

Abstract
Prostate cancer (PCa) is the second most common cancer and constitutes about 14.7% of total cancer cases. PCa is highly prevalent and more aggressive in African-American (AA) men than in European-American (EA) men. PCa tends to be highly heterogeneous, and its complex biology is not fully understood. We use metabolomics to better understand the mechanisms behind PCa progression and disparities in its clinical outcome. Adenosine deaminase (ADA) is a key enzyme in the purine metabolic pathway; it was found to be upregulated in PCa and is associated with higher-grade PCa and poor disease-free survival. The inosine-to-adenosine ratio, which is a surrogate for ADA activity was high in PCa patient urine and higher in AA PCa compared to EA PCa. To understand the significance of high ADA in PCa, we established ADA overexpression models and performed various in vitro and in vivo studies. Our studies have revealed that an acute increase in ADA expression during later stages of tumor development enhances in vivo growth in multiple pre-clinical models. Further analysis revealed that mTOR signaling activation could be associated with this tumor growth. Chronic ADA overexpression shows alterations in the cells’ adhesion machinery and a decrease in cells’ ability to adhere to the extracellular matrix in vitro. Losing cell-matrix interaction is critical for metastatic dissemination which suggests that ADA could potentially be involved in promoting metastasis. This is supported by the association of higher ADA expression with higher-grade tumors and poor patient survival. Overall, our findings suggest that increased ADA expression may promote PCa progression, specifically tumor growth and metastatic dissemination.
Keywords: Prostate cancer, tumor metabolism, adenosine deaminase, cell adhesion, tumor progression
Introduction
Prostate cancer (PCa) is the most commonly diagnosed cancer in American men. One in eight men are diagnosed with PCa in their lifetime; ~300,000 men are estimated to have PCa in 2023; and about 3.1 million men are living with PCa in the United States today [1]. Prostate tumors have a complex biology and both aggressive and non-aggressive tumors have highly differential features. PCa tumor heterogeneity is not fully understood, and significant knowledge gaps exist regarding tumor characteristics and progression.
Our group is working to identify the metabolic pathways and associated molecular mechanisms that drive PCa progression. Our goal is to identify the metabolic alterations associated with PCa and to understand their functions in the tumor setting. Previously, we studied PCa patients’ metabolic landscapes and reported alterations within the methionine-homocysteine pathway in African American (AA) PCa, which tends to be clinically aggressive compared to European American (EA) PCa [2-5]. Methionine is an essential amino acid; its metabolism leads to homocysteine/cysteine and adenosine/inosine production. In our current study, we examine the enzyme adenosine deaminase (ADA, EC 3.5.4.4.) which regulates the adenosine-inosine axis.
ADA catalyzes adenosine’s irreversible conversion into inosine. Biologically, it is well known that ADA’s generation of inosine counteracts adenosine’s immunosuppressive effects. ADA is well-studied in the context of the immune system and in immunodeficiencies like Severe Combined Immunodeficiency (SCID). In immunodeficient diseases, ADA levels are lower, causing adenosine to build up, which in turn leads to severe DNA damage and lymphocytotoxicity [6]. In certain types of cancers, however, ADA is elevated and associated with poor survival [7]. ADA inhibition was shown to prevent breast cancer progression in mice and induce cytotoxicity in cervical cancer and malignant pleural mesothelioma cells [8-10]. However, ADA’s biological effects in any solid tumor have not been fully characterized. In our study, we sought to elucidate ADA’s effects on tumor progression in PCa. Our goal is to delineate ADA’s intrinsic tumor effects in relation to PCa development and progression. As a result, we have assessed ADA’s clinical levels in PCa and also extensively studied the effects of ADA in PCa progression using in vitro experimental models, and in vivo immunocompromised murine models. We found that ADA is elevated in PCa and is associated with higher Gleason scores and poor disease-free survival. Our studies further revealed that ADA elevation promotes tumor-advancing features like increased tumor growth and decreased cell adhesion.
Methods
Clinical samples
All clinical samples used in this study were obtained using informed consent and approval of the Institutional Review Board at Baylor College of Medicine (Protocol title: Integrative metabolomics of cancer progression; Protocol number: H-28445; Expiration date: 09/20/2026) and collaborating institutions, including the National Cancer Institute (Study PI: Dr. Stefan Ambs). The urine samples were kindly provided by Dr. Stefan Ambs (National Cancer Institute). Tissue microarrays for the RNA in situ hybridization study were obtained from Henry Ford Health. Tissue microarrays for ADA immunostaining were obtained from the Baylor College of Medicine Human Tissue Acquisition and Pathology Core. The urine samples were obtained from the NCI-Maryland Prostate Cancer case-control study. The clinical characteristics of the urine samples are described in Table 1.
Table 1.
Summary of clinical and ancestry data for PCa case-control urine cohort
| Variable | African American | European American | ||
|---|---|---|---|---|
|
|
|
|||
| Control (n=49) | PCa Cases (n=80) | Control (n=50) | PCa Cases (n=80) | |
| Age (years), ŷ | 64.6±7.5 | 61.9±8.2 | 69.3±9.4 | 62.9±8.6 |
| BMI, ŷ | 30.5±6.2 | 27.7±4.5 | 27.9±4.9 | 27.9±3.9 |
| Gleason score (n) | - | Low ≤6:40 | - | Low ≤6:40 |
| - | High ≥7:40 | - | High ≥7:40 | |
| Cases with biochemical recurrence (n) | - | 9 | - | 7 |
| PSA (ng/mL), ŷ | 7.7±5.4 | 8.5±8.8* | 8.1±6.7 | 8.8±8.1 |
| West African ancestry (ratio), ŷ | 0.7±0.1 | 0.8±0.1 | 0.1±0.0 | 0.1±0.1 |
| European ancestry (ratio), ŷ | 0.3±0.0 | 0.2±0.1 | 0.9±0.0 | 0.8±0.0 |
| Native American ancestry (ratio), ŷ | 0±0.1 | 0.1±0.1 | 0±0.0 | 0.1±0.1 |
| Genetic Ancestry tested (n) | 40 | 78 | 40 | 54 |
11 outliers removed.
Fluorescent RNA in situ hybridization
Slides were incubated at 60°C for 1 hour and deparaffinized by immersing them in xylene twice for 5 minutes each with periodic agitation. The slides were then immersed in 100% ethanol twice for 3 minutes each with periodic agitation and dried at 60°C for 5 minutes. Tissues were treated with 5-8 drops of H2O2 for 10 minutes at room temperature. Slides were washed twice in distilled water and then boiled in 1X Target Retrieval for 15 minutes. Slides were washed twice in distilled water, immersed in 100% EtOH for 3 minutes, and then dried for 5 minutes at 60°C. Tissues were marked using a hydrophobic barrier pen (cat# H-4000 Vector Labs, Burlingame, CA), allowed to dry, and then treated with Protease Plus for 30 minutes at 40°C in a HybEZ Oven (cat# 310010, Advanced Cell Diagnostics, Hayward, CA). Slides were washed twice in distilled water and then treated with the ADA probe (cat# 490141, Advanced Cell Diagnostics, Hayward, CA) for 2 hours at 40°C in the HybEZ Oven. Slides were then washed in 1X Wash Buffer (cat# 310091, Advanced Cell Diagnostics, Hayward, CA) twice for 2 minutes each. All slides were then treated with Amp 1 for 30 minutes, Amp 2 for 30 minutes, and Amp 3 for 15 minutes, all at 40°C in the HybEZ oven with two washes in 1X Wash Buffer for 2 minutes each after each step. The slides were treated with HRP-C1 for 15 minutes, Opal 520 (cat# FP487001KT, Akoya Biosciences) diluted 1:1500 in TSA Buffer for 30 minutes, HRP Blocker for 15 minutes, and HRP-C3 for 15 minutes all at 40°C in the HybEZ oven with two washes in 1X Wash Buffer for 2 minutes each after each step. DAPI was added to all slides for 30 seconds at room temperature and rinsed. We added 1-2 drops of ProLong Gold Antifade Mountant (cat# P36930, Molecular Probes, Eugene, OR) and covered the slides with coverslips. H2O2, Target Retrieval, Protease Plus, Wash Buffer, Amps 1-3, HRP-C1-3, TSA Buffer, HRP Blocker, and DAPI are all part of the RNAscope Multiplex Fluorescent Reagent Kit v2 (cat# 323100, Advanced Cell Diagnostics, Hayward, CA).
Immunohistochemistry
Slides were deparaffinized via three xylene washes and rehydrated with a graduated alcohol series. Antigen retrieval was performed at 60°C in citrate-based antigen unmasking solution diluted 1:100 (Vector Labs, Burlingame, CA) for 20 minutes. Slides were washed with 1X Phosphate Buffered Saline-Tween (PBS-T) and treated with BLOXALL endogenous blocking solution for 10 minutes and blocked with 2.5% Normal horse serum (Vector Labs, Burlingame, CA) for 20 minutes. Primary antibody incubation was performed overnight at 4°C with 0.5 mg/mL Mouse ADA antibody (MBS1752027, MyBioSource, San Diego, CA). The secondary antibody and the substrate for the detection were added from the Vectastain Universal Elite ABC Immunohistochemistry Kit (Cat# PK-8200, Vector Labs, Burlingame, CA). Slides were incubated in secondary antibody for 20 minutes and washed with PBS-T. The substrate was added after the wash. Slides were briefly rinsed in water before counterstaining with Gill’s No. 2 Hematoxylin (Cat# 10143746, VWR, Radnor, PA) for 1 min. Slides were rinsed in water and incubated with Scott’s Bluing Reagent (Cat# RC669732, VWR, Radnor, PA) for 1 min. Slides were washed and dehydrated in a graduated alcohol series followed by xylene dips before mounting. We used control slides to verify the ADA antibody’s specificity. The control slides were generated from xenografts formed in mice using control and ADA-overexpressing prostate cell lines. Colon tissues were also used as an additional positive control and the antibody was also verified using a western blot. The staining was scored by an experienced genitourinary pathologist in the core lab, who assigned each section an intensity and extent score. These two values were multiplied (intensity x extent) to produce a final score for each core sample.
Cell lines
The MDA-PCa-2a cell line was a gift from Dr. Nora Navone (MD Anderson Cancer Center). LNCaP was obtained from the Tissue Culture Core at Baylor College of Medicine. The 22Rv1 cell line was obtained from ATCC (CRL-2505). C4-2B was a gift from Dr. Nancy Weigel (Baylor College of Medicine). Prostate stromal cell line HPS-19I was a gift from Dr. David Rowley (Baylor College of Medicine). Short Tandem Repeat (STR) analysis was completed for all cell lines to verify their authenticity at the MD Anderson Cytogenetics and Cell Authentication Core. Mycoplasma tests (MycoAlertTM from Lonza, Anaheim, CA) were conducted routinely to ensure that the cells remained free of mycoplasma contamination.
Cell culture
MDA-PCa-2a cells were cultured in BRFF-HPC1 media (AthenaES, Baltimore, MD) with 20% fetal bovine serum (FBS, Hyclone Labs, Thermo Scientific, Rockford, IL) and 0.1% penicillin-streptomycin (Hyclone Labs, Thermo Scientific, Rockford, IL). MDA-PCa-2a cells were grown in plates/flasks coated with a fibronectin coating mix (AthenaES, Baltimore, MD). LNCaP, 22Rv1, and C4-2B were grown in RPMI-1640 media (Invitrogen Corp., Carlsbad, CA) supplemented with 20% fetal bovine serum and 0.1% penicillin-streptomycin. The cells were maintained at 37°C, 5% CO2, and 95% humidity. The cells were passaged or harvested when they reached about 80% confluency and treated with 0.25% Trypsin-EDTA (Gibco, Life Technologies, Grand Island, NY) for about 5-10 minutes. Once all the cells were off the substratum, culture media was added for neutralization. The cells were then counted using Trypan blue stain (Life Technologies, Eugene, OR) and CountessTM Automated Cell Counter (Life Technologies, Singapore). The cells were seeded at appropriate densities according to the culture dishes used or the experimental requirements.
Lentiviral transduction
To generate a stable ADA overexpressing system, the cells were transduced with lentiviral cDNA open reading frames (Precision LentiORFs, Horizon Discovery, Cambridge, UK). The precision lentiORF for ADA (Catalog #OHS5836-EG100, Clone: OHS5899-202616178 (PLOHS_100005311)) and a non-target ORF (Catalog #OHS5833) was used as a control. A multiplicity of infection (MOI) of 5 was used for lentiviral transduction. About 8 µg/mL of blasticidin (Gibco, Life Technologies, Grand Island, NY) was used for selection. The cells were also checked for the presence of GFP (ADA overexpression) and RFP (control) under the fluorescence microscope to verify transduction efficiency. To create a knockdown rescue in the ADA OE cells, we performed transduction with shRNA lentivectors (Gentarget Inc., San Diego, CA). shRNA targeting ADA and non-target shRNA (vector control) were used (catalog #LVS-1002). An MOI of 5 was used for lentiviral transduction, and 1 µg/mL puromycin was used for the selection of effectively transduced cells. The inducible ADA overexpression lines were developed at the Cell-Based Assay Screening Service (CBASS) at Baylor College of Medicine. The lines were generated by transducing tet-inducible lentiviral vector for ORF expression (Addgene, pInducer 20, catalog #44012). The ADA gene was cloned into the overexpression cell line and the 11th beta-strand of GFP was used as a doxycycline control (Dox-control).
Quantitative real-time polymerase chain reaction
RNA extraction from cells was performed using the Aurum total RNA Mini Kit from Biorad. For tissues, RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, Germany). Extracted RNA was quantified and its purity (absorbance at 260/280 nm) was verified using a spectrophotometric plate reader (Synergy HTX multi-mode reader, Biotek Instruments, Winooski, VT). RNA was reverse transcribed into cDNA using the cDNA Superscript Mix (Quanta Biosciences, cat# 95048-500) and RT-qPCR was conducted using SYBR green (Life Technologies, cat# 4385614). 18S or RPL30 were used as appropriate housekeeping controls. Primers used in this study are listed in Table 2.
Table 2.
Primers used in this study
| Primer | Sequence |
|---|---|
| ADA | Forward 5’-GCC TTC GAC AAG CCC AAA GTA-3’ |
| Reverse 5’-CTC TGC TGT GTT AGC TGG GAG-3’ | |
| RPL30 | Forward 5’-GAA GAC GAA AAA GTC GCT GGA-3’ |
| Reverse 5’-TGG GCA GTT GTT AGC GAG AA-3’ |
Protein extraction and estimation
Cells were washed in ice-cold PBS three times to remove residual culture media. RIPA buffer was added in appropriate amounts to the cells and incubated for 10 minutes in ice. The cells were then sonicated and spun down. The supernatant (cell lysate) was collected, and the total protein was quantified using PierceTM BCA Protein Assay Kit (Thermo Fisher, Waltham, MA).
Western blot
Cell lysates with 30 µg protein were mixed with 2× Laemmli sample buffer (Biorad, catalog #1610737) in a 1:1 ratio and heated at 95°C for 10 minutes. Then the prepared samples were loaded onto a 4-15% Mini-PROTEAN Tris PAGE gel’s lanes (Bio-rad #4561086). Electrophoresis was carried out at 100 V for about 1.5-2 hours. The separated proteins were transferred from the gel to the polyvinyl difluoride (PVDF) membrane (Immobilon-P, 0.45 µm, Merck Millipore Ltd., Ireland) at 200 mA for 1.5 hours. The transferred blot was blocked using 5% skimmed milk and incubated with the ADA antibody (MyBioSource, #MBS1752027) overnight. The blot was washed with TBST for 10 minutes three times and then incubated with the appropriate secondary antibody for 1 hour. The blot was again washed with TBST for 10 minutes three times. Finally, the blots were probed using a chemiluminescent substrate (Thermo Fisher, cat# 34579) in the Chemi-Doc Imaging system (Biorad, Hercules, CA). Polyclonal rabbit α/β-tubulin antibody (CST, #2148S) was used as an internal control.
ADA enzyme assay
ADA assay kit (DZ117A; Diazyme Laboratories, Inc., Poway, CA) was used to determine ADA activity in cell lysates. About 4 ug of protein was used for the assay. The manufacturer’s protocol was followed to assess the ADA enzyme activity.
LC/MS
Adenosine and inosine levels in urine were measured by LC/MS. Adenosine (CNLM-3806-CA-PK, Cambridge Isotope Laboratories, Inc., Andover, MA) and inosine (NLM-4264-0.01, Cambridge Isotope Laboratories, Inc., Andover, MA) standards for concentrations 5-100 ng/mL were prepared in a urine matrix (MSG5000, Golden West Diagnostics, Temecula, CA). 10 µL of urine was mixed with 90 µL of methanol:water (1:1) and internal standards (Labeled tryptophan (CNLM-2475-H-PK, Cambridge Isotope Laboratories, Inc., Andover, MA), zeatine and inosine) were spiked into the samples. Samples were then loaded into LC/MS sampler vials and injected into the mass spectrometer.
To evaluate the adenosine and inosine levels in cell lines, the cells were harvested and washed three times to remove any residual media. About 100 µl water and 5 µl internal standard (labeled adenosine and inosine) were added to the cells. The internal standards were dissolved in a 1:1 methanol-water mix by vortexing for 5 min. The cells were homogenized by probe-sonication for 30 seconds in ice. We added 50 µl DTT (500 mM-freshly prepared) and vortexed the samples for 20 seconds. The samples were then incubated at 65°C for 30 min at 300 rpm. After incubation, the samples are kept on ice for a minute. 400 µl cold methanol was added and mixed well by vortexing for 5 min. This mixture was centrifuged, and the supernatant was collected and dried. After drying, 50 µl of a 1:1 methanol-water was added. To ensure proper dissolution, samples were vortexed for 5 min and sonicated for 5 min. The samples were then centrifuged for 5 min. Finally, samples were transferred into the inserts of LC/MS vials and injected into the mass spectrometer. To separate all the metabolites in the study, we used the Agilent Zorbax SB-CN (3×100 mm; 1.8 μm) column and mobile phase, A: 0.1% formic acid in HPLC grade water, B: 0.1% formic acid in HPLC grade acetonitrile. Acetonitrile, methanol, and water for high-performance liquid chromatography (HPLC) were purchased from Burdick & Jackson (Morristown, NJ). Relative levels of adenosine and inosine were measured using the multiple reaction monitoring (MRM) transitions 268.11→136.1 and 269.1→137.2.
Adhesion assay
The cell-ECM/surface adhesion was measured using the Agilent xCELLigence Real-Time Cell Analysis (RTCA) DP (dual purpose) instrument. About 10,000 cells were seeded onto Cell invasion and migration (CIM)-Plates (G082975-Agilent, San Diego, CA). Adhesion was automatically measured by the instrument at regular time intervals for 100 hours.
In vivo experiments
Male NOD-SCID-Gamma (NSG) mice were obtained from Baylor College of Medicine, and male athymic nude mice (Strain #553) were purchased from Charles River (Frederick, MD) for these studies. The experiments started when the mice were between 4-6 weeks old. Nude mice were used to grow LNCaP tumors. All other cell line xenograft studies were conducted in NSG mice. These mice were housed in germ-free cages with a maximum of 4 mice per cage in the immunodeficient mice facility. Mice were randomized into experimental groups based on the cage. The sample size for in vivo studies was determined based on statistical power calculations and past experiments from our laboratory. All in vivo experiments were started with 10 mice per group. Animals lost to attrition were excluded from the study. The data from the remaining mice were used for evaluation. The studies were not blinded, and outliers were not excluded. The experiments were carried out and the health of the mice was monitored per the animal protocol approved by the Institutional Animal Care and Use Committee (IACUC) of Baylor College of Medicine (Protocol title: Integrative metabolomics of cancer detection and progression; Protocol number: AN-5676; Expiration date: 02/13/2026).
In vivo tumor engraftment and growth studies
To evaluate tumor engraftment and growth, cells were injected subcutaneously. Five million cells were used for MDA-PCa-2a and C4-2B and 3 million cells for 22Rv1. For LNCaP tumors, we added 500,000 19I stromal cells and 5 million LNCaP cells. All the cells were mixed with 50 µL Matrigel and 50 µL culture media. 100 µL of the cells-matrigel mixture was injected into each mouse after they were anesthetized with 2% vaporized isoflurane.
We conducted two tumor growth studies: 1) ADA was overexpressed constitutively; and 2) ADA levels were elevated via doxycycline induction after tumor engraftment. In the first study, we used the constitutive ADA OE lines from in vitro studies (MDA-PCa-2a and LNCaP) and an ADA-inducible cell line (22Rv1) that was kept under constant induction. The 22Rv1 ADA OE cell line used for this experiment is an inducible cell line that was placed under constant ADA induction while in culture, during injection, and during tumor growth via continuous doxycycline exposure. The corresponding control that we used is a doxycycline control, which, like the ADA OE line, was also maintained under constant doxycycline exposure. This eliminated any doxycycline-mediated consequences.
For the second study, to induce ADA post-tumor engraftment, we added doxycycline to the drinking water at the concentration of 0.5 mg/mL after tumors formed (~50 mm3). The water was changed twice a week and we put up a special care instruction form to indicate deviation from regular drinking water. Two controls were used in this study, an uninduced control without doxycycline exposure and a doxycycline control (dox-control, where GFP is induced instead of ADA).
The tumor dimensions were measured using a vernier caliper once every two days. The volume was calculated using the formula ½ (length × breadth × width). The tumors were resected when they reached 500 mm3. The resected tumors were cut into two halves: one half was flash-frozen in liquid nitrogen and the other was fixed using 10% formalin and later paraffin-embedded (FFPE). The frozen and FFPE tissues were used for further molecular analyses.
Reverse phase protein array (RPPA)
The RPPA core at Baylor College of Medicine conducted the RPPA analysis. Cell lysates were arranged on nitrocellulose-coated slides (Grace Bio-labs, Bend, OR) using Aushon 2470 Arrayer (Aushon BioSystems, Billerica, MA) and printed in triplicates (technical replicate). Three biological replicates were used for the study and each replicate was printed in triplicate. Immunolabeling was done on an automated slide stainer Autolink 48 (Dako, Carpinteria, CA) according to the manufacturer’s protocol (Autostainer catalyzed signal amplification (CSA) kit, Dako, Carpinteria, CA). Each slide was incubated with a single primary antibody at room temperature for 30 minutes followed by a goat anti-rabbit or mouse IgG secondary antibody. For negative control, a slide was incubated with antibody diluent instead of primary antibody. The Catalyzed Signal Amplification System kit (Dako Cytomation, Carpinteria, CA, USA) and fluorescent IRDye 680 Streptavidin (LI-COR, Lincoln, Nebraska, USA) were used for detection. The total protein for each printed spot was evaluated by staining one slide for every 20 slides with Sypro Ruby Blot Stain (Molecular Probes, Eugene, OR) according to the manufacturer’s directions. Slides were scanned using a GenePix AL4200 scanner (at 635 nm wavelength for antibody slides or 535 nm wavelength Sypro Ruby Blot-Stained slides), and the images were analyzed by GenePix Pro 7.2 software (Molecular Devices, Sunnyvale, CA). The fluorescence signal for each spot was estimated based on the fluorescence intensity after subtracting the corresponding slide background signal.
We used the following method to normalize the RPPA data. Each spot on the array had a background-subtracted foreground signal intensity (SI). If the background intensity was higher than the foreground intensity, the spot’s SI was set to a very small intensity value (1). To normalize a sample/spot’s SI for a specific antibody, we subtracted the negative control’s corresponding SI from each spot’santibosy SI and then normalized to the corresponding SI of total protein within the same group. The normalized antibody SI was calculated using this formula: N=(A - C)*M/T.
Where N is the normalized antibody SI, A is the antibody SI, C is the negative control SI, M is the median SI of the spots from the same group, and T is the SI of total protein.
The differentially expressed proteins were detected by t-test at nominal P<0.05 and FDR<0.25. We used the Benjamini & Hochberg Procedure (BH method) to correct the FDR in MDA-PCa-2a and LNCaP data. Using the differential proteins, hypergeometric enrichment analysis was conducted to identify key pathways and gene sets enriched in Hallmark and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway collections and Molecular Signatures Database (MSigDB) gene sets. A nominal P<0.01 and an FDR<0.05 were used as thresholds for determining the significance.
RNA sequencing
RNA was extracted using the RNeasy mini kit (Qiagen, Hilden, Germany). Extracted RNA was quantified and its purity (absorbance at 260/280 nm) was verified using a spectrophotometric plate reader (Synergy HTX multi-mode reader, Biotek Instruments, Winooski, VT). Library preparation and sequencing were done at the University of Michigan Advanced Genomics Core. Library preparation and selection of poly-adenylated transcripts were done using NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, E7490) and xGen Broad-range RNA Library Prep (IDT, 1009813) with xGen Normalase UDI Primers (IDT, various). This underwent 151 bp paired-end sequencing according to the manufacturer’s protocol (Illumina NovaSeq). De-multiplexed Fastq files were generated using BCL Convert Conversion Software v4.0 (Illumina). Snakemake [11] was used to manage the bioinformatics workflow and ensure reproducibility.
Quality Control (QC) and sequence alignment: The reads were trimmed using Cutadapt v2.3 [12]. The reads were evaluated with FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) (v0.11.8) to assess the quality of the data. Reads were mapped to the reference genome GRCh38 (ENSEMBL 109) using STAR v2.7.8a and count estimates were assigned to the genes with RSEM v1.3.3 [13]. ENCODE standards were followed for alignment options in RNA-seq [14]. QC metrics from several different steps in the pipeline were combined by multiQC v1.7 [15]. After filtering low-count reads, 15328 protein-coding genes were used for differential expression analysis. The differential expression was analyzed with DESeq2 [16].
To characterize biologically significant changes in molecular signaling pathways among ADA OE and control tumors, we employed GSEA [17] to identify significantly enriched concepts in both 22RV1 and C4-2B data. In GSEA, the cumulative distribution function was constructed by performing 1000 random gene set assignments (permutations) (GSEA pre-ranked method). A nominal P<0.05 and a FDR<0.25 were used to threshold the concepts. Here we focused on well-defined, large-scale biological processes, termed the Hallmark (H) and KEGG (C2, Kyoto Encyclopedia of Genes and Genomes) pathways and Molecular Signatures Database (MSigDB) gene sets. Initial Principal component analysis (PCA) indicated the two outlier samples in C4-2B and three outlier samples in 22RV1 data. After further quantifying anomalies with robust PCA methods (rPCA), those samples were removed from further analysis. RNA sequencing data has been uploaded to GEO and the accession number is GSE239575.
Statistical analyses
To compare ADA across Gleason scores, we used ANOVA and Tukey’s multiple comparisons test. The log-rank test was used to compute the p-value for survival analysis. We used the Wilcox matched pairs signed rank test to compare ADA levels detected in the benign vs cancer TMAs. For the inosine to adenosine ratio in benign vs cancer and EA vs AA, p values were computed using Student’s unpaired two-tailed t-tests. The log-rank test was also used to evaluate the significance of tumor engraftment in in vivo studies. Significance was determined using Student’s unpaired two-tailed t-test for all in vitro and in vivo studies.
Results
High ADA is associated with aggressive prostate cancer
Our studies showed ADA upregulation at both transcript and protein levels in PCa. High ADA levels are found to be associated with higher Gleason scores and poorer survival. The inosine-to-adenosine ratio, which is a metabolic readout of ADA activity, was also found elevated in PCa patients. The inosine-to-adenosine ratio was found to be higher in urine of AA PCa patients compared to EA PCa patients.
ADA levels were analyzed across different clinical samples to determine expression patterns in benign and cancerous prostate tissues. The RNA in situ hybridization that was done across five Tissue microarrays (TMA) revealed that ADA was high in prostate tumors and that levels were higher in high-grade (7+) tumors (N=345; benign (44), 3+3 (77), 3+4 (115), 4+3 (50), 8 and above (30)) (ANOVA p-value <0.00001 and Tukey’s multiple comparisons p-value: Benign Vs 3+3: 0.9, Benign Vs 3+4: <0.0001, Benign Vs 4+3: 0.0003, Benign Vs 8+: <0.0001, 3+3 Vs 3+4: 0.0002, 3+3 Vs 4+3: 0.0029, 3+3 Vs 8+: <0.0001, 3+4 Vs 4+3: ns, 3+4 Vs 8+: ns, 4+3 Vs 8+: ns) (Figure 1A).
Figure 1.
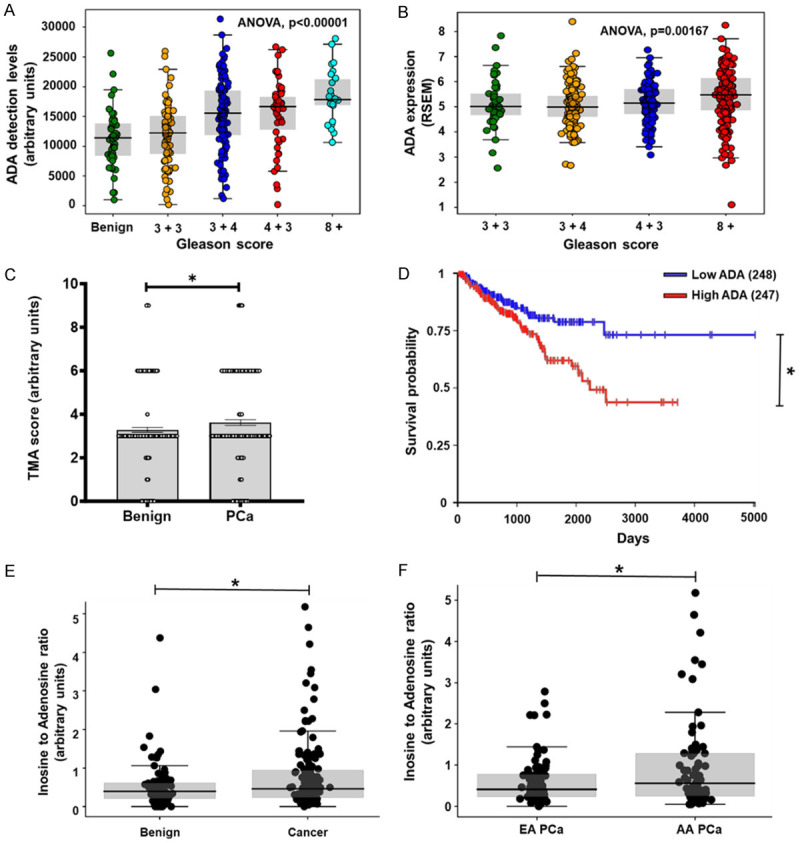
Elevated ADA in aggressive PCa. (A) RNA scope analysis showed increased ADA levels in higher Gleason tumors (N: Benign =3+3=77, 3+4=115, 4+3=50, 8+=30). (B) ADA levels were found to be elevated with high Gleason scores in the TCGA prostate cancer dataset (N: 3+3=45, 3+4=146, 4+3=101, 8+=206). Normalized RNA sequencing data is represented as RSEM (RNA-Seq by Expectation Maximization). (C) Immunostaining of TMAs for ADA showed that the ADA protein levels were high in PCa (N: Benign =79, PCa=77). (D) Survival analysis using the TCGA prostate cancer dataset revealed that high ADA in associated with poor survival in PCa (N: Low ADA-249, High ADA-248, Median =5.249 RSEM). (E) High inosine to adenosine ratio in the urine of PCa patients is indicative of high ADA enzyme activity (N: benign =98, PCa =155). The urine samples were obtained from a case-control study. (F) High inosine to adenosine ratio in AA PCa compared to EA PCa (N: AA=78, EA=77) in the case-control cohort described in (E). For comparison of ADA levels across Gleason scores, ANOVA, for Benign Vs Cancer comparison of ADA levels Wilcox matched-pairs signed rank test, for inosine to adenosine ratio in Benign Vs Cancer and EA Vs AA, Student’s unpaired two-tailed t-test were used to compute the p values. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
RNA expression analysis of RNA-Seq data from TCGA (n=498) also showed that ADA transcript levels were increased in tumors with higher Gleason scores (N=498; 3+3=45, 3+4=146, 4+3=101, 8+=206) (ANOVA p value =0.00167 and Tukey’s multiple comparisons p-value: 3+3 Vs 3+4: ns, 3+3 Vs 4+3: ns, 3+3 Vs 8+: ns, 3+4 Vs 4+3: ns, 3+4 Vs 8+: 0.001, 4+3 vs 8+: ns) (Figure 1B).
ADA protein levels were evaluated in four TMAs using immunohistochemical analysis (IHC). These TMAs consisted of prostate cancer tissues and matched benign cores from PCa patients. Our IHC analysis showed ADA to be elevated in PCa tissues (p value =0.0316) (Figure 1C).
Kaplan-Meier survival analysis of the TCGA data set revealed that high ADA expression is also associated with poor progression-free survival (N: low ADA=249, high ADA=248) (p value =0.0057) (Figure 1D). The population was divided into low ADA and high ADA based on the median value of normalized ADA levels (Normalization of RNA-Seq data by Expectation Maximization, RSEM=5.249).
As previously stated, ADA deaminates adenosine and converts it to inosine. Therefore, an increase in ADA activity will lead to increased conversion of adenosine to inosine, resulting in higher inosine and lower adenosine levels. In an independent set of urine samples from a PCa case-control study (benign, n=98 and PCa, n=155), we found a high inosine-to-adenosine ratio (p value =0.0165) (Figure 1E) in PCa patients. The inosine-to-adenosine ratio was also elevated in the urine samples from AA PCa (population with mean West African ancestry ratio >0.8 (SD=0.1)) patients compared to EA PCa (population with lower West African ancestry ratio, mean <0.1 (SD=0.1)) patients (p value =0.0201) (Figure 1F). There were 40 EA PCa patients with Gleason 6, 22 with Gleason 7, and 13 with 8+ Gleason scores. There were 40 AA PCa patients Gleason 6, 28 with Gleason 7, and 7 with 8+ Gleason scores. The samples’ other clinical characteristics are listed in Table 1.
Constitutive ADA overexpression decreases PCa cell adhesion
To understand ADA’s biological role in PCa, ADA-overexpressing (ADA OE) cell line models were established, and a thorough phenotypic and molecular examination was done. We found that ADA overexpression alters cell adhesion mechanisms and decreases cellular adhesion.
ADA OE models were developed by lentiviral transduction in MDA-PCa-2a (ancestry-verified AA cell line) and LNCaP (ancestry-verified EA cell line) cells. To verify the specificity of the overexpression vector, a knockdown of ADA (ADA OE-KD) was performed on the ADA OE cell lines using shRNA (Figure 2). Therefore, ADA OE-KD cells contain both ADA open reading frame (ORF) and shRNA and have intermediate expression levels of ADA. ADA OE-KD will rescue (reverse) the effects observed in the ADA OE, thus ensuring that the changes observed are the consequence of ADA elevation and not due to any non-target effects. Vector control was established for both ADA OE (control) and ADA OE-KD (ADA OE-NT). The overexpression and knockdown models were verified by qPCR (mRNA), western blot (protein), ADA enzyme assay (enzyme activity), and LC/MS (adenosine and inosine levels) in both cell line models (Figure 2A-H). Transduction of an empty vector (NT) into the ADA overexpression cell line (ADA OE) moderately reduced the level of ADA, possibly due to repeated transductions and selection processes affecting the cells’ functions (like transcription and translation). Irrespective of this, the expression of ADA in the ADA OE-NT cells was higher than that in ADA OE-KD cells. In addition, as expected, both ADA OE and ADA OE-NT pheno-copied each other in functional assays (Figure 3).
Figure 2.
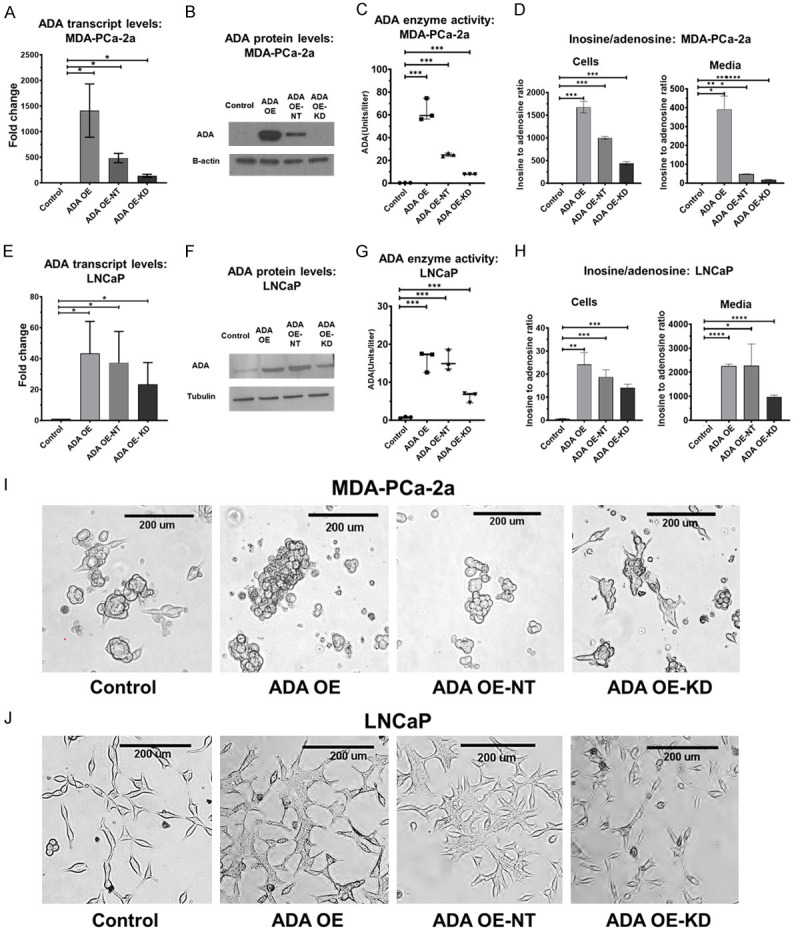
Establishing ADA OE in vitro models. Validation of established ADA OE and rescue models by qPCR, western blot, enzyme activity assay and LC/MS in (A-D) MDA-PCa-2a (AA) and (E-H) LNCaP cell lines (EA). Morphological changes in the (I) MDA-PCa-2a and (J) LNCaP cells. Student’s unpaired two-tailed t-tests was used for evaluating the statistical significance. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Figure 3.
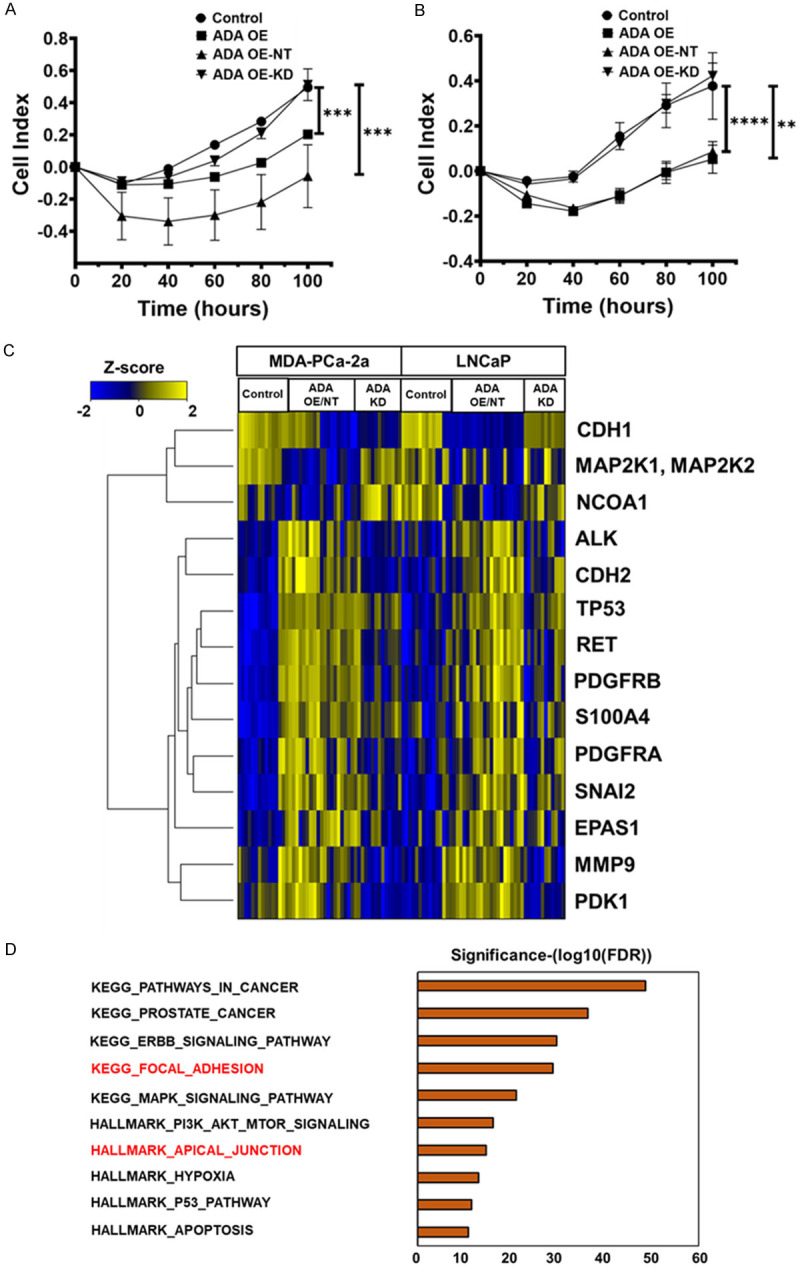
Constitutive ADA overexpression alters the cells’ adhesion potential. Decreased adhesion potential upon ADA OE observed in both (A) MDA-PCa-2a and (B) LNCaP cells (N=3/group/cell line). (C) RPPA analysis in both MDA-PCa-2A and LNCaP cells revealed alterations in several proteins. This heatmap shows proteins that are differentially expressed upon ADA OE (ADA OE and ADA OE-NT) in both MDA-PCa-2a and LNCaP cell lines (N=3/group/cell line). Heatmap is clustered by the log fold change in protein expression. Gradations of yellow and blue represent increased and reduced fold-change in the proteins, respectively. (D) Gene set enrichment analysis (GSEA) of the RPPA data revealed that adhesion machinery was among the top altered mechanisms upon ADA elevation in both MDA-PCa-2a and LNCaP cells. Pathways associated with cell adhesion are highlighted in red. All GSEA concepts listed are significant at P<0.01 and FDR<0.05. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
ADA OE cells exhibited morphological and adhesion changes compared to the control. This effect was also rescued in the ADA OE-KD cells. In MDA-PCa-2a, the cells with high ADA appeared rounded, and in LNCaP, they appeared stretched (Figure 2I, 2J). The ADA OE cells were either floating or only weakly attached to the surface. We used the Xcelligence real-time cell analysis system to quantify the changes in cell-surface adhesion. In both cell lines, cells with high ADA (ADA OE and ADA OE-NT) had decreased adhesion potential compared to the cells with lower ADA (control and ADA OE-KD) (Figure 3A, 3B).
To further pinpoint the molecular changes associated with the decreased adhesion we observed, we utilized a reverse phase protein array (RPPA) platform for targeted proteomics analysis. We probed 233 validated antibodies, including total and phosphoproteins of various pathways and functional protein groups. Cells with high ADA levels showed changes in the cadherins and cadherin-regulating proteins (CDH1, CDH2, SNAI2), an increase in extracellular matrix (ECM) remodeling proteins (MMP9, S100A4), and several other proteins that are associated with invasion and metastasis (RET, PDFGRA, PDFGRB) (Figure 3C). Gene set enrichment analysis (GSEA) was conducted using KEGG and Hallmark pathways to identify significantly enriched pathways. We found that focal adhesion and apical junction pathways were two of the top enriched pathways upon ADA overexpression (Figure 3D).
ADA overexpression delays tumor engraftment in mice
We conducted our in vivo studies in NOD-SCID-Gamma (NSG) (MDA-PCa-2a, 22Rv1) and nude mice (for LNCaP only) to understand ADA’s role in PCa progression. The cells were injected subcutaneously, and their growth was monitored. It was evident that the presence of high ADA delayed the onset of tumors. Only the control tumors with baseline ADA levels had a successful engraftment rate and developed tumors faster. This was consistent in all three cell lines (MDA-PCa-2a, LNCaP, and 22Rv1). ADA OE cells formed tumors after 14 weeks (Vs 11 weeks in control, in MDA-PCa-2a, N=6 per group, P<0.001), 40 days (Vs 14 days in control, in LNCaP, N=8 per group, P<0.0001), and 14 days (Vs 9 days in control, in 22Rv1, N=9 per group, P<0.0001, Figure 4A-C).
Figure 4.
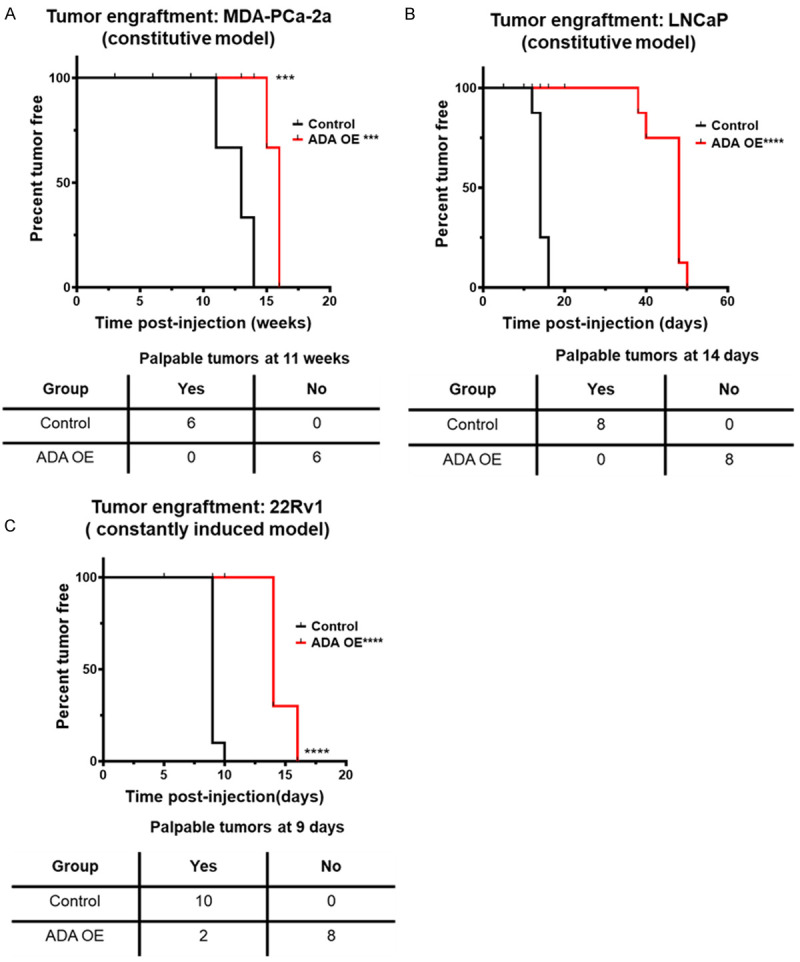
ADA OE delays tumor onset. Constitutive ADA expression delays the tumor engraftment in (A) MDA-PCa-2a (N=6/group), (B) LNCaP (N=8/group), and (C) 22Rv1 (N=9/group) cells. Palpable tumors are typically ≤10 mm3. p-values were computed using log-rank test. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
Increased ADA expression enhances prostate tumor growth
To understand ADA’s significance during the later stages of tumor progression, tet-inducible ADA-OE cell lines were used in vivo. 22Rv1 and C4-2B cell lines that are more robust and have better in vivo tumorigenicity were used for this study. The cells were injected without ADA induction and monitored until tumors were formed (approximate size: 50 mm3). ADA expression was then induced after the tumors reached 50 mm3 and the growth rate was further monitored until the tumors reached 500 mm3. Upon ADA induction, the tumors experienced a growth spurt. The ADA-OE-induced tumors grew faster than the control tumors in both the 22Rv1 and C4-2B models (Figure 5A, 5B).
Figure 5.

ADA elevation promotes prostate tumor growth. ADA elevation accelerates the tumor growth when induced post-tumor initiation in (A) 22Rv1 (N: Dox-control =7, ADA-uninduced =7, ADA induced =8) and (B) C4-2B (N: Dox-control =7, ADA-uninduced =9, ADA induced =8), ADA-inducible xenografts. Induction of ADA by doxycycline verified in by RT-qPCR and ADA enzyme assay in (C) 22Rv1 and (D) C4-2B tumors. p-values were computed using Mann-Whitney test for tumor growth and Student’s two-tailed t-tests for ADA induction levels. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
The tumor volumes were normalized to the volumes on the day of induction. The ratio of the final volume to the volume at the start of doxycycline induction was plotted as the outcome. The induction of ADA in tumors was verified by qPCR and also using ADA enzyme activity assay (Figure 5C, 5D). RNA sequencing of these tumors followed by GSEA also revealed that nutrient-sensing mTOR signaling was enriched upon ADA induction (Figure 6).
Figure 6.
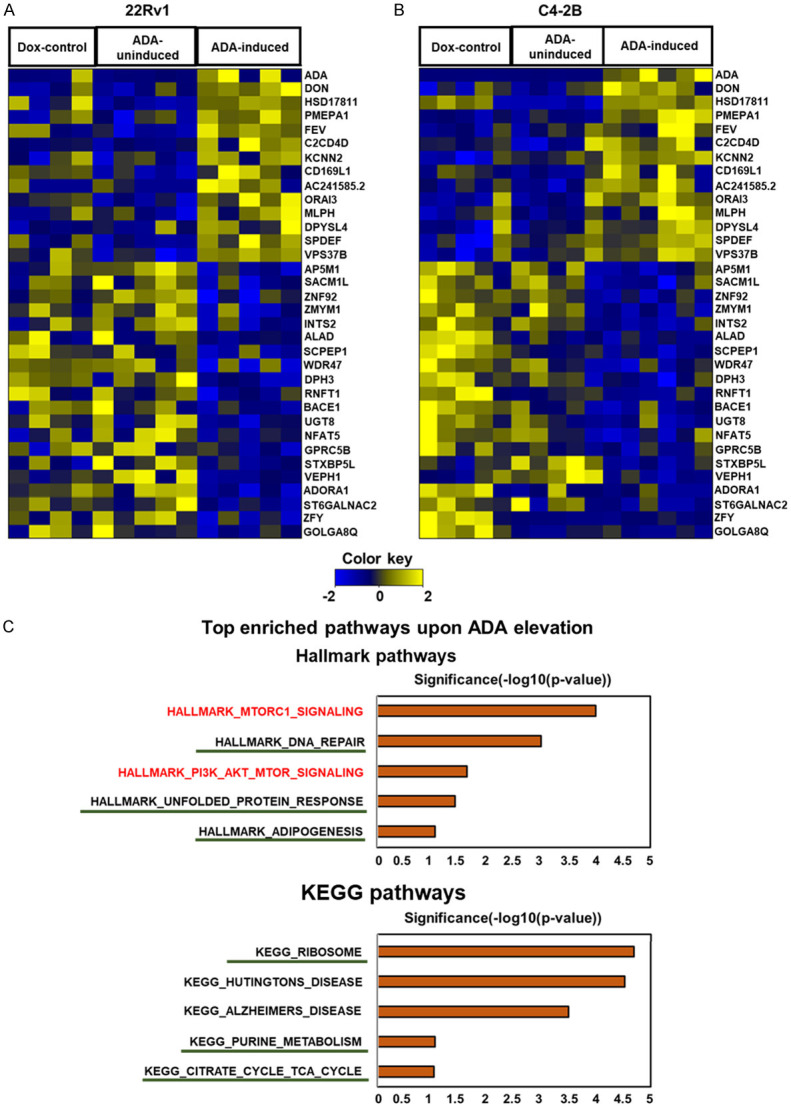
mTOR signaling is enriched in ADA-induced tumors. RNA sequencing analysis revealed several gene alterations in both (A) 22Rv1 and (B) C4-2B tumors with high ADA activity. Heatmaps are clustered by the log fold change in gene expression. Gradations of yellow and blue represent increased and reduced fold-change in the genes, respectively. (C) GSEA of RNA sequencing data obtained from these tumors shows that mTOR signaling (highlighted in red) is enriched in tumors with high ADA. Pathways regulated downstream of mTOR were also enriched (underlined). All GSEA concepts listed are significant at P<0.05 and FDR<0.25.
Discussion
We study PCa’s metabolic landscape to understand the biochemical changes associated with PCa development and progression. We analyzed the clinical samples and found that ADA was high in PCa. We also found that ADA has a significant impact on tumor growth and cell adhesion.
Our clinical data analyses show that ADA levels are elevated in PCa especially in higher Gleason tumors. Tumors with Gleason scores ≤6 are considered to be indolent and are eligible for active surveillance. Tumors with a Gleason score of 7, whether 3+4 or 4+3, are considered intermediate grade, with 4+3 being slightly worse than the 3+4 score. Tumors with scores ≥8 are considered highly aggressive, where progression and disease recurrence are highly likely [18,19]. Finding significantly elevated ADA levels in tumors with higher Gleason scores indicates that the enzyme levels are associated with aggressive PCa.
High ADA levels lead to increased conversion of adenosine to inosine. Therefore, under such conditions, the ratio of inosine to adenosine will be high. The metabolic studies we did using urine samples from a case-control study show that the inosine-to-adenosine ratio is higher in PCa patients - especially in AA PCa patients, who are most likely to have a poor clinical outcome [18,19]. These findings further support the association between elevated ADA and aggressive PCa.
The in vitro studies we conducted to delineate ADA’s role in PCa revealed that when ADA is elevated for prolonged periods, cells lose their ability to attach to the surface. Cellular adhesion machinery maintains the organization and integrity of the cells in a tissue. It maintains the cells’ apical-basal polarity and is also important for regulating inter-cellular and extracellular communications. RPPA analysis revealed alterations in focal adhesion and apical junction pathways upon constitutive ADA overexpression. Focal adhesion pathway components maintain the connections between the cell’s cytoskeleton and extracellular matrix (ECM). They sense the changes in the microenvironment and alter the cell morphology and the cell’s adhesion to the ECM/basement membrane [20,21]. The adhesion decrease observed in high ADA conditions could be a result of alterations in the focal adhesion pathway. The apical junction proteins are integral in maintaining cell-cell attachments and cell polarity. As the tumors progress and get dedifferentiated, the focal and apical junction complexes get altered significantly. The cells develop a more mesenchymal-like phenotype where they lose their polarity and adhesion potential. This makes them more motile and promotes migration and metastasis [21]. In the same RPPA study, we observed a decrease in E-cadherin levels and an increase in N-cadherin levels upon ADA OE. This cadherin switch is indicative of the epithelial-mesenchymal transition (EMT). Proteins S100A4 and SNAI2, which are upregulated in ADA OE cells, are known to repress E-cadherins and some critical junction proteins, thus promoting the EMT phenotype [22,23]. Alterations in focal and apical complexes and ECM remodeling all determine cells’ adhesion capacity. S100A4 is also secreted into the extracellular space and facilitates ECM reconstitution by deploying matrix metalloproteases like MMP9 [23]. The adhesion decrease observed upon ADA elevation could be attributed to changes in these proteins and pathways. Adhesion decrease is significant in tumor progression during the later aggressive stages, especially during metastatic dissemination. Several other proteins, like RET, PDGFRA, and PDGFRB, which are associated with invasion and metastasis [24-27], were also elevated upon ADA overexpression. These molecular alterations that we observed in vitro further support an association between ADA and aggressive PCa.
In vivo models are more relevant in determining the molecular pathophysiology of any disease. However, PCa in vivo models pose several challenges. It is extremely difficult to recapitulate the human prostate cancer landscape in animal models. The existing prostate cell lines have a successful xenograft formation only in immunodeficient mice and rarely develop spontaneous metastasis. This in turn makes it difficult to understand ADA’s role in promoting PCa metastasis and invasion. Our in vivo tumor xenograft studies with the constitutive ADA OE cell lines lead to delayed tumor engraftment. This delay might be because of the altered adhesion that prevents them from attaching and establishing the tumor niche, thereby delaying tumor formation. While adhesion decrease is critical in the later stages of tumor growth and progression, it can interfere with early-stage tumor formation in mice models. This presented another challenge in that impaired tumor formation from ADA OE cells prevented us from observing what occurs in the later stages of disease progression.
To circumvent these challenges and further investigate ADA’s role in promoting tumor growth, we generated inducible ADA OE models in more aggressive 22Rv1 and C4-2B cell lines. Using an inducible model enabled us to induce ADA after the tumor formation stage and also to have some control over the level of ADA expression. In the in vivo experiments, in which doxycycline regulated ADA levels, we observed rapid growth after the tumor engraftment. This suggests that elevated ADA promotes tumor growth in PCa’s later stages.
RNA sequencing analysis done on these tumors illuminated some potential mechanisms that ADA activates to promote tumor growth. The mTOR (mTORC1 and PI3K/AKT/mTOR) signaling was significantly enriched indicating its association with the high ADA activity. mTOR is part of the mTORC protein complex. mTOR senses the cellular metabolic patterns and regulates the cell growth/survival signaling. mTOR signaling promotes growth and survival by stimulating several biosynthetic pathways [28,29]. An important role of mTOR is to promote protein synthesis by fostering ribosome biogenesis, thereby facilitating a higher translation rate. As a consequence of high protein production caused by mTOR activity, the unfolded protein response is also often upregulated [30-32]. mTOR also promotes cell growth by regulating mitochondrial functions, whereby it facilitates TCA anaplerosis by enhancing glutamine and glucose uptake. By elevating the TCA cycle, mTOR provides substrates for purine synthesis which is vital to transcription and energy homeostasis processes that ensure cells’ sustenance and survival [33-36]. The AKT-activated mTOR pathway is critical for lipid biosynthesis and adipocyte development. It is well-established that mTORC1 positively regulates adipogenesis [37,38]. In addition to positive enrichment of the mTOR signaling we also observed the enrichment of downstream processes activated by mTOR like ribosome biogenesis, DNA repair, adipogenesis, purine synthesis, and TCA cycle in tumors with high ADA. It is well-known that inosine elevation activates mTOR and the associated downstream metabolic biosynthetic and cell growth pathways. It has been postulated that inosine activates mTOR via purinergic receptors [39,40]. As described earlier, elevated ADA expression results in increased inosine levels which could activate mTOR-driven pathways promoting tumor growth in PCa. Additional future confirmatory studies are required to confirm these findings.
Taken together, our clinical, in vitro, and in vivo findings all point toward the significance of ADA upregulation in later stages of tumor progression. ADA enzymatic levels could experience a gradual surge as the tumor progresses. The initial spike in ADA after tumor formation could potentially lead to faster growth and progression. While the initial elevation in ADA activity activates the cell growth and sustenance pathways, over time, the prolonged/chronic ADA elevation could lead to alterations in the adhesion machinery and potentially promote metastatic dissemination. The prolonged elevation allows the metabolites to alter microenvironment dynamics and instigate changes in ECM and cell adhesion.
Our study is the first to use extensive in vitro and in vivo models to demonstrate ADA’s intrinsic effects in any cancer. We have shown evidence for ADA upregulation in PCa using clinical samples at the transcript, protein, and metabolite levels. We have established both the acute and chronic effects of ADA upregulation on prostate tumors. Our work has opened the door to further evaluating inosine/adenosine’s biomarker potential, the mechanism associated with ADA’s regulation of growth/adhesion pathways, and ADA’s therapeutic potential for PCa in the future.
Acknowledgements
The authors acknowledge the Global Center for Mass Spectrometry Excellence supported by Agilent Technologies at BCM. This research was partially supported by the following grants: Prostate Cancer Foundation Challenge Award (ASK, JAJ, and MI), Dan L. Duncan Cancer Center (P30 CA125123) supporting Human Tissue Acquisition and Pathology, and Metabolomics Shared Resources, RO1CA227559 (Ask, GP), RO1CA267090 (ASK), and Translational Grant from V-Foundation (ASK). This project was also supported by CPRIT Proteomics and Metabolomics Core Facility (N.P.) (RP210227), NIH (P30 CA125123), and Dan L. Duncan Cancer Center, and partially funded by NIH grant (1R01CA282282).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73:17–48. doi: 10.3322/caac.21763. [DOI] [PubMed] [Google Scholar]
- 2.DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. CA Cancer J Clin. 2019;69:211–233. doi: 10.3322/caac.21555. [DOI] [PubMed] [Google Scholar]
- 3.Gohlke JH, Lloyd SM, Basu S, Putluri V, Vareed SK, Rasaily U, Piyarathna DWB, Fuentes H, Rajendiran TM, Dorsey TH, Ambati CR, Sonavane R, Karanam B, Bhowmik SK, Kittles R, Ambs S, Mims MP, Ittmann M, Jones JA, Palapattu G, Putluri N, Michailidis G, Sreekumar A. Methionine-homocysteine pathway in African-American prostate cancer. JNCI Cancer Spectr. 2019;3:pkz019. doi: 10.1093/jncics/pkz019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lillard JW Jr, Moses KA, Mahal BA, George DJ. Racial disparities in black men with prostate cancer: a literature review. Cancer. 2022;128:3787–3795. doi: 10.1002/cncr.34433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chowdhury-Paulino IM, Ericsson C, Vince R Jr, Spratt DE, George DJ, Mucci LA. Racial disparities in prostate cancer among black men: epidemiology and outcomes. Prostate Cancer Prostatic Dis. 2022;25:397–402. doi: 10.1038/s41391-021-00451-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitmore KV, Gaspar HB. Adenosine deaminase deficiency - more than just an immunodeficiency. Front Immunol. 2016;7:314. doi: 10.3389/fimmu.2016.00314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao ZW, Yang L, Liu C, Wang X, Guo WT, Zhang HZ, Dong K. Distinct roles of adenosine deaminase isoenzymes ADA1 and ADA2: a pan-cancer analysis. Front Immunol. 2022;13:903461. doi: 10.3389/fimmu.2022.903461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kutryb-Zajac B, Koszalka P, Mierzejewska P, Bulinska A, Zabielska MA, Brodzik K, Skrzypkowska A, Zelazek L, Pelikant-Malecka I, Slominska EM, Smolenski RT. Adenosine deaminase inhibition suppresses progression of 4T1 murine breast cancer by adenosine receptor-dependent mechanisms. J Cell Mol Med. 2018;22:5939–5954. doi: 10.1111/jcmm.13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monroy-Mora A, de Lourdes Mora-García M, Alheli Monroy Mora K, Hernández-Montes J, García-Rocha R, Don-López CA, Weiss-Steider B, Montesinos-Montesinos JJ, Monroy-García A. Inhibition of adenosine deaminase activity reverses resistance to the cytotoxic effect of high adenosine levels in cervical cancer cells. Cytokine. 2022;158:155977. doi: 10.1016/j.cyto.2022.155977. [DOI] [PubMed] [Google Scholar]
- 10.Nakajima Y, Kanno T, Nagaya T, Kuribayashi K, Nakano T, Gotoh A, Nishizaki T. Adenosine deaminase inhibitor EHNA exhibits a potent anticancer effect against malignant pleural mesothelioma. Cell Physiol Biochem. 2015;35:51–60. doi: 10.1159/000369674. [DOI] [PubMed] [Google Scholar]
- 11.Köster J, Rahmann S. Snakemake--a scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 12.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. Embnet Journal. 2011;17:10–12. [Google Scholar]
- 13.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Munjal A, Leslie SW. Gleason score. Encyclopedia of Genetics, Genomics, Proteomics and Informatics. 2022:800. [Google Scholar]
- 19.Epstein JI. Prostate cancer grading: a decade after the 2005 modified system. Mod Pathol. 2018;31:S47–63. doi: 10.1038/modpathol.2017.133. [DOI] [PubMed] [Google Scholar]
- 20.Janiszewska M, Primi MC, Izard T. Cell adhesion in cancer: beyond the migration of single cells. J Biol Chem. 2020;295:2495–2505. doi: 10.1074/jbc.REV119.007759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maziveyi M, Alahari SK. Cell matrix adhesions in cancer: the proteins that form the glue. Oncotarget. 2017;8:48471–48487. doi: 10.18632/oncotarget.17265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alves CL, Elias D, Lyng MB, Bak M, Ditzel HJ. SNAI2 upregulation is associated with an aggressive phenotype in fulvestrant-resistant breast cancer cells and is an indicator of poor response to endocrine therapy in estrogen receptor-positive metastatic breast cancer. Breast Cancer Res. 2018;20:60. doi: 10.1186/s13058-018-0988-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fei F, Qu J, Zhang M, Li Y, Zhang S. S100A4 in cancer progression and metastasis: a systematic review. Oncotarget. 2017;8:73219–73239. doi: 10.18632/oncotarget.18016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thein KZ, Velcheti V, Mooers BHM, Wu J, Subbiah V. Precision therapy for RET-altered cancers with RET inhibitors. Trends Cancer. 2021;7:1074–1088. doi: 10.1016/j.trecan.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ban K, Feng S, Shao L, Ittmann M. RET signaling in prostate cancer. Clin Cancer Res. 2017;23:4885–4896. doi: 10.1158/1078-0432.CCR-17-0528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hofer MD, Fecko A, Shen R, Setlur SR, Pienta KG, Tomlins SA, Chinnaiyan AM, Rubin MA. Expression of the platelet-derived growth factor receptor in prostate cancer and treatment implications with tyrosine kinase inhibitors. Neoplasia. 2004;6:503–512. doi: 10.1593/neo.04157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun Y, Yue L, Xu P, Hu W. An overview of agents and treatments for PDGFRA-mutated gastrointestinal stromal tumors. Front Oncol. 2022;12:927587. doi: 10.3389/fonc.2022.927587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. 2021;101:1371–1426. doi: 10.1152/physrev.00026.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang YP, Lei QY. Metabolite sensing and signaling in cell metabolism. Signal Transduct Target Ther. 2018;3:30. doi: 10.1038/s41392-018-0024-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gaudette BT, Jones DD, Bortnick A, Argon Y, Allman D. mTORC1 coordinates an immediate unfolded protein response-related transcriptome in activated B cells preceding antibody secretion. Nat Commun. 2020;11:723. doi: 10.1038/s41467-019-14032-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Szwed A, Kim E, Jacinto E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol Rev. 2021;101:1371–1426. doi: 10.1152/physrev.00026.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou Z, Tao T, Li H, Zhu X. MTOR signaling pathway and mTOR inhibitors in cancer: progress and challenges. Cell Biosci. 2020;10:31. doi: 10.1186/s13578-020-00396-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, Manning BD. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science. 2016;351:728–733. doi: 10.1126/science.aad0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danesh Pazhooh R, Rahnamay Farnood P, Asemi Z, Mirsafaei L, Yousefi B, Mirzaei H. mTOR pathway and DNA damage response: a therapeutic strategy in cancer therapy. DNA Repair (Amst) 2021;104:103142. doi: 10.1016/j.dnarep.2021.103142. [DOI] [PubMed] [Google Scholar]
- 35.De Vitto H, Arachchige DB, Richardson BC, French JB. The intersection of purine and mitochondrial metabolism in cancer. Cells. 2021;10:2603. doi: 10.3390/cells10102603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moffatt BA, Ashihara H. Purine and pyrimidine nucleotide synthesis and metabolism. Arabidopsis Book. 2002;1:e0018. doi: 10.1199/tab.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai H, Dong LQ, Liu F. Recent advances in adipose mTOR signaling and function: therapeutic prospects. Trends Pharmacol Sci. 2016;37:303–317. doi: 10.1016/j.tips.2015.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laplante M, Sabatini DM. An emerging role of mTOR in lipid biosynthesis. Curr Biol. 2009;19:R1046–R1052. doi: 10.1016/j.cub.2009.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, Ben-Sahra I, Manning BD. The mTORC1 signaling network senses changes in cellular purine nucleotide levels. Cell Rep. 2017;21:1331–1346. doi: 10.1016/j.celrep.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Niemann B, Haufs-Brusberg S, Puetz L, Feickert M, Jaeckstein MY, Hoffmann A, Zurkovic J, Heine M, Trautmann EM, Müller CE, Tönjes A, Schlein C, Jafari A, Eltzschig HK, Gnad T, Blüher M, Krahmer N, Kovacs P, Heeren J, Pfeifer A. Apoptotic brown adipocytes enhance energy expenditure via extracellular inosine. Nature. 2022;609:361–368. doi: 10.1038/s41586-022-05041-0. [DOI] [PMC free article] [PubMed] [Google Scholar]