

Abstract
Parkinson’s disease (PD) is associated with an increase in secreted S100B within the midbrain and cerebrospinal fluid. In addition, S100B overexpression in mice accelerates the loss of substantia nigra pars compacta dopaminergic (DA) neurons, suggesting a role for this protein in PD pathogenesis. We found that in the mouse SNc, S100B labeled astrocytic processes completely envelop the somata of tyrosine hydroxylase (TH) expressing DA neurons only in male mice. These data suggest that an increase in S100B secretion by astrocytes within the midbrain could play a role in DA dysfunction during early PD. We therefore asked if acute exposure to extracellular S100B alters the activity of identified TH expressing DA neurons in primary mouse midbrain cultures. Acute exposure to 50 pM S100B specifically inhibited A-type voltage-gated potassium currents in TH+, but not TH− neurons. This was accompanied by ~2-fold increases in the frequency of both intrinsic firing, as well as L-type voltage-gated calcium channel (VGCC)-mediated calcium fluxes only in TH+ neurons. Further, exposure to 100 μM 4-aminopyridine (4-AP), an A-type voltage-gated potassium channel inhibitor, mimicked the S100B mediated increase in intrinsic firing and L-type VGCC-mediated calcium fluxes in TH+ neurons. Taken together, our finding that extracellular S100B alters the activity of native DA neurons via an inhibition of A-type voltage-gated potassium channels has important implications for understanding the pathophysiology of early PD.
Keywords: astrocytes, calcium channels, neurodegeneration, Parkinson’s disease, potassium channels, S100B
1 |. INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, projected to reach pandemic proportions by 2040 (Dorsey & Bloem, 2018). Understanding the mechanisms by which dopaminergic (DA) neurons degenerate in PD is therefore vital for developing effective disease-modifying treatments that slow down or even stop the progressive loss of DA neurons.
Interestingly, patients with PD display increased levels of S100B in their cerebrospinal fluid and serum (Sathe et al., 2012; Schaf et al., 2005), as well as significantly higher S100B expression in the substantia nigra pars compacta (SNc) (Sathe et al., 2012). Several lines of evidence show that apart from being merely associated with PD, upregulated S100B can also actively contribute to the degeneration of DA neurons. Specifically, studies show that: (i) A single nucleotide polymorphism, rs9722, which is associated with increased levels of serum S100B results in an elevated risk for early onset PD (Fardell et al., 2018; Hohoff et al., 2010), (ii) Ablation of S100B in mice protects against MPTP-induced DA loss (Sathe et al., 2012), (iii) Mice overexpressing S100B develop Parkinsonian features (Liu et al., 2011; Sathe et al., 2012), and (iv) Overnight S100B elevation correlates with increased PD severity and sleep disruption (Carvalho et al., 2015). Taken together, these reports clearly converge on the idea that abnormal levels of S100B in the SNc could trigger the degeneration of DA neurons in PD patients.
During PD, midbrain astrocytes become reactive and demonstrate a pathological increase in the expression levels of astrocyte-specific proteins such as glial fibrillary acid protein (GFAP) (Batassini et al., 2015; Thannickal et al., 2007) and S100B (Sathe et al., 2012). Among the many proteins that are upregulated in reactive astrocytes, S100B is of particular interest in the context of PD because of its potential to initiate pathological processes in DA neurons during early-stage PD. Indeed, extracellular S100B has been shown to alter neuronal activity in multiple brain regions (Morquette et al., 2015; Ryczko et al., 2021), and extracellularly secreted S100B from astrocytes accelerates neurodegeneration by engaging receptor for advanced glycation endproducts (RAGE)-mediated pro-inflammatory pathways in astrocytes and microglia (Hofmann et al., 1999; Huttunen et al., 2000; Riuzzi et al., 2012). In addition to signaling through RAGE receptors, S100B and the S100 family of proteins interact intracellularly with several ion channels and receptors expressed in neurons, and these interactions result in significant biological effects such as increased neurotropism or the modulation of neuronal excitability (Hermann et al., 2012). The documented effects of S100B on neurons point to S100B as an active participant in modulating neuronal function during health and disease.
In this study, we test the hypothesis that an abnormal increase in extracellular S100B, secreted by midbrain astrocytes alters the activity of SNc DA neurons by interacting with ion channels expressed on the surface of these neurons. Using primary cocultures of mouse midbrain neurons and astrocytes, we show that exposure of midbrain cultures to picomolar concentrations of S100B specifically alters the activity of tyrosine hydroxylase expressing (TH+) DA neurons by mechanisms that include: (i) Increasing intrinsic neuronal firing via an inhibition of A-type voltage-gated potassium channels (VGKCs) and (ii) Increasing the frequency of spontaneous Ca2+ fluxes via L-type voltage-gated calcium channels (VGCCs). Since a large body of evidence now suggests that VGKCs, VGCCs, and astrocytes play central roles in the development of PD (Booth et al., 2017; Gomez et al., 2019; Ilijic et al., 2011; Iyer et al., 2017; Koyama & Appel, 2006; Lang et al., 2019; Liss et al., 2001; Martel et al., 2011; Noh et al., 2019; Olson et al., 2005; Sun et al., 2017), our results have important implications for understanding how extracellular S100B secreted by midbrain astrocytes can alter the function of voltage-gated channels expressed in DA neurons, thereby resulting in early PD.
2 |. MATERIALS AND METHODS
2.1 |. Mice
All experimental procedures with mice were approved by the Texas A&M University Institutional Animal Care and Use Committee (IACUC), animal use protocol numbers 2017-0053 and 2019-0346. Adult (3–4 month) male and female C57BL/6 wild type mice and embryos (ED14) from timed-pregnant female C57BL/6 mice were obtained from the Texas A&M Institute for Genomic Medicine (TIGM). Pregnant mice were group housed in a temperature controlled environment on a 12:12 h light:dark cycle with food and water available ad libitum.
2.2 |. Stereotaxic intracranial injections
AAV 2/5 GfaABC1D-Lck-GCaMP6f was stereotaxically injected into the SNc of adult (3–4 month) mice using previously described methods (Srinivasan et al., 2015; Srinivasan, Lu, et al., 2016). Briefly, each mouse was unilaterally injected with 1 μl of AAV 2/5 GfaABC1D-Lck-GCaMP6f (1013 genome copies/ml) (Vector Builder) with a beveled glass injection pipette using a motorized Pump 11 Pico Plus Elite pump (Harvard Apparatus), attached to a stereotaxic frame (Kopf Instruments). AAVs were injected into the SNC at a rate of 500 nl/min. Coordinates for SNc injections were 3.0 mm posterior to bregma, 1.5 mm lateral to midline, and 4.2 mm ventral to the pial surface. For all stereotaxic surgeries, mice were anesthetized using isoflurane dispensed through a SomnoSuite Low Flow Anesthesia System (Kent Scientific).
2.3 |. Immunostaining of mouse brain sections and DA cultures
Three to four-month-old male and female mice were transcardially perfused with phosphate-buffered saline (PBS, ThermoFisher) followed by 10% Formalin/PBS (VWR). Brains were postfixed in 10% Formalin/PBS for 24–48 h at 4°C, then moved to 30% sucrose (Sigma-Aldrich) in PBS for dehydration. Brains were sectioned in the coronal plane to obtain midbrain sections with 40 μm thickness using a sliding microtome (SM2010 R, Leica). Midbrain sections were permeabilized in 0.5% Triton X-100 (Sigma-Aldrich) in PBS, and blocked in 10% normal goat serum (NGS, Abcam) in PBS. Primary antibodies used were rabbit polyclonal anti-S100B (1:1000, Abcam) and chicken polyclonal anti-TH (1:1000, Abcam), mouse monoclonal anti-NeuN (1:1000, Abcam), rabbit polyclonal anti-GFP (1:1000, Abcam). Secondary antibodies used were goat polyclonal anti-rabbit Alexa Fluor 488 (1:1000, Abcam), goat polyclonal anti-chicken Alexa Fluor 594 (1:1000, Abcam), and goat polyclonal anti-mouse Alexa Fluor 405 (1:1000, Abcam). Antibodies were incubated in a 1% NGS/PBS solution. For each midbrain subregion, 2 fields of view (FOV) from 2 to 3 sections per mouse (three mice total per brain region) were used to quantify wrapping of neuronal somata in the SNc, ventral tegmental area (VTA) and substantia nigra pars reticulata (SNr) by S100B-containing astrocytic processes.
DA cultures were fixed by placing coverslips in 10% Formaldehyde/PBS for 40 min. Cultures were permeabilized in 0.01% Triton X-100/PBS and blocked in 10% NGS/PBS solution. Antibodies used in sections were also used in cultures, with the addition of mouse polyclonal anti-NeuN (1:1000, Abcam) and goat polyclonal anti-mouse Alexa Fluor 405 (1:1000, Abcam). Imaging was performed using a confocal microscope (Fluoview 1200, Olympus) with a ×60 and 1.35 numerical aperture (NA) oil-immersion objective (Olympus).
2.4 |. Primary mouse midbrain neuron and astrocyte cocultures
For co-culturing primary mouse midbrain neurons and astrocytes, neurobasal medium, DMEM + GlutaMAX medium, GlutaMAX supplement, B-27, equine serum, and penicillin–streptomycin were purchased from ThermoFisher. Deoxyribonuclease I (DNase), poly-l-lysine, poly-l-ornithine, laminin, ascorbic acid, kanamycin, and ampicillin were purchased from Sigma-Aldrich. Corning 35 mm uncoated plastic cell culture dishes were purchased from VWR, 12 mm circular cover glass No. 1 was purchased from Phenix Research Products (Candler). Papain was purchased from Worthington Biomedical Corporation (Lakewood).
Detailed methods to coculture primary mouse DA neuron and astrocytes have been previously described (Bancroft & Srinivasan, 2020; Henley et al., 2017; Zarate et al., 2021). Briefly, cultures were obtained from embryonic day (ED14) mouse embryos of mixed sexes. Timed-pregnant mice (obtained from Texas A&M Institute for Genomic Medicine) were sacrificed via cervical dislocation and embryos were removed. Embryos were decapitated and ventral midbrain was dissected using previously described methods (Bancroft & Srinivasan, 2020; Henley et al., 2017). Following dissection, cells were digested in papain for 15 min at 37°C, then cells were separated using DNase treatment and mechanical trituration in a stop solution of 10% equine serum in PBS. Cells were plated at a density between 200,000 and 300,000 cells per cover glass on 12 mm circular cover glasses triple coated with poly-l-lysine, poly-l-ornithine and laminin. After plating, cells were placed in an incubator at 37°C with 5% CO2 for 1 h, followed by addition of 3 ml of neurobasal media supplemented with GlutaMAX, B-27, equine serum, ascorbic acid and containing penicillin–streptomycin, kanamycin, and ampicillin. Culture medium was exchanged at 3 days intervals and all primary mouse midbrain neuron-astrocyte cocultures were maintained for at least 3 weeks before performing experiments.
2.5 |. Adeno-associated virus vectors
To confirm that S100B containing processes enveloping neurons in the SNc were astrocytic, we utilized adeno-associated virus (AAV) 2/5 GfaABC1D-Lck-GCaMP6 obtained as a gift from Baljit Khakh deposited to Addgene (Watertown).
To perform electrophysiology and image spontaneous Ca2+ fluxes in visually identified DA neurons from primary mouse midbrain cocultures, we utilized AAVs that reliably report TH+ DA neurons (either AAV 2/5 TH-GFP or AAV 2/5 TH-tdTomato). AAV 2/5 TH-GFP and AAV 2/5 TH-tdTomato were packaged by Vector Builder (Chicago, IL). The targeting plasmid for packaging AAV 2/5 TH-tdTomato was cloned from a pAAV-mouse THp-eGFP plasmid obtained as a gift from Dr. Viviana Gradinaru (California Institute of Technology, Pasadena, CA). The eGFP cassette in the pAAV-mouse THp-eGFP plasmid was replaced with tdTomato between restriction sites Not1 and BamH1. Spontaneous Ca2+ fluxes in midbrain neurons were recorded using either AAV 2/5 hSyn-GCaMP6f or AAV 2/5 hSyn-RCaMP, both of which were purchased from Addgene.
All AAV infections were performed at 14 days in vitro (DIV), as previously described (Bancroft & Srinivasan, 2020). For AAV infections, the culture medium was removed and 1 ml of serum-free DMEM + GlutaMAX medium containing ~1 × 1010 genome copies of each AAV was added to each dish and allowed to incubate at 37°C with 5% CO2 for 1 h. Serum-free medium containing AAVs was then removed and replaced with 3 ml of neurobasal medium supplemented with equine serum, B27, and GlutaMAX. Electrophysiological measurements and imaging experiments were performed on day 5 following infection with appropriate AAVs.
2.6 |. Electrophysiology
Whole-cell recordings in both current and voltage-clamp mode were performed on cultured mouse midbrain neurons. Whole-cell current clamp was used to measure action potential (AP) frequency while whole-cell voltage-clamp was used to measure the fast inactivating (IA) and non-inactivating (IK) voltage-gated potassium currents. All recordings were performed using a Multiclamp 700B amplifier, Digidata 1400A interface, and pClamp 10 software (Molecular Devices). All recordings utilized the extracellular gas-free recording buffer described below for Ca2+ imaging with the addition of AP5 (50 μM, Hello Bio), NBQX (10 μM, Hello Bio), and Bicuculline (10 μM, Hello Bio) to measure intrinsic action potentials in cultured mouse midbrain neurons. Voltage-clamp experiments measuring IA and IK potassium currents also utilized tetrodotoxin (TTX, 0.3 μM). For electrophysiology and Ca2+ imaging experiments, monomeric recombinant human S100B peptide was obtained from Sino Biological (cat # 10181-H07E; Wayne, PA). To denature S100B peptide, recombinant S100B was boiled at 100°C for 1 h. Extracellular recording buffers were bath perfused and solution exchanges were performed using a gravity-based feed delivery system with a perfusion rate of 2 ml/min. In all physiology experiments (electrophysiology and Ca2+ imaging), multiple bath applications of S100B were performed on the same cultures with a minimum 10 min washout period between applications. We did not observe any differences in the effect of S100B between the first and last applications. The internal pipette solution for both current and voltage-clamp recordings contained (in mM): 136 K-gluconate, 10 HEPES, 0.1 EGTA, 4 Mg-ATP, 0.1 GTP, 25 NaCl, 2 MgCl2, pH 7.2 with KOH, 290–300 mOsm. Patch pipettes were pulled from glass capillary tubing (KG-33, 1.5 mm outer diameter, King Precision Glass) using a Flaming/Brown P-87 pipette puller (Sutter Instrument) with resistance between 6 and 12 MΩ, and were fire polished prior to use.
To isolate IA currents in neurons, two voltage-step protocols were sequentially employed in whole-cell mode. The first protocol was used to record total voltage-gated potassium channel (VGKC) currents and consisted of a 100 ms prepulse (max conductance) at −90 mV followed by a 250 ms step at increasingly depolarized potentials (from −100 to 50 mV, 10 mV steps) (Figure S1A). The second protocol was used to inhibit IA currents in order to specifically isolate IK. For this protocol, the patched neuron was subjected to a 100 ms prepulse at +50 mV, followed by 250 ms steps at increasingly depolarized potentials (from −100 to +50 mV, 10 mV steps) (Figure S1B). In order to specifically visualize and measure IA in TH+ and TH− midbrain neurons, current traces from the second protocol were subtracted from the first protocol using Clampfit 11.1 (Molecular Devices) (Figure S1C).
2.7 |. Imaging of spontaneous Ca2+ fluxes in cultured midbrain neurons
The protocol for imaging spontaneous Ca2+ fluxes in primary cultures of mouse midbrain neurons has been previously published (Bancroft & Srinivasan, 2020). Briefly, cultures were placed in a gas free recording buffer containing (mM): 154 NaCl, 5 KCl, 2 CaCl2, 0.5 MgCl2, 5 D-glucose, 10 HEPES, pH adjusted to 7.4 with NaOH (all purchased from Sigma-Aldrich). Imaging was performed using a confocal microscope (Fluoview 1200, Olympus) with a ×40, 0.8 NA water-immersion objective (Olympus). We used a 488-nm laser line to excite GCaMP6f and eGFP, and a 569-nm laser line for tdTomato and RCaMP. The imaging frame was clipped to allow for a sampling rate of 1 frame per second. For all experiments, spontaneous activity was imaged for 300 s, followed by peptide/drug application. Recording buffers were bath perfused using a peristaltic pump at a rate of 2 ml/min. For each field of view imaged, a corresponding z-stack of both GCaMP6f and TH-tdTomato expression was captured and co-localization was used to identify TH+ cells. Diltiazem was purchased from Tocris (Minneapolis, MN), Mibefradil and 4-aminopyridine (4-AP) were purchased from Sigma-Aldrich, and cyclopiazonic acid (CPA) was purchased from Abcam.
2.8 |. Image processing and data analyses
Image processing was performed using ImageJ v1.52e (NIH). To quantify astrocyte wrapping of neuronal somata from immunostained mouse midbrain sections, z-stacks of TH, NeuN, and corresponding S100B labeled midbrain sections were converted to projection images using the maximum intensity projection tool in ImageJ. TH and NeuN expressing neuronal somata were traced using the polygon tool. Neuronal somata regions of interest (ROIs) were manually demarcated in this way were applied to the S100B maximum intensity projection image, followed by clearing of all S100B signals outside of the neuronal soma ROI. Then, the S100B signal from maximum intensity projections were manually thresholded to capture all S100B-labeled astrocytic processes within the neuronal ROI. A separate ROI for S100B labeled astrocytic processes within the neuronal somata ROI was created in this way, and ROI areas as well as mean gray values were separately measured for both neuronal somata and for S100B-containing astrocytic processes within the neuronal somata ROI. Astrocyte wrapping around each neuronal somata was derived as a percentage by dividing the neuronal ROI area by the S100B ROI area and multiplying by 100. This analysis was performed for neurons and astrocytes in the SNc, VTA, and SNr.
For analysis of Ca2+ fluxes in TH+ and TH− neurons from primary mouse midbrain cocultures, time stacks (t-stacks) obtained from confocal imaging of live midbrain neurons were used to create maximum intensity projections and adjusted to increase brightness in order to visualize all active neuronal somata in a field of view. Somata displaying spontaneous Ca2+ fluxes were individually traced with the polygon tool and added to the ROI manager. The ROIs were used to extract neuronal Ca2+ flux traces from t-stacks, and only somata with one or more spontaneous Ca2+ flux events were selected for analysis. Mean gray values from Ca2+ flux event traces were converted to ΔF/F values using a 10 s period with no Ca2+ flux events to obtain baseline fluorescence (F). For each XY time-series movie analyzed, a corresponding z-stack image of both hSyn-GCaMP6f/hSyn-RCaMP and TH-tdTomato/TH-GFP expression was used to create red and green fluorescence co-localized max intensity projection images. Images obtained in this way were merged, and co-localization of GCaMP6f/RCaMP with tdTomato/GFP was used to definitively identify TH+ neurons, while cells displaying GCaMP6f/RCaMP labeling without tdTomato/GFP were identified as TH− neurons. All neuronal Ca2+ flux traces obtained using the above procedures were manually analyzed with MiniAnalysis v6.0.7 (Synaptosoft) to detect and quantify Ca2+ flux frequency (events/min) and amplitude (ΔF/F). Area under the curve (AUC) measurements were quantified using Origin 2019 v9.6 (OriginLab). Traces were plotted as line graphs and the integrate tool in the gadgets menu of Origin 2019 was applied to each trace. The baseline was held constant for all traces and area under the curve was measured for individual time segments of each trace as follows: Spontaneous (0–300 s), 4-AP (300–600 s), 4-AP + diltiazem (900–1200 s). All analysis of electrophysiological data, including subtraction of VGKC traces was performed using Clampfit 11.1 (Molecular Devices). Action potentials from electrophysiological recordings were quantified using the event detection function in MiniAnalysis (Synaptosoft).
2.9 |. Statistical analyses
All statistical analyses were performed using Origin 2019 v9.6. Data presented are mean ± SEM. For each data set, normality was first determined in Origin using the Shapiro–Wilk test. Normally distributed data were analyzed via two-sample t tests or paired sample t tests when appropriate. Non-normally distributed data were analyzed with Mann–Whitney or paired sample Wilcoxon-signed rank test for between-group differences. Data were considered to be significantly different at p < .05. For statistical analysis of differences in VGKC current amplitudes, current amplitudes of control versus S100B exposed cells were subjected to mixed-design ANOVA analysis, followed by a post hoc Bonferroni test to statistically compare differences in current amplitude at specific voltages. The exact statistical test used and sample sizes are described for each experiment in the figure legends. Exact p-values comparing datasets are shown in each of the figures.
3 |. RESULTS
3.1 |. Wrapping of S100B-containing astrocytic processes around SNc DA neurons is significantly increased only in male mice
The foundational premise of this study is based on testing the hypothesis that extracellular S100B, secreted by astrocytes can alter the activity of DA neurons in the SNc. Therefore, we first assessed the extent to which S100B containing astrocytic processes envelop TH+ and TH− neurons in the SNc, VTA, and SNr of adult male and female mice.
To do this, midbrain sections of 3–4-month-old adult male and female mice were immunostained for TH, S100B, and NeuN, then imaged and quantified (Figure 1a–d). Surprisingly, we found that TH+ SNc neurons in only male mice showed a significant ~1.69-fold increase in wrapping of S100B containing astrocytic processes compared to TH+ neurons in the VTA and TH− neurons in the SNC, VTA, and SNr (SNc TH+: 26.26 ± 1.76 μm2, SNc TH−: 15.54 ± 2.71 μm2, VTA TH+: 15.49 ± 1.40 μm2, VTA TH−: 15.52 ± 1.54 μm2, SNr TH−: 19.46 ± 1.57 μm2). (Figure 1d). By contrast, female mice demonstrated largely similar astrocytic wrapping of TH+ and TH− neurons in the SNc, VTA, and SNr (SNc TH+: 19.85 ± 1.28 μm2, SNc TH−: 15.73 ± 1.36 μm2, VTA TH+: 15.69 ± 1.08 μm2, VTA TH−: 15.08 ± 1.77 μm2, SNr TH−: 18.19 ± 1.75 μm2) (Figure 1d). In addition, astrocytic wrapping of TH+ SNc neurons in male mice was significantly ~1.32-fold higher than in female mice (Figure 1d). To definitively confirm that the cells wrapping around TH+ neurons in the SNc are astrocytes, we injected the midbrain of adult male mice with an AAV expressing Lck-GCaMP6f under transcriptional control of the well-established astrocyte-specific promoter, GfaABC1D. Following 2 weeks of expression, brains were extracted, sectioned, and fixed with 10% formalin and then immunostained with an anti-GFP and anti-TH antibody to visualize GCaMP6f expression in relation to DA neuron somata. As shown in Figure 1e, GCaMP6f was robustly expressed in astrocytic processes that wrap around TH+ neuronal somata.
FIGURE 1.
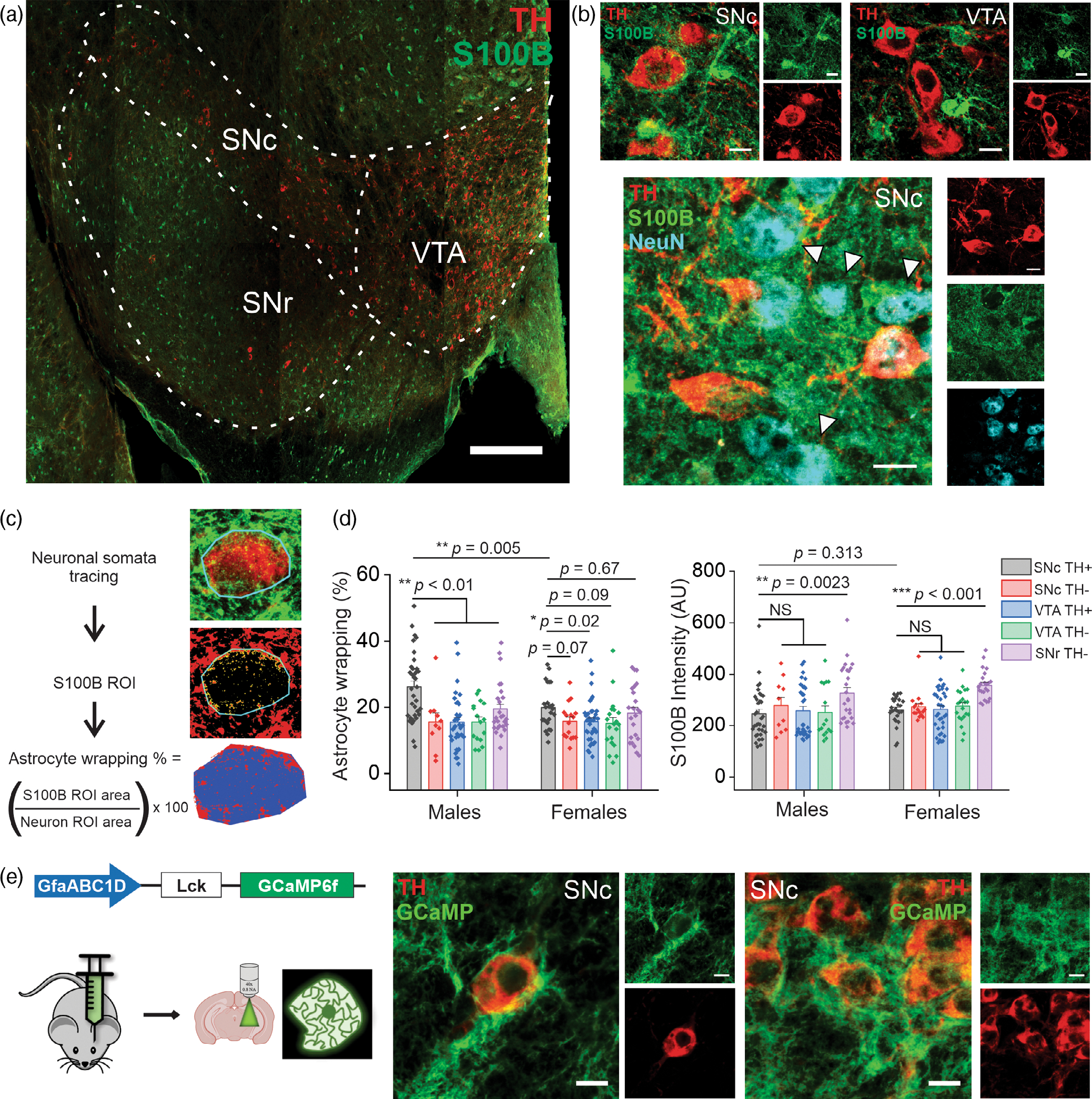
Wrapping of S100B-containing astrocytic processes around SNc DA neurons is significantly increased only in male mice. (a) A representative confocal mosaic of a mouse midbrain section immunostained for S100B (green) and TH (red), scale bar = 300 μm. Subregions of the midbrain are indicated with dotted lines (SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata; VTA, ventral tegmental area). (b) Representative high magnification confocal images of S100B and TH expression in the SNc (top left) and VTA (top right), NeuN expression in the SNc is shown in the lower panel with white arrows denoting TH− neurons, scale bar = 10 μm. (c) Schematic of astrocyte wrapping analysis, described in detail in the methods section. (d) Graphs showing astrocyte wrapping of neuronal somata (left panel) and S100B expression intensity (right panel) across specific midbrain subregions in male and female mice. n = 8 midbrain sections from three male and three female mice. All errors are SEM; p-values are based on two sample t tests except for SNc TH+, VTA TH+, and SNc TH+, SNr TH− comparisons which are Mann–Whitney tests. (e) Schematic for genetically expressing membrane bound GCaMP6f in astrocytes using AAV 2/5 GfaABC1D-Lck-GCaMP6f, including representative confocal images of astrocyte processes enveloping TH+ neurons in the SNc
These data reveal a striking region- and sex-specific difference in the extent to which S100B containing astrocytic processes envelop midbrain neurons. Specifically, we show that astrocytic wrapping of neuronal somata in the midbrain is specific to SNc DA neurons only in male mice. Thus, S100B secreted from astrocytes could potentially exert robust functional effects on SNc DA neurons of male mice.
3.2 |. Acute exposure to extracellular S100B specifically inhibits voltage-gated A-type potassium currents in TH+ but not in TH− neurons
Our finding that S100B-containing astrocytic processes display robust wrapping around the SNc DA neurons of adult male mice suggests that small increases in secreted S100B from astrocytes could alter the activity of DA neurons. To test this hypothesis, we used cultured primary mouse midbrain neurons and determined if acute extracellular exposure to picomolar concentrations of extracellular S100B peptide alters DA neuron activity.
Since VGKCs are known to play a central role in regulating the frequency of action potentials in neurons (Iyer et al., 2017; Koyama & Appel, 2006; Liss et al., 2001; Martel et al., 2011; Noh et al., 2019), we first assessed the effect of extracellular S100B on VGKC currents in TH+ DA neurons. As described previously (Bancroft & Srinivasan, 2020; Srinivasan et al., 2016; Yuan et al., 2022; Zarate et al., 2021), primary mouse ventral midbrain neurons were cultured from ED14 embryos and maintained for ~21 DIV prior to experiments (Figure 2a). Immunostaining of ~3-week old midbrain cultures showed NeuN+ neurons that were either TH+ or TH−, along with primary midbrain astrocytes expressing S100B (Figure 2b). To visually identify live TH+ neurons, mouse midbrain-astrocyte cocultures were infected with AAV 2/5 TH-GFP 5 days prior to performing electrophysiological recordings. This procedure enabled an unambiguous identification of TH+ and TH− neurons in live mouse midbrain cultures (Figure 2c). Fast inactivating A-type (IA), and non-inactivating (IK) voltage-gated potassium currents were measured using whole-cell voltage clamp recording protocols, as described in methods (Figure S1). All recordings were acquired in the presence of AP5, DNQX, bicuculline, and TTX in order to synaptically isolate neurons, and block action potentials.
FIGURE 2.
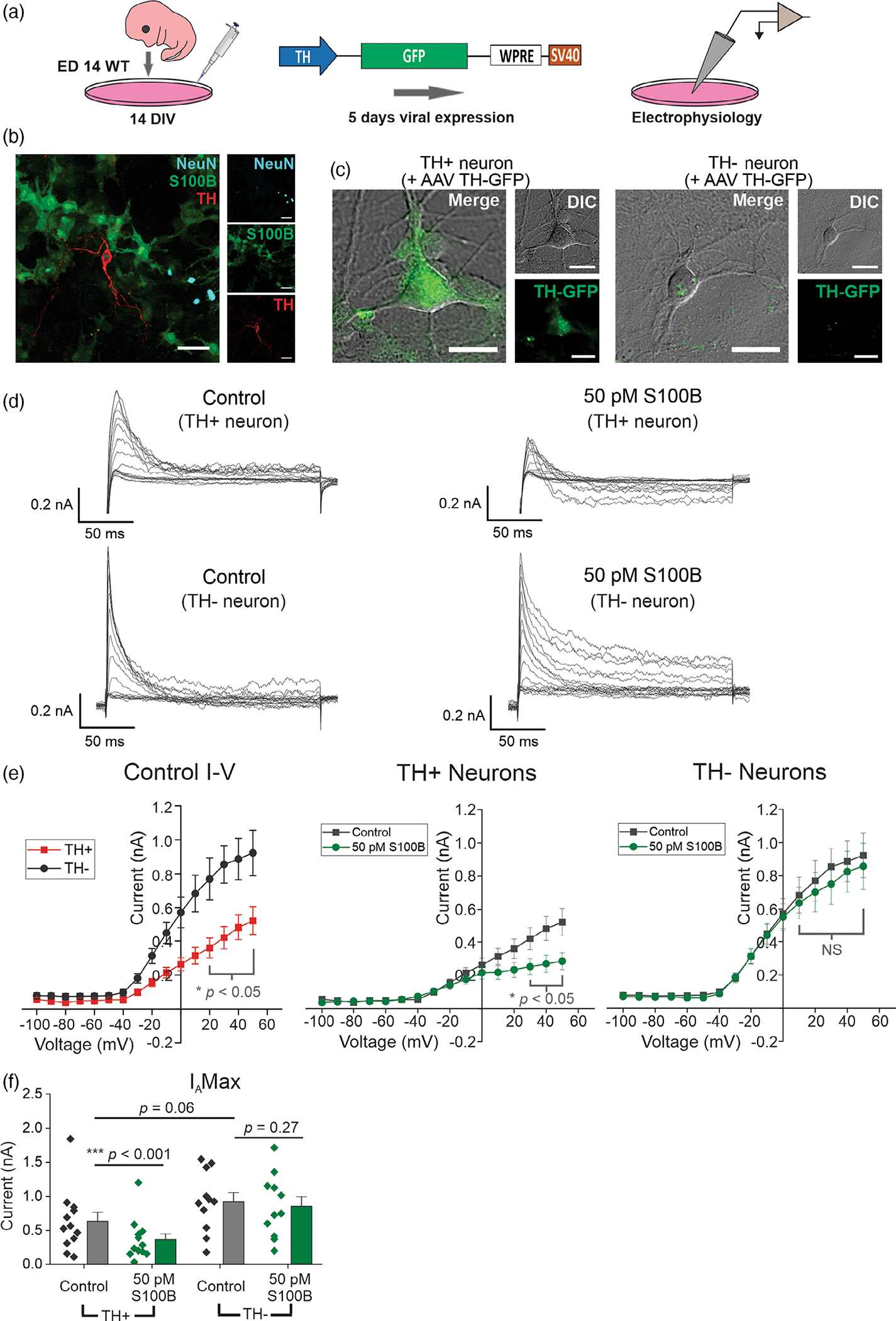
Acute exposure to S100B specifically inhibits A-type voltage-gated potassium currents (IA) in TH+ neurons. (a) Schematic for measuring spontaneous action potentials and ionic currents in primary mouse midbrain neuron cultures using AAV 2/5 TH-GFP virus. (b) Representative image of a formalin-fixed primary mouse midbrain culture stained for NeuN, S100B, and TH; scale bar = 50 μm. (c) Representative images of TH-GFP expression in TH+ and TH− neurons. (d) Representative traces of subtracted IA from TH+ and TH− neurons with (right) and without (left) 50 pM S100B. (e) Left panel, I–V curves of TH+ and TH− neurons in regular aCSF. Right panels, average I–V curves from TH+ and TH− neurons with and without S100B. (f) Comparison of max IA from the most depolarizing stimulation step in TH+ and TH− neurons. n = 12 for TH+ neurons and 11 for TH− neurons from three independent weeks of culture. All errors are SEM; p-values for I-V curves are based on mixed-design ANOVA with post hoc Bonferroni correction; p-values for max IA Wilcoxon signed rank tests are used for TH+ control, TH+ S100B, paired sample t tests for TH− control, TH− S100B, and Mann–Whitney tests for TH+ control, TH− control
In the case of fast inactivating IA currents, at depolarizing potentials of −40 to +50 mV, TH+ neurons showed significantly smaller current amplitudes than TH− neurons (Figure 2d,e). In addition, TH+ neurons showed a linear I–V relationship at all recorded potentials, which was in contrast to TH− neurons that displayed inward rectification at strongly depolarizing potentials between +20 and +50 mV (Figure 2e). Five minutes bath exposure to 50 pM S100B peptide significantly decreased IA by ~1.75-fold at +50 mV in TH+ neurons (Control: 633.17 ± 132.84 pA, S100B: 361.77 ± 89.55 pA) (Figure 2e,f). By contrast, following acute exposure to extracellular S100B, TH− neurons did not show any change IA (Control: 922.76 ± 134.10 pA, S100B: 857.310 ± 139.20 pA) (Figure 2e,f).
In the case of non-inactivating voltage-gated potassium currents (IK), both TH+ and TH+ neurons showed very similar I–V profiles, with outward rectification at strongly depolarizing voltages (Figure 3a–c). Bath exposure to 50 pM S100B peptide had no effect on IK in either TH+ or TH− neurons (Control TH+: 1600.34 ± 143.99, S100B TH+: 1529.07 ± 186.15, Control TH−: 2047.56 ± 201.12, S100B TH−: 1978.74 ± 206.81) (Figure 3b,c). Taken together, these data show that extracellular S100B inhibits only IA but not IK in TH+ DA neurons, with no effect on either IA or IK in TH− midbrain neurons.
FIGURE 3.
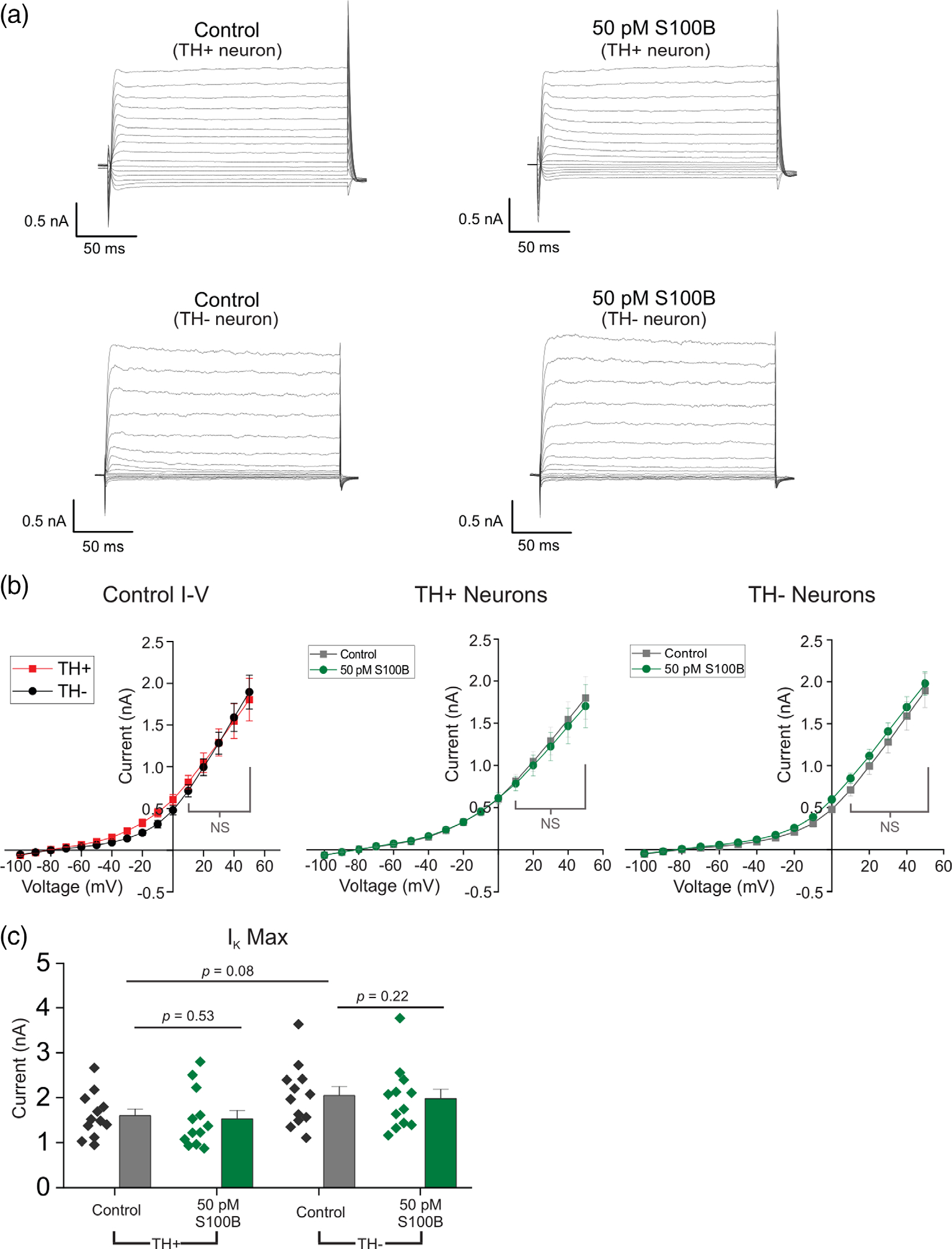
Acute S100B exposure does not inhibit non-inactivating voltage-gated potassium currents (IK) in midbrain neurons. (a) Representative traces of IK from TH+ and TH− neurons with (right) and without (left) 50 pM S100B. (b) Left panel, I–V curves of TH+ and TH− neurons in regular aCSF. Right panels, average I–V curves from TH+ and TH− neurons with and without S100B. (c) Comparison of max IK from the most depolarizing stimulation step in TH+ and TH− neurons. n = 11 for TH+ neurons and 12 for TH− neurons from three independent weeks of culture. All errors are SEM; p-values for I–V curves are based on mixed-design ANOVA with post hoc Bonferroni correction; p-values for max IK are based on paired sample t tests for TH+ control, TH+ S100B and TH− control, TH− S100B, and two sample t tests for TH+ control, TH− control
3.3 |. Acute exposure to extracellular S100B increases the frequency of intrinsic APs in TH+ neurons
Fast inactivating IA plays a central role in governing the frequency of APs in pacemaking neurons, such that inhibition of IA increases the frequency of APs (Amendola et al., 2012; Haddjeri-Hopkins et al., 2021; Tarfa et al., 2017). Having found that extracellular S100B inhibits IA only in TH+ DA neurons, we next asked if extracellular S100B exposure also alters the frequency of intrinsic firing in cultured midbrain DA neurons. As shown in Figure 2c, TH+ and TH− neurons were visually identified based on the presence or absence of TH-GFP expression as a reporter for TH+ neurons, and spontaneous APs were recorded using whole-cell current clamp (Figure 4a). To measure intrinsic firing of midbrain neurons, all recordings were acquired in the presence of synaptic blockers, AP5, DNQX, and bicuculline.
FIGURE 4.
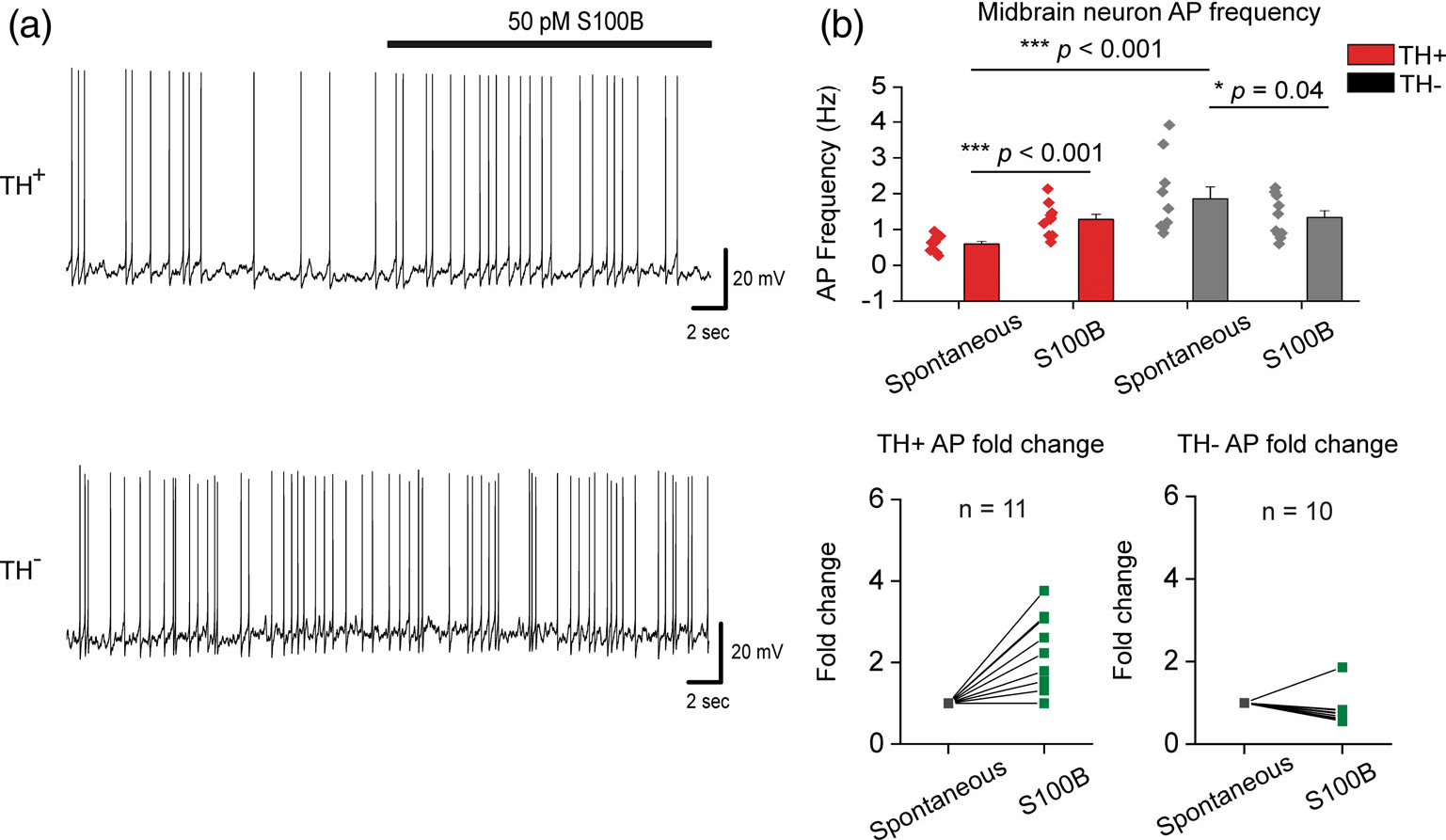
Acute S100B exposure increases intrinsic AP frequency in TH+ neurons. (a) Representative traces of AP recordings from TH+ and TH− neurons with acute application of 50 pM S100B. (b) Average AP frequency of TH+ and TH− neurons with and without S100B; fold change for each TH+ and TH− neuron is shown in the graphs below. n = 11 for TH+ neurons and 10 for TH− neurons from three independent weeks of culture. All errors are SEM; p-values for AP frequency are based on paired sample t tests for TH+ spontaneous, TH+ S100B, Wilcoxon signed rank tests for TH− spontaneous, TH− S100B, and Mann–Whitney tests for TH+ spontaneous, TH− control
We found that in the absence of S100B, TH+ neurons showed an average intrinsic AP frequency of 0.6 ± 0.07 Hz, while TH− neurons showed a significantly higher intrinsic AP frequency of 1.9 ± 0.33 Hz (Figure 4a,b). As rationalized, bath exposure to 50 pM extracellular S100B significantly increased AP frequency in TH+ neurons by 2.1-fold (from 0.6 ± 0.07 Hz to 1.28 ± 0.14 Hz), while significantly decreasing AP frequency by 1.4-fold in TH− neurons (from 1.9 ± 0.33 Hz to 1.34 ± 0.19) (Figure 4b). These data show that in TH+ DA neurons, extracellular S100B specifically increased intrinsic AP frequency, likely via the inhibition of IA.
3.4 |. Cultured mouse midbrain TH+ and TH− neurons demonstrate spontaneous Ca2+ fluxes that require L-type VGCCs
Multiple studies indicate that dysfunctional Ca2+ influx via L-type VGCCs contributes to DA neuron loss and the development of PD (Branch et al., 2014; Ilijic et al., 2011; Kang et al., 2012; Singh et al., 2016; Verma & Ravindranath, 2019; Wang et al., 2012; Wang et al., 2017). Based on these observations, we rationalized that S100B-mediated inhibition of IA could depolarize TH+ neurons, thereby increasing L-type VGCC activity in TH+ DA neurons.
We have previously shown that neurons in primary mouse midbrain cultures demonstrate robust spontaneous Ca2+ fluxes that are strongly inhibited by the L-type VGCC inhibitor, diltiazem, however, these prior studies did not differentiate between TH+ and TH− neurons (Bancroft & Srinivasan, 2020; Yuan et al., 2022). Therefore, we first conducted a series of experiments to specifically characterize spontaneous Ca2+ fluxes in TH+ and TH− neurons. Primary mouse midbrain cultures were infected with AAV 2/5 hSyn-GCaMP6f and AAV 2/5 TH-tdTomato, which enabled the measurement of spontaneous Ca2+ fluxes in midbrain neurons, along with the ability to distinguish between TH+ and TH− neurons (Figure S2A,B). We found that TH cells displayed a ~2-fold higher spontaneous Ca2+ flux frequency than TH+ neurons as well as a ~2-fold higher amplitude in spontaneous Ca2+ flux events when compared to TH+ neurons (TH+ frequency: 2.19 ± 0.19 events/min, TH− frequency: 4.61 ± 0.27 events/min, TH+ amplitude: 0.19 ± 0.02 ΔF/F, TH− amplitude: 0.55 ± 0.04 ΔF/F) (Figure S2C,D and Movie S1). Furthermore, spontaneous Ca2+ fluxes in both TH+ and TH− neurons depended on extracellular, but not intracellular Ca2+ stores (Figure S3 and Movies S2 and S3), and spontaneous Ca2+ flux events in both TH+ and TH− neurons were almost completely inhibited by the L-type VGCC blocker diltiazem (TH+ spontaneous: 3.14 ± 0.47 events/min, TH+ diltiazem: 0.55 ± 0.12 events/min, TH− spontaneous: 2.04 ± 0.28 events/min, TH− diltiazem: 0.8 ± 0.21 events/min) (Figure S4A and Movie S4), but only partially inhibited by the T-type VGCC blocker, mibefradil (TH+ spontaneous: 7.10 ± 0.44 events/min, TH+ mibefradil: 5.90 ± 0.44 events/min, TH− spontaneous: 6.75 ± 0.47 events/min, TH− mibefradil: 5.38 ± 0.51 events/min) (Figure S4B and Movie S5).
Taken together, these data show that the vast majority of spontaneous Ca2+ flux events in both TH+ and TH− neurons occur due to the activity of L-type VGCCs with some contribution from T-type VGCCs and depend on extracellular Ca2+ stores, and that TH− neurons display more robust and frequent Ca2+ fluxes than TH+ neurons.
3.5 |. Acute exposure to extracellular S100B increases spontaneous Ca2+ fluxes only in TH+ DA neurons
Having observed that TH+ and TH− neurons display robust L-type VGCC-mediated spontaneous Ca2+ fluxes, we next sought to determine if exposure to extracellular S100B alters these spontaneous Ca2+ fluxes in TH+ and TH− neurons. To do this, midbrain cultures were bath perfused with 50 pM of S100B peptide, and spontaneous Ca2+ flux frequencies and amplitudes in TH+ and TH− neurons were measured. S100B application caused a significant 2-fold increase in the frequency of Ca2+ fluxes in TH+ neurons, but not in TH− neurons (TH+ spontaneous: 2.19 ± 0.19 events/min, TH+ S100B: 3.25 ± 0.22 events/min, TH− spontaneous: 4.61 ± 0.27 events/min, TH− S100B: 4.59 ± 0.30 events/min) (Figure 5, Movie S1). The ability of extracellular S100B to specifically increase Ca2+ flux frequency only in TH+ neurons was also observed when data were plotted as average Ca2+ flux frequencies across multiple independent weeks of midbrain cultures (TH+ spontaneous: 2.17 ± 0.30 events/min, TH+ S100B: 3.28 ± 0.34 events/min, TH− spontaneous: 4.45 ± 0.45 events/min, TH− S100B: 4.37 ± 0.62 events/min) (Figure 5b), which rules out a skewing of the data as a result of a few individual midbrain cultures with abnormal S100B responses. As an additional control, bath application of denatured S100B peptide did not alter Ca2+ flux frequency in TH+ neurons (TH+ spontaneous: 5.13 ± 1.31 events/min, TH+ dS100B: 4.93 ± 1.57 events/min, TH− spontaneous: 9.88 ± 2.65 events/min, TH− dS100B: 9.00 ± 3.21 events/min) (Figure S5), thus confirming the necessity for properly folded S100B to mediate this effect.
FIGURE 5.
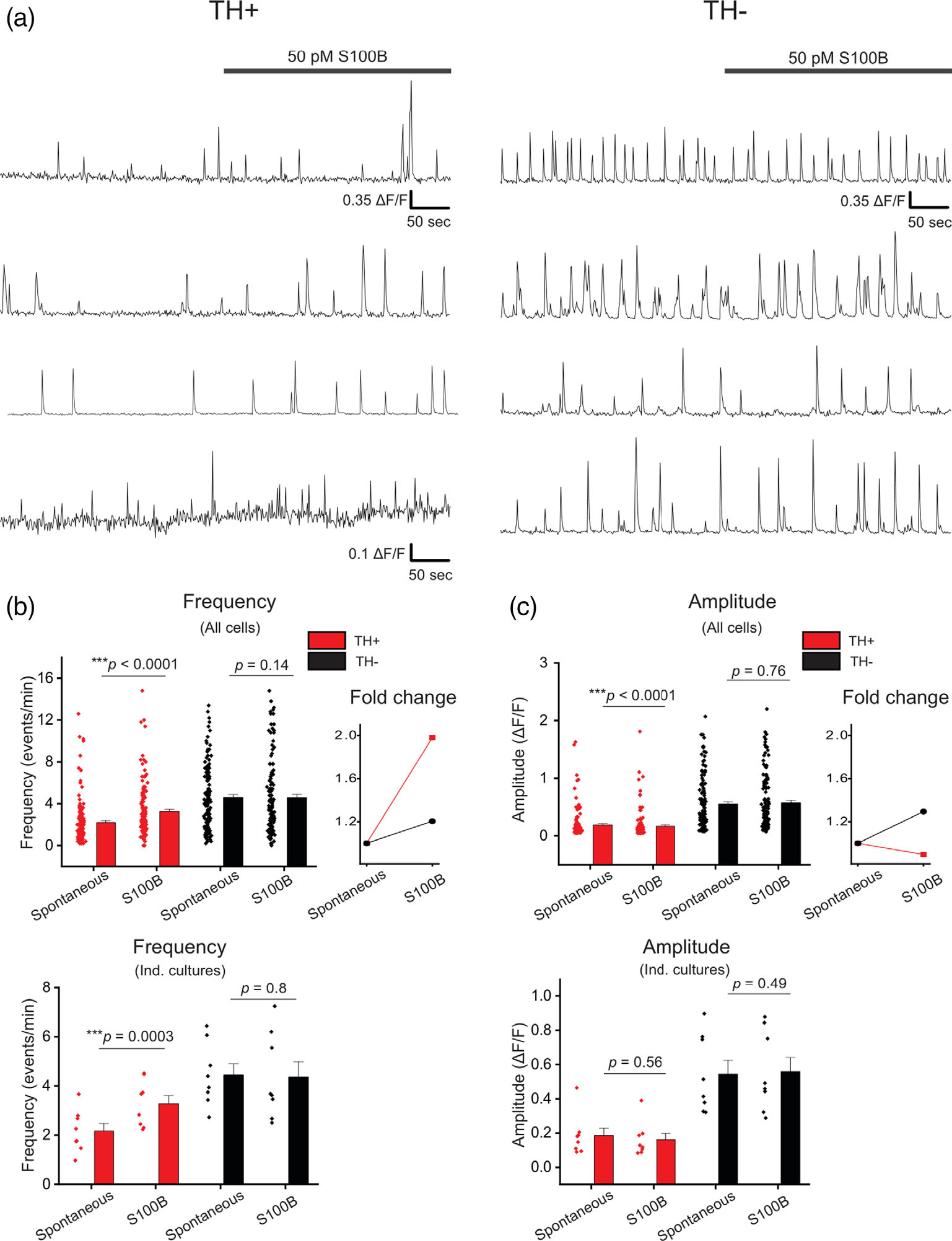
Acute exposure of primary midbrain cultures to S100B peptide increases spontaneous Ca2+ flux frequency only in TH+ neurons. (a) Multiple representative traces of spontaneous Ca2+ fluxes in TH+ and TH− neurons with acute bath application of 50 pM S100B peptide are shown. (b) A graph with average frequency of Ca2+ flux events from individual TH+ (red) and TH− (black) neurons with and without S100B peptide is shown. The line graph on the right shows the average fold change of Ca2+ flux frequency for TH+ and TH− cells following S100B application. The graph below shows the average frequency of TH+ and TH− neurons binned by week of culture. (c) A graph with average amplitude of Ca2+ events from individual TH+ (red) and TH− (black) neurons with and without S100B peptide. The line graph on the right shows the average fold change of Ca2+ flux amplitude for TH+ and TH− cells following S100B application. The graph below shows the average amplitude of TH+ and TH− neurons binned by individual culture. n = 137 for TH+ neurons and 134 for TH− neurons from eight independent cultures. All errors are SEM; p-values for all cells are based on Wilcoxon signed rank tests for TH+ spontaneous, TH+ S100B and TH− spontaneous, TH− S100B and Mann–Whitney tests for TH+ spontaneous, TH− spontaneous; p-values for individual cultures are based on paired sample t tests for TH+ spontaneous, TH+ S100B and TH− spontaneous, TH− S100B, and two sample t tests for TH+ spontaneous, TH− spontaneous
S100B caused a small statistically significant decrease in the amplitude of VGCC mediated Ca2+ fluxes in TH+ neurons (TH+ spontaneous: 0.19 ± 0.02 ΔF/F, TH+ S100B: 0.17 ± 0.02 ΔF/F) (Figure 5c). However, we did not observe a S100B induced change in amplitude when these data were plotted as average amplitudes obtained from multiple cells in the same culture (Ind. cultures, Figure 5c). By contrast, TH− neurons did not demonstrate a S100B induced change in amplitude when the data were plotted from individual cells or individual cultures (TH− spontaneous: 0.55 ± 0.04 ΔF/F, TH− S100B: 0.57 ± 0.04 ΔF/F) (Figure 5c). It should be noted that we observed a very small, statistically significant reduction in Ca2+ flux amplitudes for TH+ neurons only when the data were plotted as an average of amplitudes from individual neurons and not across independent DA cultures (Figure 5c). These data show that S100B specifically increases the frequency of Ca2+ fluxes only in TH+ midbrain neurons with no effect on TH− neurons.
3.6 |. Acute exposure to extracellular S100B increases spontaneous Ca2+ fluxes in TH+ DA neurons via L-type, but not T-type VGCCs
We next assessed the extent to which S100B requires L-type and T-type VGCCs for increasing Ca2+ flux frequency in TH+ neurons. Ca2+ fluxes in midbrain neurons were recorded following bath application of 50 pM S100B, and a subsequent co-application of 50 pM S100B with 100 μM diltiazem (Figure 6a). As shown in our prior experiment (Figure 5), acute application of S100B once again significantly increased Ca2+ flux frequency in TH+ neurons, but not TH− neurons and did not alter the amplitude of Ca2+ fluxes in either TH+ or TH− neurons (Figure 6b,c). Co-exposure of midbrain cultures to 50 pM S100B + 100 μM diltiazem completely inhibited the S100B-mediated increase of Ca2+ fluxes in TH+ neurons, with no effect on Ca2+ flux amplitude for the few remaining Ca2+ events (TH+ spontaneous: 4.68 ± 0.43 events/min, TH+ S100B: 5.32 ± 0.36 events/min, TH+ S100B + diltiazem: 2.42 ± 0.33 events/min, TH− spontaneous: 5.75 ± 0.51 events/min, TH− S100B: 5.49 ± 0.48 events/min, TH− S100B + diltiazem: 2.61 ± 0.31 events/min) (Figure 6a–c, Movie S4). To further confirm blockade of L-type VGCCs inhibits the S100B-mediated increase in Ca2+ fluxes in TH+ neurons, we reversed the order of drug applications in the experiment described above (Figure 6d–f). We observed that L-type VGCC blockade with 100 μM diltiazem prior to S100B application completely abolished the S100B-mediated increase in Ca2+ fluxes in TH+ neurons (TH+ spontaneous: 3.14 ± 0.47 events/min, TH+ diltiazem: 0.55 ± 0.12 events/min, TH+ S100B + diltiazem: 0.24 ± 0.07 events/min, TH− spontaneous: 2.04 ± 0.28 events/min, TH− diltiazem: 0.8 ± 0.21 events/min, TH− S100B + diltiazem: 0.26 ± 0.16 events/min) (Figure 6d–f). These results confirm that S100B increases Ca2+ flux frequency in TH+ neurons via L-Type VGCCs.
FIGURE 6.
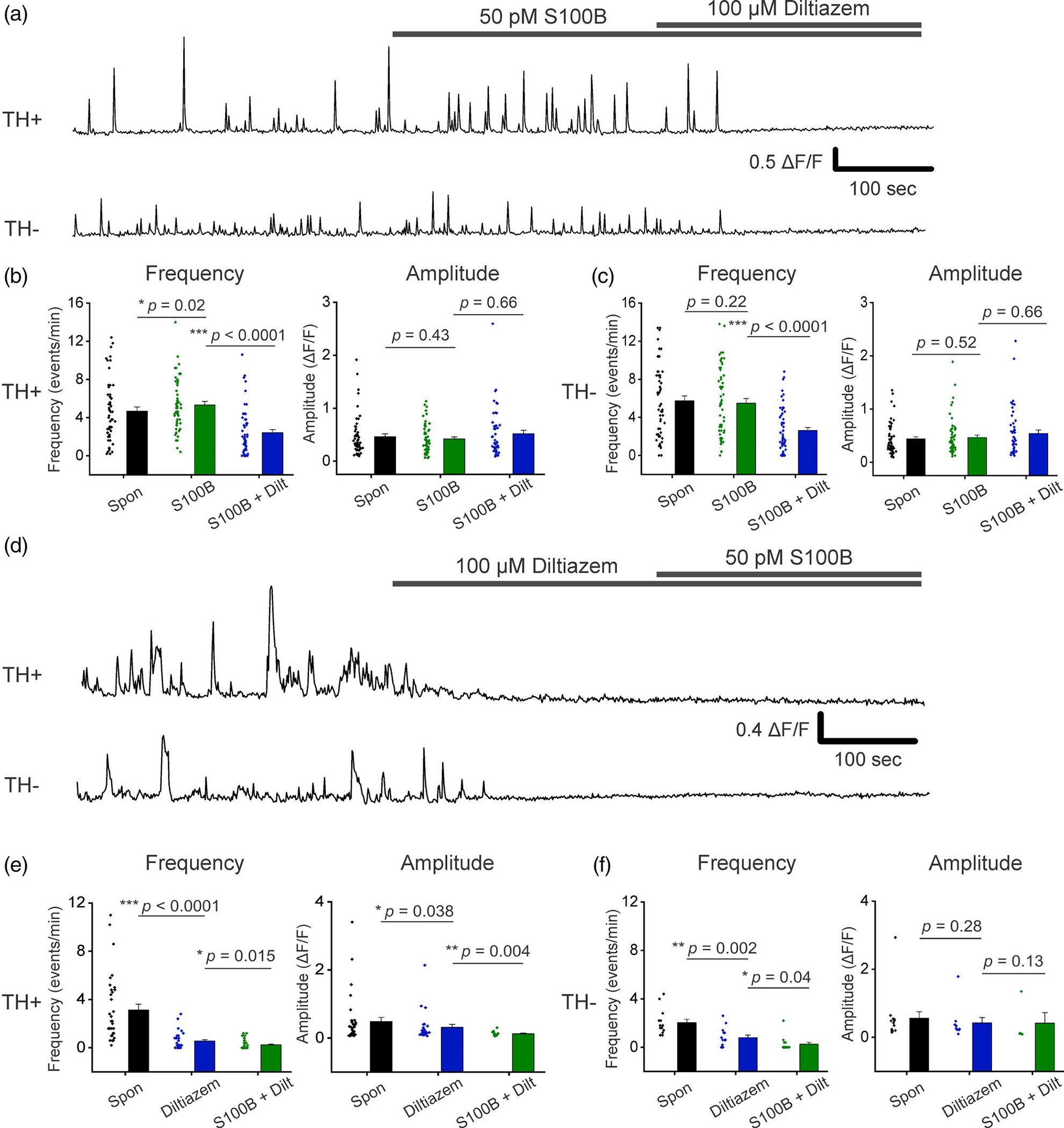
Extracellular S100B mediated increase in spontaneous Ca2+ fluxes in TH+ DA neurons require active L-type VGCCs. (a) Representative traces of spontaneous Ca2+ fluxes in a TH+ and TH− neuron with bath application of S100B, followed by co-application of S100B with diltiazem. (b) Graphs show average Ca2+ flux frequency and amplitude of TH+ neurons without any drug (black) with bath applied S100B (green), and co-applied S100B + diltiazem (blue). (c) Graphs show average Ca2+ flux frequency and amplitude of TH− neurons without drug (black) with bath applied S100B (green), and co-applied S100B + diltiazem (blue). n = 55 for TH+ neurons and 57 for TH− neurons from four independent weeks of culture. All errors are SEM; p-values for frequency and amplitude are based on Wilcoxon signed rank tests for all cases in panels b and c. (d) Representative traces of spontaneous Ca2+ fluxes in a TH+ and TH− neuron with bath application of diltiazem, followed by co-application of diltiazem with S100B. (e) Graphs show average Ca2+ flux frequency and amplitude of TH+ neurons without any drug (black) with bath applied diltiazem (blue), and co-applied diltiazem + S100B (green). (f) Graphs show average Ca2+ flux frequency and amplitude of TH− neurons without any drug (black) with bath applied diltiazem (blue), and co-applied diltiazem + S100B (green). n = 34 for TH+ neurons and 18 for TH− neurons from four independent weeks of culture. All errors are SEM; p-values for frequency and amplitude are based on Wilcoxon signed rank tests for all cases in panels e and f
To determine if T-type VGCCs are involved in S100B-mediated increases in the Ca2+ flux frequency of TH+ DA neurons, spontaneous Ca2+ fluxes were recorded in aCSF, then treated for 15 min with the T-type VGCC antagonist, mibefradil. Ca2+ fluxes in TH− and TH+ DA neurons were then recorded in the presence of 1 μM mibefradil + 50 pM S100B (Figure 7a). We found that mibefradil significantly reduced Ca2+ flux frequency in both TH+ and TH− neurons (TH+ spontaneous: 7.10 ± 0.44 events/min, TH+ mibefradil: 5.89 ± 0.44 events/min, TH− spontaneous: 6.75 ± 0.47 events/min, TH− mibefradil: 5.38 ± 0.51 events/min) (Figure 7b,c). However, co-application of S100B with mibefradil was unable to inhibit S100B-mediated increases in Ca2+ flux frequency in TH+ DA neurons (TH+ S100B + mibefradil: 6.57 ± 0.38 events/min), (Figure 7b, Movie S5). In the case of TH− neurons, we observed significant reductions in Ca2+ flux frequency following exposure to either mibefradil alone or co-exposure of mibefradil with S100B (TH− S100B + mibefradil: 5.03 ± 0.44 events/min) (Figure 7c) In addition, none of the recording conditions altered Ca2+ flux amplitudes in TH+ and TH− neurons (TH+ spontaneous: 0.44 ± 0.07 ΔF/F, TH+ mibefradil: 0.42 ± 0.06 ΔF/F, TH+ S100B + mibefradil: 0.37 ± 0.05 ΔF/F, TH− spontaneous: 0.40 ± 0.05 ΔF/F, TH− mibefradil: 0.45 ± 0.07 ΔF/F, TH− S100B +mibefradil: 0.42 ± 0.07 ΔF/F) (Figure 7b,c). When taken together, these results show that the effect of S100B-mediated increases in Ca2+ flux frequency in TH+ neurons does not depend on T-type VGCCs but requires L-type VGCCs.
FIGURE 7.
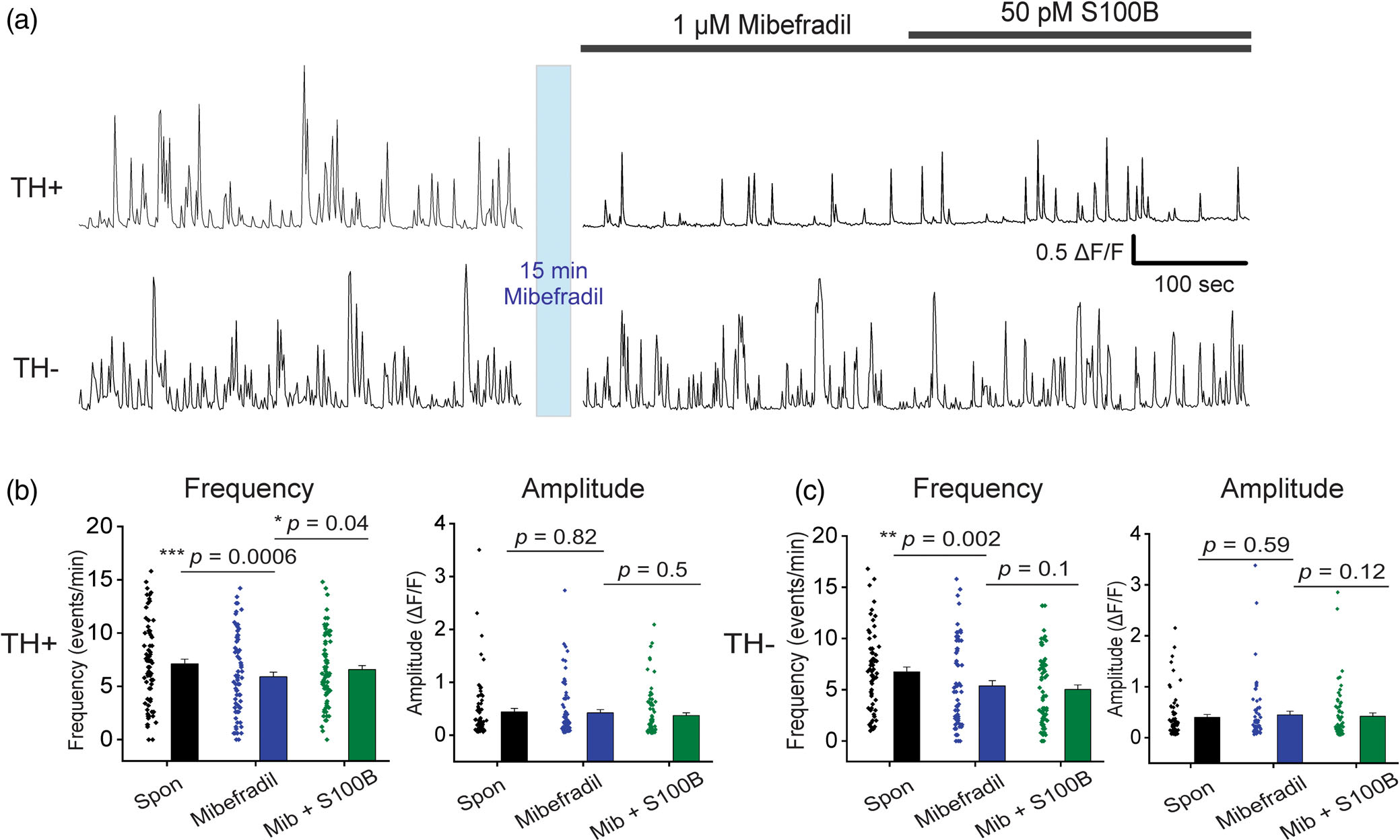
Extracellular S100B mediated increase in spontaneous Ca2+ fluxes in TH+ DA neurons does not require active T-type VGCCs. (a) Representative traces of spontaneous Ca2+ fluxes in a TH+ and TH− neuron after 15 min incubation with 1 μM mibefradil, followed by co-application of S100B with mibefradil. (b) Graphs show average Ca2+ flux frequency and amplitude of TH+ neurons without any drug (black) with bath applied mibefradil (blue), and co-applied S100B + mibefradil (green). (c) Graphs show average Ca2+ flux frequency and amplitude of TH− neurons without drug (black) with bath applied mibefradil (blue), and co-applied S100B + mibefradil (green). n = 75 for TH+ neurons and 66 for TH− neurons from four independent weeks of culture. All errors are SEM; p-values for frequency and amplitude are based on Wilcoxon signed rank tests for all cases in panels b and c
3.7 |. Inhibition of A-type VGKCs is sufficient for increasing intrinsic APs and Ca2+ flux frequency in TH+ neurons
Our results thus far show that acute extracellular exposure to S100B specifically inhibits IA only in TH+ DA neurons, and this is accompanied by an increase the frequency of L-type VGCC-mediated Ca2+ fluxes. Based on these data, we rationalized that S100B-mediated inhibition of IA in TH+ neurons underlies the increase in L-type VGCC activity. To directly test this hypothesis, we asked if inhibiting IA in TH+ neurons with 4-AP, which is a known inhibitor of A-type potassium currents (Bourdeau et al., 2007; Mei et al., 2000; Williams & Hablitz, 2015), mimics the observed S100B-mediated increase in intrinsic AP and Ca2+ flux frequency. Based on the established fact that IA governs AP frequency in neurons (Bourdeau et al., 2007; Mei et al., 2000; Williams & Hablitz, 2015), we measured the effect of 100 μM 4-AP on intrinsic AP firing in synaptically isolated TH+ and TH− neurons in the presence of AP5, DNQX, and bicuculline. Bath application of 100 μM 4-AP significantly increased AP firing frequency in TH+ and TH− neurons by ~1.8-fold (TH+ spontaneous: 0.56 ± 0.08 Hz, TH+ 4-AP: 1.07 ± 0.14 Hz, TH− spontaneous: 1.07 ± 0.11 Hz, TH− 4-AP: 1.71 ± 0.18 Hz) (Figure 8a,b).
FIGURE 8.
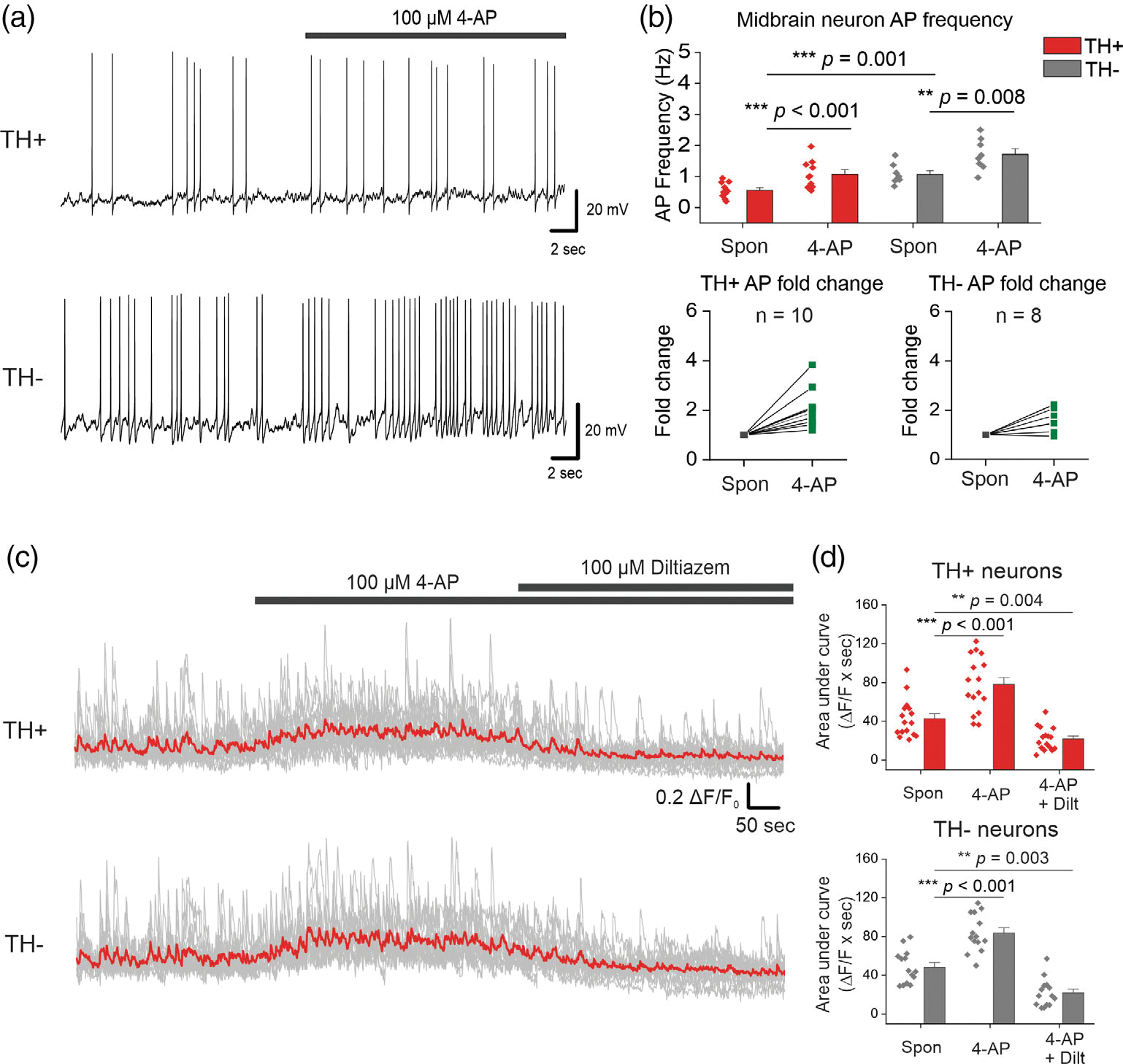
The A-type VGKC inhibitor, 4-AP mimics S100B-mediated increases in intrinsic APs and L-type VGCC-mediated Ca2+ flux frequencies in midbrain neurons. (a) Representative traces of AP recordings from TH+ and TH− neurons with acute application of 100 μM 4-AP. (b) Average AP frequency of TH+ and TH neurons with and without 4-AP; fold change for each TH+ and TH− neuron is shown in the graphs below. n = 10 for TH+ neurons and 9 for TH− neurons from two independent weeks of culture. All errors are SEM; p-values are based on paired sample t tests for TH+ spontaneous, TH+ 4-AP and TH− spontaneous, TH− 4-AP or two sample t tests for TH+ spontaneous, TH− spontaneous. (c) Representative Ca2+ traces of TH+ and TH− neurons with bath application of 4-AP, followed by co-application of 4-AP + diltiazem, the red line represents the average Ca2+ activity from all neurons of that cell type. (d) Graphs of average area under the curve with bath application of 4-AP, followed by co-application of 4-AP + diltiazem. n = 16 for TH+ neurons and 14 for TH− neurons from two independent weeks of culture. All errors are SEM; p-values for TH+ neurons are based on Wilcoxon signed-rank tests, and paired sample t tests for TH− neurons
In order to assess the effect of 4-AP mediated inhibition of IA on L-type VGCC activity, we recorded Ca2+ signals in TH+ and TH− neurons following bath application of 100 μM 4-AP, and a subsequent co-application of 100 μM 4-AP and 100 μM of the L-type VGCC blocker diltiazem (Figure 8c). AUC was used as a for TH− neurons measure of changes in Ca2+ fluxes following the application of 4-AP, and 4-AP + diltiazem. We found that 100 μM 4-AP significantly increased Ca2+ fluxes by ~1.85-fold in TH+ neurons, while co-application of 4-AP with 100 μM diltiazem completely inhibited this increase in Ca2+ fluxes (Spontaneous: 42.90 ± 5.00 ΔF/F × seconds, 4-AP: 78.10 ± 7.11 ΔF/F × seconds, 4-AP + diltiazem: 21.81 ± 2.97 ΔF/F × seconds) (Figure 8c,d). Similarly, 100 μM 4-AP significantly increased Ca2+ fluxes in TH− neurons by ~1.75-fold, while co-application of 4-AP with 100 μM diltiazem completely inhibited this increase in Ca2+ fluxes (Spontaneous: 48.44 ± 4.52 ΔF/F × seconds, 4-AP: 83.65 ± 5.12 ΔF/F × seconds, 4-AP + diltiazem: 21.80 ± 3.87 ΔF/F × seconds) (Figure 8c,d). These data correlate well with the observed 100 μM 4-AP-induced increase in intrinsic AP frequency in TH+ and TH− neurons. Taken together, these data provide clear evidence for the role of VGKC-mediated IA currents in regulating L-type VGCC activity specifically in TH+ DA neurons.
4 |. DISCUSSION
Developing effective neuroprotective therapies for PD requires an understanding of mechanisms by which abnormal astrocyte-neuron signaling in the midbrain could lead to a loss of SNc DA neurons. In this context, S100B, a ubiquitously expressed astrocytic protein, is particularly interesting because multiple reports in PD patients and animal models suggest that this protein plays an active role in the degeneration of SNc DA neurons (Carvalho et al., 2015; Fardell et al., 2018; Hohoff et al., 2010; Liu et al., 2011; Sathe et al., 2012; Schaf et al., 2005). Here, we utilize primary cultures of mouse midbrain neurons to show that acute extracellular exposure to picomolar concentrations of S100B alters the activity of A-type VGKCs, and consequently, Ca2+ fluxes via L-type VGCCs only in TH+ DA neurons. Taken together, our findings provide a novel mechanistic basis for understanding how an abnormal increase in extracellularly secreted S100B by midbrain astrocytes during the early stages of PD could specifically alter DA neuron function, thereby predisposing the midbrain to neurodegeneration.
We show a striking sex difference whereby S100B-containing astrocytic processes envelop DA neuronal somata in the SNc of only male but not female mice (Figure 1). This finding is significant because of two reasons: (i) Our reported sex difference correlates with the well-established 2-fold increased risk for clinical PD in males compared with females (Baldereschi et al., 2000), (ii) Astrocytic coverage of neuropil is a critical determinant in modulating the excitability of neurons via mechanisms such as the secretion of astrocyte-derived factors, the clearance of neurotransmitters, and extracellular K+ buffering (Verhoog et al., 2020). Thus, the observed morphological differences in interaction of astrocytic processes with SNc DA neurons, in combination with an increased S100B density in the SNc when compared to the VTA suggest that any abnormal increase in astrocytic secretion of S100B into extracellular milieu of the SNc could significantly alter the functionality of SNc DA neurons with relatively little effect on DA neurons within the VTA. In addition, the average concentration of S100B levels in the CSF of PD patients is ~3.1 μg/L (Sathe et al., 2012), and ~70% to 80% of S100B in the CSF is due to secretion from the brain and not from serum (Begcevic et al., 2016; Sathe et al., 2012). These values lead us to estimate that extracellular S100B concentrations in the SNc during PD are likely in the range of 50–100 pM. Therefore, this study utilizes a PD-relevant extracellular concentration of 50 pM S100B to assess alterations in midbrain DA neuron function.
Recent studies demonstrate that secreted S100B can buffer extracellular Ca2+, thereby altering the function of Ca2+ sensitive K+ channels in pacemaking neurons (Morquette et al., 2015; Ryczko et al., 2021). In addition, a study using the mollusk as an animal model has shown that extracellular S100B alters potassium currents thereby increasing electrical discharge in the central nervous system (Kubista et al., 1999). Inspired by these data, we asked if extracellular S100B exposure alters the function of VGKCs and intrinsic APs in cultured midbrain neurons. Experiments revealed that 50 pM S100B specifically inhibited A-type voltage-gated potassium currents (IA) in TH+, but not TH− neurons (Figure 2). Mechanistically, the inhibition of IA by S100B is not likely to involve Ca2+ buffering because picomolar concentrations of S100B cannot buffer extracellular Ca2+ to the extent of altering ion channel function. Therefore, the ability of S100B to inhibit IA likely involves a direct interaction between S100B and VGKCs. The specific inhibition of IA only in TH+ neurons suggests that TH+ and TH− neurons differ in their VGKC composition. In line with this rationale, we show that TH+ and TH− neurons display significant qualitative and quantitative differences in baseline I–V relationships for fast inactivating IA (Figure 2e). Although we do not yet know the identity of VGKC subtypes that are inhibited by extracellular S100B in TH+ DA neurons, studies have shown that the vast majority of fast inactivating IA in SNc DA neurons require the Kv4.3 subtype (Haddjeri-Hopkins et al., 2021; Hahn et al., 2003; Liss et al., 2001). Based on these reports, we infer that extracellular S100B likely inhibits Kv4.3 channels in cultured TH+ DA neurons.
We show that extracellular S100B causes a 2-fold increase in intrinsic AP frequency in TH+, and not in TH− neurons (Figure 4). This finding is in line with established roles for A-type VGKCs in regulating AP frequency (Amendola et al., 2012; Haddjeri-Hopkins et al., 2021; Putzier et al., 2009; Tarfa et al., 2017). Therefore, inhibition of IA by S100B is expected to cause an increase in AP frequency, which would result in the observed potentiating effect of S100B on intrinsic APs in TH+ neurons. Paradoxically, TH− neurons showed a decrease intrinsic AP frequency following exposure to extracellular S100B (Figure 4b). Since extracellular S100B does not affect either IA or IK in TH− neurons, our data suggest that S100B-mediated inhibition of intrinsic APs in TH− neurons likely occurs via a mechanism that is independent of VGKCs.
Our data suggest that the ability of extracellular S100B to specifically increase L-type VGCC Ca2+ fluxes only in TH+ and not TH− neurons (Figures 5 and 6) lies downstream of S100B-mediated inhibition of IA in TH+ neurons. This conclusion is strongly supported by the finding that 4-AP-mediated inhibition of IA in midbrain neurons causes a significant increase in intrinsic firing frequency and spontaneous Ca2+ fluxes in TH+ neurons (Figure 8). The idea that IA inhibition causes an increase in L-type VGCC activity in TH+ DA neurons is novel. Although a previous study has shown that inhibition of L-type VGCCs does not affect intrinsic firing in DA neurons (Guzman et al., 2009) to our knowledge, the data presented in this study are the first to show that an inhibition of IA in TH+ DA neurons increases the frequency of Ca2+ fluxes through L-type VGCCs. This idea is also supported by the finding that spontaneous L-type VGCC-mediated Ca2+ flux frequencies are significantly higher in TH− neurons when compared to TH+ cells (Figure S2D), which correlates quite well with the 2-fold higher intrinsic AP frequency in TH-neurons when compared with TH+ neurons (Figure 4). Furthermore, it is notable that extracellular S100B did not alter the activity of T-type VGCCs in TH+ neurons (Figure 7), which suggests that VGKCs and L-type VGCCs in TH+ DA neurons are specifically and functionally coupled.
In summary, this study uncovers a novel mechanism in which extracellular S100B specifically inhibits fast inactivating A-type VGKCs in TH+ DA neurons, thereby increasing intrinsic neuronal firing, which in turn causes an increase in the frequency of Ca2+ fluxes via L-type VGCCs. In this study, we do not consider the local release of S100B onto neurons from astrocytes, however, a recent study has used optogenetic stimulation for the local release of endogenous astrocytic S100B, which results in altered activity of pyramidal neurons in the visual cortex (Ryczko et al., 2021). In addition, the release of S100B from astrocytes could activate RAGE receptors expressed on astrocytes, resulting in altered gliotransmission and cytokine release (Hofmann et al., 1999; Huttunen et al., 2000; Riuzzi et al., 2012). Given the important role of L-type VGCC dysfunction in PD (Branch et al., 2014; Ilijic et al., 2011; Kang et al., 2012; Singh et al., 2016; Verma & Ravindranath, 2019; Wang et al., 2012; Wang et al., 2017), these findings are relevant to understanding how extracellular S100B alters DA neuron function, thereby initiating neurodegenerative processes during early PD. Furthermore, our finding that the interaction between extracellular S100B and VGKCs is specific to TH+ DA neurons provides a potential avenue for discovering novel neuroprotective PD drugs that specifically disrupt S100B-VGKC interactions in DA neurons. Important questions include whether or not an acute interaction between extracellular S100B and VGKCs in TH+ DA neurons leads to PD, and whether the sex-specific association of S100B with SNc DA neurons only in male mice can partially explain observed sex differences in clinical PD. In this regard, future work will focus on assessing the in vivo effects of extracellularly secreted astrocytic S100B on SNc DA function, neuronal loss, and PD-related behavioral deficits.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Texas A&M Institute for Genomic Medicine (TIGM) for providing timed pregnant mice to culture midbrain neurons. We thank Dr. Eunyoung Bang and Ms. Taylor Huntington for their assistance with Figure 1. This work was partially funded by a research grant from the American Parkinson Disease Association (APDA) to Rahul Srinivasan and a National Institutes of Health (NIH) research grant, R01NS115809 to Rahul Srinivasan.
Funding information
National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01NS115809; National Institutes of Health; American Parkinson Disease Association; Texas A&M
Footnotes
CONFLICT OF INTEREST
Authors declare no competing financial interests.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Amendola J, Woodhouse A, Martin-Eauclaire MF, & Goaillard JM (2012). Ca(2)(+)/cAMP-sensitive covariation of I(A) and I(H) voltage dependences tunes rebound firing in dopaminergic neurons. The Journal of Neuroscience, 32(6), 2166–2181. 10.1523/JNEUROSCI.5297-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldereschi M, di Carlo A, Rocca WA, Vanni P, Maggi S, Perissinotto E, Grigoletto F, Amaducci L, & Inzitari D (2000). Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. ILSA working group. Italian longitudinal study on aging. Neurology, 55(9), 1358–1363. 10.1212/wnl.55.9.1358 [DOI] [PubMed] [Google Scholar]
- Bancroft EA, & Srinivasan R (2020). Quantifying spontaneous Ca2+ fluxes and their downstream effects in primary mouse midbrain neurons. Journal of Visualized Experiments, 163, e61481. 10.3791/61481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batassini C, Broetto N, Tortorelli LS, Borsoi M, Zanotto C, Galland F, Souza TM, Leite MC, & Gonçalves CA (2015). Striatal injury with 6-OHDA transiently increases cerebrospinal GFAP and S100B. Neural Plasticity, 2015, 387028. 10.1155/2015/387028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begcevic I, Brinc D, Drabovich AP, Batruch I, & Diamandis EP (2016). Identification of brain-enriched proteins in the cerebrospinal fluid proteome by LC-MS/MS profiling and mining of the human protein atlas. Clinical Proteomics, 13, 11. 10.1186/s12014-016-9111-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HDE, Hirst WD, & Wade-Martins R (2017). The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends in Neurosciences, 40(6), 358–370. 10.1016/j.tins.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeau ML, Morin F, Laurent CE, Azzi M, & Lacaille JC (2007). Kv4.3-mediated A-type K+ currents underlie rhythmic activity in hippocampal interneurons. The Journal of Neuroscience, 27(8), 1942–1953. 10.1523/JNEUROSCI.3208-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branch SY, Sharma R, & Beckstead MJ (2014). Aging decreases L-type calcium channel currents and pacemaker firing fidelity in substantia nigra dopamine neurons. The Journal of Neuroscience, 34(28), 9310–9318. 10.1523/JNEUROSCI.4228-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho DZ, Schönwald SV, Schumacher-Schuh AF, Braga CW, Souza DO, Oses JP, Donis KC, & Rieder CR (2015). Overnight S100B in Parkinson’s disease: A glimpse into sleep-related neuroinflammation. Neuroscience Letters, 608, 57–63. 10.1016/j.neulet.2015.10.010 [DOI] [PubMed] [Google Scholar]
- Dorsey ER, & Bloem BR (2018). The Parkinson pandemic-a call to action. JAMA Neurology, 75(1), 9–10. 10.1001/jamaneurol.2017.3299 [DOI] [PubMed] [Google Scholar]
- Fardell C, Zettergren A, Ran C, Carmine Belin A, Ekman A, Sydow O, Bäckman L, Holmberg B, Dizdar N, Söderkvist P, & Nissbrandt H (2018). S100B polymorphisms are associated with age of onset of Parkinson’s disease. BMC Medical Genetics, 19(1), 42. 10.1186/s12881-018-0547-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez JA, Perkins JM, Beaudoin GM, Cook NB, Quraishi SA, Szoeke EA, Thangamani K, Tschumi CW, Wanat MJ, Maroof AM, Beckstead MJ, Rosenberg PA, & Paladini CA (2019). Ventral tegmental area astrocytes orchestrate avoidance and approach behavior. Nature Communications, 10(1), 1455. 10.1038/s41467-019-09131-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Chan CS, & Surmeier DJ (2009). Robust pacemaking in substantia nigra dopaminergic neurons. The Journal of Neuroscience, 29(35), 11011–11019. 10.1523/JNEUROSCI.2519-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddjeri-Hopkins A, Tapia M, Ramirez-Franco J, Tell F, Marqueze-Pouey B, Amalric M, & Goaillard JM (2021). Refining the identity and role of Kv4 channels in mouse substantia Nigra dopaminergic neurons. eNeuro, 8(4), ENEURO.0207–ENEU21.2021. 10.1523/ENEURO.0207-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn J, Tse TE, & Levitan ES (2003). Long-term K+ channel-mediated dampening of dopamine neuron excitability by the antipsychotic drug haloperidol. The Journal of Neuroscience, 23(34), 10859–10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley BM, Cohen BN, Kim CH, Gold HD, Srinivasan R, McKinney S, Deshpande P, & Lester HA (2017). Reliable identification of living dopaminergic neurons in midbrain cultures using RNA sequencing and TH-promoter-driven eGFP expression. Journal of Visualized Experiments, 120, e54981. 10.3791/54981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann A, Donato R, Weiger TM, & Chazin WJ (2012). S100 calcium binding proteins and ion channels. Frontiers in Pharmacology, 3, 67. 10.3389/fphar.2012.00067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, & Schmidt AM (1999). RAGE mediates a novel proinflammatory axis: A central cell surface receptor for S100/calgranulin polypeptides. Cell, 97(7), 889–901. 10.1016/s0092-8674(00)80801-6 [DOI] [PubMed] [Google Scholar]
- Hohoff C, Ponath G, Freitag CM, Kästner F, Krakowitzky P, Domschke K, Koelkebeck K, Kipp F, von Eiff C, Deckert J, & Rothermundt M (2009). Risk variants in the S100B gene predict elevated S100B serum concentrations in healthy individuals. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics, 153B(1), 291–297. 10.1002/ajmg.b.30950 [DOI] [PubMed] [Google Scholar]
- Huttunen HJ, Kuja-Panula J, Sorci G, Agneletti AL, Donato R, & Rauvala H (2000). Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. The Journal of Biological Chemistry, 275(51), 40096–40105. 10.1074/jbc.M006993200 [DOI] [PubMed] [Google Scholar]
- Ilijic E, Guzman JN, & Surmeier DJ (2011). The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiology of Disease, 43(2), 364–371. 10.1016/j.nbd.2011.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer R, Ungless MA, & Faisal AA (2017). Calcium-activated SK channels control firing regularity by modulating sodium channel availability in midbrain dopamine neurons. Scientific Reports, 7(1), 5248. 10.1038/s41598-017-05578-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang S, Cooper G, Dunne SF, Dusel B, Luan CH, Surmeier DJ, & Silverman RB (2012). CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nature Communications, 3, 1146. 10.1038/ncomms2149 [DOI] [PubMed] [Google Scholar]
- Koyama S, & Appel SB (2006). A-type K+ current of dopamine and GABA neurons in the ventral tegmental area. Journal of Neurophysiology, 96(2), 544–554. 10.1152/jn.01318.2005 [DOI] [PubMed] [Google Scholar]
- Kubista H, Donato R, & Hermann A (1999). S100 calcium binding protein affects neuronal electrical discharge activity by modulation of potassium currents. Neuroscience, 90(2), 493–508. 10.1016/s0306-4522(98)00422-9 [DOI] [PubMed] [Google Scholar]
- Lang C, Campbell KR, Ryan BJ, Carling P, Attar M, Vowles J, Perestenko OV, Bowden R, Baig F, Kasten M, Hu MT, Cowley SA, Webber C, & Wade-Martins R (2019). Single-cell sequencing of iPSC-dopamine neurons reconstructs disease progression and identifies HDAC4 as a regulator of Parkinson cell phenotypes. Cell Stem Cell, 24(1), 93–106.e6. 10.1016/j.stem.2018.10.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, & Roeper J (2001). Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and KChip3.1 transcription. The EMBO Journal, 20(20), 5715–5724. 10.1093/emboj/20.20.5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wang H, Zhang L, Xu Y, Deng W, Zhu H, & Qin C (2011). S100B transgenic mice develop features of Parkinson’s disease. Archives of Medical Research, 42(1), 1–7. 10.1016/j.arcmed.2011.01.005 [DOI] [PubMed] [Google Scholar]
- Martel P, Leo D, Fulton S, Berard M, & Trudeau LE (2011). Role of Kv1 potassium channels in regulating dopamine release and presynaptic D2 receptor function. PLoS One, 6(5), e20402. 10.1371/journal.pone.0020402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei YA, Wu MM, Huan CL, Sun JT, Zhou HQ, & Zhang ZH (2000). 4-aminopyridine, a specific blocker of K(+) channels, inhibited inward Na(+) current in rat cerebellar granule cells. Brain Research, 873(1), 46–53. 10.1016/s0006-8993(00)02469-0 [DOI] [PubMed] [Google Scholar]
- Morquette P, Verdier D, Kadala A, Fethiere J, Philippe AG, Robitaille R, & Kolta A (2015). An astrocyte-dependent mechanism for neuronal rhythmogenesis. Nature Neuroscience, 18(6), 844–854. 10.1038/nn.4013 [DOI] [PubMed] [Google Scholar]
- Noh W, Pak S, Choi G, Yang S, & Yang S (2019). Transient potassium channels: Therapeutic targets for brain disorders. Frontiers in Cellular Neuroscience, 13, 265. 10.3389/fncel.2019.00265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, & Surmeier DJ (2005). G-protein-coupled receptor modulation of striatal CaV1.3 L-type Ca2+ channels is dependent on a shank-binding domain. The Journal of Neuroscience, 25(5), 1050–1062. 10.1523/JNEUROSCI.3327-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putzier I, Kullmann PH, Horn JP, & Levitan ES (2009). Dopamine neuron responses depend exponentially on pacemaker interval. Journal of Neurophysiology, 101(2), 926–933. 10.1152/jn.91144.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riuzzi F, Sorci G, Beccafico S, & Donato R (2012). S100B engages RAGE or bFGF/FGFR1 in myoblasts depending on its own concentration and myoblast density. Implications for muscle regeneration. PLoS One, 7(1), e28700. 10.1371/journal.pone.0028700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryczko D, Hanini-Daoud M, Condamine S, Breant BJB, Fougere M, Araya R, & Kolta A (2021). S100beta-mediated astroglial control of firing and input processing in layer 5 pyramidal neurons of the mouse visual cortex. The Journal of Physiology, 599(2), 677–707. 10.1113/JP280501 [DOI] [PubMed] [Google Scholar]
- Sathe K, Maetzler W, Lang JD, Mounsey RB, Fleckenstein C, Martin HL, Schulte C, Mustafa S, Synofzik M, Vukovic Z, Itohara S, Berg D, & Teismann P (2012). S100B is increased in Parkinson’s disease and ablation protects against MPTP-induced toxicity through the RAGE and TNF-alpha pathway. Brain, 135(Pt 11), 3336–3347. 10.1093/brain/aws250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaf DV, Tort AB, Fricke D, Schestatsky P, Portela LV, Souza DO, & Rieder CR (2005). S100B and NSE serum levels in patients with Parkinson’s disease. Parkinsonism & Related Disorders, 11(1), 39–43. 10.1016/j.parkreldis.2004.07.002 [DOI] [PubMed] [Google Scholar]
- Singh A, Verma P, Balaji G, Samantaray S, & Mohanakumar KP (2016). Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochemistry International, 99, 221–232. 10.1016/j.neuint.2016.07.003 [DOI] [PubMed] [Google Scholar]
- Srinivasan R, Henley BM, Henderson BJ, Indersmitten T, Cohen BN, Kim CH, McKinney S, Deshpande P, Xiao C, & Lester HA (2016). Smoking-relevant nicotine concentration attenuates the unfolded protein response in dopaminergic neurons. The Journal of Neuroscience, 36(1), 65–79. 10.1523/JNEUROSCI.2126-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Huang BS, Venugopal S, Johnston AD, Chai H, Zeng H, Golshani P, & Khakh BS (2015). Ca2+ signaling in astrocytes from Ip3r2/mice in brain slices and during startle responses in vivo. Nature Neuroscience, 18(5), 708–717. 10.1038/nn.4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan R, Lu T-Y, Chai H, Xu J, Huang BS, Golshani P, Coppola G, & Khakh BS (2016). New Transgenic Mouse Lines for Selectively Targeting Astrocytes and Studying Calcium Signals in Astrocyte Processes In Situ and In Vivo. Neuron, 92(6), 1181–1195. 10.1016/j.neuron.2016.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Jiang M, Fu X, Cai Q, Zhang J, Yin Y, Guo J, Yu L, Jiang Y, Liu Y, Feng L, Nie Z, Fang J, & Jin L (2017). Mesencephalic astrocyte-derived neurotrophic factor reduces cell apoptosis via upregulating HSP70 in SHSY-5Y cells. Translational Neurodegeneration, 6, 12. 10.1186/s40035-017-0082-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarfa RA, Evans RC, & Khaliq ZM (2017). Enhanced sensitivity to hyperpolarizing inhibition in Mesoaccumbal relative to Nigrostriatal dopamine neuron subpopulations. The Journal of Neuroscience, 37(12), 3311–3330. 10.1523/JNEUROSCI.2969-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thannickal TC, Lai YY, & Siegel JM (2007). Hypocretin (orexin) cell loss in Parkinson’s disease. Brain, 130(Pt 6), 1586–1595. 10.1093/brain/awm097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoog QP, Holtman L, Aronica E, & van Vliet EA (2020). Astrocytes as guardians of neuronal excitability: Mechanisms underlying epileptogenesis. Frontiers in Neurology, 11, 591690. 10.3389/fneur.2020.591690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, & Ravindranath V (2019). CaV1.3 L-type calcium channels increase the vulnerability of substantia Nigra dopaminergic neurons in MPTP mouse model of Parkinson’s disease. Frontiers in Aging Neuroscience, 11, 382. 10.3389/fnagi.2019.00382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QM, Xu YY, Liu S, & Ma ZG (2017). Isradipine attenuates MPTP-induced dopamine neuron degeneration by inhibiting upregulation of L-type calcium channels and iron accumulation in the substantia nigra of mice. Oncotarget, 8(29), 47284–47295. 10.18632/oncotarget.17618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Ma Z, Wang J, & Xie J (2012). L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotoxicity Research, 21(3), 266–270. 10.1007/s12640-011-9271-x [DOI] [PubMed] [Google Scholar]
- Williams SB, & Hablitz JJ (2015). Differential modulation of repetitive firing and synchronous network activity in neocortical interneurons by inhibition of A-type K(+) channels and Ih. Frontiers in Cellular Neuroscience, 9, 89. 10.3389/fncel.2015.00089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan M, Bancroft EA, Chen J, Srinivasan R, & Wang Y (2022). Magnetic fields and magnetically stimulated Gold-coated superparamagnetic iron oxide nanoparticles differentially modulate L-type voltage-gated calcium channel activity in midbrain neurons. ACS Applied Nano Materials, 5, 205–215. 10.1021/acsanm.1c02665 [DOI] [Google Scholar]
- Zarate SM, Pandey G, Chilukuri S, Garcia JA, Cude B, Storey S, Salem NA, Bancroft EA, Hook M, & Srinivasan R (2021). Cytisine is neuroprotective in female but not male 6-hydroxydopamine lesioned parkinsonian mice and acts in combination with 17-beta-estradiol to inhibit apoptotic endoplasmic reticulum stress in dopaminergic neurons. Journal of Neurochemistry, 157(3), 710–726. 10.1111/jnc.15282 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.