

Abstract
Ref-1/APE1 (Redox Effector/Apurinic Endonuclease 1) is a multifunctional enzyme that serves as a redox factor for several transcription factors (TFs), e.g., NF-kB, HIF-1α, which in an oxidized state fail to bind DNA. Conversion of these TFs to a reduced state serves to regulate various biological responses such as cell growth, inflammation, and cellular metabolism. The redox activity involves a thiol exchange reaction for which Cys65 (C65) serves as the nucleophile. Using CRISPR editing in human pancreatic ductal adenocarcinoma (PDAC) cells, we changed C65 to Ala (C65A) in Ref-1 to evaluate alteration of Ref-1 redox dynamics as well as chronic loss of Ref-1 redox activity on cell signaling pathways, specifically those regulated by NF-kB and HIF-1α. The redox activity of Ref-1 requires partial unfolding to expose C65, which is buried in the folded structure. Labeling of Ref-1 with polyethylene glycol-maleimide (PEGm) provides a readout of reduced Cys residues in Ref-1 and thereby an assessment of partial unfolding in Ref-1. In comparing Ref-1WT vs Ref-1C65A cell lines, we found an altered distribution of oxidized versus reduced states of Ref-1. Accordingly, activation of NF-kB and HIF-1α in Ref-1C65A lines was significantly lower compared to Ref-1WT lines. The bioinformatic data revealed significant downregulation of metabolic pathways including OXPHOS in Ref-1C65A expressing clones compared to Ref-1WT line. Ref-1C65A also demonstrated reduced cell proliferation and use of tricarboxylic acid (TCA) substrates compared to Ref-1WT lines. A subcutaneous as well as PDAC orthotopic in vivo model demonstrated a significant reduction in tumor size, weight, and growth in the Ref-1C65A lines compared to the Ref-1WT lines. Moreover, mice implanted with Ref-1C65A redox deficient cells demonstrate significantly reduced metastatic burden to liver and lung compared to mice implanted with Ref-1 redox proficient cells. These results from the current study provide direct evidence that the chronic absence of Cys65 in Ref-1 results in redox inactivity of the protein in human PDAC cells, and subsequent biological results confirm a critical involvement of Ref-1 redox signaling and tumorigenic phenotype.
Keywords: Ref-1/APE1, Transcription factors (TFs), PDAC, Redox, NF-kB, HIF-1α
Graphical abstract
Highlights
-
•
Cys65 impacts the distribution of conformational states of Ref-1 in PDAC cells.
-
•
Cys65 in Ref-1 protein is critical for full activation of multiple TFs and regulation of mitochondrial metabolism.
-
•
Mutation of Cys65 in Ref-1 changed the gene expression profile of PDAC cells including downregulation of oxidative phosphorylation and upregulation of apoptosis genes as indicated by RNA sequencing.
-
•
Mutation of Cys65 in Ref-1 protein affects PDAC cell phenotype both in vivo and in vitro.
-
•
Absence of Cys65 in Ref-1 does not affect Ref-1 DNA repair endonuclease activity.
-
•
Chronic absence of Cys65 in Ref-1 results in redox inactivity of the protein in human PDAC cells.
-
•
Biological implications of altered Cys65 include a reduction in tumor growth and metastasis.
Abbreviations
- APE1
Apurinic/apyrimidinic Endonuclease 1
- Ref-1
Redox Effector Factor-1
- TFs
transcription factors
- TCA
tricarboxylic acid
- PDAC
pancreatic ductal adenocarcinoma
- Cys
Cysteine
- TME
tumor microenvironment
- OXPHOS
oxidative phosphorylation
- KI
Knock-In
- MMS
Methyl methanesulfonate
- DTT
Dithiothreitol
- WT
Wild type
- HALO
H-score calculation
- PEGm
polyethylene glycol-maleimide
- AMS
4′-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- H2O2
hydrogen peroxide
- CA9
carbonic anhydrase 9
- PCA
principal component analysis
- DEGs
differentially expressed genes
- FDR
false discovery rate
- GSEA
gene set enrichment analysis
- BER
Base excision repair
- pH3
phosphohistone H3
- GOBP
Gene Ontology Biological Process
1. Introduction
Apurinic/apyrimidinic endonuclease 1/Redox factor-1 (APE1/Ref-1 or Ref-1) possesses multiple enzymatic activities, plays a crucial role in maintaining genomic stability, and regulates cellular response to oxidative stress [1]. The endonuclease activity of APE1 is critical for DNA repair in the base excision repair pathway and cleaves the phosphodiester bond of the damaged DNA strand 5’ of an abasic site, which is subsequently acted upon to complete the restoration of the DNA strand. APE1 has also been shown to play a role in recognizing damage in RNA molecules and in modulating RNA stability and translation [2,3].
In addition to its endonuclease activity, Ref-1 has a distinct function as a redox regulatory protein that is essential for maintaining cellular redox homeostasis. There are seven cysteine (Cys) residues in mammalian Ref-1 including 65, 93, 99, 138, 208, 296, and 310. Of the seven Cys residues, three (Cys65, Cys93, and Cys99) are involved in Ref-1's redox function [4,5]. Cys65 and Cys93 are buried and surface inaccessible, whereas Cys99 is solvent accessible [6] (Fig. 1A). Cys65 serves as the nucleophilic Cys in the thiol exchange reaction required to reduce oxidized transcription factors [4]. A critical role of Cys65 in redox activity is supported by studies of zebrafish Ref-1, which has no redox activity. Upon substitution of Thr58 with a cysteine residue, the structural equivalent of Cys65 in human Ref-1, zRef-1 (Thr to Cys) became redox active [4,6,7].
Fig. 1.

Overview of the cysteines in Ref-1 and the PEGm redox assay. A) 3D structure of Ref-1 demonstrating the position of the seven cysteine residues in the protein; B–C) A Schematic of the gel-based redox shift assay examining dynamic alteration of Ref-1 redox status in the cells. A reduced Cys residue will be labeled by the PEGm reagent resulting in a 15 kDa shift as shown in B and C. The mixed conformations of Ref-1 protein generate band-shifts based on the protein size and available cysteines and were detected by Ref-1 primary antibody (D).
Data from numerous laboratories has demonstrated that Cys65 plays a critical role as a redox switch, allowing the protein to switch between an oxidized and reduced state in response to changes in the cellular redox environment. Ref-1, in its reduced state, has been shown to interact with and increase the DNA binding activity of multiple crucial oncogenic transcription factors (TFs), including HIF-1α, NF-κB, AP-1, STAT3 and others [1,8]. By regulating the activity of these transcription factors, Ref-1 plays a critical role in a wide range of cellular processes, including DNA repair, cell proliferation, differentiation, inflammation, metabolism, and apoptosis [1,9,10].
Ref-1 is expressed ubiquitously in humans at a concentration of approximately 0.35–7.0 × 106 molecules per cell, ∼3 μM in cells [4,11,12]. Numerous studies demonstrated that Ref-1 is overexpressed in human malignancies such as sarcomas, prostate, bladder, ovarian, and pancreatic cancers [[13], [14], [15]]. For instance, pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers due to a high propensity for metastasis, poor response to current treatment regimens, and lack of markers for early diagnosis, resulting in a 5-year overall survival of around 12% [8]. Oncogenic Kras mutations lead to alteration of signaling pathways that are associated with the progression and metastasis of PDAC including metabolic reprogramming, inflammation, and remodeling of the local tumor microenvironment (TME) [16]. Ref-1 redox signaling can contribute to activation of several inflammatory and hypoxia signaling pathways through its regulation of HIF-1α, NF-κB, AP-1, and STAT3 signaling, that are active in PDAC and highly expressed in the tumor and its associated TME [[16], [17], [18]]. Additionally, recent data by our laboratories have demonstrated a relationship of Ref-1 redox signaling, tumor metabolism, and altered mitochondrial function [16].
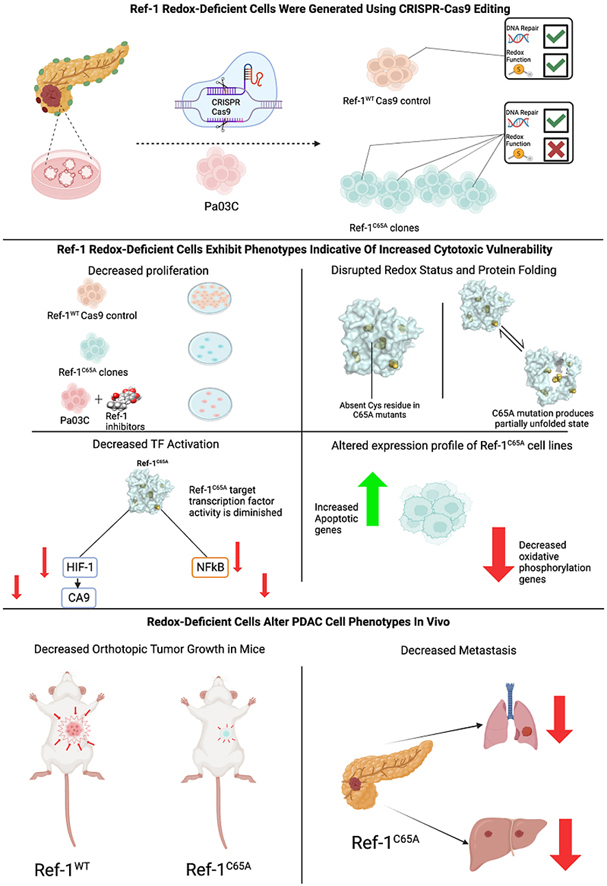
Deletion of the Ref-1 gene, which removes the redox, repair, and RNA stability and translation functions, is embryonic lethal in animals. Multiple studies have demonstrated that homozygous disruption of the Ref-1 structural gene (APEX1) in mice died very early in embryonic development [[19], [20], [21]]. Similarly, most studies in cell culture indicate that the expression of Ref-1 is essential [17]. Although numerous studies have investigated the impact on Ref-1 activity of the various Cys residues, most of the published reports, including ours, have been tested or performed in cell-free systems or in cells with overexpression of mutants on top of high levels of endogenous Ref-1 [5,[22], [23], [24]]. While these studies are informative and demonstrate effects on Ref-1 redox activity when targeted Cys residues are substituted with alternative amino acids, such as Alanine or Serine, they may underestimate the effects [24,25]. Thus, we used CRISPR editing to engineer PDAC patient-derived cells (Pa03C) to express a Ref-1 protein in which C65 was changed to Ala (C65A) and the resulting cell lines homozygous for the Ref-1C65A mutation. The use of CRISPR editing and Ref-1C65A cell lines allows us to evaluate the effects of loss of Ref-1 redox activity on cell signaling pathways. These cell lines will allow us to better understand the role Ref-1 redox signaling plays in growth and metastasis of pancreatic cancer without the confounding effects of wild type (WT) Ref-1 still being present, even at low levels. In our studies presented here, we observed less fully folded Ref-1 in the Ref-1C65A expressing cells, compared to controls suggesting that substitution of C65 (C65A) impacts overall folding of Ref-1 in addition to impairing its redox activity. Accordingly, impaired, or only partial activation of oncogenic TFs were also observed in Ref-1C65A expressing cells, as well as reduced expression of genes in the oxidative phosphorylation (OXPHOS) pathway compared to Cas9 control cells. Ref-1 and its transcriptional targets have been reported to be highly active in PDAC. In vivo studies using Ref-1-C65A-PDAC implanted tumors, both subcutaneous or orthotopic, demonstrated significant reduction in primary tumor size and metastatic lesions in the liver and lung in Ref-1C65A PDAC cells compared to the Cas9 control lines, further demonstrating the critical role of C65 in Ref-1 redox signaling and downstream pathways including proliferation and metastasis. To our knowledge, the present study provides the first direct evidence that the chronic absence of Cys65 in Ref-1 results in redox inactivity of the protein in human PDAC cells, and subsequent biological results confirm a critical involvement of Ref-1 redox signaling and tumorigenic phenotype.
2. Materials and methods
2.1. Cell culture
Low passage PDAC patient-derived cells, Pa03C, were cultured with DMEM (Invitrogen; Carlsbad, CA) supplemented with 10% FBS (Hyclone; Logan, UT) at 37 °C in 5% CO2. Authentication was performed using STR analysis, and cells were confirmed to be mycoplasma negative throughout the experiments and passaged less than ten times from thaw [26]. These cells were originally isolated from a metastatic lesion in the liver in a male pancreatic cancer patient [27]. CRISPR Cas9-mediated homozygous knock-in clones of APEX1 (Cys65 to Ala, C65A) in Pa03C cells were generated by Synthego Corporation (Redwood City, CA, USA). To generate these cells, ribonucleoproteins containing the Cas9 protein and synthetic chemically modified sgRNA produced at Synthego were electroporated into the cells along with a single-stranded oligodeoxynucleotide (ssODN) donor using a protocol optimized by Synthego. Editing efficiency is assessed 48 h post electroporation. Genomic DNA is extracted from a portion of the cells, PCR amplified and sequenced using Sanger sequencing. The resulting chromatograms are processed using Synthego Inference of CRISPR edits software (http://ice.synthego.com/Click to follow link.">ice.synthego.com).
To create monoclonal cell populations, edited cell pools are seeded at 1 cell/well using a single cell printer into 96 or 384 well plates. All wells are imaged every 3 days to ensure expansion from a single-cell clone. Clonal populations are screened and identified using the PCR-Sanger-ICE genotyping strategy described above. Clonal isolation from Pa03C cells was used to generate four clones (H4, H7, E19, J5) that express Ref-1C65A. The Cas9 cells which were used as a control in these experiments refer to a mock transfection control line in which the parental cells, Pa03C, were transfected with Cas9 and donor template without the gRNAs. The guide RNA sequences, and donor sequences are in Supplemental Table S1. The C65A knock-in (KI) clones were confirmed by sequencing and using the redox labeling assay as described below.
2.2. Cell proliferation and colony formation assay
Cell proliferation was measured with Trypan Blue excluding method (Invitrogen, Eugene, USA) as previously described. Briefly, 8x104 cells were plated in 6-well plates and counted each 24h interval up to 96h [28].
Colony formation assay: Briefly, 8000 cells were plated in 6-cm dish and allowed to grow in 10% FBS DMEM up to 8–10 days. Cells were pre-washed with cold PBS, and then stained with 0.1% methylene blue (LabChem, Cat#: LC169202) in 70% methanol (MeOH) for 1 h at room temperature. After three washes with H2O, stained colonies were imaged on Bio-Rad Chemidoc imaging program, and counted on ImageJ analyzer using the “Colony Area” plugin [29].
2.3. Drug treatment and Cytotoxicity
Pa03C cells were seeded at 2500 cells/well in 96-well plates and cell viability was measured with Alamar blue (Invitrogen, Eugene, USA) 48 h after treatment with various concentrations of methyl methanesulfonate (MMS) (Sigma, Cat#129925). Cellular response to MMS was normalized to a non-treated (media only or vehicle) control [30]. At least three replicates were performed.
For examining alteration of Ref-1 redox state, Pa03C cells were treated with different concentrations of H2O2 (Thermo Fisher, Cat#H325-500) for 30min. Dithiothreitol (DTT; reducing agent, 5 mM (Roche, Cat#38225822)) and diamide (oxidizing agent, 4 mM (Sigma, Cat#D3648)) were used as controls.
2.4. Mitochondria functional assay
S-1 Mitoplates (Biolog, Hayward, CA) were used to investigate mitochondrial function of TCA cycle substrates as previously published [16]. Assays were performed as per manufacturer's protocol. Briefly, plates were activated by adding the Assay Mix to the wells to dissolve the substrates for 60 min at 37 °C. Cas9 control cells in low glucose media (0.5 mM) were used as control and Ref-1C65A E19 clone was treated with DMSO or APX2009 (5 μM, 24 h) in low glucose media. Following treatment, cells were collected, counted, resuspended in provided buffer and plated at 5x104 cells/well. This resuspension was added to the plate, which was immediately read at 590 nm kinetically at 5 min intervals for 4h at 37 °C. Data was analyzed using Graphpad Prism 8, and statistical significance was determined using the 2-way ANOVA and p-values <0.05 were considered statistically significant.
2.5. Ref-1 apurinic/apyrimidinic endonuclease assay
Pa03C C65A mutant cell efficiency to repair DNA was measured by the APE1/Ref-1 repair activity assay using cell lysates collected across passages in the presence of an AP site mimic substrate consisting of two annealed oligonucleotides (Eurogentec Ltd.):
| 5' - (6-FAM) – GAA TCC * CCA TAC GTA TTA TAT CCA ATT CC – 3′ |
5' – GGA ATT GGA TAT AAT ACG TAT GGT GGA TTC - (DABCYL) - 3′ as has been previously described [31,32]. The substrate contained the fluorescent 6-FAM label AP site mimic, tetrahydrofuran (*), on one strand and the Dabcyl quencher on the complementary strand. Ref-1 cleavage of this AP site mimic resulted in the release of the 6-FAM portion of the oligo, and the amount of fluorescence over time was directly proportional to Ref-1 endonuclease activity.
Briefly, cells were collected across 4 passages and lysed in cold PBS containing 2.0 mM fresh DTT. Cells were then pulse-sonicated on ice, and the protein lysate was quantified using the reducing agent compatible RC DC method (Bio-Rad). Because of the differential Ref-1 expression in the C65A mutant cells, Cas9 cell lysate sampled at 100 μg/mL was set as the optimal top concentration to adjust the C65A mutant lysates by amount to equalize the Ref-1 protein level. Lysate was resuspended in 10 mM HEPES and serially diluted 1:2 in 10 mM HEPES across 5 dilutions. A master mix was made for a final amount per well of 50 nM annealed oligo, 50 mM Tris, 1 mM MgCl2, and 50 mM NaCl, pH7.5. The activity negative control consisted of cell lysate with the addition of final 50.0 mM EDTA. The fluorescence was read kinetically at 1-min intervals for 5 cycles at 37 °C on a Synergy H1Ⓡ platform (BioTek). The rate of the reaction (Vmax) was used to determine any change in APE1 repair activity of the C65A mutant lysate as compared to Cas9. 2-way ANOVA statistical analysis was performed on Prism (GraphPad).
2.6. Western blotting analysis
Cells were lysed in 1% SDS extraction buffer supplemented with protease inhibitors (Santa Cruz Biotechnology, TX, USA) [26,33]. Briefly, cell extract was heated at 95 °C for 5 min, then sonicated (4 pulses, 4 cycles) to shear the DNA in the samples as previously described [26]. Denatured samples (20–40 μg) were subjected to SDS-PAGE and proteins were transferred onto nitrocellulose membranes by electrophoretic transfer. Non-specific binding sites were blocked at room temperature for 1hr with 5% (w/v) Blotting-Grade milk (Bio-Rad Laboratories, CA, USA) in Tris-buffer saline (Boston Bio Products, MA, USA) containing 0.05% (v/v) Tween-20 (Thermo Fisher, MA, USA). Membranes were incubated overnight with the primary antibodies, and then with the peroxidase-conjugated secondary antibody for 1hr (Supplemental Table 2S). Signal was then captured by using Bio-Rad ChemiDoc imager, and band intensities were analyzed by densitometry on Image Lab (Bio-Rad Laboratories, CA, USA) or ImageJ software [8,26].
2.7. Assessment of Ref-1 protein redox status
Alteration of protein redox state was assessed using the -SulfoBiotics- Protein Redox State Monitoring Kit Plus (Dojindo, Cat#SB12) for cellular proteins in accordance with the manufacturer's instructions (Dojindo Molecular Technologies Inc., Rockville, MD, USA) [34,35]. Briefly, to study the protein redox states in Pa03C cells, samples were prepared through protein precipitation using trichloroacetic acid (TCA) (Fisher, Cat#196057) and labeled with Protein-SHifter Plus upon non-reducing conditions in accordance with the manufacturer's instructions (Fig. 1B and C). After cell extracts were subjected to SDS-PAGE, gels were exposed to UV light on a transilluminator to remove Protein-SHifter. Proteins in the gel were then electro-transferred to a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, CA, USA). The membrane was blocked with 5% non-fat dry milk and incubated with Ref-1 antibody (Novus, Cat#13B8E5C2). Signals were obtained by using a horseradish peroxidase-linked secondary antibody (Bio-Rad, Cat#1706516) and the Super Signal Chemiluminescence System (Thermo, Cat#A38554) and bands were quantitated for Western blot band intensities.
2.8. Real-time quantitative PCR (qRT-PCR)
Cells were collected and processed for RNA extraction according to the manufacturer's protocol (Qiagen, Hilden, Germany, USA) [33]. [33]. RNA concentrations were determined using a NanoDrop (Thermo Fisher, MA, USA). Subsequently, 1 μg of RNA/25-μl reaction mix was reverse-transcribed to cDNA using (Applied Biosystems, Warrington, UK). qRT-PCR was performed in 96-well plates, with a final volume of 20 μL/well using the SYBR Green PCR kit (Applied Biosystems, Foster City, CA, USA) on the CFX96 real-time PCR detection system (BioRad, Hercules, CA, USA). Primers for indicated genes are commercially available (OriGene, Technologies, MD, USA) and primers sequence are shown in supplemental data (Supplemental Data: Table S3). qRT-PCR cycling conditions were 1 min at 95 °C, 10min at 95 °C, 15 s at 95 °C and 1 min at 60 °C for 40 cycles. Relative changes in mRNA expression levels were assessed by the 2−ΔΔCT method, and changes in mRNA expression of the target gene were normalized to that of b-actin gene [8].
2.9. NF-κb/HIF-1α luciferase activity
Pa03C Cas9 Ref-1WT cells or Ref-1C65A cells were co-transfected with constructs containing luciferase driven by NF-κB or HIF-1α and a Renilla luciferase control, pRL-TK (Promega Corp., Madison, WI), in a 3:1 ratio by using Lipofectamine 2000 (Invitrogen) as previously described [36]. Sixteen hours after transfection, transfection media was exchanged to regular growth media. For HIF-1α transfected cells, one set of plates was exposed to 1% hypoxia (with 5% CO2) for HIF-1α induction, with another parallel set in normoxia for 24h. For NF-κB transfected cells, 20 ng/mL hTNFα (R&D Systems, Cat#10291-TA-020) was used for a 6h induction before assaying for luciferase activity. Firefly and Renilla luciferase activities were assayed by using the Dual Luciferase Reporter Assay System (Promega Corp, Cat# E1910). Each time, lipofectamine-only control was included, and the final data are expressed as the fold-change of the relative luciferase units compared with the Cas9 Ref-1WT uninduced control. All of the transfection experiments were performed in triplicate and repeated at least 3 times in independent experiments.
2.10. In vivo studies
NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice were obtained from the Preclinical Modeling and Therapeutics Core (IU Simon Comprehensive Cancer Center) and maintained in pathogen-free conditions. All in vivo animal studies were conducted in compliance with guidelines of National Institutes of Health and Institutional Animal Care and Use Committee of Indiana University School of Medicine. For orthotopic implant in the pancreas, each mouse was implanted with Pa03C Ref-1WT expressing Cas 9 cells or C65A expressing clones (H7 or E19 cells) at 1.3x104 cells/mouse. Following implant, the animals were monitored for five weeks for tumor progression and sacrificed after week 5. At the end of the study, all animals were euthanized, and primary tumors, livers, and lungs were collected for histology assessment. To ensure representative areas were captured, each tumor was halved, and each half was either flash frozen or fixed in 10% formalin. The left lobes of the livers were fixed in formalin for histology analysis. All tissues were stained with H&E staining.
Metastasis of the primary tumor to the liver and lungs was measured using two complimentary approaches for increased rigor. A quantitative approach using the HALO® software platform was used for scoring tumors. For determination of metastasis, the software used the HALO classifier which was trained using machine learning algorithms and digital image analysis techniques to quantify metastatic lesions present within the liver and lung tissues. The total area of metastatic lesions in each tissue was measured and reported as a percentage (%) of total tissue analyzed. Quantification of Ref-1 (Novus Biological, Cat# NB100-101) and pH3 (Cell Signaling, Cat# 9701) was conducted on whole tumor tissues using Aperio ImageScope which generated a pixel count for cells that stained positive for Ref-1 or pH3over the rest of the tissue. The brown color indicated positive staining for Ref-1 or pH3 in cells, while the rest of the tissue exhibited a blue stain [37].
2.11. Bioinformatic analysis (RNA-seq)
The RNA-seq reads were mapped to the human genome hg38 using the RNA-seq aligner STAR (v2.7.2a) [38], “--outSAMmapqUnique 60”. Uniquely mapped sequencing reads were assigned to Gencode 312 gene using featureCounts (v1.6.2) [39], with the following parameters: "–p –Q 10 –O”. Only genes were kept for further analysis if their read counts were more than 10 in at least 3 samples. The gene expression levels were normalized by the methods of trimmed mean of M values (TMM) method [39], then subjected to differential expression analysis using edgeR (v3.20.8) [40,41] by comparing C65A expressing clones, e.g., E19 or H7, and WT expressing Cas9. Differentially expressed genes (DEGs) were identified with the cutoffs, false discovery rate (FDR) less than 0.05 and |log2FC| larger than 1. The values of log2FC for all genes between mutant clone and Cas9 control were considered for gene set enrichment analysis (GSEA, version 4.3.2) [42] on hallmark and pathway gene sets.
2.12. Statistical analysis
All experiments were performed at least three independent times unless it's specified as duplicated, and the data obtained were expressed as “Mean and Standard Error.” Significance was calculated using one-way ANOVA where applicable using Graph Pad Prism Version 9 as previously described. For the orthotopic model, Wilcoxin-test was used for primary tumor as well as metastatic burden. Statistical significance was considered when p-value was <0.05.
3. Results
3.1. Characterization of Ref-1C65A expressing PDAC cells
To characterize the effects of replacing Cys65 with Alanine (C65A) in Ref-1, we first assessed expression levels of Ref-1 in the cells. There is a ∼50% reduction in both mRNA and protein levels in the four Ref-1C65A-expressing cells in comparison to Cas9 Ref-1WT control cells (Fig. 2 A, B). Interestingly, we did not observe any differential cell proliferation up to 96h time interval, suggesting that ∼50% less expression of Ref-1C65A does not alter cell proliferation (Fig. 2C). However, significant differences in the numbers of cell colonies in two of the clones Ref-1C65A (E19 and H7) were observed with respect to control lines Ref-1WT (Cas9) over the course of eight days (Fig. 2D and E).
Fig. 2.

In vitro characterization of cells with an inactivating mutation in Cys65 in Ref-1 in pancreatic cancer cells. mRNA (A) and protein levels (B) in Ref-1C65A expressing clone cells (E19, H7, J5, E14) with Cas9 Ref-1WT was used as a control line. One-way ANOVA, *p < 0.05, ***p < 0.001, Mean ± SEM, N = 3. C) The cell growth rate of Ref-1C65A expressing cells up to 96h, N = 3. Representative images of formation of colony in Ref-1C65A expressing cells (D), and quantification of number of colonies or Colony Formation Area (CFA) (E), One-way ANOVA, **p < 0.01, ***p < 0.001, Mean ± SEM, N = 3. F) Endonuclease activity of Ref-1 was assessed in Ref-1C65A expressing cells, N = 3. G) Cytotoxic effects of MMS were assessed in Ref-1C65A expressing cells, N = 3.
To determine if the mutation of C65 to Alanine impacted the DNA repair activity of Ref-1, an oligo-based endonuclease assay was used. We did not observe any significant differences on Ref-1 endonuclease activity in the C65A-expressing cells (Fig. 2F). Also confirming that the clones do not have deficiencies in DNA repair activity, C65A-expressing cells did not demonstrate differential sensitivity to Methyl methanesulfonate (MMS), a well-known alkylating agent acted upon by the BER pathway [43](Fig. 2G). These results demonstrated that ∼50% less expression of Ref-1 did not alter the DNA repair activity in the cells [43].
3.2. Cys65 impacts the distribution of conformational states of Ref-1 in PDAC cells
Determining the extent of Cys oxidation in a protein isolated from cells can provide information not only on redox status but also on folded state of the protein. In the case of a redox factor in which solvent accessible Cys residues are involved in disulfide bonding, the redox status of the protein can be determined directly using Cys labeling approaches. In the case of Ref-1, which is an unusual redox factor in that the nucleophilic Cys residue (Cys65) is a buried residue, Cys labeling of the protein isolated from cells provides information on oxidation of buried Cys residues, which in turn is related to the conformational state, partially or fully folded. Ref-1 has 7 Cys residues; 2 are solvent accessible (99 and 138) and 5 are buried (65, 93, 208, 296, and 310) as seen in crystal structures of Ref-1 and illustrated in Fig. 1A [44]. Oxidation of Cys residues might involve any of the following: disulfide bonds, glutathionylation, S-nitrosylation, addition of oxygen to form sulfenic, sulfinic, or sulfonic acid. Although no intramolecular disulfide bonding has been reported for Ref-1, intermolecular disulfide bond formation has been suggested [45]. In addition, glutathionylation and S-nitrosation have been reported for Ref-1 [46,47]. Oxidized Cys residues cannot react with the Cys modifying reagents.
To investigate the extent of oxidation of Ref-1 in live cells that express Ref-1C65A, a gel-based assay was developed and optimized. Initially, we used (4′-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid) AMS-based redox assay, which is a protein-thiol sensitive labeling reagent that detects reduced forms of the protein and labels thiol groups with a 0.5 kDa moiety per thiol group, generating a shift in the molecular weight of the protein on the gel [48]. As demonstrated in Figure S1, although we observed an increase in the molecular weight of the Ref-1 protein post-labeling, the assay was not sensitive enough to detect differences between Ref-1C65A -expressing clones and Cas9 Ref-1WT lines (7X vs 6X) due to the single Cysteine mutation [48].
Therefore, an alternative labeling reagent, polyethylene glycol-maleimide (PEGm), which following isolation and denaturation (but not thiol reduction) results in ∼15 kDa addition per thiol, was used and could label and differentiate all 7 Cys residues in Ref-1 as observed on the gel in Fig. 1, Fig. 3A and referred to as 1X-7X species. As expected, in contrast to Ref-1WT lines, there is one less band detected in Ref-1C65A clones, confirming the mutation of the Cys65 residue and the robust sensitivity of the assay in detecting reduced Cys residues (Fig. 3A). Labeling of 5X, 6X, or 7X Cys residues with PEGm in Ref-1WT or 4X, 5X, or 6X Cys residues in Ref-1C65A is consistent with folded conformations of the protein in which 2, 1, or 0 solvent-accessible Cys residues are oxidized. In this case, none of the buried Cys residues are oxidized and are therefore labeled with PEGm. Bands for 1X-4X in WT (1X-3X in C65A) likely reflect states of the protein that were partially or fully unfolded in which Cys residues are accessible and have been oxidized resulting in fewer reduced Cys residues to react with PEGm. In comparing the labeling of Ref-1WT with Ref-1C65A, two major differences are observed. First, there is less fully folded Ref-1C65A as compared to Ref-1WT if the total for 4X-6X is compared to 5X-7X relative to 1X in each sample, respectively (Fig. 3B). Partially or fully unfolded states would be represented by bands 1X-4X in WT (1X-3X in Ref-1C65A samples). Notably neither sample contains a significant 2X band. Ref-1C65A lacks a 3X band; Ref-1WT has very little 3X but has a prominent 4X band. It is possible that this 4X band in Ref-1WT corresponds to labeling of C65 in the WT sample as there is no equivalent to this in Ref-1C65A. This would be consistent with a partially unfolded state in which C65 is accessible and oxidized. In both samples, there are significant 1X bands but virtually no unlabeled Ref-1.
Fig. 3.

Cys65 is critical for maintaining Ref-1 redox function in PDAC cells. A) The impact of absence of Cys65 on Ref-1 redox status, 7X corresponds to fully labeled Ref-1WT (Parental line and Cas9), and 6X is for Ref-1C65A expressing clones, whereas 1X is least reduced forms of Ref-1, N = 6. B) Ref-1 redox status was calculated by taking the ratio of most reduced Ref-1 over most oxidized form (7X+6X+5X/1X; Ref-1WT) or (6X+5X+4X/1X; Ref-1WT), **p < 0.01 compared to Cas9 control, One-way ANOVA, Mean ± SEM, N = 3.
3.3. Dynamics of Ref-1 redox status in response to oxidative stress in PDAC cells
To investigate the responsiveness of Ref-1 redox status in PDAC cells to acute oxidative stress using hydrogen peroxide (H2O2) as a model, both Ref-1WT and Ref-1C65A-E19 cells were exposed to increasing concentrations of H2O2, and then dynamic alteration of Ref-1 redox status in the cells was assessed. Cells treated with DTT (reducing agent) and diamide (chemically relevant oxidizing agent) were used as controls (lanes 1 and 2). With the strong oxidizing agent, diamide, fully labeled 7X Ref-1WT protein is undetectable (Fig. 4A; lane 3). Similar results were found in Ref-1C65A clone cells (6X band; Fig. 4B; lane 3). Treatment with H2O2 in Ref-1WT cells resulted in a dose-dependent reduction in fully labeled 7X and increase in 5X and 4X Ref-1, as would be expected under oxidizing conditions (Fig. 4A, lanes 4–7). Quantitation of this is in Fig. 4C. In Ref-1C65A-E19 cells, a similar trend was observed with a reduction in 6X and increase in 4X and 3X bands (Fig. 4B, lanes 4–7). In this case, the 3X band likely corresponds to a partially unfolded state of Ref-1C65A that results following treatment with H2O2. Quantitation of these findings are shown in Fig. 4D. Fig. 4C shows that there is a dose-dependent decrease in the fully labeled 7X Ref-1 in cells that express Ref-1WT, and while this decrease is also observed in H2O2-treated Ref-1C65A cells (Fig. 4D), the amount of fully labeled Ref-1 is dramatically reduced compared to Cas9 cells. In both Ref-1WT and Ref-1C65A cells, there is an increase in oxidation of solvent accessible Cys residues (5X and 4X species, respectively) following treatment with diamide or increasing concentrations of H2O2. Overall, this is the first evidence demonstrating detailed as well as dynamic alterations in the oxidation of Ref-1 in response to oxidative stress.
Fig. 4.

Dynamics of Ref-1 redox status in response to oxidative stress in PDAC cells. Alteration of Ref-1 redox status in response to the acute oxidative stress induced by hydrogen peroxide (H2O2) in both Ref-1WT (A) and Ref-1C65A cells (B). Cells treated with DTT (reducing agent) and diamide (chemically relevant oxidizing agent) for 30min were used as controls, N = 2. Quantification of PEG-labeled Ref-1WTCas9 (C) or Ref-1C65A-E19 (D) after treatment with increasing concentrations of H2O2, n = 2. Fold change refers to band intensity of band of interest compared to the 7X or 6X labeled band of the control cells.
3.4. Cys65 in Ref-1 protein is critical for full activation of multiple TFs and mitochondrial metabolism
PDAC is an extremely hypoxic tumor and multiple oncogenic TFs such as HIF-1α and NF-κB respond to these stress conditions and are highly active in PDAC and the cells in its associated TME [49,50]. Both NF-κB and HIF-1α are under Ref-1 redox control [1,3,21]. To confirm that mutation at Cys65 resulted in a blockade of Ref-1 redox activity, reporter assays of both NF-κB and HIF-1α were performed in Ref-1WT-Cas9 and Ref-1C65A expressing cells. We evaluated the transcriptional activity of NF-κB in cells with and without TNFα stimulation. There were not significant differences in NF-κB activity under basal conditions. However, there was a 40–80% reduction in activation of NF-κB activity in Ref-1C65A clones (Fig. 5A). Similarly, hypoxic conditions were used to stimulate HIF-1α activity, and we observed significantly reduced HIF-1α transcriptional activity (40–90%) in the Ref-1C65A clones (Fig. 5B). Consistently, clone H7 had a stronger phenotype with respect to diminution of NF-κB or HIF-1α activity. As an additional marker of HIF-1α activity, expression levels of HIF-1 regulated gene, carbonic anhydrase 9 (CA9) which regulates intercellular pH was used as in our previous published data [51]. Both CA9 mRNA and protein levels are strongly upregulated in hypoxia conditions as expected (Fig. 5C–E). Our previous work with Ref-1 knockdown and Ref-1 redox inhibitor treatment demonstrated that CA9 regulation by HIF-1α is Ref-1 redox dependent [51]. Due to a decrease in Ref-1 redox activity owing to the mutation of C65, HIF activity was decreased which led to a decrease in CA9 expression levels under hypoxia. For example, CA9 expression levels in Ref-1C65A mutant E19 and H7 clones were 2-3-fold reduced in expression of CA9 both by mRNA and protein in comparison to Ref-1WT cells (Fig. 5C–E). Taken together, these results clearly support that mutation of Cys65A in Ref-1 failed to fully activate TFs that are under Ref-1 redox control due to the lack of the necessary Cys65 in the thiol exchange reaction to reduce the oxidized TFs.
Fig. 5.

Cys65 in Ref-1 protein is critical for full activation of multiple TFs. A) NF-κB transcriptional activity was assessed after stimulation with TNFα (20 ng/mL, 6 h) in both Ref-1WT-Cas9 and Ref-1C65A cells. Data is represented as Mean ± SEM, N = 3. p < 0.0001 is represented as ## for stimulation in Ref-1WT- Cas9 cells. p < 0.0001 is represented as ## for inhibition of NFκB in Ref-1C65A cells compared to Ref-1WT-Cas9 under stimulation. B) Ref-1WT-Cas9 and Ref-1C65A cells were exposed to normoxic or hypoxic condition (24h, 1% O2), then assayed for HIF-1 reporter transcriptional activity and the data is represented as Mean ± SEM for N = 3, ##p < 0.0001 (## represents significance for HIF1 stimulation in Ref-1WT-Cas9 cells and ## signifies HIF1 inhibition in Ref-1C65A cells compared to Ref-1WT-Cas9 under Hypoxia). CA9 mRNA levels (C) and protein levels (D, E) were assessed using qPCR and western blot analysis (*p < 0.05, **p < 0.01, ***p < 0.001, Mean ± SEM, N = 3). F) Graph showing levels of TCA cycle substrates using a mitochondria functional plate-based assay. Ref-1WT-Cas9 and Ref-1C65A clone E19 cells were grown in low glucose (0.5 mM) media, with and without treatment with APX2009 (5uM, 24 h). Data represented as Mean ± SEM for Average rate of reaction (N = 2, *p < 0.05, **p < 0.01).
A mitochondrial substrate plate-based assay was used as additional confirmation that the C65A mutation resulted in blockade of Ref-1 redox activity. We have previously published that inhibition of Ref-1 redox function dramatically decreases mitochondrial metabolism in a dose-dependent manner using this assay [52]. Based on those findings, in this study, we used Cas9 control cells and C65A cells (clone E19) that were grown in low glucose media to test for the effect of C65A mutation on TCA cycle substrate utilization (α-ketoglutaric acid, succinate, fumarate, and l-malate) (Fig. 5F). In cells that only express Ref-1C65A, there is significantly reduced uptake of succinate (by 30%) and l-malate (by 45%) thus indicating a decrease in TCA cycle utilization (Fig. 5F). This is similar to what is observed following treatment with Ref-1 redox inhibitors. Additionally, as a control, the E19 clone was treated with the Ref-1 specific redox inhibitor, APX2009 under low glucose conditions. As expected, there is no effect of APX2009 treatment on the C65A-expressing cells, strengthening the importance of this cysteine for Ref-1's redox function. These data resulted in our focus on the E19 and H7 clones for further studies.
5. Mutation of Cys65 in Ref-1 dramatically changed the gene expression profile of PDAC cells and including downregulation of oxidative phosphorylation and upregulation of apoptosis genes as indicated by RNA sequencing.
In order to better understand the effects on transcriptional regulation in cells expressing C65A, we performed RNA sequencing with the parent Pa03C cells (Ref-1WT), Cas9 control (Ref-1WT), and the E19 and H7 clones (Ref-1C65A). The plot of principal component analysis (PCA) based on gene expression indicated that WT and Cas9 samples were similar in gene expression to each other and yet different from the E19 and H7 clones (Supplemental Fig. 3). E19 showed more differences from Cas9 in the first principal component (PC1), and H7 was different in both PC1 and PC2. Given the differential analysis comparing with Cas9, we identified 1408 up- and 1073 down-regulated differentially expressed genes (DEGs) in E19 clones (Fig. 6A), as well as 946 up- and 910 down-regulated DEGs in H7 compared to Cas9 control (Fig. 6B). It is worth noting that 547 upregulated DEGs were shared by both E19 and H7 (Fig. 6C), which is 6.89-fold (fold enrichment (F.E.) = 6.89) of the number by random selection (p < 1 × 10−325). Similar significant tendency was observed for downregulated DEGs between E19 and H7 (F.E. = 5.71 with p = 1.02 × 10−174 in Fig. 6D). Interestingly, the Gene Ontology Biological Process (GOBP) by Gene set enrichment analysis (GSEA) given the transcriptome comparisons of E19 or H7 cells to Cas9 were predominantly upregulated. As Ref-1 is a transcriptional regulator, this was surprising to us. However, on closer examination some of the GOBPs that were upregulated were negative regulators of signaling pathways or migration that could lead to a reduction in proliferative capacity and migratory potential (Fig. 6E). Moreover, hallmark genes for oxidative phosphorylation (OXPHOS) and respiratory electron transport ATP synthesis pathway tended to be significantly downregulated for both E19 and H7, while hallmark genes for apoptosis or genes related to regulation of extrinsic apoptotic signaling pathway were significantly upregulated (Fig. 6F). The significant downregulation of metabolic pathways including OXPHOS further confirmed our previously published data demonstrating that Ref-1 redox small molecule inhibitors impact upon metabolism as part of their mechanism of cell death in tumor cells [16]. These results indicate that two clones with the C65A mutation in Ref-1 exhibited very similar impact on hallmark genes, e.g., OXPHOS, and biological pathways that negatively regulate signaling associated with cancer phenotypes, e.g., G-protein coupled receptor signaling, TNF superfamily cytokine production, or MAP kinase activity, from the perspective of transcriptomics profile. However, the differences between the two C65A clonal lines, E19 and H7 suggest a diverse and complex impact of Ref-1 C65A mutation in a situation where Ref-1 is missing the key Cys involved in its redox signaling mechanism that we will continue to explore. This also highlights the similarities and differences between an acute knockdown or inhibition situation compared to a chronic condition created in these studies.
Fig. 6.

Mutation of Cys65 in Ref-1 dramatically changed the gene expression profile of PDAC cells and including downregulation of oxidative phosphorylation and upregulation of apoptosis genes as indicated by RNA sequencing. DEGs (FDR <0.05 & |logFC| > 1) identified in C65A clones, (A) E19 or (B) H7 compared with Cas9. Significant overlap of (C) up-regulated DEGs or (D) down-regulated DEGs in E19 and H7 compared with Cas9. (E) Selected Gene Ontology Biological Processes (GOBPs) based on Gene Set Enrichment Analysis (GSEA) notably over-represented in upregulated genes (NES >0) in E19 (left panel) or H7 (right panel) compared with Cas9, where the bar shows NES score of each GOBP), whereas the dot on the line indicates corresponding FDR-adjusted p value in the scale of -log10. The vertical dash lines represent FDR = 0.25. (F) GSEA) plots of selected hallmark gene sets or pathway or GOBP for E19 (left panels) or H7 (right panels) compared with Cas9.
3.5. Mutation of Cys 65 in Ref-1 protein affects PDAC cell phenotype both in vivo and in vitro
To investigate the impact of mutation of Cys65 in Ref-1 on PDAC tumor growth, we established an orthotopic PDAC in vivo model and monitored tumor growth over time. Mice implanted with Ref-1C65A expressing clones demonstrated significantly smaller tumor weight compared to the mice implanted with Cas9 control cells (Ref-1WT) (p < 0.05 E19, p < 0.0001 H7, Ref-1WT vs Ref-1C65A) (Fig. 7A). Similar results were found in our initial subcutaneous xenografts in which Ref-1C65A resulted in smaller tumor volume and tumor weight compared to Ref-1WT (p < 0.05; Fig. 2S). In addition, we examined the expression levels of Ref-1 along with proliferation marker, pH3 (phosphohistone H3) in whole tumor tissue. pH3 has much stronger prognostic value than classical prognosticators for evaluating invasive tumors, such as PDAC or breast cancer [53]. Tumors from mice implanted with Ref-1C65A cells express ∼50% less Ref-1 compared to tumors that express Ref-1WT (p < 0.001; Fig. 7B, D), further confirming the in vitro data (Fig. 2B).Interestingly, the Ref-1C65A expressing clone (H7) that demonstrated the strongest phenotype on tumor growth and in in vitro assays also demonstrated significantly reduced expression of pH3 compared to tumors with Ref-1WT (p < 0.001, Fig. 7C and D). pH3 has much stronger prognostic value than classical prognosticators for evaluating invasive tumors, such as PDAC or breast cancer [53].
Fig. 7.

Mutation of Cys 65 in Ref-1 protein affects PDAC cell phenotype in vivo. A) Weights of primary tumors in orthotopic pancreatic in vivo model at sacrifice (Week 5). Quantification of Ref-1 (B) and pH3 staining (C) in primary tumors with representative images were shown in panel (D). The number of metastatic lesions in the liver (E) and lung (F) were quantified using HALO, and the representative images of H&E staining are shown in panel (G). Arrows indicate metastatic lesions within the tissues. *p < 0.05, **p < 0.01, ****p < 0.0001, Mean ± SEM, n = 10–12 mice per group.
In orthotopic implantation, the Pa03C cells spontaneously metastasize to distant sites including liver and lung making this model particularly relevant to the human disease [54,55]. Mice implanted with Ref-1C65A redox deficient cells demonstrate significantly reduced metastatic burden to liver and lung compared to mice implanted with Ref-1 redox proficient cells (Cas9 control) as shown in Fig. 7E–G (*p < 0.05, **p < 0.01) with representative images in Fig. 7G. Taken together these findings demonstrate that Ref-1 redox signaling plays a role in tumor growth, proliferation, and invasiveness in vivo as well as the ability to metastasize to a distant site.
4. Discussion
Ref-1 has long been recognized as a modulator of redox status of transcription factors in cells; however, a detailed mechanism of Ref-1 protein oxidation and reduction status has not been reported in cellular studies under endogenous conditions. Most investigations have been performed in cell-free systems or in cells with overexpression of mutant Ref-1 trans-proteins on top of high levels of endogenous Ref-1 [5,[22], [23], [24]]. We used CRISPR editing to engineer PDAC cells to express a Ref-1 protein in which C65 was changed to Ala (C65A), and the resulting cell lines homozygous for the Ref-1C65A mutation. This approach allowed us to investigate the impact of losing the critical Cysteine at position 65 and the resulting effects on downstream oncogenic TFs that Ref-1 regulates and subsequent cell signaling in PDAC cells. This is also now a system with chronic C65A Ref-1 expression in contrast to acute previous studies. We demonstrate that Cys65 in Ref-1 is critical for Ref-1 redox function and transcriptional regulation in human PDAC cells and influences tumor growth and metastasis in vivo. We also demonstrate the impact of the C65 mutation on the redox status of Ref-1 using a gel-based redox shift assay to examine redox status of Ref-1, in cells having only Ref-1C65A which has not previously been achieved.
Previous findings have demonstrated that Ref-1 endonuclease activity is independent from redox activity, but primarily these studies were performed in biochemical assays and cell free systems [1,56]. Using our Ref-1C65A cell model, we confirmed that Cys65 mutation in Ref-1 does not affect Ref-1 endonuclease activity in cells (Fig. 2F). Accordingly, none of C65A-expressing cells lines demonstrate differential sensitivity to MMS, a laboratory-based DNA alkylating agent which generates methylated DNA adducts repaired by the BER pathway of which Ref-1 is a major component (Fig. 2G) [3,43,57].
In previous studies, using an EMSA redox assay, we demonstrated that the redox activity of Ref-1 was lost when Cys65 in Ref-1 replaced with Alanine. However, these studies typically either were cell free or used overexpression of the Cys65Ala protein on top of endogenous cellular Ref-1 [5]. Additional studies demonstrated a supporting role of Cys99 and Cys93 in the redox function of Ref-1, but none of the other four Cys in Ref-1 appear to be involved [5]. Additional support for the significant role of Ref-1 in the redox signaling function was demonstrated when Zebrafish Ref-1 containing Cys in similar locations as in mammalian Ref-1 and is DNA repair active, but redox inactive. Substituting Thr58 which is structurally equivalent to mammalian C65, with a Cys resulted in gain of function redox activity of the Zebrafish Ref-1 again supporting the critical role of Cys65 in this activity [5,58]. In the current study, we examined the oxidation status of Cys residues in Ref-1 from cells expressing either Ref-1WT or Ref-1C65A. Reduced Cys residues in Ref-1 react with PEGm while oxidized Cys residues do not. Indirectly, this assay provides information on the conformational state of Ref-1 in the cell prior to precipitation with TCA and denaturation. If all 7 Cys residues are reduced, we infer that Ref-1 is fully folded in the cell. Similarly, labeling of 6 or 5 Cys residues is consistent with fully folded Ref-1 as two Cys residues are solvent accessible, while the remaining 5 Cys residues are buried in the structure of Ref-1. Labeling of a single Cys residue is consistent with a primarily unfolded state of Ref-1 in which most of the Cys residues are exposed and can be oxidized more readily. The state with 4 Cys residues labeled is consistent with partial unfolding and exposure of Cys65. This conclusion is based on the finding that there is no equivalent state in labeled Ref-1C65A. Overall, the labeling results suggest that there is less fully folded (6X, 5X, and 4X) relative to unfolded (1X) Ref-1C65A present in cells as compared to Ref-1WT (7X, 6X, 5X vs. 1X). Thus, in cells, Cys65 impacts the amount of fully folded Ref-1 in cells consistent with an earlier report characterizing Ref-1C65S. Studies have demonstrated a TRX1/Ref-1/PRDX1 redox re-cycling in the cells, in which TRX1 biochemically reduces oxidized Ref-1 and converts it back to its active form, while PRDX1 is purported to oxidize Ref-1 [8,59]. This redox cycling provides an explanation for the existence of mixed redox populations of Ref-1 in the cells. However, our interest is not only in the Ref-1 redox signature status in PDAC tumor cells, but also the impact of the loss of Cys65 in Ref-1 on the tumor cells.
It has been demonstrated that Ref-1 cannot form intramolecular disulfide bonds as the distance between cysteine residues is too great [5,22]. We can also rule out the possibilities of Ref-1 intermolecular disulfide bonds (e.g., Ref-1-Ref-1) situations at least in Pa03C cells as we did not observe any protein complexes above 150 kDa. These findings support the hypothesis that Ref-1 is highly reduced in PDAC cells, again supporting the increased activity of critical TFs, such as HIF-1α, NF-kB, STAT3, AP-1, through Ref-1 redox signaling for PDAC tumor cell growth and metastasis. Interestingly, we observed more oxidized Ref-1 in C65A expressing clones compared to Ref-1WT (Fig. 3B). To further explore this model, we tested the reactivity of Ref-1 redox status to acute oxidative stress in PDAC cells by exposing the cells to increasing concentrations of H2O2. We observed a clear dose-dependent oxidation in Ref-1 when cells were challenged with H2O2; however, such effects were more dramatic in Ref-1C65A cells, confirming that Cys65 is vital for maintaining reduced forms of Ref-1 in the cells, presumably by allowing for a protein folding configuration that is refractive to oxidation. Accordingly, Ref-1C65A redox impaired cells failed to fully activate multiple oncogenic TFs, such as NF-kB, HIF-1α as well as its downstream effector CA9 compared to Cas9 Ref-1WT redox competent cells. Similarly, we observed reduced mitochondrial function of the Ref-1C65A cells compared to Cas9 control cells. While we observed reduced TF activation and impact on mitochondrial function, the redox activation of the Ref-1 target TFs did not go away completely. This could be due to some activity provided by the presence of C93 and C99 in the Ref-1C65A protein. Previous studies have shown, albeit in cell free biochemistry assays or cellular overexpression, that a Ref-1-C93A/99A protein failed to biochemically reduce TFs and DNA binding, but to a lesser extent than a Ref-1C65A protein. It was suggested that Cys93 and Cys99 may also play supportive role in maintaining redox activity of Ref-1 and, as such, perhaps one of the reasons we still detect partial or impaired activation of TFs in the cells. An additional explanation on why the redox activation of the TFs is not reduced to zero in the Ref-1C65A cell lines could be due to the reported chaperone activity of Ref-1 [60] These previous findings demonstrate Ref-1 can facilitate the reduction of target TFs by glutathione or thioredoxin by chaperoning or carrying these reducing proteins directly to the TFs and they act to reduce the TF independent of Ref-1 redox function. This has been reported to occur at lower concentrations and independent of the redox and repair activities of Ref-1. Therefore, in cells consisting of only Ref-1C65A, the active redox function of Ref-1 would exist at such a low concentration, particularly if only relying on C93 and C99, that the chaperone activity becomes the primary redox route of Ref-1. This can be further explored using the Ref-1C65A clonal cell lines to further investigate this activity.
In vivo studies using pancreatic cancer cells that express Ref-1C65A were implanted, both subcutaneous or orthotopic, and demonstrated significant reduction in primary tumor size and metastatic lesions in the liver and lung in Ref-1C65A PDAC cells compared to the Cas9 control (WT) lines. This further supports our in vitro cellular data and the critical role of C65 in Ref-1 redox signaling and downstream pathways including proliferation and tumor metastasis or invasiveness. It also supports the translational and clinical application of blocking Ref-1 redox function with small molecule inhibitors which we are pursuing. In fact, APX3330, the first in class small molecule inhibitor of Ref-1 redox signaling completed Phase I trials (NCT03375086) with positive results and strong safety data [61,62] While the slowing of proliferation of the primary tumor is of great interest, the reduction of metastatic lesions in PDAC tumors expressing Ref-1C65A has great clinical implications. In pancreatic cancer, only about 20% of patients are eligible or have surgery to remove their primary tumor and metastatic lesions are involved in most of the patient suffering and death [63].
In summary, the data presented here support including inhibitors of Ref-1 redox signaling in neoadjuvant chemotherapy regimens could be useful in lowering the metastatic burden in PDAC patients. To our knowledge, the present study provides the first direct evidence of the nature of Ref-1 redox status and protein folding in tumor cells, that the chronic absence of Cys65 in Ref-1 results in redox inactivity of the protein in human PDAC cells, and subsequent biological results confirm a critical involvement of Ref-1 redox signaling, tumor growth and metastasis. It also suggests a role of Cys93 and Cys99 in the redox signaling activity of Ref-1 or a role of these cysteines in conjunction with the reported chaperone activity of Ref-1. CRISPR mutations for Cys93, Cys99 and combinations of Cys93/65 and Cys99/65 are being constructed and will be useful tools to compare to the Ref-1C65A cell lines presented here. Additionally, the chaperone role of Ref-1 can also be explored using these CRISRP Ref-1 Cys mutant cell lines.
Author contributions
MM performed experiments, data validation, formal analysis, writing and editing the manuscript; EK, NNC, SG and RW performed experiments and assay and data analysis. OB assisted in performing the experiments. SL, JW performed bioinformatic analysis, formal analysis, writing and editing; MMG contributed formal analysis, writing, and editing. MLF, MRK provided expertise, experimental design, analysis, funding, and writing/editing of manuscript. All authors contributed to the article and approved the submitted version.
Funding
MLF and MRK were supported by grants from the National Institute of Health and National Cancer Institute R01CA282478, R01CA167291, and R01CA254110 and NHLBI U01HL143403, R01CA264471, DOD W81XWH2210864, and U01CA274304 (MLF). Additional support was received from the Riley Children's Foundation (MLF and MRK), Pediatric Oncology Research Funding from the Tom Wood Lexus Foundation (MRK) and the IU Simon Comprehensive Cancer Center, P30CA082709 (MLF, MRK).
Declaration of competing interest
MRK is CSO and consultant of Apexian Pharmaceuticals LLC and a medical consultant with Ocuphire Pharma. Both companies have or are using clinical compounds licensed from MRK through Indiana University Research and Technology Corporation. Apexian Pharmaceuticals, and Ocuphire Pharma had no control or oversight of the studies, interpretation, writing or presentation of the data in this manuscript. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
The schematic figures were created using BioRender (BioRender.com).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2023.102977.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Mijit M., et al. APE1/Ref-1 - one target with multiple indications: emerging aspects and new directions. J Cell Signal. 2021;2(3):151–161. [PMC free article] [PubMed] [Google Scholar]
- 2.Malfatti M.C., et al. Coping with RNA damage with a focus on APE1, a BER enzyme at the crossroad between DNA damage repair and RNA processing/decay. DNA Repair. 2021;104 doi: 10.1016/j.dnarep.2021.103133. [DOI] [PubMed] [Google Scholar]
- 3.Caston R.A., et al. The multifunctional APE1 DNA repair-redox signaling protein as a drug target in human disease. Drug Discov. Today. 2021;26(1):218–228. doi: 10.1016/j.drudis.2020.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thakur S., et al. APE1/Ref-1 as an emerging therapeutic target for various human diseases: phytochemical modulation of its functions. Exp. Mol. Med. 2014;46(7):e106. doi: 10.1038/emm.2014.42. -e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luo M., et al. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2012;51(2):695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luo M., et al. Redox regulation of DNA repair: implications for human health and cancer therapeutic development. Antioxidants Redox Signal. 2010;12(11):1247–1269. doi: 10.1089/ars.2009.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georgiadis M.M., et al. Evolution of the redox function in mammalian apurinic/apyrimidinic endonuclease. Mutat. Res. 2008;643(1–2):54–63. doi: 10.1016/j.mrfmmm.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mijit M., et al. RelA is an essential target for enhancing cellular responses to the DNA repair/ref-1 redox signaling protein and restoring perturbated cellular redox homeostasis in mouse PDAC cells. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.826617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kelley M.R., Georgiadis M.M., Fishel M.L. APE1/Ref-1 role in redox signaling: translational applications of targeting the redox function of the DNA repair/redox protein APE1/ref-1. Curr. Mol. Pharmacol. 2012;5(1):36–53. doi: 10.2174/1874467211205010036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fishel M.L., et al. Anti-tumor activity and mechanistic characterization of APE1/Ref-1 inhibitors in bladder cancer. Mol. Cancer Therapeut. 2019;18(11):1947–1960. doi: 10.1158/1535-7163.MCT-18-1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D.S., Herman T., Demple B. Two distinct human DNA diesterases that hydrolyze 3'-blocking deoxyribose fragments from oxidized DNA. Nucleic Acids Res. 1991;19(21):5907–5914. doi: 10.1093/nar/19.21.5907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wisniewski J.R., et al. A "proteomic ruler" for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics. 2014;13(12):3497–3506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McIlwain D.W., et al. APE1/Ref-1 redox-specific inhibition decreases survivin protein levels and induces cell cycle arrest in prostate cancer cells. Oncotarget. 2018;9(13):10962–10977. doi: 10.18632/oncotarget.23493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi Y.-D., et al. APE1 promotes pancreatic cancer proliferation through GFRα1/Src/ERK axis-cascade signaling in response to GDNF. Int. J. Mol. Sci. 2020;21(10):3586. doi: 10.3390/ijms21103586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song H., et al. APE1 and SSRP1 is overexpressed in muscle invasive bladder cancer and associated with poor survival. Heliyon. 2021;7(4) doi: 10.1016/j.heliyon.2021.e06756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gampala S., et al. Ref-1 redox activity alters cancer cell metabolism in pancreatic cancer: exploiting this novel finding as a potential target. J. Exp. Clin. Cancer Res. 2021;40(1):251. doi: 10.1186/s13046-021-02046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah F., et al. APE1/Ref-1 knockdown in pancreatic ductal adenocarcinoma - characterizing gene expression changes and identifying novel pathways using single-cell RNA sequencing. Mol. Oncol. 2017;11(12):1711–1732. doi: 10.1002/1878-0261.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shah F., et al. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic. npj Precis. Oncol. 2017;1 doi: 10.1038/s41698-017-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ordway J.M., Eberhart D., Curran T. Cysteine 64 of ref-1 is not essential for redox regulation of AP-1 DNA binding. Mol. Cell Biol. 2003;23(12):4257–4266. doi: 10.1128/MCB.23.12.4257-4266.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue Z., Demple B. Knockout and inhibition of Ape1: roles of Ape1 in base excision DNA repair and modulation of gene expression. Antioxidants. 2022;11(9) doi: 10.3390/antiox11091817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah F., et al. Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic. npj Precis. Oncol. 2017;1:19. doi: 10.1038/s41698-017-0023-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Su D., et al. Interactions of apurinic/apyrimidinic endonuclease with a redox inhibitor: evidence for an alternate conformation of the enzyme. Biochemistry. 2011;50(1):82–92. doi: 10.1021/bi101248s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J., et al. Inhibition of apurinic/apyrimidinic endonuclease I's redox activity revisited. Biochemistry. 2013;52(17):2955–2966. doi: 10.1021/bi400179m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pramanik S., et al. The human AP-endonuclease 1 (APE1) is a DNA G-quadruplex structure binding protein and regulates KRAS expression in pancreatic ductal adenocarcinoma cells. Nucleic Acids Res. 2022;50(6):3394–3412. doi: 10.1093/nar/gkac172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ando K., et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-{kappa}B and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36(13):4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mijit M., et al. Activation of the integrated stress response (ISR) pathways in response to Ref-1 inhibition in human pancreatic cancer and its tumor microenvironment. Front. Med. 2023;10 doi: 10.3389/fmed.2023.1146115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones S., et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santini S.J., et al. SIRT1-Dependent upregulation of antiglycative defense in HUVECs is essential for resveratrol protection against high glucose stress. Antioxidants. 2019;8(9) doi: 10.3390/antiox8090346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guzman C., et al. ColonyArea: an ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS One. 2014;9(3) doi: 10.1371/journal.pone.0092444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Seo Y.R., et al. Implication of p53 in base excision DNA repair: in vivo evidence. Oncogene. 2002;21(5):731–737. doi: 10.1038/sj.onc.1205129. [DOI] [PubMed] [Google Scholar]
- 31.Bapat A., et al. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J. Pharmacol. Exp. Therapeut. 2010;334(3):988–998. doi: 10.1124/jpet.110.169128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Trilles R., et al. Discovery of macrocyclic inhibitors of apurinic/apyrimidinic endonuclease 1. J. Med. Chem. 2019;62(4):1971–1988. doi: 10.1021/acs.jmedchem.8b01529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mijit M., et al. Identification of novel pathways regulated by APE1/ref-1 in human retinal endothelial cells. Int. J. Mol. Sci. 2023;24(2):1101. doi: 10.3390/ijms24021101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Suzuki Y.J., et al. Protein redox state monitoring studies of thiol reactivity. Antioxidants. 2019;8(5) doi: 10.3390/antiox8050143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ding Q., et al. Angiotensin-converting enzyme 2 (ACE2) is upregulated in Alzheimer's disease brain. bioRxiv. 2020 doi: 10.3390/ijms22041687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Logsdon D.P., et al. Regulation of HIF1alpha under hypoxia by APE1/ref-1 impacts CA9 expression: dual targeting in patient-derived 3D pancreatic cancer models. Mol. Cancer Therapeut. 2016;15(11):2722–2732. doi: 10.1158/1535-7163.MCT-16-0253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jackson L.R., et al. Use of multimodality imaging, histology, and treatment feasibility to characterize a transgenic Rag2-null rat model of glioblastoma. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.939260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dobin A., et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30(7):923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 40.Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McCarthy D.J., Chen Y., Smyth G.K. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40(10):4288–4297. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Subramanian A., et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lirussi L., et al. Nucleolar accumulation of APE1 depends on charged lysine residues that undergo acetylation upon genotoxic stress and modulate its BER activity in cells. Mol. Biol. Cell. 2012;23(20):4079–4096. doi: 10.1091/mbc.E12-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gorman M.A., et al. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo M., et al. Characterization of the redox activity and disulfide bond formation in apurinic/apyrimidinic endonuclease. Biochemistry. 2011;51(2):695–705. doi: 10.1021/bi201034z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim Y.J., et al. S-glutathionylation of cysteine 99 in the APE1 protein impairs abasic endonuclease activity. J. Mol. Biol. 2011;414(3):313–326. doi: 10.1016/j.jmb.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qu J., et al. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35(8):2522–2532. doi: 10.1093/nar/gkl1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rudyk O., Eaton P. Biochemical methods for monitoring protein thiol redox states in biological systems. Redox Biol. 2014;2:803–813. doi: 10.1016/j.redox.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Geismann C., Arlt A. Coming in the air: hypoxia meets epigenetics in pancreatic cancer. Cells. 2020;9(11) doi: 10.3390/cells9112353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naqvi A.A.T., Hasan G.M., Hassan M.I. Investigating the role of transcription factors of pancreas development in pancreatic cancer. Pancreatology. 2018;18(2):184–190. doi: 10.1016/j.pan.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 51.Logsdon D.P., et al. Blocking HIF signaling via novel inhibitors of CA9 and APE1/Ref-1 dramatically affects pancreatic cancer cell survival. Sci. Rep. 2018;8(1):13759–13773. doi: 10.1038/s41598-018-32034-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gampala S., et al. Ref-1 redox activity alters cancer cell metabolism in pancreatic cancer: exploiting this novel finding as a potential target. J. Exp. Clin. Cancer Res. 2021;40(1):251. doi: 10.1186/s13046-021-02046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim J.Y., et al. The value of phosphohistone H3 as a proliferation marker for evaluating invasive breast cancers: a comparative study with Ki67. Oncotarget. 2017;8(39):65064–65076. doi: 10.18632/oncotarget.17775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shouksmith A.E., et al. Identification and characterization of AES-135, a hydroxamic acid-based HDAC inhibitor that prolongs survival in an orthotopic mouse model of pancreatic cancer. J. Med. Chem. 2019;62(5):2651–2665. doi: 10.1021/acs.jmedchem.8b01957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shannon H.E., et al. Longitudinal bioluminescence imaging of primary versus abdominal metastatic tumor growth in orthotopic pancreatic tumor models in NSG mice. Pancreas. 2015;44(1):64–75. doi: 10.1097/MPA.0000000000000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fishel M.L., et al. Apurinic/Apyrimidinic Endonuclease/Redox Factor-1 (APE1/Ref-1) redox function negatively regulates NRF2. J. Biol. Chem. 2014;290(5):3057–3068. doi: 10.1074/jbc.M114.621995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gampala S., et al. DNA Damage, DNA Repair and Disease. 2020. Basic, translational and clinical relevance of the DNA repair and redox signaling protein APE1 in human diseases; pp. 286–318. [Google Scholar]
- 58.Luo M., et al. Role of the multifunctional DNA repair and redox signaling protein Ape1/Ref-1 in cancer and endothelial cells: small-molecule inhibition of the redox function of Ape1. Antioxidants Redox Signal. 2008;10(11):1853–1867. doi: 10.1089/ars.2008.2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nassour H., et al. Peroxiredoxin 1 interacts with and blocks the redox factor APE1 from activating interleukin-8 expression. Sci. Rep. 2016;6 doi: 10.1038/srep29389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ando K., et al. A new APE1/Ref-1-dependent pathway leading to reduction of NF-κB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36(13):4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shahda S., et al. A phase I study of the APE1 protein inhibitor APX3330 in patients with advanced solid tumors. J. Clin. Oncol. 2019;37(15_suppl):3097. -3097. [Google Scholar]
- 62.Chu L., et al. CTC enumeration and characterization as a pharmacodynamic marker in the phase I clinical study of APX3330, an APE1/Ref-1 inhibitor, in patients with advanced solid tumors. J. Clin. Oncol. 2019;37(15_suppl) -e14531. [Google Scholar]
- 63.Oba A., et al. Neoadjuvant treatment in pancreatic cancer. Front. Oncol. 2020;10:245. doi: 10.3389/fonc.2020.00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.