

Abstract
We sought a mutation in the DNA binding domain of the arabinose operon regulatory protein, AraC, of Escherichia coli that allows the protein to bind DNA normally but not activate transcription. The mutation was isolated by mutagenizing a plasmid overproducing a chimeric leucine zipper-AraC DNA binding domain and screening for proteins that were trans dominant negative with regard to wild-type AraC protein. The mutant with the lowest transcription activation of the araBAD promoter was studied further. It proved to alter a residue that had previously been demonstrated to contact DNA. Because the overproduced mutant protein still bound DNA in vivo, it is deficient in transcription activation for some reason other than absence of DNA binding. Using the phase-sensitive DNA bending assay, we found that wild-type AraC bends DNA about 90° whereas the mutant bends DNA by a smaller amount.
The catabolite gene activator protein (CAP) and the AraC protein together stimulate the activity of the promoter of the araBAD operon, pBAD, in Escherichia coli (9, 18, 28). At this promoter, the AraC protein binds adjacent to and partially overlaps the RNA polymerase binding site while CAP binds behind AraC and a blank turn of the DNA lies between the two proteins (3, 6, 23, 24). The binding site for AraC is larger than the binding sites of many proteins, as each of the monomers of the dimeric AraC protein contacts two adjacent major-groove regions. Thus, the 41-base binding site for AraC at pBAD comprises bases −73 to −33 and the 22-base CAP binding site comprises bases −104 through −83 (Fig. 1).
FIG. 1.

Protein binding sites and likely interactions between RNA polymerase and AraC and CAP at the ara pBAD promoter. By analogy to other activators that bind in the −40 to −70 region, AraC likely interacts with the C-terminal domain of alpha, which is consistent with in vitro data (31, 32). Further, the interaction likely is not exclusively with the polymerase-proximal subunit of AraC since the presence of RNA polymerase bound at pBAD substantially alters the DNA contacts made by the polymerase-distal subunit of AraC (31). Further, the C-terminal domain of the alpha subunit of RNA polymerase likely interacts with CAP since mutations in AR1 of CAP affect pBAD activation (33).
At pBAD, both CAP and AraC likely interact with RNA polymerase via contacts made by the C-terminal domain of the alpha subunit of RNA polymerase. In the lac and gal operons, CAP is known to utilize residues in the area known as activation region 1 (AR1) or AR2 to contact the C-terminal domain of alpha (1, 4, 7, 8, 13, 22, 35). Mutations in AR1 of CAP also affect CAP’s activation of pBAD (33), suggesting the existence of the same sort of CAP-alpha interactions. Further, RNA polymerase with the C-terminal domain of alpha truncated cannot be activated by AraC at pBAD (32), suggesting but not proving the existence of an AraC interaction with the C-terminal domain of alpha. Two predictions follow from consideration of the information just given. First, it should be possible to isolate mutations in AraC that interfere with the presumed AraC-RNA polymerase interactions. Second, the long distance along the DNA from the RNA polymerase binding site to the CAP binding site would not greatly hinder CAP-alpha interactions if the intervening DNA were substantially bent, suggesting that AraC may generate a significant bend in the DNA.
Something is already known concerning the first expectation. Chimeric AraC proteins consisting of the DNA binding domain of AraC dimerized by a leucine zipper region from C/EBP are capable of fully activating pBAD (5). This suggests that all of the determinants of AraC required for activation lie in its DNA binding domain. Accordingly, mutations have been sought in the DNA binding domain that affect transcription activation but not DNA binding (25). Although 11 different mutations in six different sites were identified that produce an apparently activation-negative, DNA binding-positive phenotype, all proved to bind arabinose more weakly than the wild-type protein and not to be defective in AraC-RNA polymerase interactions. In light of the number of mutations isolated, it seems difficult to isolate AraC-RNA polymerase interaction mutations by simple scoring of activation and DNA binding properties.
In the work reported here, we have addressed two questions raised by a consideration of the above-mentioned facts. We developed and applied a general scheme for the direct selection of mutants of AraC that bind DNA normally but do not activate transcription, and we have measured the amount of DNA bending produced by AraC.
MATERIALS AND METHODS
Strains, plasmids, and media.
The plasmid used for overexpression of the leucine zipper dimerization domain-AraC DNA binding domain protein was pSE380 (Invitrogen, San Diego, Calif.), which was constructed by Bustos and Schleif (5). The AraC DNA binding domain, amino acids 174 to 291, was cloned into the BamHI and XbaI sites of pSE380, while DNA coding for amino acids 302 to 350 of the leucine zipper dimerization domain from C/EBP was cloned into the NcoI and BamHI sites. To mutagenize AraC, we transformed the plasmid containing the DNA binding domain of AraC into competent mutator cells (endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac mutD5 mutS mutT Tn10 Tetr), Epicurian coli XL1-red cells (Stratagene), which have a 5,000-times-higher rate of mutation than wild-type cells. Mutagenized plasmid DNA was isolated by miniprep, and the DNA coding for the DNA binding domain of AraC was excised with BamHI and XbaI, purified on a 0.8% agarose gel, and ligated into plasmid DNA containing the leucine zipper dimerization domain.
Transcription activation and repression by the mutagenized chimeric protein were monitored in E. coli SH288 (F′ araC102 araBAD+/Δara-leu-498 pC-lacZ Strr Δlac-74 thi-1), which contains the episome from F′102 in SH284 (10). Reduced transcription activation from pBAD will result in reduced catabolism of arabinose, yielding red colonies on tetrazolium arabinose plates, whereas cells with wild-type transcription from pBAD will appear white. Additionally, in the same cells, AraC unable to bind DNA and hence unable to repress pC produces blue colonies when grown on minimal salts–0.4% glycerol–0.4% Casamino Acids–10 μg of vitamin B1 per ml–0.002% 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) plates. If AraC can bind DNA and repress pC, the colonies are white. The trans-dominant negative phenotype was assayed in strain BS2 (F′ araC+ araBAD+/Δara-leu-498 Δlac-74 thi-1 [λ I1-I1-pBAD-lacZ] Strr), which was constructed by the method of Simons et al. (29). It carries a λ phage with the I1-I1 pBAD P5 promoter (24) fused to the lacZ gene. All mutants were sequenced by double-stranded sequencing (17).
Enzyme assays.
The plasmid containing the mutagenized chimeric AraC gene was transformed into SH288 for arabinose isomerase assays (26). Cells were grown in minimal salts–0.4% glycerol–10 μg of vitamin B1 per ml–0.4% Casamino Acids until they reached an optical density at 600 nm (OD600) of 0.4. A 3-ml volume of cells was centrifuged and assayed as described by Schleif and Wensink (26). Repression and trans dominance were assayed through β-galactosidase levels. Cells were grown in the same minimal medium used for the arabinose isomerase assay until they reached an OD600 of 0.4. A 1-ml volume of cells was withdrawn and assayed as described by Miller (21). The results reported are averages of two independent assays.
DNA migration retardation assay.
The DNA migration retardation assay was performed with wild-type and mutant chimeric AraC proteins as previously described (11). Radiolabeled pBAD DNA fragments were generated with PCR so that an I1-I1 binding site for AraC was located approximately 80 bp from each end of a 160-bp fragment. This placement of the AraC binding site allows the maximum DNA bending effect on electrophoretic mobility. To generate the DNA fragment by PCR, 100 ng of a 32P-5′-end-labeled primer (ATAATCACGGCAGAAAAGTCCA) at 106 cpm/ng was mixed with 150 ng of an unlabeled primer (GTGCGCGTGCAGCCCTTATTGCCC) and template plasmid pES51 (12) containing the I1-I2-pBAD promoter. The PCR cycle parameters used were 95°C for 1 min, 55°C for 1 min, and 72°C for 1 min for 28 cycles.
Crude cell lysates were prepared from cells overexpressing parental or mutant chimeric AraC proteins. Cells were grown to an OD600 of 0.7 in YT broth (26). A 3-ml culture volume was centrifuged and resuspended in 0.5 ml of 100 mM potassium phosphate (pH 7.4)–50 mM KCl–10% glycerol–1 mM dithioerythritol–0.1 mM ZnCl2–1 mM EDTA. The resuspended cells were lysed by sonication and centrifuged at 8,500 × g for 10 min. The supernatant was removed, and 170 μl of glycerol was added to 500 μl of the supernatant. The lysates were then stored at −70°C for up to 2 weeks. Binding reactions were carried out with 10 mM Tris-acetate (pH 7.4)–1 mM EDTA–50 mM KCl–1 mM dithiothreitol– 5% glycerol–50 ng of calf thymus DNA/μl. Protein from the lysates was added so that just 100% of 1 ng of I1-I2-32P-end-labeled DNA was bound. Samples were equilibrated for 20 min and loaded onto a nondenaturing 6% polyacrylamide gel cross-linked with 0.1% methylene-bisacrylamide.
DNA for phase-sensitive bending assay.
AraC protein was purified to homogeneity by Jeff Withey (27). Operator DNA constructs were prepared by using standard molecular biological techniques (19). The operator construct series was amplified by PCR from plasmid DNA templates by using oligonucleotides with the sequences CATCAGGAATTCGATCAG and GTAGTCGAATTCATGATG, the last 12 bases of each being complementary to the template. Each member of each series was constructed as a series of four overlapping oligonucleotides that were fused by using PCR ligation, cut with EcoRI, and then inserted into the EcoRI site of pUC19. The sequence of I1-O2 is TAGCATTTTTATCCATATCTAGAAACCAATTGTCCATA, in which the half-sites have been underlined. The complete sequence of the I1-O2 zero-base insert DNA used in the bending assay is GAATTCGATCAGACATTGTCTAGACGATCAGAC ATTGTGCACATCGATACGTAGTACGCGTAAAAACGCGCAAAAA-X-T CATATAGCATTTTTATCCATAAGAAGAAACCAATTGTCCATAAGATC TCAGACAGTAGAGTCGACACGATCAGACATTGGATCCTCAGACATG AGCTCGCATCATGAATTC, where the X marks a sequence of varying length, as described below, and the position of the operator is underlined. The I1-I1 and I1-I2 operator series were constructed similarly, differing only in the sequence of the second half-site. The following sequences substituted for the bases underlined above to make the different operators were TAGCATTTTTATCCATAAGATTAGCATTTTTATCCATA (I1-I1) and TAGCATTTTTATCCATAAGATTAGCGGATCCTACCTGA (I1-I2). Series members having extra bases between the reference bend and the operator were also made from four overlapping oligonucleotides. The additions for rotating the reference bend were made at the position marked by the X in the insert sequence above, where X was CCGCG, CCGCGG, CCCGCGG, CAGCGCGG, CAGCCGCGG, CAGCCCGCGG, or CAGCACCGCGG for the constructs with zero through six bases added, respectively. The constructs used for determination of the constant k were made in the same way. The sequences for the bent portions of those constructs (shown in boldface type), which substitute for the underlined sequence in the above insert were TAGCATTTTTATCCATAAGATTAGCGATCCTACCTGA for one bend, TAGCATTTTTATCCATTTTTATCCATA for two bends, and TAGCATTTTTATCCATTTTT TAGCATTTTTATCCATA for three bends. The operator synthesis PCR was performed with one 32P-labeled primer and the other primer in excess to minimize the amount of labeled single-stranded product.
Phase-sensitive bending assay.
The bending assay was performed essentially as previously described (36), except that the binding conditions were as follows. All assay binding reactions were performed with 20-μl volumes. The buffer used for the binding reaction contained 150 mM KCl, 10 mM Tris-acetate (pH 7.4), 1 mM EDTA, 5% glycerol, 1 mM dithioerythritol, and 0.05% (vol/vol) Nonidet P-40. Arabinose, if present, was at 1% (wt/vol). AraC protein was diluted immediately prior to use by slowly adding binding buffer to an aliquot of protein stock solution. AraC was added to a final concentration between 1 and 100 nM. DNA fragments were added to concentrations between 1 and 10 nM. Samples were incubated at 37°C for 20 min, a time sufficient to ensure that the reactions would reach equilibrium. Electrophoresis through rinsed, presoaked 6% (wt/vol) acrylamide–0.1% (wt/vol) bisacrylamide gels containing 10 mM Tris-acetate and 1 mM K-EDTA was carried out at 5 V/cm for 6 h in a horizontal apparatus with a connected recirculating pump maintaining the buffer temperature at 20°C. For the bending assays in the presence of arabinose, the running buffer included 1% (wt/vol) arabinose. Gels were vacuum dried, and the radioactive bands were visualized and quantitated by using a Molecular Dynamics PhosphorImager PC.
The constant k was determined by applying the equation given in Results to the results of the phase-sensitive assay of migration of a standard bend series comprising one, two, and three phased A5 tracts, which were assumed to bend the DNA by 18, 36, and 54° (16).
RESULTS
Isolation of the mutant.
AraC mutants defective in the ability to make specific interactions with RNA polymerase should be able to bind DNA normally and repress pBAD and pC by looping but not be able to activate transcription from pBAD. The failure of a previous screen (25) to yield such mutants could be the result of a weak phenotype of the mutation, redundant interaction sites on AraC, or an absence altogether of specific interaction sites. We therefore developed a particularly powerful screen for mutants defective in transcription activation but not defective in DNA binding. We employed an overproducing plasmid encoding a chimeric protein consisting of the DNA binding and transcription activation domain of AraC fused to the leucine zipper dimerization domain of C/EBP (5). Candidates having mutations in this gene were scored in the presence of wild-type AraC encoded by a chromosomal gene. The desired activation-defective mutants should dimerize, although not with wild-type AraC, and bind to the araI site at ara pBAD, where they will neither activate transcription nor allow wild-type AraC to activate transcription. No other plausible mutant types should display this trans-dominant negative phenotype.
To limit the screen, only the DNA coding for the DNA binding domain of the chimeric AraC was mutagenized. Fifty thousand plasmid-transformed colonies were screened for defective activation of pBAD and normal repression of pC. As described in Materials and Methods, reduced transcription activation from pBAD will result in reduced catabolism of arabinose, thus giving red colonies on arabinose tetrazolium indicator plates. Cells possessing an ara pC-lacZ fusion and containing AraC capable of repressing pC produce white colonies on X-Gal indicator plates, whereas cells containing AraC defective in DNA binding and therefore defective in pC repression produce blue colonies. One hundred candidates passing the first induction and repression screens were retransformed and also tested for their trans-dominant phenotype from the I1-I1-pBAD (24) promoter in the presence of wild-type AraC. The mutant with the strongest phenotype, H213Y, activated the pBAD promoter less than 10% as well as the parental chimeric protein but could repress transcription from pC very well (Table 1). It was characterized more fully.
TABLE 1.
Binding and inducing properties of mutant proteins
| Activator | Fold activation from:
|
Fold repression from O1-pCc | |
|---|---|---|---|
| I1-I2-pBADa | pFGHb | ||
| None (no AraC) | 1 | 1 | 1 |
| AraC | 100 | 190 | |
| H213Y AraC | 59 | 90 | |
| Parental chimera | 63 | 21 | 16 |
| H213Y chimera | 4 | 15 | 12 |
Activation from I1-I2-pBAD was quantitated with the arabinose isomerase assay. The strain without parental chimeric AraC contained 140 arabinose isomerase units, and that with the parental chimera contained 8,600 arabinose isomerase units.
Activation from pFGH-lacZ was measured by the β-galactosidase assay. The strain with parental chimeric AraC contained 4,200 Miller units, and the strain without AraC contained 200 Miller units.
Repression from pC-lacZ in cells grown in the absence of arabinose was quantitated with the β-galactosidase assay. The strain with parental chimeric AraC contained 10 Miller units, and the strain without AraC contained 160 Miller units.
Measurement of the mutant’s trans-dominant negative behavior with regard to wild-type AraC showed the dominant negative effect to be strong. Activation of I1-I1-pBAD-lacZ by wild-type AraC was 660 Miller units, whereas activation was decreased to 20 Miller units in the presence of chimeric AraC carrying the H213Y mutation. We also tested the activation effect of the H213Y chimeric AraC on the pFGH promoter and found it to be nearly as active as the parental protein (Table 1). The fact that the mutant protein induces pFGH as well as the wild type does suggests that its transcription activation defect at pBAD results from something other than modification of a region used for contact with RNA polymerase.
DNA bending by wild-type and mutant AraC.
Before examining the DNA bending properties of the mutant, it was necessary to learn the DNA bending properties of wild-type AraC. The direction and amount of bending in a DNA molecule or protein-DNA complex can be measured approximately by gel electrophoresis methods that rely on the fact that bent DNA migrates through a gel more slowly than straight DNA (14, 15, 30, 36). The phase-sensitive bending assay designed by Zinkel and Crothers (36) seems particularly well suited to careful measurement of DNA bending. It compares the amount of migration retardation among a series of protein-bound DNA fragments. The fragments differ from each other as the distance of a reference bend from the protein binding site is increased incrementally by the insertion of one, two, etc., bases, thereby rotating the reference bend with respect to the protein-derived bend. When the reference and protein-derived bends are in phase, the bending angles sum and the DNA fragment migrates most slowly, and when the bends are out of phase, a portion of the total amount of bending is negated and the fragment migrates more rapidly. The resulting data allow approximate determination of the direction of the protein-induced bend and its magnitude.
We measured the bending produced by AraC in three different operator construct series. The normal araI site from which AraC activates transcription from pBAD consists of the polymerase-distal I1 half-site and the polymerase-proximal I2 half-site. In the absence of arabinose, dimeric AraC loops between the I1 half-site and the O2 half-site located 212 bp upstream. To examine the possibility that the intrinsic bend of any of these sites is significantly different from that of the others or determine whether AraC bends each half-site similarly, we examined the bending produced by three series of constructs. Each series contained centrally placed two-half-site operators in which the first half-site was araI1 and the second was either the araI1, araI2, or araO2 half-site, therefore making the operators I1-I1, I1-I2, and I1-O2. Each series member contained a reference bend, the operator, and an intervening segment whose length increased within each series to increase the distance, and therefore the angle of rotation, of the reference bend with respect to the operator. The operators were positioned such that the portions of their major grooves that are contacted by AraC were in helical phase with the reference bend so that the reference bend was toward the major grooves when no additional bases were inserted. We used two phased A5 tracts to provide an about 36° reference bend (16).
Figure 2 shows the results of the phase-sensitive bending assay for the I1-I1, I1-I2, and I1-O2 operator constructs with arabinose present in the binding reaction mixture and running buffer. Identical data were produced in the absence of arabinose (data not shown). For each construct, the bend angle α was calculated from the following equation (14):
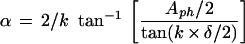 |
where Aph (amplitude of phasing) is the difference in the Rf values of the maximally and minimally retarded DNA species when their mean is taken to be an Rf of 1.0, δ is the angle of the directed reference bend, in this case 36°, and k is an empirical constant, determined in our case to be 0.7 by using known bends. We used a series of one, two, and three phased A5 tracts to determine k. All three AraC-bound operators, I1-I1, I1-I2, and I1-O2, were found to be bent about 90° toward AraC, both in the presence and in the absence of arabinose.
FIG. 2.


(Left) Representation of the DNA fragments used in the phase-sensitive DNA bending assay. The top pair shows bends in phase and out of phase, and the bottom three show the structures of the three araI sites used. The shaded region is the area in which bases were added to shift the phase of the reference bend with respect to the araI sites. The reference bend was generated by two in-phase A5 tracts. (Right) Phase-sensitive bend assays of I1-I1, I1-I2, and I1-O2 sites. The distances of migration, in millimeters, are given next to the minimally and maximally retarded species for each construct.
We attempted to use the phase-sensitive assay to measure the DNA bending generated by the parental and mutant chimeric proteins (Fig. 3). For unknown reasons, the chimeric proteins exacerbate the tendency of the DNA used in the assay to form indistinct bands. Therefore, we present the data which show that the mutant chimera bends DNA less than the parental chimera but refrained from attempting to determine actual bending angles. The top part of Fig. 4 shows the results of a simple DNA migration retardation assay in which the binding site was located near the middle of a 160-bp DNA fragment. The mutant chimeric protein retarded migration substantially less than the parental chimeric protein did. The bottom part of Fig. 4 shows that AraC containing the H213Y mutation also retarded the DNA less than wild-type AraC did.
FIG. 3.

Phase-sensitive bending assay of the parental C/EBP-AraC chimera and the H213Y chimera performed with crude cell lysates.
FIG. 4.

Migration retardation assay of DNA bending by the chimeric proteins and by AraC using DNA containing the I1-I2 binding site in the middle of a 160-bp fragment. The protein sources were 1 μl of a lysate containing either parental or H213Y chimeric AraC and 1 μl of purified wild-type (WT) AraC at 0.4 mg/ml.
DISCUSSION
We used a simple screen to identify mutations within AraC specifically defective in transcription activation. It utilizes a chimeric protein composed of the DNA binding and transcription activation domain of AraC fused to the leucine zipper dimerization domain of C/EBP. Mutations in the chimeric protein that are defective in transcription activation but not defective in DNA binding should be trans dominant negative with regard to wild-type AraC protein. We identified a number of mutants with the desired characteristics and studied the one with the strongest phenotype, H213Y, more carefully.
The H213Y mutation was previously isolated in a screen for mutants with reduced ability to activate pBAD (3). Because its alteration was found to lie within a potential recognition helix in AraC, the mutant protein was used in a missing-contact experiment and found to have lost contact with three bases in each of the half-sites of araI. This raises the question of why the mutant protein is defective in transcription activation. It certainly binds DNA because it is trans dominant negative. Although it is possible that the amino acid residues that directly contact DNA also contact RNA polymerase to help activate transcription, this possibility seems somewhat unlikely. We therefore considered the possibility that the protein is defective in DNA bending and that a change in bending interferes with the formation of some of the protein-protein contacts required for activation at pBAD. Consequently, we first measured the DNA bending produced by AraC protein at araI and at two closely related sites.
AraC bends DNA substantially, approximately 90°. H213Y AraC and the H213Y AraC-C/EBP chimeric protein bend DNA less. The reduced bending is shown both in the phase-sensitive assay and by the migration rate reduction induced by the mutant proteins when the AraC binding site is in the middle of the DNA fragment. The possibility that the H213Y chimera retards DNA less than the parental protein does because it is a monomer in vivo is not plausible. The mutation lies in a domain of the protein that does not contain determinants for dimerization. Additionally, H213Y chimeric AraC represses transcription of the promoter pC, whereas the monomeric DNA binding domain is incapable of repressing pC (34). It does not seem likely that the mutation could alter the structure of the DNA binding domain in such a way that migration retardation is altered in a phase-specific way in the phase-sensitive assay, as well as in the simple-bend assay, particularly in light of the fact that the residue that is altered apparently directly contacts DNA.
In contrast to its behavior at the pBAD promoter, the H213Y AraC-C/EBP chimera activated the pFGH promoter nearly as well as the parental chimeric protein did. Examination of the pBAD and pFGH promoter structures reveals why this might be the case (Fig. 5). At pBAD, AraC binds between RNA polymerase and CAP and the bending produced by AraC likely facilitates the formation of an RNA polymerase interaction with the rear subunit of AraC (31), as well as an RNA polymerase-CAP interaction (33). At pFGH, it is CAP that binds between RNA polymerase and AraC. As a result, DNA bending by AraC may be unnecessary for transcription activation at pFGH because no polymerase contacts are likely to be made beyond the downstream face of AraC.
FIG. 5.

Protein binding sites and likely RNA polymerase interactions with CAP and AraC at the ara pFGH promoter. At pFGH, the direct-repeat half-sites for AraC binding are reversed from their direction at pBAD.
We do not understand why the H213Y mutation has a significantly stronger effect in the context of the C/EBP-AraC chimera than when the mutation is in AraC itself. When present in the chimera, the mutation reduces activation by a factor of more than 10, but when in intact AraC, the mutation reduces activation by a factor of about 2. It is possible that a site that can function in activation lies within the dimerization domain of AraC. Another possibility is that the DNA bending produced by AraC is different from that produced by the chimeric protein.
Mutations changing the degree of DNA bending have been isolated in several other proteins. In FIS protein, the mutation R71A results in a reduced ability to bend the DNA, and yet the protein binds DNA with close to wild-type affinity (2). A mutant form of the phage φ29 P4 protein also results in reduced DNA bending with retention of nearly normal DNA binding affinity (20). These mutants may be defective in transcription activation because other required protein-protein interactions cannot form due to insufficient DNA bending. Many other studies have implicated DNA bending in the activation of promoter activity, but in such work, as was the case here, it is most difficult to determine the underlying relevant mechanism, whether it is DNA bending per se, the creation of a binding site for another protein, the creation or improvement of a binding site for the C-terminal domain of the RNA polymerase alpha subunit, or the facilitation of the formation of a multiprotein complex.
ACKNOWLEDGMENTS
This work was supported by NIH grant GM18277 to R.F.S.
We thank Jeff Withey and Beth MacDougall-Shackleton for their assistance and Richard Gourse for activating us to examine the bending of H213Y after the principal investigator had rejected the idea.
REFERENCES
- 1.Bell A, Gaston K, Williams R, Chapman K, Kolb A, Buc H, Minchin S, Williams J, Busby S. Mutations that alter the ability of the Escherichia coli cyclic AMP receptor protein to activate transcription. Nucleic Acids Res. 1990;18:7243–7250. doi: 10.1093/nar/18.24.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bokal A J, Ross W, Gaal T, Johnson R C, Gourse R L. Molecular anatomy of a transcription activation patch: FIS-RNA polymerase interactions at the Escherichia coli rrnB P1 promoter. EMBO J. 1997;16:154–162. doi: 10.1093/emboj/16.1.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brunelle A, Schleif R. Determining residue-base interactions between AraC protein and araI DNA. J Mol Biol. 1989;209:607–622. doi: 10.1016/0022-2836(89)90598-6. [DOI] [PubMed] [Google Scholar]
- 4.Busby S, Ebright R. Transcription activation at class II CAP-dependent promoters. Mol Microbiol. 1997;23:853–859. doi: 10.1046/j.1365-2958.1997.2771641.x. [DOI] [PubMed] [Google Scholar]
- 5.Bustos S A, Schleif R F. Functional domains of the AraC protein. Proc Natl Acad Sci USA. 1993;90:5638–5642. doi: 10.1073/pnas.90.12.5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carra J, Schleif R. Variation of half-site organization and DNA looping by AraC protein. EMBO J. 1993;12:35–44. doi: 10.1002/j.1460-2075.1993.tb05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebright R H. Transcription activation at class I CAP-dependent promoters. Mol Microbiol. 1993;8:797–802. doi: 10.1111/j.1365-2958.1993.tb01626.x. [DOI] [PubMed] [Google Scholar]
- 8.Eschenlauer A C, Reznikoff W S. Escherichia coli catabolite gene activator protein mutants defective in positive control of lac operon transcription. J Bacteriol. 1991;173:5024–5029. doi: 10.1128/jb.173.16.5024-5029.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Greenblatt J, Schleif R. Arabinose C protein: regulation of the arabinose operon in vitro. Nat New Biol. 1971;233:166–170. doi: 10.1038/newbio233166a0. [DOI] [PubMed] [Google Scholar]
- 10.Hahn S, Schleif R F. In vivo regulation of the Escherichia coli araC promoter. J Bacteriol. 1983;155:593–600. doi: 10.1128/jb.155.2.593-600.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hendrickson W, Schleif R F. Regulation of the Escherichia colil-arabinose operon studied by gel electrophoresis DNA binding assay. J Mol Biol. 1984;178:611–628. doi: 10.1016/0022-2836(84)90241-9. [DOI] [PubMed] [Google Scholar]
- 12.Huo L, Martin K, Schleif R F. Alternate DNA loops regulate the arabinose operon in Escherichia coli. Proc Natl Acad Sci USA. 1988;85:5444–5448. doi: 10.1073/pnas.85.15.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Igarashi K, Ishihama A. Bipartite functional map of the E. coli RNA polymerase alpha subunit: involvement of the C-terminal region in transcription activation by cAMP-CRP. Cell. 1991;65:1015–1022. doi: 10.1016/0092-8674(91)90553-b. [DOI] [PubMed] [Google Scholar]
- 14.Kerppola T K, Curran T. DNA bending by Fos and Jun: the flexible hinge model. Science. 1991;254:1210–1214. doi: 10.1126/science.1957173. [DOI] [PubMed] [Google Scholar]
- 15.Koo H S, Crothers D M. DNA bending at adenine-thymine tracts. Nature. 1986;320:501–506. doi: 10.1038/320501a0. [DOI] [PubMed] [Google Scholar]
- 16.Koo H-S, Drak J, Rice J, Crothers D M. Determination of the extent of DNA bending by an adenine-thymine tract. Biochemistry. 1990;29:4227–4234. doi: 10.1021/bi00469a027. [DOI] [PubMed] [Google Scholar]
- 17.Kraft R, Tardiff J, Krauter K S, Leinwand L A. Using mini-prep plasmid DNA for sequencing double stranded templates with Sequenase. BioTechniques. 1988;6:544–546. [PubMed] [Google Scholar]
- 18.Lee N, Wilcox G, Gielow W, Arnold J, Cleary P, Englesberg E. In vitro activation of the transcription of araBAD operon by araC activator. Proc Natl Acad Sci USA. 1974;71:634–638. doi: 10.1073/pnas.71.3.634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maniatis T, Fritsch E F, Sambrook J. Molecular cloning: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- 20.Mencia M, Monsalve M, Salas M, Rojo F. Transcriptional activator of the phage phi29 late promoter: mapping of residues involved in interaction with RNA polymerase and in DNA bending. Mol Microbiol. 1996;20:273–282. doi: 10.1111/j.1365-2958.1996.tb02616.x. [DOI] [PubMed] [Google Scholar]
- 21.Miller J H. Experiments in molecular genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1972. [Google Scholar]
- 22.Niu W, Kim Y, Tau G, Heyduk T, Ebright R H. Transcription activation at class II CAP-dependent promoters: two interactions between CAP and RNA polymerase. Cell. 1996;87:1123–1134. doi: 10.1016/s0092-8674(00)81806-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogden S, Haggerty D, Stoner C, Kolodrubetz D, Schleif R. The Escherichia colil-arabinose operon: binding sites of the regulatory proteins and a mechanism of positive and negative regulation. Proc Natl Acad Sci USA. 1980;77:3346–3350. doi: 10.1073/pnas.77.6.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reeder T, Schleif R. AraC protein can activate transcription from only one position and when pointed in only one direction. J Mol Biol. 1993;231:205–218. doi: 10.1006/jmbi.1993.1276. [DOI] [PubMed] [Google Scholar]
- 25.Saviola B, Seabold R, Schleif R. Arm-domain interactions in AraC. J Mol Biol. 1998;278:539–548. doi: 10.1006/jmbi.1998.1712. [DOI] [PubMed] [Google Scholar]
- 26.Schleif R, Wensink P. Practical methods in molecular biology. New York, N.Y: Springer-Verlag; 1981. [Google Scholar]
- 27.Schleif R F, Favreau M A. Hyperproduction of AraC protein from Escherichia coli. Biochemistry. 1982;21:778–782. doi: 10.1021/bi00533a031. [DOI] [PubMed] [Google Scholar]
- 28.Sheppard D E, Englesberg E. Further evidence for positive control of the l-arabinose system by gene araC. J Mol Biol. 1967;25:443–454. doi: 10.1016/0022-2836(67)90197-0. [DOI] [PubMed] [Google Scholar]
- 29.Simons R W, Houman F, Kleckner N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene. 1987;53:85–96. doi: 10.1016/0378-1119(87)90095-3. [DOI] [PubMed] [Google Scholar]
- 30.Thompson J F, Landy A. Empirical estimation of protein-induced DNA bending angles: applications to lambda site-specific recombination complexes. Nucleic Acids Res. 1988;16:9687–9705. doi: 10.1093/nar/16.20.9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X, Reeder T, Schleif R. Transcription activation parameters at ara pBAD. J Mol Biol. 1996;258:14–24. doi: 10.1006/jmbi.1996.0230. [DOI] [PubMed] [Google Scholar]
- 32.Zhang X. Ph.D. dissertation. Baltimore, Md: Johns Hopkins University; 1997. [Google Scholar]
- 33.Zhang X, Schleif R. Catabolite gene activator protein mutations affecting activity of the araBAD promoter. J Bacteriol. 1998;180:195–200. doi: 10.1128/jb.180.2.195-200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang, X., and R. Schleif. Unpublished data.
- 35.Zhou Y, Zhang X, Ebright R H. Identification of the activating region of catabolite gene activator protein (CAP): isolation and characterization of mutants of CAP specifically defective in transcription activation. Proc Natl Acad Sci USA. 1993;90:6081–6085. doi: 10.1073/pnas.90.13.6081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zinkel S A, Crothers D M. DNA bend direction by phase sensitive detection. Nature. 1987;328:178–181. doi: 10.1038/328178a0. [DOI] [PubMed] [Google Scholar]