

ABSTRACT
Staphylococcus aureus is a major human pathogen that causes a variety of illnesses, ranging from minor skin and soft tissue infections to more severe systemic infections. Although the primary host immune response can typically clear bacterial infections, S. aureus is uniquely resistant to inflammation. For instance, our laboratory has determined that S. aureus is highly resistant to nitric oxide (NO⋅), an important component of the innate immune response that plays a role in both immunomodulatory and antibacterial processes. Additionally, NO⋅ and its derivatives can cause damage to S. aureus DNA, more specifically, deamination and/or oxidation of DNA bases; however, regulation and repair mechanisms of DNA in S. aureus are understudied. Thus, we hypothesize that several DNA repair mechanisms may account for the replication fidelity of S. aureus and may contribute to fitness in the presence of NO⋅. Here, we show the role of several DNA repair mechanisms in S. aureus. More specifically, we found that recombinational repair genes recJ, recG, and polA may play a role in the repair of NO⋅-induced replication fork collapses. We also show the role of the base excision repair pathway protein, MutY, in reducing NO⋅-mediated mutagenesis. Overall, our results suggest that NO⋅ leads to DNA damage, which subsequently induces the activity of several DNA repair pathways, contributing to the replication fidelity and fitness of S. aureus.
IMPORTANCE
Pathogenic bacteria must evolve various mechanisms in order to evade the host immune response that they are infecting. One aspect of the primary host immune response to an infection is the production of an inflammatory effector component, nitric oxide (NO⋅). Staphylococcus aureus has uniquely evolved a diverse array of strategies to circumvent the inhibitory activity of nitric oxide. One such mechanism by which S. aureus has evolved allows the pathogen to survive and maintain its genomic integrity in this environment. For instance, here, our results suggest that S. aureus employs several DNA repair pathways to ensure replicative fitness and fidelity under NO⋅ stress. Thus, our study presents evidence of an additional strategy that allows S. aureus to evade the cytotoxic effects of host NO⋅.
KEYWORDS: DNA repair, Staphylococcus aureus, nitric oxide
INTRODUCTION
Staphylococcus aureus is a Gram-positive bacterium that is a highly invasive human pathogen. It is known to cause a variety of illnesses, ranging from superficial skin and soft tissue infections to more severe systemic infections such as endocarditis, osteomyelitis, and sepsis (1). S. aureus asymptomatically colonizes the anterior nares and skin, and an estimated 30% of the human population are natural carriers (2). The treatment of infections has become increasingly difficult due to antibiotic resistance. As such, methicillin-resistant Staphylococcus aureus (MRSA) has been the most common cause of infection since the 1960s (3). Historically, it is known that MRSA-related hospitalizations lead to severe morbidity and mortality globally. However, in recent decades, community-acquired MRSA (CA-MRSA) infections have been increasingly found in otherwise healthy populations (4). Additionally, CA-MRSA clones have been found to be phylogenetically distinct from hospital-associated MRSA and have exhibited both hypervirulence and improved transmission within the host.
One major factor contributing to S. aureus pathogenesis is resistance to the broad-spectrum antimicrobial immune radical, nitric oxide (NO⋅). NO· is an important component of the host innate immune response and plays a role in antibacterial and immunomodulatory processes (5). Although inflammatory NO⋅ is typically required for clearance of pathogenic bacterial infections, S. aureus is uniquely resistant to this immune radical, which distinguishes S. aureus from closely related coagulase-negative staphylococcal species that are unable to grow in its presence (6). During infection, NO⋅ is produced by activated phagocytes through the inducible nitric oxide synthase (iNOS) and can react directly with invading organisms in surrounding inflamed host tissues (7, 8). NO⋅ and its derivatives are known to target heme, iron-sulfur clusters, thiols, lipids, and DNA (9–11). Accordingly, under physiologically relevant concentrations of NO⋅, the reversible binding of NO⋅ to cytochrome heme centers results in the inhibition of aerobic respiration, which can be restored once NO⋅ is detoxified (12). The reactions of NO⋅ and its targets subsequently interfere with many pathways, inducing metabolic and replicative stress. S. aureus therefore must evolve mechanisms of survival under these conditions. The mechanism underlying S. aureus resistance to NO⋅ is complex and consists of several metabolically regulated gene products (5, 6, 13). With that said, we are still lacking in understanding of what makes S. aureus effective resistance to NO⋅ so unique. As previously stated, it is known that NO⋅ targets S. aureus DNA, leading to DNA damage (9, 10). More specifically, NO⋅ leads to the oxidation and deamination of DNA bases; however, the regulation and repair mechanisms of DNA in S. aureus are understudied.
In several bacteria, DNA is damaged due to several endogenous and exogenous factors such as radiation, chemicals, and environmental stress (14). This damage can, in turn, inhibit replication and downstream gene transcription, ultimately affecting cell survival and leading to the accumulation of mutations (15). DNA damage can result from replication fork collapses, single-strand breaks, or exposure to metabolic byproducts such as reactive oxygen species (ROS) or reactive nitrogen species (RNS) (16). Since S. aureus has uniquely evolved to replicate in the presence of NO⋅, we wanted to determine if there were any DNA repair pathways that enhanced the overall fitness of this pathogen in the host environment as well as contributed to replication integrity under NO⋅ stress. We know that NO⋅ exposure results in deamination and/or oxidation of DNA bases, but the regulation and repair mechanisms of this damage are unknown in S. aureus. Thus, we employed mutants from the nucleotide excision repair (NER) (e.g., uvrABC), base excision repair (BER) (e.g., nth, nfo, mutY, and ung), recombination repair (e.g., queA, topB, and sbcC), and replication fork restart pathways (e.g., polA, recG, recJ, and recQ) to determine a role under NO⋅ stress. Overall, DNA repair mechanisms may pose a target for novel therapeutics that sensitize pathogens to effectors of the host defense.
RESULTS
Mutation accumulation analysis suggests that NO⋅ stress leads to deamination and/or oxidation of DNA in S. aureus
Since we speculate that NO⋅ targets DNA and leads to deamination and/or oxidation of S. aureus DNA bases, we performed a modified mutation accumulation assay to determine if there were any specific mutations accumulated under NO⋅ stress compared to an unexposed group (17). We passaged 40 lineages for 40 days, both in the presence and absence of NO·. Upon alignment of whole genome sequencing reads to a reference genome, we found that NO· exposure increased the number of accumulated mutations by more than 25%. That is, 173 mutations were in the exposed group, and 112 were in the unexposed group (Table 1). Additionally, as with many bacteria, we found that 70% of the mutations in both groups resulted in GC→AT mutations, potentially explaining the fact that the S. aureus genome is nearly 70% AT. The mutations most frequently induced under NO⋅ stress compared to the unexposed group were C:G→T:A, G:C→A:T, C:G→A:T, and T:A→C:G, which are all products of DNA deamination and/or oxidation. Another, more rare form of mutation, tandem base substitutions, only occurred in NO·-exposed lineages (Table 1). Tandem base substitutions are thought to arise through the deamination of cytosine to uracil in DNA, followed by subsequent excision by Ung. Other rare mutations that occurred in the untreated strains were mutation 10 (Table 1), which was a loss of a small 3.125 kb plasmid, and mutation 13 (Table 1), which was the loss of the ACME and SCC-Mec islands. Together, these findings confirm that NO⋅ exposure results in the deamination and oxidation of DNA bases in S. aureus, primarily cytosine and guanine.
TABLE 1.
Mutation accumulation
| Mutation | Group − NO⋅ | Group + NO⋅ | Total |
|---|---|---|---|
| (A)7→6 | 1 | 0 | 1 |
| (A)7→8 | 2 | 0 | 2 |
| (ATT)7→6 | 1 | 1 | 2 |
| (T)7→6 | 1 | 0 | 1 |
| (T)7→8 | 4 | 1 | 5 |
| (TACAGAAACAAA)2→1 | 0 | 1 | 1 |
| 2 bp→AC | 0 | 1 | 1 |
| 2 bp→AG | 0 | 1 | 1 |
| 2 bp→TT | 0 | 1 | 1 |
| Δ3,125 bp | 2 | 0 | 2 |
| Δ4 bp | 1 | 0 | 1 |
| Δ54 bp | 0 | 1 | 1 |
| Δ54,659 bp | 1 | 0 | 1 |
| Δ69 bp × 2 | 1 | 1 | 2 |
| A→C | 1 | 5 | 6 |
| A→G | 11 | 9 | 20 |
| A→T | 5 | 8 | 13 |
| C→A | 6 | 23 | 29 |
| C→G | 23 | 22 | 45 |
| C→T | 39 | 68 | 107 |
| G→A | 75 | 88 | 163 |
| G→C | 2 | 4 | 6 |
| G→T | 22 | 9 | 31 |
| T→A | 4 | 5 | 9 |
| T→C | 4 | 12 | 16 |
| T→G | 3 | 2 | 5 |
| 209 | 263 | 472 |
Identification of DNA repair mechanisms that contribute to S. aureus control of NO⋅-induced mutagenesis
Since we have confirmed that NO⋅ targets S. aureus DNA, resulting in deamination and oxidation of DNA bases, we also wanted to determine if there were any DNA repair mechanisms involved in targeting NO⋅-mediated mutagenesis. To do so, we employed 15 DNA repair mutant strains from various DNA repair pathways: NER, BER, recombination repair, and replication fork restart. These specific strains were selected due to the recent identification of the target genes in S. aureus based on known homology in Bacillus subtilis. We performed mutation rate assays with the 15 DNA repair mutant strains alongside a WT JE2 control. We found that in the absence of NO⋅, ΔrecG, Δnth, Δnfo, Δung, and ΔmutY transposon mutants display significantly increased mutation rates compared to WT JE2 (Fig. 1A). Upon addition of NO·, the mutation rate of wild type (WT) is elevated by approximately fivefold (P = 0.001, Mann-Whitney), consistent with the mutation accumulation results (Table 1). However, the mutation rate of the ΔmutY mutant was further increased in the presence of NO⋅, suggesting elevated levels of guanine oxidation (Fig. 1B). Overall, the hypermutable phenotype we observed with the ΔmutY mutant under NO⋅ exposure suggests that MutY likely targets NO⋅-mediated mutagenesis in S. aureus. Finally, all hypermutability in the ∆mutY mutant, either in the absence or presence of NO·, was resolved upon genetic complementation (Fig. S2A and B).
FIG 1.

Elevated mutation rates suggest that MutY may play a role in modulating mutagenicity in the presence of NO⋅ in S. aureus. Mutability of S. aureus WT JE2 and 15 DNA repair transposon mutants shown either unexposed (A) or exposed (B) to a disc of 500 mM diethylenetriamine NONOate ( (n = 12). Data were analyzed via the Wilcoxon rank-sum test for nonparametric analyses (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
Identification of DNA repair mechanisms associated with maintaining replicative fitness under NO⋅ stress in S. aureus
Although we have identified a DNA repair pathway responsible for controlling NO⋅-induced mutagenesis, we also wanted to determine if there were any DNA repair mechanisms that contribute to replicative fitness under NO⋅ stress. To do so, we employed the same 15 DNA repair mutant strains screened in the previous section alongside a WT JE2 control and performed growth curves. Without the addition of NO⋅, we observed a similar growth rate across all strains (Fig. S1A). However, following the addition of a mixture of NO⋅ donors (1 mM diethylamine NONOate [DEANO] and 10 mM NOC-12) at an OD660 of 0.2, one of these strains, ΔrecG, exhibited a significant growth defect (Fig. 2A and B). Additionally, we performed growth curves in chemically defined media, exposed the cultures to a NO⋅ donor [10 mM diethylenetriamine NONOate (DETA/NO)] at inoculum, and observed average terminal optical density (OD) across all DNA repair mutant strains (Fig. 2C). The results show a significantly lower average terminal OD for ΔpolA, ΔrecJ, and ΔrecG than for WT JE2 (Fig. 2D). None of these mutants had a defect in reaching WT terminal OD in the absence of NO· (Fig. S1B). We also observed increased lag times for ∆nfo, ∆mpg, and ∆tag in PN medium; however, none of these mutants showed reduced growth rate or terminal OD (Fig. 2C and D). Taken together, these results suggest that RecG and, to a lesser extent, PolA and RecJ may play roles in maintaining fitness in S. aureus in the presence of NO⋅. Finally, the major NO· sensitivity phenotype associated with the ∆recG mutation was resolved upon genetic complementation (Fig. S3).
FIG 2.

Growth curves suggest that RecG may contribute to S. aureus ability to confer fitness under NO⋅ stress. A representative growth curve is shown for S. aureus WT JE2 and 15 DNA repair transposon mutants grown in tryptic soy broth with a mixture of 1 mM DEANO and 10 mM NOC-12 added at OD 0.2 (A) (n = 3). The amount of time it took each mutant strain to reach an OD of 0.6 with a mixture of 1 mM DEANO and 10 mM NOC-12 added at OD 0.2 (B) (n = 3). A representative growth curve is shown for S. aureus WT JE2 and 15 DNA repair transposon mutants grown in PNG with the addition of 10 mM DETA/NO at inoculum (C) (n = 3). Average terminal OD from growth curves (C) of S. aureus WT JE2 and 15 DNA repair transposon mutants grown in PNG minimal media with 10 mM DETA/NO added at inoculum (D) (n = 3). Data were analyzed via one-way analysis of variance with Dunnett’s multiple comparisons test for correction (*, P < 0.05) or Student’s two-tailed t-test (*, P < 0.05) where appropriate.
Replication fork restart DNA repair pathway contributes to NO⋅ resistance in vivo
Since the ∆recG mutant exhibited the most consistent and most profound fitness defect in the presence of NO· in vitro, we decided to test whether it had a fitness defect due to NO· in vivo. In order to confirm the role of RecG in maintaining replication integrity in the presence of NO⋅ in vivo, we infected C57BL/6J mice subcutaneously with 107 colony-forming units (CFU) of either S. aureus WT LAC or S. aureus ΔrecG mutant. The mice were assessed both 3 and 7 days post infection by monitoring lesion size and bacterial burden within the abscess. Our results show that mice infected with the ΔrecG mutant display a significant decrease in both lesion size and bacterial burden in infected abscesses compared to WT-infected mice (Fig. 3A and B). This suggests that mice infected with the ΔrecG mutant develop a less severe infection than those infected with WT S. aureus. Additionally, to determine the direct role of host NO⋅ production during infection, we treated mice with an iNOS inhibitor, N-iminoethyl-L-lysine (L-NIL), and observed the effect on ΔrecG mutant-infected mice. The L-NIL-treated mice infected with the ΔrecG mutant displayed a lesion size and bacterial burden in infected abscesses similar to WT levels, which were significantly elevated compared to untreated ΔrecG mutant-infected mice. While L-NIL-treated animals can still generate ROS, the ∆recG mutant does not display a severe growth defect in the presence of H2O2 when compared with WT, suggesting that NO· specifically necessitates RecG during infection (Fig. S4). Overall, our results suggest a role for RecG in protecting against S. aureus cell death due to host NO⋅ production during infection.
FIG 3.
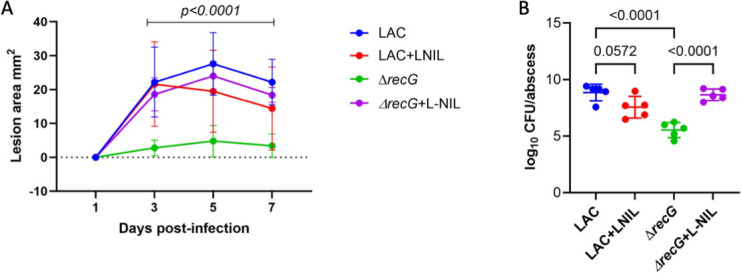
In vivo results suggest that RecG plays a role in maintaining replication integrity in S. aureus in the presence of host NO⋅ production. C57BL/6J mice were infected subcutaneously with 107 CFU of either WT LAC or the ΔrecG mutant (n = 5). A representative graph of lesion sizes measured on days 1, 3, 5, and 7 post infection is shown in A. Bacterial burden was measured by harvesting the abscess 7 days post infection and enumerating the bacteria (B). Data were analyzed via one-way analysis of variance with Tukey’s multiple comparisons test for correction.
DISCUSSION
In response to a typical S. aureus infection, activated host innate immune cells produce reactive nitrogen species that attack various targets of the cell. The most direct targets are enzymes in the respiratory chain, the blocking of which leads to increased intracellular reactive oxygen species (18). Since we know that RNS and ROS target S. aureus DNA, likely resulting in deamination and/or oxidation of DNA bases, we performed a mutation accumulation (MA) experiment to observe via sequencing analysis which mutations most frequently occur under NO⋅ stress. The MA experiment is ideal to estimate rates of spontaneous mutations that occur over the duration of the experiment, and in our case, we were able to compare the accumulation of mutations under NO⋅ exposure compared to an unexposed group (17). Additionally, we performed our experiment without selection, ensuring the elimination of selection bias on mutant frequency. Overall, we observed that the mutations that were most frequently induced by NO⋅ were all products of DNA oxidation and deamination: C→T, G→A, C→A, and T→C (19). The mutations that occurred most frequently under NO⋅ stress were C→T and G→A transitions. These mutations likely occurred due to the deamination of cytosine or guanine (9, 20). A C→T transition can arise in two mechanisms. First, deaminating guanine to xanthine will result in a C:X pair. Since xanthine typically pairs with thymine, upon further replication, the C:X would result in a T:X pair and finally be repaired to T:A. The second mechanism involving a C:G pair under NO⋅ exposure is that the cytosine is deaminated to uracil, which results in a U:G pair, followed by synthesis to a U:A pair, and upon further replication, results in a T:A pair. Another result of cytosine deamination followed by the excision of uracil by Ung is tandem base substitutions (21). This is due to the tendency of dU bases to “flip” out of the double helix. Following a round of replication and subsequent excision of uracil, two tandem base mutations can occur. Indeed, all three tandem base substitutions were detected in the NO·-treated lineages and all derived from parental C:G sites (Table 1). The C→A transversion occurred less frequently; however, it is interesting due to the bias of occurrence in the NO⋅ exposed group. A common DNA base lesion formed during replication stress is 8-oxoG, which can mismatch to adenine (22). What most likely occurs is a C:G pair oxidized to C:8-oxoG, followed by replication to an A:8-oxoG mispair. Upon replication, A:oxo-G is likely repaired to A:T, resulting in a C→A transversion. Finally, the mutation that least likely occurred is the T→C transition. This transition likely arises due to the deamination of adenine to hypoxanthine, so a T:A pair turns into T:HX. HX typically binds to cytosine, resulting in a HX:C pair, which, following another round of replication, results in a C:G pair. Ultimately, our results show that NO⋅ exposure results in the accumulation of DNA transitions over transversions. Furthermore, the overall takeaway from the MA analysis is that the exposure of WT S. aureus to NO⋅ results in accumulation of DNA damage, more specifically, deaminated and oxidized DNA. S. aureus likely relies on the BER pathway to repair these accumulated lesions, ultimately aiding the overall ability of this pathogen to modulate mutagenesis in a NO⋅-concentrated environment.
Though we have confirmed that NO⋅ exposure results in deamination and oxidation of DNA bases in S. aureus, we also wanted to know if there were any DNA repair mechanisms contributing to S. aureus ability to mitigate mutagenesis in this NO⋅-concentrated environment. We observed mutagenicity across several DNA repair transposon insertion mutants compared to WT JE2, both in the presence and absence of NO⋅. Overall, we observed increased mutation rates in ΔrecG, Δnth, Δnfo, ΔmutY, and Δung compared to WT in the absence of NO⋅, suggesting that inactivation of these genes results in the acquisition of spontaneous mutations. RecG is an ATP-dependent helicase involved in replication fork restart following double-strand breaks (16). Without RecG activity, we would expect to see an increase in mutation rate since its absence would likely result in the incomplete resolution of DNA Holliday junctions and, thus, an accumulation of mutagenic DNA. Nth, Nfo, MutY, and Ung are all members of the BER pathway involved in single-stranded DNA damage repair (23). The main function of the BER pathway is to repair nonbulky single-base DNA lesions such as oxidized or deaminated DNA bases, alkylated or abasic sites, and dUTP incorporation during DNA replication (24). The misincorporation of dUTP during DNA replication likely occurs one of two ways: either due to deamination of cytosine residues in DNA or incorporation of dUTP before the intermediate can be catalyzed to dTTP, since the replicative polymerase cannot distinguish between them (25, 26). Nth and Nfo are endonuclease III and endonuclease IV, respectively. They both play a role in resolving apurinic or apyrimidic (AP) sites left by DNA glycosylase activity when repairing damaged DNA during BER (27, 28). In inactivating these genes, we would expect to see the increase in mutability that we observe in our results since insufficient removal of the AP site would subsequently result in an interruption of this repair process. Ung and MutY are both DNA glycosylases involved in BER (29, 30). Ung excises uracil residues from DNA. MutY is an adenosine DNA glycosylase that hydrolyzes free adenine bases from 8-oxo-guanine:A mismatches (31, 32). An inactivated mutY or ung mutant strain would likely lack the capability to initiate the BER process, resulting in the accumulation of single-base lesions in DNA left unresolved, which likely explains our results. Interestingly, following the addition of NO⋅ in our mutation rate assay, we observed that ΔmutY displays a further enhanced mutation rate. Since we know that NO⋅ causes deamination and/or oxidation of DNA bases and both Ung and MutY target oxidized and/or deaminated DNA bases, the hypermutable phenotype seen with the inactivated ΔmutY strain is likely a result of the accumulation of unresolved oxidized DNA bases (33). Overall, our results suggest a role of the BER pathway, more specifically, the DNA glycosylase MutY, in targeting NO⋅-mediated mutagenesis in S. aureus.
Although we have identified a process involved in controlling NO⋅ stress-induced mutagenesis in S. aureus, we wanted to determine if there were any DNA repair mechanisms that contribute to replicative fitness under NO⋅ stress. We performed growth curves with a WT JE2 strain alongside several DNA repair transposon insertion mutant strains and compared the optical density over an elapsed time in the presence and absence of NO⋅. Under NO⋅ stress, the ΔrecG mutant displayed the most significant growth defect, suggesting that this pathway may aid S. aureus ability to confer fitness in a NO⋅-stressed environment. This phenotype was further explained by infecting mice with the ΔrecG mutant, confirming that mice develop a less severe infection than WT-infected mice. However, in mice treated with L-NIL, this phenotype is completely reversed, which suggests that the replication fork restart pathway player, RecG, plays an important role in protecting S. aureus from host production of NO⋅ during infection. RecG is an ATP-dependent helicase with 3′–5′ activity and typically works alongside RecJ, which is a single-stranded DNA-specific exonuclease with 5′–3′ activity and also showed a modest growth defect under NO· stress (Fig. 2) (16). Both play a critical role in homologous recombination and DNA repair; more specifically, they play a role in catalyzing branch migration during replication fork restart (34). In the event of DNA double-strand breaks (DSBs) and subsequently a replication fork collapse, homologous recombination is initiated. Typically, the ends of a DSB are processed by a 5′–3′ helicase-nuclease complex, RexAB, which leaves behind a 3′-ssDNA overhang (35). This allows a recombinase, RecA, to bind to the 3′-ssDNA overhang while it searches for a homologous sequence to initiate strand invasion. Following strand invasion, DNA synthesis can occur whereby the 3′ end is extended via DNA polymerase III simultaneously alongside branch migration, carried out by the 3′–5′ helicase, RecG (16). Ultimately, the Holliday junction resolution is carried out by the resolvase protein, RecU. Previously, it has been shown that NO⋅ causes replication fork collapses, which can result in DNA double-strand breaks, leading to mutagenic DNA rearrangement in S. aureus (36). In this study, our results suggest a putative role of RecG in maintaining replicative fitness under NO⋅ stress.
MRSA poses a serious threat to population health worldwide. Since S. aureus has also uniquely evolved resistance to the primary host immune defense, inflammatory NO⋅, our laboratory sought to determine the mechanisms S. aureus employs in order to thwart the activity of NO⋅. In this study, we found several DNA repair mechanisms that enhance S. aureus replicative fidelity and overall fitness in this NO⋅-concentrated environment. Thus, these DNA repair mechanisms could potentially act as a target for novel therapeutics that sensitize this pathogen to the primary host immune response. Finding alternative therapies to target this multi-drug-resistant pathogen is essential to circumventing the global burden of antibiotic resistance.
MATERIALS AND METHODS
Bacterial strains and growth conditions
S. aureus cultures were grown in brain heart infusion (BHI) medium, shaken at 250 rpm at 37°C overnight. S. aureus JE2 and LAC USA300 were used in this study as background strains (Table 2). Antibiotic selection in S. aureus was carried out using erythromycin (5 µg/mL) where appropriate. All Nebraska Tn mutations were transduced into fresh WT backgrounds and PCR verified prior to analyses. Complementation of the ∆recG and ∆mutY mutations was performed by cloning each gene and its promoter region into pOS1 to generate strains AR1762 and AR1767, respectively (Table 2).
TABLE 2.
List of strains and plasmids used in the experiment
| Strains | Genotype | Source/reference |
|---|---|---|
| S. aureus JE2 | Methicillin-resistant S. aureus laboratory strain | Laboratory strain |
| S. aureus LAC | USA300 methicillin-resistant clinical isolate; laboratory strain | Laboratory strain |
| NE11 | S. aureus JE2 ∆recJ::ermR | (37) |
| NE22 | S. aureus JE2 ∆polA::ermR | (37) |
| NE152 | S. aureus JE2 ∆topB::ermR | (37) |
| NE445 | S. aureus JE2 ∆umuC::ermR | (37) |
| NE761 | S. aureus JE2 ∆nth::ermR | (37) |
| NE888 | S. aureus JE2 ∆ung::ermR | (37) |
| NE972 | S. aureus JE2 ∆recQ::ermR | (37) |
| NE1028 | S. aureus JE2 ∆nfo::ermR | (37) |
| NE1040 | S. aureus JE2 ∆mutY::ermR | (37) |
| NE1344 | S. aureus JE2 ∆recG::ermR | (37) |
| NE1379 | S. aureus JE2 ∆queA::ermR | (37) |
| NE1451 | S. aureus JE2 ∆sbcC::ermR | (37) |
| NE1613 | S. aureus JE2 ∆mpg::ermR | (37) |
| NE1825 | S. aureus JE2 ∆tag::ermR | (37) |
| NE1866 | S. aureus JE2 ∆dinB::ermR | (37) |
| AR 1758 | S. aureus LAC ∆recG::ermR | This study |
| AR1762 | S. aureus LAC ∆recG::ermR + pAS12 (pOS1 with recG) | This study |
| AR1767 | S. aureus JE2 ∆mutY::ermR + pAS13 (pOS1 with mutY | This study |
Growth rate analysis
S. aureus cultures were grown overnight in BHI at 37°C, shaking at 250 rpm. Overnight cultures were washed three times with phosphate-buffered saline (PBS) and inoculated at a 1:200 ratio in a 96-well plate containing tryptic soy broth (200 µL/well). Cells were grown at 37°C and shaken in a BioTek microplate reader. For NO⋅ growth curves, a mixture of 10 mM NOC-12 (t1/2 = 100 min) and 1 mM DEANO (t1/2 = 2 min) was added at an OD660 of 0.20. Growth was monitored every 15 min for 24 h. This provides a rapid and acute NO· stress for a mid-exponential culture to respond to.
Mutation rate assay
S. aureus cultures were shaken at 250 rpm at 37°C overnight. Overnight cultures were serially diluted and plated on BHI agar either with or without exposure to NO⋅. The NO⋅ donor we used in this experiment was DETA/NO, which was resuspended in 0.01 N NaOH and has a half-life (t1/2) of 20 h. In cultures exposed to NO⋅, a disc was placed in the center of the plate, and 20 µL of 500 mM DETA/NO was added to the disc. Plates were incubated at 37°C. On the following day, 20 single colonies were picked from both NO⋅-exposed and NO⋅-unexposed plates and resuspended in 200 µL of PBS within a 96-well plate. Each resuspension was serially diluted and subsequently plated on BHI agar plates containing rifampicin (100 µg/mL) and BHI agar plates lacking antibiotics. Following incubation overnight at 37°C, we were able to calculate the mutation rate by dividing the number of colonies found on the BHI + rifampicin plate by the CFU/mL on BHI agar plates lacking antibiotic.
Mutation accumulation assay
S. aureus WT USA300 culture was struck out on 80 tryptic soy agar (TSA) plates. Forty plates were exposed to NO⋅ (500 mM DETA/NO), and 40 plates were unexposed. For 40 consecutive days, a single colony was picked and struck out onto a fresh TSA plate, either with or without exposure to NO⋅. For the NO⋅-exposed plates, a dot was randomly drawn near the NO⋅ disc prior to incubation at 37°C to ensure elimination of selection bias. Finally, on day 40, a single colony was picked and struck out to create a lawn on a fresh TSA plate. On the following day, the lawn was resuspended in 50% BHI and glycerol and stored at −80°C. Genomic DNA was extracted from the 80 isolates and subjected to sequencing analysis.
Whole genome sequencing analysis
At the end of the mutation accumulation experiment, we extracted genomic DNA from the 80 isolates using the Epicentre MasterPure Gram Positive DNA Purification Kit (Qiagen). DNA was sequenced using Illumina NextSeq 500. We sequenced one clone from the final time point of every evolved lineage. Sequencing reads were trimmed and quality filtered using Trimmomatic version 0.36 with the following criteria: LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 (38). Reads were aligned to a reference genome closely related to the ancestral strain (Staphylococcus aureus subsp. aureus USA300_FPR3757), and variants were called using breseq version 0.35.0 in consensus mode (39). Breseq was run with default parameters, except that a minimum of five reads from each strand were required to support variant calls. The sample read depth ranged from 91 to 355×. Each lineage was monitored for the possibility of cross-contamination.
Murine infection model
S. aureus LAC and ΔrecG were grown for 12–16 h in BHI at 37°C. Cultures were diluted to 1:200 in fresh BHI and grown until OD600 reached 2.0. Then, 1 mL of each culture was harvested at 10,000 × g and washed twice with 1 mL of PBS. Bacterial pellets were reconstituted in 200 µL of PBS and serially diluted to 1010. All dilutions were plated on BHI agar. Colonies were counted the next day, while the bacterial suspensions were stored at 4°C. Bacterial suspensions were adjusted to 5 × 108/mL based on CFU enumeration. Both male and female, 6–8-week-old C57BL/6J mice weighing 20–25 g were used in this study. Mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and housed with 14-h light cycles. On the day of infection, mice were weighed, and 12× (body weight) in µL of Avertin/2,2,2-tribromoethanol (Acros) was administered via intraperitoneal injection. The left flank on the dorsal side of each animal was shaved, and 20 µL or 107 CFU of the bacterial suspension was injected subcutaneously using a 26G needle. Animals were monitored every day, and abscesses were measured on days 3, 5, and 7. On day 7, the mice were euthanized in a CO2 chamber, followed by cervical dislocation. The abscesses were excised precisely, avoiding extra tissue, and homogenized in PBS. The homogenates were serially diluted and plated on BHI agar for CFU enumeration the next day. For L-NIL treatment, 100 µg/mL of L-NIL hydrochloride (Cayman) was added to the drinking water 24 h prior to infection. The L-NIL water was changed every 2 days from the day of infection.
ACKNOWLEDGMENTS
We would like to acknowledge funding sources for this work, namely, AIR01093613 to A.R.R.
Contributor Information
Anthony R. Richardson, Email: anthony.richardson@pitt.edu.
K. Heran Darwin, New York University School of Medicine, New York, New York, USA.
William Wiley Navarre, University of Toronto, Toronto, Canada.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/mbio.02156-23.
Growth without NO.
MutY complementation.
RecG complementation.
RecG peroxide.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, Harrison LH, Lynfield R, Dumyati G, Townes JM, Craig AS, Zell ER, Fosheim GE, McDougal LK, Carey RB, Fridkin SK, Active Bacterial Core surveillance (ABCs) MRSA Investigators . 2007. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 298:1763–1771. doi: 10.1001/jama.298.15.1763 [DOI] [PubMed] [Google Scholar]
- 2. Otto M. 2010. Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev Dermatol 5:183–195. doi: 10.1586/edm.10.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Monaco M, Pimentel de Araujo F, Cruciani M, Coccia EM, Pantosti A. 2017. Worldwide epidemiology and antibiotic resistance of Staphylococcus aureus. Curr Top Microbiol Immunol 409:21–56. doi: 10.1007/82_2016_3 [DOI] [PubMed] [Google Scholar]
- 4. Thurlow LR, Joshi GS, Richardson AR. 2012. Virulence strategies of the dominant USA300 lineage of community-associated Methicillin-resistant Staphylococcus aureus (CA-MRSA). FEMS Immunol Med Microbiol 65:5–22. doi: 10.1111/j.1574-695X.2012.00937.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richardson AR, Dunman PM, Fang FC. 2006. The nitrosative stress response of Staphylococcus aureus is required for resistance to innate immunity. Mol Microbiol 61:927–939. doi: 10.1111/j.1365-2958.2006.05290.x [DOI] [PubMed] [Google Scholar]
- 6. Richardson AR, Libby SJ, Fang FC. 2008. A nitric oxide-inducible lactate dehydrogenase enables Staphylococcus aureus to resist innate immunity. Science 319:1672–1676. doi: 10.1126/science.1155207 [DOI] [PubMed] [Google Scholar]
- 7. Lewis RS, Tamir S, Tannenbaum SR, Deen WM. 1995. Kinetic analysis of the fate of nitric oxide synthesized by Macrophages in vitro. J Biol Chem 270:29350–29355. doi: 10.1074/jbc.270.49.29350 [DOI] [PubMed] [Google Scholar]
- 8. Nalwaya N, Deen WM. 2005. Nitric oxide, oxygen, and superoxide formation and consumption in macrophage cultures. Chem Res Toxicol 18:486–493. doi: 10.1021/tx049879c [DOI] [PubMed] [Google Scholar]
- 9. Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS. 1991. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 254:1001–1003. doi: 10.1126/science.1948068 [DOI] [PubMed] [Google Scholar]
- 10. Wink DA, Mitchell JB. 1998. Chemical biology of nitric oxide: insights into regulatory, cytotoxic, and cytoprotective mechanisms of nitric oxide. Free Radic Biol Med 25:434–456. doi: 10.1016/s0891-5849(98)00092-6 [DOI] [PubMed] [Google Scholar]
- 11. Schopfer FJ, Baker PRS, Freeman BA. 2003. NO-dependent protein nitration: a cell signaling event or an oxidative inflammatory response? Trends Biochem Sci 28:646–654. doi: 10.1016/j.tibs.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 12. Kinkel TL, Roux CM, Dunman PM, Fang FC. 2013. The Staphylococcus aureus SrrAB two-component system promotes resistance to nitrosative stress and hypoxia. mBio 4:e00696–13. doi: 10.1128/mBio.00696-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Richardson AR, Payne EC, Younger N, Karlinsey JE, Thomas VC, Becker LA, Navarre WW, Castor ME, Libby SJ, Fang FC. 2011. Multiple targets of nitric oxide in the tricarboxylic acid cycle of salmonella enterica Serovar typhimurium. Cell Host Microbe 10:33–43. doi: 10.1016/j.chom.2011.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Krejci L, Altmannova V, Spirek M, Zhao X. 2012. Homologous recombination and its regulation. Nucleic Acids Res 40:5795–5818. doi: 10.1093/nar/gks270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sedgwick SG, Ho C, Woodgate R. 1991. Mutagenic DNA repair in enterobacteria. J Bacteriol 173:5604–5611. doi: 10.1128/jb.173.18.5604-5611.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ha, K. P., & Edwards, A. M.. 2021. DNA repair in Staphylococcus aureus. MMBR, 85(4), e0009121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dillon MM, Sung W, Lynch M, Cooper VS. 2015. The rate and molecular spectrum of spontaneous mutations in the GC-rich multichromosome genome of Burkholderia cenocepacia. Genetics 200:935–946. doi: 10.1534/genetics.115.176834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grosser MR, Weiss A, Shaw LN, Richardson AR. 2016. Regulatory requirements for Staphylococcus aureus nitric oxide resistance. J Bacteriol 198:2043–2055. doi: 10.1128/JB.00229-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Richardson AR, Soliven KC, Castor ME, Barnes PD, Libby SJ, Fang FC. 2009. The base excision repair system of salmonella enterica Serovar typhimurium counteracts DNA damage by host nitric oxide. PLoS Pathog 5:e1000451. doi: 10.1371/journal.ppat.1000451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Caulfield JL, Wishnok JS, Tannenbaum SR. 1998. Nitric oxide-induced deamination of cytosine and guanine in deoxynucleosides and oligonucleotides. J Biol Chem 273:12689–12695. doi: 10.1074/jbc.273.21.12689 [DOI] [PubMed] [Google Scholar]
- 21. Sepúlveda-Yáñez JH, Alvarez Saravia D, Pilzecker B, van Schouwenburg PA, van den Burg M, Veelken H, Navarrete MA, Jacobs H, Koning MT. 2021. Tandem substitutions in somatic hypermutation. Front Immunol 12:807015. doi: 10.3389/fimmu.2021.807015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shibutani S, Takeshita M, Grollman AP. 1991. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature 349:431–434. doi: 10.1038/349431a0 [DOI] [PubMed] [Google Scholar]
- 23. Krwawicz J, Arczewska KD, Speina E, Maciejewska A, Grzesiuk E. 2007. Bacterial DNA repair genes and their eukaryotic homologues: 1. mutations in genes involved in base excision repair (BER) and DNA-end processors and their implication in Mutagenesis and human disease. Acta Biochim Pol 54:413–434. [PubMed] [Google Scholar]
- 24. Krokan HE, Bjørås M. 2013. Base excision repair. Cold Spring Harb Perspect Biol 5:a012583. doi: 10.1101/cshperspect.a012583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brynolf K, Eliasson R, Reichard P. 1978. Formation of okazaki fragments in polyoma DNA synthesis caused by misincorporation of uracil. Cell 13:573–580. doi: 10.1016/0092-8674(78)90330-6 [DOI] [PubMed] [Google Scholar]
- 26. Tye BK, Chien J, Lehman IR, Duncan BK, Warner HR. 1978. Uracil incorporation: a source of pulse-labeled DNA fragments in the replication of the Escherichia coli chromosome. Proc Natl Acad Sci U S A 75:233–237. doi: 10.1073/pnas.75.1.233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Melamede RJ, Hatahet Z, Kow YW, Ide H, Wallace SS. 1994. Isolation and characterization of endonuclease VIII from Escherichia coli. Biochemistry 33:1255–1264. doi: 10.1021/bi00171a028 [DOI] [PubMed] [Google Scholar]
- 28. Demple B, Johnson A, Fung D. 1986. Exonuclease III and Endonuclease IV remove 3' blocks from DNA synthesis primers in H2O2-damaged Escherichia coli. Proc Natl Acad Sci USA 83:7731–7735. doi: 10.1073/pnas.83.20.7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ambur OH, Davidsen T, Frye SA, Balasingham SV, Lagesen K, Rognes T, Tønjum T. 2009. Genome dynamics in major bacterial pathogens. FEMS Microbiol Rev 33:453–470. doi: 10.1111/j.1574-6976.2009.00173.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Michaels ML, Cruz C, Grollman AP, Miller JH. 1992. Evidence that MutY and MutM combine to prevent mutations by an oxidatively damaged form of guanine in DNA. Proc Natl Acad Sci U S A 89:7022–7025. doi: 10.1073/pnas.89.15.7022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Banda DM, Nuñez NN, Burnside MA, Bradshaw KM, David SS. 2017. Repair of 8-oxoG:a mismatches by the MUTYH glycosylase: mechanism, metals and medicine. Free Radic Biol Med 107:202–215. doi: 10.1016/j.freeradbiomed.2017.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wallace SS. 2013. DNA glycosylases search for and remove oxidized DNA bases. Environ Mol Mutagen 54:691–704. doi: 10.1002/em.21820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Endutkin AV, Panferova EP, Barmatov AE, Zharkov DO. 2021. DNA glycosylases for 8-oxoguanine repair in Staphylococcus aureus. DNA Repair (Amst) 105:103160. doi: 10.1016/j.dnarep.2021.103160 [DOI] [PubMed] [Google Scholar]
- 34. West SC. 1997. Processing of recombination intermediates by the RuvABC proteins. Annu Rev Genet 31:213–244. doi: 10.1146/annurev.genet.31.1.213 [DOI] [PubMed] [Google Scholar]
- 35. Dillingham MS, Kowalczykowski SC. 2008. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol Mol Biol Rev 72:642–671, doi: 10.1128/MMBR.00020-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cromie GA, Connelly JC, Leach DR. 2001. Recombination at double-strand breaks and DNA ends: conserved mechanisms from Phage to humans. Mol Cell 8:1163–1174. doi: 10.1016/s1097-2765(01)00419-1 [DOI] [PubMed] [Google Scholar]
- 37. Fey PD, Endres JL, Yajjala VK, Widhelm TJ, Boissy RJ, Bose JL, Bayles KW. 2013. A genetic resource for rapid and comprehensive phenotype screening of nonessential Staphylococcus aureus genes. mBio 4:e00537–12. doi: 10.1128/mBio.00537-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible Trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. doi: 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deatherage DE, Barrick JE. 2014. Identification of mutations in laboratory-evolved Microbes from next-generation sequencing data using Breseq. Methods Mol Biol 1151:165–188. doi: 10.1007/978-1-4939-0554-6_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Growth without NO.
MutY complementation.
RecG complementation.
RecG peroxide.