

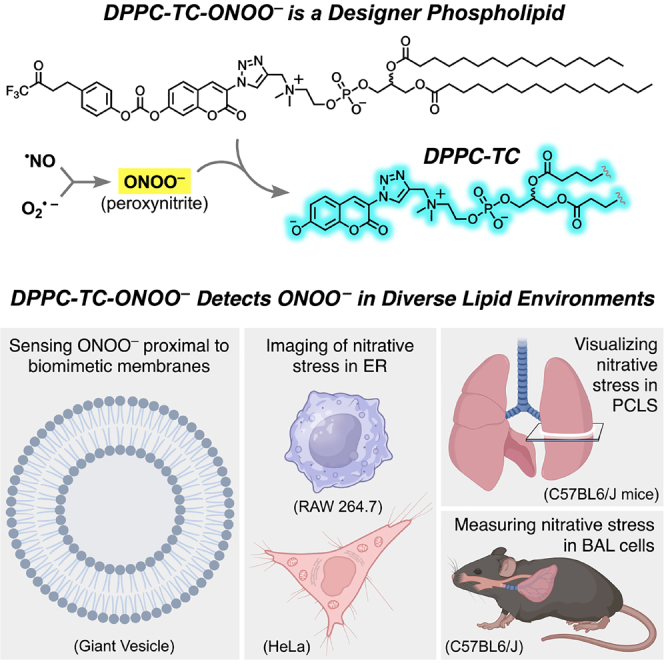
Summary
Lipid membranes and lipid-rich organelles are targets of peroxynitrite (ONOO−), a highly reactive species generated under nitrative stress. We report a membrane-localized phospholipid (DPPC-TC-ONOO–) that allows the detection of ONOO− in diverse lipid environments: biomimetic vesicles, mammalian cell compartments, and within the lung lining. DPPC-TC-ONOO– and POPC self-assemble to membrane vesicles that fluorogenically and selectively respond to ONOO−. DPPC-TC-ONOO–, delivered through lipid nanoparticles, allowed for ONOO− detection in the endoplasmic reticulum upon cytokine-induced nitrative stress in live mammalian cells. It also responded to ONOO− within lung tissue murine models upon acute lung injury. We observed nitrative stress around bronchioles in precision cut lung slices exposed to nitrogen mustard and in pulmonary macrophages following intratracheal bleomycin challenge. Results showed that DPPC-TC-ONOO– functions specifically toward iNOS, a key enzyme modulating nitrative stress, and offers significant advantages over its hydrophilic analog in terms of localization and signal generation.
Subject areas: Analytical chemistry, Biotechnology, Membranes
Graphical abstract
Highlights
-
•
Designer phospholipid detects peroxynitrite present near lipid membranes
-
•
Nitrative stress in PCLS is observable upon its exposure to nitrogen mustard
-
•
Nitrative stress due to acute lung injury in mice can be quantitatively assessed
-
•
Fluorescence emission is dependent on inducible nitric oxide synthase
Analytical chemistry; Biotechnology; Membranes
Introduction
The integrity of lipid membranes and lipid-rich organelles are essential to maintaining cellular homeostasis and physiology. These lipid environments are potential targets of reactive oxygen and nitrogen species (RONS).1,2 A major source of lipid membrane damage is peroxynitrite (ONOO−),3 which is produced through the reaction between nitric oxide (·NO) and superoxide (O2·–) at near diffusional rates (∼5 x 109 mol−1s−1).4,5 It has been proposed that biological membranes and hydrophobic tissue compartments are critical locations for the formation of ·NO-derived reactive species.6 The majority of ONOO− formation is likely to occur in proximity to the lipid portion of the cell, as ·NO is ∼10 times more soluble in hydrophobic environments.7 Superoxide is primarily produced by NOX family NADPH oxidases, which are transmembrane proteins that transport electrons across biological membranes.8 The mechanism of peroxynitrite-mediated lipid damage has been proposed to involve hydrogen abstraction by peroxynitrite derived radicals from unsaturated lipids, such as linolenic acid and arachidonic acid, generating carbon-centered lipid radicals.1,9 These radicals react with molecular oxygen (O2), forming lipid peroxyl radicals, which react with nearby unsaturated lipids, forming lipid hydroperoxides, and new carbon-centered radicals that propagate oxidative damage.3
In cells, inducible nitric oxide synthase (iNOS) catalyzes the production of ·NO from l-arginine and O2 in response to various stimuli, including bacterial or viral infections, inflammation, and proinflammatory cytokines.10 The expression and activity of iNOS must be tightly regulated, because excessive or prolonged iNOS activation is detrimental to the host, leading to tissue damage and fibrosis.11 In HeLa, peroxynitrite production through iNOS is facilitated by the combination of interferon-γ (IFN-γ), lipopolysaccharide (LPS), and phorbol myristate acetate (PMA).12 In RAW 264.7, a macrophage model, ONOO− production can be stimulated by LPS. LPS binds Toll-like receptor 4 (TLR4), which triggers the activation of nuclear factor kappa B (NF-kB) signaling, resulting in the production of iNOS13,14 and ROS.15,16 Macrophage upregulation of iNOS production in response to injury is a characteristic of pro-inflammatory activation. One such model is intratracheal bleomycin (ITB)-induced acute lung injury (ALI).17 The extent of pulmonary ITB injury has been demonstrated to be heavily iNOS dependent.18 This system, therefore, represents a relevant biological model to examine the efficacy of ONOO− detection in vivo.
Investigating the generation and reactivity of ONOO− in these biological systems is challenging due to its highly reactive nature.19 The protonated form of ONOO−, peroxynitrous acid (ONOOH, pKa = 6.8), has a chemical half-life of 0.63 s.3 The biological half-life of ONOO− and ONOOH depends on the concentration of the biological targets with which they react and has been estimated at ∼10–20 ms.2 High-performing ONOO− sensors have thus been a focus of emerging research.20,21,22,23,24 Despite the interest in developing biosensing tools to study lipid membranes,25,26,27,28,29 the available ONOO− sensors lack the ability to selectively localize and remain within lipid membranes and lipid-rich organelles or specific tissue compartments. Furthermore, investigating the reactivity of ONOO− in biological membranes or lung tissue has typically relied on indirect detection techniques, including immunohistochemistry,18 EPR (electron paramagnetic resonance),30 or bite marks, such as tyrosine nitration31 and lipid peroxidation.32 These markers are neither specific nor continuous, making the cellular visualization of ONOO− generation and reactivity challenging. Taken together, there is an unmet need for biocompatible peroxynitrite probes that localize and function specifically in lipid membranes and lipid-rich organelles and tissues. Such molecular tools could provide significant insights into pathophysiologies associated with nitrative stress.3,33,34,35,36,37,38,39
Herein, we report the direct detection of ONOO− in biomimetic and biological lipid environments with diverse origins using DPPC-TC-ONOO–, a high-fidelity probe derived from 1,2-dipalmitoyl-rac-glycero-3-phosphocholine (rac-DPPC). We demonstrated that DPPC-TC-ONOO– and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) self-assemble to giant vesicles (GVs) that fluorogenically respond to ONOO−, validating in vitro the feasibility of DPPC-TC-ONOO–. Lipid nanoparticles (LNPs) successfully delivered DPPC-TC-ONOO– into HeLa and RAW 264.7 cells. Real-time confocal imaging demonstrated the substantial fluorescence enhancement of the ER in both cell types under nitrative stress induced through endogenous stimulation. The redox selectivity of DPPC-TC-ONOO– was confirmed to be iNOS-dependent through control experiments with 1400W, a small-molecule inhibitor of iNOS.40 The biological potential of DPPC-TC-ONOO– was further demonstrated through murine models of ex vivo and in vivo ALI. For the ex vivo ALI study, we used mechlorethamine hydrochloride, a carcinogen historically categorized as nitrogen mustard (NM), which is a pulmonary toxicant known to induce nitrative stress.41 An organotypic culture of precision cut lung slices (PCLS) was used to determine the localization of DPPC-TC-ONOO– within the tissue. The iNOS-dependent nitrative stress was detected around bronchioles upon the treatment of PCLS with NM. For the in vivo investigation of ALI, immunostaining of bronchoalveolar lavage (BAL) cells, which are 95% or more macrophages,42 indicated that DPPC-TC-ONOO– responds to iNOS-dependent nitrative stress upon intratracheal bleomycin (ITB) challenge.
Results
The peroxynitrite-sensing phospholipid and its hydrophilic analog have a modular chemical design
We developed a concise synthetic route for the peroxynitrite-sensing phospholipid, DPPC-TC-ONOO– (Figure 1A), which includes rac-DPPC as the phospholipid framework, 3-triazole-coumarin as the fluorophore, and p-(4,4,4-trifluoro-3-oxobutyl)phenyl as the peroxynitrite-reactive module. We also synthesized its hydrophilic analog, TEG-TC-ONOO–, which contains triethylene glycol (TEG) in place of rac-DPPC (Figure 1A). TEG-TC-ONOO– allowed us to make systematic comparisons between the amphiphilic and hydrophilic probe designs in terms of localization. TEG-TC-ONOO– lacks the ability to localize in biological membranes as observed in our in vitro and ex vivo studies. In addition, TEG-TC-ONOO– is highly soluble in aqueous media and buffers, which aided our mass spectrometry and spectrophotometric assessments of the peroxynitrite-mediated fluorescence activation mechanism and fidelity. For both DPPC-TC-ONOO– and TEG-TC-ONOO–, the p-(4,4,4-trifluoro-3-oxobutyl)phenyl and 3-triazole-7-hydroxycoumarin groups were connected through a carbonate linkage. We reasoned that this connection could mask 7-O of the coumarin, diminishing its fluorescence intensity. In the presence of ONOO−, the p-(4,4,4-trifluoro-3-oxobutyl)phenyl moiety would undergo an oxidative spirocyclization, which is in agreement with the previously reported transformation of a dichlorofluorescein caged at its xantane oxygen as diaryl ether.43 We hypothesized that for our probe design the spirocyclization should generate a zwitterionic carbonate that reacts with water, eliminating the oxa-spiro[4,5] decenone 6, uncaging the coumarin motif via decarboxylation, subsequently forming DPPC-TC or TEG-TC (Figure 1B).
Figure 1.

DPPC-TC-ONOO– and TEG-TC-ONOO– as peroxynitrite probes
(A) The chemical synthesis of DPPC-TC-ONOO– and TEG-TC-ONOO–, each starting from 1,1-trifluoro-4-(4-hydroxyphenyl)butan-2-one (1).
(B) Peroxynitrite-mediated conversion of TEG-TC-ONOO– and DPPC-TC-ONOO– to their fluorogenically active products DPPC-TC and TEG-TC.
Both DPPC-TC-ONOO– and TEG-TC-ONOO– were synthesized through convergent routes in which the longest linear sequence required three chemical steps. First, 1,1,1-trifluoro-4-(4-hydroxyphenyl)butan-2-one (1) was converted to its chloroformate using triphosgene and successively connected with 3-azido-7-hydroxycoumarin (2) to furnish the carbonate 3 in 60% yield over two steps. This compound served as a common intermediate toward DPPC-TC-ONOO– and TEG-TC-ONOO–. Through copper-catalyzed azide-alkyne cycloaddition ("click") reaction, the carbonate 3 was conjugated with either the propargylated rac-DPPC 4 or propargyl-TEG-OH 5 to afford DPPC-TC-ONOO– (43%) or TEG-TC-ONOO– (57%), respectively. Of note, the click reactions of 3-azido-7-hydroxycoumarin with a large number of diverse alkynes have been reported to provide 1,4-substituted triazoles exclusively.44 To obtain the alkynyl lipid 4, we converted 1,2-dipalmitoyl-rac-glycerol to its cyclic phosphate triester using ethylene chlorophosphate and opened the subsequent ring by the nucleophilic addition of 3-dimethylamino-1-propyne (see supplemental information for details).
Sensing occurs via oxidative uncaging of the coumarin motif and is specific to peroxynitrite
To gain a mechanistic insight into the reaction of the probes with ONOO−, we examined the products by liquid chromatography followed by high-resolution mass spectrometry (LC-HRMS) (Figure S1). The amphiphilic probe, DPPC-TC-ONOO–, displayed a poor electrospray ionization profile, while TEG-TC-ONOO– provided evidence for the formation of its oxidative decarboxylation product, TEG-TC, upon mixing with ONOO−. This mass spectrometry data does not provide direct evidence of a reactive carbonate intermediate, and the identification of such is outside the scope of this project. However, we observed the progressive formation of the oxa-spiro[4,5] decenone 6, which provides support to a potential spirocyclization/elimination cascade. We then determined the pKa of TEG-TC to be 7.1 ± 0.1 using UV-Vis spectrophotometric measurements (Figure S2). Under our confocal imaging conditions (pH 7.5–8.5 for cell-free assays, pH 7.4 for live cells), a substantial population of the 7-hydroxycoumarin motif should exist in its fluorogenically active aryloxide (deprotonated) form. Indeed, measuring fluorescence (405/475 nm) of TEG-TC for different pHs (5.8–8.8) showed that the signal intensities from the samples at pHs 7.5–8.8 were similar and 18- to 20-fold higher than that at pH 5.8 (Figure S3). We assessed the change in triazole-coumarin fluorogenicity upon uncaging the coumarin by measuring the relative fluorescence quantum yield (ΦFrel) values (Table 1) (for absorbance and emission spectra, see Figure S4). As the standard, we used coumarin 343, whose absolute fluorescence quantum yield (ΦF) is 0.63 in ethanol.45 The ΦFrel values of TEG-TC-ONOO– (entry 1) and DPPC-TC-ONOO– (entry 3) were measured as 0.08 and 0.09, respectively. Each with a low ΦFrel and molar extinction coefficient, these probes were validated to exhibit weak brightness due to the derivatization of their coumarin 7-O atom as a carbonate group. The ΦFrel values for TEG-TC and DPPC-TC were measured as 0.66 (entry 2) and 0.64 (entry 4), respectively. For these measurements, each compound was independently synthesized from its respective starting material and purified (see supplemental information for details). Taken together, these results suggested that the conversion of the caged coumarins to their uncaged forms enhances the fluorescence quantum yield by ≥ 7-fold and brightness by 30-to-35-fold.
Table 1.
Characterization of the peroxynitrite probes and their uncaged forms
| Entry | Compound | Max Ex (nm) | Max Em (nm) | Extinction Coefficient (M−1 cm−1) | Quantum Yield | Brightness | Relative brightness |
|---|---|---|---|---|---|---|---|
| 1 | TEG-TC-ONOO– | 400.0 | 472.0 | 4475 | 0.08 | 358 | 1.4 |
| 2 | TEG-TC | 397.0 | 471.4 | 15613 | 0.66 | 10305 | 41.1 |
| 3 | DPPC-TC-ONOO– | 395.0 | 471.2 | 2784 | 0.09 | 251 | 1.0 |
| 4 | DPPC-TC | 398.0 | 472.0 | 13749 | 0.64 | 8799 | 35.1 |
The fluorogenic response rate and magnitude of the probe were evaluated by a series of fluorescence measurements with TEG-TC-ONOO– (Figures 2A–2C). Physiologically normal ONOO− production rate in cells has been estimated to be in the single-digit μM/s range.3,46 Guided by this information, and in agreement with the fluorescence titration conditions employed for cytosolic resorufin-based peroxynitrite sensors,47 we reasoned that administering 600 μM of ONOO− into a pH 7.5 medium would provide an effective level of ONOO− exposure equivalent to a biological flux rate of ∼10–20 μM/s.2,3 The existence of ONOO− at this pH was validated through UV-Vis spectrophotometry (Figure S5). We tested pH dependance of fluorescence generation from TEG-TC-ONOO– using buffers with different pHs, including 6.8, which is the pKa of ONOOH.3 Addition of ONOO− (600 μM) led to a significant increase in fluorescence at pH 7.8 or 8.8, which continued until 30 min, reaching up to 6.5 or 8 times of the untreated samples (t = 0 min) (Figure 2A). This trend of increasing fluorescence output was mirrored in the level of conversion of TEG-TC-ONOO– into TEG-TC at pH 7.5, based on UV-Vis spectrophotometric measurements (Figure S6). In contrast, those at lower pHs (5.8 and 6.8) showed little or no fluorescence generation, largely due to the predominant background consumption of ONOO− (e.g., protonation to ONOOH)3 or that surviving ONOOH did not lead to probe activation. It is also worth noting that for pH 5.8 or 6.8, each being lower than the pKa of TEG-TC (7.1), the fluorescence output from TEG-TC is weakened (cf. Figure S3), likely due to the decreased ratio of the deprotonated coumarin over its protonated form. The interesting observation that fluorescence increases over 30 min suggests a multi-step mechanism for the formation of TEG-TC from TEG-TC-ONOO–. To gain a new chemical insight, we conducted a competitive oxidation assay the ketone 1 (Figure 2B). The ketone 1 (600 μM) generated no fluorescence when treated with ONOO− (600 μM) (gray square). If the starting mixture contained TEG-TC-ONOO– (1 μM) and excess of 1 (600 μM), ONOO− (600 μM) addition generated only weak fluorescence (orange triangle), indicating that 1 can largely scavenge ONOO−. When 1 (600 μM) was introduced after 5 min of incubation of TEG-TC-ONOO– (1 μM) pre-mixed with ONOO− (600 μM) (inverted blue triangle), fluorescence generation became substantial (∼4-fold over 4 min, >6-fold over 15 min), which follows a trend similar to that for TEG-TC-ONOO– (1 equiv) treated with ONOO− (600 μM) (red circle). These results show that the reaction between the probe and ONOO− might be rapid for the tested condition, yet fluorescence generation from the probe is relatively slow, supporting the proposition of a complex, multistep mechanism.
Figure 2.

Peroxynitrite-specific reactivity and redox selectivity of TEG-TC-ONOO–
(A) The time-dependent change in relative fluorescence intensity at 405/475 nm excitation/emission for TEG-TC-ONOO– (1 μM) treated with ONOO− (600 μM) at various pHs: 5.8, 6.8, 7.8, and 8.8. Buffer: 50 mM Tris (pH 6.8, 7.8, or 8.8) or MES (pH 5.8).
(B) The time-dependent change in relative fluorescence intensity at 405/475 nm excitation/emission for TEG-TC-ONOO– and/or 1,1,1-trifluoro-4-(4-hydroxyphenyl)butan-2-one (the ketone 1), treated with ONOO−. TEG-TC-ONOO– (1 μM), 1 (600 μM), and ONOO− (600 μM). The solutions were prepared using Tris 7.5 (50 mM) buffer.
(C) Selectivity of TEG-TC-ONOO– against RONS. TEG-TC-ONOO– (1 μM) was dissolved in Tris buffer (50 mM, pH 7.5) and subjected to RONS with an estimated initial concentration of 600 μM. After the addition of ONOO− (600 μM) into Tris buffer (50 mM), pH increased to 8.7. EDTA (600 μM) was added to chelate a transition metal (e.g., copper) that leads to superoxide dismutation. Data represent F/F0 measurements of samples incubated for 60 min. F and F0 are defined as the fluorescence with and without the redox agent. (a–c) Ex/Em: 405/475 nm. Error bars represent standard deviation, n = 3.
The peroxynitrite-specificity of the sensor moiety was assessed by subjecting TEG-TC-ONOO– to other redox species (Figures 2C and S7). Change in fluorescence intensity following excitation at 405 nm was measured over 60 min of incubation of TEG-TC-ONOO– with each species and presented based on the signal-to-background ratio F/F0, where F and F0 are defined as the fluorescence with and without the redox agent. In the absence of a redox species, F/F0 for TEG-TC-ONOO– increased only 13% (F/F0 = 1.13) over 60 min, likely due to slow background hydrolysis or oxidation. Treatment of TEG-TC-ONOO– with ONOO− gave rise to ∼570% increase in fluorescence (F/F0 = 6.69). Notably, incubating TEG-TC-ONOO– with species other than ONOO− resulted in only negligible levels of change in fluorescence. Among those, ·NO had essentially no effect on fluorescence intensity (F/F0 = 1.03). The most significant fluorescence enhancement was observed for hydrogen peroxide (H2O2), with an intensity 16% stronger than that of the background (F/F0 = 1.16). These results indicate that the relative fluorescence enhancement of TEG-TC-ONOO– is nearly 36-fold for ONOO− compared to the other RONS tested in this study.
Giant vesicles respond to peroxynitrite by lighting up at the membrane
Biomimetic membranes can be represented by GVs, which have dimensional properties (1–30 μm in diameter) relevant to those of mammalian cells (10–100 μm), display physical characteristics feasible for confocal fluorescence imaging, and are utilized for the bottom-up construction of biomimetic systems.48,49 Therefore, we sought to study GVs for the cell-free evaluation of the utility and performance of DPPC-TC-ONOO– in membranes (Figure 3). We set the pH of GV imaging buffers to 8.5 to extend the lifetime of ONOO−, because the sample handling for confocal microscopy requires a longer timescale than that for a plate reader fluorescence measurement.
Figure 3.

Performance of DPPC-TC-ONOO– in biomimetic membranes
(A) Conceptual illustration of vesicle membrane sensing ONOO−.
(B) Confocal images acquired for untreated (top row) and peroxynitrite-treated (middle and bottom rows) GVs.
(C) Confocal images acquired for GVs treated with other RONS. (B,C) Vesicles were prepared from POPC (98.5%, molar ratio) and DPPC-TC-ONOO– (1%) via electroformation. Liss-Rhod PE (red, 560 nm excitation) was used at 0.5 mol % to label vesicle membranes. In the absence of ONOO− or in the presence of other RONS (100 μM), vesicles emit minimal fluorescence following excitation at 405 nm (DPPC-TC channel). GVs emit strong fluorescence only when ONOO− is present, not when other RONS are used. Fluorescence images are shown for each field of view to demonstrate the localization of the fluorescent phospholipid product DPPC-TC with respect to the vesicle membranes. Scale bar = 5 μm.
We produced GVs from POPC and DPPC-TC-ONOO– (∼99:1 M ratio) using electroformation50 (Figure S8). To label vesicle membranes, we employed 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (18:1 Liss-Rhod PE) in 0.5 mol percent of the total lipid composition. Sucrose solution (400 mM) was used to hydrate the lipid films. A high solute concentration of sucrose facilitated the formation and stabilization of GVs.51 Following electroformation, solutions of GVs were used within 24 h. Vesicle size distribution was analyzed by dynamic light scattering (DLS), confirming the presence of GVs: 91% of vesicles had diameters ranging from ∼5 to 20 μm, with 48% having an average size of 5 μm in diameter (Figure S9). GVs were then imaged using fluorescence confocal microscopy: they were first treated with the redox species, then mounted directly onto a microscope slide. Although significant vesicle motility was observed, the population density was sufficiently high to locate some static vesicles. Addition of ONOO− (estimated initial concentration of 100 μM) into GVs led to an observable fluorescence (405 nm excitation, “DPPC-TC channel”) throughout the vesicle membrane after 5 min of incubation (Figure 3B, middle row). A potential conclusion one can draw from this observation is that DPPC-TC-ONOO– (weak fluorescence) and DPPC-TC (strong fluorescence) stay in the bilayer and display a near homogeneous distribution.
Interestingly, confocal imaging of a selected multilamellar vesicle system (Figure 3B, bottom row) showed partial fluorescence light-up following ONOO− addition. While the outermost vesicle system (either a single or two vesicles being fused) interacts with ONOO−, the smaller vesicles entrapped within exhibited no observable fluorescence at 405 nm excitation. This observation can be rationalized by the following possibilities: (a) ONOO− may have poor membrane permeability in our imaging buffer conditions and/or (b) most of the available ONOO− has been consumed by reacting with the probe in the outermost vesicle system.
The qualitative confocal imaging showed that GVs exhibited selectivity toward ONOO−. Incubation (5 min) with other redox species resulted in no observable fluorescence generation at 405 nm excitation (Figure 3C). Taken together, these results indicate that (i) DPPC-TC-ONOO– can localize into the vesicle membranes made predominantly of a natural phospholipid, (ii) it induces no observable macroscopic alteration to the membrane upon reaction with ONOO−, and (iii) the detection of ONOO− in the membranes can be achieved with high redox selectivity.
Lipid nanoparticle-assisted delivery of DPPC-TC-ONOO– allows for the direct imaging of the endoplasmic reticulum targeted by peroxynitrite in live mammalian cells
With the utility of DPPC-TC-ONOO– proven in biomimetic membranes, we sought to incorporate it into live HeLa and RAW 264.7 cells, which are relevant to redox-induced stress and have well-recognized pathways for RONS production. We utilized LNPs obtained from 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) to deliver DPPC-TC-ONOO–, which circumvented its limited solubility due to the relatively hydrophobic butanone-coumarin motif. LNPs were obtained via sonication from DOTMA, DOPE, and DPPC-TC-ONOO– (47.5 : 47.5: 5.0 mole ratio), which were hydrated with a 300 mM sucrose solution. DOTMA and DOPE were chosen as the carrier lipids due to their cationic/zwitterionic natures, which has been postulated to enhance cellular uptake through the cell membrane.52
The impact of different LNP compositions and incubation times on the cellular incorporation of DPPC-TC-ONOO– was assessed. Initial trials indicated that the molar concentration of DPPC-TC-ONOO– in LNPs influenced both the homogeneity of the LNPs and the fluorescence signal generation within stimulated cells. Specifically, LNPs containing greater than 10 mol % DPPC-TC-ONOO– were considerably more heterogeneous in size (up to 20 μm observed, in contrast to ∼100 nm). Those with less than 4 mol % DPPC-TC-ONOO– led to insufficient fluorescence signal. To balance these counteracting factors, 5 mol % DPPC-TC-ONOO– was used, with 47.5 mol % DOTMA and DOPE, each. In these initial assays, cells were incubated with LNPs for up to 15 h to complete the uptake and intracellular localization of DPPC-TC-ONOO–. Shorter incubation times (3-to-5 h) led to its uptake but not subcellular localization. To facilitate extended incubation time frames, LNPs were prepared in DMEM (Dulbecco’s Modified Eagle Medium) instead of HBSS (Hanks’ Balanced Salt Solution), which we observed to be deleterious to the cells for incubations exceeding 3 h. However, high serum concentration in DMEM may have caused interference between LNPs and serum-protein, resulting in aggregate formation as observed by microscopy. To overcome this obstacle, we used LNPs (50 μM) in a mixture of 2/3 volumetric ratio of sucrose solution and Opti-MEM, a reduced serum medium known to increase the efficiency of lipofection.53
With a feasible LNP preparation and delivery protocol established for live cell imaging, we then turned our attention to assessing the cytocompatibility of the LNPs, with or without DPPC-TC-ONOO–, DPPC-TC, and the oxa-spiro[4,5] decenone 6. We employed a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell viability assay in HeLa and RAW 264.7 cells treated with these molecules (8–250 μM) over a 24-h period. The dose-response curves (Figure S10) provided high IC50 values for each compound: DPPC-TC-ONOO– (≥100 μM), DPPC-TC (≥50 μM), and 6 (≥70 μM) (Table S1). These results suggested that the probe-containing LNPs (50 μM) and the byproducts of the reaction from peroxynitrite (DPPC-TC and 6) do not induce cytotoxicity under our cell imaging conditions.
The biological utility of DPPC-TC-ONOO– was first demonstrated by imaging live HeLa cells that are under oxidative stress (by PMA) and nitrosative stress (by IFN-γ/LPS) (Figures 4 and S11). For a physiologically relevant stress model, we employed the standard stimulation condition involving IFN-γ/LPS/PMA.12 Treatment of cells with IFN-γ/LPS leads to the endogenous production of ·NO, and PMA provokes increased levels of O2·–. These two reactive species combine to form ONOO−.4,5 HeLa cells treated with LNPs that contain DPPC-TC (positive control) exhibited strong fluorescence at 405/475 nm (Figure S11A). In contrast, those treated with LNPs containing DPPC-TC-ONOO– and left unstimulated (negative control) generated no observable signal (Figure S11B). Upon endogenous ONOO− generation through stimulation with IFN-γ/LPS/PMA, we observed a substantial increase in fluorescence at 405/475 nm (Figure 4A). Notably, the intracellular fluorescence became observable just after 3 min post PMA addition following the overnight IFN-γ/LPS treatment. This indicated that, in the background of stimulated iNOS function, PMA instantly leads to ONOO− production (by activating NADPH oxidase directly) and that DPPC-TC-ONOO– can rapidly sense proximal ONOO−. The change in signal intensity after 30 min was negligible, suggesting the possibility of probe consumption and/or lack of proximal ONOO−. We also evaluated the intracellular redox selectivity of DPPC-TC-ONOO– using N-(3-(aminomethyl)benzyl)-acetamidine, known as 1400W, which inhibits the production of ·NO by irreversibly binding to iNOS.40 The cells that were treated with 1400W only (Figure 4B) or treated with both 1400W and IFN-γ/LPS/PMA (Figure 4C) generated no observable signal at 405/475 nm. These controls showed that DPPC-TC-ONOO– selectively senses peroxynitrite generated from iNOS in response to nitrosative but not oxidative stress alone. To determine whether DPPC-TC-ONOO– localizes and functions at a specific subcellular compartment in HeLa, we carried out a comprehensive colocalization study with dyes specific for lipid-rich and membrane-enclosed organelles, including the ER, Golgi apparatus, mitochondrion, and lysosome (Figures 4D, 4E, and S11C–S11F). In addition, we used actin stain to gain a visual insight into the cytoskeletal structure of the whole cell. To assess the degree of colocalization, we determined the Pearson’s correlation coefficients (PCC) of the signals from DPPC-TC overlapping with that from the selected organelle tracker dye. The DPPC-TC signal overlapped almost perfectly (PCC = 0.938) with the perinuclear signal of ER-Tracker Green, suggesting a high statistical correlation and preference of DPPC-TC-ONOO– to localize in the ER. Cells stained with dyes specific for Golgi, mitochondria, and lysosomes displayed less significant correlation.
Figure 4.

Confocal imaging of lipid environments targeted by ONOO− in live HeLa cells
(A–E) Cells were incubated with LNPs (50 μM) and stained with organelle trackers and/or actin dye (CellMask Deep Red Actin Tracking Stain) prior to imaging. LNPs obtained from a 47.5: 47.5:5.0 M ratio of DOTMA, DOPE, and DPPC-TC-ONOO–. (A) Cells treated with LNPs, stimulated with IFN-γ/LPS/PMA. Time coarse images acquired upon PMA treatment. (B) Cells treated with LNPs and 1400W. (C) Cells treated with LNPs, then with 1400W and IFN-γ/LPS/PMA. (D and E) Quantitative colocalization study of cells treated with LNPs and stimulated with IFN-γ/LPS/PMA. Organelle trackers: ER-Tracker, MitoTracker, CellLight Golgi-RFP, LysoTracker. DPPC-TC channel: 405/475 nm. Scale bars: (A–D) 20 μm, (E) 5 μm.
We expanded our investigations to include murine-derived macrophage model, RAW 264.7 (Figures 5 and S12). RNS play important roles in macrophage activation and differentiation as part of the inflammatory response.54 Treatment of RAW 264.7 cells with LPS leads to the expression of iNOS, which produces ·NO and an increasing O2·– formation from multiple sources, ultimately generating ONOO−. RAW 264.7 cells were treated with LNPs containing either DPPC-TC (positive control, Figure S12A) or DPPC-TC-ONOO– (Figures 5 and S12A–S12F). Those treated with LNPs containing DPPC-TC-ONOO– and left unstimulated generated negligible levels of signal at 405/475 nm (Figure S12A). Endogenous ONOO− production through stimulation with LPS led to a substantial increase in fluorescence intensity over 16 h (Figure 5A). The intracellular redox selectivity of DPPC-TC-ONOO– was evaluated using 1400W. The cells treated with 1400W only (Figure 5B) or treated with 1400W and LPS (Figure 5C) generated little-to-no signal in the DPPC-TC channel. Assessment of DPPC-TC-ONOO– localization in RAW 264.7 cells was carried out using the ER, Golgi, mitochondrion, and lysosome trackers (Figures 5D, 5E, and S12C–S12F). The PCC values for the merged images of ER (0.946) and Golgi (0.729) suggested that DPPC-TC-ONOO– localizes primarily in the ER and then Golgi. This observation suggests that the probe localization in the ER is predominant but not exclusive, likely due to the abundance of ER-Golgi intermediate compartment (ERGIC) in RAW 264.7 cells and increased prevalence in cross-talk between the ER and Golgi.55 For both cell types, the use of LNPs appears to be critical for the probe localization. Our control experiments with LNPs containing Liss-Rhod PE showed high statistical correlations (PCC of 0.76–0.94) with the ER tracker (Figures S13A and S13B). Additionally, stimulated cells showed overlapping signals of Liss-Rhod PE and DPPC-TC (Figures S13C and S13D). In contrast to the observations with DPPC-TC-ONOO–, its hydrophilic counterpart, TEG-TC-ONOO–, displayed inferior specificity in regard to subcellular localization (Figure S14). It is worth noting that the use of LNPs proved unsuccessful for cellular uptake of either TEG-TC or TEG-TC-ONOO–. Therefore, cells were directly treated with TEG-TC (positive control, Figures S14A, S14B, S14E, and S14F) or TEG-TC-ONOO– (Figures S14C, S14D, S14G, and S14H) and incubated for 20 min. Those treated with TEG-TC-ONOO– were then stimulated via the methods described above. Both the positive controls and stimulated cells displayed TEG-TC signal throughout the cells, including the nucleus. In the case of RAW 264.7 cells, the intracellular homogeneity of the signals were especially substantial (Figures S14E and S14G).
Figure 5.

Confocal imaging of lipid environments targeted by ONOO− in live RAW246.7 cells
(A–E) Cells were seeded into glass microscope dishes, incubated with LNPs (50 μM) and stained with organelle trackers and/or actin dye prior to imaging. LNPs obtained from a 47.5: 47.5:5.0 M ratio of DOTMA, DOPE, and DPPC-TC-ONOO–. (A) Cells treated with LNPs, stimulated with LPS. Time coarse images acquired after the addition of LPS. (B) Cells treated with LNPs and 1400W. (C) Cells treated with LNPs, then with 1400W and LPS. (D and E) Quantitative colocalization study of cells treated with LNPs and stimulated with LPS. Scale bars: (A–D) 20 μm or (E) 5 μm.
Lipid nanoparticles carrying DPPC-TC-ONOO– allow for detecting nitrative stress in lung tissue murine models of ex vivo and in vivo acute lung injury
After successfully imaging the iNOS-dependent ONOO− activity in live mammalian cells, we sought to explore the potential of DPPC-TC-ONOO– for detecting ONOO− generated in lung tissue murine models of ex vivo56 and in vivo42 ALI (Figure 6). To find an optimal LNP concentration for PCLS, we performed a dose-response assay with DPPC-TC (25, 50, 75, 100, 150 μM) in unexposed PCLS (Figure S15), which served as the positive control in confocal images of PCLS (Figure S16). No cytotoxicity was observed for LNP concentrations of 25–100 μM. Based on this result, subsequent PCLS studies were performed with 75 μM LNPs (effective DPPC-TC-ONOO– concentration of ∼4 μM) (Figures 6A and S16). In control PCLS (NM-untreated) from C57BL6/J (WT) mice, LNPs carrying DPPC-TC-ONOO– led to a weak fluorescence signal (405 nm excitation) around the bronchial epithelium, presumably showing basal nitrative stress (Figure 6A, column 1). In PCLS exposed to NM, the fluorescence signal was qualitatively stronger and primarily localized to the bronchial epithelium (Figure 6A, column 2). The signal from DPPC-TC did not overlap with that from MitoTracker. Notably, in PCLS obtained from Nos2−/− mice, weak fluorescence were detected for both the control and NM-treated samples in comparison to PCLS from C57BL6/J mice (Figure 6A, columns 3 and 4). These results indicate that the fluorescence generation from DPPC-TC-ONOO– in the lung tissue is both NM-dependent and iNOS-dependent, validating that the function of DPPC-TC-ONOO– is specific to nitrative stress. To our best knowledge, this is the first example of directly detecting the in situ generation of ONOO− in PCLS.
Figure 6.

Investigation of the alveolar nitrative stress ex vivo and in vivo
(A) PCLS obtained from C57BL6/J (WT) and Nos2−/− mice were either untreated (NM(−)) or exposed to nitrogen mustard (NM(+)). After 24 h, PCLS were incubated with LNPs carrying DPPC-TC-ONOO– for 1 h and then imaged. (n = 3 PCLS). Scale bar: 100 μm.
(B) Illustration and timeline of ITB-induced ALI model and LNP instillation. Quadrant plots were constructed from flow cytometry analyses of myeloid-derived BAL cells for migratory phenotype (CD11b+) and DPPC-TC-ONOO– probe activity. Quadrant plots are shown for individual mice, which were used to quantify DPPC-TC as a measure of ONOO− generation. Gating threshold applied for each marker and DPPC-TC was based on fluorescent signals of antibody or DPPC-TC-stained and unstained BAL cells. (n = 3 per group).
The effect of the phospholipid component of DPPC-TC-ONOO– on detecting ONOO− was evaluated for both control and NM-exposed PCLS of C57BL6/J mice (Figure S16). Compared to the use of 75 μM LNPs, which have ∼4 μM DPPC-TC-ONOO–, use of 75 μM TEG-TC-ONOO– generated considerably weak signals within PCLS exposed to NM following excitation at 405 nm. This suggested that the hydrophilic nature of the TEG-TC-ONOO– may hinder its ability to localize in the tissue, which may cause its diffusion. A similar comparison can be made for the positive controls where PCLS samples were incubated with DPPC-TC or TEG-TC. The signal from those incubated with 75 μM LNPs containing ∼4 μM DPPC-TC was more localized and brighter compared to those incubated with 75 μM TEG-TC. Collectively, these control experiments show that TEG-TC-ONOO–, the lipid-free analog of DPPC-TC-ONOO–, lack the ability to localize in membranes or to generate detectable signal in PCLS under nitrative stress.
Next, we employed a previously reported ITB method, which causes a severe inflammatory response characterized by the infiltration of immune cells, which is dependent upon iNOS activation.18,42 During in vivo ALI, release of cytokines and chemokines, such as tumor necrosis factor-alpha (TNF-α) and interleukin-1 beta (IL-1β), recruit macrophages and other immune cells to the site of injury.57 During the initial phase of injury (d0–d3), pro-inflammatory macrophages are dominant, which is associated with higher levels of iNOS expression.18,42 For a generalizable and biocompatible instillation protocol, we first explored LNP concentrations ranging from 0.5 to 2.0 mM. We used LNPs carrying DPPC-TC (5 mol % of the lipid content) in C57BL6/J mice and analyzed the viability of the cells from BAL via flow cytometry (Figure S17). Results indicated 94% viability at 0.5 mM LNP concentration, which we employed for the subsequent experiments. On d0, mice (C57BL6/J or Nos2−/−) were anesthetized and received an intratracheal instillation of either PBS (control) or bleomycin (Figure 6B). To detect the ONOO− generation during the early inflammatory response, mice were again anesthetized and instilled with 0.5 mM LNPs containing DPPC-TC-ONOO– (5 mol % of the lipid content) on day 3. The animals were sacrificed, and BAL was collected for immunostaining 3 h-post instillation. Flow staining58 was used to define myeloid derived cells (CD45+), migratory (CD11b+) and pulmonary in nature (CD11c+) (Figure S18). We observed a significant increase in recruited population as a result of ITB for both WT (13 ± 2.4%) and Nos2−/− (17 ± 6.1%) compared to healthy WT (2 ± 0.8%) (Figure S19). This increase was also observed for the migratory, and hence activated, population due to ITB in WT (28 ± 6.6%) and Nos2−/− (20 ± 8.2%), compared to control WT (2 ± 0.5%) (Figure S19). The migratory population was further analyzed for DPPC-TC signal quantification (Figure 6B; Table 2). As indicated by the increase in migratory macrophages in both C57BL6/J and Nos2−/− mice following ITB, inflammatory activation was not significantly altered by the lack of ·NO generation. The degree of DPPC-TC fluorescence generated in response to ITB in Nos2−/− (entry 3) was no different from the control (entry 1). However, the fluorescence output was increased (∼30%) in C57BL6/J with ITB (entry 2). These data indicate that DPPC-TC-ONOO– was successfully delivered via LNPs to the BAL cells in vivo and was able to directly quantify the production of ONOO− in ALI.
Table 2.
Flow cytometry analysis of activated alveolar macrophages
| Entry | Treatment | DPPC-TC p:n ratio |
|---|---|---|
| 1 | WT–PBS | 0.55 ± 0.021 |
| 2 | WT–ITB | 0.72 ± 0.008 |
| 3 | Nos2−/−ITB | 0.56 ± 0.018 |
Characterization of the DPPC-TC positive-to-negative (p:n) ratio in CD11b+ macrophage population from BAL cells (n = 3 per group, values are defined as mean± standard error of mean).
Discussion
Lipid membranes and lipid-rich organelles are targets of peroxynitrite (ONOO−), a highly reactive species generated under nitrative stress. Detection of ONOO− in lipid environments could significantly advance our understanding of nitrative stress at the tissue, cellular, and subcellular levels, but has remained to be an unsolved challenge. In this work, we describe the development and diverse applications of DPPC-TC-ONOO–, a membrane-localized, biocompatible probe that allows for the investigation of lipid environments targeted by ONOO−.
DPPC-TC-ONOO– and POPC self-assemble to biomimetic membranes in the form of GVs that respond to ONOO− by lighting up, remain physically intact, and show excellent selectivity against other RONS. DPPC-TC-ONOO– was successfully delivered into HeLa (Figures 4 and S11) and RAW 264.7 (Figures 5 and S12) via LNPs made of DOTMA and DOPE. These cell types can generate both NOS-derived ·NO and increased oxidative stress upon stimulation. Within HeLa cells, the production of these reactive species can be separately stimulated. NOS activation is dependent upon IFN-γ/LPS treatment, while NADPH oxidase is stimulated to produce O2·– by PMA administration. This was clearly demonstrated via the detection of fluorescence signal within just 3 min of PMA stimulation following overnight treatment with IFN-γ/LPS (Figure 4A). The iNOS-mediated production of ONOO− upon LPS treatment results in significant subcellular fluorescence enhancement in RAW 264.7 cells. Importantly, in macrophages oxidative stress results from multiple sources and, thus, the LPS-mediated induction of ·NO production is sufficient to produce ONOO− (Figure 5A). Comprehensive colocalization studies indicated that DPPC-TC-ONOO– localizes primarily in the ER for each cell type. Of significant note, LNP-mediated delivery plays a predominant role for the localization of DPPC-TC-ONOO– to the ER, which was supported through the use of LNPs carrying Liss-Rhod PE, the fluorescent control lipid (Figure S13). Additionally, control samples using the iNOS inhibitor 1400W indicated that DPPC-TC-ONOO– is selective to iNOS.
TEG-TC-ONOO–, the hydrophilic analog of DPPC-TC-ONOO–, exhibits poor site-specificity, especially in RAW 264.7 (Figure S14), lacks the ability to localize in membranes or to generate detectable signal in PCLS under nitrative stress. Furthermore, both DPPC-TC and DPPC-TC-ONOO– can localize in the lung lining of bronchioles and parenchyma (Figures 6A and S16). Unlike TEG-TC-ONOO–, which diffuses non-selectively throughout the lung lining, DPPC-TC-ONOO– allows for the localized visualization of nitrative stress due to ALI. A potential fluorescence activation of DPPC-TC-ONOO– due to hydrolysis, or another mechanism we currently do not know, is possible during cellular targeting, but is minor as shown in control samples (Figures S11B and S12B). DPPC-TC-ONOO– partitions into lipid membranes, thus, its relatively nonpolar sensor head group may bend into the bilayer and have a limited contact with water.
Qualitative confocal imaging of PCLS that were incubated with LNPs revealed nitrative stress primarily around bronchioles upon NM exposure. Finally, flow cytometry analysis of activated alveolar macrophages from a murine model of ALI demonstrated ONOO− production upon ITB challenge via fluorogenic activation of DPPC-TC-ONOO– (Figure 6B; Table 2). Through the use of C57BL6/J and Nos2−/− mice, both of the ex vivo and in vivo experiments validated that the function of DPPC-TC-ONOO– is iNOS-specific. Of note, distinguishing the presence of ONOO− from any other possible source of probe activation at low flux of ·NO or inadequate levels of oxidative stress is important and warrants future investigations.
Interest in expanding the chemical toolbox of designer lipids is surging and demand for applying these tools in both basic research and biomedicine is substantial.59,60,61,62,63,64 Our work opens a platform for the development of lipid self-assemblies capable of sensing a chemical cue, offering a new design opportunity for synthetic protocell compartments65,66,67,68 and a purely chemical perspective to cell-free sensing.69 In addition, delivery of designer lipids to cells and tissues via LNPs expands the current application landscape of nanocarriers, which are typically employed for delivering small molecules, sugars, peptides, proteins, and nucleic acids.70 DPPC-TC-ONOO– enables the real-time imaging of the ER during nitrative stress. The ER, which functions in protein synthesis and folding,71 must maintain its redox homeostasis to facilitate disulfide bridge formation in proteins.72 Additionally, the ER membrane is a key site for lipid peroxidation in ferroptosis,73 a non-apoptotic cell death mechanism characterized by the iron-dependent accumulation of lipid peroxides.74,75 These accumulating knowledge highlight the importance of ER-specific redox probing in live mammalian cells. Finally, compared to the widely used techniques that measure redox species indirectly, the use of DPPC-TC-ONOO– offers a facile and direct approach for investigating nitrative stress in lipid-rich tissues both ex vivo and in vivo. Modular design, convergent synthesis, and LNP-mediated delivery of DPPC-TC-ONOO– can serve as a blueprint for lipid-based probes to investigate other redox species within and proximal to lipid environments. The knowledge gained from this methodology can advance the understanding of lipid-specific redox biochemistry at the tissue, cellular, and subcellular levels.
Limitations of the study
Our study used LNPs without size or homogeneity optimizations. Microscopy imaging of cells was carried out with standard cell lines. For more clinical relevance, primary cell cultures must be used. Furthermore, we have not characterized the phenotypes of cells and tissue compartments that fluoresce around bronchioles in PCLS upon NM exposure. This work used murine models of ALI and is currently not applicable to humans or other animal models.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-mouse/human CD11b antibody | BioLegend | Cat # 101206; RRID: AB_312789 |
| Alexa Fluor® 647 anti-mouse CD11c antibody | BioLegend | Cat # 117312; RRID: AB_389328 |
| Alexa Fluor® 700 anti-mouse CD45 antibody | BioLegend | Cat # 103128; RRID: AB_493715 |
| TruStainFcXanti-mouse CD16/32 | BioLegend | Cat # 101320; RRID: AB_1574975 |
| Chemicals, peptides, and recombinant proteins | ||
| Bleomycin | Santa Cruz Biotechnology | Cat # sc-200134B |
| 1-2-dipalmitoyl-rac-glycerol | Millipore-Sigma | CAS # 40290-32-2 |
| 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | CAS # 26853-31-6 |
| 1,2-palmitoyl-sn-glycero-3-phosphocholine (DPPC) | Avanti Polar Lipids | CAS # 63-89-8 |
| 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) | Avanti Polar Lipids | CAS # 384833-00-5 |
| eFluor780-conjugated fixable viability dye | Invitrogen | Cat # 65-0865-14 |
| ER-Tracker™ Green | ThermoFischer Scientific | Cat # E34251 |
| MitoTracker Deep Red FM | ThermoFischer Scientific | Cat # M22426 |
| CellLightTM Golgi-RFP, | ThermoFischer Scientific | Cat # C10593 |
| LysoTrackerTM Deep Red | ThermoFischer Scientific | Cat # L12492 |
| CellMask Deep Red Actin dye | ThermoFischer Scientific | Cat # A57248 |
| Opti-MEM | ThermoFischer Scientific | Cat # 11058021 |
| Critical commercial assays | ||
| (MTT) cell proliferation assay kit | ATCC | 30-1010K |
| LDH kit | Roche Applied Science | 06343767001 |
| WST-1 reagent | Roche Applied Science | 11644807001 |
| Experimental models: Cell lines | ||
| HeLa | ATCC | CRM-CCL-2 |
| RAW 264.7 | ATCC | TIB-71 |
| Experimental models: Organisms/strains | ||
| C57BL6/J WT mice | Jackson Laboratories | Strain # 000664 |
| NOS2-/- mice | Jackson Laboratories | Strain # 002609 |
| Software and algorithms | ||
| ChemDraw 22.2.0 | PerkinElmer | v22.2.0 |
| GraphPad Prism | GraphPad | v8 |
| ImageJ | National Institutes of Health and the Laboratory for Optical and Computational Instrumentation | https://imagej.nih.gov/ij/ |
| Adobe Illustrator | Adobe | v2021 |
| Imarius x64 | Oxford Instruments | v9.0.1 |
| BioRender | BioRender | https://www.biorender.com/ |
| Kaluza 2.1 | Beckmann Coulter | https://www.beckman.com/flow-cytometry/software/ |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Enver Cagri Izgu (ec.izgu@rutgers.edu).
Materials availability
Compounds generated in this study will be made available upon reasonable request.
Data and code availability
-
•
Any primary data in this study can be requested from the lead contact.
-
•
This study did not generate any new code.
-
•
Any additional information to reanalyze the data can be requested from the lead contact.
Experimental model and study participant details
HeLa cells (ATCC, Cat #CRM-CCL-2) and RAW 264.7 (ATCC, Cat # TIB-71) were used for the cell-based study with the probe. 6- to 8-week-old C57BL6/J (WT) and Nos2–/– mice bred in-house but originally derived from Jackson Laboratories (Bar Harbor, ME, USA) were used in this study for in vivo and ex vivo study.
Method details
Materials
1-2-dipalmitoyl-rac-glycerol, copper (II) sulfate (CuSO4), 2-{2-[2-(2-propynyloxy)-ethoxy]-ethoxy}ethanol, 1,1,1-trifluoro-4-(4-hydroxyphenyl)butan-2-one, ethylene chlorophosphite, coumarin 343 were purchased from Millipore-Sigma. 3-azido-7-hydroxycoumarin was purchased from Biosynth International Inc. Tris(3-hydroxypropyltriazolyl-methyl)amine (THPTA) was purchased from Click Chemistry Tools. 1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-palmitoyl-sn-glycero-3-phosphocholine (DPPC), and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (ammonium salt) were purchased from Avanti Polar Lipids. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) cell proliferation assay kit was purchased from ATCC. Dulbecco's modified Eagle’s medium/Nutrient Mixture F-12 (DMEM/F-12 HAM), low-gelling temperature agarose, and gentamicin were purchased from Millipore Sigma, penicillin/streptomycin was obtained from (Gibco), LDH kit and WST-1 reagent were purchased from Roche. Bleomycin sulphate was purchased from Santa Cruz Biotechnology, ketamine and xylazine from Fort Dodge Animal Health, Fort Dodge, IA) and eFluor 780-conjugated fixable viability dye from Invitrogen. TruStain FcX anti-mouse CD16/32 antibody (Fc Block), FITC anti-mouse/human CD11b antibody, Alexa Fluor® 647 anti-mouse CD11c antibody, Alexa Fluor® 700 anti-mouse CD45 antibody were purchased from BioLegend. All other chemical materials, including the common reagents for synthesis, buffers, and media, were obtained from commercial sources as detailed in Supporting Information. Milli-Q® water was used in dilutions and preparations of buffers and stock solutions unless otherwise specified.
General synthetic methods
All reactions were performed under a dry nitrogen atmosphere unless otherwise stated. All glassware was oven-dried before use. Purification of the synthesized compounds was performed using a Büchi Reveleris® flash chromatography system equipped with a FlashPure EcoFlex Diol (50 μm spherical) column or a silica (50 μm irregular) column. Nuclear magnetic resonance (NMR) spectroscopic analyses was carried out using a Bruker Avance Neo 500 MHz spectrometer. NMR data is provided for new compounds. 1H NMR spectra were acquired at 500 MHz, 13C NMR spectra were acquired at 126 MHz, 31P NMR spectra were acquired at 202 MHz, and 19F NMR spectra were acquired at 471 MHz. Chemical shifts (δ) for 1H NMR spectra were referenced to (CH3)4Si at δ = 0.00 ppm or to CHCl3 at δ = 7.26 ppm. 13C NMR spectra were referenced to CDCl3 at δ = 77.23 ppm. 31P NMR spectra were referenced externally to Ph3PO dissolved in CDCl3 at δ = 29.06 ppm. The external reference solution was placed in a co-axial NMR tube (∼0.1 mL effective volume), which was then inserted into the NMR tube containing the analyte solution (∼0.4 mL). The following abbreviations are used to describe 1H NMR resonances: s (singlet), d (doublet), t (triplet), m (multiplet), dd (doublet of doublets), br (broad), and app (apparent). Coupling constants (J) are reported in Hz. Liquid chromatography followed by high-resolution mass spectrometry (LC-HRMS) analysis from electrospray ionization (ESI) was carried out on a Waters Acquity-Xevo G2-XS QTof instrument. Low-resolution mass spectrometry (LRMS) analysis was performed using a Finnigan LCQ™ DUO mass spectrometer. Both fluorescence and UV-Vis absorbance measurements were conducted using a Spectra Max ID3 instrument (Molecular Devices). Dynamic light scattering (DLS) measurements were carried out on a Zetasizer Nano C (Malvern Panalytical). Confocal fluorescence microscopy was performed on a Leica TCS SP8 microscope equipped with a 63x, 1.40 NA oil immersion objective and a Tokai Hit stage-top incubator. Images were processed using LasX Lightning deconvolution. All statistical analyses were performed using GraphPad Prism software version 9.
Preparation procedures and characterization data for small molecules
Synthesis of 3-azido-2-oxo-2H-chromen-7-yl (4-(4,4,4-trifluoro-3-oxobutyl)phenyl) carbonate (compound 3)

To a dry 5-mL round-bottom flask was added triphosgene (41 mg, 0.140 mmol, 0.60 equiv) and 0.5 mL of toluene under a nitrogen-rich atmosphere. The solution was cooled to 0°C and pyridine (28 μL, 0.340 mmol, 1.5 equiv) in 0.5 mL of toluene, followed by 1,1,1-trifluoro-4-(4-hydroxyphenyl)butan-2-one (50 mg, 0.230 mmol, 1.0 equiv) in 1.0 mL of toluene were added dropwise. The reaction mixture was allowed to stir at room temperature (rt) and monitored by TLC until completion (3 hours). The resulting mixture was concentrated under reduced pressure, affording the chloroformate intermediate S1. The thick residue was re-dissolved in 1.0 mL of CH2Cl2 and cooled to 0°C while being stirred vigorously and purged with nitrogen gas continuously. To this mixture, 3-azido-7-hydroxycoumarin (2) (32 mg, 0.160 mmol, 1.0 equiv) in 0.5 mL CH3CN and pyridine (25 μL, 0.460 mmol, 2.0 equiv) in 0.5 mL of CH2Cl2 were added dropwise. The reaction was allowed to stir at rt for 4 h. The crude mixture was then concentrated to an oil under reduced pressure and purified by silica gel column chromatography (mobile phase: CH2Cl2 / MeOH, step gradient from 0 to 2% MeOH). The product fractions were collected and concentrated under reduced pressure, providing the carbonate 3 (43 mg, 60%) as a white solid.
1H NMR (500 MHz, CDCl3): δ 7.47-7.45 (d, J = 8.6 Hz, 1H), 7.33 (d, J = 2.3 Hz, 1H), 7.28 (s, 1H), 7.27–7.21 (m, 5H), and 3.06–3.02 (m, 4H).
13C NMR (126 MHz, CDCl3): δ 190.3 (C=O), 157.0 (C=O), 151.8, 151.6, 151.4, 149.5, 137.7, 129.6, 129.5, 128.0, 126.4, 124.9, 121.2, 118.3, 117.5, 109.6, 37.9, and 27.7.
19F NMR (471 MHz, CDCl3): δ -79.2 (s).
HRMS (ESI) m/z: Calculated for C20H11F3N3O6–, [M – H]–, requires 446.0605; found 446.0668.
Synthesis of 3-(4-((2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)-2-oxo-2H-chromen-7-yl (4-(4,4,4-trifluoro-3-oxobutyl)phenyl) carbonate (TEG-TC-ONOO–)

All of the solutions were bubbled with N2 gas before and after being mixed. To a stirred solution of the propargyl-TEG-OH (5) (3.2 mg, 0.017 mmol, 1.5 equiv) and the carbonate 3 (5 mg, 0.011 mmol, 1.0 equiv) in 2.0 mL of THF was added a mixture of copper (II) sulfate (1.0 mg, 0.006 mmol, 0.5 equiv), THPTA (4.8 mg, 0.011, 1.0 equiv) in 0.1 mL of water. To the resulting mixture was added sodium ascorbate (1.1 mg, 0.006 mmol, 0.5 equiv) in 0.1 mL of water. The reaction was stirred at rt for 24 hours under a nitrogen atmosphere. The crude product was concentrated under reduced pressure and purified by silica gel column chromatography (mobile phase: CH2Cl2 / MeOH, step gradient from 0 to 5% MeOH). The product fractions were collected and concentrated under reduced pressure, providing TEG-TC-ONOO– (4 mg, 57 %) as a clear film.
1H NMR (500 MHz, CDCl3): δ 8.70 (s, 1H), 8.60 (s, 1H), 7.74–7.72 (d, J = 2.4 Hz, 2H), 7.45 (d, J = 8.5 Hz, 1H), 7.38–7.36 (dd, J = 8.7, 2.2 Hz, 1H), 7.29–7.23 (m, 4H), 4.80 (s, 2H), 3.76–3.62 (m, 12H), and 3.07–3.03 (m, 4H)
13C NMR (126 MHz, CDCl3): δ 190.3, 155.4, 153.8, 153.1, 151.1, 149.4, 145.5, 137.8, 132.4, 129.9, 129.6, 129.5, 123.9, 122.8, 121.0, 118.8, 116.2, 109.7, 72.5, 70.7, 70.6, 70.4, 69.9, 64.4, 61.8, 37.9, and 27.7.
19F NMR (471 MHz, CDCl3): δ -79.2 (s).
LRMS (ESI) m/z: Calculated for C29H27F3N3O10–, [M – H]–, requires 634.17; found 634.19.
Synthesis of 3-((2-oxido-1,3,2-dioxaphospholan-2-yl)oxy)propane-1,2-diyl dipalmitate (compound S2)

To a dry 25-mL round-bottom flask was added 1-2-dipalmitoyl-rac-glycerol, (500 mg, 0.850 mmol, 1.0 equiv) in 5.0 mL of benzene under a dry nitrogen atmosphere. The mixture was brought to 0°C and TEA (0.50 mL, 3.75 mmol, 4.4 equiv) was added dropwise. After 15 minutes of stirring, ethylene chlorophosphate (0.75 mL, 8.16 mmol, 9.6 equiv) was added dropwise. The reaction was allowed stirred at rt for 24 hours. The solution was quenched with saturated NaHCO3 and washed with EtOAc three times. The collected organic layers were dried with sodium sulfate and concentrated under reduced pressure. The cyclic phosphate triester product S2 (44 mg, 93%) was obtained as a white solid and used without further purification.
1H NMR (500 MHz, CDCl3): δ 5.19–5.17 (p, J = 4.8 Hz, 1H), 4.39–4.35 (dd, J = 11.8, 3.9 Hz, 2H), 4.34–4.12 (m, 5H), 4.11–4.08 (dd, J = 11.5, 6.0 Hz, 1H), 2.28 (t, J = 7.5, Hz, 2H), 2.25 (t, J = 7.5, Hz, 2H), 1.55–1.54 (br s, 4H), 1.21–1.19 (br s, 48H), and 0.82–0.80 (app t, J = 6.7 Hz, 6H).
13C NMR (126 MHz, CDCl3): δ 173.4 (C=O), 173.1 (C=O), 69.5, 69.4, 66.8, 66.7, 66.22 (overlapped), 66.20 (overlapped), 61.5, 34.3, 34.2, 32.1, 29.9 (overlapped), 29.83, 29.82, 29.80, 29.78, 29.65, 29.64, 29.5, 29.45, 29.43, 29.3, 29.2, 25.0, 24.9, 24.8, 22.9, and 14.3.
31P NMR (202 MHz, CDCl3): δ 17.74 (s).
LRMS (ESI) m/z: Calculated for C37H71O8PNa+, [M + H]+, requires 697.48; found 697.50.
Synthesis of 2,3-bis(palmitoyloxy)propyl (2-(dimethyl(prop-2-yn-1-yl)ammonio)ethyl) phosphate (the alkynyl lipid 4)

In a dry 10-mL pressure tube, S2 (100 mg, 0.160 mmol, 1 equiv) in 2.0 mL of dry CH3CN was added. 3-Dimethylamino-1-propyne (540 mg, 6.40 mmol, 40 equiv) in 3 mL of dry CH3CN were added to a dry 5 mL round bottom with sodium sulfate, bubbled with nitrogen and stirred for 30 minutes. The mixture was then transferred to the solution containing S2 and sealed and stirred for 60 h under a dry nitrogen environment at 65°C. The reaction was diluted in CHCl3, filtered, and concentrated to an oil under reduced pressure. The crude product was purified by silica gel chromatography with a diol column (mobile phase: CHCl3 / MeOH / 7 M NH4OH, step gradient from 0 to 25% MeOH / 7 M NH4OH mixture, 6:1 v/v). The product fractions were collected and concentrated under reduced pressure, providing the alkynyl lipid 4 (49 mg, 58%) as a white solid.
1H NMR (500 MHz, CDCl3): δ 5.22 (m, 1H), 4.69 (s, 2H), 4.42–4.39 (dd, J = 12.0, 3.1 Hz, 1H), 4.33 (br s, 1H), 4.14 (dd, J = 12.0, 7.1 Hz, 1H), 3.96–3.92 (br s, 1H), 3.90–3.87 (br s, 2H), 3.43 (s, 6H), 2.93 (s, 1H), 2.30 (t, J = 7.5 Hz, 2H), 2.28 (t, J = 7.5 Hz, 2H), 1.52–1.51 (br s, 4H), 1.18 (br s, 48H), and 0.88 (app t, J = 7.0 Hz, 6H).
13C NMR (126 MHz, CDCl3): δ 173.8 (C=O), 173.4 (C=O), 81.5, 72.3, 70.8, 64.4, 63.7, 63.1, 59.4, 55.5, 51.8, 34.6, 34.4, 32.2, 30.0 (overlapped), 29.92, 29.89, 29.78, 29.76, 29.59 (overlapped), 29.56, 29.41, 29.38, 27.8, 25.2, 25.1, 22.9, and 14.3.
31P NMR (202 MHz, CDCl3): δ -0.73 (s).
HRMS (ESI) m/z: Calculated for C42H81NO8P+, [M + H]+, requires 758.5694; found 758.5727.
Synthesis of the DPPC-TC-ONOO–

All of the solutions were bubbled with N2 gas before and after being mixed. To a stirred solution of the the alkynyl lipid 4 (15.4 mg, 0.034 mmol, 1.3 equiv) and the coumarin azide 3 (20.0 mg, 0.026 mmol, 1.0 equiv) in 2.0 mL of THF was added a mixture of copper sulfate (2.0 mg, 0.013 mmol, 0.5 equiv) and THPTA (5.6 mg, 0.013, 0.5 equiv) in 0.1 mL of water. To the resulting mixture was added sodium ascorbate (5.2 mg, 0.026 mmol, 1.0 equiv) in 0.1 mL of water. After being stirred for 24 hours, the reaction mixture was concentrated under reduced pressure and the crude product was purified by silica gel chromatography with a diol column (mobile phase: CH2Cl2 / MeOH / H2O, step gradient from 0 to 35% MeOH / H2O mixture, 30:1 v/v). The product fractions were collected and concentrated under reduced pressure, providing DPPC-TC-ONOO– (8.3 mg, 43%) as a clear film.
1H NMR (500 MHz, CDCl3 / CD3OD, 3:1 v/v) δ 9.11 (s, 1H), 8.67 (s, 1H), 7.86–7.85 (d, J = 8.4 Hz, 1H), 7.55 (s 1H), 7.44 (s, 1H), 7.31–7.29 (d, J = 8.5 Hz, 2H), 7.24–7.22 (d, J = 8.3 Hz, 2H), 5.30–5.18 (m, 1H), 4.86 (s, 2H), 4.43 (dd, J = 12.1, 3.2 Hz, 1H), 4.36 (bs, 2H), 4.17 (dd, J = 12.0, 6.9 Hz, 1H), 4.04 (bs, 2H), 3.66 (bs, 2H), 3.41–3.39 (m, 4 H), 3.25 (s, 6H), 2.32 (m, 4H), 1.59 (bs, 4H), 1.26 (bs, 48H), and 0.88 (t, J = 7.0 Hz, 6H).
13C NMR (126 MHz, CDCl3 / CD3OD, 3:1 v/v) δ 177.9, 177.5, 159.5, 158.3, 157.4, 155.3, 152.9, 144.2, 139.6, 138.3, 134.3, 133.4, 133.1, 126.3, 124.6, 123.0, 119.9, 113.7, 74.3, 68.2, 67.5, 66.6, 63.1, 62.4, 55.2, 39.5, 38.1, 37.9, 35.8, 33.53 (overlapped), 33.51, 33.49 (overlapped), 33.38, 33.36, 33.19 (overlapped), 33.15, 32.98, 32.96, 31.9, 28.8, 28.7, 26.5, and 17.7.
31P NMR (202 MHz, CDCl3 / CD3OD, 3:1 v/v) δ -4.38 (s).
19F NMR (471 MHz, CDCl3 / CD3OD, 3:1 v/v): δ -77.6 (s).
HRMS (ESI) m/z: Calculated for C62H93F3N4O14P+, [M + H]+, requires 1205.6373; found 1205.6379.
Synthesis of 7-hydroxy-3-(4-((2-(2-(2-hydroxyethoxy)ethoxy)ethoxy)methyl)-1H-1,2,3-triazol-1-yl)-2H-chromen-2-one (TEG-TC)

All of the solutions were bubbled with N2 gas before and after being mixed. To a stirred solution of the propargyl-TEG-OH (5) (83.2 mg, 0.44 mmol, 1.50 equiv) and 3-azido-7-hydroxycoumarin (2) (60.0 mg, 0.295 mmol, 1.00 equiv) in 10.0 mL of THF was added a mixture of copper sulfate (29.0 mg, 0.15 mmol, 0.50 equiv) and THPTA (65.1 mg, 0.15 mmol, 0.50 equiv) in 0.5 mL of water. To the resulting mixture was added sodium ascorbate (59.4 mg, 0.3 mmol, 1.00 equiv) in 0.5 mL of water. After being stirred for 12 hours, the reaction mixture was concentrated under reduced pressure and the crude product was purified by silica gel column chromatography (mobile phase: CH2Cl2 / MeOH, step gradient from 0 to 5% MeOH). The product fractions were collected and concentrated under reduced pressure, providing TEG-TC (63 mg, 55%) as a clear film.
1H NMR (500 MHz, CD3OD) δ 8.57 (s, 1H), 8.49 (s, 1H), 7.64 (d, J = 8.6 Hz, 1H), 6.89 (dd, J = 8.5, 2.3 Hz, 1H), 6.81 (d, J = 2.2 Hz, 1H), 4.73 (s, 2H), 3.79–3.61 (m, 10H), and 3.55 (m, 2H)
13C NMR (126 MHz, CD3OD) δ 163.5, 156.8, 155.3, 144.6, 135.9, 130.5, 124.6, 119.2, 114.4, 110.4, 102.1, 72.3, 70.2, 70.1, 70.0, 69.4, 63.5, and 60.8.
HRMS (ESI) m/z: Calculated for C18H20N3O7–, [M – H]–, requires 390.1307; found 390.1308.
Synthesis of DPPC-TC

All of the solutions were bubbled with N2 gas before and after being mixed. To a stirred solution of the alkynyl lipid 4 (42.0 mg, 0.055 mmol, 1.0 equiv) and 3-azido-7-hydroxycoumarin (2) (14.6 mg, 0.072 mmol, 1.3 equiv) in 4.0 mL of THF was added a mixture of copper sulfate (4.4 mg, 0.027 mmol, 0.5 equiv) and THPTA (12.8 mg, 0.027 mmol, 0.5 equiv) in 0.2 mL of water. To the resulting mixture was added sodium ascorbate (11.0 mg, 0.055 mmol, 1.0 equiv) dissolved in 0.2 mL of water. After being stirred for 12 hours, the reaction mixture was concentrated under reduced pressure and the crude product was purified by silica gel chromatography with a diol column (mobile phase: CH2Cl2 / MeOH / H2O, step gradient from 0 to 35% MeOH / H2O mixture, 30:1 v/v). The product fractions were collected and concentrated under reduced pressure, providing DPPC-TC (30.1 mg, 57%) as a brown solid.
1H NMR (500 MHz, CDCl3 / CD3OD, 3:1 v/v) δ 9.00 (s, 1H), 8.50 (s, 1H), 7.57 (d, J = 8.7 Hz, 1H), 6.94 (dd, J = 8.6, 2.4 Hz, 1H), 6.89 (d, J = 2.4 Hz, 1H), 5.30–5.18 (m, 1H), 4.84 (s, 2H), 4.43 (dd, J = 12.1, 3.2 Hz, 1H), 4.36 (bs, 2H), 4.17 (dd, J = 12.0, 6.9 Hz, 1H), 4.03 (t, J = 6.3 Hz, 2H), 3.65 (bs, 2H), 3.25 (s, 6H), 2.32 (m, 4H), 1.59 (bs, 4H), 1.26 (bs, 48H), and 0.88 (t, J = 7.0 Hz, 6H)
13C NMR (126 MHz, CDCl3 / CD3OD, 3:1 v/v) δ 177.9, 177.5, 167.4, 160.6, 159.2, 140.2, 139.2, 134.5, 132.8, 122.5, 119.0, 114.2, 106.8, 74.2, 68.2, 67.6, 66.6, 63.3, 62.8, 55.2, 38.1, 38.0, 35.8, 33.6, 33.5, 33.39, 33.37, 33.21, 33.17, 33.0, 32.98, 28.8, 28.7, 26.5, and 17.8.
31P NMR (202 MHz, CDCl3 / CD3OD, 3:1 v/v) δ -0.60 (s).
HRMS (ESI) m/z: Calculated for C51H84N4O11P–, [M – H] –, requires 959.5880; found 959.5889.
Preparation and usage of reactive nitrogen and oxygen species
The information given here concerns the fluorescence plate reader measurements and GV-based redox sensing. All of the reactive nitrogen and oxygen species were prepared under oxygen-free conditions by bubbling high-purity N2 gas into the reagent solutions and buffers. The stock solutions of ONOO–, ·NO, H2O2, and O2·– were prepared separately, while 1O2 and HO· were generated in situ in the presence of the probe (TEG-TC-ONOO– or the GVs containing DPPC-TC-ONOO–). The stock solutions were used immediately upon preparation. ONOO– solution was prepared according to a previously reported protocol.76 In a 20 mL vial, 50 mM of NaNO2 (0.5 mL) and 50 mM of H2O2 (0.5 mL) were stirred vigorously. To this mixture was rapidly added 1 M hydrochloric acid solution (1 mL), which was immediately followed by the addition of 1.5 M NaOH solution (1 mL). The resulting pH depends on how fast the NaOH solution is added following the addition of HCl. The concentration of ONOO– was determined by UV-Vis spectrophotometry, using the previously reported extinction coefficient (ε302) of 1670 M-1cm-1.77 ·NO solution (1.8 mM) was prepared based on a previously reported protocol.47 Specifically, 2.0 g NaNO2 was dissolved in 3 mL of water, followed by slow addition of 20 mL of an aqueous H2SO4 solution (2 M). The evolving gas was passed through a Tygon® tubing filled with NaOH pellets and bubbled into deoxygenated Tris buffer (50 mM, pH 7.5). H2O2 solution (8 mM) was prepared by diluting a commercial H2O2 solution (30 w/w%, 9.8 M). O2·– solution (8 mM) was prepared from KO2 dissolved in DMSO. The KO2 powder (2.6 mg) was added into DMSO (2 mL) and the resulting mixture was sonicated until the powder fully dissolved and the solution turned clear. The resulting solution (∼18 mM) was then diluted as necessary. HO· was generated in situ, with the probe being present, following a previously reported protocol.47 Specifically, we added 2.5 μL of aqueous Fe(ClO4)2 solution (18 mM) to 147.5 μL mixture of Tris buffer (50 mM, pH 7.5), TEG-TC-ONOO– (1 μM), and H2O2 (36 mM). For confocal microscopy of GVs, 5 μL of H2O2 (16 mM) and 5 μL of iron perchlorate (8 mM) were mixed, which was used as the stock solution used for cell-free assays. 1O2 was generated in situ, with the probe being present, according to a previously reported protocol.47 Specifically, we added 2.5 μL of aqueous NaOCl (18 mM) to a 147.5 μL mixture of Tris buffer (50 mM, pH 7.5), TEG-TC-ONOO– (1 μM), and H2O2 (36 mM).
Determination of relative fluorescence quantum yield
The relative fluorescence quantum yields for TEG-TC-ONOO–, TEG-TC, DPPC-TC-ONOO–, and DPPC-TC were determined using a previously reported equation78 shown below:
where Qs is the quantum yield of the unknown, Qr is the quantum yield of a known dye, ms and mr are gradient of the plot of integrated fluorescence intensity against absorbance of the unknown and known dyes, respectively, and lastly, n (s and r) is the index of refraction. Absorbance (405 nm) and fluorescence intensity (λex/em of 405/475 nm) were measured. Coumarin 343 was used as the reference standard for Qr (0.63) in ethanol which was found directly from Sigma Aldrich. Ethanol was used for both the standard and the samples; the refractive index ratio is equal to 1. The quantum yields for TEG-TC-ONOO–, TEG-TC, DPPC-TC-ONOO–, and DPPC-TC were 0.08, 0.66, 0.09, and 0.64, respectively.
Microplate fluorescence measurements
1.5 μL of TEG-TC-ONOO– in DMSO (100 μM) and 5 μL of the stock solution of the redox-active species were added to a 96-well plate containing Tris buffer (50 mM, pH 7.5), for a total volume of 150 μL. At this final volume, the concentrations of TEG-TC-ONOO– and the redox-active species were predicted to be 1 μM and 600 μM, respectively. Fluorescence intensity (λex/em 405/475 nm) of the samples were measured on a microplate reader (Molecular Devices SpectraMax iD3 or Tecan Infinite Pro 2000) at the 1 and 60-minute marks. For peroxynitrite, additional measurements were taken at 3, 5, 10, 20, 30, 40, and 50 minutes. This protocol was also employed for TEG-TC (positive control). F/F0 was calculated by taking the final fluorescent measurements at the 60-minute mark over the initial fluorescence measurements at t0.
Preparation, usage, and confocal imaging of giant vesicles
GVs were prepared from stock solutions of POPC (5 mM), DPPC-TC-ONOO– (0.25 mM), and Liss-Rhod PE dye (45 μM) in CHCl3. A mixture of POPC, DPPC-TC-ONOO–, and Liss-Rhod PE (98.5 : 1.0 : 0.5 molar ratio) was taken into a gas tight Hamilton syringe and spread on the conductive side of an indium tin oxide (ITO) coated glass slide (only the central area). The glass slide was then placed under high vacuum for 1 hour, resulting in complete removal of CHCl3. The electroformation chamber (Figure S8) was assembled by the following steps: A short piece of copper tape was attached to each side of the silicon gasket, which was then placed on the conductive side of an untreated ITO-coated glass. The lipid-containing ITO-coated glass slide was taken from the high vacuum and placed on the top of the silicon gasket. The chamber was clamped with three binder clippers (two for the sides and one for the bottom) to ensure proper sealing. The chamber was then filled with a hydrating buffer while avoiding air bubbles. 250 μL of aqueous sucrose solution (400 mM) was added and the chamber was sealed with a small piece of clay. The sealed chamber was connected through its copper tapes to a function generator using alligator clippers, which completed the electroformation circuit. Electroformation process was carried out at 2 V and 10 Hz for 3 hours. Upon completion, the two side binder clippers were removed, and vesicles were harvested with a needle syringe. 50 μL aliquots of the vesicle solution (pH 8.5) were treated with 0.5 μL of the stock solution of the redox agent of choice. The reaction mixture was left undisturbed for 5 minutes. A 5 μL sample of the resulting mixture was then collected and mounted between a cover glass and cover slip (1.2 mm thickness), which was subsequently sealed using a clear nail polish. Z-stack images were acquired in Lightning mode using two tracks: DPPC-TC (405/475 nm) and rhodamine (560/610 nm) channels.
Protocols for Cell Studies
100k HeLa or RAW264.7 cells were seeded into a 35 mm glass microscope dish and cultured overnight in DMEM with 10% FBS at 37°C with 5% CO2. A mixture of DPPC-TC-ONOO– (5%), DOTMA (47.5%) and DOPE (47.5%) was dried under reduced pressure, then hydrated with 300 mM sucrose to get a final lipid concentration of 125 μM. Sonication of this mixture over 60 minutes provided LNPs. The LNP solution was then diluted with Opti-MEM™ to a final LNP concentration of 50 μM. This mixture was directly added to live cells grown in glass-bottomed dishes and the cells were incubated at 37°C for 3 hours for the complete uptake of LNPs. After 3 hours, the cell media was replaced with fresh Opti-MEM media containing appropriate stimulants, and cells were incubated overnight at 37°C with 5% CO2. For endogenous generation of peroxynitrite through stimulation in HeLa cells, a solution of IFN-γ (100 ng/mL) and LPS (1 mg/mL) in Opti-MEM (250 μL) was first added to HeLa cells. The resulting samples were incubated for 12 hours, then the media was decanted. Prior to imaging, a solution of PMA (10 nM) in HBSS (250 μL) was added and the resulting samples were incubated for 60 minutes at 37°C. For RAW cells, LPS (100 ng/mL) in Opti-MEM (250 μL) was added to the cells and the resulting samples were incubated for 16 hours overnight. For iNOS inhibition in cells, 1400W (20 μM) was introduced into Opti-MEM. Cells were then washed and the organelle tracker, dissolved in HBSS, was added.
Staining cells with organelle trackers or actin dye
ER-Tracker™ Green (1 μM, 200 μL, 510/540 nm excitation/emission, ER stain) applied at 37°C for 10 min; MitoTracker Deep Red FM (250 nM, 200 μL, 644/665 nm, Mitochondrion stain) applied at room temperature for 5 min; CellLight™ Golgi-RFP, BacMam 2.0 (15 μL in Opti-MEM media, 555/584 nm, Golgi stain) applied at 37°C, overnight; LysoTracker™ Deep Red (100 nM, 200 μL, 647/670 nm, lysosome stain) applied at 37°C for 60 min; CellMask Deep Red Actin dye (1 μM, 200 μL, 669/710 nm, actin stain) applied at 37°C for 10 min. After the stated incubation time, the dye solution was removed, cells were washed 3 times with PBS, and 1 mL of HBSS was added prior to imaging.
MTT assay for HeLa and RAW 264.7
Cytotoxicity of the probe DPPC-TC-ONOO–, its uncaged analog DPPC-TC, the oxa-spiro[4,5] decenone 6, and LNPs against HeLa and RAW 264.7 cells was determined using a standard MTT assay. DPPC-TC-ONOO– and DPPC-TC were each mixed with DOPE and DOTMA to form LNPs as described below. HeLa (10,000 cells/well) or RAW 264.7 (25,000 cells/well) were seeded in a 96-well tissue culture plate and cultured overnight to allow the attachment of the cells to the surface. The cell media was then replaced with 100 μL DMEM with 10% FBS containing the liposome or compound (6 or tamoxifen) of interest. After 24 hours, 10 μL of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT reagent) was added to each well and the plate was incubated at 37°C for 3 hours to yield formazan crystals. To these wells, 100 μL of “detergent reagent” (from the MTT assay kit) was added and the plate was incubated in dark at rt for 2 hours to allow the dissolution of the formazan crystals. The absorbance of each well was recorded at 570 nm on a plate reader. The OD570 value for the sample containing cell-free media (DMEM with 10% FBS) was subtracted from each reading. The OD570 values were then normalized to the value for the sample containing untreated cells. Using these values, we plotted the dose-response curves of HeLa (Figure S10A) or RAW 264.7 cells (Figure S10B), which provided IC50 values (Table S1) through a non-linear regression fitting model.
Live cell imaging with TEG-TC or TEG-TC-ONOO– probe
Both HeLa and RAW 264.7 cells were seeded into a 35 mm glass microscope dish and cultured overnight in DMEM with 10% FBS, 37°C, 5% CO2. A 20 μM solution of either TEG-TC or TEG-TC-ONOO– was directly added to live cells grown in glass-bottomed dishes and the cells were incubated at 37°C for 20 minutes. For endogenous stimulation of peroxynitrite in HeLa cells, a solution of IFN-γ (100 ng/mL) and LPS (1 mg/mL) in Opti-MEM (250 μL) was added to the cells. The sample was incubated for 12 hours, then the media was decanted. Prior to imaging, a solution of PMA (10 nM) in HBSS (250 μL) was added and the resulting sample was incubated for 60 minutes at 37°C. For endogenous generation of peroxynitrite through stimulation in RAW cells, LPS (100 ng/mL) in Opti-MEM (250 μL) was added to the cells and the resulting samples were incubated for 16 hours overnight.
Animal use, PCLS, instillation, and BAL extraction
All experiments were conducted with 6- to 8-week-old C57BL6/J (WT) and Nos2–/– mice bred in-house but originally derived from Jackson Laboratories (Bar Harbor, ME, USA). Food and water were provided ad libitum and mice were housed under standard conditions. All experiments were performed in compliance with Rutgers University Institutional Animal Care and the biosafety protocols approved by the institutional biosafety committee following the guidelines by U.S. National Institutes of Health Guide for the Care and Use of Laboratory Animals.
To prepare PCLS, mice were first anesthetized with an intraperitoneal injection of ketamine (135 mg/kg) and xylazine (30 mg/kg) and euthanized via exsanguination. The trachea was cannulated, and 1.5% low-melting point agarose was instilled into the lung and allowed to congeal. The lungs were separated and further embedded in additional agarose for stability when slicing. Slicing was performed using a Krumdieck Tissue Slicer (Alabama Research & Development) to achieve a slice thickness of 300 μm. PCLS were cultured in DMEM/F-12 HAM supplemented with 100 U/mL penicillin/streptomycin and 50 μg/mL gentamicin at 37°C and 5% CO2. PCLS were exposed to NM (50 μM mechlorethamine hydrochloride in DMEM/F-12 HAM or DMEM/F-12 HAM alone (control) for 1 hour at 37°C. After 1 day, PCLS were incubated with LNPs (75 μM) for 1 hour, subsequently stained with MitoTracker Red for 2 minutes, and imaged on a Leica TCS SP8 confocal microscope. PCLS with airways of comparable sizes were selected to be imaged. PCLS viability after incubation with LNPs was determined using established lactate dehydrogenase (LDH) and water-soluble tetrazolium salt-1 (WST-1) assays56 PCLS were prepared from both C57BL6/J and Nos2–/– mice.
Mice were anesthetized with isoflurane and received a single 50 μL intratracheal instillation of bleomycin (ITB, 3 U/kg in DMSO/PBS (6:94 v/v)) or DMSO/PBS (6:94 v/v) (control).79 To ensure complete dose retention after the instillation, mice were observed for 10 minutes post-recovery. Mice were re-anesthetized 72 hours later (d3), and received a 50 μL intratracheal instillation of LNPs (0.5 mM) or sucrose (300 mM, control). The optimal LNP concentration was determined to be 0.5 mM based on a cell viability assay (Figure S17). After 3 hours, mice were sacrificed by a single intraperitoneal injection of xylazine (30 mg/kg) and ketamine (135 mg/kg) and exsanguination. Following exposure of the abdominal cavity and thoracotomy, cardiac perfusion was performed (3 mL 1x PBS). 5x1 mL of ice-cold PBS was instilled through a 20-gauge canula to collect BAL fluid. The collected fluid was centrifuged at 300g for 8 minutes to pellet cells for flow cytometry.
Flow cytometry
Cells collected from the BAL fluid were diluted in staining buffer (5% FBS in 1x PBS, 0.2% sodium azide) to a volume of 100 μL. samples were treated for 10 minutes at 4°C with TruStain FcX anti-mouse CD16/32 (Fc Block, 1:100) to inhibit non-specific binding during staining and analysis. Samples were treated with the mixture containing the following antibodies (1:100) for 30 minutes at 4°C: CD11b, CD11c, and CD45 in dark. Samples were centrifuged for 6 minutes at 400 g, staining buffer was used to wash the cells. Cells were then stained with eFluor 780-conjugated fixable viability dye for 30 minutes at 4°C. After being fixed with 3% paraformaldehyde for 20 minutes at 4°C and cells were washed with cold PBS and resuspended in 100 μL PBS. Using a Gallios 10-color flow cytometer, cells were examined. Using Kaluza software, cells were initially sorted based on size and complexity, screened for viability and myeloid-derived origin. They were analyzed for CD11b and CD11c expression (Figure S19) and categorized for different phenotypes. CD11b+ population was used for DPPC-TC signal quantification (Figure 6B; Table 2).
Quantification and statistical analysis
Statistical analyses for the plate reader data were performed using GraphPad Prism 8 software. PCC values for microscopy experiments were calculated using the Imarius software (v9.0.1). The value of n represents the number of replicates in each experiment. The results were reported as either mean ± SD or mean ± SEM, these statistical parameter details are mentioned in figure or table legends.
Acknowledgments
We thank Prof. Gene Hall for allowing access to the fluorometer. We thank Prof. Ki-Bum Lee for allowing access to the microplate reader and we acknowledge Meizi Chen and Liming Wang for their assistance with fluorescence assays. We thank Kern Hast, and Mark Dresel for useful discussions. This work was supported by the US National Institutes of Health/National Institute of Biomedical Imaging and Bioengineering, Trailblazer Award (EB029548), the Rutgers Center for Lipid Research, the American Cancer Society, Institutional Research Grant Early Investigator Award, and the Rutgers Cancer Institute of New Jersey NCI Cancer Center Support Grant (P30CA072720) (to E.C.I.); Steven A. Cox Scholarship for Cancer Research (to T.A.); National Heart, Lung, and Blood Institute (HL086621) (to A.J.G.); National Institute of Environmental Health Sciences (NIEHS) (training grant ES007148 for E.R.S. and program grant ES005022), National Institute of Arthritis and Musculoskeletal and Skin Diseases (U54AR055073), NIEHS (R01ES033698 and R01ES004738) (to D.L.L.).
Author contributions
E.C.I. conceptualized and supervised the project. B.G. and H.E. conducted the syntheses, mass spectrometry and spectrophotometric analyses of the probes. B.G. established protocols for the preparation of giant vesicles. B.G., T.A., and M.R.L.S. performed confocal imaging. T.A., M.R.L.S., and C.G. conducted cell work. T.A. performed the pKa measurement, MTT assay, and flow cytometry (with assistance from E.R.S.). A.B. and T.A. performed ex vivo work under the guidance of A.J.G. and D.L.L. E.A. and E.R.S. performed in vivo experiments under the guidance of A.J.G. All of the authors contributed to the interpretation of data. E.C.I. wrote the article with input from all authors.
Declaration of interests
E.C.I., B.G., and H.E. are co-inventors of a provisional patent application filed by Rutgers University on the subject of this work.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: November 23, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.108567.
Supplemental information
References
- 1.Rubbo H., Trostchansky A., O’Donnell V.B. Peroxynitrite-mediated lipid oxidation and nitration: Mechanisms and consequences. Arch. Biochem. Biophys. 2009;484:167–172. doi: 10.1016/j.abb.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Radi R. Oxygen radicals, nitric oxide, and peroxynitrite: Redox pathways in molecular medicine. Proc. Natl. Acad. Sci. USA. 2018;115:5839–5848. doi: 10.1073/pnas.1804932115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szabó C., Ischiropoulos H., Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007;6:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein S., Czapski G. The reaction of NO· with O2·− and HO2·−: A pulse radiolysis study. Free Radic. Biol. Med. 1995;19:505–510. doi: 10.1016/0891-5849(95)00034-u. [DOI] [PubMed] [Google Scholar]
- 5.Koppenol W.H. The basic chemistry of nitrogen monoxide and peroxynitrite. Free Radic. Biol. Med. 1998;25:385–391. doi: 10.1016/s0891-5849(98)00093-8. [DOI] [PubMed] [Google Scholar]
- 6.Liu X., Miller M.J., Joshi M.S., Thomas D.D., Lancaster J.R. Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc. Natl. Acad. Sci. USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw A.W., Vosper A.J. Solubility of nitric oxide in aqueous and nonaqueous solvents. J. Chem. Soc., Faraday Trans. 1. 1977;73:1239–1244. [Google Scholar]
- 8.Bedard K., Krause K.H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 9.Squadrito G.L., Pryor W.A. Oxidative chemistry of nitric oxide: the roles of superoxide, peroxynitrite, and carbon dioxide. Free Radic. Biol. Med. 1998;25:392–403. doi: 10.1016/s0891-5849(98)00095-1. [DOI] [PubMed] [Google Scholar]
- 10.Alvarez M.N., Piacenza L., Irigoín F., Peluffo G., Radi R. Macrophage-derived peroxynitrite diffusion and toxicity to Trypanosoma cruzi. Arch. Biochem. Biophys. 2004;432:222–232. doi: 10.1016/j.abb.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 11.Zamora R., Vodovotz Y., Billiar T.R. Inducible Nitric Oxide Synthase and Inflammatory Diseases. Mol. Med. 2000;6:347–373. [PMC free article] [PubMed] [Google Scholar]
- 12.Sun X., Xu Q., Kim G., Flower S.E., Lowe J.P., Yoon J., Fossey J.S., Qian X., Bull S.D., James T.D. A water-soluble boronate-based fluorescent probe for the selective detection of peroxynitrite and imaging in living cells. Chem. Sci. 2014;5:3368–3373. [Google Scholar]
- 13.Sharif O., Bolshakov V.N., Raines S., Newham P., Perkins N.D. Transcriptional profiling of the LPS induced NF-κB response in macrophages. BMC Immunol. 2007;8:1. doi: 10.1186/1471-2172-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arias-Salvatierra D., Silbergeld E.K., Acosta-Saavedra L.C., Calderon-Aranda E.S. Role of nitric oxide produced by iNOS through NF-κB pathway in migration of cerebellar granule neurons induced by Lipopolysaccharide. Cell. Signal. 2011;23:425–435. doi: 10.1016/j.cellsig.2010.10.017. [DOI] [PubMed] [Google Scholar]
- 15.Das S., Alhasson F., Dattaroy D., Pourhoseini S., Seth R.K., Nagarkatti M., Nagarkatti P.S., Michelotti G.A., Diehl A.M., Kalyanaraman B., Chatterjee S. NADPH Oxidase–Derived Peroxynitrite Drives Inflammation in Mice and Human Nonalcoholic Steatohepatitis via TLR4-Lipid Raft Recruitment. Am. J. Pathol. 2015;185:1944–1957. doi: 10.1016/j.ajpath.2015.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pfeiffer S., Lass A., Schmidt K., Mayer B. Protein tyrosine nitration in cytokine-activated murine macrophages. Involvement of a peroxidase/nitrite pathway rather than peroxynitrite. J. Biol. Chem. 2001;276:34051–34058. doi: 10.1074/jbc.M100585200. [DOI] [PubMed] [Google Scholar]
- 17.Hay J., Shahzeidi S., Laurent G. Mechanisms of bleomycin-induced lung damage. Arch. Toxicol. 1991;65:81–94. doi: 10.1007/BF02034932. [DOI] [PubMed] [Google Scholar]
- 18.Golden T.N., Venosa A., Gow A.J. Cell Origin and iNOS Function Are Critical to Macrophage Activation Following Acute Lung Injury. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.761496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrer-Sueta G., Radi R. Chemical Biology of Peroxynitrite: Kinetics, Diffusion, and Radicals. ACS Chem. Biol. 2009;4:161–177. doi: 10.1021/cb800279q. [DOI] [PubMed] [Google Scholar]
- 20.Cheng P., Zhang J., Huang J., Miao Q., Xu C., Pu K. Near-infrared fluorescence probes to detect reactive oxygen species for keloid diagnosis. Chem. Sci. 2018;9:6340–6347. doi: 10.1039/c8sc01865k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mao Z., Xiong J., Wang P., An J., Zhang F., Liu Z., Seung Kim J. Activity-based fluorescence probes for pathophysiological peroxynitrite fluxes. Coord. Chem. Rev. 2022;454 [Google Scholar]
- 22.Knewtson K.E., Rane D., Peterson B.R. Targeting Fluorescent Sensors to Endoplasmic Reticulum Membranes Enables Detection of Peroxynitrite During Cellular Phagocytosis. ACS Chem. Biol. 2018;13:2595–2602. doi: 10.1021/acschembio.8b00535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang X., Zhang D., Ye Y., Zhao Y. Recent advances in multifunctional fluorescent probes for viscosity and analytes. Coord. Chem. Rev. 2022;453 [Google Scholar]
- 24.Liu J., Zhao M., Zhao F., Song X., Ye Y. ONOO--triggered fluorescence H2S donor for mitigating drug-induced liver injury. Sens. Actuators, B. 2023;378 [Google Scholar]
- 25.Soh N., Makihara K., Ariyoshi T., Seto D., Maki T., Nakajima H., Nakano K., Imato T. Phospholipid-linked Coumarin: A Fluorescent Probe for Sensing Hydroxyl Radicals in Lipid Membranes. Anal. Sci. 2008;24:293–296. doi: 10.2116/analsci.24.293. [DOI] [PubMed] [Google Scholar]
- 26.Danylchuk D.I., Jouard P.-H., Klymchenko A.S. Targeted Solvatochromic Fluorescent Probes for Imaging Lipid Order in Organelles under Oxidative and Mechanical Stress. J. Am. Chem. Soc. 2021;143:912–924. doi: 10.1021/jacs.0c10972. [DOI] [PubMed] [Google Scholar]
- 27.Dadina N., Tyson J., Zheng S., Lesiak L., Schepartz A. Imaging organelle membranes in live cells at the nanoscale with lipid-based fluorescent probes. Curr. Opin. Chem. Biol. 2021;65:154–162. doi: 10.1016/j.cbpa.2021.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Aparin I.O., Yan R., Pelletier R., Choi A.A., Danylchuk D.I., Xu K., Klymchenko A.S. Fluorogenic Dimers as Bright Switchable Probes for Enhanced Super-Resolution Imaging of Cell Membranes. J. Am. Chem. Soc. 2022;144:18043–18053. doi: 10.1021/jacs.2c07542. [DOI] [PubMed] [Google Scholar]
- 29.Klymchenko A.S. Fluorescent Probes for Lipid Membranes: From the Cell Surface to Organelles. Acc. Chem. Res. 2023;56:1–12. doi: 10.1021/acs.accounts.2c00586. [DOI] [PubMed] [Google Scholar]
- 30.Swartz H.M., Khan N., Khramtsov V.V. Use of Electron Paramagnetic Resonance Spectroscopy to Evaluate the Redox State In Vivo. Antioxid. Redox Signal. 2007;9:1757–1771. doi: 10.1089/ars.2007.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ischiropoulos H., al-Mehdi A.B., Fisher A.B. Reactive species in ischemic rat lung injury: contribution of peroxynitrite. Am. J. Physiol. 1995;269:L158–L164. doi: 10.1152/ajplung.1995.269.2.L158. [DOI] [PubMed] [Google Scholar]
- 32.Rubbo H., Radi R., Trujillo M., Telleri R., Kalyanaraman B., Barnes S., Kirk M., Freeman B.A. Nitric oxide regulation of superoxide and peroxynitrite-dependent lipid peroxidation. Formation of novel nitrogen-containing oxidized lipid derivatives. J. Biol. Chem. 1994;269:26066–26075. [PubMed] [Google Scholar]
- 33.Paxinou E., Chen Q., Weisse M., Giasson B.I., Norris E.H., Rueter S.M., Trojanowski J.Q., Lee V.M., Ischiropoulos H. Induction of α-Synuclein Aggregation by Intracellular Nitrative Insult. J. Neurosci. 2001;21:8053–8061. doi: 10.1523/JNEUROSCI.21-20-08053.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pacher P., Schulz R., Liaudet L., Szabó C. Nitrosative stress and pharmacological modulation of heart failure. Trends Pharmacol. Sci. 2005;26:302–310. doi: 10.1016/j.tips.2005.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaki M.H., Okamoto T., Sawa T., Fujii S., Akaike T. Nitrative stress in respiratory inflammation caused by influenza virus infection. Clin. Exp. All. Rev. 2007;7:19–26. [Google Scholar]
- 36.Pacher P., Beckman J.S., Liaudet L. Nitric Oxide and Peroxynitrite in Health and Disease. Physiol. Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sugiura H., Ichinose M. Oxidative and Nitrative Stress in Bronchial Asthma. Antioxid. Redox Signal. 2008;10:785–797. doi: 10.1089/ars.2007.1937. [DOI] [PubMed] [Google Scholar]
- 38.Sugiura H., Ichinose M. Nitrative stress in inflammatory lung diseases. Nitric Oxide. 2011;25:138–144. doi: 10.1016/j.niox.2011.03.079. [DOI] [PubMed] [Google Scholar]
- 39.Wang F., Yuan Q., Chen F., Pang J., Pan C., Xu F., Chen Y. Fundamental Mechanisms of the Cell Death Caused by Nitrosative Stress. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.742483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Garvey E.P., Oplinger J.A., Furfine E.S., Kiff R.J., Laszlo F., Whittle B.J., Knowles R.G. 1400W Is a Slow, Tight Binding, and Highly Selective Inhibitor of Inducible Nitric-oxide Synthase in Vitro and in Vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 41.Sunil V.R., Vayas K.N., Abramova E.V., Rancourt R., Cervelli J.A., Malaviya R., Goedken M., Venosa A., Gow A.J., Laskin J.D., Laskin D.L. Lung injury, oxidative stress and fibrosis in mice following exposure to nitrogen mustard. Toxicol. Appl. Pharmacol. 2020;387 doi: 10.1016/j.taap.2019.114798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevenson E.R., Wilkinson M.L., Abramova E., Guo C., Gow A.J. Intratracheal Administration of Acyl Coenzyme A Acyltransferase-1 Inhibitor K-604 Reduces Pulmonary Inflammation Following Bleomycin-Induced Lung Injury. J. Pharmacol. Exp. Ther. 2022;382:356–365. doi: 10.1124/jpet.122.001284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang D., Wang H.-L., Sun Z.-N., Chung N.-W., Shen J.-G. A Highly Selective Fluorescent Probe for the Detection and Imaging of Peroxynitrite in Living Cells. J. Am. Chem. Soc. 2006;128:6004–6005. doi: 10.1021/ja0603756. [DOI] [PubMed] [Google Scholar]
- 44.Sivakumar K., Xie F., Cash B.M., Long S., Barnhill H.N., Wang Q. A Fluorogenic 1,3-Dipolar Cycloaddition Reaction of 3-Azidocoumarins and Acetylenes. Org. Lett. 2004;6:4603–4606. doi: 10.1021/ol047955x. [DOI] [PubMed] [Google Scholar]
- 45.Reynolds G.A., Drexhage K.H. New coumarin dyes with rigidized structure for flashlamp-pumped dye lasers. Opt Commun. 1975;13:222–225. [Google Scholar]
- 46.Carballal S., Bartesaghi S., Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta. 2014;1840:768–780. doi: 10.1016/j.bbagen.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Q., Zhu Z., Zheng Y., Cheng J., Zhang N., Long Y.-T., Zheng J., Qian X., Yang Y. A Three-Channel Fluorescent Probe That Distinguishes Peroxynitrite from Hypochlorite. J. Am. Chem. Soc. 2012;134:18479–18482. doi: 10.1021/ja305046u. [DOI] [PubMed] [Google Scholar]
- 48.Vance J.A., Devaraj N.K. Membrane Mimetic Chemistry in Artificial Cells. J. Am. Chem. Soc. 2021;143:8223–8231. doi: 10.1021/jacs.1c03436. [DOI] [PubMed] [Google Scholar]
- 49.Göpfrich K., Haller B., Staufer O., Dreher Y., Mersdorf U., Platzman I., Spatz J.P. One-Pot Assembly of Complex Giant Unilamellar Vesicle-Based Synthetic Cells. ACS Synth. Biol. 2019;8:937–947. doi: 10.1021/acssynbio.9b00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stein H., Spindler S., Bonakdar N., Wang C., Sandoghdar V. Production of Isolated Giant Unilamellar Vesicles under High Salt Concentrations. Front. Physiol. 2017;8:63. doi: 10.3389/fphys.2017.00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kajii K., Shimomura A., T Higashide M., Oki M., Tsuji G. Effects of Sugars on Giant Unilamellar Vesicle Preparation, Fusion, PCR in Liposomes, and Pore Formation. Langmuir. 2022;38:8871–8880. doi: 10.1021/acs.langmuir.2c00989. [DOI] [PubMed] [Google Scholar]
- 52.Felgner P.L., Gadek T.R., Holm M., Roman R., Chan H.W., Wenz M., Northrop J.P., Ringold G.M., Danielsen M. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA. 1987;84:7413–7417. doi: 10.1073/pnas.84.21.7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Young A.T.L., Moore R.B., Murray A.G., Mullen J.C., Lakey J.R.T. Assessment of different transfection parameters in efficiency optimization. Cell Transplant. 2004;13:179–185. [PubMed] [Google Scholar]
- 54.Alvarez M.N., Peluffo G., Piacenza L., Radi R. Intraphagosomal Peroxynitrite as a Macrophage-derived Cytotoxin against Internalized Trypanosoma cruzi Consequences for Oxidative Killing and Role of Microbial Peroxiredoxins in Infectivity. J. Biol. Chem. 2011;286:6627–6640. doi: 10.1074/jbc.M110.167247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Y., James S.J., Wyllie D.H., Wynne C., Czibula A., Bukhari A., Pye K., Bte Mustafah S.M., Fajka-Boja R., Szabo E., et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proc. Natl. Acad. Sci. USA. 2019;116:16479–16488. doi: 10.1073/pnas.1901090116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Herbert J., Kelty J.S., Laskin J.D., Laskin D.L., Gow A.J. Menthol flavoring in e-cigarette condensate causes pulmonary dysfunction and cytotoxicity in precision cut lung slices. Am. J. Physiol. Lung Cell Mol. Physiol. 2023;324:L345–L357. doi: 10.1152/ajplung.00222.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laskin D.L., Malaviya R., Laskin J.D. Role of Macrophages in Acute Lung Injury and Chronic Fibrosis Induced by Pulmonary Toxicants. Toxicol. Sci. 2019;168:287–301. doi: 10.1093/toxsci/kfy309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaynagetdinov R., Sherrill T.P., Kendall P.L., Segal B.H., Weller K.P., Tighe R.M., Blackwell T.S. Identification of Myeloid Cell Subsets in Murine Lungs Using Flow Cytometry. Am. J. Respir. Cell Mol. Biol. 2013;49:180–189. doi: 10.1165/rcmb.2012-0366MA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klymchenko A.S., Kreder R. Fluorescent Probes for Lipid Rafts: From Model Membranes to Living Cells. Chem. Biol. 2014;21:97–113. doi: 10.1016/j.chembiol.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Flores J., White B.M., Brea R.J., Baskin J.M., Devaraj N.K. Lipids: chemical tools for their synthesis, modification, and analysis. Chem. Soc. Rev. 2020;49:4602–4614. doi: 10.1039/d0cs00154f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mukhopadhyay T.K., Morstein J., Trauner D. Photopharmacological control of cell signaling with photoswitchable lipids. Curr. Opin. Pharmacol. 2022;63 doi: 10.1016/j.coph.2022.102202. [DOI] [PubMed] [Google Scholar]
- 62.Lou J., Sagar R., Best M.D. Metabolite-Responsive Liposomes Employing Synthetic Lipid Switches Driven by Molecular Recognition Principles. Acc. Chem. Res. 2022;55:2882–2891. doi: 10.1021/acs.accounts.2c00446. [DOI] [PubMed] [Google Scholar]
- 63.Bumpus T.W., Baskin J.M. Greasing the Wheels of Lipid Biology with Chemical Tools. Trends Biochem. Sci. 2018;43:970–983. doi: 10.1016/j.tibs.2018.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Farley S., Laguerre A., Schultz C. Caged lipids for subcellular manipulation. Curr. Opin. Chem. Biol. 2021;65:42–48. doi: 10.1016/j.cbpa.2021.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kamat N.P., Tobé S., Hill I.T., Szostak J.W. Electrostatic Localization of RNA to Protocell Membranes by Cationic Hydrophobic Peptides. Angew. Chem. 2015;54:11735–11739. doi: 10.1002/anie.201505742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Izgu E.C., Björkbom A., Kamat N.P., Lelyveld V.S., Zhang W., Jia T.Z., Szostak J.W. N-Carboxyanhydride-Mediated Fatty Acylation of Amino Acids and Peptides for Functionalization of Protocell Membranes. J. Am. Chem. Soc. 2016;138:16669–16676. doi: 10.1021/jacs.6b08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lou J., Best M.D. A General Approach to Enzyme-Responsive Liposomes. Chem. Eur J. 2020;26:8597–8607. doi: 10.1002/chem.202000529. [DOI] [PubMed] [Google Scholar]
- 68.Lyu Y., Peng R., Liu H., Kuai H., Mo L., Han D., Li J., Tan W. Protocells programmed through artificial reaction networks. Chem. Sci. 2019;11:631–642. doi: 10.1039/c9sc05043d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Boyd M.A., Kamat N.P. Designing Artificial Cells towards a New Generation of Biosensors. Trends Biotechnol. 2021;39:927–939. doi: 10.1016/j.tibtech.2020.12.002. [DOI] [PubMed] [Google Scholar]
- 70.Tenchov R., Bird R., Curtze A.E., Zhou Q. Lipid Nanoparticles–From Liposomes to mRNA Vaccine Delivery, a Landscape of Research Diversity and Advancement. ACS Nano. 2021;15:16982–17015. doi: 10.1021/acsnano.1c04996. [DOI] [PubMed] [Google Scholar]
- 71.Schwarz D.S., Blower M.D. The endoplasmic reticulum: structure, function and response to cellular signaling. Cell. Mol. Life Sci. 2016;73:79–94. doi: 10.1007/s00018-015-2052-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bhattarai K.R., Riaz T.A., Kim H.-R., Chae H.-J. The aftermath of the interplay between the endoplasmic reticulum stress response and redox signaling. Exp. Mol. Med. 2021;53:151–167. doi: 10.1038/s12276-021-00560-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.von Krusenstiern A.N., Robson R.N., Qian N., Qiu B., Hu F., Reznik E., Smith N., Zandkarimi F., Estes V.M., Dupont M., et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat. Chem. Biol. 2023;19:719–730. doi: 10.1038/s41589-022-01249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stockwell B.R., Friedmann Angeli J.P., Bayir H., Bush A.I., Conrad M., Dixon S.J., Fulda S., Gascón S., Hatzios S.K., Kagan V.E., et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beckman J.S., Chen J., Ischiropoulos H., Crow J.P. Oxidative chemistry of peroxynitrite. Methods Enzym. 1994;233:229–240. doi: 10.1016/s0076-6879(94)33026-3. [DOI] [PubMed] [Google Scholar]
- 77.Hughes M.N., Nicklin H.G. The chemistry of pernitrites. Part I. Kinetics of decomposition of pernitrous acid. J. Chem. Soc. A. 1968;0:450–452. [Google Scholar]
- 78.Würth C., Grabolle M., Pauli J., Spieles M., Resch-Genger U. Relative and absolute determination of fluorescence quantum yields of transparent samples. Nat. Protoc. 2013;8:1535–1550. doi: 10.1038/nprot.2013.087. [DOI] [PubMed] [Google Scholar]
- 79.Guo C., Atochina-Vasserman E., Abramova H., George B., Manoj V., Scott P., Gow A. Role of NOS2 in pulmonary injury and repair in response to bleomycin. Free Radic. Biol. Med. 2016;91:293–301. doi: 10.1016/j.freeradbiomed.2015.10.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Any primary data in this study can be requested from the lead contact.
-
•
This study did not generate any new code.
-
•
Any additional information to reanalyze the data can be requested from the lead contact.