. In preparation for flow cytometry, cells and EBs were trypsinized to obtain single cell suspension, resuspended in 300 μl of phosphate-buffered saline (PBS) supplemented with 1% FCS, and fixed by the dropwise addition of 900 μl of ice-cold 95% ethanol. After being vortexed and chilled on ice for 10 min, cells were collected by centrifugation (3000 × g, 5 min) and washed twice in 700 μl of cold PBS. Cells were resuspended in PBS and stained by the addition of 2.5 mg/ml propidium iodide (Sigma-Aldrich) and 0.5 mg/ml RNase A and then analyzed on a Beckman Coulter flow cytometer with WinMDi software.
Generation of Stable Cell Lines
pSRα-p107-HA and pSRα-p107ΔS/T-P-HA (Ashizawa et al., 2001) were used to clone p107wt and p107ΔS/T-P as XhoI fragments into pCAGiP (Pratt et al., 2000). Ten micrograms of linearized pCAGp107-HA and pCAGp107ΔS/T-P-HA were electroporated into 3–5 × 107 ES cells by using a gene pulser (200 V, 500 μF; Bio-Rad, Hercules, CA). Stable ES clones expressing p107wt or p107ΔS/T-P were selected by addition of 1 μg/ml puromycin to the medium 24 h after transfection. pGL3promoter-4 × CCC luciferase reporter and pGL3promoter-4 × CAT luciferase reporter were transfected with the pCA-GiP vector (Cartwright et al., 1998). Clones were selected by the addition of 1 μg/ml puromycin and screened by luciferase assay.
Antibodies, Immunoblotting, Kinase Assays, and Gel Mobility Shift Assays
Rabbit polyclonal antibodies recognizing cyclin E (M-20), cyclin A2 (C-19), cyclin B1 (H-433), Cdk2 (M-2), p107 (C-18), p27Kip1 (C-19), Oct4 (N-19), E2F-4 (C-20), and cdc25a (144) were from Santa Cruz Biotechnology (Santa Cruz, CA), and p21Cip1 (HJ21) antibodies from were from NeoMarkers (Fremont, CA). Mouse monoclonal antibodies recognizing hemagglutinin (HA) were from Roche Diagnostics (Indianapolis, IN). Rat anti-β-tubulin (MCA78G) was from Serotec (Oxford, United Kingdom). Antibodies were routinely used at 1 μg/ml for immunoblotting, except anti-β-tubulin, which was used at 0.2 μg/ml. Horseradish peroxidase-conjugated secondary antibodies used were sheep anti-mouse, donkey anti-rabbit (Amersham Biosciences, Piscataway, NJ), and rabbit anti-rat (DakoCytomation California, Carpinteria, CA). Whole cell lysate preparation, immunoblotting, kinase assays, and gel mobility shift assays were as described previously (Stead et al., 2002).
Northern Analysis
Preparation of total RNA and Northern blotting were conducted as described previously (Stead et al., 2002). Constructs used to generate DNA fragments have been described previously (Rathjen et al., 1999), with the exception of cyclin E1 obtained from N. Dyson (Massachusetts General Hospital, Boston, MA).
Fractionation of Whole Cell Protein by Gel Filtration
Column, buffers, and equipment were equilibrated at 4°C. The Superdex 200 HR 10/30 column (Amersham Biosciences) was calibrated using standards from an HMV calibration kit (10 μg of each protein—thyroglobulin 669 kDa, ferritin 440 kDa, catalase 232 kDa, lactate dehydrogenase 140 kDa, bovine serum albumin (BSA) 67 kDa; Amersham Biosciences) at a flow rate of 0.5 ml/min. The void volume was 8.0 ml. Whole cell lysates were prepared as described previously (Stead et al., 2002) and then ultracentrifuged at 5 × 104 rpm for 30 min at 4°C. Seven milligrams (total protein) of lysate was fractionated in gel filtration buffer (50 mM HEPES, pH 7.5, 10 mM MgCl2, and 150 mM NaCl) at a flow rate of 0.5 ml/min, collecting 500-μl fractions. Fractions were snap frozen and stored at –80°C. Every second fraction was analyzed. The column was stripped overnight at room temperature with 1 mg/ml trypsin in 50 mM Tris-HCl, pH 7.5, and 20 mM CaCl2. Trypsin was washed from the column with 50 ml of water at a flow rate of 0.7 ml/min. At the same flow rate, the column was washed with 50 ml of 1 M NaOH, 50 ml of H2O, 30 ml of 0.1 N HCl, and 60 ml of milliQ water. The column was stored in 20% ethanol.
Luciferase Assays
For transient transfections, ES cells were seeded at 7 × 104 cells/ml (3.5 × 104 cells/well of a 24-well tray) and grown for 8–10 h. Triplicate wells were transfected with 100 ng of pGL3TATAbasic-6 × E2F luciferase reporter, pGL3promoter-4 × CCC luciferase reporter, or pGL3promoter-4 × CAT luciferase reporter and 25 ng pRL-TK by using the FuGene transfection method following the manufacturer's instructions. When expression vectors were included in transfections, the concentration of transfected DNA was equalized with empty pCAGiP vector (Pratt et al., 2000). After 40 h, cells were assayed for luciferase activity by using the DLR luciferase kit. For stably integrated reporters, cell pellets were lysed with luciferase buffer (1% Triton-X 100, 25 mM glycyl glycine, pH 7.8, 15 mM MgSO4, 4 mM EGTA, and 1 mM dithiothreitol [DTT]). Fifty microliters of cleared lysate was assayed with 50 μl of Complete luciferase buffer (48 mM phosphate buffer, 6 mM ATP, and 3 mM DTT in luciferase buffer) and 50 μl of 1× luciferin. Luciferase readings were equalized according to the protein concentration of each sample.
ChIP Assays
Protein–protein and protein–DNA complexes were cross-linked by addition of 1% formaldehyde to tissue culture medium and incubated for 10 min at room temperature. The reaction was terminated by addition of glycine to 0.125 M for 5 min at room temperature. After washing twice with cold PBS, adherent cells were scraped from the plate and embryoid bodies collected by aspiration. Cells were lysed with SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris, pH 8.0, 0.5 mM phenylmethylsulfonyl fluoride [PMSF], 50 μg/ml N-tosyl-l-phenylalanine chloromethyl ketone [TPCK], 50 μg/ml N-tosyl-l-lysine chloromethyl ketone [TLCK], 1 μg/ml leupeptin, and 1 mM DTT) for 10 min at 4°C. Samples were then diluted with the addition of 2 ml of chromatin immunoprecipitation (ChIP) buffer (0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris, pH 8.0, 176 mM NaCl, 0.5 mM PMSF, 50 μg/ml TPCK, 50 μg/ml TLCK, 1 μg/ml leupeptin, and 1 mM DTT). Sonication was optimized at seven times for 30 s at half maximum power (Sonifier cell disruptor B-30; Branson, Danbury, CT). Chromatin was separated from cell debris and stored at –20°C until used. Chromatin was equalized according to DNA concentrations, determined using a Varian Cary 3 Bio UV-visible spectrophotometer. Protein A-Sepharose (PAS; Amersham Biosciences) was solubilized and washed according to manufacturer's instructions. PAS slurry (50%) was incubated with 0.5 mg/ml sonicated herring sperm DNA and 1 mg/ml BSA overnight at 4°C. Blocked PAS was stored at 4°C. Chromatin extracts (700 μg) were precleared with 60 μl of blocked PAS for 30 min at 4°C and then incubated with 2 μg of primary antibody overnight at 4°C. p107 (C-18) and cdc25A (C-20) (nonspecific immunoprecipitation) rabbit polyclonal antibodies were from Santa Cruz Biotechnology. Immune complexes were collected with 60 μl of blocked PAS for 1 h at 4°C. Immune complexes bound to PAS were washed rocking at 4°C (7 min each) once with low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris, pH 8.0, 150 mM NaCl, 0.5 mM PMSF, 50 μg/ml TPCK, 50 μg/ml TLCK, 1 μg/ml leupeptin, and 1 mM DTT), once with high salt wash buffer (low salt buffer with NaCl adjusted to 500 mM), once with LiCl wash buffer (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris, pH 8.0, 0.5 mM PMSF, 50 μg/ml TPCK, 50 μg/ml TLCK, 1 μg/ml leupeptin, and 1 mM DTT), and twice with TE (10 mM Tris, pH 8.0, and 0.1 mM EDTA). Immune complexes were eluted with 250 μl of 0.1 M NaHCO3 in 1% SDS twice for 15 min each at room temperature. To reverse the cross-links, NaCl (final concentration 0.2 M) was added to combined eluants and incubated at 65°C overnight. Proteins were then degraded by addition of 10 μl of 0.5 M EDTA, 20 μl of 1 M Tris-HCl, pH 6.5, and 2 μl of 10 mg/ml proteinase K and incubation at 45°C for 1 h. After a phenol/chloroform extraction, DNA was precipitated with ethanol and 20 μg of glycogen. Immunoprecipitated DNA was resuspended in 30 μl of MQ. Input samples were resuspended at 0.1 μg/μl. Polymerase chain reactions (PCRs) were conducted on 1 μl of DNA in a 20-μl reaction containing 10× PCR buffer [250 mM Tris-HCl, pH 9.1, and 160 mM (NH4)2SO4], 50 ng of each primer, 200 μM dNTPs (Roche Diagnostics), 2.5 mM MgCl2 (cyclin E1 primers) or 1.5 mM MgCl2 (albumin primers), 1 M betaine (Sigma-Aldrich), and 1.5 U of Taq polymerase (Geneworks, Adelaide, Australia). PCR conditions were as follows: 1 cycle: 95°C, 5 min; 60°C, 5 min; 72°C, 3 min and then 30–39 cycles: 95°C, 1 min; 60°C, 2 min; 72°C, 1.5 min. To ensure the PCR was exponentially increasing, PCRs were conducted for 30, 33, 36, and 39 cycles with the first and last sample of the differentiation before conducting PCRs on all precipitated products. PCR products were separated by electrophoresis on 1.5% Tris borate-EDTA-agarose gels and visualized with ethidium bromide. The sequences for the primers used are as follows: cyclin E1 1st: 5′-dCGTAAAAGAACACGCCCCCCG-3′; cyclin E1 2nd: 5′-dAAGCTGTGTCCGCCGCAGGCAGGCG-3′; albumin 1st: 5′-dGGTAAAGCTCCCGGGATCCGCCAAT-3′; and albumin 2nd: 5′-dGTGGACTGTCACTTTGGTGGCTGGC-3′.
RESULTS
Changes in Cell Cycle Dynamics during ES Cell Differentiation Are Associated with the Establishment of Cell Cycle-regulated Cdk Activities
ES cells and EPL cells divide rapidly, in part because they spend only a short amount of time in G1 before committing to undergo DNA replication (Savatier et al., 1994; Stead et al., 2002). We predicted that during differentiation, increased cell division times would be reflected by an increase in the length of G1. To test this idea, we differentiated EPL cells as EBs and evaluated cell cycle structure changes by flow cytometry of propidium iodide (PI)-stained cells. EPL cells were used as a starting point for these experiments because they differentiate more synchronously than EBs derived directly from ES cells (Lake et al., 2000), making it possible to define temporal events more accurately. To confirm our prediction that differentiation is associated with changes in cell cycle structure, we assessed changes in flow cytometry profiles of PI-stained cells during EB differentiation. Differentiation was monitored by evaluating levels of marker transcripts, including Rex1 (ES/ICM), Oct4 (pluripotent cells), Fgf5 (primitive ectoderm), and Brachyury (nascent mesoderm; Figure 1A). Although the cell cycle structure of ES/EPL cells and EPL EBs to day 2 are comparable (Figure 1B), the proportion of cells in G1 phase increased markedly at day 3 onward (Figure 1B). These changes were coincident with loss of mRNAs associated with pluripotency and increased levels of transcripts associated with differentiation (Figure 1A).
Figure 1.

Changes in Cdk activity accompany restructuring of the cell cycle during ES cell differentiation. (A) EPL cells derived directly from ES cells were grown as EBs for 5 d in the absence of LIF. At each day, transcript levels for Rex1, Oct4, Fgf5, Brachyury, and GAPDH were evaluated by Northern blot. Quantitation of mRNA levels was by phosphorimaging and is shown relative to GAPDH levels. (B) Flow cytometry of PI-stained cells from ES cells, EPL cells, and EBs at the times indicated. (C) Cdk2, cyclin A2, B1, and E1 immunoprecipitates from whole cell lysates (100 μg of protein) were assayed for histone H1 activity and then quantitated by phosphorimaging. (D) Lysates (20 μg of protein) from ES cells, EPL cells, MEFs (passage 3), NIH 3T3 fibroblasts, and EBs (day 1–5) were subject to immunoblot analysis by probing for cyclin E1, p21Cip1, p27Kip1, and β-tubulin (load control).
Cdk activity is a major factor that influences the rate of cell cycle progression and the length of cell cycle phases (Bremner et al., 1995; Resnitzky and Reed, 1995; Leone et al., 1999). We previously established a mechanism for rapid ES/EPL cell division by demonstrating that pluripotent cells of embryonic origin exhibit unusually high levels of Cdk activity, regardless of cell cycle position (Stead et al., 2002). To address the molecular mechanisms regulating changes in cell cycle structure during ES cell differentiation, we analyzed the activity of Cdk-associated complexes in differentiating, asynchronous cell populations. We first evaluated Cdk activities by immunoprecipitating Cdk or cyclin subunits from cell lysates and performed in vitro kinase assays by using histone H1 as a substrate (Stead et al., 2002). As expected, high levels of Cdk2-, cyclin E1-, cyclin A2-, and cyclin B1-associated H1 kinase activity were observed in ES and EPL cells and early EBs (Figure 1C). By day 4, however, Cdk2-, cyclin E1-, and cyclin B1-associated kinase activity had decreased by approximately fourfold (Figure 1C). The timing of decreased cyclin E1 and Cdk2 kinase activity coincided with the lengthening of G1 phase (Figure 1B). Because cyclin E1-associated kinase activity is rate limiting for the G1/S phase transition (Ohtsubo and Roberts, 1993; Resnitzky et al., 1994), the collapse of Cdk2/cyclin E1 kinase activity can account for expansion of the G1 phase during differentiation. Decreased Cdk2–cyclin E1 activity was paralleled by a decline in cyclin E1 protein levels and increased levels of the Cdk inhibitors p21Cip1 and p27Kip2 (Figure 1D). Although we do not provide direct evidence, we predict that cyclin E1 availability and Cdk inhibition are the major factors influencing the down-regulation of Cdk2 activity during ES cell differentiation.
Establishment of Cell Cycle-regulated Cdk Activity during ES Cell Differentiation
With the exception of Cdk1-cyclin B1 kinase, there is a general absence of cell cycle-dependent Cdk activity in pluripotent cells of embryonic origin (Stead et al., 2002). To investigate the possible relationship between changes in cell cycle structure and Cdk activity in pluripotent and differentiating cells, we asked whether Cdk activities become cell cycle dependent during differentiation. Because differentiation of EBs is a dynamic process, synchronization experiments at each time point could not be performed. Instead, we used nocodazole trap experiments (Pathan et al., 1996; Stead et al., 2002) and compared Cdk activity in G2/M-enriched cells at each stage of differentiation to that of asynchronous cells. Although Cdk2, cyclin A2, and cyclin E1 kinase activities are cell cycle independent in ES/EPL cells, by day 2 or 3 of EB differentiation they become cell cycle regulated (Figure 2). Therefore, as a function of differentiation, Cdk activities switch from being cell cycle independent to cell cycle regulated. This correlated with the establishment of cell cycle-regulated cyclin A2 and cyclin E1 protein levels, implicating cyclin availability as a rate-limiting factor. Cyclin B1 retained cell cycle regulation in EBs, consistent with our previous reports that cyclin B1-associated H1 kinase activity is already cell cycle regulated in pluripotent cells (Stead et al., 2002).
Figure 2.
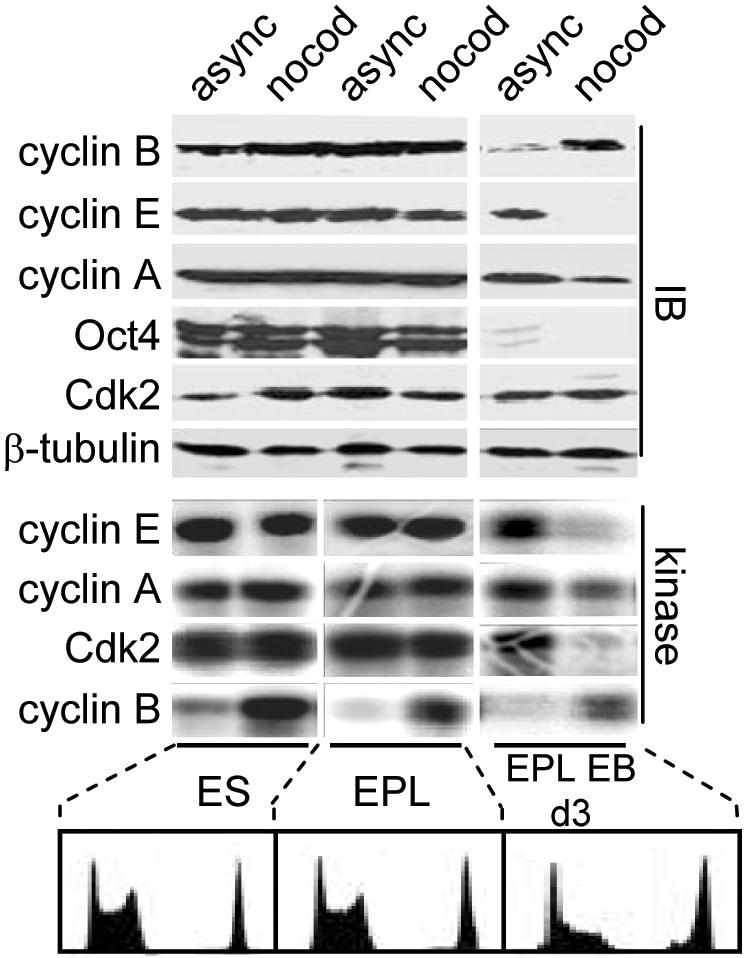
Establishment of cell cycle-regulated Cdk activity during EB differentiation. Asynchronous (async) or nocodazole (nocod)-blocked ES cells, EPL cells, or day 3 EBs were harvested and subject to immunoblot analysis (IB) or assayed for H1 kinase activity. Cell cycle profiles of PI-stained cells are shown (bottom).
Redistribution of Cdk Activities into Higher Order Complexes during Differentiation
To investigate the underlying biochemical changes associated with differentiation, restructuring of the cell cycle, and changes in Cdk activity, we used gel filtration analysis to compare the composition of Cdk2–cyclin complexes in ES cells and differentiated cells. Changes in the complexity of Cdk–cyclin complexes have been previously reported to be associated with various biological changes in different cell types (Sweeney et al., 1998) and in response to different stimuli (Steiner et al., 1995; Prall et al., 1997; Musgrove et al., 1998; Prall et al., 1998; McConnell et al., 1999; Parry et al., 1999; Swarbrick et al., 2000). We therefore asked whether part of the mechanism underpinning changes in cell cycle parameters during differentiation could be associated with the redistribution of Cdk–cyclins into different functional complexes.
To address this question, we initially compared Cdk–cyclin complexes in ES cells and MEFs by fractionation on a Superdex 200 size exclusion column. Equal volumes of protein fractions were analyzed by Western blot or subjected to immunoprecipitation by using antibodies directed against cyclin or Cdk subunits. Immunoprecipitates were then assayed for kinase activity in vitro by using histone H1 as a substrate. In ES cell lysates, the major peak of cyclin A2 protein and cyclin A2 kinase activity coeluted in complexes with the 160-kDa marker (Figure 3A). The elution profile of active cyclin A2 complexes was broader in MEFs and was not necessarily associated with fractions where the majority of cyclin A2 eluted (Figure 3A). Consistent with other differentiated cell types (Prall et al., 1997; Musgrove et al., 1998), the majority of cyclin E1 and Cdk2 protein eluted in lower molecular mass fractions (<160-kDa marker) in MEFs, whereas kinase activity eluted in higher molecular mass fractions (>160-kDa marker) (Figure 3A). In contrast, the majority of cyclin E1 and Cdk2 kinase activity eluted in low molecular mass fractions (<160 kDa marker) in ES cells (Figure 3A), overlapping with both cyclin E1 and Cdk2 protein elution profiles. Levels of Cdk2- and cyclin E1-associated kinase activity eluting in high-molecular-mass fractions (>160-kDa marker) were significantly greater in ES cells compared with MEFs (Figure 3A). These observations suggest that in ES cells, the majority of Cdk2/cyclin E1 is assembled into active complexes that span a lower molecular mass range than active Cdk2–cyclin E1 complexes in MEFs. Similar trends also were seen for cyclin A2 complexes. These observations demonstrate significant differences in the activity and composition of active Cdk2–cyclin complexes in ES cells and MEFs.
Figure 3.

Redistribution of active Cdk activity into higher order complexes during differentiation. (A) Whole cell lysates from ES cells and MEFs (passage 3) were fractionated by size exclusion chromatography. Cdk2, cyclin A2, and cyclin E1 H1 kinase activities and protein levels were evaluated in alternate fractions. Gel filtration columns were calibrated using molecular mass markers. Lysates from ES cells, EPL cells, and EBs (day 2–5) were fractionated as described in A and assayed for cyclin E1 (B) or Cdk2 activity (C). Corresponding fractions also were subject to immunoblot analysis. To highlight differences in elution profiles, equivalent exposures are not represented; ES, EPL, and EPLEB days 2 and 3 are similar exposures, whereas EBs days 4 and 5 are longer exposures. Note that immunoblots and kinase assays in A were exposed for longer periods for analysis of MEF lystates to allow a more direct comparison of elution profiles with ES cell lysates.
Differences between ES and MEF gel filtration profiles suggested that the composition and complexity of Cdk–cyclin complexes could change during differentiation. To address this question further, we compared the elution profiles of cyclin E1- and Cdk2-associated kinase activity in ES cells, EPL cells, and EPL EBs. Cyclin E1-associated Cdk activity in ES cells, EPL cells, and day 2 EBs eluted in similar size fractions after gel filtration, with the majority of active Cdk2–cyclin E1 eluting after the 160-kDa marker (Figure 3B).
Active cyclin E1 complexes in cells differentiated for 3–5 d shifted into higher order complexes with a concomitant decrease in activity, even though the bulk of cyclin E1 protein levels did not shift significantly into larger complexes. By day 5 of EB differentiation, the size range of active cyclin E1 complexes in EBs overlapped with that of MEFs (Figure 3B). These data show that differentiation of ES cells is associated with the redistribution of active Cdk2–cyclin E1 into high-molecular-mass complexes, of similar size typically seen in differentiated cell types (Prall et al., 1997; Musgrove et al., 1998). In contrast to ES cells, only a small fraction of the total pool of cyclin E1 is assembled into active Cdk complexes, indicating that during differentiation, qualitative changes in the proportion of active cyclin E1-containing complexes occur. The similarity in the elution profile for cyclin E1-associated and Cdk2 kinase activity (Figure 3C) suggests that the majority of active Cdk2 is bound to cyclin E1.
Down-Regulation of Cyclin E1 Transcription and Activation of the pRb–E2F Pathway during Differentiation
Changes in Cdk2 activity can, in part, be accounted for by a decline in cyclin E1 protein (Figure 1D) and mRNA (Figure 4A). During differentiation, transcription of the cyclin E1 gene switches from being cell cycle independent to cell cycle regulated (Figure 4B). Cyclin E1 protein stability does not change significantly during differentiation (Faast and Dalton, unpublished data), indicating that changes in transcription rates are likely to be a major determinant of cyclin E1 protein levels and Cdk2–cyclin E1 activity in ES cells and during differentiation. Because cyclin E1 transcription is dependent on regulation by the E2F and pocket proteins, we asked whether E2F-dependent transcription also was elevated in pluripotent cells and decreased during differentiation. This was evaluated by establishing stable cell lines carrying a luciferase reporter construct driven by four canonical E2F binding sites (Cartwright et al., 1998) and comparing reporter activity in EPL cells to that in day 5 EBs. This analysis showed that general E2F-dependent transcription is ∼12-fold higher in pluripotent (EPL) cells compared with EBs (Figure 4C). Mutation of E2F binding sites in the reporter decreased reporter activity, indicating that transcription from the reporter in the wild-type construct is E2F dependent. These results are consistent with our previous observation that cyclin E1 transcript levels decrease during differentiation (Figure 4A). This is likely to be due, in part at least, to establishment of cell cycle-regulated E2F–pRb activity in contrast to ES cells where E2F is constitutively active (Stead et al., 2002). The major E2F activity defined by gel mobility shift assays in ES/EPL cells is E2F4 (Humbert et al., 2000; Stead et al., 2002). To formally show that E2F4 binds the cyclin E1 promoter in ES cells in vivo, we performed ChIP assays (Figure 4D). In contrast to the albumin promoter, we show that E2F4 binds the cyclin E1 promoter in vivo consistent with it having a role in driving cyclin E transcription.
Figure 4.

Down-regulation of cyclin E1 mRNA due to the collapse of E2F transcriptional activity during EB differentiation. (A) Cyclin E1 and GAPDH mRNA levels in cell samples (as in Figure 1) were evaluated by Northern blotting. Quantitation of transcript levels was determined by PhosphorImager analysis relative to the GAPDH control. (B) Cyclin E1 and GAPDH mRNA levels in asynchronous or nocodazole-blocked ES cells and day 1 and day 3 EBs. (C) Clonal ES cell lines with stably integrated luciferase reporters driven by E2F or mutated E2F sites were differentiated into EPL or day 5 EBs. Protein harvested from cells and bodies was analyzed for luciferase activity. Luciferase units were normalized using protein concentrations. Data represents a typical experiment. (D) ChIP analysis of E2F4 binding activity on the cyclin E1 and albumin promoters in ES cells. Cross-linked chromatin (700 μg) from asynchronous ES cells was incubated with E2F4 or cdc25a (nonspecific) antibodies. Immunoprecipitated DNA was analyzed by PCR by using primers specific for different promoters. PCR controls included analysis of 0.03% of the total input chromatin (input) and the addition of water instead of input DNA (– template).
Recruitment of p107 to the Cyclin E1 Promoter Corresponds to Changes in Cyclin E1 Transcription during Differentiation
Changes in cyclin E1 transcription that occur during differentiation are likely to be a consequence of altered pocket protein regulation after the establishment of cell cycle-regulated Cdk activity. This would predict that although pocket proteins are not assembled into complexes with E2Fs in ES cells, E2F–pocket protein complexes will be formed upon the establishment of cell cycle-dependent Cdk activities. In this study, our focus was on p107 because it is the most prevalent E2F-associated pRb family member in ES cells (Stead et al., 2002), but the studies do not preclude similar regulation of the other pRb family members. Our hypothesis was that although p107 and pRb are expressed in ES cells, they are held in a biochemically inactive state by precocious, cell cycle-independent Cdk activity (Stead et al., 2002). This is supported by our observations that during differentiation, E2F complexes become assembled into higher order complexes with pocket proteins as judged by gel mobility shift assays (Figure 5A).
Figure 5.

Recruitment of p107 to the cyclin E1 promoter corresponds to changes in cyclin E1 mRNA during differentiation. (A) E2F ternary complexes form during ES cell differentiation. Band shift analysis using extracts from asynchronous ES, EB day 4, and NIH 3T3 cells. (B) ChIP analysis of p107 binding activity on the cyclin E1 and albumin promoters in EPL cells and during differentiation. Cross-linked chromatin (700 μg) from asynchronous EPL cells and EPLEBs was incubated with p107 or cdc25a (nonspecific) antibodies. Immunoprecipitated DNA was analyzed by PCR by using primers specific for the cyclin E1 and albumin promoters. To ensure equal concentrations of chromatin was analyzed, PCRs on 0.03% of the total input chromatin (input) were conducted. (C) Ectopic p107 represses endogenous cyclin E1 transcription in ES cells. ES cells were stably transfected with pCAG vector (vector), pCAG-p107wt or pCAG-p107ΔS/T-P. Stably integrated forms of p107, from different clonal lines, were detected via HA tags by immunoblot analysis. Cyclin E1 and GAPDH mRNA levels in cell samples were evaluated by Northern blotting. Quantitation of transcript levels was determined by phosphorimager analysis relative to the GAPDH control. (D) Ectopic p107 represses endogenous E2F activity in ES cells. ES cells were transfected with the E2F- or mutated E2F-luciferase reporter, a Renilla control, and 50 ng of empty pCAG expression vector (Vector), wild-type p107 (p107wt), or mutant p107 (p107ΔS/T-P). Firefly luciferase activity was normalized to Renilla luciferase activity, and data are presented as relative activity. Experiments were performed in triplicate, and data represent a typical experiment.
Collectively, these data indicate that pocket proteins are not recruited to endogenous E2F target promoters in ES cells, hence accounting for constitutive E2F-dependent transcriptional activity throughout the cell cycle (Stead et al., 2002). To address this question, we asked whether p107 binds the cyclin E1 promoter in vivo by using ChIP assays. In pluripotent (EPL) cells, p107 recruitment was low but became elevated during EB differentiation (Figure 5B). These results indicate that recruitment of p107, and possibly other pocket proteins, to E2F target genes occurs as Cdk2 activity collapses and becomes cell cycle dependent during differentiation.
To establish that the absence of pocket protein function explains the unrestrained E2F activity in ES cells, we asked whether a form of p107 that could evade Cdk action could regulate cyclin E1 transcription and E2F reporter gene activity. This hypothesis was tested by expressing a mutant form of p107 that evades Cdk regulation through multiple amino acid substitutions in key phosphorylation sites (Ashizawa et al., 2001; Kondo et al., 2001). The first approach to address this question involved the generation of stable ES cell lines expressing wild-type p107 (p107wt) or a mutant that evades Cdk inhibition (p107ΔS/T-P; Figure 5C). Although levels of p107ΔS/T-P expressed in each cell line were significantly lower than p107wt, the mutant had a more potent effect on suppressing endogenous cyclin E1 transcription. This was a general trend between several independent clonal cell lines tested. As a second readout, transient transfections were performed where an E2F-luciferase reporter was cotransfected with p107wt or p107ΔS/T-P expression plasmids (Figure 5D). In this assay, expression of both p107wt and p107ΔS/T-P suppressed activation of the reporter gene by endogenous E2F. Enforced p107 activity therefore inhibits E2F-dependent transcription in both assays. In endogenous E2F target genes, p107ΔS/T-P is a more potent regulator because of its ability to evade regulation by Cdk activity.
DISCUSSION
The fundamental mechanisms of cell cycle control are conserved in somatic cells and involve the cell cycle-regulated activity of cyclin-dependent kinases. Before specification into definitive cell types, pluripotent ES cells cycle at unusually rapid rates involving cell cycle mechanisms driven by cell cycle-independent Cdk activities (Stead et al., 2002; Faast et al., 2004). In this report, we set out to define the molecular events underpinning changes in cell cycle dynamics during ES cell differentiation.
A basic requirement for cell cycle progression is that replication origins are licensed to fire once in each S phase by a mechanism that requires low Cdk activity in the preceding G1 period (reviewed by Blow and Hodgson, 2002). Low Cdk activity in G1 establishes conditions where prereplicative complexes (pre-RCs) can assemble at origins. Once a Cdk signal is generated, pre-RCs are activated and generate a single bidirectional replication fork. On face value, constitutive Cdk activity would not be consistent with such a model because it would be expected that under these conditions, pre-RC formation would be inhibited. There are several possible explanations to reconcile this. It is possible that our previous study (Stead et al., 2002) failed to detect a subtle decrease in Cdk2 activity that would be expected during G1 for origins to license. In early zebrafish and Xenopus development, Cdk2 activity oscillates weakly in comparison with Cdk1–cyclin B (Rempel et al., 1995; Yarden and Geiger, 1996), consistent with this possibility. Alternatively, decreased Cdk1–cyclin B1 activity may suffice to enable pre-RCs to form in G1, even when Cdk2 activity remains elevated. The explanation that we favor, however, is that a pool of Cdk2 involved in origin regulation is inactive in G1, but this went undetected using standard assays that measure total Cdk activity in whole cell lysates. It is possible that a subset of Cdk2 complexes are in fact inactivated during G1, allowing for pre-RC assembly, but this is not detected in the background of elevated Cdk2 activity.
Deregulation of Cdk2 activity is often associated with accelerated entry into S phase (Duronio et al., 1996; Hua et al., 1997; Krude et al., 1997) and suggests a mechanism for how pluripotent ES cells divide rapidly by truncating the length of G1 (Stead et al., 2002). Before these studies began, however, it was not clear how the embryonic mode of cell cycle regulation switched to that of a typical somatic mechanism involving cell cycle-dependent Cdk activities. Our results clearly show that the acquisition of a full G1 phase during differentiation is associated with the collapse of Cdk2 activity. Moreover, Cdk2 activity becomes cell cycle dependent for the first time in development of embryonic cells. The major levels of control governing the establishment of Cdk2 activity are likely to include transcriptional control of cyclin synthesis, because cyclin A2 and E1 levels are clearly rate limiting in controlling Cdk2 activity in differentiation.
Up-regulation of Cdk inhibitors such as p21Cip1 and p27Kip1 also may be important for the collapse of Cdk2 activity that occurs during ES cell differentiation. All of these activities are absent in pluripotent cells. We previously reported the absence of INK family members such as p16INK4a in ES cells (Faast et al., 2004), suggesting that pluripotent cells of embryonic origin do not express the main Cdk inhibitory proteins. Another factor that may contribute to the overall change in cell cycle control could be the recruitment of Cdk2–cyclin into higher order complexes. Rapid proliferation in pluripotent cells may be generated by the high levels of discrete, active Cdk2–cyclin E1 complexes, enabling phosphorylation of transient substrates or rapid association with newly synthesized substrates. Our data suggest that the majority of Cdk2 is assembled into low-molecular-mass active complexes in pluripotent cells but as differentiation proceeds, the size of active complexes increases. This is likely to indicate their recruitment into different types of complexes and a change in their subcellular localization.
Cell cycle-regulated Cdk activity is normally required for the temporal inactivation of pRb family members and activation of E2F-dependent transcription during G1 phase. The constitutive activity of pRb/p107 kinases in ES cells predicts that pocket proteins will be held in a biochemically active state. The elevated transcription of E2F target genes throughout the cell cycle is consistent with this scenario (Stead et al., 2002). We focused on E2F4 as an example of how general E2F activity could be regulated in this system because it has been previously been reported to be the major E2F activity in ES cell (Humbert et al., 2000; Stead et al., 2002). Other E2F family members such as E2F1, 2, and 3 also may be important in ES cells, but because E2F activities were previously defined in whole cell extracts, their significance may have been overlooked because E2F1, 2, and 3 activity is more pronounced in nuclear extracts. Our data establish that constitutive transcription of canonical E2F target genes are regulated by E2F4-Dp activity and are not subject to p107-mediated repression. Although we did not specifically evaluate the status of pRb and p130 in these studies, it is likely that they will behave similarly to p107 in terms of them being inactive in ES cells and cell cycle regulated at some point during differentiation by cyclical Cdk activity. We hypothesized that the establishment of cell cycle-dependent Cdk activities would mark the time when the R-point machinery, including pRb family members, would become activated for the first time during development. This would establish a major transition point between pluripotency and the differentiated state. Our findings indicate that loss of pluripotency is associated with decreased Cdk activity, activation of the pRb pathway, and periodicity in transcription in E2F-dependent genes such as cyclin E1. Each of these elements are interrelated and conceivably work through a self-regulating feedback loop (Figure 6). Although cell cycle-dependent Cdk activity, cyclin E1 transcription and activation of pRb pathway regulate each other, the initiating step that triggers the transition from constitutive to periodic regulation was not defined by these studies. Breaking the self-sustaining loop of constitutive cyclin E1 transcription seems to be caused by the establishment of transcriptional periodicity and up-regulation of inhibitory proteins, but how this is established is unresolved. To determine whether the establishment of periodicity could be imposed by pocket protein activation per se, we used enforced expression of a constitutively active p107 mutant (p107ΔS/T). No major changes in periodicity were observed in these experiments, although E2F transcription was suppressed (Figure 5). It is likely that pocket protein activity needs to be itself cell cycle regulated to impose periodicity of E2F target genes. The experiments performed therefore shed no light on how periodicity is imposed.
Figure 6.

Establishment of G1 regulatory (R-point) controls through cell cycle-regulated Cdk2 activity during ES cell differentiation. Pluripotent ES cells exhibit elevated, constitutive Cdk activities, pRb family members are inactive and E2F-dependent transcription is cell cycle independent. During differentiation, Cdk activities collapse and become cell cycle regulated. As part of the establishment of cell cycle regulation, p107 and other pocket proteins (such as pRb and p130) become active and are able to repress E2F target genes during G1. Which of these events occurs first is unknown.
In Drosophila, establishment of G1 phase, after mitosis 16, requires cell cycle regulation of cyclin E expression, Cdk2–cyclin E activity, and the activity of pRb family members (Duronio and O'Farrell, 1994; Knoblich et al., 1994; Richardson et al., 1995; Secombe et al., 1998; Du and Dyson, 1999; Li et al., 1999). Accordingly, we propose that incorporation of G1/S regulation during mouse embryogenesis may require down-regulation of Cdk2–cyclin E activity and activity of pRb family members, as indicated by our ES cell-based studies. This would signify a major transition that has not been previously described in mammalian development. It has been previously reported that decreased cyclin E-CDK2 activity is required for differentiation in both Drosophila and Xenopus embryos (Knoblich et al., 1994; Howe et al., 1995; Rempel et al., 1995; Richardson et al., 1995; Hartley et al., 1996, 1997; Secombe et al., 1998; Li et al., 1999). This is consistent with our observations that document for the first time, the dynamics and molecular regulation of cell cycle regulation during early mammalian development by using ES cells as a model.
Regulation of the cell cycle in pluripotent cells of embryonic origin sets a precedent in terms of mammalian biology. This raises the question as to whether the molecular regulation of cell division in self-renewing stem cells is mechanistically coupled to self-renewal itself. Several reports have described that inactivation of the three pocket proteins pRb, p107, and p130 causes cell immortalization, suggesting a link between the cell cycle machinery and immortalized/self-renewing states (Dannenberg et al., 2000; Sage et al., 2000). The activity of Cdks and inactivity of pocket proteins in ES cells are consistent with this possibility. It will be intriguing to determine whether self-renewal in stem cells is related to their unusual mode of cell cycle regulation.
Acknowledgments
We thank Anne Chapman-Smith for advice and assistance with chromatography. This work was supported by grants from the National Health and Medical Research Council of Australia and from the Australian Research Council through the Centre for Molecular Genetics of Development. E. S. and J. W. were supported by Australian Postgraduate Research Awards.
This article was published online ahead of print in MBC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E04-12-1056) on February 9, 2005.
Abbreviations used: Cdk, cyclin-dependent kinase; ChIP, chromatin immunoprecipitation; dpc, days postcoitum; EB, embryoid body; EPL, early primitive ectoderm-like; ES, embryonic stem.
References
- Ashizawa, S., Nishizawa, H., Yamada, M., Higashi, H., Kondo, T., Ozawa, H., Kakita, A., and Hatakeyama, M. (2001). Collective inhibition of pRB family proteins by phosphorylation in cells with p16INK4a loss or cyclin E overexpression. J. Biol. Chem. 276, 11362-11370. [DOI] [PubMed] [Google Scholar]
- Blow, J. J., and Hodgson, B. (2002). Replication licensing-defining the proliferative state? Trends Cell Biol. 12, 72-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breeden, L. L. (2003). Periodic transcription: a cycle within a cycle. Curr. Biol. 13, R31-R38. [DOI] [PubMed] [Google Scholar]
- Bremner, R., Cohen, B. L., Sopta, M., Hamel, P. A., Ingles, C. J., Gallie, B. L., and Phillips, R. A. (1995). Direct transcriptional repression by pRB and its reversal by specific cyclins. Mol. Cell. Biol. 15, 3256-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartwright, P., Muller, H., Wagener, C., Holm, K., and Helin, K. (1998). E2F-6, a novel member of the E2F family is an inhibitor of E2F-dependent transcription. Oncogene 17, 611-623. [DOI] [PubMed] [Google Scholar]
- Dannenberg, J. H., van Rossum, A., Schuijff, L., and te Riele, H. (2000). Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev. 14, 3051-3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doetschman, T. C., Eistetter, H., Katz, M., Schmidt, W., and Kemler, R. (1985). The in vitro development of blastocyst-derived embryonic stem cell lines: formation of visceral yolk sac, blood islands and myocardium. J. Embryol. Exp. Morphol. 87, 27-45. [PubMed] [Google Scholar]
- Du, W., and Dyson, N. (1999). The role of RBF in the introduction of G1 regulation during Drosophila embryogenesis. EMBO J. 18, 916-925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duronio, R. J., Brook, A., Dyson, N., and O'Farrell, P. H. (1996). E2F-induced S phase requires cyclin E. Genes Dev. 10, 2505-2513. [DOI] [PubMed] [Google Scholar]
- Duronio, R. J., and O'Farrell, P. H. (1994). Developmental control of a G1-S transcriptional program in Drosophila. Development 120, 1503-1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, B. A., and Schubiger, G. (1986). Parameters controlling transcriptional activation during early Drosophila development. Cell 44, 871-877. [DOI] [PubMed] [Google Scholar]
- Edgar, B. A., Sprenger, F., Duronio, R. J., Leopold, P., and O'Farrell, P. H. (1994). Distinct molecular mechanism regulate cell cycle timing at successive stages of Drosophila embryogenesis. Genes Dev. 8, 440-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faast, R., White, J., Cartwright, P., Crocker, L., Sarcevic, B., and Dalton, S. (2004). Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a). Oncogene. 23, 491-502. [DOI] [PubMed] [Google Scholar]
- Foe, V. E., and Alberts, B. M. (1983). Studies of nuclear and cytoplasmic behaviour during the five mitotic cycles that precede gastrulation in Drosophila embryogenesis. J. Cell Sci. 61, 31-70. [DOI] [PubMed] [Google Scholar]
- Harbour, J. W., Luo, R. X., Dei Santi, A., Postigo, A. A., and Dean, D. C. (1999). Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98, 859-869. [DOI] [PubMed] [Google Scholar]
- Hartley, R. S., Rempel, R. E., and Maller, J. L. (1996). In vivo regulation of the early embryonic cell cycle in Xenopus. Dev. Biol. 173, 408-419. [DOI] [PubMed] [Google Scholar]
- Hartley, R. S., Sible, J. C., Lewellyn, A. L., and Maller, J. L. (1997). A role for cyclin E/Cdk2 in the timing of the midblastula transition in Xenopus embryos. Dev. Biol. 188, 312-321. [DOI] [PubMed] [Google Scholar]
- Hogan, B., Beddington, R., Constantini, F., and Lacy, E. (1994). Manipulating the Mouse Embryo. A Laboratory Manual, Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
- Howe, J. A., Howell, M., Hunt, T., and Newport, J. W. (1995). Identification of a developmental timer regulating the stability of embryonic cyclin A and a new somatic A-type cyclin at gastrulation. Genes Dev. 9, 1164-1176. [DOI] [PubMed] [Google Scholar]
- Hua, X. H., Yan, H., and Newport, J. (1997). A role for Cdk2 kinase in negatively regulating DNA replication during S phase of the cell cycle. J. Cell Biol. 137, 183-192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert, P. O., et al. (2000). E2F4 is essential for normal erythrocyte maturation and neonatal viability. Mol. Cell 6, 281-291. [DOI] [PubMed] [Google Scholar]
- Knoblich, J. A., Sauer, K., Jones, L., Richardson, H., Saint, R., and Lehner, C. F. (1994). Cyclin E controls S phase progression and its down-regulation during Drosophila embryogenesis is required for the arrest of cell proliferation. Cell 77, 107-120. [DOI] [PubMed] [Google Scholar]
- Kondo, T., Higashi, H., Nishizawa, H., Ishikawa, S., Ashizawa, S., Yamada, M., Makita, Z., Koike, T., and Hatakeyama, M. (2001). Involvement of pRB-related p107 protein in the inhibition of S phase progression in response to genotoxic stress. J. Biol. Chem. 276, 17559-17567. [DOI] [PubMed] [Google Scholar]
- Krude, T., Jackman, M., Pines, J., and Laskey, R. A. (1997). Cyclin/Cdk-dependent initiation of DNA replication in a human cell-free system. Cell 88, 109-119. [DOI] [PubMed] [Google Scholar]
- Lake, J., Rathjen, J., Remiszewski, J., and Rathjen, P. D. (2000). Reversible programming of pluripotent cell differentiation. J. Cell Sci. 113, 555-566. [DOI] [PubMed] [Google Scholar]
- Leone, G., DeGregori, J., Jakoi, L., Cook, J. G., and Nevins, J. R. (1999). Collaborative role of E2F transcriptional activity and G1 cyclin dependent kinase activity in the induction of S phase. Proc. Natl. Acad. Sci. USA 96, 6626-6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Q. J., Pazdera, T. M., and Minden, J. S. (1999). Drosophila embryonic pattern repair: how embryos respond to cyclin E-induced ectopic division. Development 126, 2299-2307. [DOI] [PubMed] [Google Scholar]
- McConnell, B. B., Gregory, F. J., Stott, F. J., Hara, E., and Peters, G. (1999). Induced expression of p16(INK4a) inhibits both CDK4- and CDK2-associated kinase activity by reassortment of cyclin-CDK-inhibitor complexes. Mol. Cell. Biol. 19, 1981-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, A. W., and Kirschner, M. W. (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275-280. [DOI] [PubMed] [Google Scholar]
- Musgrove, E. A., Swarbrick, A., Lee, C. S., Cornish, A. L., and Sutherland, R. L. (1998). Mechanisms of cyclin-dependent kinase inactivation by progestins. Mol. Cell. Biol. 18, 1812-1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport, J., and Kirschner, M. (1982). A major developmental transition in early Xenopus embryos: I. Characterization and timing of cellular changes at the midblastula stage. Cell 30, 675-686. [DOI] [PubMed] [Google Scholar]
- Newport, J. W., and Kirschner, M. W. (1984). Regulation of the cell cycle during early Xenopus development. Cell 37, 731-742. [DOI] [PubMed] [Google Scholar]
- Nigg, E. A. (1995). Cyclin-dependent protein kinases: key regulators of the eukaryotic cell cycle. Bioessays 17, 471-480. [DOI] [PubMed] [Google Scholar]
- Ohtsubo, M., and Roberts, J. M. (1993). Cyclin-dependent regulation of G1 in mammalian fibroblasts. Science 259, 1908-1912. [DOI] [PubMed] [Google Scholar]
- Parry, D., Mahony, D., Wills, K., and Lees, E. (1999). Cyclin D-CDK subunit arrangement is dependent on the availability of competing INK4 and p21 class inhibitors. Mol. Cell. Biol. 19, 1775-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathan, N. I., Geahlen, R. L., and Harrison, M. L. (1996). The protein-tyrosine kinase Lck associates with and is phosphorylated by Cdc2. J. Biol. Chem. 271, 27517-27523. [DOI] [PubMed] [Google Scholar]
- Pelton, T. A., Sharma, S., Schulz, T. C., Rathjen, J., and Rathjen, P. D. (2002). Transient pluripotent cell populations during primitive ectoderm formation: correlation of in vivo and in vitro pluripotent cell development. J. Cell Sci. 115, 329-339. [DOI] [PubMed] [Google Scholar]
- Prall, O. W., Rogan, E. M., Musgrove, E. A., Watts, C. K., and Sutherland, R. L. (1998). c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Mol. Cell. Biol. 18, 4499-4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall, O. W., Sarcevic, B., Musgrove, E. A., Watts, C. K., and Sutherland, R. L. (1997). Estrogen-induced activation of Cdk4 and Cdk2 during G1-S phase progression is accompanied by increased cyclin D1 expression and decreased cyclin-dependent kinase inhibitor association with cyclin E-Cdk2. J. Biol. Chem. 272, 10882-10894. [DOI] [PubMed] [Google Scholar]
- Pratt, T., Sharp, L., Nichols, J., Price, D. J., and Mason, J. O. (2000). Embryonic stem cells and transgenic mice ubiquitously expressing a tau-tagged green fluorescent protein. Dev. Biol. 228, 19-28. [DOI] [PubMed] [Google Scholar]
- Rathjen, J., Lake, J. A., Bettess, M. D., Washington, J. M., Chapman, G., and Rathjen, P. D. (1999). Formation of a primitive ectoderm like cell population, EPL cells, from ES cells in response to biologically derived factors. J. Cell Sci. 112, 601-612. [DOI] [PubMed] [Google Scholar]
- Rempel, R. E., Sleight, S. B., and Maller, J. L. (1995). Maternal Xenopus Cdk2-cyclin E complexes function during meiotic and early embryonic cell cycles that lack a G1 phase. J. Biol. Chem. 270, 6843-6855. [DOI] [PubMed] [Google Scholar]
- Resnitzky, D., Gossen, M., Bujard, H., and Reed, S. I. (1994). Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol. 14, 1669-1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky, D., and Reed, S. I. (1995). Different roles for cyclins D1 and E in regulation of the G1-to-S transition. Mol. Cell. Biol. 15, 3463-3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson, H., O'Keefe, L. V., Marty, T., and Saint, R. (1995). Ectopic cyclin E expression induces premature entry into S phase and disrupts pattern formation in the Drosophila eye imaginal disc. Development 121, 3371-3379. [DOI] [PubMed] [Google Scholar]
- Sage, J., Mulligan, G. J., Attardi, L. D., Miller, A., Chen, S., Williams, B., Theodorou, E., and Jacks, T. (2000). Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 14, 3037-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savatier, P., Huang, S., Szekely, L., Wiman, K. G., and Samarut, J. (1994). Contrasting patterns of retinoblastoma protein expression in mouse embryonic stem cells and embryonic fibroblasts. Oncogene 9, 809-818. [PubMed] [Google Scholar]
- Savatier, P., Lapillonne, H., van Grunsven, L. A., Rudkin, B. B., and Samarut, J. (1996). Withdrawal of differentiation inhibitory activity/leukemia inhibitory factor up-regulates D-type cyclins and cyclin-dependent kinase inhibitors in mouse embryonic stem cells. Oncogene 12, 309-322. [PubMed] [Google Scholar]
- Secombe, J., Pispa, J., Saint, R., and Richardson, H. (1998). Analysis of a Drosophila cyclin E hypomorphic mutation suggests a novel role for cyclin E in cell proliferation control during eye imaginal disc development. Genetics 149, 1867-1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snow, M.H.L. (1977). Gastrulation in the mouse: growth and regionalization of the epiblast. J. Embryol. Exp. Morphol. 42, 293-303. [Google Scholar]
- Solter, D., Skreb, N., and Damjanov, I. (1971). Cell cycle analysis in the mouse EGG-cylinder. Exp. Cell Res. 64, 331-334. [DOI] [PubMed] [Google Scholar]
- Stead, E., White, J., Faast, R., Conn, S., Goldstone, S., Rathjen, J., Dhingra, U., Rathjen, P., Walker, D., and Dalton, S. (2002). Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene 21, 8320-8333. [DOI] [PubMed] [Google Scholar]
- Steiner, P., Philipp, A., Lukas, J., Godden-Kent, D., Pagano, M., Mittnacht, S., Bartek, J., and Eilers, M. (1995). Identification of a Myc-dependent step during the formation of active G1 cyclin-cdk complexes. EMBO J. 14, 4814-4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiffler, L. A., Ji, J. Y., Trautmann, S., Trusty, C., and Schubiger, G. (1999). Cyclin A and B functions in the early Drosophila embryo. Development 126, 5505-5513. [DOI] [PubMed] [Google Scholar]
- Swarbrick, A., Lee, C. S., Sutherland, R. L., and Musgrove, E. A. (2000). Cooperation of p27(Kip1) and p18(INK4c) in progestin-mediated cell cycle arrest in T-47D breast cancer cells. Mol. Cell. Biol. 20, 2581-2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney, K. J., Swarbrick, A., Sutherland, R. L., and Musgrove, E. A. (1998). Lack of relationship between CDK activity and G1 cyclin expression in breast cancer cells. Oncogene. 16, 2865-2878. [DOI] [PubMed] [Google Scholar]
- Tyers, M., and Jorgensen, P. (2000). Proteolysis and the cell cycle: with this RING I do thee destroy. Curr. Opin. Genet. Dev. 10, 54-64. [DOI] [PubMed] [Google Scholar]
- Yarden, A., and Geiger, B. (1996). Zebrafish cyclin E regulation during early embryogenesis. Dev. Dyn. 206, 1-11. [DOI] [PubMed] [Google Scholar]
- Zhang, H. S., Gavin, M., Dahiya, A., Postigo, A. A., Ma, D., Luo, R. X., Harbour, J. W., and Dean, D. C. (2000). Exit from G1 and S phase of the cell cycle is regulated by repressor complexes containing HDAC-Rb-hSWI/SNF and Rb-hSWI/SNF. Cell 101, 79-89. [DOI] [PubMed] [Google Scholar]