

Abstract
A functional module can be defined as a spatially or chemically isolated set of functionally associated components that accomplishes a discrete biological process. Modularity is a key attribute of cellular systems, but the mechanisms that underlie the evolution of functional modules are largely unknown. Duplication of modules has been shown to be an efficient mechanism for the generation of functional innovation in the field of artificial intelligence, but has not been studied in biological networks. Therefore, we ask whether module duplication occurs in cellular networks. We developed a generic framework for the analysis of module duplication, and use it in a large-scale analysis of Saccharomyces cerevisiae protein complexes. Protein complexes are well defined, experimentally derived, functional modules. We observe that at least 6%–20% of the protein complexes have strong similarity to other complexes; thus a considerable fraction has evolved by duplication. Our results indicate that many complexes evolved by step-wise partial duplications. We show that duplicated complexes retain the same overall function, but have different binding specificities and regulation, revealing that duplication of these modules is associated with functional specialization.
A functional module can be defined as a spatially or chemically isolated set of functionally associated biological components that accomplishes a discrete biological process (Hartwell et al. 1999; Ravasz et al. 2002). Modularity is a key attribute of cellular systems (Hartwell et al. 1999; Snel and Huynen 2004), such as the transcriptional (Ihmels et al. 2002), metabolic (Ravasz et al. 2002) and the protein interaction networks (Rives and Galitski 2003; Wuchty et al. 2003; Pereira-Leal et al. 2004). Here, we study protein complexes as functional modules in the protein interaction network.
The mechanisms that underlie the evolution of functional modules are largely unknown. Theoretical simulations in neural networks have shown that duplication and specialization of entire modules is an effective mode of network growth (Calabretta et al. 1998, 2000). In biological systems duplication is observed at different scales, such as genes, chromosomal segments, and whole genomes. For example in genomes, duplication of individual genes is a major mechanism of evolution (Teichmann et al. 1998) and represents a source of both functional novelty and specialization (Prince and Pickett 2002).
New modules could evolve either by duplications of their components, or by evolution of a novel interface between existing components. The second scenario must have occurred frequently, as there are many functional modules in biology that are dissimilar to each other, such as the ribosome and RNA polymerase. However, we also know of many examples in which functional modules consist of similar components. For these cases, the components must have evolved by duplication, but it is unclear how duplication of individual genes could contribute to the duplication of functional modules. The observation that yeast has undergone a complete genome duplication (Wolfe and Shields 1997; Dujon et al. 2004; Kellis et al. 2004), in conjunction with the dosage balance theory (Papp et al. 2003a; Veitia 2003) suggests that duplication of complete modules might have occurred in the yeast protein interaction network. At the same time, incremental evolution is known to be important for evolution of metabolic pathways (Teichmann et al. 2001) and transcriptional regulatory networks (Conant and Wagner 2003b; Teichmann and Babu 2004). This suggests that incremental, stepwise duplications should also occur for functional modules. The relative contribution of these duplication mechanisms to the generation of new modules in yeast is unclear.
Protein complexes are one well-defined type of functional module in cells, as they represent physical modules in the protein–protein interaction network (Dezso et al. 2003). The observation that some complexes have similarities to other protein complexes in terms of component composition lends further support to the hypothesis that duplication of functional modules occurs in biological systems. Examples are the complexes involved in tethering and fusion of intracellular vesicles (Whyte and Munro 2001, 2002), and the snRNP complexes (Salgado-Garrido et al. 1999). These examples are found across many eukaryotes, suggesting that duplication of functional modules, specifically of protein complexes, may be a widespread evolutionary phenomenon. So far, there has not been a comprehensive assessment of this phenomenon, to our knowledge. Here we address the extent to which duplication of protein complexes has occurred in the yeast, Saccharomyces cerevisiae, the mechanisms involved in the duplication of a protein complex, and the functional consequences of such duplication. Understanding how new complexes are generated, and how homologous complexes differ in their functions has implications for the prediction of three-dimensional structure and protein engineering of complexes, as well as functional assignment of uncharacterized complexes identified in large-scale experiments (Gavin et al. 2002; Ho et al. 2002).
Results and Discussion
Are protein complexes in yeast duplicated?
In order to determine if protein complexes can undergo duplication, we need to define the possible duplication scenarios. In the first scenario, some but not all components of a complex duplicate, which gives rise to a new complex that consists of some of the same components as the ancestral complex, as well as a few components that are not identical, but are similar to the ancestral components. We call this situation a “partial duplication” that results in “concurrent complexes.” These concurrent complexes have some shared, identical components and other similar, homologous components, as illustrated in Figure 1A. The second scenario consists of duplication of all the components of a complex, which we call a “complete” duplication. After sequence divergence of the components such that each set of components specifically recognizes themselves only, there will be two complexes with similar, but no shared components, as shown in Figure 1A. We call such complexes “parallel complexes.” Both parallel and concurrent complexes are homologous complexes. In addition to partial or complete duplications of components, unrelated components can be gained or lost in complexes. We can ascertain whether duplication has occurred by identifying cases where pairs of complexes have similar and/or shared sets of components. We call such protein complexes homologous, because they are evolutionarily related through duplication of one or more of the components in the complexes.
Figure 1.

Detection of duplicated proteins complexes. (A) Schematic illustrating two types of duplication of protein complexes. In the top panel on the left, we show a partial duplication in which one of the proteins in the complex has duplicated, and the other protein is part of both resulting complexes. On the right, a duplication of both components of the complex is illustrated, which we term `complete duplication' of a complex. In the bottom panel, we give an example of yeast protein complexes that follow the two duplication scenarios. The heterodimeric complexes farnesyl transferase (FTase), genranylgeranyl transferase I (GGTase I) illustrate a partial duplication. They share one subunit (α subunit, shown in yellow), but each has a distinct β subunit, coded by paralogous genes (shown in shades of blue) (Casey and Seabra 1996). GGTase I and Rab Geranylgeranyl Transferase (RabGGTase) illustrate a complete duplication. Paralogous genes code for the α and β subunits of both complexes (Casey and Seabra 1996). (B) Schematic representation of the method used to identify duplicated complexes. Each pair of complexes is compared in terms of their components to ascertain whether they are homologous, having evolved by partial or complete duplication. Any components that are shared, in other words that are identical proteins, are counted, as represented by the variable I. Any components that are homologous according to their domains as assigned by the Pfam or SUPERFAMILY databases, or by sequence similarity, are counted as similar components, represented by the variable S. For each pair of complexes, a score based on S, I, and the sizes of the two complexes (C) is calculated. If the score exceeds a certain threshold, we consider the two as duplicate (or homologous) complexes. If the two complexes have similar as well as identical components, they have evolved by partial duplication, and if only similar components are present, they have evolved by complete duplication.
In order to determine whether and to what extent any form of complex duplication occurs in cellular networks, we looked for homologous protein complexes within three independent data sets for the budding yeast S. cerevisiae (Gavin et al. 2002; Ho et al. 2002; Mewes et al. 2002). The most accurate, but also smallest data set are the manually curated protein complexes provided by the MIPS/CYGD database (Mewes et al. 2002). These represent predominantly stable protein complexes, whose components tend to be permanently associated in the cell. This data set was used to develop and calibrate a homology detection method for duplication events across complexes. The two other data sets of protein complexes are from large-scale purification and mass spectrometry experiments by TAP (Gavin et al. 2002) and HMS-PCI (Ho et al. 2002). These data sets have a significant proportion of false positives, and represent a combination of permanent and transient protein interactions. Many of the complexes in these two data sets are slightly different versions of the same complex. This was an important factor for our method of determining homologous complexes.
In order to quantify the level of similarity between two complexes, we developed a simple scoring system that takes into account the number of similar and shared proteins, as well as complex size. (Please refer to Fig. 1B and the Methods section for details.) This is a generic methodology and can be used to analyze other types of functional modules as well. Due to the nature of the large-scale data sets of protein complexes identified by mass spectrometry, we were conservative in allowing shared components (see Methods). This means that our estimates for the extent of duplication among protein complexes are a lower bound, but ensures that our analysis will not be contaminated by false positives. This is important as we wish to investigate the mechanisms and consequences of module duplication.
With these conservative parameters, we observe several instances of homologous complexes in all three data sets. Seven percent (7%) of the MIPS complexes and 6% of the TAP complexes have homologous complexes (Fig. 2). In the HMS-PCI complexes, the fraction is higher: 20%. This may be due to a bias towards signal transduction and DNA damage response (Ho et al. 2002) in the proteins studied in this data set, while in the two other data sets, functional classes are more homogeneously distributed. In all three data sets, these levels of duplication are significantly higher than could be expected by chance, with P-values less than 10–3 based on 1,000 experiments with random shuffling of the complex components. These results show that duplication of modules is an important mechanism for creation of new protein complexes, albeit not the predominant one. Duplicated complexes occur in all functional categories and subcellular localizations, and the components of the complexes are not necessarily coexpressed. A more extensive discussion of this is available in the Supplemental material. Note that these numbers provide lower estimates for the contribution of complex duplication in cellular networks, and that the true contribution is likely higher. For example, the complexes discussed in the introduction are not contained in any of the data sets, and as such were not used in this quantification. However, for the functional and evolutionary analysis presented below, it is critical that we do not have false positive homologies.
Figure 2.

Extent of duplication of complexes in the three data sets and in pairwise protein interactions. (A) Proportion of complexes which have at least one homologous complex in each of the data sets. This includes both partial and complete duplications. The PPI column represents the proportion of pairwise protein interactions that are partial or complicate duplicates of another pairwise interaction. For both the protein complexes and pairwise protein–protein interactions, each complex or interaction is counted as a partial duplicate if it has both shared and homologous components to another complex or pairwise interaction. A complex is classified as a complete duplicate only if there is no partial duplicate to it. Expected values are shown as white bars. (B) Proportion of the complexes with homologs that can be classified as a partial (blue) or complete (red) duplication for the three protein complex data sets, and the pairwise protein–protein interactions (PPI). Expected values are shown as light-shaded bars. (C) Network representation of the subset of the yeast protein–protein interaction network where interactions have been produced by duplication, colored according to the type of duplication. Red edges indicate interactions that have completely duplicated, and blue edges indicate interactions where only one of the components has duplicated and inherited the interaction (partial duplication). (D) Network representation of duplicated complexes in the MIPS data set. Nodes correspond to complexes and edges to homologies between complexes. Blue edges represent concurrent complexes (partial duplication), whereas red edges denote parallel complexes (complete duplication). Network layout performed with Java BioLayout (Enright and Ouzounis 2001).
How do complexes duplicate?
The groups of homologous complexes we have identified have clearly evolved by duplication of some or all of the components of the complexes. On the one hand, it seems unlikely that several components of a complex would duplicate even roughly simultaneously, unless they are clustered within one chromosomal region, or they are the result of a complete genome duplication as observed in S. cerevisiae (Wolfe and Shields 1997; Dujon et al. 2004; Kellis et al. 2004). On the other hand, duplication of some but not all components of a complex will create a dosage imbalance that may have deleterious effects (Papp et al. 2003a; Veitia 2003), and would not be expected if complete genome duplication is the main route by which duplication of protein complexes occurs.
Concerted duplication of all components would result in parallel complexes, such as GGTase I and Rab GGTase (Fig. 1A). The outcome of a stepwise process would be the existence of concurrent complexes, with some shared and some homologous components, such as FTase and GGTase I (Fig. 1A). Figure 2 summarizes the extent of partial and complete duplications within the three data sets. In these experimentally derived physical modules, concurrent complexes predominate (67% MIPS, 70% TAP, 96% HMS-PCI). In 1000 random shuffling experiments, we find that concurrent complexes are significantly more frequent than expected by chance (P < 10–3 in all cases). The duplication of subunits of duplicated complexes could occur in three ways. First, duplication of several or all components could occur if the genes are clustered on a chromosome and are duplicated as the result of segmental chromosomal duplications. Secondly, duplication of parallel complexes could occur as the results of a complete genome duplication, and concurrent complexes could also evolve in this way if complete genome duplication was followed by extensive gene loss. Third, protein complexes (both concurrent and parallel) could duplicate in a partial, stepwise manner. We will discuss the three possibilities below.
For complete simultaneous duplication to occur as a result of a segmental duplication, one would expect components of modules with duplicates to be adjacent on a chromosome, as it is difficult to envisage simultaneous duplication of distinct chromosomal segments. In order to test this hypothesis, we asked whether genes coding for proteins in duplicated modules are more likely to be adjacent in yeast chromosomes than those of singleton modules. As noted before (Teichmann and Veitia 2004), we observe that the components of protein complexes are significantly more likely to cluster in the chromosome than are random gene pairs. However, we fail to observe any increased chromosomal clustering in genes coding for components of homologous complexes (see Supplemental material).
One alternative hypothesis would be that the generation of these duplicated complexes originated from the complete genome duplication observed in yeast (Wolfe and Shields 1997; Dujon et al. 2004; Kellis et al. 2004), and that concurrent complexes would be the result of gene loss, rather than stepwise duplication. However, only a very small proportion of the paralogous gene pairs in duplicated complexes can be traced to complete genome duplication (Kellis et al. 2004): one out of 39 in MIPS, one out of 62 in TAP, and 12 out of 158 in HMS-PCI. Furthermore, most of the duplicated complexes listed in Table 1 exist in duplicated forms in eukaryotes from entirely different phylogenetic branches, implying that their duplication occurred prior to the divergence of fungi and metazoa. An example is the AP-complexes, which exist in all eukaryotes that have a completely sequenced genome. The two dehydrogenase complexes even exist in prokaryotes. This indicates that the duplication of these complexes occurred long before the complete genome duplication in S. cerevisiae. In fact we find that more extensive duplication of individual complexes is observed in other organisms, and the numbers of duplicates do not concur with complete genome duplications known for these organisms. For example there is further duplication of some subunits of the AP-1 and AP-2 complexes in mammals, giving rise to alternate forms of these complexes (concurrent complexes), as well as a complete duplication giving rise to the AP-4 complex. These two observations indicate that the complete genome duplication followed by extensive gene loss that happened in S. cerevisiae was not a major contributor to the duplication of protein complexes. We cannot however discount a role for more ancient whole genome duplications, which we cannot resolve given the completely sequenced genomes currently available. Considering all our results, the most plausible explanation is that partial, stepwise duplications of individual components of protein complexes is prevalent over simultaneous duplications of many components.
Table 1.
Homologous complexes identified in the manually curated protein complex data set, with a description of the general function and specificity of each complex
| Complex name | Function | Specific to |
|---|---|---|
| Pyruvate dehydrogenase | Carbohydrate oxidation in TCA | Pyruvate |
| 2-Oxoglutarate dehydrogenase | Carbohydrate oxidation in TCA | 2-Oxo-glutarate |
| Ftase | Protein prenytransferase | C15 isoprenoid, Rho and Ras small, GTPases, laminin, heterotrimeric G proteins, etc. |
| GGTase I | ″ | C20 isoprenoid, Rho and Ras small GTPases |
| RabGGTase | ″ | C20 isoprenoid, Rab small GTPases |
| SBF complex | Transcriptional activation during cell cycle progression | Transcription of G1 cyclins, cell wall biosynthesis genes, etc. |
| MBF complex | ″ | Transcription of S-phase cyclins, genes required for DNA synthesis, etc. |
| SWI/SNF complex | Transcriptional activation and repression | Not essential for growth. Transcription of genes involved in mating type switching, sucrose fermentation |
| Chromatin remodelling | ||
| RSC complex | ″ | Essential for mitotic growth. Active in chromosomal segregation |
| Organization of cytoskeleton. Transcription from RNA | ||
| Pol. II promoters (uncertain) | ||
| MSH2/MSH3 | DNA mismatch repair | Loops with two to eight unpaired bases, one-base insertion/deletion loops |
| MSH2/MSH6 | ″ | Base-base mismatches, one-base insertion/deletion loops |
| AP-1 complex | Coat protein binding | Clathrin, endosome to Golgi transport |
| Ap-2 complex | ″ | Clathrin, endocytosis at plasma membrane |
| AP-3 complex | Unknown | Unknown, transport to lysosome |
| B-COPI subcomplex | Coat protein binding | F-COPI coat |
Further complexes from this data set that have some resemblance, but are below the score threshold considered by us, are discussed in the Supplementary Material. RabGGTase, the AP-complexes and COPI do not share components with any other complex, and as such are the result of complete duplications. All other complexes share at least one component and are the result of partial duplications.
In order to gain more insight into the extent of partial and complete duplications among protein complexes, we analyzed the simplest unit of the protein interaction network–binary interactions. We applied the same methodology of homology detection to the binary protein–protein interactions (excluding interactions determined by yeast–two-hybrid assays). The network of protein–protein interactions is often represented as a graph of interactions between pairs of proteins (Jeong et al. 2001; Wagner 2001), encompassing both transient and permanent interactions. Sixty percent (60%) of the interactions in this network have arisen by duplication, of which the majority are partial duplications (92%; Fig. 2). Thus, although we do observe some instances of complete duplication, partial duplication is clearly the predominant mechanism for creating new protein interactions.
In summary, our results point to partial, stepwise duplications being a frequent route of duplication of protein complexes. It is important to note that in the three data sets, a proportion of the duplication events correspond to parallel complexes, i.e., complete duplications. Are these complete duplications the result of a simultaneous duplication of the different components, or are they the final outcome of a series of partial duplications? Very little information on the evolution of protein complexes is available, but anecdotal examples like the proposed evolutionary route of the AP-complexes suggest the latter. A hypothesis proposed for the evolution of these complexes is that three out of the four subunits duplicated initially, while the fourth subunit remained as a single copy and was shared between the AP1 and AP2 tetramers. This is the constellation observed in C. elegans and D. melanogaster. Later, the subunit duplicated as well, so that all four components are homologous in AP1 and AP2, and no components are shared. This is observed in budding yeast, mouse and human (Boehm and Bonifacino 2001).
According to the gene dosage balance hypothesis (Papp et al. 2003a; Veitia 2003) there is a dosage imbalance immediately after duplication if some, but not all, of the components of a complex duplicate. This is because the relative concentrations of the components change, upsetting the stoichiometry and binding equilibrium of the complex. This may have a deleterious effect, and if this is the case it would be selected against. The situation is exacerbated by the essential nature of many of the complexes detected as duplicates. For example, protein prenylation is an essential process in eukaryotes, but is mediated by three complexes, representing both partial and complete duplication scenarios. As suggested by Papp et al. (2003a) “if imbalance were deleterious (the balance hypothesis) we would expect adaptations to minimize the degree of imbalance.” Such adaptations could occur if the gene duplicate diverges rapidly (Lynch and Conery 2000) and/or is incompletely copied (Katju and Lynch 2003; Papp et al. 2003b; Lynch and Katju 2004). Divergence of the protein coding sequence could alter specificities of binding within the complex and to external components. For example, in the adaptin complexes (AP-1,2,3) subfunctionalization has occurred such that the different duplicates mediate distinct vesicular trafficking steps as a result of interactions with distinct components in these pathways. Furthermore, divergence at the level of the control of gene expression, namely by incomplete duplication of regulatory elements, could also minimize dosage imbalance.
What are the functional consequences of module duplication?
Having established that duplication of protein complexes is a feature of the protein interaction network of the budding yeast, we now ask what are the differences in function between homologous complexes. When an individual gene is duplicated, one of the copies is often lost due to asymmetrical selection (Prince and Pickett 2002; Conant and Wagner 2003a). However, when both copies are conserved, then either the duplicate acquires a different function (neofunctionalization), or there is a specialization or division of the original function between the two duplicates (subfunctionalization) (Prince and Pickett 2002). In artificial, man-made systems such as neuronal networks, duplication of modules is associated with functional specialization. Thus we hypothesize that in cellular networks, duplication of protein complexes is used to achieve functional specialization.
According to this hypothesis, homologous complexes should have the same general functionality. In order to test this, we determined the likelihood of two homologous complexes having the same general function, using the GO (Ashburner et al. 2000) definition of biological processes and subcellular localization (Huh et al. 2003). In all the data sets, the majority of pairs of homologous complexes are assigned to the same GO biological process and subcellular localization, and these numbers are significantly larger than expected by chance (Fig. 3). These results imply that duplicated modules tend to retain the same general functionality. Detailed analysis of all the duplications in the manually curated complexes lends further support to this, and also reveals that duplication leads to complexes with different specificities in their activities, according to the concept of subfunctionalization (Table 1).
Figure 3.

Functional consequences of complex duplication. Proportion of pairs of duplicated complexes belonging to the same GO biological process and to the same subcellular localization, compared with 10,000 random samplings of equal size of the data sets. In all cases pairs of duplicated complexes are significantly more likely to have the same functional assignment than could be expected by chance (zMIPS = 8.0, zTAP = 13.1, zHMS-PCI = 35.0, in all cases P ≤ 10–4). For similar subcellular localization, the proportion of complexes is significantly larger than could be expected by chance (zMIPS = 2.15 P ≤ 10–2, zHMS-PCI = 9.6 P ≤ 10–4) in both the MIPS and HMS-PCI data sets, and the TAP data set does not show any significant deviation from the random expectation. In an independent experiment controlling for shared proteins in duplicated complexes, we observed that in all data sets, and for both classification schemes, the proportion of homologous complexes with the same classification is significantly higher than expected by chance (P < 10–4).
All the groups of homologous complexes identified by us in the MIPS data set of manually curated complexes are listed in Table 1 along with our description of their functions and functional similarities and differences. The SWI/SNF and RSC complexes provide a good example of the phenomenon of functional specialization. Both are involved in transcriptional regulation and chromatin remodelling and can act as activators and repressors. However, their yeast mutant phenotypes are different, and there is some evidence to suggest that they act on different target genes (Martens and Winston 2003), illustrating duplication with retention of function but different specificities.
This is true even for those cases that are classified to different GO cellular processes or subcellular localizations. For example, the heterodimeric enzymes farnesyl transferase and geranylgeranyl transferase shown in Figure 1A are classified in two distinct biological processes, protein modification and signal transduction respectively. However, both enzymes mediate the covalent modification of proteins with isoprenylgroups, the main difference between them lying in their lipid and protein substrates (Casey and Seabra 1996).
One example of modules with different subcellular localizations comes from the AP-2 and COPI coat proteins. The AP-2 complex is well characterized, mediating the targeting and nucleation of clathrin coats, operating in the endocytic pathway between the plasma membrane and early endosomes (Boehm and Bonifacino 2001). In contrast COPI, active in the early stages of the secretory pathway, is composed of two subcomplexes: F-COPI and B-COPI. B-COPI lacks any detectable similarity to clathrin, but is described as a “coat-like” complex (Boehm and Bonifacino 2001). F-COPI contains four subunits, all homologous to the four AP-2 subunits. The pattern of subunit interactions is similar in AP-complexes and the F-COPI complex (Takatsu et al. 2001), and recent structural analysis of the γ-subunit of the F-COPI complex reveals strong similarity to the α and β subunits of AP-2 complex (Hoffman et al. 2003). Thus despite very remote homology and distinct subcellular localizations, the AP-complexes and the F-COPI complex are both adaptor complexes that bind different coat proteins (clathrin or B-COPI) in a similar way.
The two examples mentioned in the Introduction, which are not included in the data sets studied here, further support this trend. The exocyst and the COG are two related tethering complexes that attach vesicles to the destination membrane prior to fusion. They perform the same function in distinct target membranes (the plasma membrane and the Golgi, respectively, Whyte and Munro 2001, 2002). The snRNP family of protein complexes includes the canonical Sm complex, which binds the small nuclear RNAs (snRNA) U1, U2, U4, U5 and the related LSm complex (subunits LSm2 to LSm8), which binds the snRNA U6 and P RNA. Both complexes are involved in splicing (Salgado-Garrido et al. 1999). Interestingly, if one subunit of the LSm complex is changed (LSm1 instead of LSm8), this new complex, which includes two extra proteins (Xrn1 and Pat1) no longer binds SnRNAs, and becomes active in decapping of RNAs in the general degradation pathway. All these complexes form a heteroheptameric ring, which binds RNAs; the specificity to the RNA and of the pathway involved being determined by the subunit composition (Salgado-Garrido et al. 1999; Bouveret et al. 2000).
These results strongly support the view that complex duplication is a mechanism by which evolution generates functional specialization. This extends what has been found previously for duplication of individual genes (Prince and Pickett 2002), and is in agreement with the behavior of artificial systems such as neural networks (Calabretta et al. 2000).
Conclusions
In conclusion, we observe that a considerable fraction of yeast protein complexes have evolved by duplication of components. We suggest that stepwise, partial duplication is a more common evolutionary route to module duplication than concerted duplication of all components. Our analysis indicates that duplication is accompanied by evolution of novel specificities, with retention of general function. These findings have implications for structural and functional annotation of uncharacterized protein complexes, and also to the engineering of new protein interactions.
How general are these results for other biological interactions? In metabolic pathways, serial duplication of multiple enzymes is observed rarely, if at all. Duplicate enzymes are distributed across the metabolic network without any coherence, because substrate specificity can change rapidly in evolution (Teichmann et al. 2001). Therefore, duplication is unlikely to play a role in evolution of metabolic modules. In transcriptional regulatory networks, duplicate transcription factors and target genes frequently inherit regulatory interactions. So although entire regulatory network motifs are not duplicated (Conant and Wagner 2003b; Teichmann and Babu 2004), it is possible that duplication could play a role for regulatory modules if defined in a different way from network motifs. In other words, for protein interactions and transcriptional regulatory interactions, the duplicated gene frequently remains associated with its ancestor, i.e., it retains a direct functional association. In contrast, in small-molecule metabolism, duplicated enzymes do not retain an association with the ancestral gene. The reason may be that the link between consecutive enzymes in a pathway is via a small molecule, and the binding pockets for these molecules are small and flexible in evolution. In contrast, transcriptional regulatory interactions occur via protein–DNA binding, involving a larger interface, and protein–protein interactions are mediated by even larger surfaces, which must evolve most slowly of all three types of interactions.
The results presented here lend further support to the idea that general principles observed in artificial organized systems also occur in the biological organization of cells (Jeong et al. 2000). Modularity has long been recognized as a feature of engineered systems. However, it is unclear whether it contributes to the ability of organisms to respond to selective challenge (Hansen 2003). Our results add to this debate and strongly support the view that, as in engineered systems, modularity provides relatively isolated units, which can be readily reconfigured and duplicated to adapt to novel circumstances. However, the stepwise duplication mechanism we propose is not typical of engineered systems. Thus, even though evolution accomplishes logical and efficient designs (Alon 2003), it acts as a “tinkerer” rather than an “engineer” (Jacob 1977).
Methods
Data sets
Three yeast protein complex data sets were used in this study: the manually curated MIPS/CYGD (Mewes et al. 2002) catalog of complexes (1185 proteins, 216 complexes), and two sets of complexes identified in large-scale proteomic experiments: TAP (Gavin et al. 2002) and HMS-PCI (Ho et al. 2002) (589 and 741 protein complexes respectively, involving 1474 and 1578 proteins, respectively). Pairwise protein interaction data for yeast were obtained from the physical interactions table of MIPS/CYGD (Mewes et al. 2002), excluding interactions determined by yeast–two-hybrid, comprising 745 proteins and 991 interactions.
Detecting module duplication
We define two modules as homologous if the majority of their components are similar. We searched for instances of module homology by pairwise comparison of all components within protein complex data sets. Component similarity was determined based on one or more of three criteria: domain architecture, i.e., the N-to-C terminal series of domains, determined from the domain assignments in the SUPERFAMILY database (Madera et al. 2004) or the Pfam database (Bateman et al. 2004), or FASTA (Pearson 1990) all-against-all comparisons within the yeast proteome, at a threshold of E < 10–2. These criteria maximize sensitivity and coverage of the protein space. The use of SUPERFAMILY and Pfam provide high sensitivity searches and explicit consideration of domain architectures, either based on SCOP structural domains in Superfamily (Madera et al. 2004) or sequence domains in Pfam (Bateman et al. 2004). The incomplete coverage of these two resources was complemented by the use of FASTA comparisons, which although providing lower sensitivity, provides complete coverage of the yeast genome.
We processed the domain assignments from SUPERFAMILY and Pfam as follows: we ignore gaps and tandem domain duplications. Because some domains and domain architectures are very common among the domain assignments in each data set, we eliminate the top ∼1% most frequent domain architectures among the SUPERFAMILY assignments. This excludes single domain proteins from the P-loop ATP hydrolase, ARM repeat, WD-40 repeat, Protein Kinase-like, histone fold, and RNA-binding domain superfamilies. We eliminate the ∼0.5% most frequent Pfam domain architectures, which excludes the Protein kinase family, the WD40 family, the TCP1/Cpn60 chaperonin family, the RNA recognition motif family, and the Proteosome A and B type families.
Quantifying the degree of module similarity
In order to quantify the degree of similarity between two complexes, we calculate a score that takes into account similar and identical components. Our scoring scheme for comparison and quantification of protein complex similarity is generic and can be used to compare other types of modules. The expression for the score is:
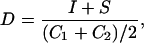 |
where I is the number of identical (i.e., shared components), S is the number of similar components, and C1 and C2 the sizes of the complexes compared.
For S, only one similarity count is considered if there are two or more homologous components in one complex that all match a single component in the other complex. The use of
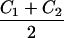 |
as a normalizing factor avoids artificially high or low scores when a small complex is compared to a large one.
The D score is bound between zero and one, where D = 0 indicates no similarity between the components of the two complexes, and D = 1 indicates that all components are similar or shared. In order to avoid considering cases where most components are shared (which are likely to represent different instances of the same complex in the two data sets determined by high-throughput methods), we demand that the S ≥ I and that S + I > 2.
Using the manually curated MIPS complexes as a gold standard, we determined a threshold of D ≥ 0.5 by manual inspection of the matched complexes. Above this threshold, there are no erroneous matches between unrelated complexes (false positives) according to our manual inspection of the MIPS complexes, but a few homologous complexes (false negatives) are not identified as similar. As our primary goal is to ascertain definitively whether duplication of functional modules occurs at all in cellular systems, our priority is to avoid false positives. We thus aim for maximum precision (i.e., finding module duplications with high reliability) and not for large coverage (i.e., finding all possible cases of duplication but also including false positives).
The significance of the observed level of duplication of complexes was assessed by 10,000 random shuffling experiments in which the sizes of the complexes in terms of number of components was kept constant, while the identity of the components was randomized. P-values were calculated as the fraction of these experiments that had more duplicates than observed in the original data set. The same procedure was used to calculate the significance of partial and complete duplications.
Functional analysis
To determine the function of a complex, we used the GO functional classifications (Ashburner et al. 2000) at the biological process level as implemented in the GoSlim annotation of the yeast proteome, obtained from SGD (Christie et al. 2004). In this scheme, there are 33 different functional categories, and one protein can be assigned to multiple categories. The complexes are assigned to functional categories based on a majority-voting scheme, and at least half of the proteins in the complex have to belong to the most frequent category. Subcellular localization information for twenty-two cellular compartments in yeast determined in a large-scale study (Huh et al. 2003) was used to assign localization to complexes in the same way as the functional categories. The significance of the observed functional similarity in homologous complexes was assessed by comparison to the results obtained by two types of random shuffling experiments. The first experiment, shown in Figure 3 involves 10,000 random samplings of proteins from the data set, with the same size and distribution as the detected homologous complexes. The second experiment (not shown) was designed to control for the fact that many complexes share components. It involved shuffling the functional annotation of the proteins 10,000 times. This means that the functions of homologous complexes are random, while the fraction of shared proteins is conserved.
Analysis of pairwise protein interactions
The pairwise physical interactions table from MIPS/CYGD without the interactions determined by yeast–two-hybrid (downloaded on the 12th August 2003) consists of 991 interactions involving 745 proteins. Homology was assessed the same way as described for components of protein complexes. Any two interactions that have one protein in common and homology between the other proteins are counted as partial duplicates in Figures 2B,C. Any two interactions for which both proteins are homologous (but not identical) are considered complete duplicates.
Acknowledgments
We thank Kiyoshi Nagai, Tony Crowther, and members of the Theoretical and Computational Biology Group at the MRC Laboratory of Molecular Biology for critical reading of the manuscript. This work was supported by the MRC Laboratory of Molecular Biology.
E-mail jleal@mrc-lmb.cam.ac.uk; fax +44 (0) 1223 213 556.
E-mail sat@mrc-lmb.cam.ac.uk; fax +44 (0) 1223 213 556.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.3102105.
Footnotes
[Supplemental material is available online at www.genome.org.]
References
- Alon, U. 2003. Biological networks: The tinkerer as an engineer. Science 301: 1866-1867. [DOI] [PubMed] [Google Scholar]
- Ashburner, M., Ball, C.A., Blake, J.A., Botstein, D., Butler, H., Cherry, J.M., Davis, A.P., Dolinski, K., Dwight, S.S., Eppig, J.T., et al. 2000. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25: 25-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman, A., Coin, L., Durbin, R., Finn, R.D., Hollich, V., Griffiths-Jones, S., Khanna, A., Marshall, M., Moxon, S., Sonnhammer, E.L., et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32: D138-D141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm, M. and Bonifacino, J.S. 2001. Adaptins: The final recount. Mol. Biol. Cell. 12: 2907-2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouveret, E., Rigaut, G., Shevchenko, A., Wilm, M., and Seraphin, B. 2000. A Sm-like protein complex that participates in mRNA degradation. EMBO J. 19: 1661-1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabretta, R., Nolfi, S., Parisi, D., and Wagner, G.P. 1998. Emergence of functional modularity in robots. In From animals to animats 5 (eds. R. Pfeifer et al.), pp. 497-504. MIT Press, Cambridge, MA.
- ———. 2000. Duplication of modules facilitates the evolution of functional specialization. Artif. Life 6: 69-84. [DOI] [PubMed] [Google Scholar]
- Casey, P.J. and Seabra, M.C. 1996. Protein prenyltransferases. J. Biol. Chem. 271: 5289-5292. [DOI] [PubMed] [Google Scholar]
- Christie, K.R., Weng, S., Balakrishnan, R., Costanzo, M.C., Dolinski, K., Dwight, S.S., Engel, S.R., Feierbach, B., Fisk, D.G., Hirschman, J.E., et al. 2004. Saccharomyces genome database (SGD) provides tools to identify and analyze sequences from Saccharomyces cerevisiae and related sequences from other organisms. Nucleic Acids Res. 32: D311-D314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conant, G.C. and Wagner, A. 2003a. Asymmetric sequence divergence of duplicate genes. Genome Res. 13: 2052-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ———. 2003b. Convergent evolution of gene circuits. Nat. Genet. 34: 264-266. [DOI] [PubMed] [Google Scholar]
- Dezso, Z., Oltvai, Z.N., and Barabasi, A.L. 2003. Bioinformatics analysis of experimentally determined protein complexes in the yeast Saccharomyces cerevisiae. Genome Res. 13: 2450-2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon, B., Sherman, D., Fischer, G., Durrens, P., Casaregola, S., Lafontaine, I., De Montigny, J., Marck, C., Neuveglise, C., Talla, E., et al. 2004. Genome evolution in yeasts. Nature 430: 35-44. [DOI] [PubMed] [Google Scholar]
- Enright, A.J. and Ouzounis, C.A. 2001. BioLayout—An automatic graph layout algorithm for similarity visualization. Bioinformatics 17: 853-854. [DOI] [PubMed] [Google Scholar]
- Gavin, A.C., Bosche, M., Krause, R., Grandi, P., Marzioch, M., Bauer, A., Schultz, J., Rick, J.M., Michon, A.M., Cruciat, C.M., et al. 2002. Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature 415: 141-147. [DOI] [PubMed] [Google Scholar]
- Hansen, T.F. 2003. Is modularity necessary for evolvability? Remarks on the relationship between pleiotropy and evolvability. Biosystems 69: 83-94. [DOI] [PubMed] [Google Scholar]
- Hartwell, L.H., Hopfield, J.J., Leibler, S., and Murray, A.W. 1999. From molecular to modular cell biology. Nature 402: C47-C52. [DOI] [PubMed] [Google Scholar]
- Ho, Y., Gruhler, A., Heilbut, A., Bader, G.D., Moore, L., Adams, S.L., Millar, A., Taylor, P., Bennett, K., Boutilier, K., et al. 2002. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature 415: 180-183. [DOI] [PubMed] [Google Scholar]
- Hoffman, G.R., Rahl, P.B., Collins, R.N., and Cerione, R.A. 2003. Conserved structural motifs in intracellular trafficking pathways: Structure of the γCOP appendage domain. Mol. Cell 12: 615-625. [DOI] [PubMed] [Google Scholar]
- Huh, W.K., Falvo, J.V., Gerke, L.C., Carroll, A.S., Howson, R.W., Weissman, J.S., and O'Shea, E.K. 2003. Global analysis of protein localization in budding yeast. Nature 425: 686-691. [DOI] [PubMed] [Google Scholar]
- Ihmels, J., Friedlander, G., Bergmann, S., Sarig, O., Ziv, Y., and Barkai, N. 2002. Revealing modular organization in the yeast transcriptional network. Nat. Genet. 31: 370-377. [DOI] [PubMed] [Google Scholar]
- Jacob, F. 1977. Evolution and tinkering. Science 196: 1161-1166. [DOI] [PubMed] [Google Scholar]
- Jeong, H., Tombor, B., Albert, R., Oltvai, Z.N., and Barabasi, A.L. 2000. The large-scale organization of metabolic networks. Nature 407: 651-654. [DOI] [PubMed] [Google Scholar]
- Jeong, H., Mason, S.P., Barabasi, A.L., and Oltvai, Z.N. 2001. Lethality and centrality in protein networks. Nature 411: 41-42. [DOI] [PubMed] [Google Scholar]
- Katju, V. and Lynch, M. 2003. The structure and early evolution of recently arisen gene duplicates in the Caenorhabditis elegans genome. Genetics 165: 1793-1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellis, M., Birren, B.W., and Lander, E.S. 2004. Proof and evolutionary analysis of ancient genome duplication in the yeast Saccharomyces cerevisiae. Nature 428: 617-624. [DOI] [PubMed] [Google Scholar]
- Lynch, M. and Conery, J.S. 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151-1155. [DOI] [PubMed] [Google Scholar]
- Lynch, M. and Katju, V. 2004. The altered evolutionary trajectories of gene duplicates. Trends Genet. 20: 544-549. [DOI] [PubMed] [Google Scholar]
- Madera, M., Vogel, C., Kummerfeld, S.K., Chothia, C., and Gough, J. 2004. The SUPERFAMILY database in 2004: Additions and improvements. Nucleic Acids Res. 32: D235-D239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens, J.A. and Winston, F. 2003. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr. Opin. Genet. Dev. 13: 136-142. [DOI] [PubMed] [Google Scholar]
- Mewes, H.W., Frishman, D., Guldener, U., Mannhaupt, G., Mayer, K., Mokrejs, M., Morgenstern, B., Munsterkotter, M., Rudd, S., and Weil, B. 2002. MIPS: A database for genomes and protein sequences. Nucleic Acids Res. 30: 31-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp, B., Pal, C., and Hurst, L.D. 2003a. Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194-197. [DOI] [PubMed] [Google Scholar]
- ———. 2003b. Evolution of cis-regulatory elements in duplicated genes of yeast. Trends Genet. 19: 417-422. [DOI] [PubMed] [Google Scholar]
- Pearson, W.R. 1990. Rapid and sensitive sequence comparison with FASTP and FASTA. Methods Enzymol. 183: 63-98. [DOI] [PubMed] [Google Scholar]
- Pereira-Leal, J.B., Enright, A.J., and Ouzounis, C.A. 2004. Detection of functional modules from protein interaction networks. Proteins 54: 49-57. [DOI] [PubMed] [Google Scholar]
- Prince, V.E. and Pickett, F.B. 2002. Splitting pairs: The diverging fates of duplicated genes. Nat. Rev. Genet. 3: 827-837. [DOI] [PubMed] [Google Scholar]
- Ravasz, E., Somera, A.L., Mongru, D.A., Oltvai, Z.N., and Barabasi, A.L. 2002. Hierarchical organization of modularity in metabolic networks. Science 297: 1551-1555. [DOI] [PubMed] [Google Scholar]
- Rives, A.W. and Galitski, T. 2003. Modular organization of cellular networks. Proc. Natl. Acad. Sci. 100: 1128-1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salgado-Garrido, J., Bragado-Nilsson, E., Kandels-Lewis, S., and Seraphin, B. 1999. Sm and Sm-like proteins assemble in two related complexes of deep evolutionary origin. EMBO J. 18: 3451-3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel, B. and Huynen, M.A. 2004. Quantifying modularity in the evolution of biomolecular systems. Genome Res. 14: 391-397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatsu, H., Futatsumori, M., Yoshino, K., Yoshida, Y., Shin, H.W., and Nakayama, K. 2001. Similar subunit interactions contribute to assembly of clathrin adaptor complexes and COPI complex: Analysis using yeast three-hybrid system. Biochem. Biophys. Res. Commun. 284: 1083-1089. [DOI] [PubMed] [Google Scholar]
- Teichmann, S.A. and Babu, M.M. 2004. Gene regulatory network growth by duplication. Nat. Genet. 36: 492-496. [DOI] [PubMed] [Google Scholar]
- Teichmann, S.A. and Veitia, R.A. 2004. Genes encoding subunits of stable complexes are clustered on the yeast chromosomes. Genetics (in press). [DOI] [PMC free article] [PubMed]
- Teichmann, S.A., Park, J., and Chothia, C. 1998. Structural assignments to the Mycoplasma genitalium proteins show extensive gene duplications and domain rearrangements. Proc. Natl. Acad. Sci. 95: 14658-14663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann, S.A., Rison, S.C., Thornton, J.M., Riley, M., Gough, J., and Chothia, C. 2001. The evolution and structural anatomy of the small molecule metabolic pathways in Escherichia coli. J. Mol. Biol. 311: 693-708. [DOI] [PubMed] [Google Scholar]
- Veitia, R.A. 2003. Nonlinear effects in macromolecular assembly and dosage sensitivity. J. Theor. Biol. 220: 19-25. [DOI] [PubMed] [Google Scholar]
- Wagner, A. 2001. The yeast protein interaction network evolves rapidly and contains few redundant duplicate genes. Mol. Biol. Evol. 18: 1283-1292. [DOI] [PubMed] [Google Scholar]
- Whyte, J.R. and Munro, S. 2001. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell 1: 527-537. [DOI] [PubMed] [Google Scholar]
- ———. 2002. Vesicle tethering complexes in membrane traffic. J. Cell. Sci. 115: 2627-2637. [DOI] [PubMed] [Google Scholar]
- Wolfe, K.H. and Shields, D.C. 1997. Molecular evidence for an ancient duplication of the entire yeast genome. Nature 387: 708-713. [DOI] [PubMed] [Google Scholar]
- Wuchty, S., Oltvai, Z.N., and Barabasi, A.L. 2003. Evolutionary conservation of motif constituents in the yeast protein interaction network. Nat. Genet. 35: 176-179. [DOI] [PubMed] [Google Scholar]