

Abstract
Nonalcoholic steatohepatitis (NASH) is emerging as the most common cause of liver disease. Although many studies in mouse NASH models have suggested therapies, translation to humans is poor, with no approved drugs for NASH. One explanation may lie in differences between mouse and human hepatocytes. We used NASH diet-fed chimeric mice reconstituted with human hepatocytes (hu-liver mice) to test a mechanism-based hepatocyte-targeted small interfering RNA (siRNA), GalNAc-siTaz, shown previously to block the progression to fibrotic NASH in mice. Following ablation of endogenous hepatocytes, male mice were reconstituted with human hepatocytes from a single donor with the rs738409-C/G PNPLA3 risk variant, resulting in ∼95% human hepatocyte reconstitution. The mice were then fed a high-fat choline-deficient l-amino acid–defined diet for 6 weeks to induce NASH, followed by six weekly injections of GalNAc-siTAZ to silence hepatocyte-TAZ or control GalNAc-siRNA (GalNAc-control) while still on the NASH diet. GalNAc-siTAZ lowered human hepatic TAZ and IHH, a TAZ target that promotes NASH fibrosis. Most important, GalNAc-siTAZ decreased liver inflammation, hepatocellular injury, hepatic fibrosis, and profibrogenic mediator expression versus GalNAc-control, indicating that GalNAc-siTAZ decreased the progression of NASH in mice reconstituted with human hepatocytes. In conclusion, silencing TAZ in human hepatocytes suppresses liver fibrosis in a hu-liver model of NASH.
Keywords: hepatocyte-humanized mice; MASH; liver fibrosis; WWTR1; GalNAc-siRNA; NASH/MASH therapy, NASH, TAZ, Indian hedgehog; IHH
Graphical abstract
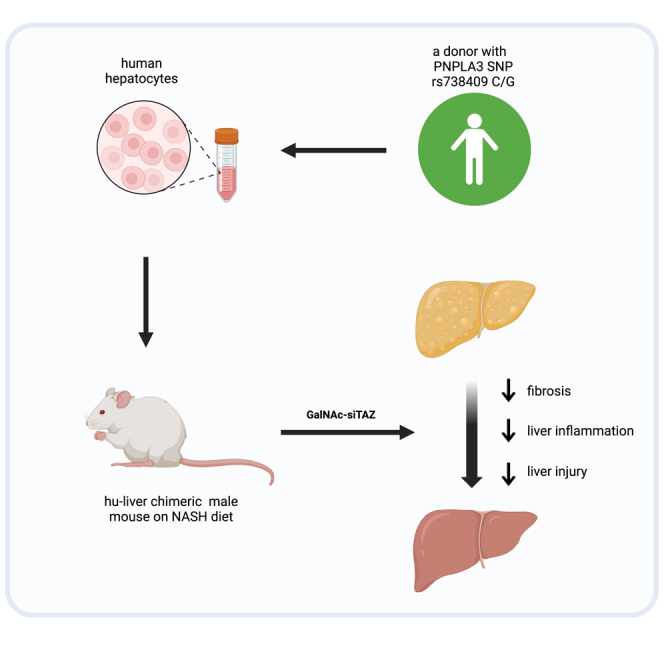
Tabas and colleagues show that TAZ-silencing therapy using a human-relevant platform (GalNAc-siTAZ) prevented nonalcoholic steatohepatitis (NASH) progression in diet-induced NASH mice whose liver cells were replaced with human liver cells. These findings support further investigation into developing this therapy for humans with NASH, a disease for which no therapy exists.
Introduction
Nonalcoholic fatty liver disease (NAFLD, also known as metabolic dysfunction–associated steatotic liver disease), is characterized by hepatosteatosis (fatty liver), and in ∼20%–30% of cases, NAFLD progresses to fibrotic nonalcoholic steatohepatitis (NASH, also known as metabolic dysfunction-associated steatohepatitis).1 NASH is emerging as the leading cause of chronic liver disease worldwide, including hepatocellular carcinoma and other comorbidities.1 Disease progression is caused by multiple insults acting on steatotic hepatocytes that result in liver inflammation, hepatocellular death, and, most important, liver fibrosis, which correlates best with clinical outcome in NASH.2 No effective therapies exist to treat NASH,3 in part due to an incomplete understanding of profibrotic mechanisms in NASH as well as a lack of human-relevant preclinical models of NASH fibrosis, which include mostly mouse models.4
In regard to mechanisms of NASH fibrosis, we have previously shown that the transcriptional coactivator with PDZ-binding motif (TAZ; encoded by WW domain-containing transcription regulator 1 [WWTR1]) is upregulated in hepatocytes in mouse and human fibrotic NASH but not steatosis.5,6 TAZ induces Indian hedgehog (IHH), which activates hepatic stellate cell-derived myofibroblasts (HSCs) and liver fibrosis. Knockdown of hepatocyte TAZ or IHH by experimental genetic methods dampened steatosis-to-fibrotic NASH progression.5,6 In this context, during the last 2 decades we have witnessed the development of therapies that can silence genes specifically in hepatocytes using small interfering RNA (siRNA) conjugated with hepatocyte-specific targeting motifs, notably, N-acetylgalactosamine (GalNAc).7 Most important, several hepatocyte-targeted siRNAs have been approved recently by the US Food and Drug Administration for specific disease indications.8,9,10,11 Reasoning that GalNAc-siTAZ may have potential as a mechanism-based therapy to suppress the progression to NASH fibrosis, we reported that its administration to mice with early fibrotic NASH suppressed the progression of hepatic inflammation, injury, and fibrosis and lowered the expression of HSC-derived profibrogenic mediators.12
Because previous studies focused on the effect of GalNAc-siTAZ in mouse hepatocytes, we became interested in a type of chimeric mouse model in which mouse hepatocytes are replaced by human hepatocytes (hu-liver mice).13,14 In a recent study, hu-liver mice were fed a NASH-inducing diet and then used to test a systemically administered drug that targets a liver metabolic pathway.15 We reasoned that a mouse NASH model whose livers are populated with human hepatocytes would be particularly valuable in testing hepatocyte-targeted siRNA therapies. Accordingly, we report here on the use of hu-liver NASH mice to investigate the efficacy of GalNAc-siTAZ in preventing the progression of diet-induced NASH fibrosis.
Results
Hu-liver mice were placed on the high-fat choline-deficient l-amino acid (HF-CDAA)-NASH diet for 6 weeks, followed by weekly subcutaneous injections with 5 mg/kg GalNAc-siTAZ or GalNAc-control RNA, with continued diet feeding (Figure 1A). The siTAZ used in this study, which targets both mouse and human WWTR1, was chosen based on screening experiments in human hepatocytes and mouse NASH models, similar to our previously published strategy for screening GalNAc-siTaz for use in mice.12 The body weights between the two groups were not statistically different (Figures 1B and S1A). Immunostaining using an antibody against the human-specific cytoplasmic protein STEM121/SC12116 indicated that ∼90% of the hepatocytes were of human origin (Figure 1C). At 12 weeks, GalNAc-siTAZ efficiently lowered the mRNA and protein contents of both liver TAZ and its downstream profibrotic mediator IHH,5 with no change in the TAZ-related protein YAP (Figure 1D). GalNAc-siTAZ also lowered the expression of genes related to liver inflammation and hepatic stellate cell activation and profibrotic processes (Figure 1E). Most important, although the livers from the GalNAc-control-treated hu-liver NASH mice accumulated mononuclear cells and demonstrated advanced fibrosis, as assessed by H&E and Sirius red staining, respectively, both parameters were decreased by GalNAc-siTAZ (Figure 1F). These changes were accompanied by a 60% decrease in anti-α-smooth muscle actin–positive (αSMA+) cells in the livers (Figure 1G). Human ALT and AST were also lower in the plasma of GalNAc-siTAZ-treated versus control mice (Figure 1H). In contrast, GalNAc-siTAZ did not significantly affect lipid droplet area in the livers and (Figure 1I). Although the reduction in IHH by GalNAc-siTAZ is a likely mechanism for the decrease in liver fibrosis in GalNAc-siTAZ-treated hu-liver NASH mice,5 we considered another possible mechanism. Both preclinical and human data suggest an important role for hepatocyte Notch activation in NASH fibrosis,17,18 and work in other settings has shown that YAP/TAZ can induce NOTCH and its ligands.19 We therefore examined the livers for NOTCH1 and HES1, a gene marker of Notch activation. Both NOTCH1 and HES1 were substantially lower in the livers of GalNAc-siTAZ-treated versus control hu-liver NASH mice (Figure 1J), raising the possibility that GalNAc-siTAZ decreases liver fibrosis in NASH by lowering both IHH and Notch signaling.
Figure 1.

Treatment of hu-liver NASH mice with GalNAc-siTAZ lowers liver inflammation and fibrosis
(A) Experimental scheme. Humanized mice were fed the HF-CDAA diet for 6 weeks and then injected once weekly for 6 additional weeks with 5 mg/kg GalNAc-siTAZ (siTAZ) or control GalNAc construct (control) while still on the diet. At 12 weeks, the 2 cohorts were analyzed for systemic and liver endpoints. (B) Body weight. (C) Livers were immunostained with anti-STEM121, and the percentages of hepatocytes that were STEM121+ were quantified; liver from a mouse fed the HF-CDAA diet for 12 weeks was used as a negative control for anti-STEM121 staining. Scale bar, 100 μm. (D) Liver WWTR1 and IHH mRNA and immunoblots of YAP, TAZ, IHH, and β-actin; a longer exposure (long) of the TAZ immunoblot is included. The densitometric data for IHH is shown. (E) Livers were assayed for the inflammatory mRNAs Cxcl9, Tnfa, Emr1, Tgfb1, and Mcp1 and the fibrosis-related mRNAs Spp1, Acat2, and Col1a1 mRNAs. (F) Livers were stained with H&E (upper row) and Sirius red (lower row) and then quantified for inflammatory cells per field and percentage of Sirius red area. Scale bar, 200 μm. (G) Livers were immunostained with anti-αSMA and quantified for the αSMA+ area, which was normalized for cellularity by dividing by the number of DAPI+ cells. Scale bar, 200 μm. (H) Plasma ALT and AST. (I) Percentage of lipid droplet area in the livers. (J) Liver NOTCH1 and HES1 mRNA. Values shown for all of the graphs are means ± SEM; n = 6 control mice and 7 siTAZ mice. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The data were analyzed using the Student’s t test.
During this experiment, the mice experienced vitamin C deficiency, and some of the mice died before the end of the experiment. Hu-liver mice often develop hypovitaminosis due to their inability to synthesize ascorbic acid.20 To counteract this, the mice are typically provided with a diet containing a small quantity of vitamin C and supplemented with additional vitamins. In our initial study, the vitamin supplement was added to the HF-CDAA diet. However, it was noted that some of the mice avoided the vitamin supplement, possibly due to a shift in dietary preference induced by the HF-CDAA diet. These mice exhibited symptoms consistent with vitamin C deficiency, including low body temperature, reduced activity, and hemorrhage. Upon recognizing these issues, the surviving animals in the study were subsequently provided with vitamin C through their drinking water, commencing on day 32 of the experiment. Although there was no difference in morbidity or mortality between the control and siTAZ cohorts in this experiment, we conducted a repeat experiment in which vitamin C deficiency was avoided. The human hepatocytes for this experiment came from the same donor as the first experiment. As with the first experiment, the livers contained ∼95% human hepatocytes (Figure 2A), and GalNAc-siTAZ efficiently lowered hepatic WWTR1 and IHH (Figure 2B), TAZ+ human hepatocytes (Figure 2C), and hepatic inflammation, fibrosis, and plasma ALT, without altering hepatosteatosis and cell proliferation (Figures 2D–2L). In this experiment, although the two cohorts looked to be equally healthy based on overall appearance and activity level, the mean body weight of the GalNAc-siTAZ cohort was modestly but significantly lower than that of the control cohort (Figures 2M and S2B). We therefore examined a subpopulation of mice in each group whose weights were similar to one another and found that liver fibrosis, as assessed by Sirius red staining, was decreased by GalNAc-siTAZ to a degree similar to that in the full cohort (Figure 2N). Finally, as in the first experiment, the livers of GalNAc-siTAZ-treated hu-liver NASH mice had a markedly lower expression of NOTCH and HES1 as well as the Notch downstream effector gene EPHB2, which has been implicated in NASH progression,21 compared with control mice (Figure 2O). Thus, in two separate cohorts of hu-liver NASH mice harboring ∼95% human hepatocytes, GalNAc-siTAZ dampened the progression to fibrotic NASH.
Figure 2.

A repeat experiment showing that GalNAc-siTAZ lowers NASH progression in hu-liver mice
A second cohort of hu-liver mice were treated with GalNAc-siTAZ or control GalNAc construct exactly as depicted in Figure 1A. (A) Livers were immunostained with anti-STEM121, and the percentages of hepatocytes that were STEM121+ were quantified; liver from a mouse fed the HF-CDAA diet for 12 weeks was used as a negative control for anti-STEM121 staining. Scale bar, 100 μm. (B) Liver WWTR1 and IHH mRNA and immunoblots of TAZ, IHH, and β-actin. (C) Livers were immunostained with anti-TAZ and anti-STEM121 and then quantified for the percentage of TAZ+cells among STEM121+cells. Scale bar, 200 μm. (D) Livers were stained with H&E (upper row) and Sirius red (lower row) and then quantified for inflammatory cells per field and percentage of Sirius red area. Scale bar, 200 μm. (E) Livers were immunostained with anti-αSMA and quantified as in Figure 1G. Scale bar, 200 μm. (F) Hydroxyproline content. (G) Livers were assayed for the inflammatory mRNAs Emr1, Mcp1, Tnfa, Cxcl9, and Tgfb1 and the fibrosis-related mRNAs Timp1, Col1a1, Col3a1, Col1a2, and Acta2. (H) Livers were immunostained with anti-Mac2 and anti-Clec4f and quantified for the percentage positive area of each. Scale bar, 200 μm. (I) Livers were immunostained with anti-F4/80 and quantified for the percentage of positive area. Scale bar, 200 μm. (J) Plasma ALT. (K) Percentage of lipid droplet area in the livers. (L) Livers were immunostained with anti-Ki67 and quantified for the percentage of positive area. Scale bar, 200 μm. (M) Body weight. (N) The livers of a subgroup mice from each cohort (5 control, 4 siTAZ) with similar body weights were assayed for percentage of Sirius red+. (O) Liver NOTCH1, HES1, and EPHB2 mRNA. Values shown for all of the graphs are means ± SEM; n = 8 mice per group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. The data were analyzed using the Student’s t test.
Discussion
Creating therapies to block the progression of fibrotic NASH has proven to be extremely challenging,3 which is particularly alarming given the fact that NASH is emerging as the leading cause of chronic liver disease worldwide.1 The lack of any approved therapies for NASH is likely due to multiple issues, including an incomplete understanding of the mechanisms that promote liver fibrosis in NASH, the possible need for targeting more than one process in NASH (combination therapy), and difficulties in predicting success in humans based on studies in preclinical models.4 The latter problem is due in part to inherent differences between hepatocytes in mice, by far the most common species for the initial testing of new therapeutic ideas for NASH, and hepatocytes in humans.22
To overcome the latter issue, investigators have developed chimeric mice with humanized livers by the transplantation of human hepatocytes into mice whose endogenous hepatocytes have been killed.13,14 The mice have a hepatocyte gene profile that is 82% similar to hepatocytes isolated from human liver, making it an effective preclinical model to study the potential therapeutic efficacy of novel drugs in vivo.13,14 Our study was based on a previous report showing that when hu-liver mice are fed an HF-CDAA diet, they display similar histological changes to human NASH, including ballooning, inflammation, apoptosis, regeneration of human hepatocytes, fibrosis, and increases in inflammatory and profibrotic genes in the liver.15 In this report, the authors showed that orally administered elafibranor, a dual peroxisome proliferator-activated receptor α/δ agonist, improved steatosis and markers of liver injury, but not fibrosis, which is consistent with the results of a previous clinical trial.23 Another study showed that hu-liver mice reconstituted with hepatocytes from a human with a genetic NASH-risk variant, PNPLA3-148M, and then fed an HF or Western-type diet had more advanced NASH features compared with HF-fed hu-mice reconstituted with nonrisk PNPLA3-148I hepatocytes.24
Advances over the last decade in the area of hepatocyte-targeted siRNA therapy have led to the approval of four drugs for use in humans: patisiran, which targets TTR to treat familial amyloid polyneuropathy8; givosiran, which targets ALAS1 to treat acute porphyria9; lumasiran, which targets HAO1 to treat primary hyperoxaluria type 110; and inclisiran, which targets PCSK9 to treat hypercholesterolemia.11 Moreover, several other hepatocyte-targeted siRNAs are in clinical trials for a variety of other diseases. We reasoned that for the testing of future hepatocyte-targeted siRNAs, hu-liver NASH mice would help bridge the transition from pure mouse studies to human clinical trials. We chose GalNAc-siTAZ for this purpose given the need for mechanism-based therapies that target proteins involved specifically in NASH liver fibrosis. A combination of in-depth mechanistic studies, analyses of liver tissue and plasma from NASH subjects, molecular-genetic causation studies in NASH mice, and the results of a study testing GalNAc-siTAZ in mouse NASH have pointed to the promise of this approach.5,6,12,25 Furthermore, we showed that silencing hepatocyte TAZ blocks the progression to hepatocellular carcinoma in NASH mice.26 Accordingly, the data here show that in mice with mostly human hepatocytes, GalNAc-siTAZ blocks fibrotic NASH progression. One mechanism for the GalNAc-siTAZ-induced decreases in fibrosis, inflammation, and liver injury suggested by our previous studies is lowering of the HSC activator IHH,5 and new data here suggest that suppression of the hepatocyte Notch pathway by GalNAc-siTAZ in NASH may be another human-relevant mechanism.17,18 Previous work has shown that hepatocyte Notch signaling contributes to NASH fibrosis by promoting Ephb2 expression,21 because knockdown of Ephb2 in hepatocytes ameliorated inflammation and fibrosis in a mouse model of NASH. In regard to inflammation, our data are consistent with GalNAc-siTAZ blocking inflammation in infiltrating monocyte-derived macrophages, which have an inflammatory phenotype in NASH liver.27 Given that only the hepatocytes were of human origin in the mice and that GalNAc-siTAZ targets hepatocytes, not macrophages, the findings suggest that the interaction between human hepatocytes and mouse macrophages remains functional in this model.
In our mouse studies thus far,5,6,12 as in the study here, we have not observed overt adverse effects of silencing or deleting hepatocyte TAZ, which may be explained in part by the very low expression of TAZ in normal liver and possibly by the ability of the TAZ-related protein YAP to compensate for the loss of TAZ.28 Although targeting hepatocyte TAZ led to improvements in liver fibrosis, decreased mononuclear cells in the liver, and lowered plasma ALT, our previous work showed that these parameters did not fully return to the levels of chow-fed mice,5 which may be due to residual pro-NASH mechanisms but could also be an effect of the relatively short treatment period of 8–12 weeks. Our study, by design, intervened only after steatosis had developed. However, it will be important in future studies to intervene after early NASH fibrosis has developed, as we did in our mouse NASH study.12 In that study, we subjected mice to the NASH diet for 16 weeks to induce early-stage NASH fibrosis and subsequently administered PBS or GalNAc-siTaz weekly for an additional 12 weeks while maintaining the mice on the NASH diet. GalNAc-siTAZ treatment significantly ameliorated both inflammation and fibrosis, and there was also an increase in matrix metalloproteinase activity in the livers of the GalNAc-siTAZ-treated mice, suggesting that siTAZ may also enhance collagen degradation during NASH regression.
As with all preclinical models, hu-liver NASH mice have limitations, including the SCID background of the mice and the fact that the nonparenchymal liver cells are of mouse origin. The SCID background may affect inflammation in NASH, but the data show that GalNAc-siTAZ reduces the expression of inflammatory genes in a manner that corresponds with a reduction of Mac2+ but not resident Clec4f+ macrophages, suggesting an effect on infiltrating macrophages.27 These findings indicate that the interaction between human hepatocytes, the site of action of GalNAc-siTAZ, and mouse macrophages remains functional in this model, making it a potentially suitable platform for investigating NASH-related inflammation. Moreover, given a major mechanism by which silencing TAZ in hepatocytes blocks liver fibrosis in NASH5 (i.e., by preventing the induction of the HSC activator IHH), the efficacy of GalNAc-siTAZ in hu-liver NASH mice suggest intact cross-talk between human hepatocytes and mouse HSCs. We conducted this study by using male mice, which are the standard in NASH studies due to male sex being a risk factor for NASH fibrosis in mice and humans,29 and female sex in mice can affect metabolic parameters.30 Nonetheless, future studies in female mice are warranted. In addition, this study used the HF-CDAA model, but our previous GalNAc-siTaz study used the fructose-palmitate-cholesterol diet model, and the results were similar.12 Finally, we think it is noteworthy that the donor human hepatocytes that were used in this study were heterozygous for the potent PNPLA3 NASH-risk variant at rs738409.31 The success of GalNAc-siTAZ in hu-mice with rs738409-C/G hepatocytes, which is known to exacerbate NASH in hu-liver mice,24 may add to the evidence suggesting the promise of this therapy. Future studies will be needed to assess GalNAc-siTAZ in hu-mice made from donor hepatocytes without this genetic risk variant.
In conclusion, this study illustrates the potential value of hu-liver mice to test new NASH therapies, especially nucleic acid–based therapies targeting human hepatocytes, with the goal of prioritizing therapeutic candidates for testing in humans. This concept would seem to apply particularly to NASH fibrosis, given its rapidly growing prevalence and devastating nature and the absence to date of any approved therapies.
Materials and methods
Hu-liver NASH mice
At PhoenixBio (Hiroshima, Japan), cryopreserved human hepatocytes (lot BD195: 2 year old Hispanic female, 1 × 106 hepatocytes, BD Biosciences, Ann Arbor, MI) were transplanted under anesthesia into 2- to 4-week-old hemizygous cDNA-uPA/SCID mice through the spleen. Male hu-liver mice (15–18-weeks of age, PXB-mice) were randomized into GalNAc-control and GalNAc-siTAZ groups based on mean body weight and blood human albumin concentration, which was assayed by latex agglutination immunonephelometry (LZ Test Eiken U-ALB, Eiken Chemical, Tokyo, Japan). Based on blood albumin measurements,14 the average replacement index was >90%, which was then verified using anti-STEM121 immunohistochemistry (see below). All of the experiments used a single donor whose hepatocytes were genotyped as C/G at rs738409, indicating they were heterozygous for the PNPLA3 NASH-risk variant.31 The experiments were carried out under procedures and guidelines approved by the Animal Ethics Committee of PhoenixBio, where the in vivo studies were conducted. The mice were housed in a sterile animal facility with a 12-h dark/light cycle and free access to food and water. In the first study, the drinking water was supplemented with 0.3 mg/mL ascorbic acid solution (FUJIFILM Wako Pure Chemical, Osaka, Japan) starting on day 32 of the study. In the second study, the drinking water was supplemented with ascorbic acid solution for the entire duration of the experiment. NASH was induced by placing the mice on a HF-CDAA32 (no. A06071302, Research Diets, New Brunswick, NJ) for 6 weeks, followed by weekly subcutaneous injections of GalNAc-siTAZ or GalNAc-control for another 6 weeks with continued diet feeding. The study was terminated at the end of week 12.
GalNAc-siRNA
The method of conjugating synthetic triantennary GalNAc ligand targeting the asialoglycoprotein receptor with chemically modified siRNA33 was applied to control (nontargeting control) and siTAZ sequences to achieve robust and durable RNAi-mediated TAZ silencing in hepatocytes with the siTAZ conjugate after subcutaneous administration. For control: sense 5′-3′: UAUAUCGUACGUACCGUCGUAU, antisense 5′-3′: AUACGACGGUACGUACGAUdtdt. For siTAZ: sense 5′-3′: CCUCGAAGCCCUCUUCAACUA, antisense 5′-3′: UAGUUGAAGAGGGCUUCGAGGAG.
Histopathologic analysis
Inflammatory cells in H&E-stained liver section images were quantified as the number of mononuclear cells per field (20× objective). For other parameters involving various stains, computerized image analysis (ImageJ, NIH, Bethesda, MD) was used to quantify the area stained. The same threshold settings were used for all of the analyses. For all of the analyses, we quantified six randomly chosen fields per section per mouse. Liver fibrosis was assessed by quantifying Picrosirius (Sirius) red-stained area (no. 24901, Polysciences, Warrington, PA). For STEM121 immunohistochemistry, paraffin sections were rehydrated and subjected to antigen retrieval by placing in a pressure cooker for 10 min in Antigen Unmasking Solution (H-3300, Vector, Newark, CA). The slides were then treated with 3% hydrogen peroxide for 10 min and blocked with Serum-Free Protein Block (X0909, Dako, Santa Clara, CA) for 30 min. Sections were incubated overnight with mouse anti-STEM121 primary antibody (Y40410, Takara, San Jose, CA) and then developed with a DAB substrate kit (no. 8059, Cell Signaling Technology, Danvers, MA).
Immunofluorescence microscopy
Paraffin sections were rehydrated, subjected to antigen retrieval by placing in a pressure cooker for 10 min in Antigen Unmasking Solution (H-3300, Vector), and blocked with serum. Sections were labeled with αSMA (F3777, Sigma, Burlington, MA), anti-TAZ (HPA007415, Millipore Sigma, Burlington, MA), anti-Mac2 (no. CL8942AP, Cedarlane, Burlington, NC), anti-Clec4f (no. AF2784, R & D, Minneapolis, MN), anti-F4/80 (no. 70076, Cell Signaling) or anti-Ki67 (no. AF7617, R & D) antibody overnight, followed by incubation with a fluorophore-conjugated secondary antibody for 1 h. The stained sections were mounted with DAPI-containing mounting medium (P36935, Life Technologies, Carlsbad, CA) and then viewed by fluorescence microscopy. ImageJ was used to quantify the stained area, with consistent threshold settings applied for each analysis. Three randomly selected fields were quantified per section per mouse for image analyses.
Plasma transaminase assays
Plasma ALT and AST concentrations were assayed using a kit from TECO Diagnostics (no. A526-120, Anaheim, CA) and Biorbyt (orb668836, Durham, NC).
Immunoblotting
Liver protein was extracted using RIPA buffer (no. 89900, Thermo Fisher, Waltham, MA), and the protein concentration was measured by a BCA assay (no. 23227, Thermo Fisher). Proteins were heated at 100°C for 5 min, separated by electrophoresis on 4%–20% Tris gels (EC60285, Life Technologies), and transferred to nitrocellulose membranes (no. 1620115, Bio-Rad, Hercules, CA). The membranes were blocked for 30 min at room temperature in Tris-buffered saline and 0.1% Tween 20 containing 5% (w/v) nonfat milk. The membranes were then incubated in the same buffer with anti-TAZ and anti-β-actin antibody (no. 8418, no. 5125, Cell Signaling Technology) or anti-IHH antibody (no. 13388-1-AP, Proteintech, Rosemont, IL) at 4°C overnight, using a 1:1,000 dilution. The protein bands were detected with horseradish peroxidase–conjugated secondary antibodies and Supersignal West Pico enhanced chemiluminescent solution (no. 34080, Thermo Fisher).
qRT-PCR
Total RNA was extracted from liver tissue using the PureLink RNA Mini Kit (12183018A, Invitrogen, Carlsbad, CA). The quality and concentration of the RNA was assessed by absorbance at 260 and 280 nm using a Thermo Scientific NanoDrop spectrophotometer. cDNA was synthesized from 1 μg total RNA using oligo(dT) and Superscript II (Invitrogen). qPCR was performed with a 7500 Real-Time PCR system (Applied Biosystems, Carlsbad, CA) using SYBR Green Master Mix (no. 4367659, Life Technologies). The primer sequences are listed in Table S1.
Measurement of hydroxyproline content of liver tissue
The content of hydroxyproline in livers was measured using the Hydroxyproline Colorimetric Assay Kit from Elabscience (E-BC-K061-S, Houston, TX). Briefly, liver tissue was hydrolyzed by incubating with the hydrolysis solution at 95°C for 1 h. After pH adjustment, liver hydrolysates were clarified using activated carbon and then incubated with the reaction reagent for color development. The absorbance was recorded at 550 nm, and hydroxyproline concentrations were determined by comparing them to standards processed simultaneously.
Statistical analysis
Data were analyzed using a Student’s t test for data, with significance set at p < 0.05.
Data and code availability
Further information and requests for resources, reagents, and data should be directed to the lead contacts, X.W. (xw2279@columbia.edu) and I.T. (iat1@columbia.edu).
Acknowledgments
This work was supported by a research grant from the Takeda Pharmaceutical Company as part of an academic research alliance between Columbia University and Takeda. The authors thank the Takeda-Drug Discovery Unit Chemistry and Liver Disease teams and the Takeda-Oligochemistry teams for material support and technical input. The Takeda scientists involved in this project included Drs. Bernard Allan, Maria Deato, Jason Pickens, Tony Gibson, Brian Ko, Regina Lemus, and Pawan Kumar.
Author contributions
X.W., M.P.M., I.T., Y.M., S.K.D., and D.R.R. were involved in the study conception and experimental design. X.W., M.P.M., and H.S. conducted liver analyses and plasma and serum assays. X.W., M.P.M., and I.T. drafted the manuscript. All of the authors edited the manuscript and approved the submitted version.
Declaration of interests
The laboratory of I.T. received research funding, material support, and technical input from the Takeda Pharmaceutical Company to study siTAZ therapeutics, for which Columbia University holds a patent.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.101165.
Contributor Information
Xiaobo Wang, Email: xw2279@columbia.edu.
Ira Tabas, Email: iat1@columbia.edu.
Supplemental information
and Table S1
References
- 1.Younossi Z.M., Golabi P., Paik J.M., Henry A., Van Dongen C., Henry L. The global epidemiology of nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH): a systematic review. Hepatology. 2023;77:1335–1347. doi: 10.1097/HEP.0000000000000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanyal A.J., Van Natta M.L., Clark J., Neuschwander-Tetri B.A., Diehl A., Dasarathy S., Loomba R., Chalasani N., Kowdley K., Hameed B., et al. Prospective study of outcomes in adults with nonalcoholic fatty liver disease. N. Engl. J. Med. 2021;385:1559–1569. doi: 10.1056/NEJMoa2029349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harrison S.A., Allen A.M., Dubourg J., Noureddin M., Alkhouri N. Challenges and opportunities in NASH drug development. Nat. Med. 2023;29:562–573. doi: 10.1038/s41591-023-02242-6. [DOI] [PubMed] [Google Scholar]
- 4.Santhekadur P.K., Kumar D.P., Sanyal A.J. Preclinical models of non-alcoholic fatty liver disease. J. Hepatol. 2018;68:230–237. doi: 10.1016/j.jhep.2017.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X., Zheng Z., Caviglia J.M., Corey K.E., Herfel T.M., Cai B., Masia R., Chung R.T., Lefkowitch J.H., Schwabe R.F., Tabas I. Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 2016;24:848–862. doi: 10.1016/j.cmet.2016.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X., Cai B., Yang X., Sonubi O.O., Zheng Z., Ramakrishnan R., Shi H., Valenti L., Pajvani U.B., Sandhu J., et al. Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab. 2020;31:969–986.e7. doi: 10.1016/j.cmet.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gardin A., Remih K., Gonzales E., Andersson E.R., Strnad P. Modern therapeutic approaches to liver-related disorders. J. Hepatol. 2022;76:1392–1409. doi: 10.1016/j.jhep.2021.12.015. [DOI] [PubMed] [Google Scholar]
- 8.Rizk M., Tüzmen S. Patisiran for the treatment of patients with familial amyloid polyneuropathy. Drugs Today. 2019;55:315–327. doi: 10.1358/dot.2019.55.5.2958475. [DOI] [PubMed] [Google Scholar]
- 9.Honor A., Rudnick S.R., Bonkovsky H.L. Givosiran to treat acute porphyria. Drugs Today. 2021;57:47–59. doi: 10.1358/dot.2021.57.1.3230207. [DOI] [PubMed] [Google Scholar]
- 10.Scott L.J., Keam S.J. Lumasiran: first approval. Drugs. 2021;81:277–282. doi: 10.1007/s40265-020-01463-0. [DOI] [PubMed] [Google Scholar]
- 11.Giordano S., Polimeni A., Esposito G., Indolfi C., Spaccarotella C. Inclisiran: present and future perspectives of a new effective LDL cholesterol-lowering agent. Curr. Opin. Lipidol. 2023;34:133–140. doi: 10.1097/MOL.0000000000000877. [DOI] [PubMed] [Google Scholar]
- 12.Wang X., Sommerfeld M.R., Jahn-Hofmann K., Cai B., Filliol A., Remotti H.E., Schwabe R.F., Kannt A., Tabas I. A therapeutic silencing RNA targeting hepatocyte TAZ prevents and reverses fibrosis in nonalcoholic steatohepatitis in mice. Hepatol. Commun. 2019;3:1221–1234. doi: 10.1002/hep4.1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grompe M., Strom S. Mice with human livers. Gastroenterology. 2013;145:1209–1214. doi: 10.1053/j.gastro.2013.09.009. [DOI] [PubMed] [Google Scholar]
- 14.Tateno C., Kawase Y., Tobita Y., Hamamura S., Ohshita H., Yokomichi H., Sanada H., Kakuni M., Shiota A., Kojima Y., et al. Generation of novel chimeric mice with humanized livers by using hemizygous cDNA-uPA/SCID mice. PLoS One. 2015;10:e0142145. doi: 10.1371/journal.pone.0142145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kisoh K., Sugahara G., Ogawa Y., Furukawa S., Ishida Y., Okanoue T., Kohara M., Tateno C. Estimating drug efficacy with a diet-induced NASH model in chimeric mice with humanized livers. Biomedicines. 2021;9:1647. doi: 10.3390/biomedicines9111647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kelly S., Bliss T.M., Shah A.K., Sun G.H., Ma M., Foo W.C., Masel J., Yenari M.A., Weissman I.L., Uchida N., et al. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc. Natl. Acad. Sci. USA. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu C., Kim K., Wang X., Bartolome A., Salomao M., Dongiovanni P., Meroni M., Graham M.J., Yates K.P., Diehl A.M., et al. Hepatocyte Notch activation induces liver fibrosis in nonalcoholic steatohepatitis. Sci. Transl. Med. 2018;10:eaat0344. doi: 10.1126/scitranslmed.aat0344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conway J., Pouryahya M., Gindin Y., Pan D.Z., Carrasco-Zevallos O.M., Mountain V., Subramanian G.M., Montalto M.C., Resnick M., Beck A.H., et al. Integration of deep learning-based histopathology and transcriptomics reveals key genes associated with fibrogenesis in patients with advanced NASH. Cell Rep. Med. 2023;4:101016. doi: 10.1016/j.xcrm.2023.101016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Totaro A., Castellan M., Di Biagio D., Piccolo S. Crosstalk between YAP/TAZ and Notch signaling. Trends Cell Biol. 2018;28:560–573. doi: 10.1016/j.tcb.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Touchette E.K., Bates M.C., Johnson M.C., O'Brien T.C., Melton R.J., Long K.R., Kakuni M., Baginski M., Radiloff D.R., Sagartz J.E. Characterization and prevention of hypovitaminosis c in chimeric mice with humanized livers. Comp. Med. 2022;72:355–363. doi: 10.30802/AALAS-CM-22-000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao Y., Batmanov K., Hu W., Zhu K., Tom A.Y., Guan D., Jiang C., Cheng L., McCright S.J., Yang E.C., et al. Hepatocytes demarcated by EphB2 contribute to the progression of nonalcoholic steatohepatitis. Sci. Transl. Med. 2023;15:eadc9653. doi: 10.1126/scitranslmed.adc9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Godoy P., Hewitt N.J., Albrecht U., Andersen M.E., Ansari N., Bhattacharya S., Bode J.G., Bolleyn J., Borner C., Böttger J., et al. Recent advances in 2D and 3D in vitro systems using primary hepatocytes, alternative hepatocyte sources and non-parenchymal liver cells and their use in investigating mechanisms of hepatotoxicity, cell signaling and ADME. Arch. Toxicol. 2013;87:1315–1530. doi: 10.1007/s00204-013-1078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ratziu V., Harrison S.A., Francque S., Bedossa P., Lehert P., Serfaty L., Romero-Gomez M., Boursier J., Abdelmalek M., Caldwell S., et al. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016;150:1147–1159.e1145. doi: 10.1053/j.gastro.2016.01.038. [DOI] [PubMed] [Google Scholar]
- 24.Kabbani M., Michailidis E., Steensels S., Fulmer C.G., Luna J.M., Le Pen J., Tardelli M., Razooky B., Ricardo-Lax I., Zou C., et al. Human hepatocyte PNPLA3-148M exacerbates rapid non-alcoholic fatty liver disease development in chimeric mice. Cell Rep. 2022;40:111321. doi: 10.1016/j.celrep.2022.111321. [DOI] [PubMed] [Google Scholar]
- 25.Moore M.P., Wang X., Shi H., Meroni M., Cherubini A., Ronzoni L., Parks E.J., Ibdah J.A., Rector R.S., Valenti L., et al. Circulating indian hedgehog is a marker of the hepatocyte-TAZ pathway in experimental NASH and is elevated in humans with NASH. JHEP. Rep. 2023;5:100716. doi: 10.1016/j.jhepr.2023.100716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X., Zeldin S., Shi H., Zhu C., Saito Y., Corey K.E., Osganian S.A., Remotti H.E., Verna E.C., Pajvani U.B., et al. TAZ-Induced Cybb in hepatocytes contributes to early liver tumorigenesis in experimental non-alcoholic steatohepatitis (online) J. Hepatol. 2021;76:910–920. doi: 10.1016/j.jhep.2021.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seidman J.S., Troutman T.D., Sakai M., Gola A., Spann N.J., Bennett H., Bruni C.M., Ouyang Z., Li R.Z., Sun X., et al. Niche-specific reprogramming of epigenetic landscapes drives myeloid cell diversity in nonalcoholic steatohepatitis. Immunity. 2020;52:1057–1074.e7. doi: 10.1016/j.immuni.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Plouffe S.W., Lin K.C., Moore J.L., 3rd, Tan F.E., Ma S., Ye Z., Qiu Y., Ren B., Guan K.L. The Hippo pathway effector proteins YAP and TAZ have both distinct and overlapping functions in the cell. J. Biol. Chem. 2018;293:11230–11240. doi: 10.1074/jbc.RA118.002715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonardo A., Nascimbeni F., Ballestri S., Fairweather D., Win S., Than T.A., Abdelmalek M.F., Suzuki A. Sex differences in nonalcoholic fatty liver disease: state of the art and identification of research gaps. Hepatology. 2019;70:1457–1469. doi: 10.1002/hep.30626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griffin C., Lanzetta N., Eter L., Singer K. Sexually dimorphic myeloid inflammatory and metabolic responses to diet-induced obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016;311:R211–R216. doi: 10.1152/ajpregu.00136.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romeo S., Kozlitina J., Xing C., Pertsemlidis A., Cox D., Pennacchio L.A., Boerwinkle E., Cohen J.C., Hobbs H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008;40:1461–1465. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wei G., An P., Vaid K.A., Nasser I., Huang P., Tan L., Zhao S., Schuppan D., Popov Y.V. Comparison of murine steatohepatitis models identifies a dietary intervention with robust fibrosis, ductular reaction, and rapid progression to cirrhosis and cancer. Am. J. Physiol. Gastrointest. Liver Physiol. 2020;318 doi: 10.1152/ajpgi.00041.2019. G174-g188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Debacker A.J., Voutila J., Catley M., Blakey D., Habib N. Delivery of oligonucleotides to the liver with GalNAc: from research to registered therapeutic drug. Mol. Ther. 2020;28:1759–1771. doi: 10.1016/j.ymthe.2020.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
and Table S1
Data Availability Statement
Further information and requests for resources, reagents, and data should be directed to the lead contacts, X.W. (xw2279@columbia.edu) and I.T. (iat1@columbia.edu).