

Abstract
Background:
Dilated Cardiomyopathy (DCM) is a major cause of heart failure and carries a high mortality rate. Myocardial recovery in DCM-related heart failure patients is highly variable, with some patients having little or no response to standard drug therapy. A genome-wide association study (GWAS) may agnostically identify biomarkers and provide novel insight into the biology of myocardial recovery in DCM.
Methods:
A GWAS for change in left ventricular ejection fraction (LVEF) was performed in 686 Caucasian subjects with recent onset DCM who received standard pharmacotherapy. GWAS signals were subsequently functionally validated and studied in relevant cellular models to understand molecular mechanism(s) that may have contributed to the change in LVEF.
Results:
The GWAS identified a highly suggestive locus that mapped to the 5’-flanking region of the CUB domain containing protein 1 (CDCP1) gene (rs6773435, p=7.12×10−7). The variant allele was associated with improved cardiac function, and decreased CDCP1 transcription. CDCP1 expression was significantly up-regulated in human cardiac fibroblasts (HCFs) in response to the platelet-derived growth factor (PDGF) signaling, and knock-down (KD) of CDCP1 significantly repressed HCF proliferation and decreased AKT phosphorylation. Transcriptomic profiling after CDCP1 KD in HCFs supported the conclusion that CDCP1 regulates HCF proliferation and mitosis. In addition, CDCP1 KD in HCFs resulted in significantly decreased expression of soluble ST2, a prognostic biomarker for heart failure and inductor of cardiac fibrosis.
Conclusions:
CDCP1 may play an important role in myocardial recovery in recent onset DCM and mediates its effect primarily by attenuating cardiac fibrosis.
Keywords: Dilated Cardiomyopathy, Heart Failure, Cardiac Fibrosis, CDCP1, PDGF Signaling, sST2
Subject Terms: Biomarkers, Genetic, Association Studies, Precision Medicine, Cardiomyopathy, Heart Failure
INTRODUCTION
The prevalence of heart failure is projected to increase by 46% from 2012 to 2030 and will affect over 8 million people in the United States by 2030.1,2 Targeting the neuro-hormonal axis has led to improved survival in heart failure with reduced ejection fraction (HFrEF), however, newer therapies based on this approach have had diminishing returns, and morbidity and mortality for this disease remain high.3 Among patients with HFrEF, dilated cardiomyopathy (DCM) is the most common cause of heart transplantation, accounting for 30–40% of all cases of HFrEF. Treatment response is highly variable among individuals with DCM, with some patients having little or no clinical response to standard drug therapy.4 Change in left ventricular ejection fraction (LVEF) that occurs during drug therapy correlates significantly with mortality and, therefore, it serves as a reliable surrogate measurement for mortality in DCM and for assessing myocardial recovery in response to pharmacotherapy.5
Genome-wide association studies (GWAS), by adopting an agnostic approach, could provide insight into novel biological pathways that, in turn, could affect drug utilization and the development of novel drug therapy. An analysis of GWAS-identified genes for drug repositioning demonstrated that a large number of these genes are targeted by drugs that have indications closely related to the GWAS trait or have indications different from the GWAS trait and, thus, can be repositioned as immediately translational opportunities.6 The genomic revolution, however, has had limited therapeutic success in HFrEF. Although case-control GWAS studies have identified DCM susceptibility genes,7 there have been no genomic studies assessing treatment response in DCM which, if performed, could potentially have direct therapeutic implications.
In the present study, we first performed a discovery GWAS for change in LVEF in 686 Caucasian subjects with recent-onset DCM who received standard pharmacotherapy. The GWAS identified a single-nucleotide polymorphism (SNP) signal mapping to the 5’-flanking region of CDCP1, a gene which had previously been reported to play a role in pulmonary fibrosis and bone marrow fibroblasts,8,9 but its possible function in cardiac fibrosis is unknown. Cardiac fibrosis is a histologic hallmark of DCM and surpasses LVEF as a prognostic marker in heart failure.10 Therefore, in follow-up of the GWAS, we performed extensive functional genomic studies which demonstrated that CDCP1 plays a role in regulating human cardiac fibroblast (HCF) proliferation through PDGF signaling and mitosis. Finally, RNA-seq analysis after CDCP1 knock-down (KD) in HCFs made it possible to identify transcriptome-wide differentially expressed genes, including significant down regulation of the interleukin 1 receptor-like 1 (IL1RL1) gene which encodes sST2, an inductor of heart failure and cardiac fibrosis.11,12 In summary, the application of a discovery GWAS followed by in-depth functional genomic studies—a strategy that we have applied previously with repeated success13–16—identified CDCP1 as a novel target that is associated with myocardial recovery in DCM and which may modulate cardiac fibrosis.
METHODS
Data availability:
The RNA-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number: GSE222695.
DCM population.
Patients were recruited in multiple medical institutions including participant institutions of the Intervention in Myocarditis and Acute Cardiomyopathy (IMAC)-2 study4, the Mayo Clinic, University of Colorado School of Medicine, University of Trieste (Italy), Mario Negri Institute for Pharmacological Research (Italy), CHU de Lille (France)17,18, and St. Anne’s University Hospital (Czech Republic). All subjects had DCM with LVEF ≤ 50% in whom coronary artery disease (CAD) or other secondary causes of HFrEF were excluded and who were diagnosed within 6 months of developing symptoms. The detailed inclusion and exclusion criteria are outlined in Supplementary Figure S1. Patients had echocardiograms performed within 30 days of their diagnosis at baseline and follow-up echocardiograms were performed after a median of 6 months of treatment with angiotensin-converting enzyme (ACE) inhibitors or angiotensin 2 receptor blocker (ARB), and β-blockers. All patients provided written informed consent to have blood samples drawn for DNA extraction and analyses. The study was reviewed and approved by the Mayo Clinic Institutional Review Board (IRB) and the IRBs of the respective participating institutions.
GWAS analysis.
Patient DNA samples were genotyped using Illumina Human 610-Quad BeadChips, as described previously.13–15 Change in LVEF after drug treatment was used as a phenotype. Approximately 7.87 million observed and imputed SNPs were analyzed using PLINK linear regression, with adjustment of co-variates (baseline LVEF, sex, age, time to follow-up echocardiogram and recruiting site). See Supplementary Methods for details.
Fine-mapping of the chromosome 3 SNP signal.
SNP function was annotated using public multi-omics datasets. The expression quantitative trait locus (eQTL) information was obtained from the Gene-Tissue Expression (GTEx) Project19. The datasets for annotation of regulatory DNA elements were obtained from the Encyclopedia of DNA Elements (ENCODE) Project20. Those sequence-based data were visualized using the interactive genome viewer (IGV).21 The sources of these sequencing datasets and other resources used in this study are listed in the Major Resources Table.
SNP/Gene functional studies in HCFs.
See Supplementary Methods for detailed methods. Briefly, the effect of the GWAS-identified SNP locus on gene transcriptional activity was tested by reporter gene assay. Genes of interest were knocked down (KD) in HCFs by siRNA and were overexpressed by cDNA construct transfection. HCF cell proliferation was measured by MTS assays after KD or growth factor treatment. RT-qPCR was used for quantification of mRNA levels and Western blots (WB) for proteins. Immunofluorescence (IF) staining was performed to visualize protein targets in HCFs. RNA sequencing (RNA-seq) was performed for quantification of transcriptome-wide gene expression level in HCFs. CDCP1-related differentially expressed genes (DEGs) were identified by comparing RNA-seq results of control to that of the CDCP1 KD group. Those DEGs were then subjected to pathway analysis for annotation of CDCP1 function.
RESULTS
DCM patient characteristics
A total of 686 patients with recent onset DCM were enrolled in the final GWAS analysis. Their clinical characteristics are outlined in Supplementary Table S1. After a period of pharmacotherapy, follow-up LVEF measurements were obtained at a median time of 6 months, demonstrating an average improvement in LVEF of 43%. LVEF measured at baseline (V1) and after pharmacotherapy (V2) is plotted in Figure 1A, demonstrating a variable myocardial recovery in response to pharmacotherapy. Change in LVEF (V2-V1) in these 686 patients showed a Gaussian distribution (Figure 1B) and was used as the phenotype for the GWAS analysis after adjustment for co-variates.
Figure 1. Myocardial Recovery in Recent Onset DCM Measured by LVEF.
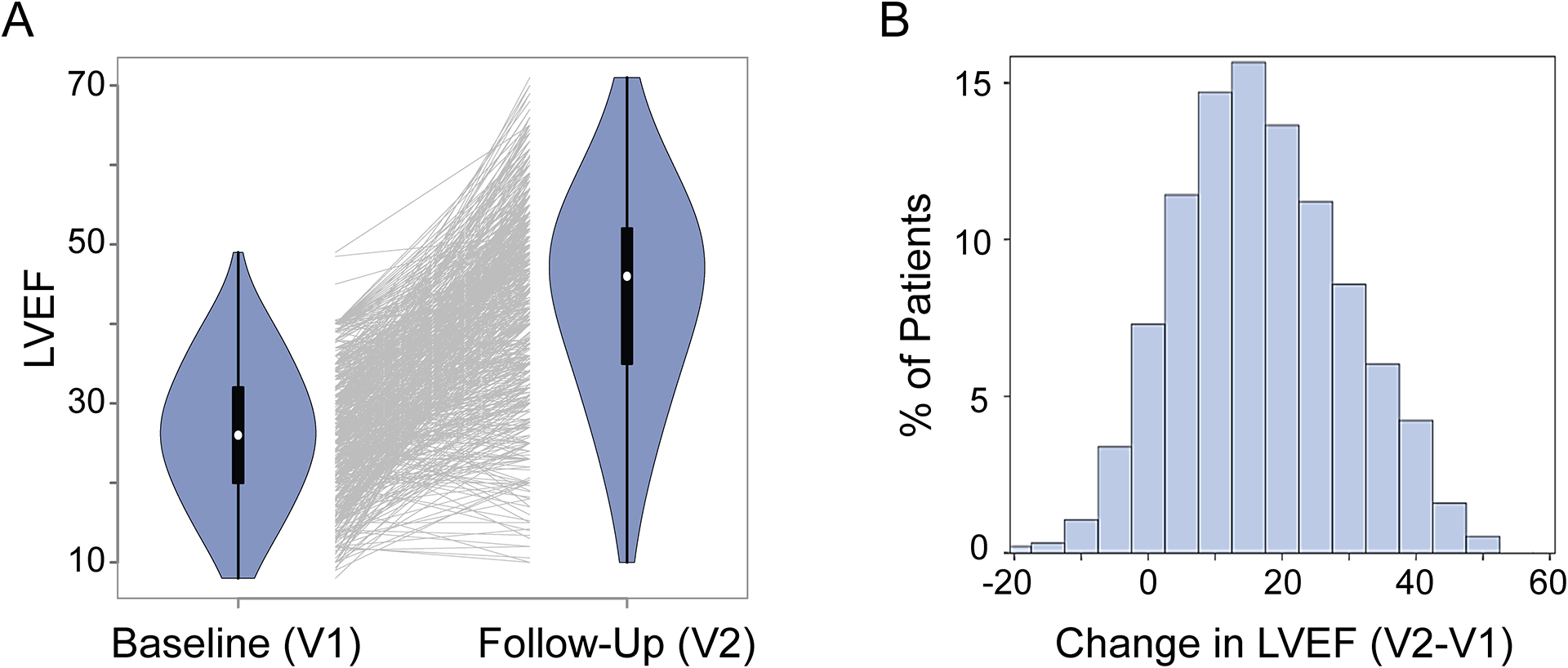
(A) Violin plots that show left ventricle ejection fraction (LVEF) data for 686 DCM patients at baseline (V1) and at follow-up after pharmacotherapy (V2). Each line in the middle of the plot links LVEF data for an individual patient at V1 and V2. Many of patients had their LVEFs increase, but for some patients LVEF values did not change or decreased after pharmacotherapy. (B) Distribution of changes in LVEFs in these 686 patients after pharmacotherapy.
GWAS for changes in LVEF in DCM patients
The Manhattan plot showing SNP associations with changes in LVEF after drug treatment is presented in Figure 2A. The Q-Q plot for the GWAS analysis did not show evidence of genomic inflation (lambda = 0.991) and is shown in the Supplementary Figure S2A. SNPs with suggestive significant associations (P < 10−5) are listed in Supplementary Table S2. Although none of SNPs reached genome-wide significant associations (P < 5.0×10−8), there were two SNPs with lowest P-values which were highly suggestive (P < 10−6) (Figure 2A). A locus zoom plot for the chromosome 3 SNPs showed that the “top” SNP, rs6773435 (G>T), mapped 5’-upstream of TMEM158, and linked SNPs covered a DNA region that included the TMEM158 gene and the 5’-flanking region of a nearby gene, CDCP1 (Figure 2B). The other highly suggestive SNP (rs11105445) on chromosome 12 mapped to an intergenic region (Supplementary Figure S2B). The GWAS analysis also showed that the minor alleles for both the chromosome 3 and chromosome 12 SNPs were associated with positive BETA values (Supplementary Table S2), indicating that the variant alleles for both SNPs were associated with increased LVEF or myocardial recovery after drug treatment. More significant genotype-dependent changes in LVEF were observed when both SNP genotypes were combined, i.e., compared with patients who had “wildtype” genotypes for both SNPs (n=206), patients who had heterozygous genotypes for both SNPs (n=82) were estimated to have 6.07 (95% Confidence Interval: 3.81 to 8.34) units improvement in LVEF (Supplementary Table S3).
Figure 2. GWAS for Changes in LVEF in DCM.
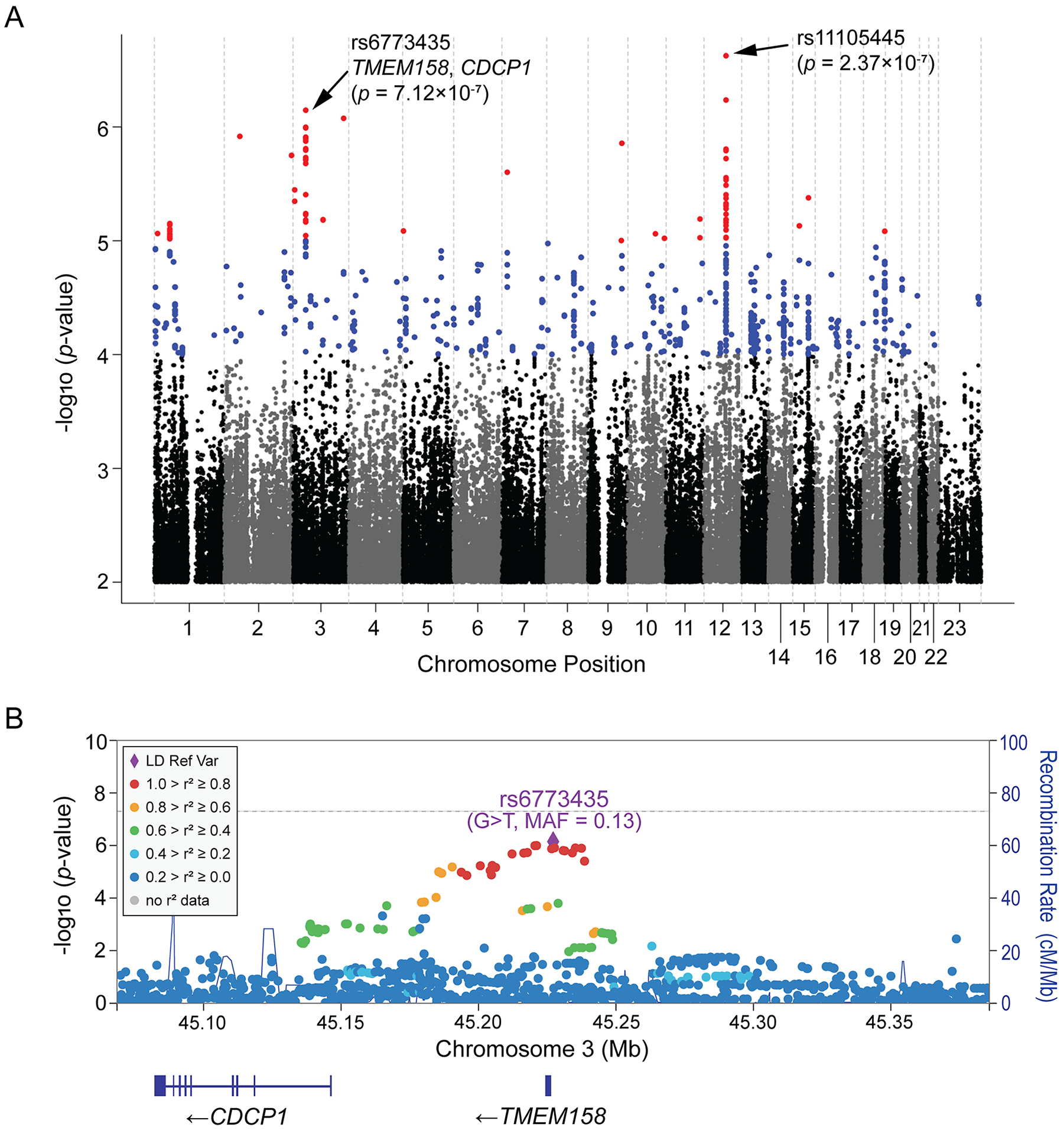
(A) Manhattan plots for the GWAS of changes in LVEFs in 686 DCM patients who received pharmacotherapy. Two “top” SNPs on chromosome 12 and chromosome 3 have been highlighted. (B) Regional association (Locus Zoom) plot for the chromosome 3 SNP signal. The color of each SNP represents its’ linkage disequilibrium (LD) in a European population (the 1000 Genomics Project) with the reference SNP, rs6773435, which is colored purple. MAF = minor allele frequency.
Although the SNP signals identified in the GWAS are highly suggestive while not genome-wide significant, it is well known that the value of biological insight gained from GWAS is not necessarily proportional to the strength of statistical association. This concept has been proven by prior GWAS reported by us13–16 and other groups,22,23 in which SNP loci with highly suggestive associations have revealed important biological mechanisms related to the GWAS phenotype with the pursuit of functional genomic studies. To further validate our GWAS findings, we performed functional genomic studies starting with the chromosome 3 SNP signal since it mapped close to a gene-coding region (Figure 2B). As described subsequently, we found that TMEM158 is a pseudogene, and that its DNA sequence includes an enhancer which influences CDCP1 transcription.
Fine-mapping of the chromosome 3 SNP signal revealed a potential role for CDCP1 in cardiac fibrosis
The chromosome 3 SNP locus, which covers the entire TMEM158 gene (Figure 2B), has been annotated as a candidate cis-regulatory DNA element (cCREs) in heart left ventricular tissue and human cardiac fibroblasts (HCFs) by ENCODE20 (Figure 3A). Specifically, the SNP locus has been annotated as an accessible chromatin site (by the ATAC-seq or DNase-seq data), promoter (by H3K4me3 ChIP-seq), enhancer (by H3K27ac ChIP-seq), and/or chromatin looping site (by CTCF ChIP-seq) (Figure 3A), indicating that this locus might modulate gene transcriptional regulation. The “top” SNP, rs6773435, is an expression quantitative trait locus (eQTL) for both TMEM158 and CDCP1 in several tissues and cell lines19 (Supplementary Figure S3), but whether it might affect gene transcription in human heart cells requires further functional studies.
Figure 3. Functional annotation of the chromosome 3 SNP locus.
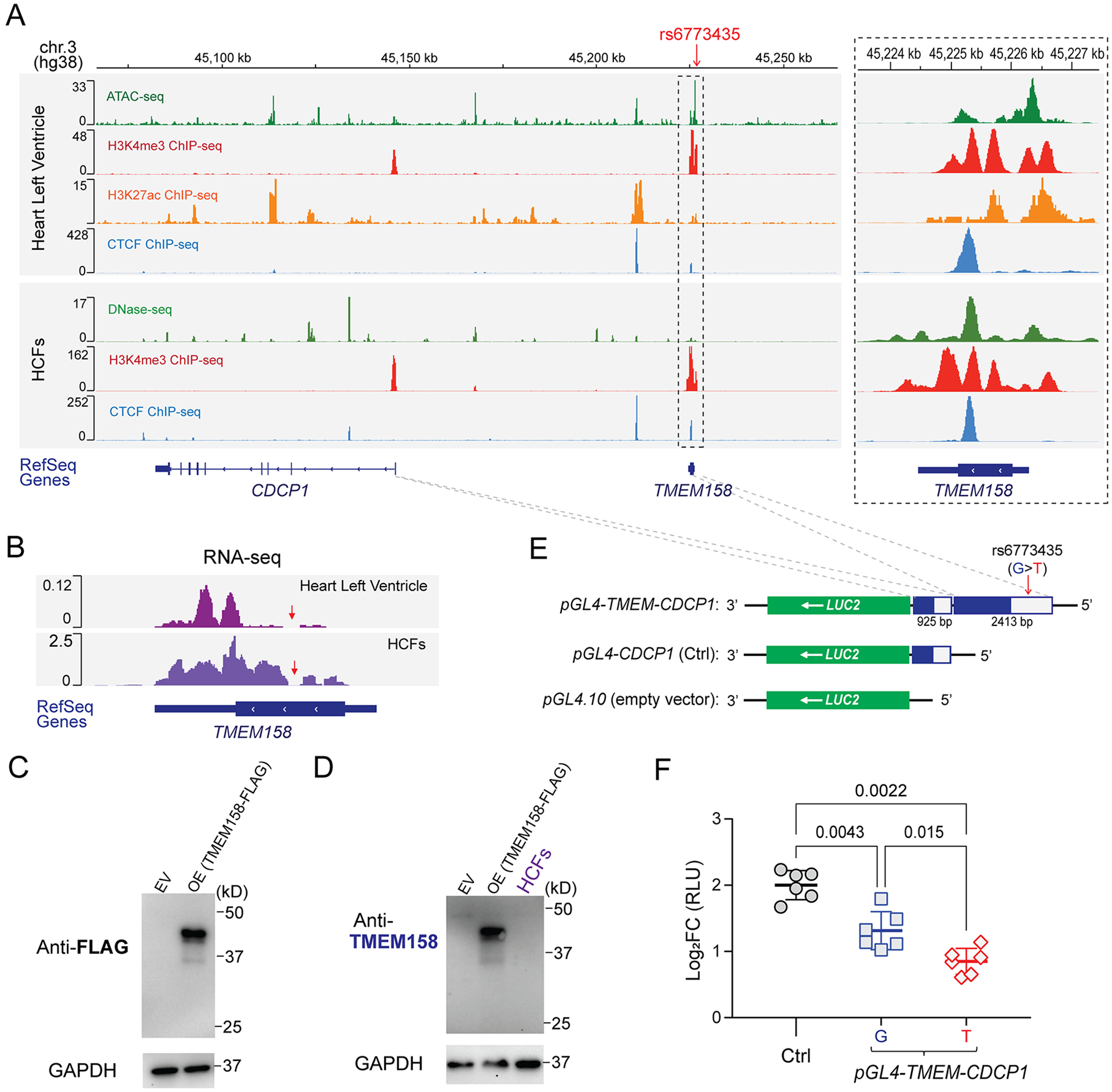
(A) Visualization of the ENCODE epigenomic dataset in the interactive genome viewer (IGV). Panels (from top to bottom) show the physical position of the rs6773435 SNP locus on chromosome 3 based on human genome assembly (hg38); ATAC-seq to assess genome-wide chromatin accessibility, ChIP-seq for H3K4me3 (promoter marker), for H3K27ac (enhancer and promoter marker), and for CTCF (chromatin looping marker) that were generated using human left ventricle tissues; DNase-seq to assess genome-wide chromatin accessibility, ChIP-seq for H3K4me3, and for CTCF that were generated using HCFs. (ATAC-seq and H3K27ac ChIP-seq data are not available for the HCFs). Genes were annotated by the NCBI human genome RefSeq. Both the TMEM158 and CDCP1 genes are transcribed from the negative DNA strand. The dashed box on the right panel is a “zoom-in” of the SNP locus. Sequencing peaks are map to the SNP locus, which indicates that the locus is likely a transcriptional activating site. (B) RNA-seq reads generated from human heart left ventricular tissue and HCFs which map to the TMEM158 gene annotated by RefSeq. The TMEM158 open reading frame (ORF) and untranslated region (UTR) were depicted by thick and thin lines, respectively. Red arrows indicate a region of the TMEM158 ORF where no RNA-seq reads were mapped. (C) Western blots using an anti-FLAG antibody for the putative TMEM158-FLAG fusion protein after overexpression (OE) in HEK-293T cells. Empty vector (EV) control sample was obtained from cells transfected with pCMV-Entry plasmid. (D) Western blot using anti-TMEM158 antibody for endogenous TMEM158 protein in HCFs. Overexpressed TMEM158-FLAG fusion protein sample, as same as which is blotted in (C) with anti-FLAG antibody, was blotted with the anti-TMEM158 antibody, which successfully detects overexpressed TMEM158-FLAG fusion protein. However, no endogenous TMEM158 protein was detected in protein lysates from HCFs. (E) Construction of reporter gene plasmids. The pGL4.10 plasmid which includes a LUC2 reporter gene was used as the backbone construct and served as an empty vector control. The CDCP1 promoter region (925 bp) was cloned into the 5′-end of the LUC2 reporter gene and was used as positive control plasmid (pGL4-CDCP1) for CDCP1 transcriptional activity. To test the effect of the rs6773435 SNP locus on CDCP1 transcription, DNA fragments that included the rs6773435 SNP/TMEM158 locus (2413 bp) were cloned into the 5’-end of the pGL4-CDCP1 (pGL4-TMEM-CDCP1). The pGL4-TMEM-CDCP1 plasmid containing the rs6773435 SNP “G” allele was compared with that containing the “T” allele. (F) Comparison of luciferase activities in HCFs transfected with pGL4.10 (empty vector), pGL4-CDCP1 (Ctrl), pGL4-TMEM-CDCP1 “G” and pGL4-TMEM-CDCP1 “T”. Data are log2 fold change (FC) in relative light units (RLUs) when compared to pGL4.10 alone (empty vector) from biological replicates (n=6) showing as mean values ± SD. Mann-Whitney test was used to calculate the presented p-values.
We first excluded the TMEM158 gene from our functional studies since we found that TMEM158 is a pseudogene which does not encode protein. Specifically, RNA-seq data generated by the GTEx Project19 demonstrated that, even though TMEM158 RNA is expressed in multiple human tissues and cell lines, intact TMEM158 mRNA is not found in any human tissues or cell lines. More specifically, a region of the TMEM158 open reading frame (ORF) has no RNA-seq reads that have been observed in heart left ventricle tissue, in HCFs (Figure 3B, red arrows) or in tissues/cells in which TMEM158 RNA is most highly expressed (Supplementary Figure S4, red arrows). These RNA-seq read mapping results suggest that an intact TMEM158 mRNA does not exist, thus it cannot be translated to protein. We also attempted to detect the putative TMEM158 protein by Western blot. To generate a positive control for the Western blot assay, a “FLAG-tagged” TMEM158 fusion protein was overexpressed and confirmed by anti-FLAG antibody (Figure 3C). Control samples were then loaded with protein lysates from HCFs and blotted with the anti-TMEM158 antibody (Figure 3D). As expected, no TMEM158 protein was detectable in HCFs (Figure 3D). It is possible that the TMEM158 RNA is an enhancer RNA (eRNA) which is transcribed at active enhancers24 since the entire TMEM158 locus has been annotated as enhancers (Figure 3A). As a result of these observations, we focused our attention on the CDCP1 gene and tested the hypothesis that its expression might be regulated by the “TMEM158” and rs6773435 SNP locus.
To determine whether the rs6773435 SNP (G>T) might affect CDCP1 transcription in HCFs, DNA fragments containing that SNP locus, and the CDCP1 promotor were cloned into the pGL4.10 luciferase reporter gene plasmid (Figure 3E). A construct with the CDCP1 promotor was cloned and served as control. Those plasmids were then transfected into HCFs, and luciferase activity was quantified and normalized to that of the pGL4.10 plasmid (empty vector). Increased luciferase activity was observed in HCFs transfected with those plasmids when compared to empty vector (Figure 3F, log2FC > 0). The control plasmid which contained only the CDCP1 promoter displayed the greatest increase in luciferase activity compared with plasmids containing the rs6773435 SNP locus, suggesting that the SNP locus negatively regulates CDCP1 transcription. Importantly, we also observed that plasmids containing the rs6773435 SNP “T” allele resulted in smaller increases in luciferase activity (Figure 3F), indicating that the variant “T” allele may result in less CDCP1 transcription as compared to the common “G” allele.
CDCP1 encodes a single-pass transmembrane glycoprotein, CUB domain-containing protein 1, which has been studied extensively in cancer25–30 and immune-related diseases.31–33 We reviewed the expression of CDCP1 in various tissues included in the GTEx portal.19 The results indicated that CDCP1 had minimal expression in myocardial tissue but is expressed in cultured fibroblasts (Supplementary Figure S5). CDCP1 has also been reported to modulate pulmonary fibrosis,8 but its role in cardiac fibrosis is unknown. We also consulted the phenotype-wide association study (PheWAS) based on the UK Biobank datasets (https://pheweb.org/UKB-Neale/) which showed that genetic variants in/near the CDCP1 gene have been significantly associated with heart failure mortality (Supplementary Figure S6), and with death due to thoracic aneurysm rupture (Supplementary Figure S7). In summary, this series of studies of the chromosome 3 SNP signal formed the basis for our investigation of the role of CDCP1 in myocardial fibrosis and recovery.
CDCP1 is required for human cardiac fibroblast proliferation
CDCP1 expression is known to be significantly up-regulated in a variety of cancers and appears to drive cancer cell growth and metastasis.25–30 Inspired by the molecular function of CDCP1 in cancer, we set out to determine whether CDCP1 might affect HCF cell growth. CDCP1 KD in primary HCFs (Figure 4A left, 4B) can significantly suppress cell proliferation (Figure 4C). We also demonstrated that TMEM158 KD (Figure 4A right) had no effect on HCF proliferation (Figure 4C), likely because TMEM158 protein is not translated from its RNA (Figure 3B, 3D). To further validate this observation, we performed CDCP1 overexpression (OE) experiments in HCFs and found that it promotes HCF proliferation (Supplementary Figure S8), an expected opposite direction observed in the CDCP1 KD experiments, confirming the role of CDCP1 in HCF proliferation.
Figure 4. CDCP1 is required for human cardiac fibroblast (HCF) proliferation.
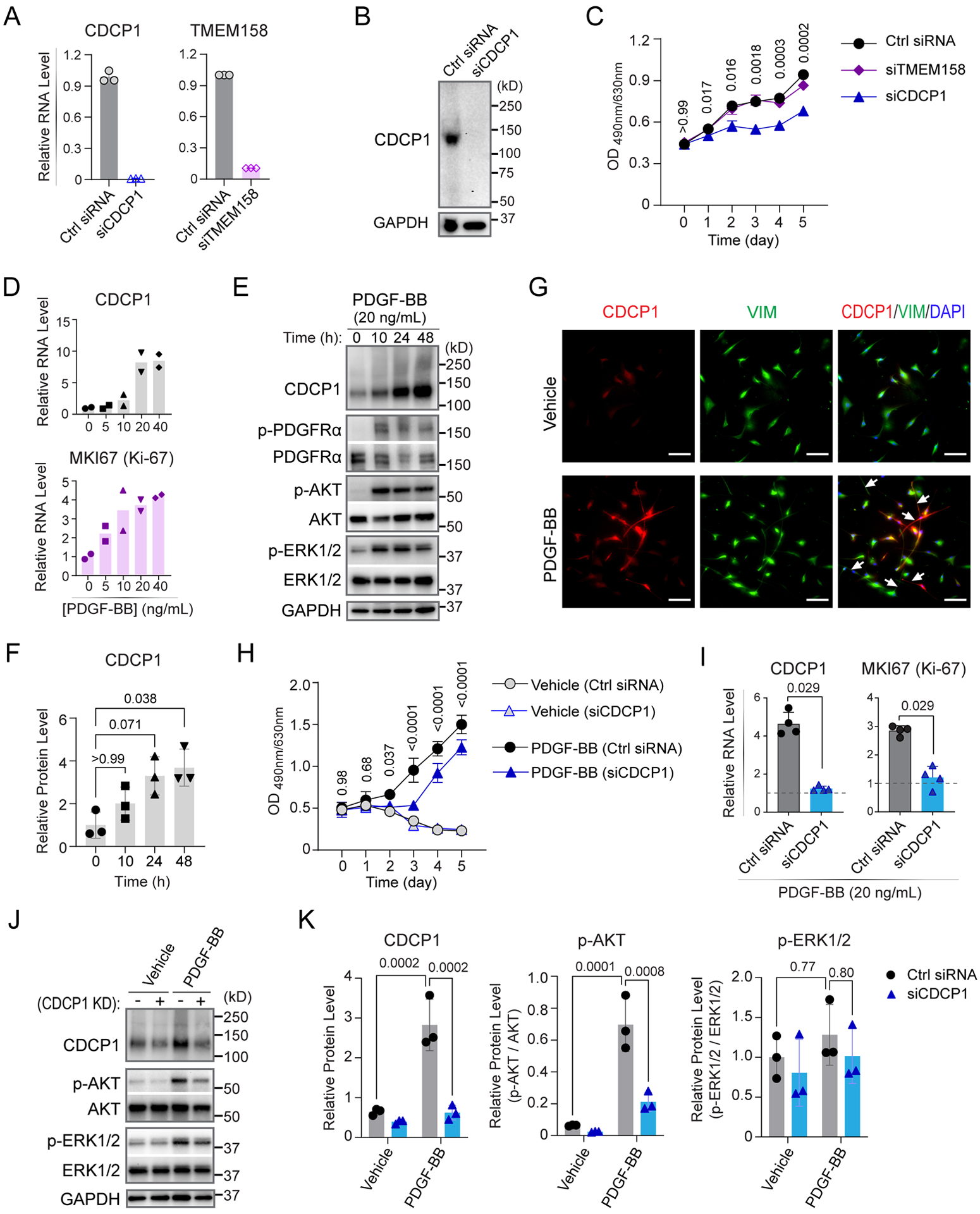
(A) RT-qPCR quantification of CDCP1 and TMEM158 mRNA levels in HCFs 48 hours after knock-down by specific siRNAs. GAPDH mRNA level were quantified as internal control. Data showing are triplicate quantification of same samples. (B) Western blot assay for CDCP1 in HCFs transfected with CDCP1 siRNA (siCDCP1). GAPDH was blotted as an internal control. (C) HCF cell proliferation assays. Cells were transfected with siRNAs at day 0 and cell viability was measured by an MTS assay every 24 hours until day 5. Each dot is a mean value for three independent experiments (n=3). Error bars represent standard deviations. Statistical analysis by 2-way ANOVA with Tukey’s multiple comparisons test. P-values are shown for comparisons of Ctrl siRNA to siCDCP1 groups. (D) CDCP1 and MKI67 mRNA levels were quantified by RT-qPCR in HCFs after 24-hour treatments with PDGF-BB at different concentrations. Plots are showing duplicate RT-qPCR assays using GAPDH as internal control. (E) Western blot assay for CDCP1 in HCFs incubated with 20 ng/mL of PDGF-BB at different time points. Phosphorylated PDGFRα (p-PDGFRα) was blotted as control of PDGF-BB treatment. Two known PDGFR downstream signaling proteins, phosphorylated AKT (p-AKT) and phosphorylated ERK1/2 (p-ERK1/2) were also blotted. GAPDH was blotted as internal control. (F) CDCP1 protein level quantified from independent Western blot experiments (n=3) as represented in (E). CDCP1 bands were normalize to that of GAPDH, and then control treatment (0 h). P-values were calculated by Kruskal-Wallis test with Dunn’s multiple comparisons to 0 h of PDGF-BB treatment. (G) Immunofluorescent staining of CDCP1 and HCF marker, vimentin (VIM), in HCFs after 24-hour treatments with vehicle or PDGF-BB. HCFs morphology was changed with long “fibers” (arrow heads) can be seen after PDGF-BB treatment. Scale bars represent 200 μm. (H) HCF cell proliferation in “starving” (serum-deprived) media supplied without (Vehicle) or with 20 ng/mL of PDGF-BB. Cells were transfected with siRNAs at day 0 and cell viability was measured by an MTS assay every 24 hours until day 5. Each dot is a mean value for three independent assays. Error bars represent standard deviation. Statistical analysis by 2-way ANOVA with Tukey’s multiple comparisons test. P-values are shown for comparisons of Ctrl siRNA to siCDCP1 in the PDGF-BB treatment groups. (I) CDCP1 and MKI67 mRNA levels were quantified by RT-qPCR in HCFs 48-hour after CDCP1 knock-down and PDGF-BB treatment, with GAPDH mRNA level as internal control. RNA levels were normalized to vehicle treatment (dashed line). Data are mean values ± S.D. for independent experiments (n=4). Mann-Whitney test was used to calculate the presented p-values. (J) Western blot assay for CDCP1 and PDGFR downstream signaling proteins (p-AKT and p-ERK1/2) in HCFs with CDCP1 knock-down (KD) and 20 ng/mL of PDGF-BB treatment. GAPDH was blotted as internal control. (K) Protein levels quantified from three independent samples by Western blot assays as represented in (J). Protein bands were normalized to that of GAPDH. Levels of p-AKT and p-ERK1/2 were showed as ratios to their total protein level. P-values were calculated by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
We also noted that baseline CDCP1 expression is low in primary HCFs, e.g., based on the RT-qPCR assay, CDCP1 mRNA level is about 0.2~2% of GAPDH and it required a significantly longer exposure time to see CDCP1 than GAPDH bands in Western blots of HCFs (Figure 4B). However, CDCP1 is known to be induced by activation of platelet-derived growth factor (PDGF) signaling in cancer cells27 and PDGF signaling is also known to induce cardiac fibrosis.34–37 To determine whether CDCP1 might also be induced in HCFs, those cells were “starved” in serum-deprived media and were then treated with platelet-derived growth factor subunit B homodimer (PDGF-BB) which activates both PDGF receptors (PDGFR) α and β. We found that the mRNA levels of CDCP1, as well as the marker of proliferation Ki-67 (MKI67), were significantly induced in HCFs after PDGF-BB treatment in a dose dependent manner (Figure 4D). CDCP1 protein was also increased after PDGF-BB treatment (20 ng/mL) in a time dependent manner, with a significantly higher CDCP1 protein level observed after 48 hours of treatment (Figures 4E, 4F). As expected, phosphorylated PDGFRα (p-PDGFRα), a marker of PDGF signaling activation, as well as downstream PDGF signaling proteins, phosphorylated AKT (p-AKT) and ERK1/2 (p-ERK1/2), were significantly up-regulated after PDGF-BB treatment (Figure 4E). CDCP1 induction by PDGF-BB was also visualized in HCFs by co-immunostaining with the fibroblast marker, vimentin (VIM), which showed that longer “fibers” were stretched out from HCFs after PDGF-BB treatment (Figure 4G). Meanwhile, proliferation of HCFs cultured in serum-deprived media was significantly promoted by PDGF-BB treatment, compared with vehicle treatment which, as expected, did not proliferate due to lack of growth factors in serum-deprived media (Figure 4H). Of importance, KD of CDCP1 repressed PDGF-BB stimulated HCF proliferation (Figure 4H), as well as the transcription of MKI67 (Figure 4I), supporting the hypothesis that CDCP1 induction is required for HCF proliferation stimulated by PDGF-BB. We also found that CDCP1 KD significantly decreases p-AKT, which promotes cell proliferation and cell cycle38 in HCFs with or without PDGF-BB treatment (Figures 4J, 4K), suggesting that the decreased HCF proliferation after CDCP1 KD could be linked to attenuated AKT phosphorylation.
In addition to proliferation, cardiac fibroblast-to-myofibroblast transdifferentiation is an important step in the development of cardiac fibrosis.34–36 After HCFs were treated with PDGF-BB, there was no change in expression of ACTA2 which encodes alpha-smooth muscle actin (α-SMA, a marker of myofibroblasts) (Supplementary Figure S9A), indicating that PDGF-BB treatment has little or no effect on myofibroblast transdifferentiation. Consistent with this finding, CDCP1 KD had no effect on α-SMA expression after PDGF-BB treatment (Supplementary Figure S9B–9D), despite significant repression of MKI67 (Figure 4I), indicating that CDCP1 plays an important role in HCF proliferation but it may not influence cardiac myofibroblast transdifferentiation.
CDCP1 has a limited role in TGF-β1-induced cardiac fibroblast-to-myofibroblast transdifferentiation
Fibroblast-to-myofibroblast transdifferentiation is well-known to be stimulated by transforming growth factor beta (TGF-β) signaling.34,35 To further investigate whether CDCP1 plays a role in TGF-β1-induced cardiac fibroblast-to-myofibroblast transdifferentiation, HCFs were stimulated by TGF-β1 in serial concentrations (Figure 5A) or time courses (Figure 5B). ACTA2 expression, as expected, increased after TGF-β1 treatment while CDCP1 expression, as well as that of MKI67, were decreased (Figure 5A, 5B). The decrease in CDCP1 expression after TGF-β1 treatment has also been observed in human lung fibroblasts.8 This decrease in CDCP1 expression is most likely a result of repressed cell proliferation, as indicated by decreased MKI67 expression, which would be expected when transdifferentiation occurs after TGF-β1 treatment. It is known that myofibroblast transdifferentiation stops fibroblast proliferation, e.g. the marker of myofibroblast, α-SMA, is also named “cell growth-inhibiting gene 46 protein”. Finally, CDCP1 KD had no effect on the protein expression of phosphorylated SMAD2 (p-SMAD2), α-SMA, COL1A1, FN-1, COL5A1, and MMP2 (matrix metallopeptidase 2), or the percentage of α-SMA positive (α-SMA+) cells, which were significantly upregulated by 48-hour of TGF-β1 treatment (Supplementary Figure S10), indicating that CDCP1 has no effect on TGF-β1-mediated myofibroblast transdifferentiation.
Figure 5. CDCP1 and TGF-β1-induced cardiac fibroblast-to-myofibroblast transdifferentiation.
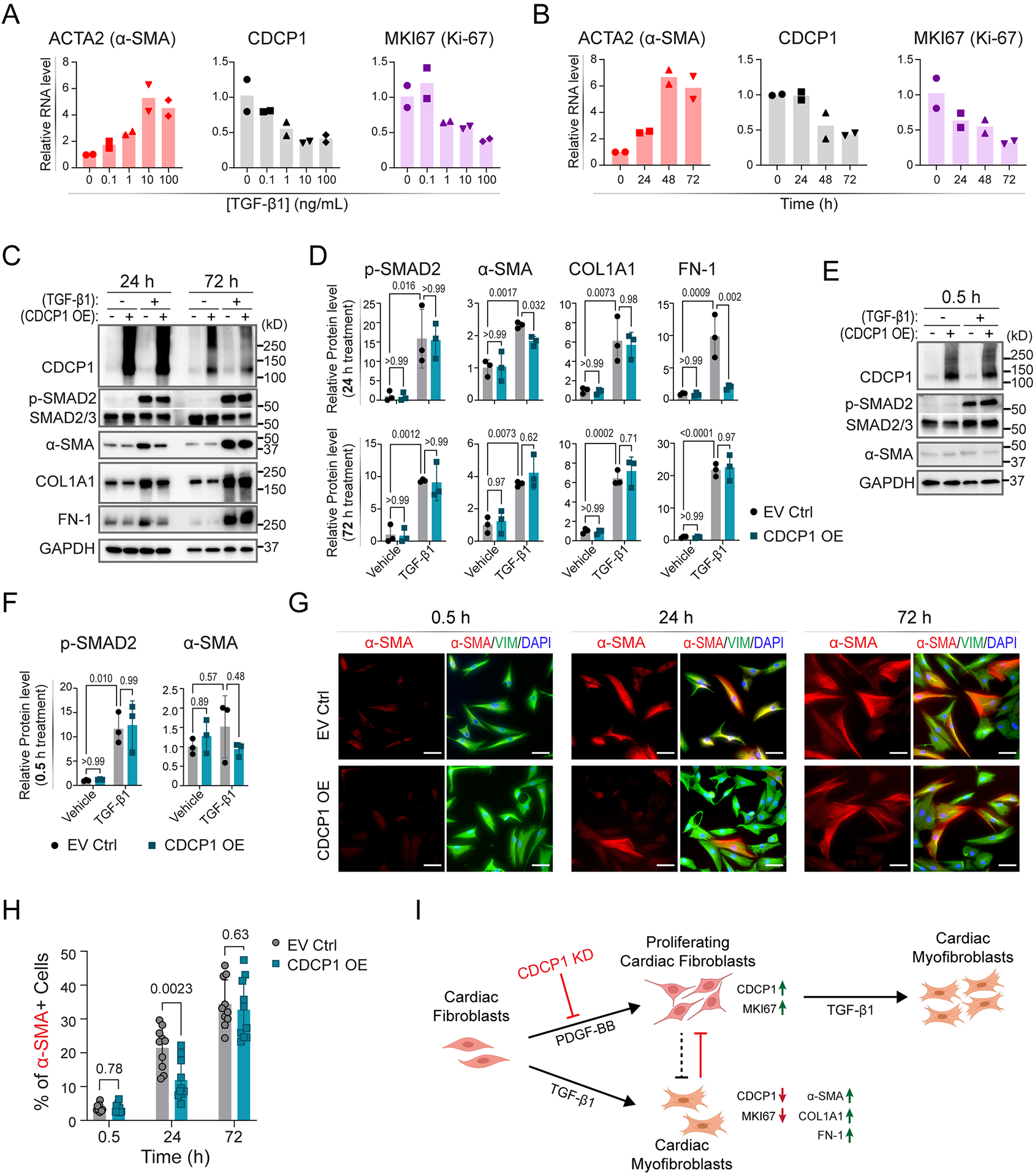
(A) Relative mRNA levels of ACTA2, CDCP1 and MKI67 in HCFs after 48-hour treatment of TGF-β1 at different concentrations, and (B) after 10 ng/mL of TGF-β1 treatment at different time (hours). RNA levels were quantified by RT-qPCR with GAPDH level as internal control from duplicate assays. (C) Western blot for CDCP1 and other protein markers in HCFs with or without CDCP1 overexpression (OE), and with or without TGF-β1 treatment (10 ng/mL) at two time points (24 and 72 hours of treatment). Phosphorylated SMAD2 (p-SMAD2) and α-SMA were blotted for control of TGF-β1 treatment and myofibroblast transdifferentiation, respectively. Extracellular matrix (ECM) proteins including collagen type I alpha 1 chain (COL1A1), and fibronectin 1 (FN-1) were also blotted. GAPDH were blotted as internal control. (D) Relative protein level quantified from three independent samples by Western blot assays as represented in (C). Protein levels were normalized to GAPDH level and then to vehicle and empty vector control (EV Ctrl) sample. Error bars represent standard deviations of triplicate assays. P-values were calculated by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (E) Western blot for CDCP1, p-SMAD2 and α-SMA in HCFs with or without CDCP1 overexpression (OE), and with or without half hour of TGF-β1 treatment (10 ng/mL). (F) Relative protein level quantified from three independent samples by Western blot assays as represented in (E). Protein levels were normalized to GAPDH level and then to vehicle and empty vector control (EV Ctrl) sample. Error bars represent standard deviations of triplicate assays. P-values were calculated by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (G) Immunofluorescent (IF) staining of α-SMA and vimentin (VIM) in HCFs transfected with empty vector (EV Ctrl) and CDCP1 cDNA plasmid (CDCP1 OE) and TGF-β1 treatment (10 ng/mL) at 0.5, 24 and 72 hours. Scale bars represent 100 μm. (H) Percentage of α-SMA positive (α-SMA+) cells based on the IF staining as represented in (G). Total cell number was counted by DAPI staining. For each condition, α-SMA+ cells were counted from 10 different fields in triplicate independent wells. P-values were calculated by multiple unpaired t tests. (I) CDCP1 function in cardiac fibroblasts. PDGF-BB stimulates cardiac fibroblasts proliferation, as well as upregulation of the Maker of Proliferation Ki-67 (MKI67). Expression of CDCP1, was upregulated in PDGF-BB-stimulated proliferating cardiac fibroblasts. TGF-β1 stimulates cardiac fibroblasts-to-myofibroblasts transdifferentiation which, meanwhile, stops cardiac fibroblasts proliferation, as indicated by downregulation of MKI67 and upregulation of α-SMA (also known as Cell Growth-Inhibiting Gene 46 Protein) in cardiac myofibroblasts. Extracellular matrix genes, including COL1A1 and FN-1, were up-regulated in myofibroblasts. CDCP1 knock-down (KD) inhibits cardiac fibroblasts cell proliferation which could lead to less transdifferentiated myofibroblasts.
Because CDCP1 baseline expression in HCFs is relatively low and is decreased after TGF-β1 treatment, we performed CDCP1 overexpression (OE) experiments in HCFs to validate observations made with CDCP1 KD and TGF-β1 treatment (Supplementary Figure S10). To capture a dynamic change in fibroblast-to-myofibroblast transdifferentiation, protein samples were collected from HCFs with CDCP1 OE at both 24- and 72-hours (h) of TGF-β1 treatment. As expected, protein levels of p-SMAD2, α-SMA, COL1A1, and FN-1 were significantly up-regulated in those cells with 24 h of TGF-β1 treatment (Figure 5C). When comparing the CDCP1 OE to empty vector (EV) control groups, a small decrease in α-SMA and FN-1 protein levels was observed in CDCP1 OE cells at 24h but not after 72h of TGF-β1 treatment (Figure 5C, 5D). However, the p-SMAD2 level, a marker of TGF-β signaling activation, did not differ from CDCP1 OE to EV control groups at both 24h and 72h of TGF-β1 treatment, suggesting that CDCP1 has no effect on TGF-β signaling activation.
Phosphorylation of SMAD2/3 often responds immediately to TGF-β stimulation. To further investigate whether CDCP1 affects TGF-β1 signaling in HCFs, cells with CDCP1 OE were treated with TGF-β1 for only 0.5 hour which is sufficient to induce SMAD2 phosphorylation (Figure 5E). However, CDCP1 OE had no effect on p-SMAD2 levels induced at 0.5 hour by TGF-β1 treatment (Figure 5E, 5F), a result that once again supports the observation that CDCP1 has no effect on TGF-β1 signaling. No induction of α-SMA was observed within this time period (0.5 hour) of TGF-β1 treatment (Figure 5E, 5F).
Finally, cells with CDCP1 OE were co-immunostained with α-SMA and vimentin, and the number of α-SMA+ cells was counted. Consistent with the Western blot result, the percentage of α-SMA+ cells in the CDCP1 OE group was less at 24-hour but did not differ after 72-hours of TGF-β1 treatment (Figure 5G, 5H). These observed decreases in α-SMA level and in percentage of α-SMA+ cells at 24h of TGF-β1 treatment in the CDCP1 OE group are likely a result of CDCP1 OE-induced HCF proliferation (Supplementary Figure S8) that precedes and hence delays myofibroblast transdifferentiation.
In summary, we demonstrated that CDCP1 is significantly induced in HCFs after PDGF-BB treatment and is required for HCF proliferation (Figures 4, 5I) but that it might have little or no effect on cardiac fibroblast-to-myofibroblast transdifferentiation stimulated by TGF-β1 (Figure 5).
Transcriptomic profiling of CDCP1 knock-down in HCFs revealed its role in regulation of sST2 expression
To further investigate CDCP1 molecular mechanism(s), we performed RNA-seq assays after the knock-down (KD) of CDCP1 in HCFs. Transcriptome-wide differentially expressed genes (DEGs) were identified (Figures 6A), followed by pathway enrichment analyses using those DEGs (Figures 6B). A total of 1,061 DEGs with fold changes (FC) of more than 2 (|Log2FC|>1.0, FDR<0.05) after CDCP1 KD were identified (Supplementary Table S4). When those DEGs were subjected to Gene Ontology (GO) pathway enrichment analysis, as expected, “regulation of cell population proliferation” was identified as one of the “top” Biological Process pathways (Figures 6B), a pathway that helps explain our observation that CDCP1 is required for HCF proliferation (Figure 4). Consistently, pathways related to cell cycle and mitosis, which play important roles during cell proliferation, were also enriched by CDCP1-mediated DEGs (Figures 6B). The GO enrichments for Molecular Function and Cellar Component pathways support a role for CDCP1 in the regulation of mitosis, e.g., the “top” enriched Molecular Function is “microtubule binding” and the “top” enriched Cellular Component is “spindle” (Supplementary Figures S11).
Figure 6. Transcriptome profiling of CDCP1 knock-down (KD) in HCFs.
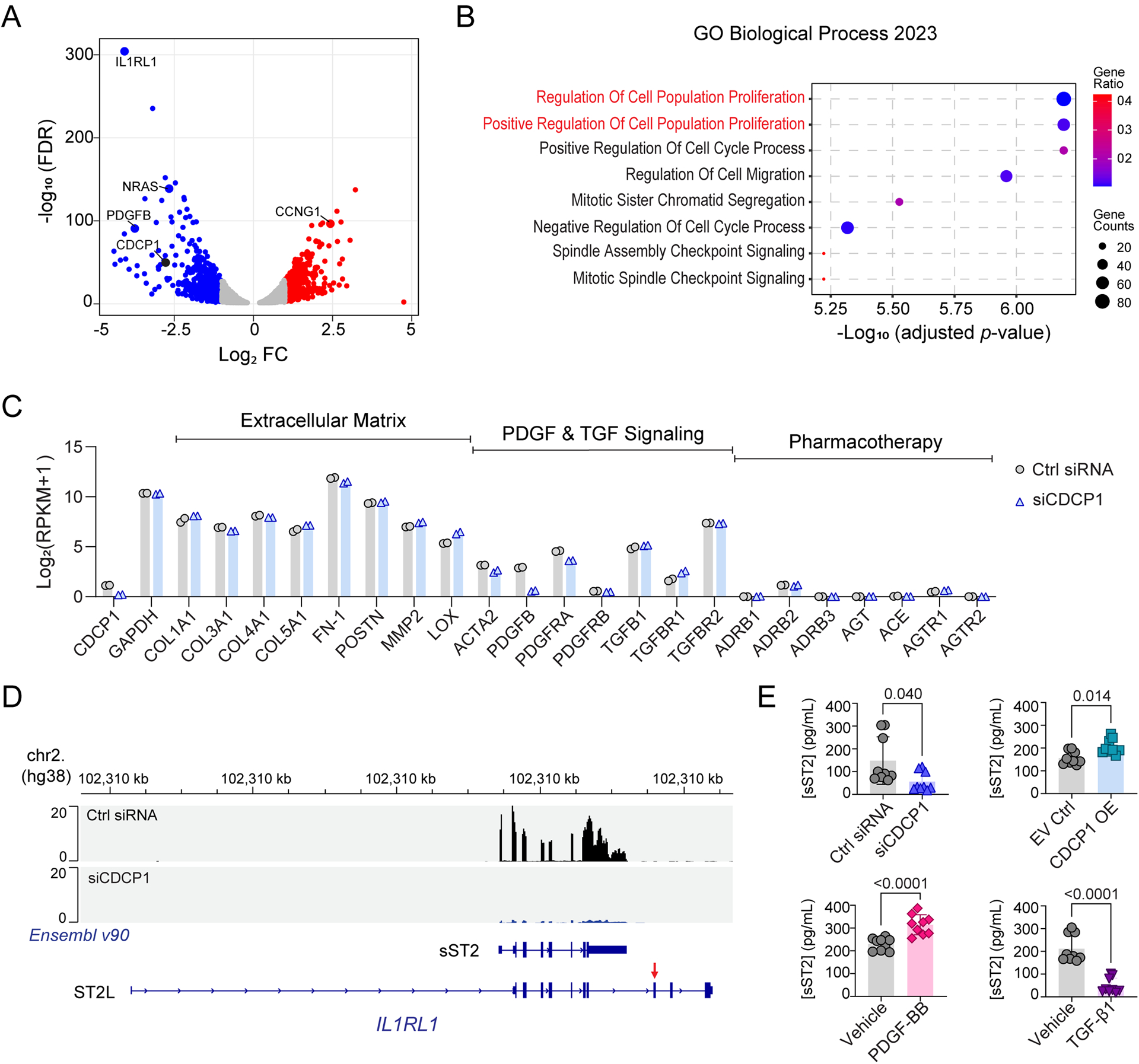
(A) Volcano plot for RNA-seq identified differentially expressed genes (DEGs) in HCFs after CDCP1 KD. The X-axis is log2 fold change (FC) in RNA level when comparing the HCFs transfected with CDCP1 siRNAs (siCDCP1) to the non-target control siRNAs (Ctrl siRNA). The Y-axis is −log10 false discovery rate (FDR) calculated from duplicates. Two biological replicate RNA samples from each experiment groups were sequenced. Each dot represents a gene quantified by the RNA-seq. (B) Top pathways enriched by DEGs after CDCP1 KD (FC > 2.0; FDR < 0.05) in the Gene Ontology (GO) enrichment analysis of Biological Process. The p-value for pathway enrichment was computed from the Fisher exact test and adjusted by using the Benjamini-Hochberg method for correction for multiple hypotheses testing. (C) Gene expression level in HCFs quantified by RNA-seq. RNA level of CDCP1, GAPDH, and genes function in extracellular matrix, PDGF and TGF signaling, and genes involved in the mechanism of actions of standard DCM pharmacotherapy were plotted. RPKM = Reads Per Kilobase of transcript, per Million mapped reads. (D) RNA-seq reads mapping to the IL1RL1 gene in HCFs. Panels (from top to bottom) are physical position of the IL1RL1 gene on chromosome 2 based on the human genome assembly hg38; RNA-seq reads mapped to the IL1RL1 gene in HCFs transfected with non-targeting control (Ctrl siRNA) and CDCP1 siRNA (siCDCP1); Two Ensembl (v90) annotated IL1RL1 transcript variants which encode soluble ST2 (sST2) and membrane-bound ST2 (ST2L). The red arrow indicates the exon which encodes the transmembrane domain of ST2L. RNA-seq reads mapped to the IL1RL1 exons which encode sST2, and those RNA-seq reads are significantly less in CDCP1 KD (siCDCP1) sample. (E) sST2 protein levels in HCF culture media quantified by ELISA assay. HCFs were transfected with siCDCP1 for KD (up left), CDCP1 cDNA plasmid for OE (up right), and treated with 20 ng/mL of PDGF-BB (down left), or 10 ng/uL of TGF-β1 (down right) for 48, 72 and 96 hours. For each group, three biological replicates were quantified at three time points (n=9). Data are presented as mean values ± S.D. Mann-Whitney test was used to calculate the presented p-values.
In addition to pathway enrichment analysis, our RNA-seq data made it possible to identify specific genes for which expression was affected by CDCP1 KD in HCFs. One of the significantly down-regulated genes was NRAS (Figures 6A) which regulates mitosis and cell proliferation through the Ras-Raf-MEK-ERK signaling pathway.39 KD of CDCP1 also led to an increase in the expression of CCNG1 (Figure 6A), which encodes a cell cyclin protein that is associated with G2/M phase arrest and the inhibition of cell proliferation.40 Interestingly, PDGFB expression was also significantly decreased in HCFs after CDCP1 KD (Figure 6A), a result suggesting a positive feedback loop between CDCP1 and PDGF signaling since PDGF-BB treatment induces CDCP1 expression in HCFs (Figure 4). These RNA-seq results provided more evidence that CDCP1 play an important role in PDGF-BB induced HCF proliferation.
The RNA-seq data also made it possible to compare the expression level of genes of interest in HCFs, e.g., baseline expression of CDCP1 is relatively low compared to that of genes involved in extracellular matrix deposition (Figure 6C). CDCP1 KD did not affect expression of those extracellular matrix genes as well as TGF-β signaling genes, including TGFB1, TGFBR1, and TGFBR2 (Figure 6C), results which support the conclusion that CDCP1 has no direct impact on TGF-β1 signaling (Figure 5). PDGFRA expression was higher than PDGFRB which was minimally expressed in HCFs (Figure 6C), suggesting a dominant role of PDGFRα in PDGF-BB stimulated HCF proliferation (Figure 4). In addition, genes that encode standard DCM drug targets such as ADRB1 (adrenoceptor β1), ACE (angiotensin I converting enzyme) and ATGR1 (angiotensin II receptor type 1) were not or only very minimally expressed in HCFs (Figure 6C), which explains the absence of a change in CDCP1 expression in HCFs when exposed to standard DCM drugs (Supplementary Figure S12).
Finally, the most significantly down-regulated gene after CDCP1 KD was IL1RL1 (Figure 6A), which encodes both membrane-bound ST2 (ST2L) and soluble ST2 (sST2), dependent on different IL1RL1 transcript variants (Figures 6D). Our RNA-seq data demonstrated that, in HCFs, IL1RL1 was transcribed to the mRNA variant which encodes sST2, e.g., RNA-seq reads mapped to the IL1RL1 transcript variant which encodes sST2 but not ST2L (Figure 6D). This observation was validated by CDCP1 OE which resulted in a significant increase in IL1RL1 as well as PDGFB mRNA levels in HCFs after 48 hours of CDCP1 cDNA transfection (Supplementary Figure S13). A significant decrease or increase in secreted sST2 protein levels in HCF culture media was also observed after CDCP1 KD or OE, respectively (Figure 6E, up panel). The sST2 level in HCF media was tightly associated with CDCP1 expression, i.e., it was significantly increased with PDGF-BB treatment (Figure 6E, down-left) which up-regulates CDCP1, and it was significantly decreased with TGF-β1 treatment (Figure 6E, down-right) which represses CDCP1. sST2 is a well-established prognostic biomarker for heart failure.11,12 Specifically, elevated circulating sST2 level is associated with poor prognosis and is considered to be an inductor of heart failure and collagen synthesis.12 These results indicate that KD of CDCP1, which leads to a decrease in sST2 expression in HCFs, may have anti-fibrotic and protective roles in myocardial recovery. This result is also consistent with our GWAS result that the rs6773435 SNP variant allele was associated with decreased CDCP1 expression and improved cardiac function.
DISCUSSION
DCM is a major cause of heart failure and drug treatment response among individual patients is highly variable (Figure 1). In an attempt to discover genetic marker(s) that might contribute to this variable response, we applied a stepwise strategy that began with a discovery GWAS for changes in LVEF in 686 patients with recent onset DCM. That GWAS identified a “top” SNP, rs6673435 (G>T), mapping to the 5’-flanking region of the CDCP1 gene on chromosome 3 (Figure 2). The rs6673435 SNP variant allele, “T”, was associated with improved LVEF in those DCM patients (Supplementary Table S2). We then performed in-depth functional genomic studies to demonstrate that the “T” allele was associated with decreased CDCP1 transcription in HCFs (Figure 3E), and that CDCP1 expression was required for HCF proliferation (Figure 4) but not for fibroblast-to-myofibroblast transdifferentiation (Figure 5). KD of CDCP1 resulted in a significant downregulation of sST2 (Figure 6), elevated levels of which are associated with increased heart failure mortality and cardiac fibrosis. In summary, the application of this strategy supported the conclusion that reduced CDCP1 expression may contribute to myocardial recovery in DCM by attenuating cardiac fibrosis.
Cardiac fibroblast migration to and proliferation at injury sites is an initial step in response to myocardial injury.34–36 This process can be stimulated by PDGFs secreted from platelets which accumulate at these injury sites. Those cardiac fibroblasts are then transdifferentiated to myofibroblasts leading to ECM deposition and cardiac fibrosis.34–36 We demonstrated that CDCP1 KD represses HCF proliferation induced by PDGF-BB treatment (Figures 4, 5I). Those heart-resident proliferating fibroblasts are the major origin of cardiac myofibroblasts which secrete extracellular matrix proteins,41,42 resulting in interstitial myocardial fibrosis, a histologic hallmark of DCM.43 Cardiac fibrosis impedes transmission of force and mechano-electric coupling of cardiomyocytes, decreases myocardial oxygen diffusion, impairs coronary flow reserve by perivascular fibrosis and by paracrine mechanisms which cause myocardial dysfunction.44,45 The presence and extent of cardiac fibrosis assessed by magnetic resonance imaging, after adjustment for LVEF and other prognostic factors, is independently and incrementally associated with cardiovascular mortality in DCM46. In fact, cardiac fibrosis surpasses LVEF as a prognostic marker in heart failure.10 Importantly, the extent of cardiac fibrosis, that may be reduced with decreased HCF proliferation as a result of decreased CDCP1 expression (Figure 4), predicts the extent of myocardial recovery in DCM,47,48 and absence of cardiac fibrosis is a strong and independent predictor of improvement in LVEF and LV end-diastolic volumes in DCM patients irrespective of severity of symptoms and initial LVEF.49
Standard heart failure therapy was used in this study population (Supplementary Table S1). Although current pharmacotherapy may also have a beneficial effect by inhibiting fibroblast activation and collagen deposition,50,51 this effect is unlikely to be mediated by its direct action on cardiac fibroblasts since those drug-targeting genes are not expressed in HCFs (Figure 6C). Unlike current pharmacotherapy, directly targeting cardiac fibroblasts by chimeric antigen receptor (CAR) T cells has improved heart function in preclinical models and can be a new therapeutic strategy.52 Therefore, the identification of a novel gene associated with clinical myocardial recovery in our GWAS and the demonstration of its role in cardiac fibroblast physiology, cardiac fibrosis and HFrEF could have important implications for the development of new heart failure therapies. CDCP1 expression in normal heart tissue (Supplementary Figure S5) and in HCFs (Figure 6C) is low. However, we have demonstrated that CDCP1 is significantly up-regulated and contributes to HCFs proliferation after activation of PDGF signaling, a profibrotic growth factor that promotes cardiac fibrosis.34–37 The PDGF-BB stimulated HCF proliferation (Figure 4) was predominantly mediated by PDGFRα which is more significantly expressed than PDGFRβ in HCFs (Figure 6C). PDGFRα is essential for cardiac fibroblast survival53 and reduction of PDGFRα positive fibroblasts in murine hearts resulted in improvement in cardiac function after injury.54 We demonstrated that CDCP1 KD reduced HCF proliferation stimulated by PDGF-BB (Figure 4), suggesting a possible role for targeting CDCP1 to modulate cardiac fibrosis and function. An important and relevant finding was that CDCP1 KD significantly reduced sST2 expression (Figure 6). Plasma sST2 level is a well-established prognostic biomarker for heart failure.11,12 HCFs express sST2 but not its transmembrane form (Figure 6D), indicating that HCFs could potentially contribute directly to plasma sST2 levels, levels which have been associated with cardiac fibrosis, adverse cardiac remodeling, and worse cardiovascular outcomes in heart failure patients.11,12
The role of CDCP1 that we have identified in HCFs and the fact that it is inducible by PDGF-BB and regulates cell proliferation (Figure 4) is consistent with its known role in cancer. CDCP1 has been found to be significantly up-regulated in a variety of cancers in which it promotes cancer cell growth and metastasis.25–30 In addition to the fact that CDCP1 is a single-pass cytoplasm membrane glycoprotein,25 those findings make CDCP1 an intriguing potential druggable target for cancer therapy.25–30 Several therapeutic reagents have been developed that target CDCP1 for cancer and have shown promising efficacy and toxicity tolerance in animal models.25,30 CDCP1 (previously named CD318) was also found to be up-regulated in immune cells and other disease states31–33 including heart failure comorbid with inflammatory disease.31
Finally, we should point out the limitations of our studies, beginning with the fact that the P-values for association of those SNPs in our GWAS did not reach the generally accepted threshold for genome-wide significance (p≤5×10−8) (Figure 2). Validation of those associations will be required in a larger cohort when that is possible—something that has proven to be very difficult for recent onset DCM patients. The retrospective design of our GWAS limited our ability to investigate the CDCP1 SNP/gene function in HCFs derived from the same patient cohort. Although we demonstrated that the rs6773435 SNP affects CDCP1 transcriptional activity in HCFs (Figure 3F), the molecular mechanism by which the transcriptional activity is affected has not been identified and requires further studies. A SNP mapping to non-coding cCRE, such as rs6773435, could affect gene transcription through different mechanisms including SNP-dependent chromatin modification, chromatin looping and transcription factor binding55,56. We used HCFs to study CDCP1 function because the function annotation of SNP locus supports a role for CDCP1 in HCFs (Figure 3) and CDCP1 has been reported to play role in pulmonary fibrosis8. However, CDCP1 also can be upregulated in immune and epithelial cells. Whether CDCP1 functions in other cell types/tissues that might contribute to myocardial recovery is unknown and should be the subject of future studies. Obviously, molecular function based on cell line models is not able to provide a comprehensive view of physiological processes underlying clinical phenotypes. The generation of Cdcp1 knock-out mice is currently ongoing in our laboratory to make it possible to further pursue the observations reported here.
In summary, we performed a discovery GWAS to assess genetic determinants of change in LVEF in DCM patients which identified a SNP signal mapping to the 5’-flanking region of the CDCP1 gene. Functional genomic studies demonstrated that decreased CDCP1 expression reduces HCF proliferation stimulated by PDGF signaling. Our results support the hypothesis that decreased CDCP1 expression may contribute to the recovery of heart function in recent onset DCM patients by attenuating cardiac fibrosis
Supplementary Material
Novelty and Significance:
What is Known?
Dilated cardiomyopathy (DCM) accounts for 30–40% of all cases of heart failure with reduced ejection fraction and is the most common cause of heart transplantation.
Myocardial recovery in recent onset DCM after standard pharmacotherapy is highly variable among individuals.
Cardiac fibrosis is one of the most important predictors of myocardial recovery in DCM.
What New Information Does This Article Contribute?
We performed a genome-wide association study (GWAS) for “changes in left ventricular ejection fraction (LVEF)” as the phenotype in 686 recent onset DCM patients, which identified a highly suggestive SNP signal mapping to the 5’-flanking region of the CDCP1 gene.
The variant allele of the GWAS-identified SNP was significantly associated with decreased CDCP1 expression and improvement in LVEF, implicating the possible role of CDCP1 in myocardial recovery of DCM patients.
CDCP1 knockdown (KD) significantly repressed human cardiac fibroblasts (HCFs) proliferation stimulated by the PDGF signaling.
To our knowledge, this is the first GWAS for change in LVEF in recent onset DCM, which identified a highly suggestive SNP mapping to the 5’-flanking region of the CUB domain containing protein 1 (CDCP1) gene. We also found that genetic variation in/near CDCP1 was significantly associated with heart failure mortality in the UK Biobank dataset. Given that CDCP1 is highly variably expressed in human fibroblasts among individuals, and since cardiac fibrosis is an important determinant of myocardial recovery in DCM, we pursued its role in cardiac fibrosis. We demonstrated that the SNP variant allele, which was associated with improved LVEF in the GWAS, resulted in decreased CDCP1 expression in HCFs. Of importance, CDCP1 KD significantly decreased HCF proliferation stimulated by PDGF signaling, a signaling pathway for which activation is known to promote cardiac fibrosis. Transcriptome-wide, the gene that was most significantly down regulated in HCF after CDCP1 KD was IL1RL1 which encodes sST2, corroborating the potential beneficial effect of CDCP1 KD on myocardial recovery and fibrosis. Our study raises the possibility of targeting CDCP1 to decrease cardiac fibrosis with potential for improvement in myocardial function and with implications for novel DCM pharmacotherapy.
Acknowledgments:
We thank Dr. Leila Jones and Ms. Svetlana Bornschlegl from the Mayo Clinic Center for Individualized Medicine for their administration and management of this project. We want to thank all the participants who donated their blood samples for this study and the investigative groups from all patient-recruiting sites who helped with DNA sample collection. We want to thank the GTEx Project, the UK Biobank, and the ENCODE Consortium for generating the datasets that have been cited in this study.
Sources of Funding:
This study was supported by the Mayo Clinic Department of Cardiovascular Medicine, and Center for Individualized Medicine through internal fundings. M. Wang was supported by the NIH T32 Training Grant in Clinical Pharmacology (GM08685). The Intervention in Myocarditis and Acute Cardiomyopathy (IMAC)-2 study was funded by the NIH R01 Grant (HL075038).
Nonstandard Abbreviations and Acronyms
- DCM
dilated cardiomyopathy
- LVEF
left ventricular ejection fraction
- GWAS
genome-wide association study
- SNP
single-nucleotide polymorphism
- CDCP1
CUB domain containing protein 1
- HCF
human cardiac fibroblast
- KD
knock down
- OE
overexpression
- EV
empty vector
- PDGF
platelet-derived growth factor
- PDGF-BB
PDGF subunit B homodimer
- TGF-β
transforming growth factor beta
- α-SMA
alpha-smooth muscle actin
- ECM
extracellular matrix
- DEG
differentially expressed gene
- IL1RL1
interleukin-1 receptor-like 1
- sST2
soluble ST2
Footnotes
Disclosures:
Dr. Weinshilboum is a co-founder of and stockholder in OneOme, LLC. The other authors report no conflicts of interest.
Supplemental Materials
Online Supplementary Figures S1–S13
Online Supplementary Tables S1–S4
Name of IMAC Investigators
REFERENCES
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al. Heart Disease and Stroke Statistics—2014 Update: A Report From the American Heart Association. Circulation. 2014;129:e28–e292. doi: 10.1161/01.cir.0000441139.02102.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenbaum AN, Agre KE, Pereira NL. Genetics of dilated cardiomyopathy: practical implications for heart failure management. Nat Rev Cardiol. 2020;17:286–297. doi: 10.1038/s41569-019-0284-0 [DOI] [PubMed] [Google Scholar]
- 3.McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, Rouleau JL, Shi VC, Solomon SD, Swedberg K, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. 2014;371:993–1004. doi: 10.1056/NEJMoa1409077 [DOI] [PubMed] [Google Scholar]
- 4.McNamara DM, Starling RC, Cooper LT, Boehmer JP, Mather PJ, Janosko KM, Gorcsan J 3rd, Kip KE, Dec GW, Investigators I. Clinical and demographic predictors of outcomes in recent onset dilated cardiomyopathy: results of the IMAC (Intervention in Myocarditis and Acute Cardiomyopathy)-2 study. J Am Coll Cardiol. 2011;58:1112–1118. doi: 10.1016/j.jacc.2011.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kramer DG, Trikalinos TA, Kent DM, Antonopoulos GV, Konstam MA, Udelson JE. Quantitative evaluation of drug or device effects on ventricular remodeling as predictors of therapeutic effects on mortality in patients with heart failure and reduced ejection fraction: a meta-analytic approach. J Am Coll Cardiol. 2010;56:392–406. doi: 10.1016/j.jacc.2010.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, Cardon LR, Mooser V. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30:317–320. doi: 10.1038/nbt.2151 [DOI] [PubMed] [Google Scholar]
- 7.Meder B, Ruhle F, Weis T, Homuth G, Keller A, Franke J, Peil B, Lorenzo Bermejo J, Frese K, Huge A, et al. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. Eur Heart J. 2014;35:1069–1077. doi: 10.1093/eurheartj/eht251 [DOI] [PubMed] [Google Scholar]
- 8.Noskovicova N, Heinzelmann K, Burgstaller G, Behr J, Eickelberg O. Cub domain-containing protein 1 negatively regulates TGF-beta signaling and myofibroblast differentiation. Am J Physiol Lung Cell Mol Physiol. 2018;314:L695–L707. doi: 10.1152/ajplung.00205.2017 [DOI] [PubMed] [Google Scholar]
- 9.Iwata M, Torok-Storb B, Wayner EA, Carter WG. CDCP1 identifies a CD146 negative subset of marrow fibroblasts involved with cytokine production. PLoS One. 2014;9:e109304. doi: 10.1371/journal.pone.0109304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schelbert EB, Piehler KM, Zareba KM, Moon JC, Ugander M, Messroghli DR, Valeti US, Chang CC, Shroff SG, Diez J, et al. Myocardial fibrosis quantified by extracellular volume is associated with subsequent hospitalization for heart failure, death, or both across the spectrum of ejection fraction and heart failure stage. J Am Heart Assoc. 2015;4. doi: 10.1161/jaha.115.002613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kakkar R, Lee RT. The IL-33/ST2 pathway: therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–840. doi: 10.1038/nrd2660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aimo A, Januzzi JL Jr., Bayes-Genis A, Vergaro G, Sciarrone P, Passino C, Emdin M. Clinical and Prognostic Significance of sST2 in Heart Failure: JACC Review Topic of the Week. J Am Coll Cardiol. 2019;74:2193–2203. doi: 10.1016/j.jacc.2019.08.1039 [DOI] [PubMed] [Google Scholar]
- 13.Gupta M, Neavin D, Liu D, Biernacka J, Hall-Flavin D, Bobo WV, Frye MA, Skime M, Jenkins GD, Batzler A, et al. TSPAN5, ERICH3 and selective serotonin reuptake inhibitors in major depressive disorder: pharmacometabolomics-informed pharmacogenomics. Mol Psychiatry. 2016;21:1717–1725. doi: 10.1038/mp.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu D, Ray B, Neavin DR, Zhang J, Athreya AP, Biernacka JM, Bobo WV, Hall-Flavin DK, Skime MK, Zhu H, et al. Beta-defensin 1, aryl hydrocarbon receptor and plasma kynurenine in major depressive disorder: metabolomics-informed genomics. Transl Psychiatry. 2018;8:10. doi: 10.1038/s41398-017-0056-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasching PA, Liu D, Scully S, Ingle JN, Lyra PC, Rack B, Hein A, Ekici AB, Reis A, Schneeweiss A, et al. Identification of Two Genetic Loci Associated with Leukopenia after Chemotherapy in Patients with Breast Cancer. Clin Cancer Res. 2022;28:3342–3355. doi: 10.1158/1078-0432.CCR-20-4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu D, Zhuang Y, Zhang L, Gao H, Neavin D, Carrillo-Roa T, Wang Y, Yu J, Qin S, Kim DC, et al. ERICH3: vesicular association and antidepressant treatment response. Mol Psychiatry. 2021;26:2415–2428. doi: 10.1038/s41380-020-00940-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de Groote P, Helbecque N, Lamblin N, Hermant X, Amouyel P, Bauters C, Dallongeville J. Beta-adrenergic receptor blockade and the angiotensin-converting enzyme deletion polymorphism in patients with chronic heart failure. Eur J Heart Fail. 2004;6:17–21. doi: 10.1016/j.ejheart.2003.09.006 [DOI] [PubMed] [Google Scholar]
- 18.Lemesle G, Maury F, Beseme O, Ovart L, Amouyel P, Lamblin N, de Groote P, Bauters C, Pinet F. Multimarker proteomic profiling for the prediction of cardiovascular mortality in patients with chronic heart failure. PLoS One. 2015;10:e0119265. doi: 10.1371/journal.pone.0119265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.TheGoTExConsortium. The Genotype-Tissue Expression (GTEx) project. Nat Genet. 2013;45:580–585. doi: 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.EncodeProjectConsortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider BP, Shen F, Gardner L, Radovich M, Li L, Miller KD, Jiang G, Lai D, O’Neill A, Sparano JA, et al. Genome-Wide Association Study for Anthracycline-Induced Congestive Heart Failure. Clin Cancer Res. 2017;23:43–51. doi: 10.1158/1078-0432.CCR-16-0908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, Cupples LA, Dehghan A, Lumley T, Rosamond WD, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–266. doi: 10.1161/CIRCGENETICS.109.895763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet. 2016;17:207–223. doi: 10.1038/nrg.2016.4 [DOI] [PubMed] [Google Scholar]
- 25.Khan T, Kryza T, Lyons NJ, He Y, Hooper JD. The CDCP1 Signaling Hub: A Target for Cancer Detection and Therapeutic Intervention. Cancer Res. 2021;81:2259–2269. doi: 10.1158/0008-5472.CAN-20-2978 [DOI] [PubMed] [Google Scholar]
- 26.Alajati A, D’Ambrosio M, Troiani M, Mosole S, Pellegrini L, Chen J, Revandkar A, Bolis M, Theurillat JP, Guccini I, et al. CDCP1 overexpression drives prostate cancer progression and can be targeted in vivo. J Clin Invest. 2020;130:2435–2450. doi: 10.1172/JCI131133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forte L, Turdo F, Ghirelli C, Aiello P, Casalini P, Iorio MV, D’Ippolito E, Gasparini P, Agresti R, Belmonte B, et al. The PDGFRbeta/ERK1/2 pathway regulates CDCP1 expression in triple-negative breast cancer. BMC Cancer. 2018;18:586. doi: 10.1186/s12885-018-4500-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karachaliou N, Chaib I, Cardona AF, Berenguer J, Bracht JWP, Yang J, Cai X, Wang Z, Hu C, Drozdowskyj A, et al. Common Co-activation of AXL and CDCP1 in EGFR-mutation-positive Non-smallcell Lung Cancer Associated With Poor Prognosis. EBioMedicine. 2018;29:112–127. doi: 10.1016/j.ebiom.2018.02.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu H, Ong SE, Badu-Nkansah K, Schindler J, White FM, Hynes RO. CUB-domain-containing protein 1 (CDCP1) activates Src to promote melanoma metastasis. Proc Natl Acad Sci U S A. 2011;108:1379–1384. doi: 10.1073/pnas.1017228108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lim SA, Zhou J, Martinko AJ, Wang YH, Filippova EV, Steri V, Wang D, Remesh SG, Liu J, Hann B, et al. Targeting a proteolytic neoepitope on CUB domain containing protein 1 (CDCP1) for RAS-driven cancers. J Clin Invest. 2022;132. doi: 10.1172/JCI154604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahlers MJ, Lowery BD, Farber-Eger E, Wang TJ, Bradham W, Ormseth MJ, Chung CP, Stein CM, Gupta DK. Heart Failure Risk Associated With Rheumatoid Arthritis-Related Chronic Inflammation. J Am Heart Assoc. 2020;9:e014661. doi: 10.1161/JAHA.119.014661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gruber CN, Patel RS, Trachtman R, Lepow L, Amanat F, Krammer F, Wilson KM, Onel K, Geanon D, Tuballes K, et al. Mapping Systemic Inflammation and Antibody Responses in Multisystem Inflammatory Syndrome in Children (MIS-C). Cell. 2020;183:982–995 e914. doi: 10.1016/j.cell.2020.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lun Y, Borjini N, Miura NN, Ohno N, Singer NG, Lin F. CDCP1 on Dendritic Cells Contributes to the Development of a Model of Kawasaki Disease. J Immunol. 2021. doi: 10.4049/jimmunol.2001406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Travers JG, Kamal FA, Robbins J, Yutzey KE, Blaxall BC. Cardiac fibrosis: The fibroblast awakens. Circ Res. 2016;118:1021–1040. doi: 10.1161/CIRCRESAHA.115.306565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. 2021;117:1450–1488. doi: 10.1093/cvr/cvaa324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schreibing F, Anslinger TM, Kramann R. Fibrosis in Pathology of Heart and Kidney: From Deep RNA-Sequencing to Novel Molecular Targets. Circ Res. 2023;132:1013–1033. doi: 10.1161/CIRCRESAHA.122.321761 [DOI] [PubMed] [Google Scholar]
- 37.Gallini R, Lindblom P, Bondjers C, Betsholtz C, Andrae J. PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp Cell Res. 2016;349:282–290. doi: 10.1016/j.yexcr.2016.10.022 [DOI] [PubMed] [Google Scholar]
- 38.Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017;169:381–405. doi: 10.1016/j.cell.2017.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimura SH, Ikawa M, Ito A, Okabe M, Nojima H. Cyclin G1 is involved in G2/M arrest in response to DNA damage and in growth control after damage recovery. Oncogene. 2001;20:3290–3300. doi: 10.1038/sj.onc.1204270 [DOI] [PubMed] [Google Scholar]
- 41.Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;7:12260. doi: 10.1038/ncomms12260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest. 2014;124:2921–2934. doi: 10.1172/JCI74783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.González A, Schelbert EB, Díez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. 2018;71:1696–1706. doi: 10.1016/j.jacc.2018.02.021 [DOI] [PubMed] [Google Scholar]
- 44.Dai Z, Aoki T, Fukumoto Y, Shimokawa H. Coronary perivascular fibrosis is associated with impairment of coronary blood flow in patients with non-ischemic heart failure. J Cardiol. 2012;60:416–421. doi: 10.1016/j.jjcc.2012.06.009 [DOI] [PubMed] [Google Scholar]
- 45.Villari B, Campbell SE, Hess OM, Mall G, Vassalli G, Weber KT, Krayenbuehl HP. Influence of collagen network on left ventricular systolic and diastolic function in aortic valve disease. J Am Coll Cardiol. 1993;22:1477–1484. doi: 10.1016/0735-1097(93)90560-n [DOI] [PubMed] [Google Scholar]
- 46.Gulati A, Jabbour A, Ismail TF, Guha K, Khwaja J, Raza S, Morarji K, Brown TD, Ismail NA, Dweck MR, et al. Association of fibrosis with mortality and sudden cardiac death in patients with nonischemic dilated cardiomyopathy. JAMA. 2013;309:896–908. doi: 10.1001/jama.2013.1363 [DOI] [PubMed] [Google Scholar]
- 47.Azevedo CF, Nigri M, Higuchi ML, Pomerantzeff PM, Spina GS, Sampaio RO, Tarasoutchi F, Grinberg M, Rochitte CE. Prognostic significance of myocardial fibrosis quantification by histopathology and magnetic resonance imaging in patients with severe aortic valve disease. J Am Coll Cardiol. 2010;56:278–287. doi: 10.1016/j.jacc.2009.12.074 [DOI] [PubMed] [Google Scholar]
- 48.Yamada T, Fukunami M, Ohmori M, Iwakura K, Kumagai K, Kondoh N, Minamino T, Tsujimura E, Nagareda T, Kotoh K, et al. Which subgroup of patients with dilated cardiomyopathy would benefit from long-term beta-blocker therapy? A histologic viewpoint. J Am Coll Cardiol. 1993;21:628–633. doi: 10.1016/0735-1097(93)90094-h [DOI] [PubMed] [Google Scholar]
- 49.Masci PG, Schuurman R, Andrea B, Ripoli A, Coceani M, Chiappino S, Todiere G, Srebot V, Passino C, Aquaro GD, et al. Myocardial fibrosis as a key determinant of left ventricular remodeling in idiopathic dilated cardiomyopathy: a contrast-enhanced cardiovascular magnetic study. Circ Cardiovasc Imaging. 2013;6:790–799. doi: 10.1161/circimaging.113.000438 [DOI] [PubMed] [Google Scholar]
- 50.De Mello WC, Specht P. Chronic blockade of angiotensin II AT1-receptors increased cell-to-cell communication, reduced fibrosis and improved impulse propagation in the failing heart. J Renin Angiotensin Aldosterone Syst. 2006;7:201–205. doi: 10.3317/jraas.2006.038 [DOI] [PubMed] [Google Scholar]
- 51.Zannad F, Alla F, Dousset B, Perez A, Pitt B. Limitation of excessive extracellular matrix turnover may contribute to survival benefit of spironolactone therapy in patients with congestive heart failure: insights from the randomized aldactone evaluation study (RALES). Rales Investigators. Circulation. 2000;102:2700–2706. doi: 10.1161/01.cir.102.22.2700 [DOI] [PubMed] [Google Scholar]
- 52.Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, Scholler J, Monslow J, Lo A, Han W, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573:430–433. doi: 10.1038/s41586-019-1546-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ivey MJ, Kuwabara JT, Riggsbee KL, Tallquist MD. Platelet-derived growth factor receptor-alpha is essential for cardiac fibroblast survival. Am J Physiol Heart Circ Physiol. 2019;317:H330–H344. doi: 10.1152/ajpheart.00054.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kuwabara JT, Hara A, Bhutada S, Gojanovich GS, Chen J, Hokutan K, Shettigar V, Lee AY, DeAngelo LP, Heckl JR, et al. Consequences of PDGFRalpha(+) fibroblast reduction in adult murine hearts. Elife. 2022;11. doi: 10.7554/eLife.69854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen TTL, Gao H, Liu D, Philips TJ, Ye Z, Lee JH, Shi GX, Copenhaver K, Zhang L, Wei L, et al. Glucocorticoids unmask silent non-coding genetic risk variants for common diseases. Nucleic Acids Res. 2022;50:11635–11653. doi: 10.1093/nar/gkac1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu D, Nguyen TTL, Gao H, Huang H, Kim DC, Sharp B, Ye Z, Lee JH, Coombes BJ, Ordog T, et al. TCF7L2 lncRNA: a link between bipolar disorder and body mass index through glucocorticoid signaling. Mol Psychiatry. 2021;26:7454–7464. doi: 10.1038/s41380-021-01274-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–1283. doi: 10.1038/ng.3643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Das S, Forer L, Schonherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48:1284–1287. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated in this study have been deposited in NCBI’s Gene Expression Omnibus (GEO) and are accessible through GEO Series accession number: GSE222695.