

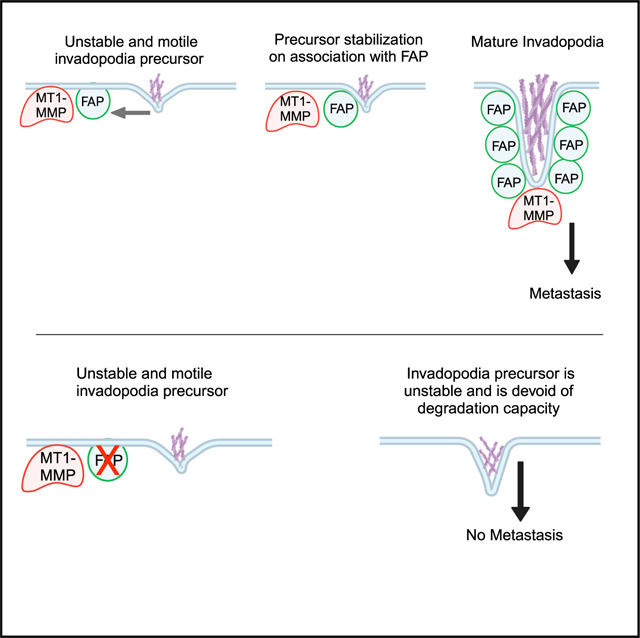
SUMMARY
During metastasis, tumor cells invade through the basement membrane and intravasate into blood vessels and then extravasate into distant organs to establish metastases. Here, we report a critical role of a trans-membrane serine protease fibroblast activation protein (FAP) in tumor metastasis. Expression of FAP and TWIST1, a metastasis driver, is significantly correlated in several types of human carcinomas, and FAP is required for TWIST1-induced breast cancer metastasis to the lung. Mechanistically, FAP is localized at invadopodia and required for invadopodia-mediated extracellular matrix degradation independent of its proteolytic activity. Live cell imaging shows that association of invadopodia precursors with FAP at the cell membrane promotes the stabilization and growth of invadopodia precursors into mature invadopodia. Together, our study identified FAP as a functional target of TWIST1 in driving tumor metastasis via promoting invadopodia-mediated matrix degradation and uncovered a proteolytic activity-independent role of FAP in stabilizing invadopodia precursors for maturation.
In brief
Bukhari et al. report a functional role of the transmembrane serine protease FAP in tumor metastasis. FAP is required for invadopodia-mediated matrix degradation independent of its proteolytic activity. FAP localizes at invadopodia and promotes invadopodia precursor stabilization. These results indicate a structural role of FAP at invadopodia to promote tumor metastasis.
Graphical Abstract
INTRODUCTION
Tumor cells need to gain invasive and migratory abilities to intravasate into the lymph and blood systems, travel through the vasculature, and extravasate from the vessel into the distant organ, where they eventually form macrometastases.1,2 Tumor cells can invade through the formation of membrane protrusions to localize various proteases to cell-matrix contact points for extracellular matrix (ECM) degradation.3,4 These structures, termed invadopodia, are actin-rich cell protrusions that contain a number of actin regulatory proteins such as cortactin, Arp2/3, and N-WASP.3–6 In addition, Tks4 and Tks5 are important adaptor proteins that regulate invadopodia structure and function.7,8 Invadopodia are also shown to contain numerous matrix proteases, including metalloproteinases (matrix metalloproteinases [MMPs]) MMP2, MMP9, membrane-type 1 (MT1)-MMP, and the disintegrin and metalloproteinases (ADAMs).6,9–12 Being distinct from other actin-based protrusions such as lamellipodia and filopodia, invadopodia are mostly found in invasive cell types: macrophages, osteoclasts, and invasive tumor cells.13 Using various imaging techniques on metastatic tumor xenografts, a number of studies have demonstrated invadopodia formation during both the intravasation and extravasation steps of tumor metastasis.14–17 In addition, suppressing invadopodia function by inhibiting essential invadopodia components such as cortactin, Tks5, or MT1-MMP prevents breast cancer metastasis in vivo.18–20 Altogether, these data suggest that invadopodia-mediated ECM degradation is essential for tumor invasion and metastasis.
Fibroblast activation protein (FAP) is a type II integral serine protease whose expression is restricted to the embryonic mesenchyme and is not observed in normal adult tissues. Interestingly, FAP is highly expressed in cancer-associated fibroblasts,21,22 and it has also been shown to be expressed in epithelial tumor cells.23 FAP was previously shown to be associated with α3β1 integrin, DPPIV, MMP-2, MT1-MMP, and uPA at invadopodia in human tumor cells.24 However, whether FAP is involved in invadopodia formation and/or ECM degradation and whether FAP expression in tumor cells promotes tumor invasion and metastasis is unknown.
The transcription factor TWIST1 promotes breast cancer metastasis and induces epithelial-mesenchymal transition (EMT).25 TWIST1 expression is associated with metastasis and poor survival rates in melanoma, neuroblastoma, prostate cancers, breast cancers, and gastric cancers.26 Mechanistic studies demonstrate that TWIST1 also induces invadopodia formation and ECM degradation to drive breast cancer metastasis.20 In search of proteins induced by TWIST1 to drive tumor invasion and metastasis, we explored the involvement of FAP in coordinating invadopodia formation and function and its role in tumor metastasis.
RESULTS
Expression of TWIST1 and FAP is tightly correlated in various human cancer types
Previous studies identified a critical role of TWIST1 in driving tumor metastasis by promoting invadopodia-mediated ECM degradation.20,25,27 To understand how TWIST1 drives ECM degradation and to identify therapeutic targets of metastasis, we expressed an inducible TWIST1 (Twist-ER) construct28,29 in human mammary epithelial human mammary epithelial cells (HMLE) to identify genes that are induced by TWIST1 and have been associated with human cancer. We found that a transmembrane serine protease FAP was induced upon TWIST1 induction and reached 8-fold at the mRNA level after 12 days (Figure 1A). FAP protein expression also increased 3 days after TWIST1 induction (Figure 1B). FAP is best known for its expression in activated cancer-associated fibroblasts, but it is also observed in tumors of epithelial origin.23,30–33 To determine whether the expression of TWIST1 and FAP is correlated in human epithelial cancers, we examined their expression in The Cancer Genome Atlas data from 1.084 human breast cancer samples, 523 head and neck cancer samples, 566 lung cancer samples, and 184 pancreatic cancer samples (Figures 1C–1F). In all cases, the spearman correlation coefficient rages from 0.58 to 0.69 with a p value of < e−15, indicating a strong correlation between TWIST1 and FAP expression in various human epithelial cancers. Collectively, these data show that FAP is upregulated downstream of TWIST1 and their expression levels are tightly correlated in various human carcinomas.
Figure 1. Expression of TWIST1 and FAP is tightly correlated in various human cancer types.

(A) qRT-PCR measured FAP mRNA levels in HMLE-TwistER cells undergoing Twist1 induction (red), and in un-induced HMLE-GFP cells (blue). Error bars represent the SEM.
(B–F) (B) Lysates from HMLE-TwistER induced cells probed for FAP and β-actin over the course of 6 days. The Cancer Genome Atlas (TCGA) data showing correlation between Twist1 and FAP in human breast cancer (C), head and neck cancer (D), lung cancer (E), and pancreatic cancer (F). Expression data were retrieved from cBioPortal. Both Spearman correlation coefficients with p values and Pearson correlation coefficients with p values are presented.
FAP is required for TWIST1-induced breast tumor metastasis
Given the strong correlation between TWIST1 and FAP expression in human carcinoma, we sought to determine whether FAP plays a role in TWIST1-induced breast cancer metastasis in vivo. HMLE-TWIST1 cells expressing either control short hairpin RNA (shRNA) or two independent shRNAs targeting FAP (Figures 2A and 2B) were labeled with GFP, transformed with oncogenic Ras, and injected into female mice. FAP knockdown did not affect the primary tumor growth rate or the primary tumor weight (Figures 2C and 2D). Strikingly, while HMLER-TWIST1 tumors expressing a control shRNA generated hundreds of GFP-positive micrometastases throughout the lungs, knockdown of FAP reduced lung metastases by an average of 400-fold (Figures 2E and 2F). These data demonstrate that FAP functions downstream of TWIST1 in promoting breast tumor cells to metastasize to the lung.
Figure 2. FAP is required for TWIST1-induced breast cancer metastasis.

(A) qRT-PCR measurement of FAP mRNA levels in HMLER-TWIST1 cells expressing shRNAs targeting FAP.
(B) Western blotting analysis on lysates obtained from HMLER-TWIST1 cells as indicated.
(C) Nude mice were injected subcutaneously with GFP + HMLER-TWIST1 cells expressing a control vector (shCtrl) or shRNAs targeting FAP. Tumor diameters were measured as indicated. Once the tumors reached 2 cm in diameter, they were harvested and weighed as shown in (D).
(E) Quantification of GFP-positive cells in each individual lung. N = 10 mice per group. **p < 0.01, Student’s t-test. Error bars in (A, C, D and E) represent the SD.
(E) Representative images of lungs of nude mice injected with HMLER-TWIST1 cells expressing the indicated shRNAs. Scale bar, 1 mm. See also Figure S1.
FAP plays a key role in invadopodia-mediated matrix degradation
We have previously shown that TWIST1 induces invadopodia-mediated ECM degradation to promote metastasis.20 Given that FAP has been associated with invadopodia,23,34–36 we next tested whether FAP plays a role in invadopodia-mediated ECM degradation. Control and FAP-knockdown HMLE-TWIST1 cells were seeded onto Oregon green (OG) gelatin-coated coverslips, and the degree of matrix degradation was imaged and quantified. Knockdown of FAP reduced matrix degradation by 80%–90% when compared with the control cells (Figures 3A–3C). TWIST1 is a key transcription factor that induces EMT.25,27 It is important to note that HMLE-TWIST1 cells expressing shRNAs against FAP exhibited similar mesenchymal phenotypes as the control cells (Figure S1). This indicates that suppression of FAP does not inhibit cells from undergoing TWIST1-induced EMT; instead, FAP is specifically required for TWIST1-induced matrix degradation.
Figure 3. FAP is required for invadopodia-mediated ECM degradation.

(A–L) OG gelatin degradation assay was performed on HMLE-TWIST1 cells expressing FAP shRNAs (A–C) plated for 16 h; SUM1315 cells expressing FAP shRNAs (D–F) were plated for 6 h; SCC61 cells expressing FAP shRNAs (G–I) were plated for 16 h; and BxPC3 cells expressing FAP shRNAs were plated for 8 h (J– L). FAP gene knockdown levels are measured by qRT-PCR (B, E, H, and K). F-actin was stained with phalloidin (red) and nuclei with DAPI (blue). Areas of degradation appear as punctate black areas beneath the cells.
(M) SCC61 cells expressing FAP shRNAs were plated on Matrigel-coated transwell chambers. Cells that invaded through the Matrigel were stained using crystal violet.
(N) Quantification of invasion normalized over cells expressing a control shRNA. Error bars in (B, E, H, and K) represent the SD. Error bars in (C, F, I, and L) represent the SEM. ***p < .001, **p < .01, *p < .05, Student’s t-test. Scale bars in (A, D, G, and J), 10 μm; scale bar in (M), 100 μm. See also Figure S2.
To further demonstrate a critical role of FAP in invadopodia-mediated matrix degradation, we tested whether FAP knockdown impacts ECM degradation in several human carcinoma cell lines, including human breast cancer cells SUM1315, MDA-MB-231-Src cells, head and neck cancer cell SCC61, and pancreatic cancer cell BxPC3 because of the strong correlation of FAP and TWIST1 expression in these human cancer types. Similar to HMLE-TWIST1 cells, knockdown of FAP significantly reduced matrix degradation in all four human cancer cell lines (Figures 3D−3L and S2A–2C). Next, we determined whether the defect in ECM degradation observed in FAP-knockdown cells would impede cells from invading through Matrigel-coated trans-wells. Indeed, knocking down FAP in SCC61 cells strongly reduced the ability of the cells to invade through the Matrigel (Figures 3M and 3N). In contrast, knockdown of FAP did not affect the ability of the cells to migrate and close the wound in the wound scratch assay (Figures S2D and S2E). Together, these data strongly indicate that FAP plays an essential role in driving ECM degradation and cell invasion in various human cancer cells.
FAP is required for ECM degradation independent of its proteolytic activity
FAP belongs to the dipeptidylpeptidase (DPP) 4 protein family (DPP4, FAP, DPP8, and DPP9), which hydrolyzes a prolyl bond that is two amino acids from the N terminus of a protein.37 FAP and DPP4 are the only two transmembrane serine proteases in the DPP4 family with more than 68% homology23 and have been reported to form both homodimeric and heterodimeric complexes.23,34 To assess whether FAP functions in matrix degradation in complex with DPP4, we knocked down DPP4 in SUM1315 cells and found that knockdown of DPP4 had no effect on ECM degradation (Figures S3A–S3C), in contrast with what was observed in cells with FAP knockdown. These results demonstrate that FAP is uniquely required for ECM degradation among the DPP4 protease family.
To determine whether the proteolytic activity of FAP is required for ECM degradation, we utilized M83, a specific FAP inhibitor with a greater than 1,000-fold specificity against FAP over DPP438 to inhibit FAP protease activity. To ensure that M83 indeed abrogated the FAP catalytic activity, we used a prolyl-specific substrate C95 that emits fluorescence upon cleavage to measure FAP enzymatic activity.39 M83 treatment completely inhibited C95 cleavage, as shown by the lack of fluorescence increase during the experimental time course, in contrast with the drastic increase in C95 cleavage observed in cells treated with the vehicle control (Figure 4A). We then treated both SUM1315 and SCC61 cells with M83 and performed matrix degradation assays. Surprisingly, M83-treated cells displayed similar levels of matrix degradation as cells treated with the vehicle alone in both cell types (Figures 4B, 4C, S3D, and S3E). In contrast, the pan-MMP inhibitor GM6001 completely inhibited ECM degradation in both cell types (Figures 4B, 4C, S3D, and S3E), which is consistent with the role of MMPs in invadopodia-mediated ECM degradation.40 These data suggest that the proteolytic activity of FAP is not required for its function in ECM degradation.
Figure 4. FAP promotes ECM degradation independent of its proteolytic activity.

(A) SUM1315 cells, seeded at 80%–90% confluency in a 96-well plate, were treated with PBS or 10 μM of the FAP inhibitor, M83, followed by 125 μM of the FAP-specific substrate, C95. FAP-mediated C95 cleavage fluorescence was measured over time.
(B) SUM1315 cells were seeded onto OG-labeled gelatin in the presence of PBS, M83, or GM6001 for 6 h and subsequently stained for F-actin (red) and nuclei (blue).
(C) Quantification of degradation of OG labeled gelatin by SUM1315 cells from (B).
(D) FAP-mediated C95 cleavage was measured in 293T cells expressing catalytically inactive FAP mutants.
(E) SUM1315 cells with FAP knockdown were transfected with the indicated constructs, seeded onto OG-labeled gelatin, and evaluated for ECM degradation relative to SUM1315 cells expressing a control shRNA.
(F) SUM1315 cells expressing FAP sh3 and transfected with the constructs indicated in (E) were seeded onto OG-labeled gelatin for 6 h. (H) Quantification of OG gelatin degradation from (F).
(G) SUM1315 cells expressing FAP sh5 and transfected with the constructs indicated in (E) were seeded onto OG-labeled gelatin for 6 h.
(I) Quantification of OG gelatin degradation from (G). Error bars in (A and D) represent the SD. Error bars in (C, H, and I) represent the SEM. ***p < .001, **p < .01, *p < 0.05. ns, not significant (p > 0.5), Student’s t-test. N = 150 cells per group. Scale bar, 10 μm. See also Figure S3.
Among all DPP4 family serine proteases, FAP is unique in that it has both exopeptidase and endopeptidase activities, while the rest of family members only present exopeptidase activity.39,41 To further determine whether the serine protease activity of FAP is required for its function in ECM degradation, we evaluated the exopeptidase vs. endopeptidase activity of FAP in ECM degradation. We mutated two key residues in the catalytic domain of FAP: S624, which is part of the catalytic triad and vital for both exo- and endopeptidase activity, and A657, which controls the geometry of the active site and is only important for the endopeptidase activity.24,41 Using C95 as the substrate, we verified that the S624A mutant displayed no protease activity, whereas the A657S mutant presented a partial protease activity (Figure 4D), which is consistent with previous data.41,42 We then expressed either a control vector, wild-type FAP, or individual FAP mutants into FAP-knockdown SUM1315 cells to assess their roles in ECM degradation (Figure 4E). We found that expression of wild-type FAP, the S624A protease-dead mutant, or the A657S endopeptidase-dead mutant all rescued the ECM degradation defect caused by knockdown of FAP (Figures 4F–4I). Collectively, these data demonstrate that FAP plays a unique and critical role in driving cancer cell ECM degradation independent of its proteolytic activity.
FAP localizes at invadopodia and is required for invadopodia stabilization
Given that FAP is required for ECM degradation and metastasis in vivo and that FAP is associated with invadopodia in endothelial and melanoma cells,34,35,43 we set out to examine whether FAP localizes to invadopodia. We focused on two human carcinoma cell lines: the head and neck cancer cell line SCC61 and the pancreatic cancer cell line BxPC3 because both have been widely used for invadopodia imaging studies. We found that FAP is required for ECM degradation in both cancer cell types (Figures 3G −3L). Colocalization of F-actin and the actin-binding protein cortactin is used to mark invadopodia protrusions.44 We found that F-actin, cortactin, and FAP colocalize in discrete punctate structures (Figure 5A), indicating that FAP localizes to the invadopodia protrusions. We further examined the distribution of FAP at the invadopodia by Z-sectioning, and found that F-actin, cortactin, and FAP colocalize at structures that protrude from the basal surface of the cells (Figures 5B and 5C), further highlighting the specificity of FAP localization at invadopodia protrusions. To determine whether FAP is present at mature invadopodia characterized by their matrix degradation capability, we performed total internal reflection fluorescence (TIRF) microscopy on cells growing on fluorescent OG gelatin and found that FAP colocalized with F-actin on the black degradation spots (Figure S4A).
Figure 5. FAP localizes at invadopodia in various human cancer cells.
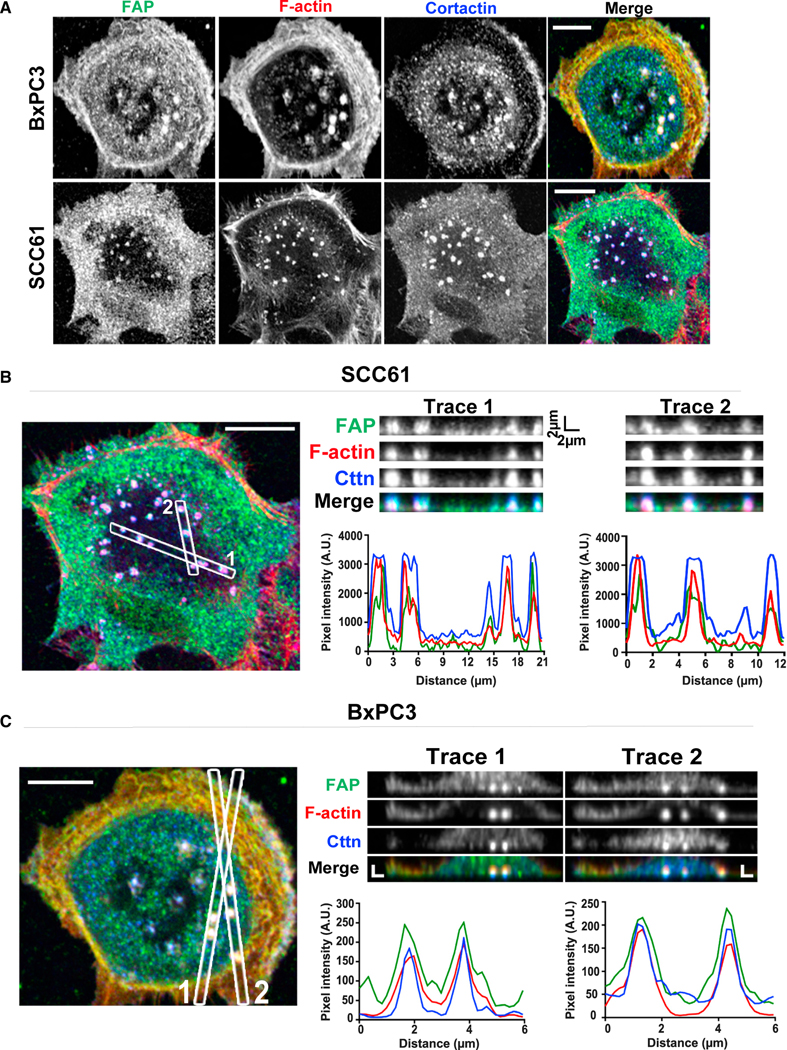
(A–C) (A) BxPC3 and SCC61 cells were seeded onto gelatin-coated coverslips for 48 h and labeled for FAP (green), F-actin (red), and cortactin (blue). Confocal image of SCC61 (B) and BxPC3 (C). Traces 1 and 2 are z axis sectioning of invadopodia in each cell line; associated graphs represent pixel intensities along the dotted lines to show the colocalization of FAP, F-actin, and cortactin. Scale bar, 10 μm. See also Figure S4.
Invadopodia are thought to assemble in a series of discrete steps.3,4,6,45 Initially, the actin structural components, actin, and cortactin along with other actin-binding proteins, converge to form the initial actin protrusions. During this time, the invadopodium is referred to as an invadopodium precursor. Following the recruitment of Tks5, a core invadopodia scaffolding protein, the invadopodium precursor begins to mature. After this subcellular protrusion has formed, proteases such as MT1-MMP localize to the maturing invadopodium, leading to ECM degradation. Given that FAP is physically localized at invadopodia and is required for invadopodia-mediated ECM degradation in a protease-independent manner, we hypothesized that FAP plays a key structural role in invadopodia formation or maturation. First, we counted F-actin+ invadopodia precursor numbers in SCC61 cells and HMLE-TWIST1 cells expressing the control shRNA or shRNAs against FAP and found that colocalization of F-actin and cortactin at invadopodia remained intact upon FAP knockdown (Figures 6A and S4B). We also did not observe any differences in the number of F-actin+ invadopodia precursors in the control and FAP-knockdown cells or in the percentage of cells with more than five invadopodia protrusions (Figures 6B and S4D), suggesting that FAP suppression does not affect invadopodium precursor assembly. Interestingly, we did observe a decrease in F-actin signal intensity at invadopodia precursors in the knockdown cells compared with the control cells (Figure 6D), despite the cell body surface area of the control and knockdown cells remaining similar (Figure S4E). Furthermore, the intensity of cortactin signals at invadopodia precursors is also significantly reduced in FAP-knockdown HMLE-TWIST1 cells compared with the control cells (Figures 6D, S4B, and S4C). We next performed immunostaining to determine whether enrichment of the invadopodia markers, Tks5 and MT1-MMP, were affected by FAP knockdown. Both Tks5 and MT1-MMP signals displayed a significant reduction at F-actin+ invadopodia in FAP-knockdown cells (Figures 6E–6H). These results suggest that FAP is required for the formation of mature invadopodia.
Figure 6. FAP knockdown blocks invadopodia precursor stabilization and subsequent maturation.
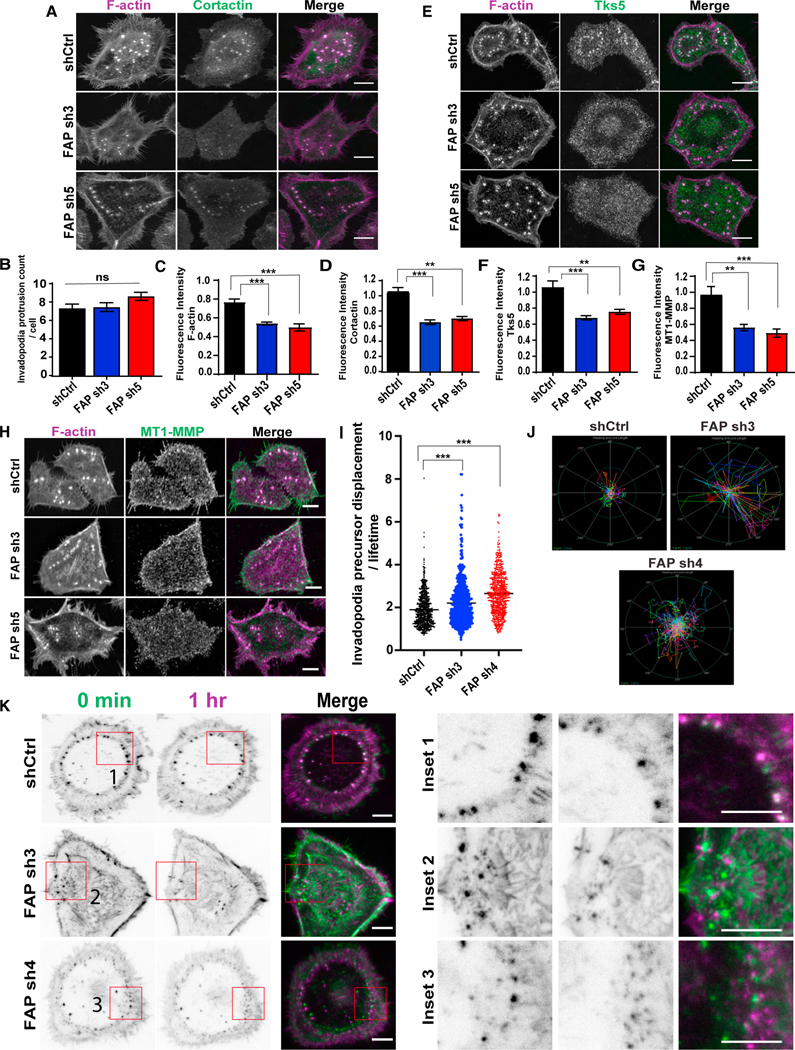
(A, D, E, H, F, G) SCC61 cells expressing the indicated FAP shRNAs were seeded onto gelatin-coated coverslips for 48 h, and stained for F-actin (magenta), and cortactin (green) (A), Tks5 (green) (E), MT1-MMP (green) (H). N = >20 cells per group. Scale bar, 10 μm. Quantification of normalized fluorescence intensity of cortactin (D), Tks5 (F), and MT1-MMP (G) at invadopodia relative to control shRNA.
(B) Quantification of invadopodia protrusion count per cell in SCC61 cells expressing FAP shRNAs.
(C) Quantification of F-actin intensity in SCC61 cells expressing different FAP shRNAs.
(I) Quantification of average invadopodia protrusion displacement divided by its lifetime (number of frames) (pixels/min) in Life-Act mCherry expressing SCC61 cells with FAP shRNAs. Each dot in the graph represents individual precursors. N > 100 precursors per group.
(J) Polar graph indicating invadopodia precursor trajectories in a representative SCC61 cell expressing shRNAs targeting FAP. The outer ring (green) represents the distance of 4 μm and inner circle (blue) represents distance of 2 μm. Tracks were measured at 20 s/frame for 30 min.
(K) Time lapse pictures of live SCC61 cells expressing F-actin and FAP shRNAs captured using TIRF microscopy at 100× magnification at time 0 (green) and at 1 h (magenta). Insets show zoomed in view of invadopodia displacement over time. Scale bar, 10 μm. Error bars are the SEM. ***p < .001, **p < .01, *p < .05. ns, not significant (p > 0.5). Student’s t-test. See also Figure S4A and Videos S1−S3.
To further dissect the role of FAP in invadopodia assembly and maturation, we expressed LifeACT-mCherry46 to label F-actin in SCC61 cells expressing the control shRNA or shRNAs against FAP and performed live imaging to examine protrusion dynamics. Interestingly, we found that F-actin+ invadopodia were significantly more dynamic and migrated significantly longer distances in SCC61 cells with FAP knockdown compared with the control cells, (Figures 6I−6K and Videos S1, S2, and S3). These results indicate that FAP plays a critical role in maintaining invadopodia precursor stability.
FAP functions upstream of MT1-MMP recruitment to promote invadopodia precursor stability
Our data show that FAP does not affect the invadopodia actin protrusion formation, but is specifically required for invadopodia precursor stabilization. Because deletion of MT1-MMP is reported to cause defects in invadopodia maturation and stability,47 we also knocked down MT1-MMP in SCC61 cells to compare the phenotypes. Consistent with previous reports,45 knockdown of MT1-MMP drastically suppressed invadopodia-mediated ECM degradation (Figures S5A–S5C). Immunostaining analysis shows that, similar to FAP, MT1-MMP knockdown did not affect the number of F-actin+ invadopodia precursor numbers per cell, but reduced both cortactin and Tks5 signal intensities at invadopodia (Figures S6A–S6E). Furthermore, FAP staining shows that knockdown of MT1-MMP did not affect FAP localization at invadopodia (Figures S6F and S6G). Together, these data suggest that, while both FAP and MT1-MMP are required for stabilizing invadopodia precursors, they likely play distinct roles: FAP is recruited to invadopodia before MT1-MMP during invadopodia stabilization.
To further examine the role of FAP in MT1-MMP recruitment, we overexpressed MT1-MMP fused with the pH-sensitive pHluorin fluorescent protein48 in SCC61 cells expressing control or shRNAs against FAP (Figures S5D and S5E). While our initial plan was to perform live imaging to observe how FAP deletion affect MT1-MMP recruitment at invadopodia, we found that high levels of MT1-MMP expression rescued the ECM degradation defect caused by knockdown of FAP (Figures S5F and S5G). Further analyses of Tks5 and cortactin localization at invadopodia also revealed that overexpression of MT1-MMP rescued both Tks5 and cortactin enrichment at invadopodia in cells with FAP knockdown (Figures S6H–S6K). Together, these data suggest that one major function of FAP in tumor invasion and metastasis is to facilitate the recruitment of MT1-MMP to invadopodia to stabilize invadopodia precursors.
Association of invadopodia precursors with FAP on the cell membrane is a necessary step to stabilize invadopodia precursors
To examine the timing of FAP recruitment onto invadopodia precursors, we performed live cell TIRF imaging on SCC61 cells expressing ECFP tagged FAP, as well as MT1-MMP pHuji49 and SPY650-FastAct for actin labeling. Our data show that FAP and MT1-MMP puncta pre-exist on the cell membrane (Figure 7A, left). When actin protrusions reach such puncta, these actin protrusions become more stable with a longer life span, often growing into larger actin protrusions with more FAP and MT1-MMP (Figure 7A and Video S4). These live imaging data are consistent with the data presented in Figure 6C, showing that the actin signal intensity is weaker in cells with FAP knockdown.
Figure 7. The association of FAP with invadopodia precursors promotes invadopodia precursor stabilization.

(A) SCC61 cells expressing ECFP-FAP (green) and MT1-MMP pHuji (red) were labeled for F-actin with SPY650 FastAct (magenta) for 2 h prior to live cell imaging. The images were clarified and 2D deconvolved using NIS-Elements. The inset shows following stages of invadopodia lifespan: (I) FAP/MT1-MMP enriched membrane areas prior to invadopodia precursor docking, (II) mature invadopodia after F-actin elongation, and (III) remnants of FAP/MT1-MMP enriched membrane spot post F-actin disassembly. Live cell imaging on SCC61 cells expressing ECFP-FAP (green) and labeled with SPY650 FastAct shows that invadopodia precursors dock with FAP, which results in F-actin stabilization and growth (B and D). In contrast, invadopodia precursors not associated with FAP disassemble quickly (C and E). The images were clarified and 2D deconvolved using NIS elements. (D and E) are fluorescence intensities normalized to the last frame before actin protrusion appeared.
(F) Persistence of invadopodia precursors that are associated with FAP (black) and those that are not associated with FAP (gray). Student’s t-test was used to compare each group from three cells with 10 precursors per cell in each group. Error bars are the SEM. ***p < .001, **p < .01, *p < .05. ns, not significant (p > 0.5). Student’s t-test.
(F) Polar graph showing path lengths and trajectories of FAP-associated invadopodia precursors (left) and non-FAP-associated precursors (right) in an SCC61 cell expressing ECFP-FAP over a period of 30 min (100 precursors in each group). The outer circle (green) represents the distance of 4.8 μm from the center and the inner circle (blue) represents the distance of 2.4 μm from the center. Scale bars 2 μm. See also Figures S5, S6, and Videos S4−S8.
We further analyzed the time course of FAP recruitment at invadopodia precursors and found that initial actin protrusions docking with FAP resulted in F-actin elongation alongside further enrichment of FAP at protrusions (Figures 7B, 7D, and Videos S5 and S6) and formation of more persistent invadopodia. In contrast, actin protrusions that could not dock with FAP could grow further (Figures 7C, 7E, Videos S7, and S8). Lifetime analysis of these two populations of actin protrusions showed that almost all non-FAP-associated actin protrusions could not persist for more than 20 min, while the majority of the FAP-associated invadopodia persisted for more than 1 hour (Figure 7F). Consistent with the FAP knockdown results, we also observed that FAP-associated invadopodia were significantly less motile compared with non-FAP associated actin protrusions (Figure 7G). These results strongly support a model in which F-actin+ invadopodia precursors need to attach to FAP to stabilize themselves and further grow into mature invadopodia for ECM degradation.
DISCUSSION
In this report, we identified FAP as a critical metastasis gene by promoting invadopodia-mediated ECM degradation in various human carcinoma cells. We show that TWIST1 induces FAP expression in various carcinoma cells and the expression levels of TWIST1 and FAP are significantly correlated in various human carcinoma types, including breast cancer, head and neck cancer, lung cancer, and pancreatic cancer. One unique feature of FAP is its restricted expression to adult tissues undergoing wound healing and epithelial cancers,24,50 which is in sharp contrast with the ubiquitous expression of its close DPP family member DPP4 in all tissues. In human and mouse tumors, FAP has been shown to be expressed in stromal fibroblasts, carcinoma cells, and immune cells.51–54 Previous studies of FAP in cancer largely focused on cancer-associated fibroblasts. Importantly, we show that FAP is required for primary tumor cells to disseminate and metastasize into the lung in a human breast cancer xenograft model. Together, these data strongly support a role of FAP in mediating tumor invasion and metastasis in a tumor cell autonomous fashion in various human carcinoma. Importantly, given the critical role of TWIST1 in inducing EMT and invasion, our results suggest that FAP functions downstream of the EMT program to mediate matrix degradation and invasion in carcinoma cells.
The recruitment of MT1-MMP is a key step marking invadopodia maturation. However, previous studies show that MT1-MMP has a critical role in directing invadopodia assembly independent of its proteolytic activity.47 Our study shows that knockdown of FAP resulted in highly motile and immature invadopodia with reduced Tks5, cortactin, and MT1-MMP recruitment. In contrast, knockdown of MT1-MMP led to non-functional invadopodia precursors presenting intact FAP localization. Furthermore, the live imaging results show that FAP/MT1-MMP could pre-exist on the cell membrane. When actin protrusions reach such FAP-positive areas, these actin protrusions become more stable with a longer life span and stable localization and often grow into larger actin protrusions with more FAP and MT1-MMP. While it has been established that proteases localize to invadopodia late in the assembly of these structures,4,45 El Azzouzi et al.55 reported that MT1-MMP islets act as memory devices to provide structural functions for podosome reemergence. In agreement with this report, our studies suggest that FAP plays a critical role in stabilizing invadopodia precursors to allow invadopodia maturation.
One surprising finding from our work is that the function of FAP in invadopodia-mediated ECM degradation is independent of its proteolytic activity, adding another dimension of FAP functions in biology. Previous studies suggest that its gelatinase activity may contribute to the overall degradation activity of invadopodia. Christiansen et al.56 found that FAP could digest collagen I into smaller fragments following initial cleavage by MMP-1 in vitro, suggesting that FAP works together with other proteases to cleave partially degraded ECM components.56 Other studies have reported that FAP can promote wound healing, tumor growth, and cell invasion through non-enzymatic functions.42,57 Genetic deletion of FAP inhibited tumor growth in a K-Rasdriven model of endogenous lung cancer and in a mouse model of colon cancer.58 We show that FAP plays a structural role in invadopodia stabilization. While our study shows that the proteolytic activity of FAP is not required for its function at invadopodia to promote matrix degradation, it is still possible that the protease activity of FAP is important for cleaving other substrates that promote tumor development. Indeed, Jackson et al.59 reported that the FAP inhibitor M83 could inhibit angiogenesis and suppress human colon tumor growth in mice.
Previous attempts to target FAP for therapeutic intervention have proved to be challenging. Inhibitors targeting the FAP protease activity, which we show here to be ineffective at abrogating ECM degradation, showed minimal clinical activity.60–62 In 2003, phase I/II clinical trials for the humanized FAP monoclonal antibody sibrotuzumab failed to demonstrate measurable therapeutic activity in patients with metastatic colorectal cancer.63 However, this antibody has not been shown to block any cellular or protease function of FAP, which might explain the lack of therapeutic effects. A better tactic would be to use monoclonal antibodies to target the region of FAP responsible for protein-protein interactions, or the region required for its potential function in MT1-MMP recruitment. Even though the development of anti-metastasis therapeutics is still in its infancy, targeting critical regulators of invadopodia, which could prevent the formation of deadly secondary tumors, is a promising place to start.
Limitations of the study
While our data strongly support the model that FAP plays a critical role in stabilizing invadopodia precursors to allow invadopodia to grow and mature, the biochemical basis of FAP function at invadopodia is not fully understood. Knockdown of FAP or MT1-MMP led to similar defects in reduced invadopodia stability, suggesting that they are both important for invadopodia stabilization and maturation. While we observed that membrane areas enriched with FAP and MT1-MMP are the anchoring sites for invadopodia precursor attachment, extensive FAP pull-down and co-immunoprecipitation experiments did not detect convincing interactions between FAP and MT1-MMP. Because of the small size and the dynamic nature of invadopodia, it is possible that immunoprecipitation approaches cannot capture such dynamic and possibly indirect interactions between FAP and MT1-MMP that were observed by live imaging. Additional biochemical approaches such as Bio-ID proteomics are needed to further analyze such potential interactions. Also, although overexpression of MT1-MMP could rescue the defects caused by FAP knockdown, we are mindful in interpreting this result since such strong MT1-MMP overexpression could override endogenous regulation mechanisms controlling invadopodia assembly and function. Our ongoing work in identifying novel FAP-interacting proteins will further shed light on the molecular mechanism of FAP at invadopodia.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Jing Yang (jingyang@ucsd.edu).
Materials availability
Plasmids generated in this study have been deposited to Addgene [pWZL-Blast FAP S624A, Cat# 207402]; [pWZL-Blast FAP A657S, Cat#207403]
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cells, human mammary epithelial cells (HMLE), HMLER, and HMLE-Twist-ER, and SUM1315 cells were obtained from Dr. Robert Weinberg. SUM1315 cells were cultured in Ham’s F12 Nutrient mix supplemented with 5% fetal bovine serum (FBS), 5 μg/mL insulin, 10 ng/mL hEGF and 10 mM HEPES. The HMLE cell line was cultured in mammary epithelial growth media (MEGM) mixed 1:1 with DMEM/F12 supplemented with 10 μg/mL insulin, 10 ng/mL hEGF, 0.5 μg/mL hydrocortisone. To induce Twist expression, HMLE-Twist-ER cells were treated with 20 nM 4-hydroxy tamoxifen (4-OHT) for the indicated number of days. The MDA-MB-231 SRCY530F and 293T cell lines were cultured in DMEM supplemented with 10% FBS. The SCC61 cell line was obtained from the laboratory of Dr. Alissa Weaver and cultured in DMEM supplemented with 10% FBS and 0.5 μg/mL hydrocortisone. The BxPC3 cell line was obtained from the laboratory of Dr. Mark McNiven and cultured in RPMI supplemented with 10% FBS. All cell lines also had 1% penicillin and streptomycin in the culture media and were cultured at 37°C in the incubator supplied with 5% of CO2. All cell lines were subjected to regular mycoplasma testing using MycoAlert mycoplasma detection kit (Lonza) and authenticated by short tandem repeat (STR) DNA Profiling Analysis (Genetica).
All breast cancer cell lines used in this study, including HMLE cell series, SUM1315, MDA-MA-231 cells are female. The BxPC3 cells are isolated from a female pancreatic cancer patient according to ATCC. The sex of 293T cells and SCC61 cells are unknown. The sex of the cell line is not known to regulate invadopodia formation or function.
Animal studies
All animal care and experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California, San Diego. 2 million HMLE-Twist ER- EGFP cells resuspended in 50% Matrigel were injected into the left and right flanks of 8-week-old female nude mice (NU/J) and allowed to grow to approximately 2 cm in diameter before mice were sacrificed. Primary tumor size was measured every 4 days using a caliper and tumor volumes were calculated as mm = length x width x width. Primary tumors were isolated and tumor weight was calculated. Lungs were harvested and imaged for GFP-positive tumor cells to determine the number of lung metastasis.
METHOD DETAILS
Plasmid construction
Control shRNAs in the pLKO.1 vector64 were obtained from the Sabatini lab (Addgene). shRNAs targeting FAP and MT1-MMP were purchased from Sigma. shRNAs against DPP4 were cloned into the pSP108 vector.
The MT1-MMP pHluorin and pECFP-FAP-C1 plasmids have been previously described.48,57 The MT1-MMP-pHuji pENTR-20 plasmid was kindly provided by Dr. Camilla Raiborg. Both MT1-MMP-pHuji and ECFP-FAP genes were then subcloned into pLV puro plasmid. The LifeAct EGFP lentiviral vector was kindly provided by Dr. Keith Mostov and mCherry was then subcloned in to place EGFP. The EGFP pRRL plasmid is a gift from Dr. Robert Weinberg’s lab and has been previously described.20 The SRC Y530F pWZL-Blast construct was kindly provided by Dr. Sara Courtneidge.7
In order for the FAP point mutants to be used in overexpression rescue experiments while avoiding being targeted by shRNAs, the wild-type FAP sequence was mutated while maintaining the amino acid sequences, using site-directed mutagenesis as previously described664, so that it would not be targeted by either shRNA. Using these mutants as a template, FAP sh3-and FAP sh5-resistant forms of the S624A and A657S point mutants were constructed. All primers used to construct the point mutants are listed in Table S1.
Viral production and infection
Stable cell lines were generated via infection of target cells using lentiviruses, which were produced by co-transfection of viral vector with packaging (pCMVΔ8.2R) and envelope (pVSV-G) plasmids into HEK293T cells using TransIT-LT1 (Mirus) as previously described.20 After 18hr, fresh media was added to the transfected 293T cells. Virus-containing supernatants were collected at 48hr and 72hr post-transfection, passed through a 0.45 μM filter and added to the target cells with 6 μg/mL protamine sulfate for 6hr. After 24hr, SCC61, HMLE, MDA-MB-231 SRC Y530F and SUM1315 cells were selected with 2 μg/mL puromycin, BxPC3 cells were selected with 1 μg/mL puromycin, MDA-MB-231 were selected with 10 μg/mL blasticidin (for SRC Y530F overexpression).
Quantitative Real-time PCR
Total RNA from cells at 80–90% confluency was isolated using the NucleoSpin RNA II kit (Macherey-Nagel), and reverse-transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The resulting cDNAs were analyzed in triplicate using the SYBR-Green PCR Mix (Biorad). All primers used for qPCR are listed in Table S1. Relative mRNA levels were determined by 2-(Ct−Cc), where Ct and Cc are the mean threshold cycle differences after normalizing to HPRT values. The data presented are the mean of three biological replicates.
Western blotting
Cells at 80–90% confluency were washed with cold PBS and lysed in lysis buffer (50 mM Tris-HCl at pH 7.5, 150 mM NaCl, 10 mM NaF, 1% Triton X-100), with 100 μM Na3VO4, 100 μM PMSF, and the Protease Inhibitor Cocktail Set III (Calbiochem) diluted 1:200. Protein concentrations were measured using DC protein assay (Biorad). Equal amounts of protein were boiled in 50mM DTT LDS sample buffer at 70°C for 10 min, ran on 4–12% pre-cast gels (PAGEgel) and transferred to PVDF membranes. After transfer, membranes were blocked with 5% BSA in TBST (1X Tris-buffered saline+0.1% Tween 20) for 1hr and incubated with the primary antibody diluted in 3% BSA in PBST (1X PBS+0.1% Tween 20) + Sodium Azide at 4°C overnight. The following day, membranes were washed 3 times with 1X TBST and incubated with the secondary antibody conjugated with horseradish peroxidase diluted in 3% non-fat dry milk in TBST. The bands were visualized with chemiluminescence substrate (Cytiva). The following antibodies were used for western blotting: FAP (D8) (1:500, Vitatex), GAPDH (1:10000, GeneTex), MT1MMP (EP12644) (1:500, Abcam) and β-actin (1:10000, GeneTex).
FAP enzymatic activity assay
SUM1315 cells were plated in 96-well plates 48 h prior to assay and washed 3 times with 1X PBS. The wash solution was replaced with fresh PBS, and either additional PBS or 10 μM M83 inhibitor was added. 125 μM C95 substrate was added to all wells, and then fluorescence emission was measured at 360/460 nm excitation/emission wavelengths.
ECM degradation assay
ECM degradation assay was carried out as previously described.20,65,66 In brief, 12 mm coverslips were incubated in 20% nitric acid overnight and the day after they were washed with H2O for 4 h. Coverslips were incubated with 50 mg/mL poly-L-lysine/PBS for 20 min followed by PBS washes before 0.15% glutaraldehyde/PBS was added for 15 min, followed by PBS washes. Coverslips were inverted onto 20 μL droplets of 1:9 0.1% Oregon Green 488 gelatin from pig skin (Invitrogen): 0.2% porcine skin gelatin type A (Sigma-Aldrich) for 5 min. Coverslips were washed in PBS and then incubated for 15 min in 5 mg/mL NaBH4. Coverslips were rinsed in PBS and incubated at 37°C in 10% fetal bovine serum/DMEM for 2 h. The duration of the assay and cell confluency were adjusted based on the cell line used. For SCC61 and HMLE-Twist cells, twenty thousand cells were seeded on each coverslip and incubated for 16 h. SUM1315 cells were seeded on OG-gelatin coated coverslips for 6 h, MDA-MB-231 SRCY530F cells were seeded on each coverslip for 4 h and BxPC3 cells were seeded for 8 h. All the cell lines were then fixed with 4% paraformaldehyde (PFA) in 1X PBS and permeabilized with 0.05% Triton X-100 in 1X PBS for 10 min. The slides were imaged by using Keyence BZX710 fluorescent microscope to detect the nuclei (DAPI-stained), gelatin matrix (Oregon Green 488-stained), and the F-actin cytoskeleton (Alexa Fluor 546 Phalloidin-stained). 10 fields for each coverslip were randomly imaged using a 40x object for a total of approximately 150 cells per sample. Gelatin degradation was quantified using ImageJ software. To measure the percentage of degraded area in each field, identical signal threshold for the Oregon Green 488-gelatin fluorescence are set for all images in an experiment and the degraded area with Oregon Green 488 signal below the set threshold was measured by ImageJ. The resulting percentage of degradation area was further normalized to the total cell number (counted by DAPI staining for nuclei) in each field as previously described.67 The final gelatin degradation index is the average percentage degradation per cell obtained from all ten fields. Each experiment was performed at least three times. The cells in Figures 4B, 4C, S3D, and S3E were treated with 10mM GM6001 inhibitor as previously described.20
Immunofluorescence
Matrix substrates were prepared using unlabeled 0.2% porcine skin gelatin type A (Sigma-Aldrich), as for the ECM degradation assays. Cells were seeded on the matrix for 2 days and then fixed in 4% paraformaldehyde in Krebs + sucrose fixative (4% PKS) for 15 min at 37°C. Cells were washed 3 times with 1X PBS, permeabilized with 0.1% Triton X-100 in 1X PBS for 5 min, and then blocked with 1% BSA in 1X PBS for 1 h. Samples were incubated with primary antibodies overnight at 4°C. The day after, coverslips were washed 3 times with 1% BSA/1X PBS blocking solution and blocked again with the same solution for 1hr. Samples were incubated with secondary antibodies for 1 h at room temperature along with Alexa Fluor 546 or 647-conjugated phalloidin (Invitrogen). Samples were then mounted with VECTASHIELD (Vector Laboratories) containing DAPI for nuclear staining prior to imaging. Primary antibodies used for IF include: cortactin (4F11) (1:100, Millipore), FAP (D28) (1:100, Vitatex), Tks5 (13H6.3) (1:100, Millipore, and Santa Cruz Biotechnology), MT1-MMP (EP12644) (1:100, Abcam). Secondary antibodies used include Alexa Fluor 488, 546 and 647 (1:200, Jackson Labs).
Confocal images were acquired using an Olympus FV1000 with 405, 488, 555, and 647 laser lines. Briefly, quantification of target protein at invadopodia was done using integrated density values of target protein at invadopodia denoted by F-actin positive puncta in ImageJ software. Following this, corrected total cell fluorescence (CTFC) values were obtained by using the formula: Integrated Density – (Area of the cell x mean fluorescence background of the cell). CTFC values were finally divided by the total number of invadopodia in a cell as previously described.68 The data presented are the mean of three biological replicates.
Transwell invasion assay
Transwell invasion assay was performed as described previously.20 In brief, 50 mg of Matrigel was overlayed on Transwell permeable supports (Costar), dried overnight, and reconstituted with DMEM without FBS. The lower chamber was filled with DMEM supplemented with 10%FBS and 40,000 cells were resuspended in serum-free media, plated into the Transwell insert in triplicates and incubated for 48 h. Cells were fixed with 4% PFA, washed extensively with PBS, stained with 0.1% crystal violet and dried. Crystal violet was released with 50 μL 10% acetic acid and the absorbance was measured at 580 nm. Each experiment was performed in triplicate.
Wound healing assay
SCC61 cells were grown to confluency in 6 well plates. A scratch was made using a 1000mL tip creating a ‘wound’ in the confluent cell monolayer and fresh media supplemented with 1% FBS was added to block cell proliferation. Phase contrast images with 5× magnification were acquired every 6hr using a Zeiss Invertoskop 40C microscope (Jena, Germany). The area of the scratch was quantified using ImageJ software.
Live cell imaging
Stable SCC61 FAP knockdown and shRNA control cell lines were further infected with lentivirus containing Life-Act mCherry for F-actin visualization. Prior to cell seeding, 35 mm glass bottom dishes (No. 1.5, MatTak corporation) were coated with 100μg/ml rat tail collagen I (Corning) diluted in 1X DPBS for 1 h at 37°C, then washed once in DPBS. Cells were seeded onto the coated dishes for 24 h, followed by TIRF imaging using 100× objective on Nikon A1R confocal TIRF STORM microscope (Tokyo, Japan) fitted with a stage top incubator set at 37°C and 5% CO2 for live cell imaging. Time-lapse fluorescent images were acquired at frame rate of 2 min/frame for 2 h using TIRF microscopy to capture the depth of 100 nm from the bottom of the cell. Quantification of invadopodia precursor motility was done using ImageJ software, as described previously.69 Briefly, invadopodia precursor tracks were generated using ‘invadopodia tracker’ ImageJ plugin. Following this, displacement of individual invadopodium precursor per frame was calculated using the formula:
SQRT((difference in X-coordinate per frame)2 + (difference in Y-coordinate per frame)2).
For recruitment dynamics of FAP and MT1-MMP on invadopodia precursors, Time-lapse fluorescent images using TIRF microscopy with penetration depth of 100 nm were obtained at frame rate of 20 s/frame for 2 h. Fluorescent intensity changes over time, as well as track paths were measured using NIS-Elements AR version 5.30.04. For these set of measurements, F-actin was stained with SPY650-FastAct for 2 h prior to imaging.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis for each experiment was performed as described in the corresponding figure legends. All statistical analyses were carried out using GraphPad Prism 8 software. Both Spearman correlation and Pearson correlation were performed to determine gene expression correlation in patient tumor samples. Student’s t-test was performed to determine statistical significance between two test groups. All quantitative data are presented as mean ± SEM or SD, and p < 0.05 is considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Rat monoclonal anti-FAP (clone D8) | Vitatex | Cat#MABS1001 |
| Rabbit polyclonal anti-GAPDH | GeneTex | Cat#GTX100118; RRID:AB_1080976 |
| Rabbit monoclonal anti-MT1-MMP (clone EP1264Y) |
Abcam | Cat#ab51074; RRID:AB_881234 |
| Rabbit polyclonal anti-β-actin | GeneTex | Cat#GTX109639; RRID:AB_1949572 |
| Mouse monoclonal anti-cortactin (clone 4F11) | Millipore Sigma | Cat#05–180-I |
| Rat monoclonal anti-FAP (clone D28) | Vitatex | Cat#MABS1002 |
| Mouse monoclonal anti-Tks5 (clone 13H6.3) | Millipore Sigma | Cat#MABT336 |
| Rabbit polyclonal anti-Tks5 | Santa Cruz Biotechnology | Cat#sc-30122; RRID:AB_2254551 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Fetal Bovine Serum | Sigma-Aldrich | Cat#F0926 |
| Insulin | Gemini | CAt#800–112P |
| human Epidermal Growth Factor (hEGF) | Sigma-Aldrich | Cat#E9644 |
| Hydrocortisone | Sigma-Aldrich | Cat#H0888 |
| Gibco HEPES 1M | Fisher Scientific | Cat#15630080 |
| TransIT-LT1 Transfection reagent | Mirus Bio | Cat#MIR2306 |
| Protamine Sulfate Salt from salmon | Sigma-Aldrich | Cat#P4020 |
| Puromycin Dihydrochloride | Gemini | Cat#400–128P |
| Blasticidin HCl | Gemini | Cat#400–165P |
| Protease Inhibitor Cocktail Set III-EDTA free | Calbiochem | Cat#539134 |
| RunBlue LDS Sample Buffer (4X Concentrate) | Expedeon | Cat#NXB31010 |
| C95 substrate | Provided by Dr. Patrick McKee (Edosada et al.)39 | N/A |
| M83 inhibitor | Provided by Dr. Patrick McKee (Lee et al.)38 | N/A |
| GM6001 inhibitor | Sigma-Aldrich | Cat#364205 |
| Gelatin from pig skin Oregon green 488 conjugated | Invitrogen | Cat#G13186 |
| Gelatin from porcine skin, Type A | Sigma-Aldrich | Cat#G2500 |
| VECTASHIELD antifade mounting media with DAPI | Vector Laboratories | Cat#H-1200 |
| Alexa Fluor 546 Phalloidin | Life technologies | Cat#A22283 |
| Alexa Fluor 647 phalloidin | Life technologies | Cat#A22287 |
| Cultrex Reduced Growth Factor | R&D systedm | Cat#3533–010-02 |
| Basement Membrane Extract, Type 2 | ||
| Rat tail collagen type I | Corning | Cat#354236 |
| SPY650-FastAct | Cytoskeleton | Cat# CY-SC505 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| MycoAlert mycoplasma detection kit | Lonza | Cat#LT07–703 |
| NucleoSpin RNA II kit (Macherey-Nagel) | Takara | Cat#740984 |
| High-Capacity cDNA Reverse Transcription | Applied Biosystems | Cat#4368813 |
| iTaq Universal SYBR Green Supermix | Biorad | Cat#1725124 |
| DC Protein assay | Biorad | Cat#5000111 |
| ECL Prime western blotting detection reagent | Cytiva | Cat#RPN2236 |
|
| ||
| Experimental Models: Cell Lines | ||
|
| ||
| Human: HEK293T cells | Provided by Dr. Robert Weinberg (Yang et al.)25 | N/A |
| Human: HMLE cells | Provided by Dr. Robert Weinberg (Yang et al.)25 | N/A |
| Human: HMLER cells | Provided by Dr. Robert Weinberg (Yang et al.)25 | N/A |
| Human: HMLE-Twist-ER cells | Provided by Dr. Robert Weinberg (Mani et al.)28 | N/A |
| Human: SUM1315 cells | Provided by Dr. Robert Weinberg (Yang et al.)25 | N/A |
| Human: MDA-MB-231 cells | ATCC | CRM-HTB-26 |
| Human: SCC61 cells | Provided by Dr. Alissa Weaver (Clark et al.)40 | N/A |
| Human: BxPC3 | ATCC | CRL-1687 |
|
| ||
| Experimental Models: Organisms/Strains | ||
|
| ||
| Nu/J mice | UC San Diego ACP | Strain: Nude/nude |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S1 for primers used in this study | Eton Bioscience | N/A |
| DPP4 shRNA#1: GGTCACCAGTGGGTCATAAAT |
Sigma-Aldrich | TRCN0000372047 |
| DPP4 shRNA#3: GCTGTGAATCCAACTGTAAAG |
N/A | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| pLKO.1 control shRNA | Provided by Dr. David Root (Moffat et al.)64 | Addgene plasmid #10879 |
| pLKO.1 FAP shRNA #3 | Sigma-Aldrich | TRCN0000006805 |
| pLKO.1 FAP shRNA #4 | Sigma-Aldrich | TRCN0000006806 |
| pLKO.1 FAP shRNA #5 | Sigma-Aldrich | TRCN0000355737 |
| pLKO.1 MT1-MMP shRNA #1 | Sigma-Aldrich | TRCN0000050853 |
| pLKO.1 MT1-MMP shRNA #3 | Sigma-Aldrich | TRCN0000050855 |
| pECFP-FAP-C1 plasmid | Provided by Dr. Mark Gorrell (Wanget al)56 | N/A |
| MT1-MMP-pHluorin plasmid | Provided by Dr. Philippe Chavrier (Lizárraga et al.)48 | N/A |
| pENTR-20 MT1-MMP-pHuji | Provided by Dr. Camilla Raiborg (Pedersen et al.)49 | N/A |
| pWZL-Blast FAP S624A | this manuscript | Addgene Cat#207402 |
| pWZL-Blast FAP A657S | this manuscript | Addgene Cat#207403 |
| LifeAct-EGFP plasmid | Provided by Dr. Keith Mostov | N/A |
| pRRL EGFP | Gift from Dr. Robert Weinberg (Eckert et al.)20 | N/A |
| pWZL-Blast SRC Y530F | Provided by Dr. Sara Courtneidge7 |
N/A |
|
| ||
| Software and Algorithms | ||
|
| ||
| GraphPad Prism 8 | GraphPad |
https://www.graphpad.com/; RRID:SCR_002798 |
| ImageJ | NIH |
https://ImageJ.nih.gov/ij/; RRID:SCR_003070 |
| cBioPortal | MSK | https://www.cbioportal.org/ |
| NIS-Elements AR Version 5.30.04 | Nikon Instruments inc. | https://www.nikonusa.com |
Highlights.
FAP promotes breast tumor metastasis
FAP expression is significantly correlated with TWIST1 in various human cancers
FAP is required for matrix degradation independent of its proteolytic activity
FAP localizes at invadopodia and promotes invadopodia precursor stabilization
ACKNOWLEDGMENTS
We thank members of the Yang Lab for technical advice and helpful discussions. We thank Dr. Alissa Weaver for the SCC61 cells, Dr. Mark McNiven for the BxPC3 cells, Dr. Mark Gorrell for the pECFP-FAP-C1 plasmid, Dr. Philippe Chavrier for the MT1-MMP-pHluorin plasmid, Dr. Camilla Raiborg for the MT1-MMP-pHuji plasmid, Dr. Keith Mostov for the LifeAct plasmid, and Dr. Sara Courtneidge for the pWZL-Blast SRC Y530F plasmid. We acknowledge the support of the UCSD Cancer Center Microscopy Shared Facility Specialized Support Grant P30 CA23100–28. This work was supported by grants from the NCI (RO1CA262794, R01CA174869, R01CA206880, and R01CA236386), California Tobacco-Related Disease Research Program (28IP-0023), AACR-Bayer Innovation and Discovery (Grant PC #847562), and UCSD Moores Cancer Center Gleiberman Head and Neck Cancer Pilot Grant to J.Y.; N.P. and L.J. were supported by NIH Pre-doctoral Training grant T32GM007752. M.S. was supported by a postdoctoral NRSA fellowship from NCI (F32CA206227) and an American Cancer Society postdoctoral fellowship (PF-17–116-01-CSM). R.F. was supported by the American-Italian Cancer Foundation post-doctoral research fellowship and is supported by an NCI training grant T32CA009523. L.J. was supported by a pre-doctoral NRSA fellowship from NCI (F31CA200271). The graphical abstract was created with Biorender.com.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science.
Footnotes
DECLARATION OF INTERESTS
The authors declare no conflict of interest.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113302.
REFERENCES
- 1.Lambert AW, Pattabiraman DR, and Weinberg RA (2017). Emerging Biological Principles of Metastasis. Cell 168, 670–691. 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai JH, and Yang J. (2013). Epithelial-mesenchymal plasticity in carcinoma metastasis. Genes Dev. 27, 2192–2206. 10.1101/gad.225334.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paz H, Pathak N, and Yang J. (2014). Invading one step at a time: the role of invadopodia in tumor metastasis. Oncogene 33, 4193–4202. 10.1038/onc.2013.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy DA, and Courtneidge SA (2011). The “ins” and “outs” of podosomes and invadopodia: characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 12, 413–426. 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Linder S. (2007). The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 17, 107–117. 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Castro-Castro A, Marchesin V, Monteiro P, Lodillinsky C, Rossé C, and Chavrier P. (2016). Cellular and Molecular Mechanisms of MT1-MMP-Dependent Cancer Cell Invasion. Annu. Rev. Cell Dev. Biol. 32, 555–576. 10.1146/annurev-cellbio-111315-125227. [DOI] [PubMed] [Google Scholar]
- 7.Seals DF, Azucena EF Jr., Pass I, Tesfay L, Gordon R, Woodrow M, Resau JH, and Courtneidge SA (2005). The adaptor protein Tks5/Fish is required for podosome formation and function, and for the protease-driven invasion of cancer cells. Cancer Cell 7, 155–165. 10.1016/j.ccr.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Buschman MD, Bromann PA, Cejudo-Martin P, Wen F, Pass I, and Courtneidge SA (2009). The novel adaptor protein Tks4 (SH3PXD2B) is required for functional podosome formation. Mol. Biol. Cell 20, 1302–1311. 10.1091/mbc.E08-09-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poincloux R, Lizárraga F, and Chavrier P. (2009). Matrix invasion by tumour cells: a focus on MT1-MMP trafficking to invadopodia. J. Cell Sci. 122, 3015–3024. 10.1242/jcs.034561. [DOI] [PubMed] [Google Scholar]
- 10.Albrechtsen R, Stautz D, Sanjay A, Kveiborg M, and Wewer UM (2011). Extracellular engagement of ADAM12 induces clusters of invadopodia with localized ectodomain shedding activity. Exp. Cell Res. 317, 195–209. 10.1016/j.yexcr.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 11.Eckert MA, Santiago-Medina M, Lwin TM, Kim J, Courtneidge SA, and Yang J. (2017). ADAM12 induction by Twist1 promotes tumor invasion and metastasis via regulation of invadopodia and focal adhesions. J. Cell Sci. 130, 2036–2048. 10.1242/jcs.198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seals DF, and Courtneidge SA (2003). The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 17, 7–30. 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 13.Paterson EK, and Courtneidge SA (2018). Invadosomes are coming: new insights into function and disease relevance. FEBS J. 285, 8–27. 10.1111/febs.14123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leong HS, Robertson AE, Stoletov K, Leith SJ, Chin CA, Chien AE, Hague MN, Ablack A, Carmine-Simmen K, McPherson VA, et al. (2014). Invadopodia are required for cancer cell extravasation and are a therapeutic target for metastasis. Cell Rep. 8, 1558–1570. 10.1016/j.celrep.2014.07.050. [DOI] [PubMed] [Google Scholar]
- 15.Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, and Condeelis J. (2012). N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J. Cell Sci. 125, 724–734. 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stoletov K, Kato H, Zardouzian E, Kelber J, Yang J, Shattil S, and Klemke R. (2010). Visualizing extravasation dynamics of metastatic tumor cells. J. Cell Sci. 123, 2332–2341. 10.1242/jcs.069443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bayarmagnai B, Perrin L, Esmaeili Pourfarhangi K, Graña X, Tüzel E, and Gligorijevic B. (2019). Invadopodia-mediated ECM degradation is enriched in the G1 phase of the cell cycle. J. Cell Sci. 132. 10.1242/jcs.227116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y, Tondravi M, Liu J, Smith E, Haudenschild CC, Kaczmarek M, and Zhan X. (2001). Cortactin potentiates bone metastasis of breast cancer cells. Cancer Res. 61, 6906–6911. [PubMed] [Google Scholar]
- 19.Perentes JY, Kirkpatrick ND, Nagano S, Smith EY, Shaver CM, Sgroi D, Garkavtsev I, Munn LL, Jain RK, and Boucher Y. (2011). Cancer cell-associated MT1-MMP promotes blood vessel invasion and distant metastasis in triple-negative mammary tumors. Cancer Res. 71, 4527–4538. 10.1158/0008-5472.CAN-10-4376. [DOI] [PubMed] [Google Scholar]
- 20.Eckert MA, Lwin TM, Chang AT, Kim J, Danis E, Ohno-Machado L, and Yang J. (2011). Twist1-induced invadopodia formation promotes tumor metastasis. Cancer Cell 19, 372–386. 10.1016/j.ccr.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niedermeyer J, Kriz M, Hilberg F, Garin-Chesa P, Bamberger U, Lenter MC, Park J, Viertel B, Püschner H, Mauz M, et al. (2000). Targeted disruption of mouse fibroblast activation protein. Mol. Cell Biol. 20, 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niedermeyer J, Garin-Chesa P, Kriz M, Hilberg F, Mueller E, Bamberger U, Rettig WJ, and Schnapp A. (2001). Expression of the fibroblast activation protein during mouse embryo development. Int. J. Dev. Biol. 45, 445–447. [PubMed] [Google Scholar]
- 23.Chen WT, and Kelly T. (2003). Seprase complexes in cellular invasiveness. Cancer Metastasis Rev. 22, 259–269. [DOI] [PubMed] [Google Scholar]
- 24.O’Brien P, and O’Connor BF (2008). Seprase: An overview of an important matrix serine protease. Biochim. Biophys. Acta 1784, 1130–1145. 10.1016/j.bbapap.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 25.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, and Weinberg RA (2004). Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939. [DOI] [PubMed] [Google Scholar]
- 26.Peinado H, Olmeda D, and Cano A. (2007). Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat. Rev. Cancer 7, 415–428. [DOI] [PubMed] [Google Scholar]
- 27.Tsai JH, Donaher JL, Murphy DA, Chau S, and Yang J. (2012). Spatiotemporal regulation of epithelial-mesenchymal transition is essential for squamous cell carcinoma metastasis. Cancer Cell 22, 725–736. 10.1016/j.ccr.2012.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al. (2008). The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Casas E, Kim J, Bendesky A, Ohno-Machado L, Wolfe CJ, and Yang J. (2011). Snail2 is an essential mediator of Twist1-induced epithelial mesenchymal transition and metastasis. Cancer Res. 71, 245–254. 10.1158/0008-5472.CAN-10-2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monsky WL, Lin CY, Aoyama A, Kelly T, Akiyama SK, Mueller SC, and Chen WT (1994). A potential marker protease of invasiveness, seprase, is localized on invadopodia of human malignant melanoma cells. Cancer Res. 54, 5702–5710. [PubMed] [Google Scholar]
- 31.Kelly T, Kechelava S, Rozypal TL, West KW, and Korourian S. (1998). Seprase, a membrane-bound protease, is overexpressed by invasive ductal carcinoma cells of human breast cancers. Mod. Pathol. 11, 855–863. [PubMed] [Google Scholar]
- 32.Jin X, Iwasa S, Okada K, Mitsumata M, and Ooi A. (2003). Expression patterns of seprase, a membrane serine protease, in cervical carcinoma and cervical intraepithelial neoplasm. Anticancer Res. 23, 3195–3198. [PubMed] [Google Scholar]
- 33.Iwasa S, Jin X, Okada K, Mitsumata M, and Ooi A. (2003). Increased expression of seprase, a membrane-type serine protease, is associated with lymph node metastasis in human colorectal cancer. Cancer Lett. 199, 91–98. [DOI] [PubMed] [Google Scholar]
- 34.Ghersi G, Zhao Q, Salamone M, Yeh Y, Zucker S, and Chen W-T (2006). The protease complex consisting of dipeptidyl peptidase IV and seprase plays a role in the migration and invasion of human endothelial cells in collagenous matrices. Cancer Res. 66, 4652–4661. 10.1158/0008-5472.CAN-05-1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghersi G, Dong H, Goldstein LA, Yeh Y, Hakkinen L, Larjava HS, and Chen WT (2002). Regulation of fibroblast migration on collagenous matrix by a cell surface peptidase complex. J. Biol. Chem. 277, 29231–29241. 10.1074/jbc.M202770200. [DOI] [PubMed] [Google Scholar]
- 36.Chen WT (1996). Proteases associated with invadopodia, and their role in degradation of extracellular matrix. Enzyme Protein 49, 59–71. 10.1159/000468616. [DOI] [PubMed] [Google Scholar]
- 37.Rosenblum JS, and Kozarich JW (2003). Prolyl peptidases: a serine protease subfamily with high potential for drug discovery. Curr. Opin. Chem. Biol. 7, 496–504. [DOI] [PubMed] [Google Scholar]
- 38.Lee KN, Jackson KW, Christiansen VJ, Dolence EK, and McKee PA (2011). Enhancement of fibrinolysis by inhibiting enzymatic cleavage of precursor alpha2-antiplasmin. J. Thromb. Haemostasis 9, 987–996. 10.1111/j.1538-7836.2011.04195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edosada CY, Quan C, Tran T, Pham V, Wiesmann C, Fairbrother W, and Wolf BB (2006). Peptide substrate profiling defines fibroblast activation protein as an endopeptidase of strict Gly(2)-Pro(1)-cleaving specificity. FEBS Lett. 580, 1581–1586. 10.1016/j.febslet.2006.01.087. [DOI] [PubMed] [Google Scholar]
- 40.Clark ES, Whigham AS, Yarbrough WG, and Weaver AM (2007). Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia. Cancer Res. 67, 4227–4235. 10.1158/0008-5472.CAN-06-3928. [DOI] [PubMed] [Google Scholar]
- 41.Meadows SA, Edosada CY, Mayeda M, Tran T, Quan C, Raab H, Wiesmann C, and Wolf BB (2007). Ala657 and conserved active site residues promote fibroblast activation protein endopeptidase activity via distinct mechanisms of transition state stabilization. Biochemistry 46, 4598–4605. 10.1021/bi062227y. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y, Simms AE, Mazur A, Wang S, León NR, Jones B, Aziz N, and Kelly T. (2011). Fibroblast activation protein-alpha promotes tumor growth and invasion of breast cancer cells through non-enzymatic functions. Clin. Exp. Metastasis 28, 567–579. 10.1007/s10585-011-9392-x. [DOI] [PubMed] [Google Scholar]
- 43.Artym VV, Kindzelskii AL, Chen WT, and Petty HR (2002). Molecular proximity of seprase and the urokinase-type plasminogen activator receptor on malignant melanoma cell membranes: dependence on beta1 integrins and the cytoskeleton. Carcinogenesis 23, 1593–1601. [DOI] [PubMed] [Google Scholar]
- 44.Bowden ET, Onikoyi E, Slack R, Myoui A, Yoneda T, Yamada KM, and Mueller SC (2006). Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp. Cell Res. 312, 1240–1253. 10.1016/j.yexcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 45.Artym VV, Zhang Y, Seillier-Moiseiwitsch F, Yamada KM, and Mueller SC (2006). Dynamic interactions of cortactin and membrane type 1 matrix metalloproteinase at invadopodia: defining the stages of invadopodia formation and function. Cancer Res. 66, 3034–3043. 10.1158/0008-5472.CAN-05-2177. [DOI] [PubMed] [Google Scholar]
- 46.Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, et al. (2008). Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607. 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ferrari R, Martin G, Tagit O, Guichard A, Cambi A, Voituriez R, Vassilopoulos S, and Chavrier P. (2019). MT1-MMP directs force-producing proteolytic contacts that drive tumor cell invasion. Nat. Commun. 10, 4886. 10.1038/s41467-019-12930-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lizárraga F, Poincloux R, Romao M, Montagnac G, Le Dez G, Bonne I, Rigaill G, Raposo G, and Chavrier P. (2009). Diaphanous-Related Formins Are Required for Invadopodia Formation and Invasion of Breast Tumor Cells. Cancer Res. 69, 2792–2800. 10.1158/0008-5472.CAN-08-3709. [DOI] [PubMed] [Google Scholar]
- 49.Pedersen NM, Wenzel EM, Wang L, Antoine S, Chavrier P, Stenmark H, and Raiborg C. (2020). Protrudin-mediated ER-endosome contact sites promote MT1-MMP exocytosis and cell invasion. J. Cell Biol. 219, e202003063. 10.1083/jcb.202003063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bušek P, Malík R, and Šedo A. (2004). Dipeptidyl peptidase IV activity and/or structure homologues (DASH) and their substrates in cancer. Int. J. Biochem. Cell Biol. 36, 408–421. 10.1016/S1357-2725(03)00262-0. [DOI] [PubMed] [Google Scholar]
- 51.Chen D, Kennedy A, Wang J-Y, Zeng W, Zhao Q, Pearl M, Zhang M, Suo Z, Nesland JM, Qiao Y, et al. (2006). Activation of EDTA-Resistant Gelatinases in Malignant Human Tumors. Cancer Res. 66, 9977–9985. 10.1158/0008-5472.CAN-06-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Henry LR, Lee H-O, Lee JS, Klein-Szanto A, Watts P, Ross EA, Chen W-T, and Cheng JD (2007). Clinical Implications of Fibroblast Activation Protein in Patients with Colon Cancer. Clin. Cancer Res. 13, 1736–1741. 10.1158/1078-0432.CCR-06-1746. [DOI] [PubMed] [Google Scholar]
- 53.Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, Gopinathan A, Tuveson DA, and Fearon DT (2010). Suppression of Antitumor Immunity by Stromal Cells Expressing Fibroblast Activation Protein–A, p. 330. [DOI] [PubMed] [Google Scholar]
- 54.Puré E. (2009). The road to integrative cancer therapies: emergence of a tumor-associated fibroblast protease as a potential therapeutic target in cancer. Expert Opin. Ther. Targets 13, 967–973. 10.1517/14728220903103841. [DOI] [PubMed] [Google Scholar]
- 55.El Azzouzi K, Wiesner C, and Linder S. (2016). Metalloproteinase MT1-MMP islets act as memory devices for podosome reemergence. J. Cell Biol. 213, 109–125. 10.1083/jcb.201510043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christiansen VJ, Jackson KW, Lee KN, and McKee PA (2007). Effect of fibroblast activation protein and a2-antiplasmin cleaving enzyme on collagen Types I, III, and IV. Arch. Biochem. Biophys. 457, 177–186. 10.1016/j.abb.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang XM, Yu DMT, McCaughan GW, and Gorrell MD (2005). Fibroblast activation protein increases apoptosis, cell adhesion, and migration by the LX-2 human stellate cell line. Hepatology 42, 935–945. 10.1002/hep.20853. [DOI] [PubMed] [Google Scholar]
- 58.Santos AM, Jung J, Aziz N, Kissil JL, and Puré E. (2009). Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 119, 3613–3625. 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jackson KW, Christiansen VJ, Yadav VR, Silasi-Mansat R, Lupu F, Awasthi V, Zhang RR, and McKee PA (2015). Suppression of Tumor Growth in Mice by Rationally Designed Pseudopeptide Inhibitors of Fibroblast Activation Protein and Prolyl Oligopeptidase. Neoplasia 17, 43–54. 10.1016/j.neo.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eager RM, Cunningham CC, Senzer N, Richards DA, Raju RN, Jones B, Uprichard M, and Nemunaitis J. (2009). Phase II trial of talabostat and docetaxel in advanced non-small cell lung cancer. Clin. Oncol. 21, 464–472. 10.1016/j.clon.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 61.Narra K, Mullins SR, Lee H-O, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM, et al. (2007). Phase II trial of single agent Val-boroPro (Talabostat) inhibiting Fibroblast Activation Protein in patients with metastatic colorectal cancer. Cancer Biol. Ther. 6, 1691–1699. 10.4161/cbt.6.11.4874. [DOI] [PubMed] [Google Scholar]
- 62.Wolf BB, Quan C, Tran T, Wiesmann C, and Sutherlin D. (2008). On the edge of validation–cancer protease fibroblast activation protein. Mini Rev. Med. Chem. 8, 719–727. 10.2174/138955708784567449. [DOI] [PubMed] [Google Scholar]
- 63.Hofheinz R-D, al-Batran S-E, Hartmann F, Hartung G, Jäger D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, and Stehle G. (2003). Stromal Antigen Targeting by a Humanised Monoclonal Antibody: An Early Phase II Trial of Sibrotuzumab in Patients with Metastatic Colorectal Cancer. Oncol. Res. Treat. 26, 44–48. 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 64.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 65.Artym VV, Yamada KM, and Mueller SC (2009). ECM Degradation Assays for Analyzing Local Cell Invasion. In Extracellular Matrix Protocols Methods in Molecular Biology, Even-Ram S. and Artym V, eds. (Humana Press; ), pp. 211–219. 10.1007/978-1-59745-413-1_15. [DOI] [PubMed] [Google Scholar]
- 66.Fontana R, and Yang J. (2021). Matrix Degradation Assay to Measure the Ability of Tumor Cells to Degrade Extracellular Matrix. Methods Mol. Biol. 2294, 151–163. 10.1007/978-1-0716-1350-4_11. [DOI] [PubMed] [Google Scholar]
- 67.Heckman KL, and Pease LR (2007). Gene splicing and mutagenesis by PCR-driven overlap extension. Nat. Protoc. 2, 924–932. 10.1038/nprot.2007.132. [DOI] [PubMed] [Google Scholar]
- 68.Bora P, Gahurova L, Mašek T, Hauserova A, Potěšil D, Jansova D, Susor A, Zdráhal Z, Ajduk A, Pospíšek M, and Bruce AW (2021). p38-MAPK-mediated translation regulation during early blastocyst development is required for primitive endoderm differentiation in mice. Commun. Biol. 4, 788. 10.1038/s42003-021-02290-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma VP, Entenberg D, and Condeelis J. (2013). High-resolution live-cell imaging and time-lapse microscopy of invadopodium dynamics and tracking analysis. Methods Mol. Biol. 1046, 343–357. 10.1007/978-1-62703-538-5_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.