

Abstract
We present an improved version of the ABSINTH implicit solvation model and forcefield paradigm (termed c-ABSINTH) by incorporating a grid-based term that bootstraps against experimentally derived and computationally optimized conformational statistics for blocked amino acids. These statistics provide high-resolution descriptions of the intrinsic backbone dihedral angle preferences for all twenty amino acids. The original ABSINTH model generates Ramachandran plots that are too shallow in terms of the basin structures and too permissive in terms of dihedral angle preferences. We bootstrap against the reference optimized landscapes and incorporate CMAP-like residue-specific terms that help us reproduce the intrinsic dihedral angle preferences of individual amino acids. These corrections that lead to c-ABSINTH are achieved by balancing the incorporation of the new residue-specific terms with the accuracies inherent to the original ABSINTH model. We demonstrate the robustness of c-ABSINTH through a series of examples to highlight the preservation of accuracies as well as examples that demonstrate the improvements. Our efforts show how the recent experimentally derived and computationally optimized coil library landscapes can be used as a touchstone for quantifying errors and making improvements to molecular mechanics forcefields.
1. INTRODUCTION
Solvent-mediated interactions make substantial contributions to the conformational and binding equilibria of proteins. In molecular simulations, the effects of solvation can be treated using either explicit representation of solvent molecules 1 or through implicit models that are based on mean-field descriptions of solvation 2. Implicit models are of considerable interest because they can enable efficient sampling and simulations of large biomolecular systems 3. Over the years, several implicit solvation models have been developed and improved upon 4 to describe solvation effects on conformational and binding equilibria 5.
The conceptual foundations of implicit solvation models rest on a formal thermodynamic dissection of the solvation process, which is readily described for a rigid solute. Formally, the free energy of solvation for a rigid solute is defined as the change in the free energy associated with the transfer of the solute from vacuum to the solvent 6. For biomolecules, the solvent of interest is an aqueous solution with a finite concentration of solution ions. The free energy of solvation can be computed as shown in Equation (1)6:
| (1) |
Here, the overall free energy solvation is written as the sum of two composite terms: , the polar / electrostatic term, is the difference between the charging free energies for the rigid solute in solvent versus vacuum , and is the nonpolar term that accounts for mean field estimates of the dispersive, attractive interactions between the solute and solvent () as well as the free energy cost associated with cavitation (). In the Poisson framework, captured via the Poisson-Boltzmann model 7 and an approximation known as the Generalized Born model 8, computations of polar and nonpolar terms are decoupled from each other. The main differences between the two approaches center on the treatment of the polar / electrostatic term and both models can be combined with empirical approaches for modeling nonpolar interactions 9.
An alternative to the Poisson framework was proposed by Lazaridis and Karplus 10, who introduced an effective energy function (EEF1) method wherein one estimates the contribution of distinct solvation groups to by using experimentally measured free energies of solvation 11 for small molecules as reference free energies. In EEF1 the overall solvation process is modeled as the sum of a single direct mean-field interaction (DMFI) term and a screening term :
| (2) |
Here, is the free energy of solvation of group i within the solute. A polyatomic solute can be thought of as a concatenation of different solvation groups, where each solvation group corresponds to a chemical moiety. Since solvent can be occluded from sites occupied by atoms of other solvation groups of the polyatomic solute, the true for solvation group i can be calculated by estimating the degree to which atoms in group i are occluded from the solvent.
The ABSINTH implicit solvation model and forcefield paradigm 12, which was introduced roughly a decade ago, is a generalization of the EEF1 paradigm. This solvation model is interoperable with standard, non-polarizable molecular mechanics forcefields. Over the years, the ABSINTH model has been deployed, with considerable success 13, in a variety of settings. Particular successes have been achieved in modeling of conformationally heterogeneous systems such as intrinsically disordered proteins (IDPs) 14, disordered regions tethered to folded domains 15, and unfolded proteins 16. The ABSINTH model has been applied in other settings including the modeling of experimental data to fit electron density maps 17 and simulations of viral proteins 18, systems undergoing order-disorder transitions 19, systems that undergo phase separation 20, transactivation domains of transcription factors 21, disordered tails of microtubule-forming tubulin proteins 22, and nanostructures formed by oligopeptides 23. Improvements 24 and generalizations 25 have been incorporated into the model since it was first introduced. A majority of the reported simulation results deploy the ABSINTH model in conjunction with novel Metropolis-based Monte Carlo (MMC) sampling methods and enhanced sampling methods 26 that achieve statistical rigor in sampling 24, 27. Recent advances have enabled the use of ABSINTH with torsional molecular dynamics (TMD) and hybrid TMD-MMC methods 19, 28.
In the ABSINTH model 12, each polyatomic solute is parsed into a set of solvation groups. Each of these groups corresponds to a model compound for which the free energy of solvation has been experimentally measured. The choice of solvation groups is different from the EEF1 model. The ABSINTH framework stays anchored to solvation groups for which data have been experimentally measured. This avoids the parsing of solvation groups based on empirical dissections of the free energies of solvation 29. The total solvent-mediated energy associated with a rigid polyatomic solute that includes the biopolymer and solution ions is written as:
| (3) |
Here, Wsolv represents the DMFI and captures the free energy change associated with transferring the polyatomic solute into a mean field solvent while accounting for the modulation of the reference free energy of solvation for each solvation group due to occlusion from the solvent by atoms of the polyatomic solute. This is an intrinsically many-body effect. Further modulations to the free energy of solvation of the solute due to screened interactions with charged sites on the polyatomic solute are accounted for by the Wel term, wherein the effects of dielectric inhomogeneities are accounted for without making explicit assumptions regarding the distance or spatial dependencies of dielectric saturation. The term ULJ is a standard 12-6 Lennard-Jones potential that models the joint contributions of steric exclusion and London dispersion, whereas Ucorr models specific torsion and bond angle-dependent stereoelectronic effects that are not captured by the ULJ term. The ABSINTH paradigm has optimal interoperability23, 30 with OPLS-AA/L31 and the CHARMM family of forcefields 32 because of the parameterization paradigm used in these forcefields, especially with regard to the assignment of partial charges to sites on neutral groups. Interoperability of the ABSINTH model with standard non-polarizable molecular mechanics forcefields requires the strict enforcement of neutral groups framework for electrostatic interactions, treating long-range electrostatics explicitly without cutoffs or tapering, reducing the numbers of ad hoc torsional potentials, and imposing a standard set of parameters for Lennard-Jones potentials across all forcefields.
In the accompanying manuscript 33, we presented experimentally derived and computationally optimized backbone dihedral angle preferences for all twenty naturally occurring amino acids. To summarize, we extracted residue-specific distributions of backbone dihedral angles from high-resolution coil library structures, and determined the populations of different basins by a gradient-based clustering method. This information was compared against the population information obtained by spectroscopic measurements on blocked amino acids, which revealed significant discrepancies. Accordingly, we optimized the backbone dihedral angle statistics to conform to those of blocked amino acids. This procedure produces the experimentally derived and computationally optimized high-resolution landscapes of dihedral angle preferences of blocked amino acids. These conformational statistics provide experimentally derived touchstones for calibrating the accuracies of molecular mechanics forcefields. Here, we assess the accuracy of the ABSINTH paradigm, made to be interoperable with the OPLS-AA/L forcefield, vis-à-vis the newly established coil-library based landscapes for all twenty amino acids. Based on mismatches between conformational basins generated using the ABSINTH model and the experimentally derived touchstone 33, we incorporate residue-specific CMAP-style 5a, 34 correction terms to the ABSINTH energy function. These terms, which are appropriately balanced to maintain the known accuracies of the ABSINTH model, lead to an improved version of ABSINTH that we refer to as c-ABSINTH. We illustrate the utility of the improvements by assessing the accuracy of c-ABSINTH for a cross-section of problems including the recapitulation of experimentally measured local conformational equilibria, preservation of established accuracies, and improved descriptions of homopolypeptides.
2. ASSESSMENT OF THE ABSINTH MODEL VIS-À-VIS THE OPTIMIZED COIL LIBRARY LANDSCAPE
We assessed the performance of ABSINTH by comparing results from simulations for blocked amino acids to conformational statistics from the optimized coil library landscapes for each of the twenty naturally occurring amino acids. These simulations were performed using the ABSINTH model combined with the OPLS-AA/L parameters as implemented in the abs3.2_opls.prm parameter set that is part of version 2.0 of the CAMPARI simulation package (http://campari.sourceforge.net).
In each simulation, the system contains a blocked amino acid enclosed in a spherical soft-wall droplet of radius 100 Å. For charged residues, we introduced a counterion: Na+ for Asp and Glu, and Cl− for Lys and Arg. His was modeled by protonating the -nitrogen atom. The simulation temperature was set to 300 K. Each simulation involved 9.8 × 107 steps of MMC sampling. Information regarding the backbone and dihedral angles was gathered from each simulation once every 2 × 103 steps. For each blocked amino acid, we performed ten independent simulations and a total of 4.9 × 105 data points were analyzed.
Figure 1 shows the Ramachandran plots for all twenty amino acids. The calculated ABSINTH-based landscapes, translated as potentials of mean force (PMFs) in terms of and , are broader and shallower when compared to the optimized coil-library landscapes 33. An important difference between the ABSINTH-derived landscapes and the optimized coil library landscapes is the significant population in the region of the (, )-space, which is centered on and . It is present in the original Ramachandran plots for Ala 35 that are based on hard-sphere potentials. However, even for potentials based on purely hard-sphere repulsions, the region becomes proscribed due to non-nearest neighbor steric interactions 36. It is noteworthy though that the region is not populated in the optimized coil library, which should be directly applicable to blocked amino acids. This suggests that considerations that go beyond steric interactions cause a destabilization of the region for all non-Gly amino acids. The persistence of the region in ABSINTH is the result of smaller van der Waals radii when compared to most standard molecular mechanics forcefields and the absence of a steric envelope for the backbone polar hydrogen. This adjustment was necessary to enable the formation of hydrogen-bonded structures such as regular -helices, -sheets, and hairpin turns 30.
Figure 1: ABSINTH PMF landscapes for all 20 naturally occurring amino acids.
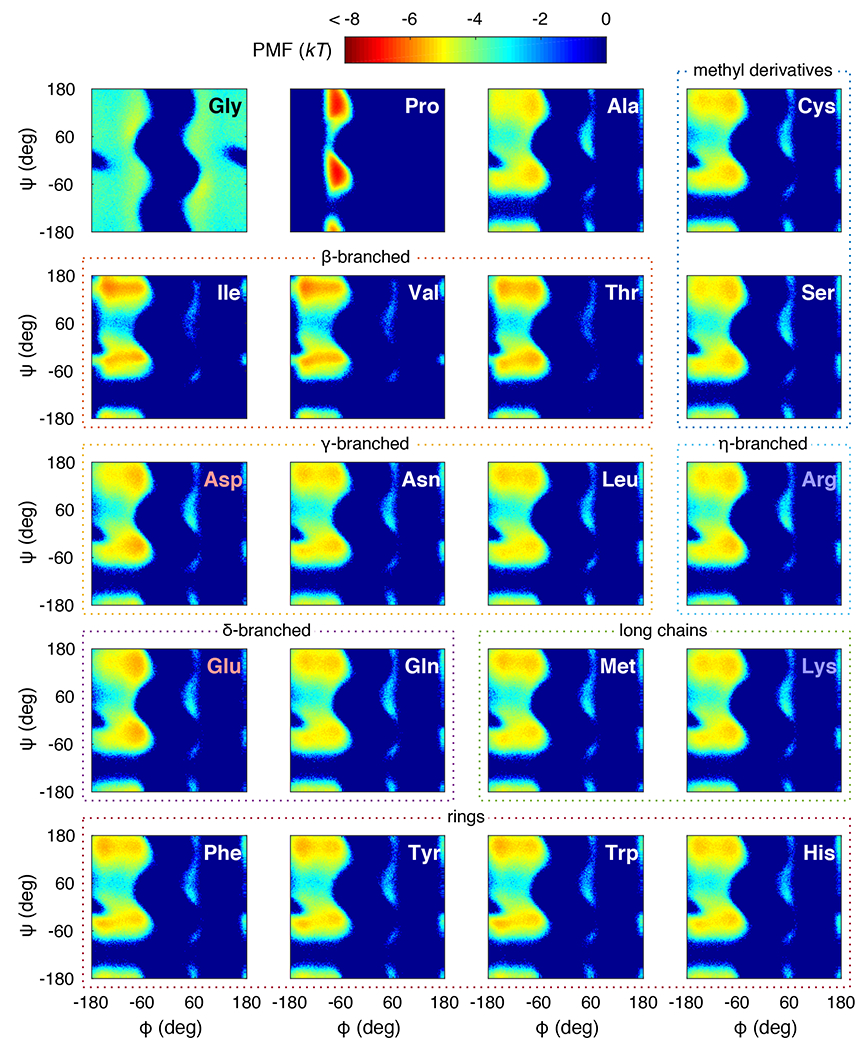
The color bar annotating the landscapes in units of kT is shown at the top. Each panel shows a PMF of a blocked amino acid labeled at the upper right corner. Red labels indicate acidic amino acids, and blue labels indicate basic ones. The different amino acids are grouped according to their stereochemistry (dotted lines). For histidine, we employed the -nitrogen protonated form (HIE).
3. QUANTIFYING DEVIATIONS AND INCORPORATION OF CORRECTION TERMS INTO ABSINTH
We quantified the magnitudes of the deviations between the ABSINTH-derived and optimized coil library landscapes. This was used to develop CMAP-style correction terms into ABSINTH. Based on the data shown in Figure 1 and the optimized coil library landscapes 33, we designed amino-acid specific correction terms using the following approach. For each amino acid, we quantified correction terms for each pixel in the Ramachandran space as follows:
| (4) |
Here, is defined in terms of the PMFs from the optimized coil library landscape (PMFOCLL) and ABSINTH (PMFABSINTH):
| (5) |
The parameter k is a non-negative scaling factor. The determination of k is explained later in this section. If a pixel is accessible in the ABSINTH-based simulations but the population is zero or negligible for the corresponding pixel in the optimized coil library landscape, then the value of Ucorr is set to the largest positive value of plus 5 kT to inhibit access to the pixel in question. Conversely, if a pixel is not accessible in the ABSINTH-based simulation, but density within the pixel is non-zero for the coil library landscape, then the correction term is set to the largest negative value of . If both the ABSINTH and target populations have no data for a specific pixel, we set the correction term to zero.
For each residue type, the prescription above discretizes the values of Ucorr across the pixels. In order to obtain a continuous surface from the discrete set of correction values, we employed a non-interpolating cardinal B-spline. This approach is based on piecewise polynomial functions that are generally used for approximation and graphic design 37. It uses a linear combination of polynomials, whose ranges depend on the locations of the data points (known as knots) and whose values depend on the given values at the knots (known as control points).
For a given sequence of knots with constant separation , a cardinal B-spline of zeroth order, denoted as , is defined as follows:
| (6) |
A cardinal B-spline of order m, denoted , is defined recursively as:
| (7) |
In Equation (7):
| (8) |
The spline curve of order m for a given set of control points has the form:
| (9) |
In the current work, the knots are on the (, )-space, and we set and N = 144. We assumed that the discrete values are the values at the center of each bin. Accordingly, and . Note that higher values for m “dilute” original values of control points by using more control points.
For the incorporation of residue-specific correction terms into ABSINTH, we have two parameters to tune, viz., the order of B-spline m and the global scaling factor k. We performed a series of ABSINTH simulations for blocked amino acids with various combinations of m and k, and for each combination of m and k, we generated PMFs for all twenty amino acids from the simulation results. The number of MMC steps was reduced to 107 and we performed ten independent simulations for each combination of m and k. All other settings were identical to those for unmodified ABSINTH simulations. We quantified the performance of the parameter combinations in terms of an average absolute error from the target population. The combination of m = 2 and k = 0.6 generates PMFs that are most similar to the target coil library landscapes (Figure 2a). We also tested other measures for performance, such as root-mean-squared errors and correlation coefficients, and they too showed similar results (Supplementary Figure S1). It should be noted that the choice of m = 2 yields the most optimal results and it provides the most conservative surface among the values of m that generate a smooth surface (m > 1). Hence, we set m = 2, to avoid losing accuracy associated with original values for the control points.
Figure 2: Effects of the spline degree m and global scaling factor k.
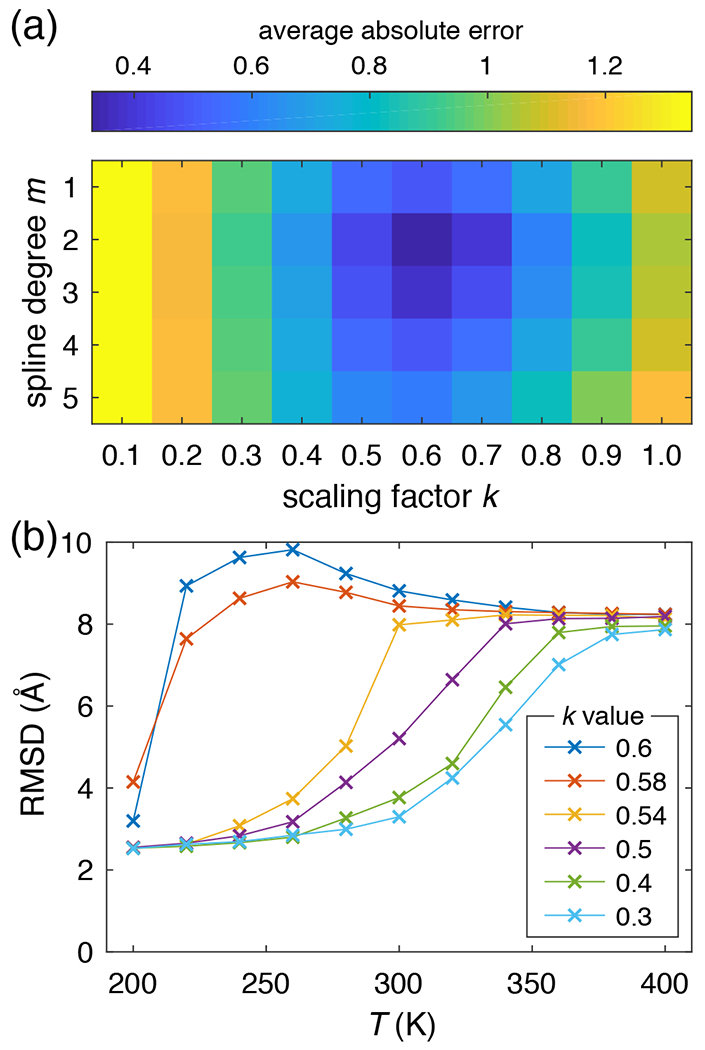
(a) Average absolute errors between corrected ABSINTH and target populations, averaged over all residue types. Different colors indicate the errors averaged over different amino acids (see color bar). (b) Folding curves of the FS-peptide generated by corrected ABSINTH with several k values from 0.3 to 0.6. The abscissa shows simulation temperatures, and the ordinate shows the root-mean-squared distance (RMSD) to the canonical helix.
Our goal was to refine and / or improve ABSINTH to obtain better agreement with the optimized coil library landscapes while preserving the established accuracies of this solvation model and forcefield paradigm. The optimized coil library landscape was achieved, in part, by a diminution of the right-handed alpha-helical basin designated as . This is a valid adjustment for blocked amino acids since a regular -helix requires a minimum of three consecutive (, ) pairs being in the basin. However, our incorporation of the Ucorr term is likely to weight the overall forcefield too strongly toward the intrinsic conformational preferences of amino acids. We tested for this possibility by simulating the temperature-dependent helix-to-coil transitions of the FS peptide 38, whose sequence is N-acetyl-A5(AAARA)3-N′-methylamide. The FS peptide is a useful target for forcefield calibration and optimization 39. It was used as a test case in the original development of the ABSINTH model and for generalizing the Lifson-Roig theory for helix-coil transitions 19.
We performed simulations of the FS peptide in a soft-wall spherical droplet of radius 45 Å with neutralizing counterions and an excess of ~15 mM NaCl background. The solute ions were modeled explicitly using the optimized parameters of Mao and Pappu 40. The initial structure of the FS peptide was generated at random and each simulation consisted of 107 MMC steps. Data were collected for analysis once every 5 × 103 steps after discarding the first 2.5 × 106 steps of simulation data. We performed simulations at eleven different temperatures ranging from 200 K to 400 K with a spacing of 20 K. We employed thermal replica exchange 41 to enhance the sampling at each temperature. We calculated the root-mean-squared distance to the canonical -helix as an order parameter for folding.
As shown in Figure 2b, simulations that use a corrected ABSINTH model with k = 0.6 fail to recover a two-state melting curve for the FS peptide. This is because the correction term over-stabilizes extended -strand conformations at low temperatures. However, by down weighting the correction term, which is achieved by setting k = 0.5, we reproduce the correct melting curve for the FS peptide (Figure 2b). In fact, for k = 0.5, the melting temperature from simulations is in close agreement with the experimentally derived value of ~ 303 K 42.
Figure 3 shows the PMFs for blocked alanine and compares the original ABSINTH with different values of k. Figure 3a shows the original ABSINTH result (k = 0.0), while Figures 3b and 3c show the results from corrected ABSINTH simulations with k = 0.5 and k = 0.6, respectively. Although k = 0.6 produces a PMF that is most similar to the optimized landscape population, we find that k = 0.5 also has a strong bias toward the target population in the coil library landscape (see Figure 2a and Supplementary Figure S1). Importantly, the region is absent from the PMFs obtained using both versions of corrected ABSINTH model. Given the preservation of the helix-to-coil transition of the FS peptide with m = 2 and k = 0.5, we assigned this model to be the new corrected ABSINTH or c-ABSINTH model. Here, the c stands for coil library-based and CMAP-like. One could argue that we used one set of data points, viz., temperature-dependent helix-to-coil transitions of the FS peptide and that our parameterization might be overly sensitive to this choice. However, it is worth emphasizing that the choice of the FS peptide was governed exclusively by the diminution of the alpha-helical basin in the coil library landscape. Using the FS peptide system, we were able to balance the incorporation of intrinsic conformational preferences while ensuring the emergence of sequence-specific secondary structure preferences. A range of parameters in the vicinity of m = 2 and k = 0.5 would work equally well, although there is a sharp change when k approaches 0.6. This might raise concerns about the robustness of basing the c-ABSINTH model on the choice of m = 2 and k = 0.5, but as shown below, the specific parameter choice for the incorporation of residue-specific terms into Ucorr proves to be robust across a range of problems that were not used in the parameterization of c-ABSINTH.
Figure 3: PMFs for alanine dipeptide simulations obtained using different versions of ABSINTH parameters.
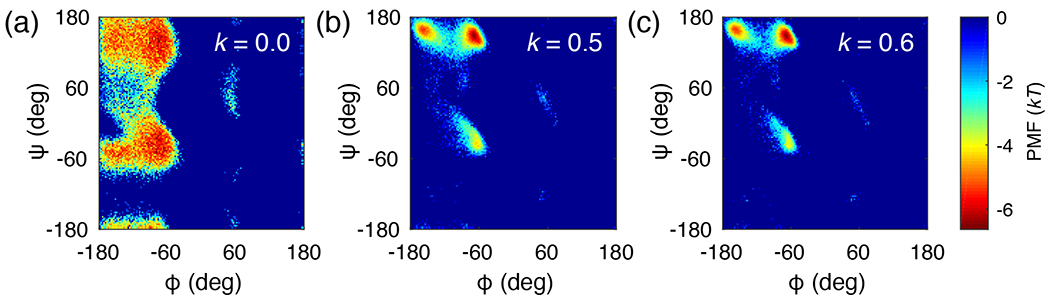
(a) Original ABSINTH. We randomly picked 50,000 data points among 490,000 points to use the same population size with other simulations. (b) Corrected ABSINTH with m = 2 and k = 0.5. (c) Corrected ABSINTH with m = 2 and k = 0.6.
Figure 4 shows the PMFs generated using the new c-ABSINTH model for each of the twenty naturally occurring amino acids modeled as blocked amino acids. We did not refine the parameters for Pro (i.e., Ucorr = 0 for all and ) because these were worked upon extensively in previous work 24. PMFs generated using c-ABSINTH deviate minimally from the optimized coil library PMFs (mean absolute error of three major basin populations = 3.56%, see Supplementary Figures S2 and S3). For each amino acid, we compared the populations of different basins between the original ABSINTH and c-ABSINTH simulations (Figure 5). This analysis follows the basin analysis method described in the accompanying manuscript 33. The c-ABSINTH model diminishes the population in the basin and eliminates population of the basin. As a result, there is a higher population in the and PII basins when compared to the PMFs based on the unmodified ABSINTH. It should be emphasized at this juncture that the distributions of sidechain dihedral angles and are essentially unchanged with the c-ABSINTH correction term (Supplementary Figures S4 and S5).
Figure 4: c-ABSINTH PMF landscapes for all 20 naturally occurring amino acids.
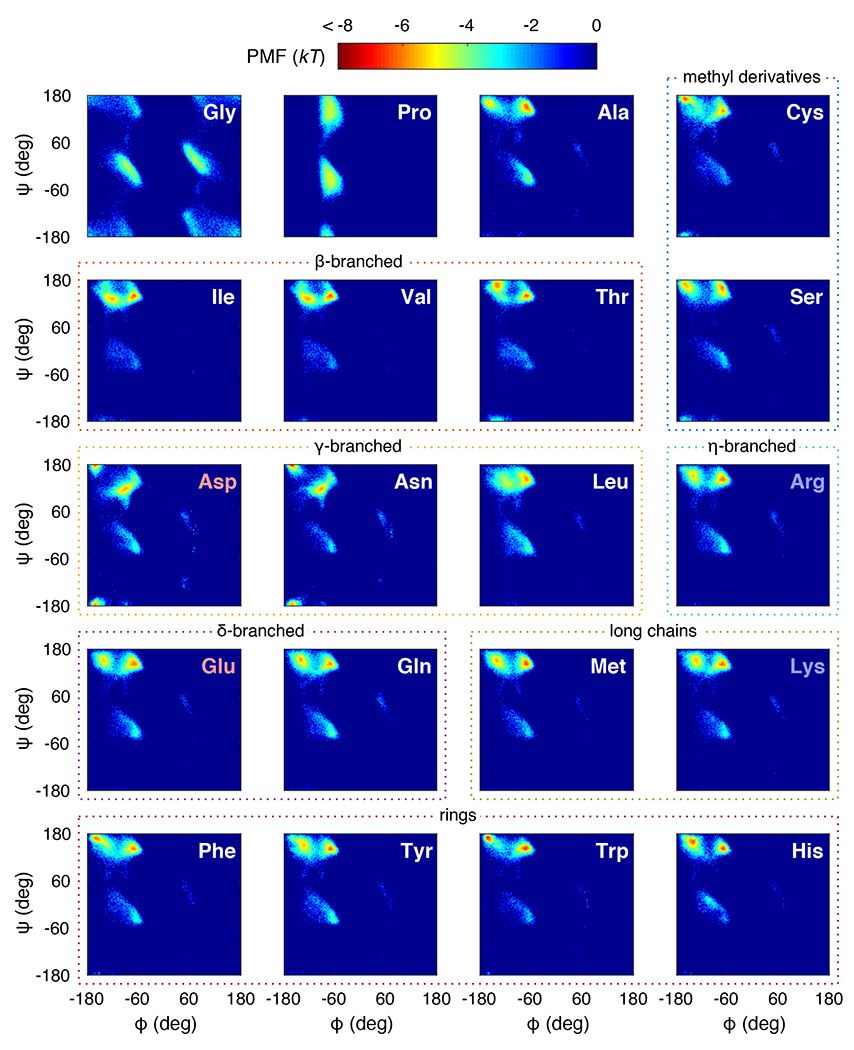
The color bar annotating the landscapes in units of kT is shown at the top. Each panel shows a PMF of a blocked amino acid labeled at the upper right corner. Red labels indicate acidic amino acids, and blue labels indicate basic ones. The different amino acids are grouped according to their stereochemistry (dotted lines). For histidine, we employed the -nitrogen protonated form.
Figure 5: Relative populations of three major basins (, PII, and ) and an unphysical basin from original ABSINTH (ABS) and c-ABSINTH (c-ABS) simulations for 18 amino acids except glycine and proline.
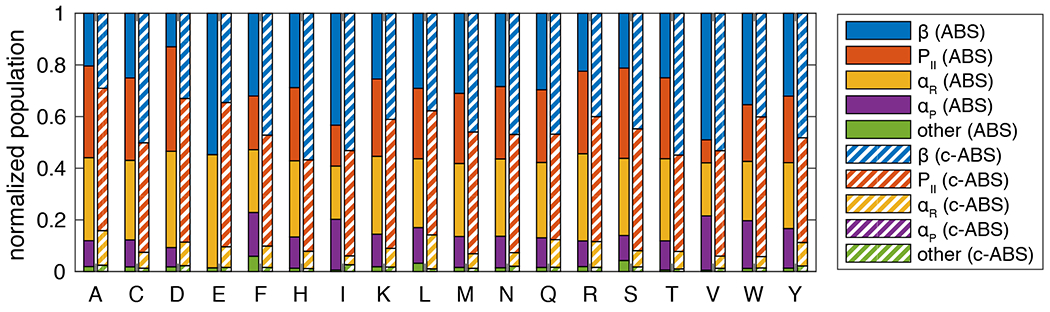
ABSINTH simulations for glutamate (E) show a single merged basin for and PII (shown in blue), and another merged basin for and (shown in yellow).
We also calculated the scalar coupling constants, 3J(HN,), using results from the original ABSINTH and the c-ABSINTH models and compared these to experimental data for blocked amino acids. As shown in Figure 6, c-ABSINTH shows better agreement with the experimental data. This is quantified in terms of a higher Pearson correlation coefficient and reduction of the systematically higher values for the coupling constants. Overall, by bootstrapping against the coil library landscape and using a CMAP-like strategy, we were able to incorporate a suitable correction term for backbone conformational statistics. Importantly, we balanced the incorporation of the correction term with maintenance of features that were accurate in the original ABSINTH model and are not part of the intrinsic preferences manifest in the coil library landscape.
Figure 6: Experimentally determined scalar coupling constants (Jexp) 74 versus the calculated values from the simulation data using the Karplus equation (Jcalc).
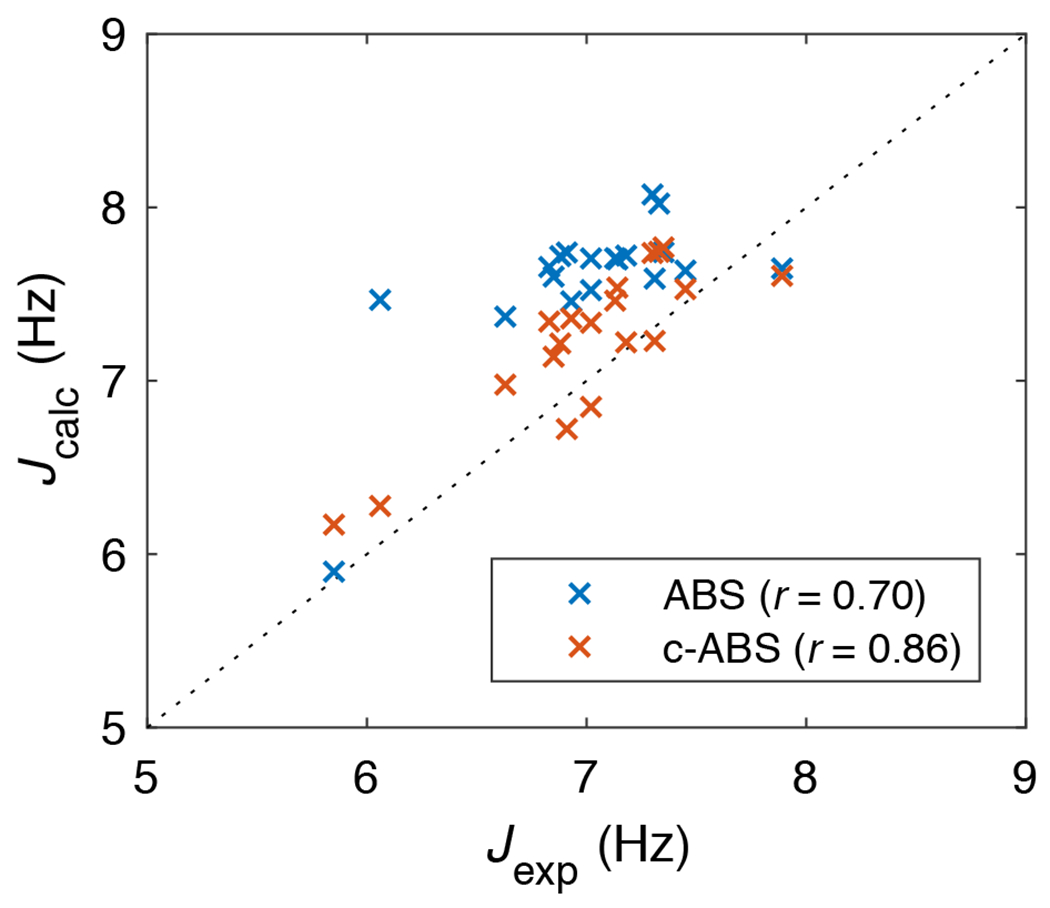
Blue crosses indicate the data from original ABSINTH simulations, and red crosses indicate those from c-ABSINTH simulations.
4. TRANSFERABILITY OF c-ABSINTH CORRECTIONS FOR PEPTIDES THAT ARE USED AS MODELS OF RANDOM COILS
We tested the accuracy of c-ABSINTH for larger systems that have served as experimental touchstones for canonical random coils. The peptides of interest were Acetyl-(Gly)2-Xaa-(Gly)2-NH2, where Xaa is the one of the eighteen residues excluding Gly and Pro. In all of the simulations of these systems, the initial structure for the peptide of interest was randomly generated, and the peptide was encapsulated in a soft-wall spherical shell of radius 100 Å. For each system, we performed twenty independent MC simulations, setting the simulation temperature to be 300 K. Of the twenty simulations, ten were based on the original ABSINTH model and the other ten used the c-ABSINTH model. The c-ABSINTH simulations differ from the original ABSINTH model only for the Ucorr term where c-ABSINTH uses the new CMAP-like corrections. Each simulation consists of 107 MMC steps, and data were accumulated once every 5 × 103 steps. We converted the computed dihedral angle data into scalar coupling constants 3J(HN,) as before to compare with experimentally measured values 43, and found that c-ABSINTH provides better agreement with the experimental data (Supplementary Figure S6).
We also simulated two oligo-alanine peptides, (Ala)5 and (Ala)7 for which accurate NMR J-coupling data are available44. We performed 20 independent simulations each starting with randomly generated initial structures. Each peptide is then situated in a soft-wall spherical droplet with radius 100 Å. Each simulation consists of 107 steps, and the first 5 × 105 steps were discarded. The atomic coordinates were registered every 5 × 103 steps. We converted the dihedral angle information into two different types of scalar coupling constants, 3J(HN,) and 2J(,N) by using the Karplus equation and the optimized parameters determined by Hu and Bax 45. For 2J(,N) calculations we used the parameters of Ding and Gronenborn 46. We compared the simulation results with the experimentally determined values (Supplementary Figure S7a–S7d). For (Ala)5, mean errors of 3J(HN,) values are 1.70 Hz (ABS) and 0.56 Hz (c-ABS), and mean errors of 2J(,N) values are −0.41 Hz (ABS) and −0.15 Hz (c-ABS). For (Ala)7, mean errors of 3J(HN,) values are 1.55 Hz (ABS) and 0.48 Hz (c-ABS), and mean errors of 2J(,N) values are −0.40 Hz (ABS) and −0.06 Hz (c-ABS). Taken together, we conclude that the better performance of c-ABSINTH on dihedral angle distributions is transferable to short peptides.
The issue of transferability was further investigated via simulations on -synuclein, which is an archetypal intrinsically disordered protein. We performed separate sets of simulations using ABSINTH and c-ABSINTH. For each model, we performed ten separate replica exchange simulations and for each replica exchange simulation we deployed 26 different simulation temperatures from 200 to 450 K. The salt conditions mimicked 20 mM NaCl solution with electroneutrality. The initial structure was randomly generated and was located in a spherical droplet with radius of 100 Å. Each replica exchange simulation consists of 5 × 106 steps per replica for a total of 1.3 × 108 per simulation. Information regarding the atomic coordinates was stored every 5 × 103 steps. The first half of simulation data for each of the replicas was discarded as part of the equilibration process.
We analyzed the simulated ensembles at 300 K, and calculated scalar coupling constants 3J(HN,), 2J(,N) and 1J(,N) using the Karplus equation and the parameters of Hu and Bax parameterization 45 as well as Ding and Gronenborn 46. Then we compared these to the experimental data of Mantsyzov et al. 47 . Overall, c-ABSINTH yields predictions for coupling constants that are more consistent with experiment than the original ABSINTH model (Supplementary Figure S7e–S7g). For 3J(HN,), mean errors are 0.52 Hz (ABS) and 0.21 Hz (c-ABS). For 2J(,N), the mean errors are −0.32 Hz (ABS) and 0.00 Hz (c-ABS). Finally for 1J(,N), the mean errors are −0.71 Hz (ABS) and −0.11 Hz (c-ABS). Diminution of mean errors for each of the three distinct scalar coupling constants indicates that simulations based on c-ABSINTH lead to a systematic reduction of the errors encountered using the original ABSINTH model, even for much longer coil-like peptides than blocked amino acids.
5. PERFORMANCE OF c-ABSINTH IN MAINTAINING THE STABILITY OF PEPTIDES WITH WELL FOLDED STRUCTURES
Next, we tested c-ABSINTH for its ability to preserve the known folds of model peptides and small proteins. These tests were performed using ten independent sets of replica exchange simulations. Each set consists of 26 different temperatures ranging from 200 K to 450 K with equal spacing of 10 K. In all, each simulation, for each of the systems consists of 107 MMC steps. Data were accumulated once every 5 × 103 steps and the first 2.5 × 106 steps were discarded. For each system, we performed simulations using the original ABSINTH and the new c-ABSINTH models.
The 35-residue fragment of the chicken villin headpiece (HP35) is a fragment of an -helical protein 48. This system has served as an important calibration for forcefields and as a model system for modeling reversible folding in molecular simulations 1, 49. We used this system to test the differences – if any – between the original ABSINTH and the new c-ABSINTH in thermal unfolding simulations. The peptide was enclosed in a spherical droplet using a soft-wall potential and the droplet radius was set to 45 Å. Solution ions mimicking 15 mM NaCl concentration were included while maintaining electroneutrality. The starting structure for all simulations was based on coordinates derived from the crystal structure (PDB ID 1YRF) from the protein data bank. This structure was equilibrated to match the fixed bonds and angles used in the ABSINTH model. All of the simulations were initiated from a common starting structure, albeit with different random seeds. To test for the convergence of the simulation results, a particular concern given the choice of a common starting structure for all simulations, we monitored root mean square deviation (RMSD) and overall ABSINTH energy values as a function of the MC step number. Both quantities reach steady state values after ~106 MC steps and this is true for each of the simulation temperatures (Supplementary Figure S8). Trajectories for low-temperatures show an initial spike and a subsequent decline of the RMSD value, indicating that the system is first relaxed and then slowly converges to the equilibrated structure ensemble.
Figure 7 shows the unfolding curves from original ABSINTH and c-ABSINTH simulations. The two models generate similar thermal unfolding profiles for HP35. This is illustrated in terms of the backbone RMSD with respect to the folded structure of HP35 equilibrated at 200 K (Figure 7a) and the loss of secondary structure as a function of temperature (Figure 7c). The secondary structure contents were calculated using the DSSP algorithm 50, which allows us to follow the changes to the helical (DSSP-H) and beta sheet / strand (DSSP-E) contents. The two models predict similar trends for the temperature dependent decreases of -helical and -sheet contents.
Figure 7: Results from original ABSINTH (ABS) and c-ABSINTH (c-ABS) thermal unfolding simulations for HP35 and NTL9 systems.
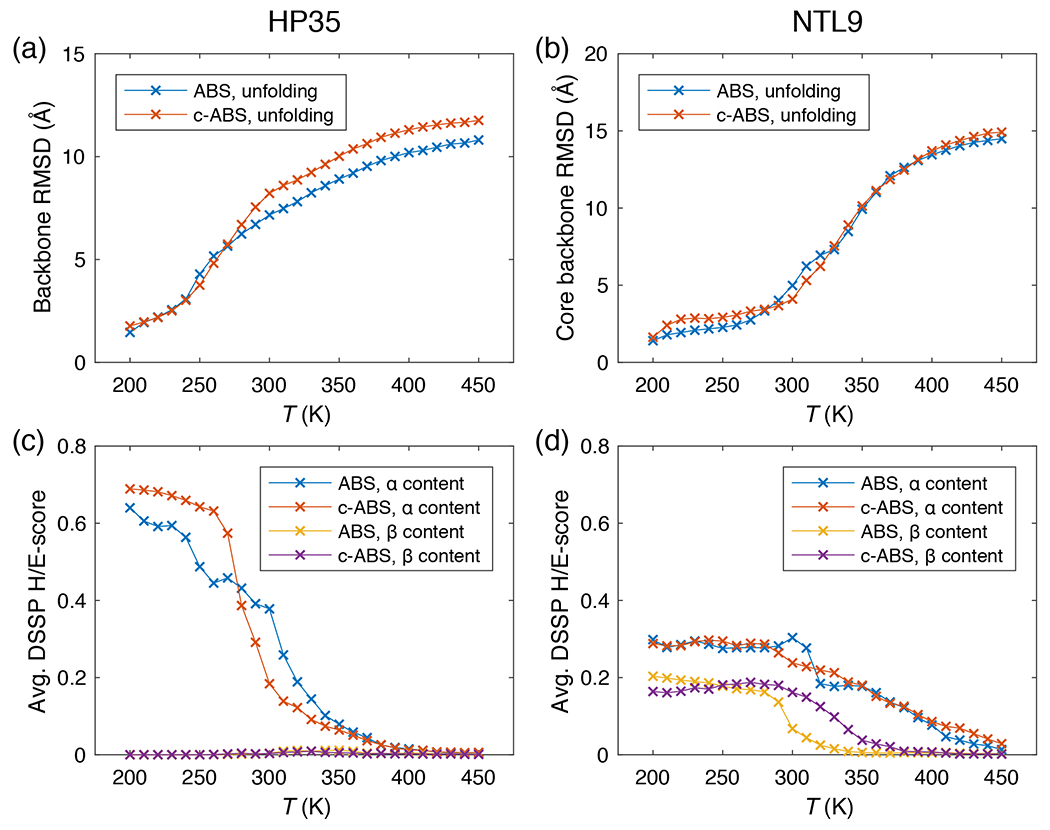
Unfolding curves of (a) HP35 and (b) NTL9 systems, generated by ABS and c-ABS simulations. The ordinate shows backbone RMSD with respect to the equilibrated structure at 200 K. For NTL9, we used backbone RMSD of the core part (residues 1-42). The and contents of (c) HP35 and (d) NTL9 systems in ABS and c-ABS simulations, quantified by average H and E scores from DSSP analysis (ordinate). Error bars are omitted for clarity.
We also studied thermal unfolding of the N-terminal domain of the L9 ribosomal protein (NTL9). This 56-residue protein has a mixed - structure and is known to fold / unfold via an apparent two-state mechanism 51. We performed simulations of NTL9 by enclosing the protein in a soft-wall spherical droplet of radius 45 Å with the neutralizing counterions in a 25 mM NaCl background. The initial structure for the simulations was based on the crystal structure (PDB ID 2HBB) from the protein data bank. We investigated the melting curve (Figure 7b, see also Supplementary Figure S9) by following the backbone RMSD of the core region (residues 1–42) referenced to the equilibrated folded structure of NTL9 at a simulation temperature of 200 K. The simulation results based on ABSINTH and c-ABSINTH generate similar melting curves. Larger fluctuations around the folded state generated using c-ABSINTH appear to derive from abrogation of the non-physical region from c-ABSINTH. The -helical and -sheet contents, calculated using DSSP, show similar trends between the forcefields, although there is a minor increase in the regular -sheet content (i.e., the DSSP-E score) with c-ABSINTH (Figure 7d). Overall, the thermal unfolding profiles for HP35 and NTL9 suggest that the intrinsic stability of ABSINTH is maintained with the incorporation of CMAP-like corrections into c-ABSINTH. The incorporation of local correction terms, which are based on experimentally derived and computationally optimized coil library landscapes, does not force destabilization of folded domains under folding conditions.
6. PERFORMANCE OF c-ABSINTH FOR MODELING THE REVERSIBLE FOLDING OF A MODEL PEPTIDE
A network of interactions involving a cluster of Trp residues stabilizes the folded state of the tryptophan zipper peptide 52. This has served as a helpful model system for calibrating the accuracies of molecular mechanics forcefields because of the interplay between backbone topology and sidechain interactions that are required to fold / unfold this system. Moreover, since we tuned c-ABSINTH to reproduce correct helical propensity of the FS peptide, it was important to ensure that the tuning did not weaken the ability to form -sheeted structures. Of the different tryptophan zippers, we chose the -hairpin peptide SWTWEGNKWTWK-NH2 based on the crystal structure (PDB ID 1LE0) from the protein data bank. We performed simulations of unfolding, starting from the PDB coordinates, and folding, starting from randomly generated conformations, in a soft-wall spherical droplet of radius 45 Å with the neutralizing counterions in a 20 mM NaCl background. Figure 8a shows the “folding” and “unfolding” curves from both models in terms of the backbone RMSD with respect to the equilibrated structure at the lowest MC temperature (see also Supplementary Figure S10). Both models capture the correct melting curves, regardless of choice of the initial structure, even though the unmodified ABSINTH model provides larger RMSDs for folded states. Indeed, investigation of -helical and -sheet contents (Figures 8c and 8e) reveals that while c-ABSINTH predicts no helical structures for the whole temperature range and some structures for low temperatures, the original ABSINTH predicts the formation of helix and loss of structure for the low temperature range (< 250 K). This is presumably the source of the large RMSD values. Importantly, the data show that c-ABSINTH performs reversible folding and unfolding of a tryptophan zipper peptide with minimal hysteresis. Therefore, it appears that the use of melting curves for the FS peptide as a restraint on the parameterization of c-ABSINTH does not compromise the ability to reversibly fold / unfold a purely -sheeted system. In this regard, it is also worth noting that c-ABSINTH is an improvement over the original ABSINTH model in terms of preserving the native hairpin structure of the tryptophan zipper under folding conditions.
Figure 8: Results from original ABSINTH (ABS) and c-ABSINTH (c-ABS) simulations on tryptophan zipper and GB1 peptide systems.
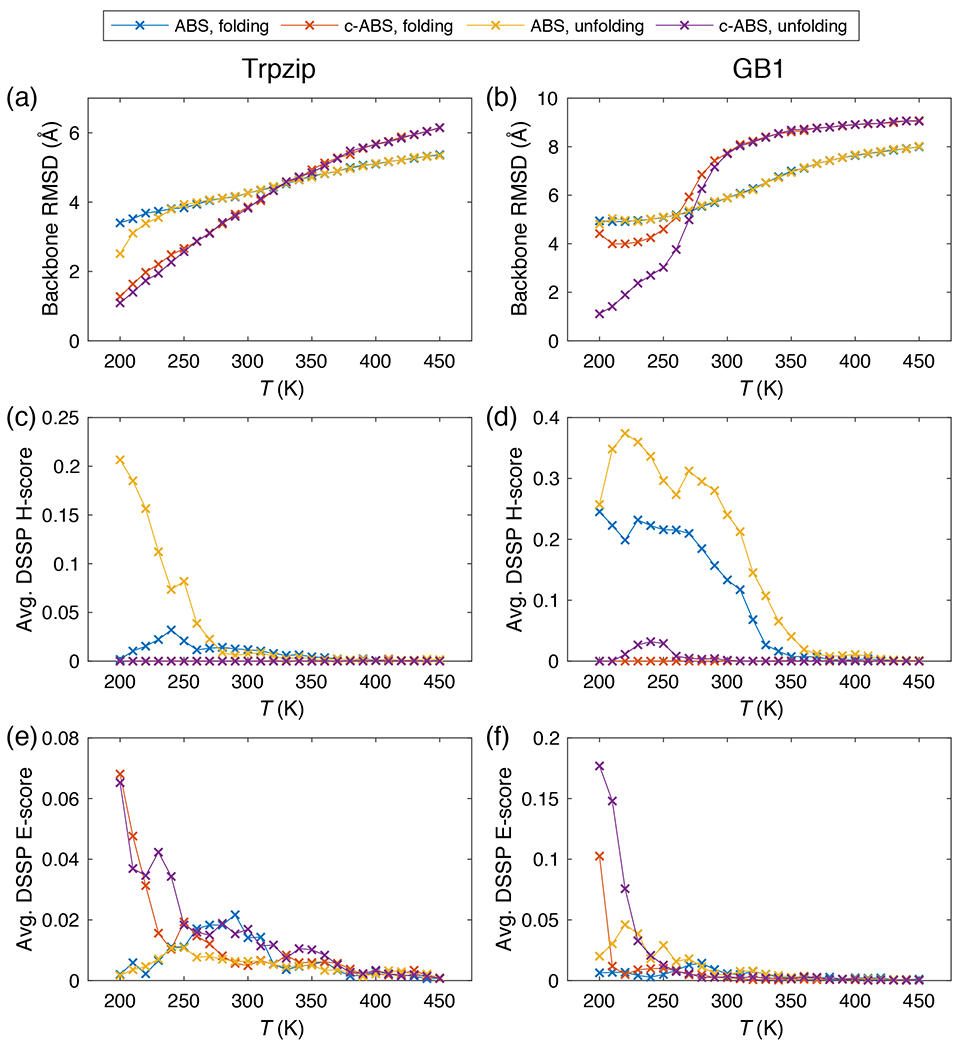
Folding and unfolding curves of (a) the tryptophan zipper and (b) the GB1 peptide system, generated by ABS and c-ABS simulations. The ordinate shows backbone RMSD with respect to the equilibrated structure at 200 K. (c, d) The and (e, f) the contents of tryptophan zipper and GB1 peptide systems in ABS and c-ABS simulations, respectively quantified by average H and E scores from DSSP analysis (ordinate). Error bars are omitted for clarity.
We also assessed the accuracy of c-ABSINTH by performing simulations of another prototypical structure, namely the C-terminal domain of the GB1 hairpin. We used the 16-residue sequence GEWTYDDATKTFTVTE and extracted the native structure from the NMR structure of the full-length GB1 protein (PDB ID 1GB1) from the protein data bank. While the tryptophan zipper has a glycine residue in its turn, the GB1 peptide has a lysine residue in the corresponding position, and consequently the effect of c-ABSINTH correction may be shown more clearly. We performed unfolding and folding simulations with both original ABSINTH and c-ABSINTH in the similar simulation conditions as above. The general trends for unfolding simulations hold here; c-ABSINTH leads to smaller RMSDs for folded states when compared to ABSINTH (Figure 8b). Also, c-ABSINTH predicts negligible -helical contents and significant -sheet contents, while ABSINTH predicts the opposite (Figures 8d and 8f). However, we observe hysteresis when comparing the folding vs. unfolding traces. On the refolding arm, the simulations based on c-ABSINTH converge on an alternate native state with high -sheet content (Figure 8b, red curve). This metastable state is higher in energy than the native -sheeted structure (Supplementary Figure S11). Clearly, the barrier between the near-native metastable state and the stable native state proves to be formidable to overcome using the amount sampling deployed here. The combination of improved move sets and enhancements using biased sampling methods should help in overcoming sampling bottlenecks due to metastable states. However, the hysteretic behavior and being trapped in the metastable state is not due to errors within the preferred ground states encoded by the c-ABSINTH energy function.
7. COMPARATIVE ASSESSMENTS OF THE COIL-TO-GLOBULE TRANSITIONS FOR DIFFERENT HOMOPOLYPEPTIDES
There is growing interest in low complexity sequences that are also intrinsically disordered 20a. The ultimate low complexity sequences are homopolypeptides. Modeling intrinsically disordered homopolypeptides is challenging because it raises the bar on the ability of a forcefield to capture, with accuracy, the interplay between intra-chain and chain-solvent interactions without relying on serendipitous cancelation of errors.
We compared the profiles obtained using ABSINTH and c-ABSINTH for coil-to-globule transitions as a function of decreasing temperature for poly-Gln, poly-Asn, poly-Ser and poly-Gly. We investigated homopolypeptides with different chain lengths (n = 10-50), except for poly-Ser, where we employed an eicosamer (S20). A single peptide of interest was placed in a soft-wall spherical cell of radius 150 Å, and its starting structure was randomly generated. As before, we employed ten independent sets of replica exchange simulations. Each set consists of 26 different temperatures with equal spacing of 10 K (temperature ranges vary with systems), and each simulation consists of 5 × 106 MMC steps per replica. Data were collected once every 5 × 103 steps while the first 1 × 106 steps were discarded.
Fluorescence correlation spectroscopy (FCS) experiments showed that poly-Gln molecules form globules in aqueous solutions at room temperature 53. This was inferred from the scaling of translational diffusion times as a function of poly-Gln length. For poly-Gln, destabilizing the region leads to a destabilization of the collapse of poly-Gln as reflected in the lower density of the globules (higher Rg) in the collapse regime, narrower temperature range for the persistence of globules, and a downshift in the apparent theta temperature (Figure 9; see also Supplementary Figures S12 and S13). These observations are in improved accord with experiments, which suggest that the room temperature ensembles are in better agreement with ABSINTH ensembles of ~330 K implying the need for a downshift of ~30 K in the theta temperature for poly-Gln 14a. The properties of poly-Ser and poly-Asn remain unclear, although tentative evidence suggests that Ser-rich and Asn-rich sequences may prefer more expanded coil-like ensembles at room temperature 14d, 14f. In fact, published data and unpublished accounts suggest that Asn-rich sequences are fundamentally different from Gln-rich sequences 14f, 54. The former are expected to be considerably more expanded, forming coil-like ensembles at room temperature, and promoting the formation of regular beta-sheeted structures in contrast to poly-Gln and Gln-rich sequences 54–55. The collapse transition is significantly destabilized for poly-Asn (Figure 9; see also Supplementary Figures S12 and S13) and this behavior seems to be in better agreement with extant experimental data when compared to the results generated using the uncorrected ABSINTH model. The key observation is that at room temperature, poly-Asn samples more expanded, coil-like ensembles in c-ABSINTH when compared to the collapsed conformations observed using ABSINTH. DSSP analysis (Supplementary Figures S14 and S15) shows that while original ABSINTH generates similar DSSP patterns for poly-Asn and poly-Gln (small amount of both and contents), c-ABSINTH distinguishes the two systems, by showing that poly-Asn does not sample -helical structures at all whereas there is a discernible preference for poly-Gln to form short stretches of -helices. These observations are concordant with published NMR data for poly-Gln stretches within the exon 1 encoded region of huntingtin 14l.
Figure 9: Normalized mean squared radii of gyration of poly-Gln (Qn) and poly-Asn (Nn) chains with different lengths (top legend) as a function of simulation temperature.
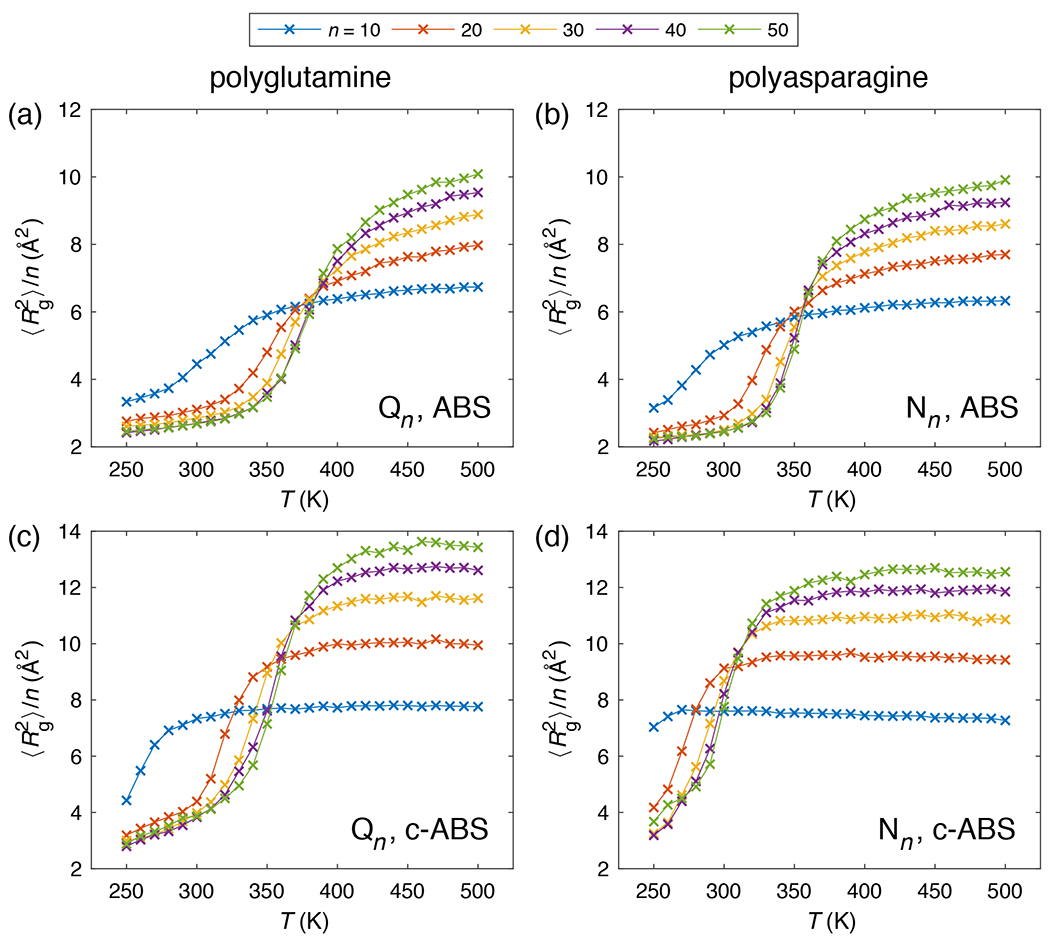
Results from ABSINTH simulations for Qn chains (a) and Nn chains (b). (c) Results from c-ABSINTH simulations for Qn chains (c) and Nn chains (d). Error bars are omitted for clarity.
For poly-Ser (S20), simulations based on the original ABSINTH model show a pathological preference for forming regular -helical rods at room temperature (Figure 10a, blue curve). To the best of our knowledge, there is no evidence to support this observation and it highlights the amplification of errors that can occur in simulations of homopolypeptides. Although definitive experiments regarding poly-Ser are lacking, the data on Ser-rich sequences suggest that these sequences form expanded, coil-like ensembles 56. This feature is thought to be due in part to the high intrinsic local preference for polyproline II conformations that is combined with the short methanol sidechain and hydrogen bond donor moiety. The anomalous helicity of poly-Ser is eliminated in the c-ABSINTH data (Figure 10a, red curve). Further, the coil-to-globule transition points as quantified by radius of gyration (Figure 10b) and heat capacity (Figure 10c) curves show that c-ABSINTH predicts expanded, coil-like ensembles for poly-Ser at room temperature and beyond. This behavior of poly-Ser appears to be in accord with the observations for Ser-rich sequences and their prevalence in IDPs.
Figure 10: Original ABSINTH (ABS) and c-ABSINTH (c-ABS) simulations on a poly-Ser (S20) chain.
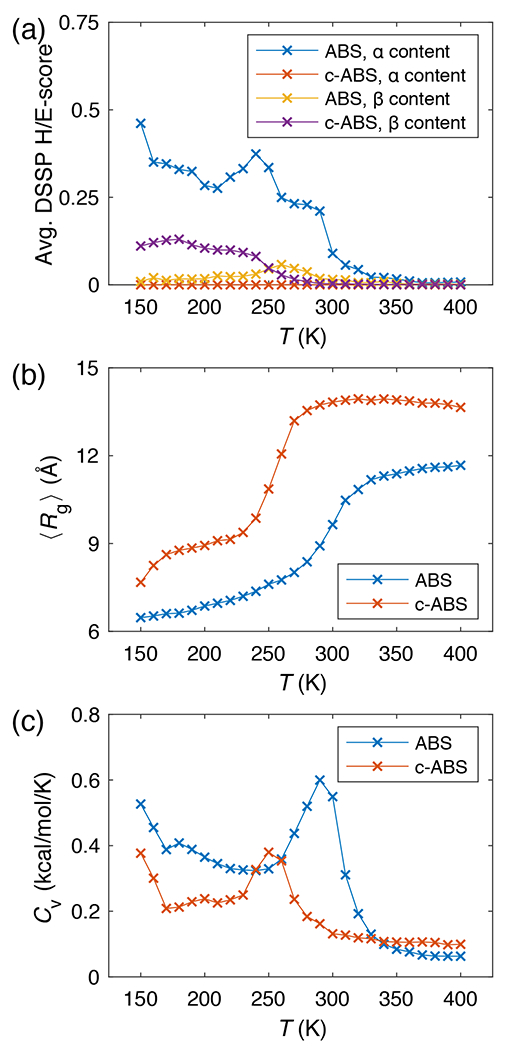
(a) The and contents of a S20 peptide quantified by average H and E scores from DSSP analysis, from ABS and c-ABS simulations. (b) The radii of gyration and (c) the heat capacities of a S20 peptide as a function of simulation temperature. Error bars are omitted for clarity.
Poly-Gly presents an interesting and unusually challenging problem for simulations. This is partly due to continued equivocation about whether or not water, at 25°C and 1 atm, is truly a poor solvent for polypeptide backbones thus driving poly-Gly to forms compact globules. Poly-Gly is among the least soluble homopolypeptides 57. Simulations based on the OPLS-AA/L forcefield and the TIP3P water model show that poly-Gly forms globules in water at 298 K 57–58. FCS experiments, performed in ultra-dilute solutions, show that the scaling of translational diffusion times, a useful proxy for the scaling of hydrodynamic sizes, consistent with that poly-Gly molecules forming heterogeneous ensembles of globular conformations in aqueous solvents at room temperature 57. These experiments also show that the intrinsic collapse of poly-Gly is not readily overridden in either 8 M urea or 7 M GdmCl 57. In simulations based on the OPLS-AA/L forcefield, interruptions of Gly-rich sequences by polar and charged residues readily destabilize the preference for collapsed conformations 20a, 57, 59. One might argue that observations for poly-Gly based on the OPLS-AA/L forcefield might be artefacts of the over-compaction that appears to be an anomalous feature of this forcefield 60. Interestingly, Pettitt and coworkers have reported poly-Gly compaction 61, albeit with different stabilities, using the Charmm22, Charmm27, and AMBER ff12sb forcefields 62. The preference for globules over expanded, rod-like conformations is thought to be the result of favorable enthalpic interactions among networks of CO dipoles rather than hydrogen bonding 61a, 63. Merlino et al. 64 have proposed that the collapse of poly-Gly might be the result of low solvent excluded volume in globules that is offset by the gain in translational entropy of the solvent. Teufel et al.65 used FCS measurements to assess the translational diffusion coefficients of a series of peptides and proteins in aqueous buffers and in 8 M GdmCl. For G20 they converted the translational diffusion coefficients to hydrodynamic radii (Rh) and obtained values of 1.04 nm and 1.18 nm, respectively in buffer vs. 8 M GdmCl for Rh. Based on these measurements, Teufel et al. 65 proposed that collapse in aqueous solutions is an intrinsic property of polypeptide backbones.
How do ABSINTH and c-ABSINTH simulations perform in terms of predicting the dimensions of poly-Gly molecules? Simulations based on the refined c-ABSINTH model show that poly-Gly forms more semi-compact conformations that lie in the transition regime between globules and random coils at room temperatures (Figure 11; see also Supplementary Figure S16). Although this behavior seems like an improvement over the original ABSINTH model, assessments of precision will require more unequivocal adjudications from experiments. We used HullRad, a method developed by Fleming and Fleming 66 to estimate Rh from the simulated ensembles. The temperature dependencies of the calculated Rh values obtained using ABSINTH and c-ABSINTH for G20 are shown in Figure 11a. The c-ABSINTH estimate for Rh at 300 K is 9.2 Å or 0.92 nm, which is on a par with the estimates made by Teufel et al. for G20 based on their FCS measurements 65, while original ABSINTH predicts Rh of 9.9 Å at 300 K. We also calculated the ratio of Rg to Rh. For globules, modeled as monodisperse spheres, the ratio of Rg to Rh would be 0.77. In a theta solvent, for a perfect Gaussian chain, the ratio would be unity and for self-avoiding walks, the ratio would be approximately 1.3. At 300 K, the ratio of Rg to Rh is 0.95 and 1.00 for the c-ABSINTH and original ABSINTH models, respectively (Figure 11b). The new results appear to be more consistent with the hydrodynamic measurements of Teufel et al.65 Detailed investigations are necessary to work out the precise interpretation of the scaling behavior that has emerged from hydrodynamic measurements. Unfortunately, the extreme insolubility of poly-Gly 57 makes it difficult to use small angle X-ray scattering to assess Rg values. However, novel experiments that combine direct measurements of Rh with multiple inter-residue distance measurements might provide a way to overcome the difficulties of obtaining non-hydrodynamic assessments of poly-Gly size as a function of temperature. Pending these measurements, and a clear adjudication of the poly-Gly collapse issue, we choose not to pursue any further refinements to c-ABSINTH.
Figure 11: Chain dimensions of a poly-Gly (G20) chain from original ABSINTH (ABS) and c-ABSINTH (c-ABS) simulations.
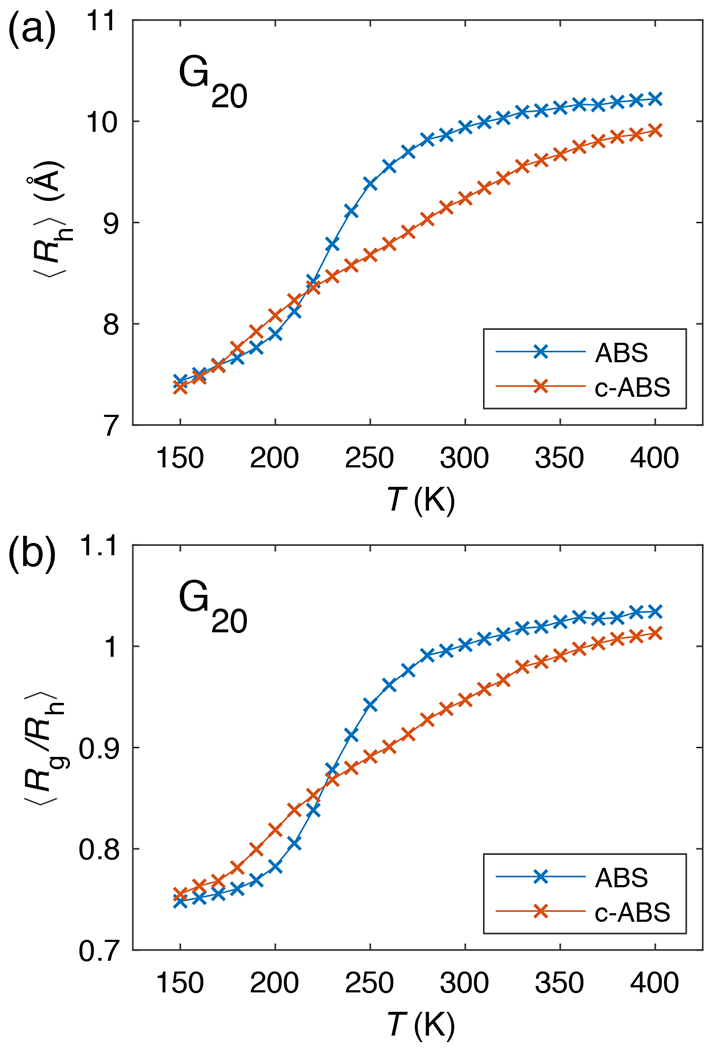
(a) The hydrodynamic radii and (b) the ratios of the radius of gyration to the hydrodynamic radius. Error bars are omitted for clarity.
8. DISCUSSION AND CONCLUSIONS
In this work, we show that the ABSINTH model can be improved upon by using information from the optimized coil library landscapes for blocked amino acids 33. We achieved this using a CMAP-like correction term and pursued a balanced approach that leads to the new c-ABSINTH model. The addition of residue-specific CMAP-like correction terms does not materially increase the computational cost associated with simulations based on c-ABSINTH versus the original ABSINTH. This is important because the computational efficiency of ABSINTH-based simulations has been significant in enabling high-throughput assessments of sequence-to-conformational relationships of IDPs 59, 67. The residue-specific correction terms in c-ABSINTH help us reproduce quantitative features of optimized coil library landscapes while also preserving the reversible foldability of a model -helix forming system.
Overall, the refinements that lead to c-ABSINTH preserve the ability to maintain stable folded forms of -helical and folds and enable the reversible folding of a model -hairpin peptide in a manner that improves upon the original ABSINTH model. Further, the CMAP terms do not compromise the stability of -hairpins with non-Gly residues occupying the basin. We also tested the c-ABSINTH model on a set of homopolypeptides. The simulations are in accord with extant experimental data for poly-Gln and recapitulate known trends for Asn-rich sequences. The c-ABSINTH model predicts absence of secondary structure preferences and coil-like ensembles for poly-Ser.
The corrections in c-ABSINTH prove inadequate to generate collapsed globular ensembles for poly-Gly at room temperatures. However, as noted in the previous section, the verdict on poly-Gly is not yet unequivocal. Irrespective of the final verdict, it is worth noting that poly-Gly presents a unique challenge for mean field models such as ABSINTH and c-ABSINTH. Specifically, context-dependent solvation effects, which refer to modulations of the intrinsic reference free energies of solvation for Gly residues in poly-Gly 68 might need to be incorporated to account for the experimental observations. Preliminary results suggest that this is an appropriate empirical route to pursue 69. It is also possible that the long lifetimes of hydrogen bonds 70 and / or interactions involving CO dipoles might contribute to anomalies in hydrodynamic measurements. These issues need to be resolved before setting an experimentally derived touchstone for the behavior of poly-Gly in water.
Recent attention has focused on the so-called over-collapse of IDPs, particularly in simulations with explicit representation of solvent molecules 60, 71. Various remedies have been prescribed to overcome this problem 72. It is noteworthy, however, that with a few exceptions 60, the over-collapse has not been an issue for the ABSINTH implicit solvation model and forcefield paradigm 56. This is mainly because the energy scales for solvation-desolvation processes are set up in ABSINTH by bootstrapping the reference free energies of solvation of sidechain and backbone moieties to that of the homologous model compound data that are derived directly from experimental measurements. As shown in all of the simulations reported here, the introduction of CMAP-like corrections into c-ABSINTH does not promote over-collapse. If anything, the collapse transition is weakened in a manner that is more consistent with experimental data.
Homopolypeptides provide a challenge for atomistic simulations based on molecular mechanics forcefields because one cannot rely on fortuitous cancelation of errors in the description of interactions that can arise as the result of sequence heterogeneity in heteropolymeric sequences. Instead, the forcefield has to include an appropriate balancing of terms that yields the correct emergent properties as a function of homopolypeptide length. Further, our investigations of archetypal homopolypeptides based on c-ABSINTH suggest that there is more to the coil-to-globule transitions of IDPs and low complexity domains than just their charge contents as has been proposed by extrapolations from findings based on Gln-rich sequences and a small number of polar tracts 67, 73. The sidechain-specific nuances appear to become more obvious with c-ABSINTH than with the original ABSINTH model. For example, there is a clearer distinction between Gln and Asn in poly-Gln versus poly-Asn, respectively in simulations based on c-ABSINTH compared to the original ABSINTH model. Similarly, the purported preference of poly-Ser for significant conformational heterogeneity is realized with c-ABSINTH as opposed to the original ABSINTH model. Therefore, investigating the nuances of specific polar sidechains, which we plan to probe using a combination of high-throughput c-ABSINTH based simulations and experiments directed at a range of IDPs enriched polar residues and deficient in charges, will likely help in arriving at a more accurate description of the diagram-of-states for IDPs than the purely charge-content based ones 67, 73. Additionally, a systematic assessment of c-ABSINTH versus ABSINTH for all problems / systems studied to date is also in the offing. Preliminary assessments suggest that these head-to-head comparisons might require at least two additional improvements, namely the incorporation of local sequence context dependent modulations to the reference free energies of solvation for model compounds and the inclusion of move sets that enable charge regulation whereby the protonation states of ionizable groups change in the simulation.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grant 5R01-NS05611410 from the National Institutes of Health. We are grateful to Martin Fossat, Alex Holehouse, and Kiersten Ruff for useful discussions.
REFERENCES
- 1.Shaw DE; Maragakis P; Lindorff-Larsen K; Piana S; Dror RO; Eastwood MP; Bank JA; Jumper JM; Salmon JK; Shan Y; Wriggers W, Atomic-level characterization of the structural dynamics of proteins. Science 2010, 330 (6002), 341–346. [DOI] [PubMed] [Google Scholar]
- 2.Mongan J; Simmerling C; McCammon JA; Case DA; Onufriev A, Generalized Born model with a simple, robust molecular volume correction. Journal of Chemical Theory and Computation 2007, 3 (1), 156–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Baker NA, Improving implicit solvent simulations: a Poisson-centric view. Current opinion in structural biology 2005, 15 (2), 137–43 [DOI] [PubMed] [Google Scholar]; (b) Dong F; Wagoner JA; Baker NA, Assessing the performance of implicit solvation models at a nucleic acid surface. Physical Chemistry Chemical Physics 2008, 10 (32), 4889–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Gallicchio E; Levy RM, AGBNP: an analytic implicit solvent model suitable for molecular dynamics simulations and high-resolution modeling. Journal of Computational Chemistry 2004, 25 (4), 479–499 [DOI] [PubMed] [Google Scholar]; (b) Gallicchio E; Paris K; Levy RM, The AGBNP2 Implicit Solvation Model. Journal of Chemical Theory and Computation 2009, 5 (9), 2544–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Deng N; Zhang BW; Levy RM, Connecting free energy surfaces in implicit and explicit solvent: an efficient method to compute conformational and solvation free energies. Journal of Chemical Theory and Computation 2015, 11 (6), 2868–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Chen J; Im W; Brooks CL, Balancing Solvation and Intramolecular Interactions: Toward a Consistent Generalized Born Force Field. Journal of the American Chemical Society 2006, 128 (11), 3728–3736 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ren P; Chun J; Thomas DG; Schnieders MJ; Marucho M; Zhang J; Baker NA, Biomolecular electrostatics and solvation: a computational perspective. Quarterly Reviews in Biophysics 2012, 45 (4), 427–91 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen J; Brooks CL 3rd, Implicit modeling of nonpolar solvation for simulating protein folding and conformational transitions. Physical Chemistry Chemical Physics 2008, 10 (4), 471–481 [DOI] [PubMed] [Google Scholar]; (d) Chen J; Brooks CL 3rd; Khandogin J, Recent advances in implicit solvent-based methods for biomolecular simulations. Current opinion in structural biology 2008, 18 (2), 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ganguly D; Chen J, Modulation of the disordered conformational ensembles of the p53 transactivation domain by cancer-associated mutations. PLoS computational biology 2015, 11 (4), e1004247. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lee KH; Chen J, Optimization of the GBMV2 implicit solvent force field for accurate simulation of protein conformational equilibria. Journal of Computational Chemistry 2017, 38 (16), 1332–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ben-Naim A, Solvent effects on protein association and protein folding. Biopolymers 1990, 29 (3), 567–96. [DOI] [PubMed] [Google Scholar]
- 7.Baker NA, Improving implicit solvent simulations: a Poisson-centric view. Curr Opin Struct Biol 2005, 15 (2), 137–43. [DOI] [PubMed] [Google Scholar]
- 8.(a) Chen J; Brooks CL 3rd; Khandogin J, Recent advances in implicit solvent-based methods for biomolecular simulations. Curr Opin Struct Biol 2008, 18 (2), 140–8 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen J; Im W; Brooks CL 3rd, Balancing solvation and intramolecular interactions: toward a consistent generalized Born force field. J Am Chem Soc 2006, 128 (11), 3728–36 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Feig M; Brooks CL 3rd, Recent advances in the development and application of implicit solvent models in biomolecule simulations. Curr Opin Struct Biol 2004, 14 (2), 217–24 [DOI] [PubMed] [Google Scholar]; (d) Sigalov G; Fenley A; Onufriev A, Analytical electrostatics for biomolecules: beyond the generalized Born approximation. J Chem Phys 2006, 124 (12), 124902. [DOI] [PubMed] [Google Scholar]; (e) Tanizaki S; Feig M, A generalized Born formalism for heterogeneous dielectric environments: application to the implicit modeling of biological membranes. J Chem Phys 2005, 122 (12), 124706. [DOI] [PubMed] [Google Scholar]
- 9.Levy RM; Zhang LY; Gallicchio E; Felts AK, On the nonpolar hydration free energy of proteins: surface area and continuum solvent models for the solute-solvent interaction energy. Journal of the American Chemical Society 2003, 125 (31), 9523–9530. [DOI] [PubMed] [Google Scholar]
- 10.Lazaridis T; Karplus M, Effective energy function for proteins in solution. Proteins 1999, 35 (2), 133–52. [DOI] [PubMed] [Google Scholar]
- 11.Makhatadze GI; Privalov PL, Energetics of protein structure. Advances in Protein Chemistry, Vol 47 1995, 47, 307–425. [DOI] [PubMed] [Google Scholar]
- 12.Vitalis A; Pappu RV, ABSINTH: a new continuum solvation model for simulations of polypeptides in aqueous solutions. Journal of computational chemistry 2009, 30 (5), 673–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cumberworth A; Bui JM; Gsponer J, Free energies of solvation in the context of protein folding: Implications for implicit and explicit solvent models. Journal of Computational Chemistry 2016, 37 (7), 629–40. [DOI] [PubMed] [Google Scholar]
- 14.(a) Warner JB; Ruff KM; Tan PS; Lemke EA; Pappu RV; Lashuel HA, Monomeric Huntingtin Exon 1 Has Similar Overall Structural Features for Wild-Type and Pathological Polyglutamine Lengths. Journal of the American Chemical Society 2017, 139 (41), 14456–14469 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sherry KP; Das RK; Pappu RV; Barrick D, Control of transcriptional activity by design of charge patterning in the intrinsically disordered RAM region of the Notch receptor. Proceedings of the National Academy of Sciences USA 2017, 114 (44), E9243–E9252 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fuertes G; Banterle N; Ruff KM; Chowdhury A; Mercadante D; Koehler C; Kachala M; Estrada Girona G; Milles S; Mishra A; Onck PR; Gräter F; Esteban-Martín S; Pappu RV; Svergun DI; Lemke EA, Decoupling of size and shape fluctuations in heteropolymeric sequences reconciles discrepancies in SAXS vs. FRET measurements. Proceedings of the National Academy of Sciences 2017, 114 (31), E6342–E6351 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Martin EW; Holehouse AS; Grace CR; Hughes A; Pappu RV; Mittag T, Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. Journal of the American Chemical Society 2016, 138 (47), 15323–15335 [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Das RK; Huang Y; Phillips AH; Kriwacki RW; Pappu RV, Cryptic sequence features within the disordered protein p27Kip1 regulate cell cycle signaling. Proceedings of the National Academy of Sciences USA 2016, 113 (20), 5616–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Kozlov AG; Weiland E; Mittal A; Waldman V; Antony E; Fazio N; Pappu RV; Lohman TM, Intrinsically Disordered C-Terminal Tails of E. coli Single-Stranded DNA Binding Protein Regulate Cooperative Binding to Single-Stranded DNA. Journal of Molecular Biology 2015, 427 (4), 763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Vitalis A; Lyle N; Pappu RV, Thermodynamics of -Sheet Formation in Polyglutamine. Biophysical Journal 2009, 97 (1), 303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Metskas LA; Rhoades E, Conformation and Dynamics of the Troponin I C-Terminal Domain: Combining Single-Molecule and Computational Approaches for a Disordered Protein Region. Journal of the American Chemical Society 2015, 137 (37), 11962–9 [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Gibbs EB; Showalter SA, Quantification of Compactness and Local Order in the Ensemble of the Intrinsically Disordered Protein FCP1. Journal of Physical Chemistry B 2016, 120 (34), 8960–8969 [DOI] [PubMed] [Google Scholar]; (j) Mao AH; Crick SL; Vitalis A; Chicoine CL; Pappu RV, Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proceedings of the National Academy of Sciences 2010, 107 (18), 8183–8188 [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Das RK; Crick SL; Pappu RV, N-Terminal Segments Modulate the -Helical Propensities of the Intrinsically Disordered Basic Regions of bZIP Proteins. Journal of Molecular Biology 2012, 416 (2), 287–299 [DOI] [PubMed] [Google Scholar]; (l) Newcombe EA; Ruff KM; Sethi A; Ormsby AR; Ramdzan YM; Fox A; Purcell AW; Gooley PR; Pappu RV; Hatters DM, Tadpole-like Conformations of Huntingtin Exon 1 Are Characterized by Conformational Heterogeneity that Persists regardless of Polyglutamine Length. Journal of Molecular Biology 2018, 430 (10), 1442–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mittal A; Holehouse AS; Cohan MC; Pappu RV, Sequence-to-Conformation Relationships of Disordered Regions Tethered to Folded Domains of Proteins. Journal of Molecular Biology 2018. [DOI] [PubMed] [Google Scholar]
- 16.Meng W; Lyle N; Luan B; Raleigh DP; Pappu RV, Experiments and simulations show how long-range contacts can form in expanded unfolded proteins with negligible secondary structure. Proceedings of the National Academy of Sciences 2013, 110 (6), 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vitalis A; Caflisch A, Equilibrium sampling approach to the interpretation of electron density maps. Structure (London, England : 1993) 2014, 22 (1), 156–167. [DOI] [PubMed] [Google Scholar]
- 18.(a) Chatterjee S; Luthra P; Esaulova E; Agapov E; Yen BC; Borek DM; Edwards MR; Mittal A; Jordan DS; Ramanan P; Moore ML; Pappu RV; Holtzman MJ; Artyomov MN; Basler CF; Amarasinghe GK; Leung DW, Structural basis for human respiratory syncytial virus NS1-mediated modulation of host responses. Nature Microbiology 2017, 2, 17101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu W; Edwards MR; Borek DM; Feagins AR; Mittal A; Alinger JB; Berry KN; Yen B; Hamilton J; Brett TJ; Pappu RV; Leung DW; Basler CF; Amarasinghe GK, Ebola virus VP24 targets a unique NLS binding site on karyopherin alpha 5 to selectively compete with nuclear import of phosphorylated STAT1. Cell host & microbe 2014, 16 (2), 187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vitalis A; Caflisch A, 50 Years of Lifson–Roig Models: Application to Molecular Simulation Data. Journal of Chemical Theory and Computation 2012, 8 (1), 363–373. [DOI] [PubMed] [Google Scholar]
- 20.(a) Wei M-T; Elbaum-Garfinkle S; Holehouse AS; Chen CC-H; Feric M; Arnold CB; Priestley RD; Pappu RV; Brangwynne CP, Phase behaviour of disordered proteins underlying low density and high permeability of liquid organelles. Nature Chemistry 2017, 9, 1118. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pak Chi W.; Kosno M; Holehouse Alex S.; Padrick Shae B.; Mittal A; Ali R; Yunus Ali A.; Liu David R.; Pappu Rohit V.; Rosen Michael K., Sequence Determinants of Intracellular Phase Separation by Complex Coacervation of a Disordered Protein. Molecular Cell 2016, 63 (1), 72–85 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Harmon TS; Holehouse AS; Rosen MK; Pappu RV, Intrinsically disordered linkers determine the interplay between phase separation and gelation in multivalent proteins. eLife 2017, 6, e30294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Staller MV; Holehouse AS; Swain-Lenz D; Das RK; Pappu RV; Cohen BA, A High-Throughput Mutational Scan of an Intrinsically Disordered Acidic Transcriptional Activation Domain. Cell systems 2018, 6 (4), 444–455.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoo TY; Choi J-M; Conway W; Yu C-H; Pappu RV; Needleman DJ, Measuring NDC80 binding reveals the molecular basis of tension-dependent kinetochore-microtubule attachments. eLife 2018, 7, e36392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arnon ZA; Vitalis A; Levin A; Michaels TCT; Caflisch A; Knowles TPJ; Adler-Abramovich L; Gazit E, Dynamic microfluidic control of supramolecular peptide self-assembly. Nature Communications 2016, 7, 13190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radhakrishnan A; Vitalis A; Mao AH; Steffen AT; Pappu RV, Improved atomistic Monte Carlo simulations demonstrate that poly-L-proline adopts heterogeneous ensembles of conformations of semi-rigid segments interrupted by kinks. Journal of Physical Chemistry B 2012, 116 (23), 6862–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wuttke R; Hofmann H; Nettels D; Borgia MB; Mittal J; Best RB; Schuler B, Temperature-dependent solvation modulates the dimensions of disordered proteins. Proceedings of the National Academy of Sciences USA 2014, 111 (14), 5213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mittal A; Lyle N; Harmon TS; Pappu RV, Hamiltonian Switch Metropolis Monte Carlo Simulations for Improved Conformational Sampling of Intrinsically Disordered Regions Tethered to Ordered Domains of Proteins. Journal of Chemical Theory and Computation 2014, 10 (8), 3550–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vitalis A; Pappu RV, Methods for Monte Carlo Simulations of Biomacromolecules. In Annual Reports in Computational Chemistry, Ralph AW, Ed. Elsevier: 2009; Vol. Volume 5, pp 49–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitalis A; Pappu RV, A simple molecular mechanics integrator in mixed rigid body and dihedral angle space. Journal of Chemical Physics 2014, 141 (3), 034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Makhatadze GI; Lopez MM; Privalov PL, Heat capacities of protein functional groups. Biophysical Chemistry 1997, 64 (1–3), 93–101 [DOI] [PubMed] [Google Scholar]; (b) Makhatadze GI; Privalov PL, Hydration effects in protein unfolding. Biophysical Chemistry 1994, 51 (2-3), 291–309. [DOI] [PubMed] [Google Scholar]
- 30.Vitalis A; Pappu RV, ABSINTH: A new continuum solvation model for simulations of polypeptides in aqueous solutions. Journal of Computational Chemistry 2009, 30 (5), 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaminski GA; Friesner RA; Tirado-Rives J; Jorgensen WL, Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. Journal of Physical Chemistry B 2001, 105 (28), 6474–6487. [Google Scholar]
- 32.(a) Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M, CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry 1983, 4 (2), 187–217 [Google Scholar]; (b) MacKerell AD; Bashford D; Bellott M; Dunbrack RL; Evanseck JD; Field MJ; Fischer S; Gao J; Guo H; Ha S; Joseph-McCarthy D; Kuchnir L; Kuczera K; Lau FTK; Mattos C; Michnick S; Ngo T; Nguyen DT; Prodhom B; Reiher WE; Roux B; Schlenkrich M; Smith JC; Stote R; Straub J; Watanabe M; Wiórkiewicz-Kuczera J; Yin D; Karplus M, All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. The Journal of Physical Chemistry B 1998, 102 (18), 3586–3616 [DOI] [PubMed] [Google Scholar]; (c) Brooks BR; Brooks CL; MacKerell AD; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M, CHARMM: The Biomolecular Simulation Program. Journal of computational chemistry 2009, 30 (10), 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) MacKerell AD; Brooks B; Brooks CL; Nilsson L; Roux B; Won Y; Karplus M, CHARMM: The Energy Function and Its Parameterization. In Encyclopedia of Computational Chemistry, von Ragué Schleyer P; Allinger NL; Clark T; Gasteiger J; Kollman PA; Schaefer HF; Schreiner PR, Eds. 2002. [Google Scholar]
- 33.Choi J-M; Pappu Rohit V., Experimentally derived and computationally optimized backbone conformational Statistics for blocked amino acids. Journal of Chemical Theory and Computation 2018, Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackerell AD; Feig M; Brooks CL, Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. Journal of Computational Chemistry 2004, 25 (11), 1400–1415. [DOI] [PubMed] [Google Scholar]
- 35.Ramachandran GN; Ramakrishnan C; Sasisekharan V, Stereochemistry of polypeptide chain configurations. Journal of Molecular Biology 1963, 7 (1), 95–99. [DOI] [PubMed] [Google Scholar]
- 36.Pappu RV; Srinivasan R; Rose GD, The Flory isolated-pair hypothesis is not valid for polypeptide chains: Implications for protein folding. Proceedings of the National Academy of Sciences 2000, 97 (23), 12565–12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bærentzen JA; Gravesen J; Anton F; Aanæs H, Splines. In Guide to Computational Geometry Processing: Foundations, Algorithms, and Methods, Springer London: London, 2012; pp 99–117. [Google Scholar]
- 38.Lockhart D; Kim P, Internal stark effect measurement of the electric field at the amino terminus of an alpha helix. Science 1992, 257 (5072), 947–951. [DOI] [PubMed] [Google Scholar]
- 39.Best RB; Hummer G, Optimized Molecular Dynamics Force Fields Applied to the Helix–Coil Transition of Polypeptides. The Journal of Physical Chemistry B 2009, 113 (26), 9004–9015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao AH; Pappu RV, Crystal lattice properties fully determine short-range interaction parameters for alkali and halide ions. Journal of Chemical Physics 2012, 137 (6), 064104. [DOI] [PubMed] [Google Scholar]
- 41.Sugita Y; Okamoto Y, Replica-exchange molecular dynamics method for protein folding. Chemical Physics Letters 1999, 314 (1), 141–151. [Google Scholar]
- 42.Thompson PA; Eaton WA; Hofrichter J, Laser Temperature Jump Study of the Helix⇌Coil Kinetics of an Alanine Peptide Interpreted with a ‘Kinetic Zipper’ Model. Biochemistry 1997, 36 (30), 9200–9210. [DOI] [PubMed] [Google Scholar]
- 43.Shi Z; Chen K; Liu Z; Ng A; Bracken WC; Kallenbach NR, Polyproline II propensities from GGXGG peptides reveal an anticorrelation with -sheet scales. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (50), 17964–17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Graf J; Nguyen PH; Stock G; Schwalbe H, Structure and Dynamics of the Homologous Series of Alanine Peptides: A Joint Molecular Dynamics/NMR Study. Journal of the American Chemical Society 2007, 129 (5), 1179–1189. [DOI] [PubMed] [Google Scholar]
- 45.Hu J-S; Bax A, Determination of and Angles in Proteins from 13C–13C Three-Bond J Couplings Measured by Three-Dimensional Heteronuclear NMR. How Planar Is the Peptide Bond? Journal of the American Chemical Society 1997, 119 (27), 6360–6368. [Google Scholar]
- 46.Ding K; Gronenborn AM, Protein Backbone and Residual Dipolar and J Couplings: New Constraints for NMR Structure Determination. Journal of the American Chemical Society 2004, 126 (20), 6232–6233. [DOI] [PubMed] [Google Scholar]
- 47.Mantsyzov AB; Maltsev AS; Ying J; Shen Y; Hummer G; Bax A, A maximum entropy approach to the study of residue-specific backbone angle distributions in -synuclein, an intrinsically disordered protein. Protein Science 2014, 23 (9), 1275–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiu TK; Kubelka J; Herbst-Irmer R; Eaton WA; Hofrichter J; Davies DR, High-resolution x-ray crystal structures of the villin headpiece subdomain, an ultrafast folding protein. Proceedings of the National Academy of Sciences of the United States of America 2005, 102 (21), 7517–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.(a) Wickstrom L; Okur A; Song K; Hornak V; Raleigh DP; Simmerling CL, The unfolded state of the villin headpiece helical subdomain: computational studies of the role of locally stabilized structure. Journal of Molecular Biology 2006, 360 (5), 1094–107 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Piana S; Lindorff-Larsen K; Shaw DE, Protein folding kinetics and thermodynamics from atomistic simulation. Proceedings of the National Academy of Sciences USA 2012, 109 (44), 17845–17850 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Beauchamp KA; Ensign DL; Das R; Pande VS, Quantitative comparison of villin headpiece subdomain simulations and triplet-triplet energy transfer experiments. Proceedings of the National Academy of Sciences of the United States of America USA 2011, 108 (31), 12734–12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kabsch W; Sander C, Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22 (12), 2577–2637. [DOI] [PubMed] [Google Scholar]
- 51.Kuhlman B; Luisi DL; Evans PA; Raleigh DP, Global analysis of the effects of temperature and denaturant on the folding and unfolding kinetics of the N-terminal domain of the protein L911Edited by P. E. Wright. Journal of Molecular Biology 1998, 284 (5), 1661–1670. [DOI] [PubMed] [Google Scholar]
- 52.Cochran AG; Skelton NJ; Starovasnik MA, Tryptophan zippers: Stable, monomeric -hairpins. Proceedings of the National Academy of Sciences 2001, 98 (10), 5578–5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crick SL; Jayaraman M; Frieden C; Wetzel R; Pappu RV, Fluorescence correlation spectroscopy shows that monomeric polyglutamine molecules form collapsed structures in aqueous solutions. Proceedings of the National Academy of Sciences 2006, 103 (45), 16764–16769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halfmann R; Alberti S; Krishnan R; Lyle N; O Donnell, Charles W; King Oliver D.; Berger B; Pappu Rohit V.; Lindquist S, Opposing Effects of Glutamine and Asparagine Govern Prion Formation by Intrinsically Disordered Proteins. Molecular Cell 2011, 43 (1), 72–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu X; Murphy RM, Asparagine Repeat Peptides: Aggregation Kinetics and Comparison with Glutamine Repeats. Biochemistry 2015, 54 (31), 4784–4794. [DOI] [PubMed] [Google Scholar]
- 56.Rauscher S; Gapsys V; Gajda MJ; Zweckstetter M; de Groot BL; Grubmüller H, Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. Journal of Chemical Theory and Computation 2015, 11 (11), 5513–5524. [DOI] [PubMed] [Google Scholar]
- 57.Holehouse AS; Garai K; Lyle N; Vitalis A; Pappu RV, Quantitative Assessments of the Distinct Contributions of Polypeptide Backbone Amides versus Side Chain Groups to Chain Expansion via Chemical Denaturation. Journal of the American Chemical Society 2015, 137 (8), 2984–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tran HT; Mao A; Pappu RV, Role of Backbone–Solvent Interactions in Determining Conformational Equilibria of Intrinsically Disordered Proteins. Journal of the American Chemical Society 2008, 130 (23), 7380–7392. [DOI] [PubMed] [Google Scholar]
- 59.Holehouse AS; Pappu RV, Collapse Transitions of Proteins and the Interplay Among Backbone, Sidechain, and Solvent Interactions. Annual Review of Biophysics 2018, 47 (1), 19–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mercadante D; Milles S; Fuertes G; Svergun DI; Lemke EA; Grater F, Kirkwood-Buff Approach Rescues Overcollapse of a Disordered Protein in Canonical Protein Force Fields. Journal of Physical Chemistry B 2015, 119 (25), 7975–7984. [DOI] [PubMed] [Google Scholar]
- 61.(a) Asthagiri D; Karandur D; Tomar DS; Pettitt BM, Intramolecular Interactions Overcome Hydration to Drive the Collapse Transition of Gly15. Journal of Physical Chemistry B 2017, 121 (34), 8078–8084 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Drake JA; Harris RC; Pettitt BM, Solvation Thermodynamics of Oligoglycine with Respect to Chain Length and Flexibility. Biophysical Journal 2016, 111 (4), 756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drake JA; Pettitt BM, Force field-dependent solution properties of glycine oligomers. Journal of Computational Chemistry 2015, 36 (17), 1275–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karandur D; Harris RC; Pettitt BM, Protein collapse driven against solvation free energy without H-bonds. Protein Science 2016, 25 (1), 103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Merlino A; Pontillo N; Graziano G, A driving force for polypeptide and protein collapse. Physical Chemistry Chemical Physics 2016, 19 (1), 751–756. [DOI] [PubMed] [Google Scholar]
- 65.Teufel DP; Johnson CM; Lum JK; Neuweiler H, Backbone-Driven Collapse in Unfolded Protein Chains. Journal of Molecular Biology 2011, 409 (2), 250–262. [DOI] [PubMed] [Google Scholar]
- 66.Fleming PJ; Fleming KG, HullRad: Fast Calculations of Folded and Disordered Protein and Nucleic Acid Hydrodynamic Properties. Biophysical Journal 2018, 114 (4), 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Das RK; Ruff KM; Pappu RV, Relating sequence encoded information to form and function of intrinsically disordered proteins. Current opinion in structural biology 2015, 32, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.(a) Auton M; Bolen DW, Application of the transfer model to understand how naturally occurring osmolytes affect protein stability. Methods in enzymology 2007, 428, 397–418 [DOI] [PubMed] [Google Scholar]; (b) Hu CY; Kokubo H; Lynch GC; Bolen DW; Pettitt BM, Backbone additivity in the transfer model of protein solvation. Protein Science 2010, 19 (5), 1011–22 [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Roseman MA, Hydrophobicity Of The Peptide C=O…H-N Hydrogen-Bonded Group. Journal of Molecular Biology 1988, 201 (3), 621–623 [DOI] [PubMed] [Google Scholar]; (d) Roseman MA, Hydrophilicity Of Polar Amino-Acid Side-Chains Is Markedly Reduced By Flanking Peptide-Bonds. Journal of Molecular Biology 1988, 200 (3), 513–522. [DOI] [PubMed] [Google Scholar]
- 69.Fossat MJ; Harmon TS; Posey AE; Choi J-M; Pappu RV, Increasing the Accuracy in All-Atom Simulations of Intrinsically Disordered Proteins based on the ABSINTH Model. Biophysical Journal 2018, 114 (3), 432a. [Google Scholar]
- 70.Erbas A; Horinek D; Netz RR, Viscous friction of hydrogen-bonded matter. J Am Chem Soc 2012, 134 (1), 623–30. [DOI] [PubMed] [Google Scholar]
- 71.(a) Mercadante D; Wagner JA; Aramburu IV; Lemke EA; Grater F, Sampling Long- versus Short-Range Interactions Defines the Ability of Force Fields To Reproduce the Dynamics of Intrinsically Disordered Proteins. Journal of Chemical Theory and Computation 2017, 13 (9), 3964–3974 [DOI] [PubMed] [Google Scholar]; (b) Best RB, Computational and theoretical advances in studies of intrinsically disordered proteins. Current opinion in structural biology 2017, 42, 147–154. [DOI] [PubMed] [Google Scholar]
- 72.(a) Best RB; Zheng W; Mittal J, Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. Journal of Chemical Theory and Computation 2014, 10 (11), 5113–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Robustelli P; Piana S; Shaw DE, Developing a molecular dynamics force field for both folded and disordered protein states. Proceedings of the National Academy of Sciences USA 2018, 115 (21), E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Holehouse AS; Das RK; Ahad JN; Richardson MO; Pappu RV, CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophysical Journal 2017, 112 (1), 16–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Avbelj F; Grdadolnik SG; Grdadolnik J; Baldwin RL, Intrinsic backbone preferences are fully present in blocked amino acids. Proceedings of the National Academy of Sciences 2006, 103 (5), 1272–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.