

Abstract
Cognitive impairment in the elderly features complex molecular pathophysiology extending beyond the hallmark pathologies of traditional disease classification. Molecular subtyping using large-scale -omic strategies can help resolve this biological heterogeneity. Using quantitative mass spectrometry, we measured ~8000 proteins across >600 dorsolateral prefrontal cortex tissues with clinical diagnoses of no cognitive impairment (NCI), mild cognitive impairment (MCI), and Alzheimer’s disease (AD) dementia. Unbiased classification of MCI and AD cases based on individual proteomic profiles resolved three classes with expression differences across numerous cell types and biological ontologies. Two classes displayed molecular signatures atypical of AD neurodegeneration, such as elevated synaptic and decreased inflammatory markers. In one class, these atypical proteomic features were associated with clinical and pathological hallmarks of cognitive resilience. We were able to replicate these classes and their clinicopathological phenotypes across two additional tissue cohorts. These results promise to better define the molecular heterogeneity of cognitive impairment and meaningfully impact its diagnostic and therapeutic precision.
Keywords: Biomarkers, Proteomics, Systems biology, Dementia, Alzheimer’s disease
1. Introduction
Dementia in the elderly is characterized by complex molecular pathophysiology extending beyond the hallmark neuropathologies of traditional disease classification. Several studies have confirmed that those with Alzheimer’s disease (AD), the most common cause of dementia in older individuals (Association, A.s, 2018), frequently harbor brain pathologies beyond the amyloid-beta (Aβ) plaques and tau neurofibrillary tangles (NFTs) required for diagnosis. These concurrent pathologies may include cerebrovascular disease, hippocampal sclerosis, neocortical Lewy body inclusions, and TAR DNA-binding protein 43 (TDP-43) aggregates (Schneider et al., 2007; Karanth et al., 2020; Kawas et al., 2015; Brayne et al., 2009; Kapasi et al., 2017). Yet, combined with amyloid and tau, these co-pathologies still account for less than half of the variance in cognitive trajectory (Boyle et al., 2021). Accordingly, genome wide association studies (GWAS) have linked AD pathogenesis to a variety of biological mechanisms, such as synaptic dysregulation, lipoprotein metabolism, membrane trafficking, glial-mediated inflammation, and endothelial integrity (Andrews et al., 2020; Lambert et al., 2010; Efthymiou and Goate, 2017; Saunders et al., 1993; Holtzman et al., 2012; Guerreiro et al., 2012; Jonsson et al., 2013; Bellenguez et al., 2022). These findings highlight the vast pathophysiological heterogeneity underlying cognitive impairment and the need to better define the molecular basis for diverse disease mechanisms and endophenotypes.
Molecular subtyping using large-scale -omic strategies promises to resolve this complex biological heterogeneity. Recent genomic and transcriptomic analyses have identified subtypes within AD corresponding to diverse biological mechanisms (Emon et al., 2020; Neff et al., 2021; Zheng and Xu, 2021), highlighting the utility of these large datasets in the molecular subclassification of dementia. However, proteomic subtypes of dementia have been poorly investigated. Marked spatial, temporal, and quantitative differences between mRNA and protein expression make proteomic subtyping a potential source of unique biological insights (Vogel and Marcotte, 2012; Maier et al., 2009). In addition, compared to RNA, protein abundance more strongly associates with dementia phenotypes, consistent with proteins being more proximate mediators of clinicopathological manifestations (Johnson et al., 2021; Hasin et al., 2017; Tasaki et al., 2022). Accordingly, we have previously demonstrated that the brain proteomes of those with AD neuropathological changes feature a wide range of alterations not observed in the transcriptome and that these protein alterations correlate strongly with clinical and pathological traits (Johnson et al., 2022; Higginbotham et al., 2020; Johnson et al., 2020; Seyfried et al., 2017). Yet, it remains unclear whether these proteomic alterations drive distinct molecular subtypes of disease.
To this end, we performed an unbiased proteomic subtyping analysis of brain tissues derived from cognitively impaired individuals enrolled in the Religious Orders Study or Rush Memory and Aging Project (ROSMAP) longitudinal cohorts (Bennett et al., 2005; Bennett et al., 2014; Bennett et al., 2018). Using tandem mass tag mass spectrometry (TMT-MS), we quantified nearly 8000 proteins across 610 brain tissues from individuals with clinical diagnoses of no cognitive impairment (NCI), mild cognitive impairment (MCI), and AD dementia. Unbiased clustering of the nearly 400 MCI and AD tissues by individual proteomic profiles resolved three major classes of cognitive impairment. Here, we thoroughly characterize these classes and their protein expression differences across cell types and biological ontologies. We highlight how two of the three classes harbor proteomic features atypical of molecular neurodegeneration. We also explore the relationship between these molecular signatures, genetic risk, and clinicopathologic phenotypes. In sum, our results underscore the biological heterogeneity among elderly individuals with cognitive impairment and how this translates into distinct proteomic profiles with different phenotypes of disease. Further investigation of these disease subtypes promises to meaningfully impact diagnostic, prognostic, and therapeutic precision in AD and related dementias.
2. Results
2.1. Clinical diagnostic groups feature neuropathological heterogeneity
The main objective of this study was to use TMT-MS to investigate a novel, unbiased proteomic strategy for classifying brain-based molecular alterations in a cognitively impaired elderly population. All brain tissues were derived from the dorsolateral prefrontal cortex (DLPFC) of individuals enrolled in the Religious Orders Study or Rush Memory and Aging Project (ROSMAP). These cohorts recruit older individuals without known dementia from United States religious orders, lay retirement centers, senior and subsidized housing communities, and church groups. These participants are then followed longitudinally with cognitive batteries, biospecimen collection, and finally brain autopsy (Bennett et al., 2005; Bennett et al., 2014; Bennett et al., 2018). Such community-based procedures are designed to provide a heterogenous, “real-world” representation of the dementia population found outside of tertiary care centers. Thus, ROSMAP has well-described clinical and pathological heterogeneity among its cognitively impaired participants (Kapasi et al., 2017; Bennett et al., 2014). We ultimately acquired 610 ROSMAP DLPFC tissues from 604 unique individuals. To stratify cognitively intact from cognitively impaired samples, we relied on the clinical consensus cognitive diagnosis (cogdx) assigned to each case. The cogdx is a final clinical diagnosis imparted at death by study physicians based on clinical history and detailed neuropsychological evaluation while blinded to neuropathological results (Bennett et al., 2006). ROSMAP cases can generally be assigned cogdx classifiers of NCI, MCI, AD, or Other Dementia.
Of the 610 ROSMAP tissues, we included 597 samples in our secondary analyses comprising 220 NCI, 173 MCI, and 204 AD cases. Eight tissues classified as “Other Dementia” were excluded due to concern they may represent clinical outliers. We also excluded five tissues with clinical diagnoses that were discordant with neuropsychological testing scores prior to death. Of the included cohort, there were five individuals with NCI and one with AD who each contributed two independent DLPFC samples, resulting in six same-case sample pairs. Individuals with MCI and AD featured significantly lower cognitive scores and higher levels of AD neuropathology compared to NCI tissues (Fig. 1). Yet, as expected, there was notable pathological heterogeneity among these clinical diagnostic groups. Amyloid and/or tau deposition was frequently encountered among the NCI cases (Fig. 1). Meanwhile, a subset of cognitively impaired individuals maintained negligeable levels of AD neuropathology. Finally, non-AD pathology (e.g., Lewy bodies, TDP-43 inclusions) was common among cognitively impaired cases, especially those with dementia who otherwise met neuropathological criteria for AD (Fig. 1). These results highlighted the pathological heterogeneity inherent in a cognitively impaired elderly population and underscored the need for classification schemes that extend beyond hallmark proteinopathy.
Fig. 1. Clinicopathological characteristics of clinical diagnostic groups.
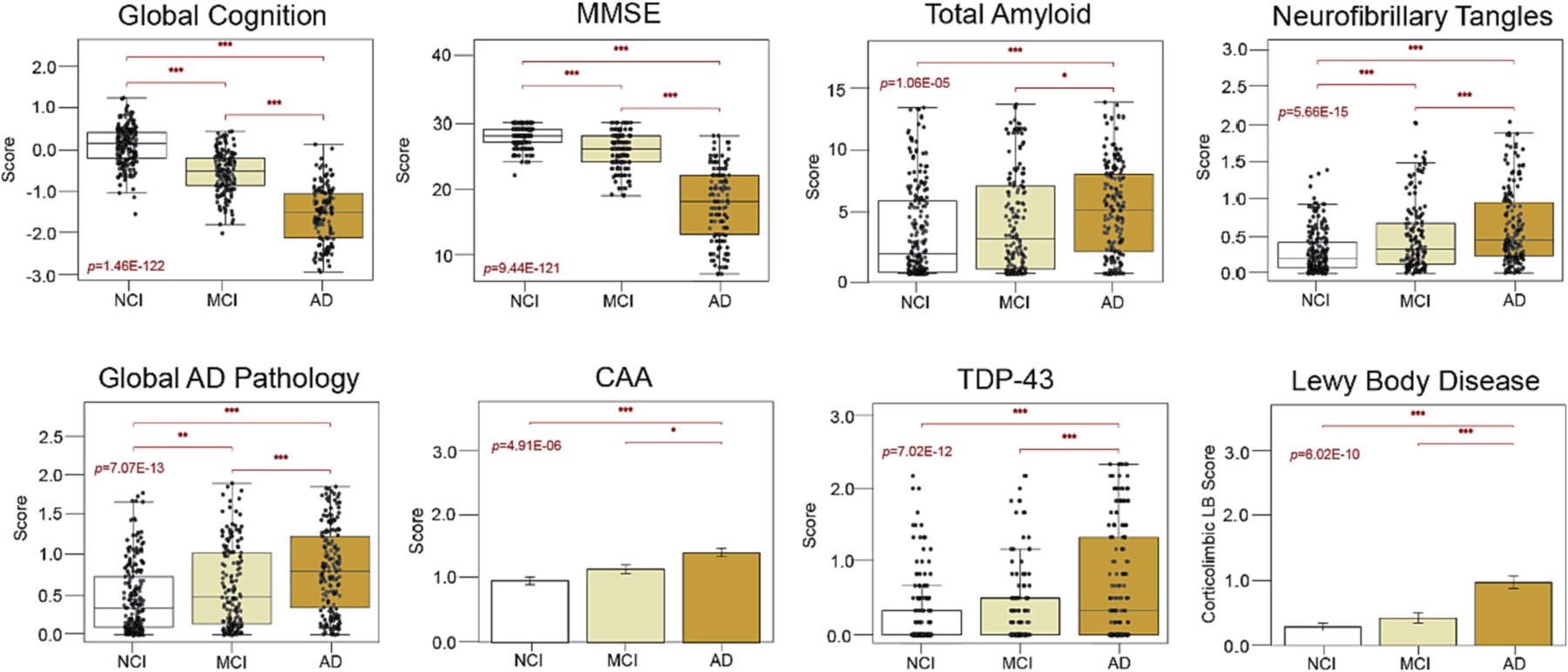
Boxplots of the cognitive and neuropathological characteristics of each clinical diagnostic group. Of the 610 ROSMAP samples analyzed by TMT-MS, we included 597 samples in our secondary analyses with clinical diagnoses of NCI (n = 220), MCI (n = 173), and AD (n = 204). MCI and AD cases featured significantly lower cognitive scores and higher levels of AD and non-AD neuropathology compared to those with NCI. Yet, there was notable pathological heterogeneity among these clinical diagnostic groups. Box plots represent the median and 25th and 75th percentiles, while data points up to 1.5 times the interquartile range from the box hinge define the extent of error bar whiskers. The ANOVA p value is provided for each boxplot with asterisks indicating statistically significant Tukey post hoc pairwise comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Abbreviations: NCI, No Cognitive Impairment; MCI, Mild Cognitive Impairment; AD, Alzheimer’s Disease; MMSE, Mini-Mental State Examination; CAA, Cerebral Amyloid Angiopathy; TDP-43, TAR DNA-Binding Protein 43.
2.2. Proteomic signatures distinguish clinical diagnostic groups with modest sensitivity and specificity
We hypothesized that congruent with this pathological heterogeneity, the clinical diagnostic groups would also feature proteomic heterogeneity. Thus, prior to unbiased clustering, we analyzed the proteomic features of our control, MCI, and AD groups (Fig. 2A). All 610 ROSMAP tissues were analyzed in two sets by TMT-MS with Set 1 containing 400 tissues and Set 2 containing 210 tissues. To harmonize quantified data within and between sets, we used a tunable median polish approach (TAMPOR) (Dammer et al., 2023). TAMPOR is a method for batch effect correction and harmonization of multiple multibatch cohorts into a single abundance data matrix suitable for systems biology analysis (Dammer et al., 2023). TAMPOR data harmonization between sets was ultimately visualized using a multidimensional scaling plot (MDS) displaying case distribution based on inter-sample variance. With successive TAMPOR iterations, set variance was diminished as shown by point convergence post-normalization (Fig. 2B) (Dammer et al., 2023). We then regressed the harmonized data for age, sex, and post-mortem interval (PMI) using an established pipeline (Johnson et al., 2022; Johnson et al., 2020).
Fig. 2. Proteomic signatures of clinical diagnostic groups.
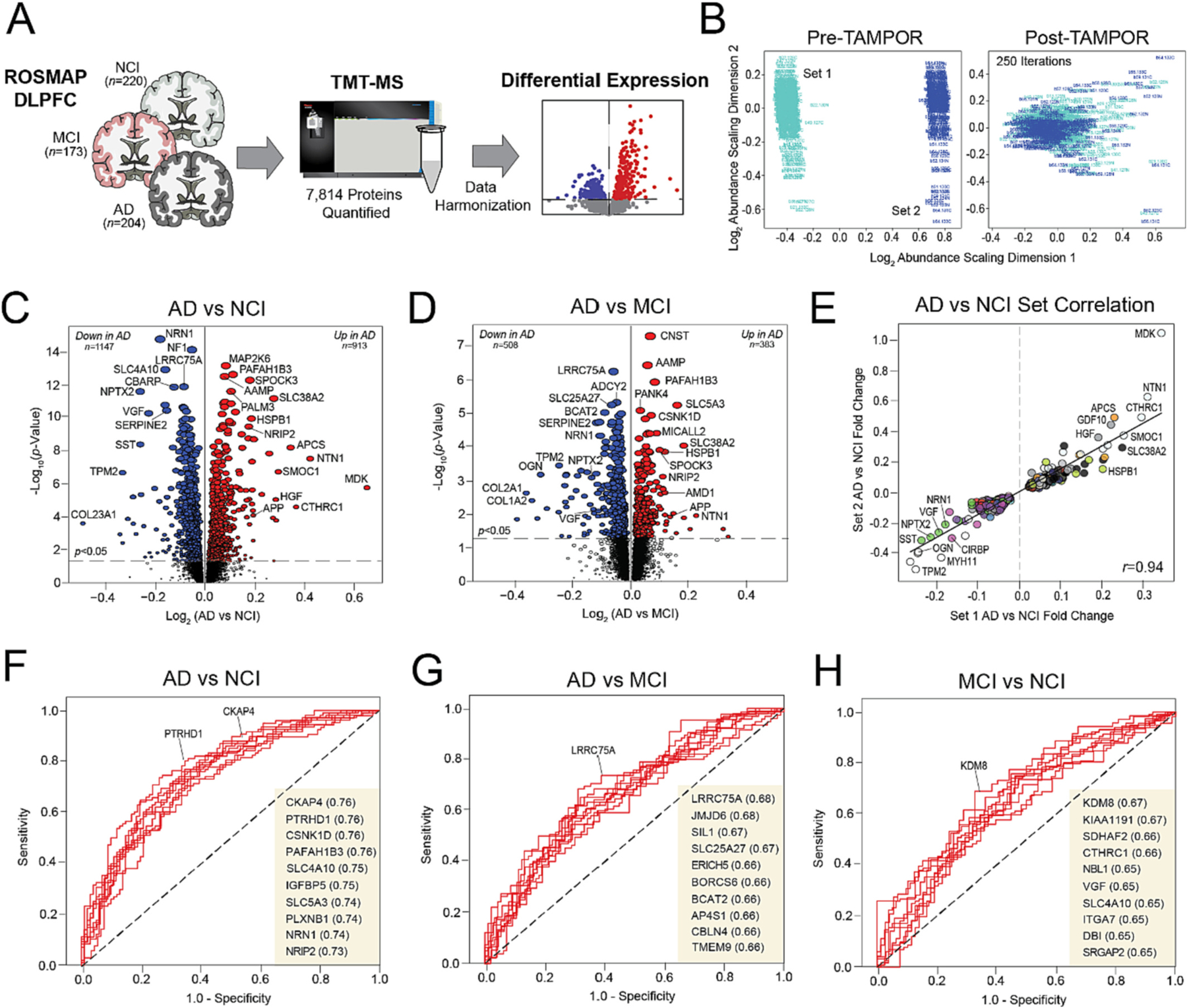
(A) Study approach for the differential expression analysis of the ROSMAP clinical diagnostic groups. (B) Multidimensional scaling plots displaying case distribution based on inter-sample variance pre- and post-TAMPOR normalization between sets. (C–D) Volcano plots displaying the log2 fold change (x-axis) against the -log10 statistical p value (y-axis) for proteins differentially expressed between pairwise comparisons of the clinical diagnostic groups. All p values across pairwise comparisons were derived by ANOVA with Tukey post-hoc correction. (E) Correlation analysis of Set 1 (x-axis) and Set 2 (y-axis) log-transformed fold changes of proteins differentially expressed (p < 0.05) between NCI and AD cases. The Pearson correlation coefficient is provided. (F–H) ROC curves of the 10 most sensitive and specific proteins for each pairwise comparison by AUC values, which are included in parentheses. Abbreviations: NCI, No Cognitive Impairment; MCI, Mild Cognitive Impairment; AD, Alzheimer’s Disease; TMT-MS, Tandem Mass Tag Mass Spectrometry; TAMPOR, Tunable Median Polish of Ratio.
TMT-MS ultimately quantified a total of 7814 proteins across all ROSMAP samples. This count included only those proteins quantified in at least 50% of samples. We then examined differential expression across pairwise comparisons of the three clinical diagnostic groups (Fig. 2C–D, Table S1). There were over 2000 significantly altered proteins between AD and NCI cases (p < 0.05), including 913 increased and 1147 decreased in disease (Fig. 2C). Markers strongly decreased in AD included several neuroprotective synaptic proteins, such as VGF nerve growth factor inducible (VGF), neuronal pentraxin 2 (NPTX2), and neuritin 1 (NRN1). All three of these proteins have been implicated in cognitive resilience and feature well-described decreases in the AD brain (Tubi et al., 2021; Cao et al., 2004; Fournier et al., 2012; Licht et al., 2011; Zacchigna et al., 2008; Gora-Kupilas and Josko, 2005; Chang et al., 2010; Lee et al., 2017; Xiao et al., 2017; Hurst et al., 2022; Yu et al., 2020). AD also featured highly significant increases in several matrisome proteins with known elevations in disease, including midkine (MDK), SPARC related modular calcium binding 1 (SMOC1), and netrin 1 (NTN1) (Johnson et al., 2022; Johnson et al., 2020). A total of 891 proteins were significantly altered between AD and MCI (Fig. 2D), while 294 distinguished NCI and MCI (not pictured). Several proteins demonstrated progressive alterations across all three pairwise comparisons (e.g., VGF, NRN1, SPOCK3), indicating expression levels highly correlated to cognitive severity. A correlation analysis demonstrated strong concordance between the differential expression of Set 1 and Set 2 samples (Fig. 2E), supporting the strength of our data harmonization.
Receiver operating characteristic (ROC) curves were generated for each of the pairwise group comparisons to determine those proteins best at distinguishing the clinical diagnostic groups (Fig. 2F–H). Each curve represented a graphical plot of the true positive rate (sensitivity) against the false positive rate (1-specificity) at various threshold settings. The resultant area under the curve (AUC), a measure of overall classifier performance between values 0 and 1, was then used to identify the strongest signatures for each comparison. Overall, even the strongest protein classifiers were only moderately sensitive and specific. AD vs NCI featured the most robust signatures (e.g., NRN1, PAFAH1B3, CKAP4), though none surpassed an AUC of 0.76 (Fig. 2F). The other pairwise comparisons involving MCI yielded even weaker AUC values, none of which surpassed 0.68. Leucine rich repeat containing 75A (LRRC75A) most strongly distinguished AD from MCI (AUC 0.68) (Fig. 2G), while the histone modifier lysine demethylase 8 (KDM8) best classified MCI from NCI cases (AUC 0.67) (Fig. 2H). Our inability to detect exceptional protein classifiers (AUC > 0.8) indicated a notable degree of proteomic heterogeneity among our clinical diagnostic groups. This fueled our hypothesis that an unbiased proteomic approach to disease classification could generate subtypes of cognitive impairment with heightened relevance to underlying molecular pathophysiology.
2.3. Unbiased proteomic classification yields three distinct classes of cognitive impairment
To generate unbiased proteomic subtypes of cognitive impairment, we sorted the 377 MCI and AD samples into proteomic classes using the well-validated algorithm MONET M1 (Fig. 3A), which groups samples by optimizing the modularity between clusters (Choobdar et al., 2019; Tomasoni et al., 2020). The parameters of MONET M1 were selected using a grid search (Fig. S1, Table S2) to minimize the percentage of cases not assigned to a class. Ultimately, 95% (n = 357) of the 377 samples were assigned to one of three classes, termed A, B, and C (Fig. 3B, Table S3). Class A (n = 128) comprised 80 MCI (62%) and 48 AD (38%) samples. Class B (n = 71) harbored 27 MCI (38%) and 44 AD (62%) samples. Finally, Class C (n = 158), the largest group, contained 56 MCI (35%) and 102 AD (65%) samples. Given each class comprised a mixture of both MCI and AD tissues, we immediately concluded symptom severity was not the only driver of class structure. There were no significant differences in the average age and sex of each class (Table S4).
Fig. 3. MONET M1 yields three disease-relevant proteomic classes of cognitive impairment.
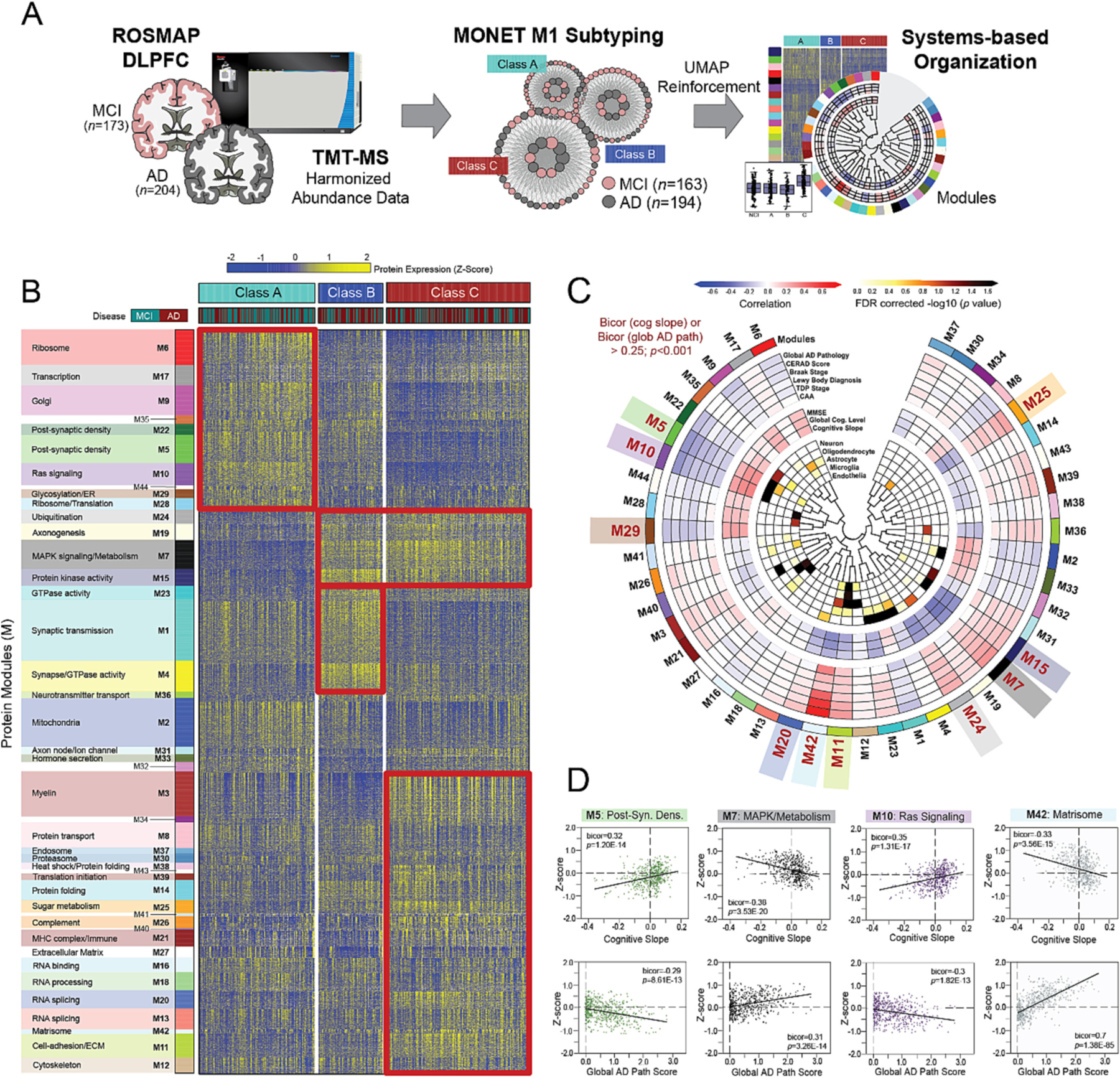
(A) Study approach for the unbiased subtyping analysis of ROSMAP MCI and AD cases. (B) Heat map of protein expression across the three proteomic classes generated by MONET M1 analysis. Classes were termed A (n = 128), B (n = 71), and C (n = 158) and each featured a mixture of MCI and AD cases. To provide biological context to the proteomic differences across classes, proteins were organized by modules (M) of co-expression informed by prior AD network analyses. Red boxes highlight modules with relatively elevated levels (yellow shading) in select classes. (C) Diagram depicting the associations of each module to cell type and ROSMAP clinicopathological traits. Modules bolded in red (M5, M10, M29, M20, M42, M11, M24, M7, M15, M25) demonstrated exceptionally strong correlations to cognitive slope and/or global AD pathology (bicor>0.25; p < 0.001). (D) Correlation plots of module abundance (z-score) to cognitive slope or global AD pathology across all analyzed cases (n = 610) for select modules with remarkably strong clinicopathological correlations. M5 and M10 demonstrated highly significant positive correlations to cognitive slope and negative correlations to global AD pathology. In contrast, M7 and M42 were negatively correlated to cognitive slope and positively correlated to global AD pathology. Bicor correlation coefficients with associated p values are shown for each correlation plot. Abbreviations: DLPFC, Dorsolateral Prefrontal Cortex; MCI, Mild Cognitive Impairment; AD, Alzheimer’s Disease; TMT-MS, Tandem Mass Tag Mass Spectrometry; FDR, False Discovery Rate; Post-Syn Dens, Post-Synaptic Density. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
To assess the reproducibility of these classes, we employed a bootstrap approach to repeatedly cluster the samples an additional 100 times (Fig. S2A–B). On each of these iterations, we applied MONET M1 to a randomly selected 80% (n ~ 300) of MCI and AD tissues. The resultant clusters generated in each bootstrap iteration were analogous to the original clustering as assessed by strong levels of overlapping class-specific samples (Fig. S2C) and highly preserved protein signatures (Fig. S2D). Thus, our unbiased classification was highly reproducible, supporting the robustness of MONET M1 in defining consistent patterns of protein expression across samples.
We then independently validated these proteomic classes by applying a high-performance dimension reduction algorithm to our ROSMAP dataset termed Uniform Manifold Approximation and Projection (UMAP). In recent studies, UMAP has proven capable of effectively reinforcing sample heterogeneity within bulk -omic datasets with clustering structures that maintain biological and clinical meaning (Yang et al., 2021). We took a supervised UMAP approach by providing the algorithm with the three sample target labels defined by MONET M1. We then ran the UMAP clustering analysis, which generated three proteomic groups nearly identical to those formed by MONET M1, reinforcing the structure of the original classes (Fig. S3). Only one of the 357 samples clustered differently between the algorithms, segregating into Class B with MONET M1 and Class C with UMAP. These results further supported the validity of our three proteomic classes of cognitive impairment.
2.4. Classes differ across a diverse range of disease-associated biological ontologies
In prior network-based studies, we have shown that the brain proteomes of those with AD neuropathological changes feature alterations in highly reproducible groups or “modules” of co-expressed proteins (Johnson et al., 2022; Higginbotham et al., 2020; Johnson et al., 2020; Seyfried et al., 2017). These disease-associated modules reflect a wide range of cell types and molecular functions and have established a biological framework for the AD brain proteome and its diverse pathophysiology. To provide biological context to the three classes, we organized their proteomic profiles by the 44 co-expression modules of our deepest AD consensus network, derived from hundreds of tissues in the early and late stages of disease (Johnson et al., 2022). Approximately 68% of the nearly 8000 proteins identified among our ROSMAP cases (n = 5290) mapped to one of these 44 modules. This mirrored previous analyses, which have shown that ~70% of proteins in any given dataset segregate into modules (Johnson et al., 2022; Higginbotham et al., 2020; Johnson et al., 2020; Seyfried et al., 2017; Dai et al., 2018; Johnson et al., 2018; Swarup et al., 2020; Umoh et al., 2018; Wingo et al., 2019; Higginbotham et al., 2019). The resultant heat map highlights module expression across the three proteomic classes (Fig. 3B). Many modules with distinct alterations across classes demonstrated strong associations to specific molecular functions (Fig. 3B), cell types (Fig. 3C), and clinicopathological traits (Fig. 3C–D). As previously described (Johnson et al., 2022; Higginbotham et al., 2020; Johnson et al., 2020), functional assignments were determined using top-ranking gene ontology (GO) terms associated with each module, and cell type enrichment was determined by analyzing module overlap with RNA sequencing (RNA-seq) and proteomic reference lists of cell type-specific markers. See Table S5 for module correlations to all traits provided for our ROSMAP cohort. Overall, these results showcased differences between classes across a diverse range of disease-relevant biological systems.
Module abundance levels (z-scores) across all cases revealed Class A proteomic signatures most closely matched those of cognitively intact individuals, distinguishing this class as the most “control-like” of the three (Fig. 4A–B, Table S6). Compared to B and C, Class A featured significantly elevated levels of modules involved in protein synthesis and transport, including M6 (ribosome), M9 (Golgi transport), and M29 (glycosylation / endoplasmic reticulum) (Fig. 3B and 4A). Modules linked to RAS signaling (M10) and the post-synaptic density (M5, M22) were also significantly increased in Class A. On the other hand, several modules featured unique decreases in Class A relative to B and C, including those linked to mitogen-activated protein kinase (MAPK) and other kinase-associated pathways (M7, M15) (Fig. 3B and 4B).
Fig. 4. Module abundances highlight class differences across a diverse range of biological ontologies.
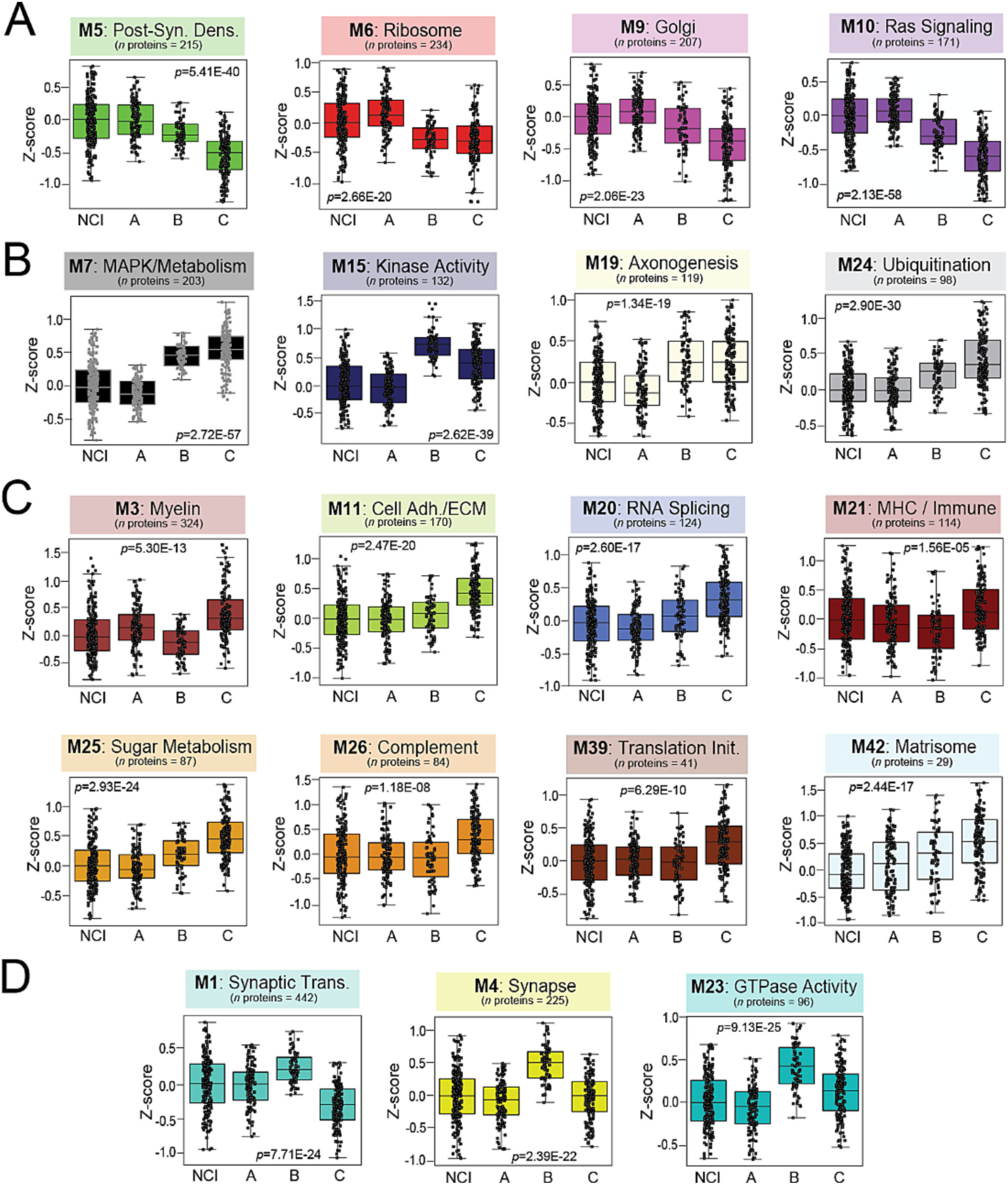
Abundance levels (z-score) of select modules across NCI cases and the three proteomic classes. ANOVA p values are provided for each abundance plot. All modules depicted were significantly altered (p < 0.001) across the four groups. Box plots represent the median and 25th and 75th percentiles, while data points up to 1.5 times the interquartile range from the box hinge define the extent of error bar whiskers. Modules relatively increased in NCI and Class A included M5, M6, M9, and M10, corresponding to post-synaptic density, ribosome, Golgi, and Ras signaling proteins, respectively (A). Kinase-associated M7 and M15 were among modules significantly decreased in NCI and Class A compared to the other two classes (B). Numerous modules were uniquely increased in Class C, most notably several linked to glial-mediated processes (M3, M11, M21, M26, M42) (C). Several synaptic modules (M1, M4, M23) were increased in Class B relative to all other cases (D). Abbreviations: NCI, No Cognitive Impairment; MCI, Mild Cognitive Impairment; AD, Alzheimer’s Disease; Cell Adh / ECM, Cell Adhesion / Extracellular Matrix; Translation Init, Translation Initiation; MHC, Major Histocompatibility Complex; Synaptic Trans, Synaptic Transmission.
Class C featured proteomic changes most consistent with the neurodegenerative trends we have previously observed in pathologically defined AD (Johnson et al., 2022; Johnson et al., 2020; Swarup et al., 2020). Compared to A and B, Class C demonstrated significantly elevated levels of numerous glial-mediated modules linked to inflammation (M26), immune function (M3, M21), and the extracellular matrix (M11, M42) (Fig. 3B and 4C). Class C was also the only class that demonstrated significant decreases in M1, a large module linked to synaptic transmission that is consistently depleted in the AD brain (Fig. 4D) (Johnson et al., 2022; Higginbotham et al., 2020; Johnson et al., 2020; Seyfried et al., 2017; Swarup et al., 2020). In contrast, Class B was distinguished from A and C by significant elevations in M1 and other neuronal modules, including M4 (synapse / GTPase activity) and M23 (GTPase activity) (Fig. 3B and 4D). These modules were largely associated with the pre-synaptic region and its associated functions (Fig. 4D). Meanwhile, post-synaptic modules (M5, M22) were significantly decreased in Classes B and C and remained relatively preserved in Class A. Collectively, these results revealed that in this heterogenous, clinically diagnosed cohort, the proteomic profiles of two-thirds of cognitively impaired cases diverged in key respects from the typical degenerative proteomic changes we have previously observed in pathologically defined tissues.
2.5. UMAP projections of cognitively intact cases support their proteomic resemblance to Class A
To better investigate the proteomic relationship of NCI cases to our three classes, we used UMAP to project the NCI cohort onto the classes based on individual proteomic profiles. Only one NCI sample from each of the five same-case pairs was included in this analysis, resulting in the projection of 215 NCI tissues. As expected, nearly 70% (n = 149) of the NCI cases clustered closely with the “control-like” Class A (NCI-A) (Fig. S4A). Of the remaining NCI cases, 12 clustered with Class B (NCI-B) and 45 with Class C (NCI-C). There were 9 cases that remained unassigned. Module expression trends observed in the MONET M1 classes were robustly replicated among the three projected NCI clusters, reflecting the signatures unique to each class (Fig. S4B–D). These NCI clusters featured no significant differences in age, sex, cognitive assessments, neuropathological burden, or APOE risk. These projection results reinforced the proteomic similarities between NCI and Class A. Yet, these data also suggested our classes of cognitive impairment may be, to some extent, predetermined during preclinical stages.
2.6. Individual protein signatures distinguish classes with high sensitivity and specificity
To identify individual proteins that best discriminate the three classes, we first performed pairwise differential expression analyses. Fig. 5 depicts these volcano plots with individual proteins colored by module membership. As expected, Classes A and C diverged the most with 3251 significantly altered proteins (p < 0.05) between them (Fig. 5A, Table S7). The “control-like” Class A featured higher levels of several M5 post-synaptic markers (VGF, SYT12, NPTX2), M10 RAS signaling molecules (RASGRF1, ARFGAP2), and M6 mitochondrial ribosome proteins (DAP3, MRPS7, MRPS9, MRPS33, MRPS34). On the other hand, Class C featured increases in numerous proteins from kinase-oriented modules (M7, M15), including MAP kinases (MAPK1, MAP2K6, MAPK3), ribosomal kinases (RPS6KA5), and diacylglycerol kinases (DGKG) (Fig. 5A). Large-fold increases in proteins linked to sugar metabolism (M25) and the extracellular matrix (M42) also distinguished Class C from A. These included several highly conserved M42 hubs (SMOC1, MDK, NTN1) strongly linked to amyloid burden and APOE-associated risk in prior studies (Johnson et al., 2022; Johnson et al., 2020). Meanwhile, Class B pairwise analyses (Fig. 5B–C) underscored its unique elevations in neuronal proteins. Several M1 and M4 members (SYN2, NPTXR, SYT17, SYNPR) were significantly increased in Class B compared to A and C.
Fig. 5. Differential expression of individual proteins reveals highly sensitive and specific classifiers.
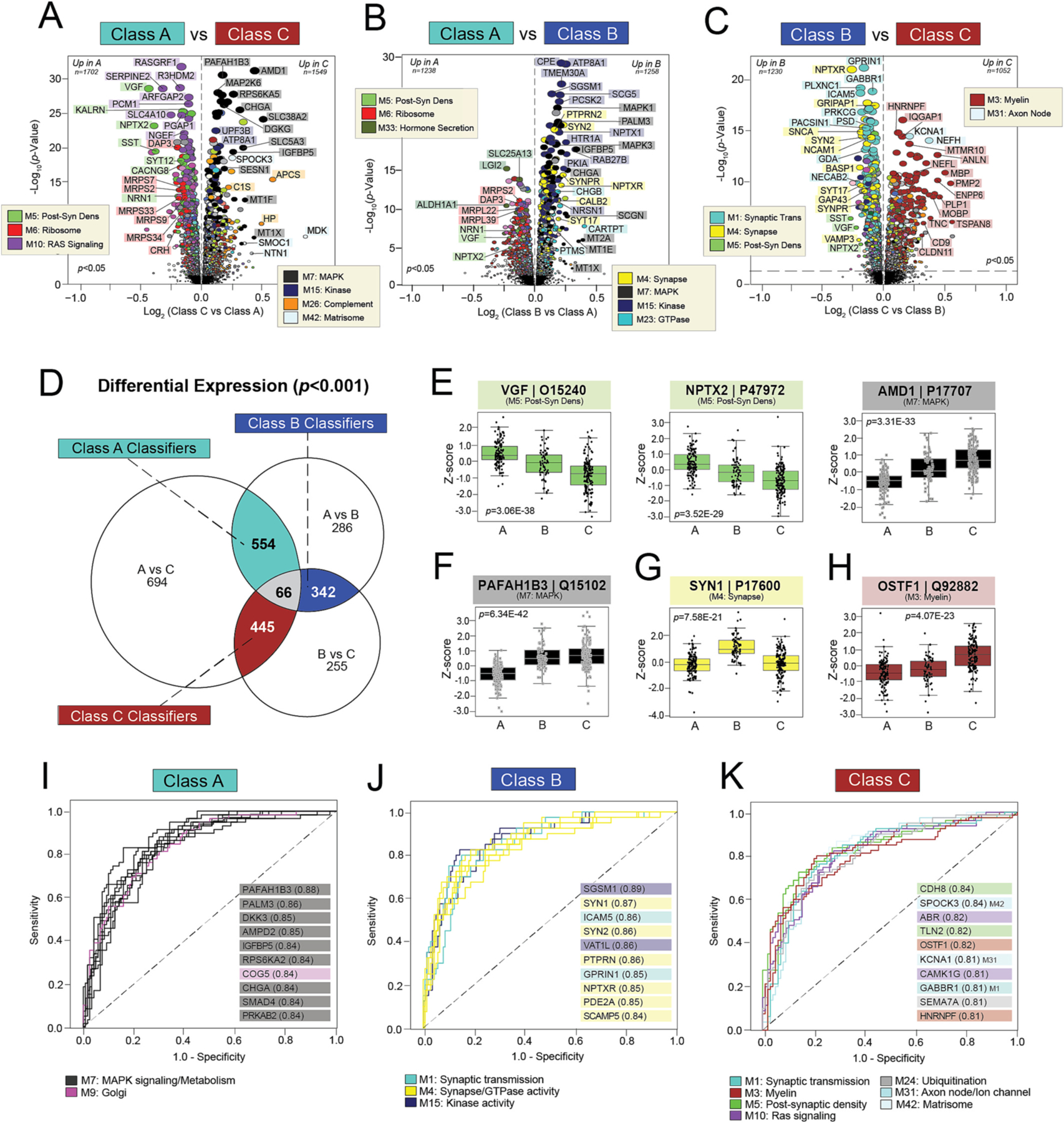
A-C) Volcano plots displaying the log2 fold change (x-axis) against the -log10 statistical p value (y-axis) for proteins differentially expressed between pairwise class comparisons. All p values across pairwise comparisons were derived by ANOVA with Bonferroni post-hoc correction. Proteins are shaded according to color of module membership. (D) Venn diagram of significantly altered proteins (p < 0.001) across pairwise class comparisons. There were 66 proteins with significant changes across all three pairwise comparisons, while hundreds of proteins were significantly altered across two of the three pairwise comparisons. The latter were deemed “classifiers”, as each was uniquely altered in one class relative to the other two. There were 554 Class A classifiers, 342 Class B classifiers, and 445 Class C classifiers. (E-H) Abundance levels (z-score) of select proteins across NCI cases and the three classes. ANOVA p values are provided for each abundance plot. Box plots represent the median and 25th and 75th percentiles, while box hinges depict the interquartile range of the two middle quartiles within a group. Data points up to 1.5 times the interquartile range from the box hinge define the extent of error bar whiskers. The 66 proteins altered across all pairwise class comparisons included neuroprotective markers with well-described links to AD (VGF, NPTX2) and those without known associations to disease (AMD1) (E). Classifiers altered in two of the three pairwise class comparisons included PAFAH1B3 for Class A, SYN1 for Class B, and OSTF1 for Class C (F-H). (I-K) ROC curves of the 10 most sensitive and specific proteins for each class by AUC values, which are included in parentheses. Proteins are shaded according to color of module membership. Abbreviations: Post-Syn Dens, Post-Synaptic Density.
A Venn diagram of significantly altered markers (p < 0.001) across pairwise class comparisons revealed 66 proteins with significant changes across all three comparisons (Fig. 5D, Table S7). These 66 markers included the neuroprotective markers VGF and NPTX2 (Tubi et al., 2021; Cao et al., 2004; Fournier et al., 2012; Licht et al., 2011; Zacchigna et al., 2008; Gora-Kupilas and Josko, 2005; Chang et al., 2010; Lee et al., 2017; Xiao et al., 2017), which both dropped significantly from Class A to B and then again from B to C (Fig. 5E). These abundance trends suggested declining neuropreservation from Class A to B to C. In contrast, there were many proteins among these 66 targets that had no known links to AD or neurodegeneration, such as adenosylmethionine decarboxylase 1 (AMD1) which significantly increased from Class A to B to C (Fig. 5E).
The Venn diagram also revealed hundreds of markers significantly altered (p < 0.001) across two of the three pairwise class comparisons (Fig. 5D, Table S8). We referred to these proteins as “classifiers”, as each was uniquely altered in one class relative to the other two. Class A featured 554 classifiers, including platelet activating factor acetylhydrolase 1b catalytic subunit 3 (PAFAH1B3) which displayed markedly decreased levels in Class A relative to B and C (Fig. 5F). Decreases in PAFAH1B3 and other members of the kinase-associated M7 and M15 comprised nearly 30% of the Class A classifiers. The remaining signatures prominently reflected the increased ribosome (M6), Golgi (M9), and Ras signaling (M10) molecules. Class B was distinguished by 342 classifiers that largely represented increases in several pre-synaptic modules (M1, M4, M23), such as synapsin (SYN1) (Fig. 5G). This neuronal protein associates closely with synaptic vesicles and plays a critical role in synaptogenesis and axon development (Cesca et al., 2010). Finally, Class C classifiers comprised 445 proteins that strongly reflected increases in proteins linked to myelin (M3) and the extracellular matrix (M11), such as osteoclast stimulating factor 1 (OSTF1) (Fig. 5H). Decreases in pre- and post-synaptic proteins (M1, M5) were also prominently featured among these Class C signatures.
We assessed the strength of these classifiers by plotting the individual ROC curve for each signature in relationship to its associated class (Fig. 5I–K, Table S8). All three classes featured numerous strong protein classifiers with AUC values above 0.8. Kinase-associated proteins (e.g., PAFAH1B3, PALM3, DKK3) were among those most sensitive and specific for Class A (Fig. 5I), while Class B was best distinguished by synaptic classifiers (e.g., SYN1, SYN2, GPRIN1, NPTXR) (Fig. 5J). Proteins with the highest AUCs for Class C reflected a more diverse set of modules, underscoring the diversity of biological dysregulation in this group. Yet, these Class C signatures still highlighted its prominent synaptic and myelin dysfunction (e.g., CDH8, TLN2, OSTF1, GABBR1, HNRNPF) (Fig. 5K). Overall, we concluded that each class featured unique protein signatures capable of distinguishing its members with high sensitivity and specificity.
2.7. Targeted mass spectrometry reinforces class-specific protein signatures
To assess the validity of our subtyping results, we acquired quantitative data from a previously published targeted MS analysis of the same cognitively impaired ROSMAP cases (Johnson et al., 2020; Yu et al., 2018). This selected reaction monitoring (SRM) analysis was performed independently by a different laboratory (Yu et al., 2018). Accordingly, the peptides targeted were chosen using criteria unrelated to the current study and not all were representative of those peptides driving our TMT protein-level alterations. Thus, we focused our validation on SRM peptides that 1) unambiguously mapped to a corresponding gene product in our TMT-MS dataset, 2) featured a gene product assigned to one of the 44 protein modules, and 3) maintained robustly significant correlations between SRM and TMT abundances (Table S9). Fig. S5A showcases the 35 validation peptides that emerged from this analysis, all maintaining SRM:TMT correlation coefficients of at least 0.3. The SRM abundances of these peptides consistently mirrored the TMT protein trends measured across classes (Fig. S5B). Class A signatures were strongly reinforced, including the notable decreases in kinase-associated proteins (M7) observed in this class compared to B and C. SRM validated two of the strongest M7 Class A signatures from our TMT ROC analysis, adenosine monophosphate deaminase 2 (AMPD2) and insulin like growth factor binding protein 5 (IGFBP5) (Fig. S5B). The uniquely increased levels of post-synaptic (M5) and RAS signaling (M10) proteins of Class A were also reinforced in the SRM dataset. Notably, SRM peptide levels of the neuroprotective marker VGF in NCI and Class A were significantly elevated compared to Classes B and C. While it was more difficult to discern the subtler differences between Classes B and C among the SRM data, certain peptides did mirror these trends (e.g., SYN3, C1QA, SPP1) (Fig. S5B). Collectively, these independent SRM results support the class-associated proteomic profiles observed in our TMT dataset.
2.8. Classes demonstrate distinct clinicopathological phenotypes
Given their robust differences in modules linked to clinicopathological traits, we hypothesized our classes would exhibit distinct clinical and pathological phenotypes. To characterize these phenotypic differences, we compared available ROSMAP disease traits directly across classes (Table S4). As expected, all three classes demonstrated significant cognitive impairment compared to NCI (Fig. 6A). Yet, Class A featured the most preserved cognition among impaired individuals. Class A also displayed the most positive cognitive slopes, indicating a slower rate of decline in these cases (Fig. 6B). Accordingly, individual proteins highly correlated to cognitive measures demonstrated starkly different levels in Class A compared to B and C (Fig. 6A–B). Post-synaptic markers of M5 (NRN1, NPTX2, OLFM1) were among those most strongly correlated to cognition and displayed precipitous declines from Class A to B and C. Several kinase-associated markers of M7 and M15 (MAP2K6, RPS6KA2, PAFAH1B3, PALM3, TMEM30A) also correlated strongly to cognitive measures but in the opposite direction, with expression levels that sharply increased from Class A to B and C. Table S10 provides a complete list of proteins significantly correlated to each trait provided for our ROSMAP cohort.
Fig. 6. Classes demonstrate different cognitive and pathological features.
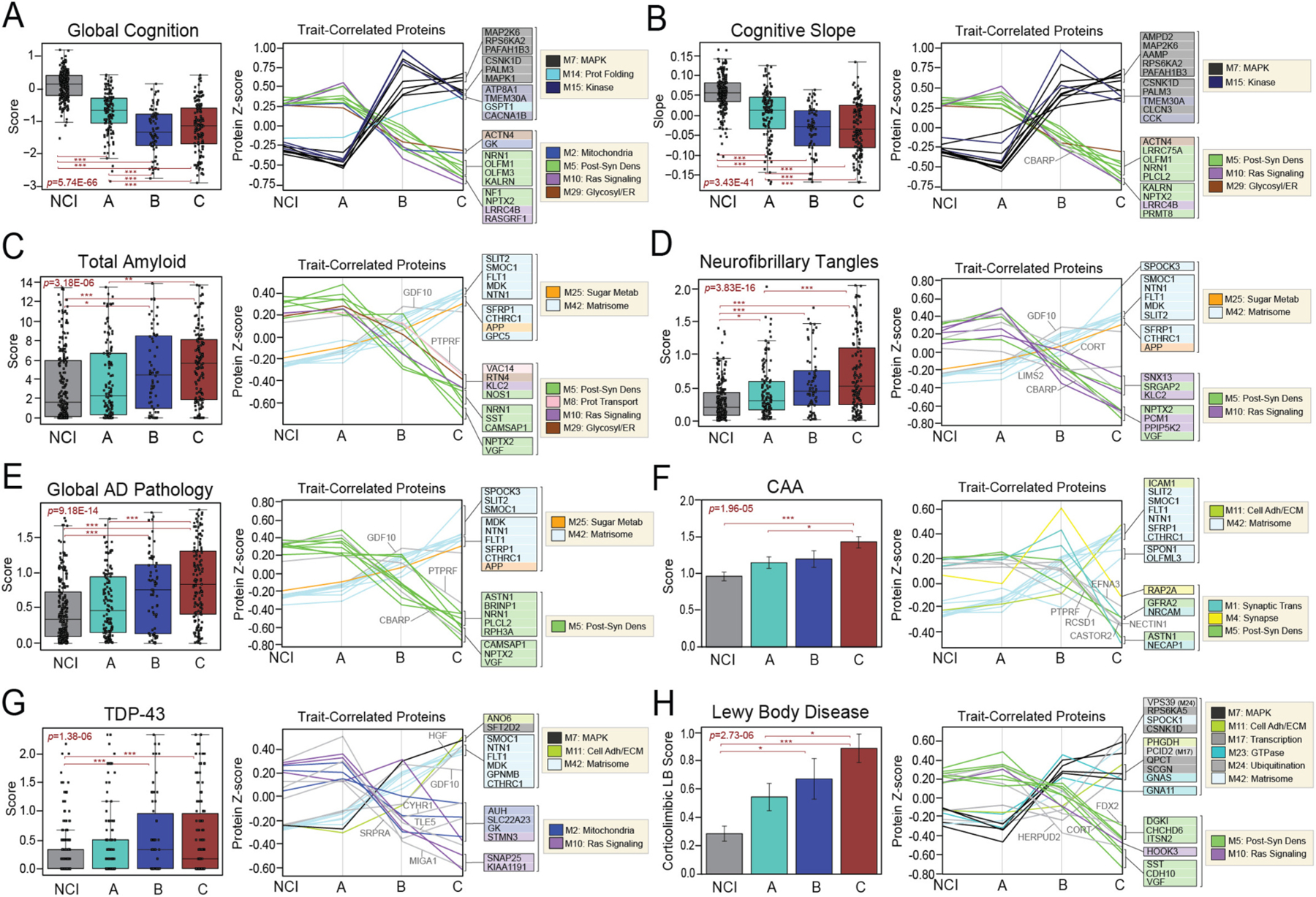
Cognitive (A-B) and neuropathological (C-H) characteristics were compared across NCI cases and the three proteomic classes. For each trait, two plots are provided. The first depicts the average scores of each trait across the four groups. The ANOVA p value across groups is provided with asterisks indicating statistically significant Tukey post hoc pairwise comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Box plots represent the median and 25th and 75th percentiles, while data points up to 1.5 times the interquartile range from the box hinge define the extent of error bar whiskers. The second plot in each panel showcases the abundance levels (z-scores) across groups of individual proteins highly correlated to that particular trait. The z-scores of the top 10 positively trait-correlated and top 10 negatively trait-correlated proteins are shown. Proteins are shaded according to color of module membership. Proteins without a module assignment are not shaded. Abbreviations: CAA, Cerebral Amyloid Angiopathy; TDP-43, TAR DNA-Binding Protein 43; Prot Folding, Protein Folding; Post-Syn Dens, Post-synaptic Density; Glycosyl, Glycosylation; ER, Endoplasmic Reticulum; Prot Transport, Protein Transport; Adh, Adhesion; ECM, Extracellular Matrix; Metab, Metabolism.
ROSMAP offers detailed pathological scoring of brain tissues using a variety of semi-quantitative and quantitative scales (Bennett et al., 2018). Fig. 6C–H showcases the levels of several pathologies across classes, including amyloid plaques, NFTs, cerebral amyloid angiopathy (CAA), TDP-43, and Lewy body inclusions. As expected, all cognitively impaired cases maintained higher levels of neuropathology compared to NCI. Of the three classes, Class A featured the lowest neuropathological burden. Its levels of amyloid, tau, and several other pathological features were on average decreased compared to Classes B and C. In contrast, Class C featured the highest burden of neuropathology among the three subtypes, surpassing A and B in average levels of NFTs, Lewy body inclusions, and CAA. Nevertheless, all three classes demonstrated a notable amount of sample-to-sample variability in AD and non-AD neuropathological burden. Proteins highly correlated to neuropathological measures were generally most altered in Class C compared to all other groups (Fig. 6C–H, Table S10). Post-synaptic (M5) and matrisome (M42) markers were strongly associated with all types of neuropathology (e.g., SST, NRN1, VGF, SMOC1, NTN1). Notably, amyloid precursor protein (APP) was among the top protein correlates of all three AD neuropathological measures, consistent with its prominent role in plaque formation.
Overall, these findings highlighted distinct clinicopathological phenotypes across our proteomic classes. Most notably, these results highlighted greater levels of cognitive stability in Class A, consistent with the robust neuroprotective trends in its proteomic profile. Meanwhile, Class C demonstrated the highest levels of neuropathology, aligning with its prominent neurodegenerative proteomic signatures.
2.9. Class C proteomic signatures strongly mirror those of high-risk ApoE4 carriers
Polymorphic alleles in the APOE gene are the strongest known genetic determinants of LOAD risk (Strittmatter et al., 1993; Corder et al., 1994; Lambert et al., 2013). Individuals carrying the E4 allele are at increased risk for AD development compared to those with the more common E3 allele. Meanwhile, a copy of the E2 allele is neuroprotective and decreases the risk of LOAD. We have previously demonstrated that APOE genotype and its associated risk strongly correlate with a variety of protein modules in the human AD brain, spanning metabolism, inflammation, synaptic activity, and other molecular functions (Dai et al., 2018). We have also shown that the expression patterns of certain modules, such as the matrisome-associated M42, are genetically regulated by the APOE locus (Johnson et al., 2022). Therefore, we hypothesized that our proteomic classes would differ in levels of APOE-related risk and associated protein signatures.
Analysis of genotype composition across classes revealed a mixture of high-risk (E3/4, E4/4), risk-neutral (E3/3), and low-risk (E2/2, E2/3) genotypes in each class. E3/3 was the most abundant genotype present throughout the dataset, comprising 60–70% of cases in each class (Fig. 7A). High-risk E4 carriers (E3/4, E4/4) were second most abundant, though less evenly distributed. Class C featured over twice as many E4 carriers (n = 46, 29%) as Classes A (n = 21, 16%) and B (n = 17, 24%). Low-risk E2 carriers (E2/2, E2/3) were notably less abundant than high-risk cases, accounting for only 12% (n = 16) of Class A, 10% (n = 8) of Class B, and 6% (n = 10) of Class C. Finally, E2/4 cases were rare and comprised no more than 3% of any class. A comparison of overall APOE-associated risk revealed all three classes featured higher risk levels compared to NCI cases. Class C displayed the highest APOE risk of the three classes, including statistically significant differences relative to Class A (Fig. 7B). A comparison of AD polygenic risk scores (PRS) across groups demonstrated similar trends, with all three classes maintaining significantly higher scores compared to NCI. Class C again featured the highest average polygenic risk of the three classes, but no longer significantly differed from Class A (Fig. 7C). After removing the APOE allele from the PRS calculation, all three classes still featured significantly higher risk compared to NCI, but the between-class risk differences became even more muted (Table S4). Collectively, these results indicated that all three classes featured significantly higher AD genetic risk relative to NCI, even independent of APOE status. Yet, risk differences between classes appeared to be driven primarily by APOE status with Class C maintaining the highest risk of the three cognitively impaired groups.
Fig. 7. Class C protein expression strongly mirrors that of ApoE4 carriers.
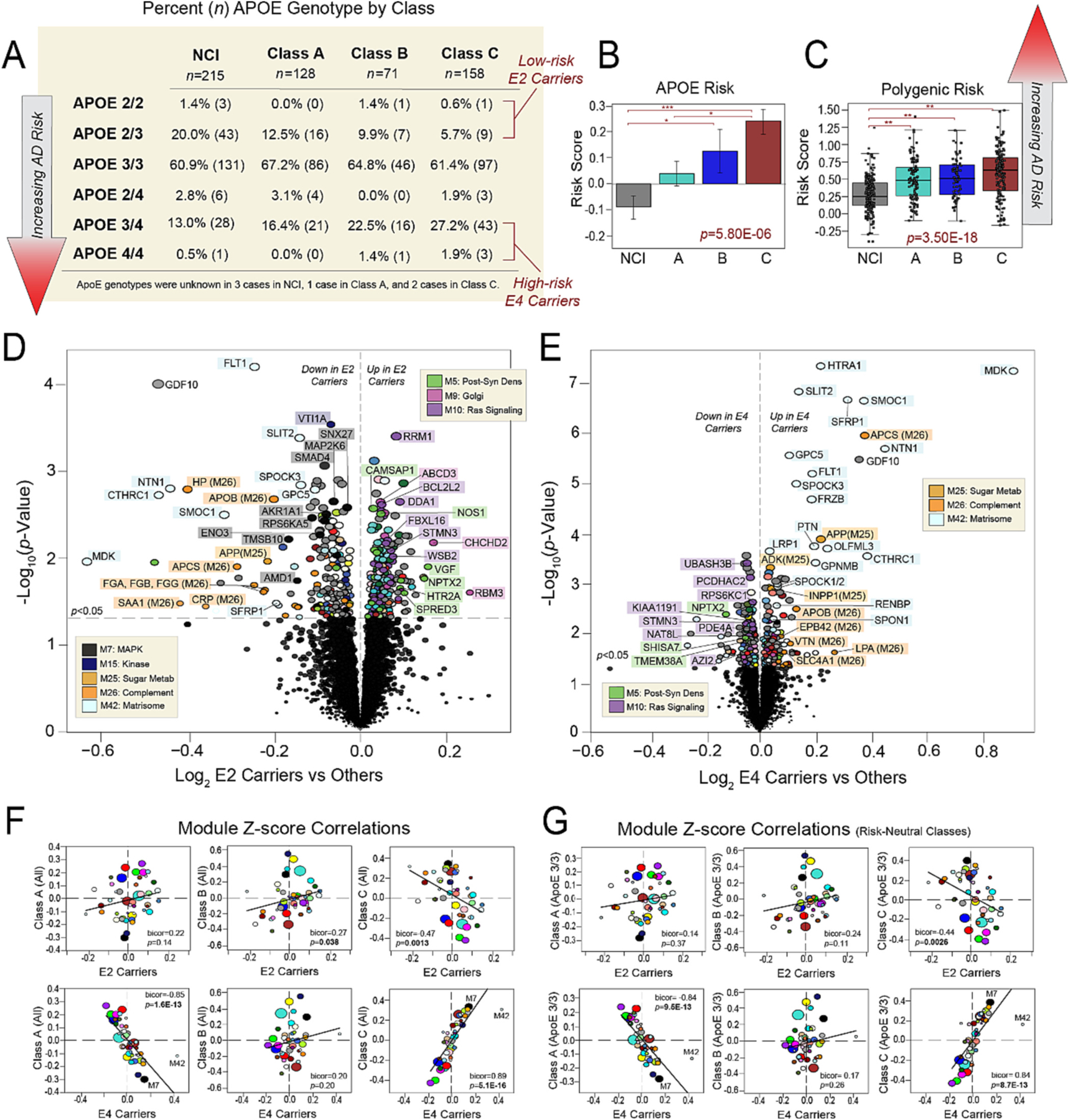
(A) Table showcasing the percentages of different APOE genotypes within NCI and each class. The corresponding number of cases with each genotype is also provided in parentheses. Cases considered low-risk E2 carriers or high-risk E4 carriers are indicated. Class C comprised twice as many high-risk E4 carriers compared to Classes A and B. (B) Comparison of average APOE risk scores across NCI and the three proteomic classes. Individual risk scores for each case were calculated by assigning −1 points to each E2 allele, 0 points to each E3 allele, and +1 points to each E4 allele. The ANOVA p value across groups is provided with asterisks indicating statistically significant Tukey post hoc pairwise comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (C) Comparison of polygenic risk scores across NCI and the three proteomic classes. The ANOVA p value across groups is provided with asterisks indicating statistically significant Tukey post hoc pairwise comparisons (*, p < 0.05; **, p < 0.01; ***, p < 0.001). (D-E) Volcano plots displaying the log2 fold change (x-axis) against the t-test-derived -log10 statistical p value (y-axis) for proteins differentially expressed in E2 carriers or E4 carriers when compared to all other cases, excepting those with E2/4 genotypes which were excluded from these analyses. Thus, (D) is a comparison of protein expression in the 34 cases with E2/2 and E2/3 genotypes to the 313 cases with E3/3, E3/4, and E4/4 genotypes, while (E) is a comparison of protein expression in the 84 cases with E3/4 and E4/4 genotypes to the 263 cases with E2/2, E2/3, and E3/3 genotypes. Proteins are shaded according to color of module membership. (F-G) Correlation plots of module abundance levels (z-scores) in E2 (E2/2, E2/3) or E4 (E3/4, E4/4) carriers to those of each proteomic class. Class-specific z-scores in (F) reflect all members of each class, while those in (G) reflect only individuals with E3/3 genotypes in each class. Bicor correlation coefficients with associated p values are shown for each correlation plot. Abbreviations: Post-Syn Dens, Post-Synaptic Density; Metab, Metabolism.
We then sought to compare risk-associated protein signatures across classes. First, we identified those protein alterations most strongly linked to APOE carrier status, regardless of class (Table S11). Fig. 7D showcases proteins significantly altered in E2 carriers (E2/2, E2/3) versus other cognitively impaired cases. E2 carriers demonstrated stark decreases in kinase related (M7, M15) proteins and increases in post-synaptic (M5), Golgi (M9), and Ras signaling (M10) markers. In contrast, E4 carriers (E3/4, E4/4) featured decreases in post-synaptic (M5) and Ras signaling (M10) proteins when compared to other cognitively impaired cases (Fig. 7E), as well as significant increases in proteins linked to sugar metabolism (M25), immune function (M26), and the matrisome (M42). As expected, hub proteins of M42 (SMOC1, MDK, NTN1) were among those markers most elevated in E4 carriers, consistent with our previous findings that this module is under control of the APOE locus (Johnson et al., 2022). Accordingly, LDL receptor related protein 1 (LRP1), another M42 member and known APOE interactor (Shinohara et al., 2017; Jaeger and Pietrzik, 2008), was also significantly elevated in E4 carriers.
To examine these risk-associated protein signatures across classes, we then correlated the proteomic profiles of E2 and E4 carriers with those of each class. E2-associated module expression was positively correlated to module expression in both Classes A and B (Fig. 7F, Table S12). However, only its correlation with Class B reached statistical significance (bicor = 0.27, p = 0.038). E2 module expression also significantly correlated to that of Class C, but in the negative direction (bicor = 0.47, p = 0.0013). In stark contrast, E4 module expression featured remarkably strong negative correlations to Class A (bicor = −0.85, p = 1.6E-13) and positive correlations to Class C module expression (bicor = 0.89, p = 5.1E-16) (Fig. 7F). E4 expression demonstrated no significant correlation to that of Class B (bicor = 0.20, p = 0.20). To ensure that these results were not driven by a minority of E2 or E4 carriers in each class, we repeated all six correlations using the module expression of only E3/3 cases in each class (Fig. 7G, Table S12). These results were nearly identical to those of the initial correlations. Notably, the strong positive association between E4 and Class C module expression was maintained (bicor = 0.84, p = 8.7E-13). Thus, we concluded that irrespective of their individual genotypes, Class C cases harbored proteomic profiles highly similar to those of high-risk E4 carriers. This supported the conclusion that with its heightened inflammatory signatures, steep cognitive slopes, and exceptionally elevated neuropathological burden, Class C reflected a high-risk state of cognitive impairment.
2.10. UMAP projections of additional cases validate proteomic classes across cohorts and brain regions
To analyze if our proteomic classes could be detected across additional cohorts and brain regions, we used previously published TMT-MS data derived from DLPFC tissues from the Banner Sun Health Research Institute and parahippocampal gyrus (PHG) Brodmann area 36 (BA36) tissues from the Mount Sinai Brain Bank (Johnson et al., 2022). In contrast to the ROSMAP tissues, detailed global cognitive assessments and clinical consensus diagnoses were not available for these cohorts. Furthermore, the available cognitive measures were not consistent across the two cohorts, with Banner limited to the Mini-Mental State Examination (MMSE) and Mount Sinai to the Clinical Dementia Rating (CDR) instrument. Thus, for consistency, we stratified the two cohorts primarily by the presence or absence of AD neuropathological change (AD-NC) (Johnson et al., 2022). Controls included cases with no significant AD-NC on neuropathological measures (CERAD 0, Braak NFT ≤ 2), while diseased cases included those with moderate to severe AD-NC (CERAD ≥ 1, Braak NFT ≥ 3). Control cases were also restricted to those with minimal to no cognitive impairment prior to death (MMSE > 24, CDR ≥ 1).
The DLPFC Banner cohort included 26 control and 150 AD-NC cases. TMT-MS across these cases resulted in the quantification 8860 proteins, of which 6087 overlapped with our ROSMAP dataset. For consistency with the ROSMAP analysis, these abundance values were regressed for age, sex, and PMI. We then employed UMAP to project the 150 AD-NC cases onto the three ROSMAP-derived classes of cognitive impairment (Fig. S6A). Most Banner cases (n = 122) segregated into Class C (Fig. S6B). Of the remaining cases, 9 were assigned to Class A and 15 to Class B. Four cases were unassigned. Module expression trends observed in the original MONET M1 classes were robustly replicated across the three projected clusters, including elevated ribosomal proteins in Class A, synaptic proteins in Class B, and inflammatory / immune proteins in Class C (Fig. S6C). The Banner classes also featured clinicopathological phenotypes consistent with those observed in the ROSMAP classes (Fig. S6D). MMSE scores were highest in Class A and lowest in Class C. As expected, all three Banner classes maintained significantly higher amyloid and tau deposition compared to controls. While CERAD scores did not significantly differ between the three Banner classes, Braak NFT scores were significantly lower in Class A compared to Class C. Finally, APOE risk trended highest in Class C but did not significantly differ between the classes.
The BA36 Mount Sinai cohort comprised 45 control and 106 AD-NC cases. TMT-MS across the Mount Sinai cohort yielded 9413 quantified proteins, of which 6245 overlapped with our ROSMAP dataset (Fig. S6E). This data was similarly regressed for age, sex, and PMI. In contrast to Banner, UMAP assigned most AD-NC Sinai cases (n = 90) to Class A, while 4 were assigned to Class B and 10 to Class C (Fig. S6F). Two cases were unassigned. Module expression trends across the projected classes replicated those of the original three cohorts (Fig. S6G). Likewise, clinicopathological traits across the three Sinai classes revealed similar trends of greater cognitive impairment, neuropathology, and APOE risk in Class C (Fig. S6H). Yet, it is notable that while Sinai Class C trended toward higher CERAD and Braak NFT scores, there were no significant differences between the three classes in these neuropathological measures. Thus, the observation that clinical measures remained significantly altered between the Sinai classes alludes to differences in cognitive resiliency that mirrored observations in the ROSMAP cases. To summarize, these findings provided further evidence that our proteomic classes and their corresponding clinicopathological profiles can be discerned not only across independent cohorts, but also across distinct brain regions.
3. Discussion
It is increasingly evident that cognitive decline in the elderly features a complex pathophysiological landscape extending beyond the hallmark pathologies of traditional disease classification. Large-scale molecular subtyping promises to resolve this heterogeneity and enhance diagnostic and therapeutic precision. To this end, we used an unbiased proteomic approach to subtype nearly 400 ROSMAP brain tissues from clinically diagnosed MCI and AD cases. We resolved three classes among these cognitively impaired cases, each driven by proteomic changes across a variety of cell types and biological ontologies. All classes featured a mix of mildly impaired and demented individuals, indicating clusters driven by more than clinical severity at death. Accordingly, further examination indicated these proteomic classes reflected differences across a combination of various clinicopathological measures. We were able to detect these proteomic classes and their corresponding clinicopathological phenotypes across independent brain cohorts and distinct brain regions, supporting the validity of our results.
The current study represented a departure from our prior brain tissue analyses, which have focused on defining the network-based proteomic signatures of AD neuropathological changes. These prior network studies were performed in carefully screened tissue cohorts, excluding cases that did not meet strict criteria for amyloid and tau deposition. Collectively, these analyses have identified highly reproducible disease-associated changes across tissue cohorts, establishing a strong global proteomic framework for the AD brain. Yet, in the current more heterogenous population of clinically stratified MCI and AD ROSMAP tissues, only a third of diseased cases (i.e., Class C) harbored signatures fully consistent with this “typical” AD-associated proteomic profile, which includes hallmarks of stark synaptic loss and heightened glial activation. In contrast, approximately two-thirds of cases in our current cognitively impaired cohort featured “atypical” proteomic features indicative of preserved or heightened synaptic transmission and homeostatic glial activity.
AD neuropathological burden did not entirely explain this typical versus atypical proteomic stratification. While Class C did harbor the greatest average levels of global AD pathology, Classes A and B also featured robust elevations in amyloid and tau compared to NCI. In addition, the differences observed in AD pathology between Classes B and C were not statistically significant. Furthermore, all three ROSMAP classes harbored quite a bit of sample-to-sample variability in AD pathological measures, each including a subset of cases with minimal amyloid and tau burden. These findings indicated that factors beyond AD neuropathological changes were contributing to typical versus atypical classification. This was also true of non-AD pathology, which was on average highest in Class C but also demonstrated significant sample-to-sample variability within classes. Classes B and C were highly comparable across various measures of non-AD inclusions, notably featuring nearly identical TDP-43 levels. Thus, while higher burdens of AD and non-AD pathology appeared to increase the odds of a “typical” Class C profile, this was not the sole determinant of this classification.
In the case of Class A, having an atypical proteomic profile correlated strongly to clinical hallmarks of resilience. Class A, which strongly mirrored NCI cases in its proteomic signatures, featured the slowest rate of cognitive decline of the three subtypes. Its strongest proteomic signatures were derived from modules associated with MAPK (M7) and RAS (M10) signaling. Of all 44 modules, these two maintained the strongest correlations to cognitive trajectory, revealing potential target pathways for disease modification. RAS signaling molecules regulate various aspects of the MAPK pathway, and both feature crosstalk with other signaling cascades to influence cellular transcription, translation, and proliferation (Santarpia et al., 2012; Degirmenci et al., 2020; Kim and Choi, 2015). Dysfunction in these pathways has been strongly linked to tumorigenesis and cancer development, highlighting their importance in maintaining cellular homeostasis. Accordingly, Class A was most strongly distinguished from B and C by diminished levels of several M7 cancer-associated signaling proteins, such as PAFAH1B3, DKK3, and AMPD2. These results suggest that preventing elevations in these signaling proteins may promote molecular resilience and milder, less aggressive cognitive phenotypes.
In contrast, the atypical proteomic profile of Class B did not reflect such a clear clinical benefit, as its cognitive trajectory was more aggressive and very similar to Class C. It is possible that Class B represents a transitional stage from A to C. Class B demonstrated clear degenerative changes relative to Class A, including increases in kinase modules (M7, M15) and decreases in RAS signaling proteins (M10). Yet, Class B lacked the hallmarks of heightened glial activation observed in Class C. The expression of known neuroprotective markers also suggested Class B was intermediate to A and C. Levels of VGF and NPTX2, neuroprotective markers that typically decrease in the degenerating brain (Tubi et al., 2021; Cao et al., 2004; Fournier et al., 2012; Licht et al., 2011; Zacchigna et al., 2008; Gora-Kupilas and Josko, 2005; Chang et al., 2010; Lee et al., 2017; Xiao et al., 2017), were highest in A and lowest in C with Class B directly in between. However, not all aspects of the Class B proteomic profile reflected this progressive continuum. Most notably, this class featured elevations distinct from A and C in several neuronal modules (M1, M4, M23). These Class B neuronal signatures strongly reflected pre-synaptic functions, including neurotransmitter transport, GTPase activity, and signal transmission. Interestingly, none of these modules correlated strongly to any genetic, clinical, or neuropathological traits. Therefore, it is unclear what function these synaptic signatures serve for Class B and whether they represent a discrete synaptic event on a transitional continuum from A to C.
Meanwhile, cluster projections of NCI cases suggested that rather than existing on a strictly progressive continuum, these proteomic classes may to some extent be preclinically determined. Most NCI cases mirrored the proteomic profiles of Class A, supporting the association between these proteomic signatures and cognitive stability. However, approximately one-fourth of NCI cases harbored Class B and C proteomic signatures, suggesting a small subset of individuals may display a proteomic predisposition for more aggressive cognitive deterioration had they lived longer. It is important to note that we regressed variability related to baseline characteristics (i.e., age, sex, PMI) from our initial proteomic dataset so that subtyping variability would focus as much as possible on disease-related alterations. Accordingly, the demographics of the three NCI clusters did not significantly differ. Yet, we also did not find any significant differences between these NCI groups with respect to clinical status, neuropathological burden, and genetic risk. This suggests that these NCI proteomic classifications may be driven by disease-related factors beyond traditional clinicopathological traits. In addition, this highlights a potentially unique role for proteomic subtyping in preclinical risk stratification.
Examination of our classes in relationship to APOE-associated proteomic signatures indicated that Class C reflects a high-risk molecular state. Class C module expression highly correlated to that of high-risk ApoE4 carriers. This was true regardless of whether the Class C member carried an E4 allele or not. We also observed strong anti-correlations between Class A and E4 carrier module expression, consistent with its milder, less aggressive disease course. Meanwhile, Class B module expression was not correlated at all to that of E4 carriers and only weakly to that of E2 carriers. Of note, it is possible that given our generally low numbers of E2 carriers among cognitively impaired cases (n 34), we were simply underpowered to detect more robust correlations to these low-risk protein signatures. This would explain why E2 proteomic signatures mirrored several trends observed in Class A (e.g., elevated RAS signaling and post-synaptic proteins) but the two failed to demonstrate statistically significant correlations.
Overall, these results align with recent transcriptomic subtyping analyses that have also identified “atypical” RNA signatures among MCI and AD brains (Neff et al., 2021; Zheng and Xu, 2021). However, several key modules differentially expressed across our proteomic classes are not observed in the AD transcriptomic network (Johnson et al., 2022). One example is M7 MAPK signaling, a module highly linked to cognitive trajectory and whose hubs were strong Class A classifiers. Despite these robust disease associations, this module is not preserved in the AD transcriptomic network (Johnson et al., 2022; Seyfried et al., 2017; Swarup et al., 2020). Another such module not reflected in the AD transcriptome is M42 matrisome (Johnson et al., 2022), which demonstrated remarkably strong neuropathologic associations and whose hubs were sensitive and specific for Class C. Thus, our results support the utility of proteomic subtyping and its ability to offer novel insights when resolving the complex molecular heterogeneity of cognitive decline.
We regionally focused our subtyping analysis primarily on the DLPFC. Evidence indicates this is one of the earliest brain regions affected in AD, as it demonstrates cortical thinning and neuropathological changes even in preclinical stages (Dickerson et al., 2009; Bakkour et al., 2009). Furthermore, in our prior network analyses of AD tissues, we have found that module structure and disease-associated alterations in the DLPFC are highly preserved across other diseased brain regions, including the temporal cortex and precuneus (Johnson et al., 2022; Johnson et al., 2020). Our validation studies in the current study support these prior findings, as we were able to identify all three classes among the temporal lobe Mount Sinai tissues. Furthermore, these temporal lobe clusters mirrored the clinicopathological phenotypes observed in the ROSMAP classes. Yet, these results do not completely rule out the possibility that region-specific variables influence proteomic classification. In future subtyping efforts, it will be important to examine multiple regions within individual subjects to better determine regional contributions to classification. In addition, while module characterizations provided insight into the cell type populations driving class distinctions, our bulk tissue approach lacked the cellular and temporal resolution to thoroughly deconstruct cell-specific contributions (Rayaprolu et al., 2021). Thus, subtyping analyses involving single cell proteomic approaches could go beyond regional differences and elucidate critical factors at the cellular level that impact disease classification.
Among other limitations, the current study was limited largely to non-Hispanic white individuals. Therefore, it is unclear if the same classes would be detected in a more racially diverse analysis. Growing evidence indicates that CSF tau and other molecular markers of AD require adjustments for race (Morris et al., 2019; Howell et al., 2017), suggesting this variable could significantly impact pathophysiological classification of disease. While we regressed variability due to age and sex from our proteomic classifications in favor of identifying distinctions driven by disease-related traits, we acknowledge that this methodology could also be viewed as a limitation. We have previously found that age and sex have limited impact on disease-associated module trends in the network AD brain proteome (Johnson et al., 2020). Nevertheless, it will be important to further explore how these demographic variables intersect with proteomic classification in future subtyping efforts. Finally, we are aware that our subtyping results may be subject to limitations inherent to our chosen clustering algorithm. We selected the MONET M1 algorithm given its reputation as a well-validated, top-performing module identification tool that excels at clustering high dimensional datasets (Choobdar et al., 2019; Tomasoni et al., 2020). Yet, to avoid the pitfalls of relying heavily on one clustering method, we ensured that our classes could also be replicated using UMAP, a dimension reduction algorithm with a differing approach. The ability of this additional tool to independently cluster nearly all ROSMAP cases identically to MONET M1 greatly enhanced the validity of our results.
Future subtyping efforts will require examining the representation of these brain-derived classes in the CSF and plasma proteomes. Our recent studies integrating the AD brain and biofluid proteomes have revealed that many key disease-associated brain modules are highly represented in CSF (Higginbotham et al., 2020; Johnson et al., 2020). In fact, we have shown that alterations in the AD CSF proteome reflect a diverse range of brain-based pathophysiology, including synaptic, vascular, inflammatory, and metabolic dysfunction (Higginbotham et al., 2020). Thus, the AD CSF proteome promises to mirror the brain with distinct classes featuring unique protein signatures and clinicopathological phenotypes. A recent subtyping analysis of AD CSF based on the levels of ~700 proteins identified subtypes of disease with distinct molecular signatures (Tijms et al., 2020). However, larger-scale integration studies of the brain and CSF proteomes are required to identify biofluid subtypes that best reflect cortical hallmarks of pathophysiology. Such efforts to refine classes with close links to brain-based molecular dysfunction will be key to meaningfully advancing diagnostic and therapeutic precision in AD.
In conclusion, our unbiased proteomic analysis revealed three classes of cognitive impairment among elderly individuals that differ across a diverse array of pathophysiologies and maintain distinct clinicopathological phenotypes. These proteomic profiles reflected elements of clinical severity, rate of deterioration, AD and non-AD pathological burden, genetic-associated risk, and additional unknown factors. Notably, these classes highlighted how each one of these clinical or pathological traits in isolation may not accurately stratify the protein-level biological heterogeneity underlying late-onset cognitive decline. Further application of unbiased proteomic subtyping promises to better define the molecular basis for the diverse disease mechanisms and endophenotypes of cognitive impairment and meaningfully impact the diagnostic, prognostic, and therapeutic precision of AD and related dementias.
4. Materials and methods
4.1. Brain tissues
4.1.1. ROSMAP tissues
A total of 610 dorsolateral prefrontal cortex (DLPFC) tissues from Brodmann area 9 (BA9) were obtained from the autopsy collections of the Religious Orders Study or Rush Memory and Aging Project (ROSMAP) (Bennett et al., 2005; Bennett et al., 2014; Bennett et al., 2018). Both studies were approved by the Institutional Review Board of Rush University Medical Center. All participants signed an informed consent, an Anatomic Gift Act, and a repository consent allowing their resources to be repurposed with appropriate inter-institutional agreements. ROSMAP features community-based cohorts, which recruit older individuals without known dementia from United States religious orders, lay retirement centers, senior and subsidized housing communities, and church groups. These participants are then followed longitudinally with cognitive batteries, biospecimen collection, and finally brain autopsy (Bennett et al., 2005; Bennett et al., 2014; Bennett et al., 2018). All participants are assigned a clinical consensus cognitive diagnosis (cogdx) at death, derived by study experts blinded to postmortem neuropathology. The cogdx scale includes values of 1 (NCI), 2 (MCI and no other cause of cognitive impairment [in addition to AD]), 3 (MCI and another cause of cognitive impairment [in addition to AD]), 4 (AD and no other cause of cognitive impairment), 5 (AD and another cause of cognitive impairment), and 6 (other dementia). All diagnoses of MCI were judged to have cognitive impairment on neuropsychology testing but did not meet criteria for dementia. All diagnoses of AD dementia met criteria for possible or probable AD based on National Institute of Neurological and Communicative Disorders and Stroke and Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) guidelines (Bennett et al., 2006; McKhann et al., 1984). ROSMAP cases are richly characterized using a variety of clinical and pathological traits that were used to phenotype the proteomic classes generated in our analysis (Bennett et al., 2005; Bennett et al., 2018). Available clinical traits included a Mini-Mental State Examination (MMSE) score (Folstein et al., 1975) and a Global Cognition score, the latter derived from the composite results of a 19-item neuropsychological battery (Wilson et al., 2015). Postmortem neuropathological traits of interest included neuritic plaque distribution, which was scored according to the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) criteria (Mirra et al., 1991), and extent of neurofibrillary tangle pathology, which was assessed with the Braak staging system (Braak and Braak, 1991). Global AD pathology scores comprised quantitative summaries of neuritic plaque, diffuse plaque, and NFT levels across several cortical brain regions (Bennett et al., 2018). Other neuropathologic scores and clinical traits were made in accordance with established criteria and guidelines as previously described (Bennett et al., 2005; Bennett et al., 2018; Bennett et al., 2006). All sample metadata are provided in https://www.synapse.org/ADsubtype. In the current study, only samples with cogdx classifiers of 1–5 were ultimately included in secondary analyses following TMT-MS. Among these were five individuals with NCI and one with AD who each contributed two independent DLPFC samples, resulting in six same-case sample pairs. Samples with cogdx classifiers of 6 were excluded from secondary analyses. We also excluded samples with cogdx classifiers that did not align with cognitive battery scores prior to death.
4.1.2. Banner and Mount Sinai tissues
Validation analyses were performed using DLPFC brain tissues derived from the Banner Sun Health Research Institute and parahippocampal gyrus (PHG) Brodmann area 36 (BA36) tissues from the Mount Sinai Brain Bank. Available clinicopathological traits for these cases were more limited compared to ROSMAP. CERAD and Braak NFT scores were available for both cohorts. Cognitive measures were limited to MMSE for Banner cases and the 5-point Clinical Dementia Rating (CDR) scale (Morris, 1993) for Mount Sinai cases. All sample metadata are provided in https://www.synapse.org/ADsubtype. Across both cohorts, control cases were defined as those with no significant AD-NC on neuropathological measures (CERAD 0, Braak NFT ≤ 2), while diseased cases included those with moderate to severe AD-NC (CERAD ≥1, Braak NFT ≥ 3). Control cases were also restricted to those with minimal to no cognitive impairment prior to death (MMSE > 24, CDR ≥ 1). As referenced in subsequent Methods sections, the details of sample preparation and TMT-MS analysis have been previously described in detail for both cohorts (Johnson et al., 2022; Bai et al., 2020). While Banner cases were analyzed by our group at Emory (Johnson et al., 2022), Mount Sinai cases were prepared and analyzed at a different center using a similar mass spectrometry pipeline (Bai et al., 2020).
4.2. Brain tissue homogenization and protein digestion
Tissue homogenization of ROSMAP cases was performed essentially as described (Seyfried et al., 2017; Ping et al., 2018). For each sample, approximately 100 mg (wet weight) of brain tissue was homogenized in 500 μL of 8 M urea lysis buffer (8 M urea, 10 mM Tris, 100 mM NaH2PO4, pH 8.5) with HALT protease and phosphatase inhibitor cocktail (ThermoFisher). Tissues were added to lysis buffer immediately after excision within Rino sample tubes (NextAdvance) supplemented with ~100 μL of stainless-steel beads (0.9 to 2.0 mm blend, NextAdvance). Tissues were then homogenized using a Bullet Blender (NextAdvance) at 4 °C with 2 full 5 min cycles. The lysates were transferred to new Eppendorf Lobind tubes and sonicated for 3 cycles comprising 5 s of active sonication at 30% amplitude, followed by 15 s on ice. Sample lysates were then centrifuged for 5 min at 15,000x g and the supernatant transferred to new tubes. Protein concentration was determined by bicinchoninic acid (BCA) assay (Pierce). For protein digestion, 100 μg of each sample was aliquoted and volumes normalized with additional lysis buffer. Samples were reduced with 1 mM dithiothreitol (DTT) at room temperature for 30 min followed by 5 mM iodoacetamide (IAA) alkylation in the dark for another 30 min. Lysyl endopeptidase (Wako) at 1:100 (w/w) was added and digestion allowed to proceed overnight. Samples were then 7-fold diluted with 50 mM ammonium bicarbonate. Trypsin (Promega) was then added at 1:50 (w/w) and digestion was carried out for another 16 h. The peptide solutions were acidified to a final concentration of 1% (vol/vol) formic acid (FA) and 0.1% (vol/vol) trifluoroacetic acid (TFA) and subsequently desalted with a 30 mg HLB column (Oasis). Prior to sample loading, each HLB column was rinsed with 1 mL of methanol, washed with 1 mL 50% (vol/vol) acetonitrile (ACN), and equilibrated with 2 × 1 mL 0.1% (vol/vol) TFA. Samples were then loaded onto the column and washed with 2 × 1 mL 0.1% (vol/vol) TFA. Elution was performed with 2 volumes of 0.5 mL 50% (vol/vol) ACN. An equal amount of peptide from each sample was aliquoted and pooled as the global internal standard (GIS), a fraction of which was TMT labeled and included in each batch as described below. Banner and Mount Sinai cases were homogenized and digested as previously described (Johnson et al., 2022; Bai et al., 2020).
4.3. Isobaric Tandem Mass Tag (TMT) peptide labeling
The 610 ROSMAP samples were labeled and analyzed in two separate sets, referred to as Set1 (n = 400) and Set2 (n = 210) throughout the Methods. Prior to TMT labeling, cases were randomized into batches by select covariates, including age, sex, PMI, and diagnosis. Set1 samples were randomized into 50 batches and peptides were labeled using TMT 10-plex kits (ThermoFisher 90,406). Set2 samples were randomized into 14 batches and peptides were labeled using TMTpro 16-plex kits (ThermoFisher 44,520). Each batch in Set1 included two TMT channels with labeled GIS standards, while each batch in Set2 comprised only one TMT channel with a labeled GIS standard. Labeling of sample peptides was performed as previously described (Johnson et al., 2018; Ping et al., 2018; Ping et al., 2020). Briefly, each sample (100 μg of peptides) was resuspended in 100 mM triethylammonium bicarbonate (TEAB) buffer (100 μL). TMT labeling reagents (5 mg) were equilibrated to room temperature and anhydrous ACN (256 μL) added to each reagent channel. Each channel was gently vortexed for 5 min. A volume of 41 μL from each TMT channel was transferred to each peptide solution and allowed to incubate for 1 h at room temperature. The reaction was quenched with 5% (vol/vol) hydroxylamine (8 μL) (Pierce). All channels were then dried by SpeedVac (LabConco) to approximately 150 μL, diluted with 1 mL of 0.1% (vol/vol) TFA, and acidified to a final concentration of 1% (vol/vol) FA and 0.1% (vol/vol) TFA. Labeled peptides were desalted with a 200 mg C18 Sep-Pak column (Waters). Prior to sample loading, each Sep-Pak column was activated with 3 mL of methanol, washed with 3 mL of 50% (vol/vol) ACN, and equilibrated with 2 × 3 mL of 0.1% TFA. After sample loading, each column was washed with 2 × 3 mL 0.1% (vol/vol) TFA followed by 2 mL of 1% (vol/vol) FA. Elution was performed with 2 volumes of 1.5 mL 50% (vol/vol) ACN. The eluates were then dried to completeness by SpeedVac. Randomization and multiplex TMT labeling of Banner and Mount Sinai cases were performed as previously described (Johnson et al., 2022; Bai et al., 2020).
4.4. High-pH off-line fractionation
High pH fractionation of all ROSMAP cases was performed essentially as described (Ping et al., 2018; Mertins et al., 2018) with slight modifications. Dried samples were resuspended in high pH loading buffer (0.07% vol/vol NH4OH, 0.045% vol/vol FA, 2% vol/vol ACN) and loaded onto an Agilent ZORBAX 300 Extend-C18 column (2.1 mm × 150 mm with 3.5 μm beads). An Agilent 1100 HPLC system was used to carry out fractionation. Solvent A comprised 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 2% (vol/vol) ACN. Solvent B comprised 0.0175% (vol/vol) NH4OH, 0.01125% (vol/vol) FA, and 90% (vol/vol) ACN. Sample elution was performed over a 58.6-min gradient at a flow rate of 0.4 mL/min with solvent B ranging from 0 to 60%. The gradient was 100% solvent A for 2 min, then 0% to 12% solvent B over 6 min, then 12% to 40% over 28 min, then 40% to 44% over 4 min, then 44% to 60% over 5 min, and then held constant at 60% solvent B for 13.6 min. A total of 96 individual equal volume fractions were collected across the gradient and subsequently pooled by concatenation (Mertins et al., 2018) into 24 fractions for Set1 and 48 fractions for Set2. The fractions were then dried to completeness by SpeedVac. Off-line fractionation of Banner and Mount Sinai cases was performed as previously described (Johnson et al., 2022; Bai et al., 2020).
4.5. Mass spectrometry analysis
4.5.1. ROSMAP Set1
MS analysis was performed as previously described (Johnson et al., 2022; Johnson et al., 2020). Briefly, fractions were resuspended in an equal volume of loading buffer (0.1% FA, 0.03% TFA, 1% ACN) and analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) essentially as previously described (Lambert et al., 2010) with slight modifications. Peptide eluents were separated on a self-packed C18 (1.9 μm, Dr. Maisch, Germany) fused silica column (25 cm × 75 μM internal diameter (ID), New Objective, Woburn, MA) by a Dionex UltiMate 3000 RSLCnano liquid chromatography system (ThermoFisher Scientific). Buffer A comprised water with 0.1% (vol/vol) FA, while buffer B comprised 80% (vol/vol) ACN in water with 0.1% (vol/vol) FA. Elution was performed over a 180-min gradient at a flow rate of 225 nL/min with buffer B ranging from 1 to 99%. The gradient was 3% to 7% buffer B over 5 min, then 7% to 30% over 140 min, then 30% to 60% over 5 min, then 60% to 99% over 2 min, then 99% buffer B for 8 min, and then back to 1% buffer B for an additional 20 min to equilibrate the column. Peptides were monitored on an Orbitrap Fusion mass spectrometer (ThermoFisher Scientific). The mass spectrometer was set to acquire in data-dependent mode using the top speed workflow with a cycle time of 3 s. Each cycle consisted of one full scan (MS1) followed by as many MS/MS (MS2) scans that could fit within the time window. MS1 scans were collected at 120,000 resolution (200 m/z) with 350–1500 m/z range, 4 × 105 automatic gain control (AGC) target, and 50-ms maximum injection time. The most intense ions were selected for MS2 higher energy collision-induced dissociation (HCD) at 30,000 resolution with 0.7 m/z isolation width, 38% collision energy, 5 × 104 AGC target, and 100-ms maximum injection time. Five of the 50 TMT batches were run on the Orbitrap Fusion mass spectrometer using synchronous precursor selection MS3-based quantitation (SPS-MS3) as previously described (Ping et al., 2018). In these 5 batches, HCD MS2 spectra were acquired at a resolution of 60,000 with 1.6 m/z isolation width, 35% collision energy, 5 × 104 AGC target, and 50 ms maximum ion time. For all batches, dynamic exclusion was set to exclude previously sequenced peaks for 20 s within a 10-ppm isolation window.
4.5.2. ROSMAP Set2
All fractions were resuspended in an equal volume of loading buffer (0.1% FA, 0.03% TFA, 1% ACN) and analyzed by LC-MS/MS as described above but with slight modifications. Peptide eluents were separated on a self-packed C18 (1.9 μm, Dr. Maisch, Germany) fused silica column (15 cm × 75 μM ID, New Objective, Woburn, MA) by an EASY-nLC 1200 liquid chromatography system (ThermoFisher Scientific). Buffers A and B were identical in composition to those used for Set1. Elution was performed over a 45-min gradient at a flow rate of 400 nL/min with buffer B ranging from 1 to 99%. The gradient was 5% to 35% buffer B over 37 min, then 35% to 99% over 1 min, then 99% buffer B for 8 min, and then back to 1% buffer B for an additional 7 min to equilibrate the column. Peptides were monitored on a Q-Exactive HFX mass spectrometer (ThermoFisher Scientific). MS1 scans were at collected at 120,000 resolution (200 m/z) with 410–1600 m/z range, 3 × 106 AGC target, and 50-ms maximum injection time. The top 20 most intense ions were selected for HCD at 45,000 resolution with 0.7 m/z isolation width, 32% collision energy, 2 × 105 AGC target, and 96-ms maximum injection time. Dynamic exclusion was set to exclude previously sequenced peaks for 20 s within a 10-ppm isolation window.
4.5.3. Banner and Mount Sinai
TMT-MS of Banner and Mount Sinai cases was performed as previously described (Johnson et al., 2022; Bai et al., 2020).
4.6. Database searches and protein quantification
All RAW files acquired from TMT-MS of the ROSMAP cases were searched against a human reference protein database using the Proteome Discoverer suite (version 2.4, ThermoFisher Scientific). MS2 spectra were searched against the UniProtKB human proteome database containing Swiss-Prot human reference protein sequences (20,338 target proteins downloaded in 2019). Due to variations in their mass tags, Set1 (TMT10) and Set 2 (TMTpro) were independently searched using the same parameters according to our previously published protocols (Johnson et al., 2022; Johnson et al., 2020; Ping et al., 2018). Briefly, Sequest HT search engine was used and parameters were specified as follows: fully tryptic specificity; maximum of two missed cleavages; minimum peptide length of 6; fixed modifications for TMT tags on lysine residues and peptide N-termini (+229.162932 Da for TMT10 or + 304.2071 Da for TMTpro) and carbamidomethylation of cysteine residues (+57.02146 Da); variable modifications for oxidation of methionine residues (+15.99492 Da), deamidation of asparagine and glutamine (+0.984 Da), and phosphorylation of serine, threonine and tyrosine (+79.9663 Da); precursor mass tolerance of 20 ppm; and fragment mass tolerance of 0.05 Da for MS2 spectra collected with the Orbitrap. A fragment mass tolerance of 0.5 Da was used for MS2 spectra acquired with the SPS-MS3 method. Percolator was used to filter peptide spectral matches (PSMs) and peptides to a false discovery rate (FDR) of <1%. Following spectral assignment, peptides were assembled into proteins and were further filtered based on the combined probabilities of their constituent peptides to a final FDR of 1%. A multi-consensus in Proteome Discoverer was then performed to achieve parsimonious protein grouping across both sets of samples. In cases of redundancy, shared peptides were assigned to the protein sequence in adherence with the principles of parsimony. As default, the top matching protein or “master protein” was the protein with the largest number of unique peptides and smallest value in the percent peptide coverage (i.e., the longest protein). Reporter ions were quantified using an integration tolerance of 20 ppm with the most confident centroid setting. Only parsimonious peptides were considered for quantification. Banner and Mount Sinai raw files were also searched and quantified using Proteome Discoverer and similar parameters as previously described (Johnson et al., 2022).
4.7. Controlling for batch-specific variance across proteomics datasets
A tunable median polish approach (TAMPOR) was used to remove technical batch variance in the proteomic data (Dammer et al., 2023). We have previously described this approach in detail (Dammer et al., 2023) and applied it to prior large, multi-cohort proteomic datasets (Johnson et al., 2022; Johnson et al., 2020). Following multi-consensus database search and protein quantification of the ROSMAP cases, TAMPOR was used to first normalize batch effects within Set1 and Set2 separately. After removal of these intra-set batch effects, all samples except set-specific GIS samples were processed jointly with TAMPOR into a single reassembled consensus sample protein matrix using the median of within-cohort pathology-free control cases as the central tendency, enforcing that the population of all log2(ratio) output for control samples within the final 610 ROSMAP samples would tend toward 0. TAMPOR was also applied separately to the Banner and Mount Sinai datasets to normalize batch effects prior to the projection validation analyses. Additional details on the data input and TAMPOR output can be found on https://www.synapse.org/#!Synapse:syn32280722.
4.8. Regression of covariates
The quantitative data from all cases in the ROSMAP, Banner, and Mount Sinai cohorts were subjected to nonparametric bootstrap regression by subtracting the trait of interest (age at death, sex, or PMI) times the median estimated coefficient from 1000 iterations of fitting for each protein in the log2(abundance) matrix as previously described (Johnson et al., 2022). Ages at death used for regression were uncensored.
4.9. Sample network generation with MONET M1 algorithm
The three top-performing methods from the Disease Module Identification DREAM Challenge were compiled in the MONET toolbox and released to the public for use (https://github.com/BergmannLab/MONET.git) (Tomasoni et al., 2020). We selected the M1 method from this toolbox to build a sample-wise network. The M1 method uses the Girvan-Newman modularity optimization method to group like features into modules (Newman and Girvan, 2004). MONET M1 has expanded on the traditional modularity optimization to a multiresolution approach by searching the network at multiple topological scales. The authors have added the resistance parameter, r, which averts genes from joining modules. If r = 0 the method returns to Newman and Girvan’s original modularity optimization; r > 0 reveals network substructure; and r < 0 network superstructure (Arenas et al., 2008). The parameter r, is fit to four user-provided hyperparameters: minimum module size, maximum module size, desired average degree, and desired average degree tolerance to produce a network described by the parameters.
After batch-correction and regression as described above, n = 610 cases were split into two groups defined by the ROSMAP cognitive diagnosis score: cognitively impaired cases (n = 377, cogdx ∈ {2, 3, 4, 5}) and non-cognitively impaired/other cases (n = 233, cogdx ∈ {1, 6}). An expression data matrix of the cognitively impaired cases n = 77 and n = 7, 723 log2 protein abundance was created (proteins with greater 50% missingness in the 377 cases were removed). The adjacency function in WeiGhted Correlation Network Analysis (WGCNA) was used to build the adjacency matrix with parameters: soft threshold power = 8, type = “signed”, corFnc = “bicor”, and the corOptions parameter set to use pairwise complete correlation (Langfelder and Horvath, 2008). The soft threshold power was determined using scale free topology analysis based on the following two guidelines: 1) The power in a plot of power (x) vs R2 (y) should be where the R2 has approached an asymptote, usually near or above 0.80, and 2) the mean and median connectivity at that power should not be exceedingly high, preferably around 100 or less. The power at which these criteria are met is a tradeoff between removing correlations due to chance and maintaining as many correlations in the data as possible for the clustering algorithm to distinguish modules. As M1 takes an edge list as input, the adjacency upper triangle correlation values were used to populate the weights of unique pairwise correlations in the edge list. No sparsification of the edge list was applied.
The hyperparameters were optimized using a grid search by varying minimum module size, i ∈ {5, 10, 25, 50}, maximum module size, j ∈ {100, 150, 200, 250, 300, 350, 376}, and desired average degree, k ∈ {25, 50}. The desired average degree tolerance was left at the default value of 0.2. The optimal parameter set was defined as the set that minimized the percentage of cases not assigned to a module and the maximum module size. The final parameters selected were i = 5, j = 200, k = 50, which built a network with 3 modules and 5.31% (n = 20) cases not assigned to a module. This final network was used in to define the three subtypes of AD observed in the ROSMAP cohort.
Using the expression data matrix and the modules assignment list, module eigenvectors were defined, using the moduleEigengenes function in WGCNA. The eigenvector is the module’s first principal component and explains covariance of all cases within each module [citation]. Using the signedKME function in WGCNA, a table of bicor correlations between each case and each module eigenvector was obtained; this module membership measure is defined as kME. Additional details of the data input and MONET M1 output can be found on https://www.synapse.org/#!Synapse:syn32567811.
4.10. MONET M1 bootstrap validation
To validate the robustness of the MONET M1 clustering, we performed 100 rounds of bootstrapped reclustering. In each iteration, we withheld a random 20% of the samples and employed MONET M1 on the remaining 80% of cases. Due to the decrease in sample number, the hyperparameters were adjusted to i = 5, j = 160, k = 50, where the maximum module size was decreased by 20%. If there are truly unique molecular subtypes, then repeated clustering should produce similar sets of clustered samples. To assess the probability of sample pairs clustering together, we calculated the rate at which each pair is assigned to them same cluster, termed the paired percentage. The average paired percentage per cluster and variance of the percentage were tracked for the 100 bootstrap steps to ensure the bootstrapping was well converged. A heatmap of the paired percentage shows that the clustering is stable and repeated rounds of MONET M1 reproduces the three observed subtypes. A hypergeometric Fisher’s exact test (FET) was performed on each of the bootstrap steps against the original MONET M1 cluster assignments and showed class-specific overlap. Along with the rate that sample pairs clustered together, the reproducibility was also assessed using the module preservation function in WGCNA. Z-summary composite preservation scores were obtained using the MONET M1 network (n = 377) as the template versus each bootstrap step, with 100 permutations. Random seed was set to 1 for reproducibility, and the quickCor option was set to 0. The summary z scores were then averaged across the 100 bootstrap steps and the standard deviation was calculated. All three subtypes were preserved across the 100 bootstrap replicates (above the blue line q = 0.05).
4.11. Uniform manifold approximation and projection (UMAP) dimension reduction
Supervised dimensionality reduction was performed using UMAP (umap-learn v0.5.2) in Python (v3.9) with the following settings: n_neighbors = 10, n_components = 2, metric = Euclidean, and min_dist = 0.1. A supervised UMAP embedding was generated for the 357 cases in the MONET M1 classes using the n = 7723 log2(protein abundance) as features and the three MONET M1 classes as target labels. The three resultant clusters mirror those of MONET M1 network analysis with only 1 case out of the 357 assigned to a cluster different from its original class. After generating the three target clusters, ROSMAP NCI, Banner AD-NC, and Mount Sinai AD-NC cases were projected separately onto the UMAP dimension 1 and UMAP dimension 2 space. Projected cases were assigned to a class by determining to which cluster the case was closet in Euclidean space. If the case was >3.5 distance away from all clusters, then it was determined to be unassigned. UMAP assignments for all cases are included with sample metadata provided in https://www.synapse.org/ADsubtype.
4.12. Biological organization of protein expression matrix
To highlight the biological differences between the three classes produced by MONET M1, the proteins used as features in the network (n = 7, 723) were grouped into the protein modules generated in our deepest AD consensus network (Johnson et al., 2022). For each protein module, proteins were sorted by kME. Protein modules were then organized into relatedness order determined by previous WGCNA analysis. This highlighted the differences in biological ontologies across the MONET M1 classes. For visualization, samples within the classes were also sorted by class-specific kME. Therefore, the first sample is the “eigensample” for the corresponding class.
4.13. ROC analyses
For each protein, a support vector machine classifier was used to classify individuals by class using a one-vs-rest strategy. Due to the multi-class nature of the model, each class identifier was binarized prior to training. To choose the model for which predictions are made, the one-vs-rest heuristically chooses the binary classifier with the highest confidence. After a model was created, the receiver operating characteristic (ROC) metric was used to evaluate each classifier. The area under the curve (AUC) was calculated for each peptide in each classifier. The peptides were then sorted based on the AUC values and the top ten were plotted for each class.
4.14. Targeted mass spectrometry analysis
Targeted MS validation was performed using previously published SRM results of the same ROSMAP DLPFC tissues (Yu et al., 2018). This SRM analysis was performed independently by a different laboratory. Detailed descriptions of sample preparation, SRM-MS analysis, and data quantitation are described elsewhere (Johnson et al., 2020; Yu et al., 2018). We examined those SRM peptides that 1) unambiguously mapped to a corresponding gene product in our TMT-MS dataset, 2) featured a gene product assigned to one of the 44 protein modules, and 3) maintained robustly significant correlations between SRM and TMT abundances. A biweight midcorrelation value and p-value statistic was calculated for each SRM:TMT pair using the WGNCA R package. Box plots were generated of the SRM log2(Light/Heavy) ratio data by assigning the samples to their MONET M1 classes and plotting the distribution in each. The class wise distribution of log2(abundance) values of the corresponding gene product in our TMT data was visualized as box plots.
4.15. Protein correlation to traits
To assess which proteins most strongly correlated to clinicopathological traits, the WGCNA R netScreen function was used. All 7723 proteins were correlated to the provided ROSMAP traits across the 357 clustered samples. The top 10 positive and top 10 negatively correlated proteins were manually filtered from the analysis. The z-scores for these proteins were plotted across the NCI group and each of the three classes.
4.16. Polygenic risk score calculation
AD polygenic risk scores (PRS) were calculated for 530 of the 610 ROSMAP participants using: a) summary statistics from a genome-wide association study (GWAS) on 21,982 AD cases and 41,944 cognitively normal controls (Kunkle et al., 2019) and b) TOPMed imputed genotype data from the 530 ROSMAP participants. Polygenic risk scoring was accomplished with continuous shrinkage (PRS-cs-auto: https://github.com/getian107/PRScs), a method that uses a Bayesian approach to define effect weights rather than discrete SNP selection. The global shrinkage parameter was learnt from the data, as the source GWAS was sufficiently powered. A second set of risk scores were also calculated excluding the APOE region (Chr19:45,016,911-46,418,605; GRCh37). A 100 kb buffer region was applied to either end of the region as defined in the source GWAS. Any variants on chromosome 19 located in this region were removed from the summary statistics prior to processing. New Bayesian posteriors were then estimated for the remaining variants. The distribution of PRS across classes was visualized as boxplots.
4.17. Other statistics
Statistical analyses were performed in Python v3.7 and visualized using matplotlib package v3.5.1. Correlations were performed using the biweight midcorrelation function as implemented in the WGCNA R package. Comparisons between two groups were performed by t-test. Comparisons among three or more groups were performed with one-way ANOVA with Tukey or Bonferroni pairwise comparison of significance. P values were adjusted for multiple comparisons by false discovery rate (FDR) correction where indicated. Box plots represent the median and 25th and 75th percentiles, while data points up to 1.5 times the interquartile range from the box hinge define the extent of error bar whiskers. Data points outside this range are identified as outliers.
Supplementary Material
Funding
This study was supported by the following National Institutes of Health funding mechanisms: NIA AG063755, NIA AG068024, R01AG061800, and U01AG061357 (N.T.S). ROSMAP is supported by NIA grants P30AG10161, P30AG72975, R01AG15819, R01AG17917, U01AG46152, and U01AG61356 (D.A.B.). ROSMAP resources can be requested at https://www.radc.rush.edu. D.F. is supported by the Krembil Foundation, the Koerner New Scientist Award, the Canadian Institutes of Health Research, the CAMH Foundation, and the Canadian Foundation for Innovation. E.ST. is supported by the Ontario Graduate Scholarship.
Footnotes
CRediT authorship contribution statement
Lenora Higginbotham: Conceptualization, Methodology, Investigation, Writing – original draft, Writing – review & editing. E. Kathleen Carter: Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft, Writing – review & editing. Eric B. Dammer: Conceptualization, Methodology, Investigation, Formal analysis, Writing – original draft. Rafi U. Haque: Methodology, Formal analysis. Erik C.B. Johnson: Conceptualization, Writing – review & editing. Duc M. Duong: Methodology, Investigation. Luming Yin: Investigation. Philip L. De Jager: Resources. David A. Bennett: Resources. Daniel Felsky: Formal analysis. Earvin S. Tio: Formal analysis. James J. Lah: Conceptualization, Supervision. Allan I. Levey: Conceptualization, Writing – review & editing, Funding acquisition, Supervision. Nicholas T. Seyfried: Conceptualization, Methodology, Investigation, Writing – original draft, Writing – review & editing, Funding acquisition, Supervision.
Declaration of Competing Interest
A.I.L, N.T.S., and D.M.D. are co-founders of Emtherapro Inc. The authors declare no conflicts of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2023.106286.
Data availability
Raw mass spectrometry data from the ROSMAP dorsolateral prefrontal cortex tissues can be found at https://www.synapse.org/#!Synapse:syn17015098. Pre- and post-processed protein expression data and case traits related to this manuscript are available at https://www.synapse.org/ADsubtype. The algorithm used for batch correction is fully documented and available as an R function, which can be downloaded from https://github.com/edammer/TAMPOR. The results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AMP-AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the AMP-AD Target Discovery Program and other programs supported by the National Institute on Aging to enable open-science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/#/DataAccess/Instructions). Additional ROSMAP resources can be requested at www.radc.rush.edu.
References
- Andrews SJ, Fulton-Howard B, Goate A, 2020. Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet. Neurol 19 (4), 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenas A, Fernandez A, Gómez S, 2008. Analysis of the structure of complex networks at different resolution levels. New J. Phys 10 (5), 053039. [Google Scholar]
- Association, A.s, 2018. Alzheimer’s disease facts and figures. Alzheimer’s & Dementia 14 (3), 367–429, 2018. [Google Scholar]
- Bai B, et al. 2020. Deep multilayer brain proteomics identifies molecular networks in Alzheimer’s disease progression. Neuron 105 (6), 975–991 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkour A, Morris JC, Dickerson BC, 2009. The cortical signature of prodromal AD: regional thinning predicts mild AD dementia. Neurology 72 (12), 1048–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellenguez C, et al. 2022. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet 54 (4), 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, et al. 2005. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology 25 (4), 163–175. [DOI] [PubMed] [Google Scholar]
- Bennett DA, et al. 2006. Decision rules guiding the clinical diagnosis of Alzheimer’s disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 27 (3), 169–176. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Yu L, De Jager PL, 2014. Building a pipeline to discover and validate novel therapeutic targets and lead compounds for Alzheimer’s disease. Biochem. Pharmacol 88 (4), 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett DA, et al. 2018. Religious orders study and rush memory and aging project. J. Alzheimers Dis 64 (s1), S161–S189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle PA, et al. 2021. To what degree is late life cognitive decline driven by age-related neuropathologies? Brain 144 (7), 2166–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82 (4), 239–259. [DOI] [PubMed] [Google Scholar]
- Brayne C, et al. 2009. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge City over-75s Cohort (CC75C) study. J. Alzheimers Dis 18, 645–658. [DOI] [PubMed] [Google Scholar]
- Cao L, et al. 2004. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat. Genet 36 (8), 827–835. [DOI] [PubMed] [Google Scholar]
- Cesca F, et al. 2010. The synapsins: key actors of synapse function and plasticity. Prog. Neurobiol 91 (4), 313–348. [DOI] [PubMed] [Google Scholar]
- Chang MC, et al. 2010. Narp regulates homeostatic scaling of excitatory synapses on parvalbumin-expressing interneurons. Nat. Neurosci 13 (9), 1090–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choobdar S, et al. 2019. Assessment of network module identification across complex diseases. Nat. Methods 16 (9), 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, et al. 1994. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet 7 (2), 180–184. [DOI] [PubMed] [Google Scholar]
- Dai J, et al. 2018. Effects of APOE genotype on brain proteomic network and cell type changes in Alzheimer’s disease. Front. Mol. Neurosci 11, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammer EB, Seyfried NT, Johnson ECB, 2023. Batch correction and harmonization of -omics datasets with a tunable median polish of ratio. Front Syst. Biol 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirmenci U, Wang M, Hu J, 2020. Targeting aberrant RAS/RAF/MEK/ERK signaling for cancer therapy. Cells 9 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson BC, et al. 2009. The cortical signature of Alzheimer’s disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb. Cortex 19 (3), 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efthymiou AG, Goate AM, 2017. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener 12 (1), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emon MA, et al. 2020. Clustering of Alzheimer’s and Parkinson’s disease based on genetic burden of shared molecular mechanisms. Sci. Rep 10 (1), 19097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR, 1975. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res 12 (3), 189–198. [DOI] [PubMed] [Google Scholar]
- Fournier NM, et al. 2012. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology 63 (4), 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gora-Kupilas K, Josko J, 2005. The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol 43 (1), 31–39. [PubMed] [Google Scholar]
- Guerreiro R, et al. 2012. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med 368 (2), 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasin Y, Seldin M, Lusis A, 2017. Multi-omics approaches to disease. Genome Biol 18 (1), 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham L, et al. 2019. Network analysis of a membrane-enriched brain proteome across stages of Alzheimer’s disease. Proteomes 7 (3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higginbotham L, et al. 2020. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci. Adv 6 (43). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Herz J, Bu G, 2012. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med 2 (3), a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell JC, et al. 2017. Race modifies the relationship between cognition and Alzheimer’s disease cerebrospinal fluid biomarkers. Alzheimers Res. Ther 9 (1), 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurst C, et al. 2022. Integrated Proteomics Identifies Neuritin (NRN1) as a Mediator of Cognitive Resilience to Alzheimer’s Disease bioRxiv, 2022.06.15.496285. [DOI] [PMC free article] [PubMed]
- Jaeger S, Pietrzik CU, 2008. Functional role of lipoprotein receptors in Alzheimer’s disease. Curr. Alzheimer Res 5 (1), 15–25. [DOI] [PubMed] [Google Scholar]
- Johnson ECB, et al. 2018. Deep proteomic network analysis of Alzheimer’s disease brain reveals alterations in RNA binding proteins and RNA splicing associated with disease. Mol. Neurodegener 13 (1), 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, et al. 2020. Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat. Med 26 (5), 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ECB, et al. 2021. Large-Scale Deep Multi-Layer Analysis of Alzheimer’s Disease Brain Reveals Strong Proteomic Disease-Related Changes Not Observed at the RNA Level bioRxiv, 2021.04.05.438450. [DOI] [PMC free article] [PubMed]
- Johnson ECB, et al. 2022. Large-scale deep multi-layer analysis of Alzheimer’s disease brain reveals strong proteomic disease-related changes not observed at the RNA level. Nat. Neurosci 25 (2), 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, et al. 2013. Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med 368 (2), 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapasi A, DeCarli C, Schneider JA, 2017. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol 134 (2), 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karanth S, et al. 2020. Prevalence and clinical phenotype of quadruple misfolded proteins in older adults. JAMA Neurol 77 (10), 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawas CH, et al. 2015. Multiple pathologies are common and related to dementia in the oldest-old: the 90+ study. Neurology 85 (6), 535–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ, 2015. Compromised MAPK signaling in human diseases: an update. Arch. Toxicol 89 (6), 867–882. [DOI] [PubMed] [Google Scholar]
- Kunkle BW, et al. 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet 51 (3), 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J-C, et al. 2010. Implication of the immune system in Alzheimer’s disease: evidence from genome-wide pathway analysis. J. Alzheimers Dis 20, 1107–1118. [DOI] [PubMed] [Google Scholar]
- Lambert JC, et al. 2013. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet 45 (12), 1452–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Horvath S, 2008. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformat 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, et al. 2017. Presynaptic neuronal pentraxin receptor organizes excitatory and inhibitory synapses. J. Neurosci 37 (5), 1062–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licht T, et al. 2011. Reversible modulations of neuronal plasticity by VEGF. Proc. Natl. Acad. Sci. U. S. A 108 (12), 5081–5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier T, Güell M, Serrano L, 2009. Correlation of mRNA and protein in complex biological samples. FEBS Lett 583 (24), 3966–3973. [DOI] [PubMed] [Google Scholar]
- McKhann G, et al. 1984. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34 (7), 939–944. [DOI] [PubMed] [Google Scholar]
- Mertins P, et al. 2018. Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography-mass spectrometry. Nat. Protoc 13 (7), 1632–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, et al. 1991. The consortium to establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41 (4), 479–486. [DOI] [PubMed] [Google Scholar]
- Morris JC, 1993. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43 (11), 2412–2414. [DOI] [PubMed] [Google Scholar]
- Morris JC, et al. 2019. Assessment of racial disparities in biomarkers for Alzheimer disease. JAMA Neurol 76 (3), 264–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff RA, et al. 2021. Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci. Adv 7 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MEJ, Girvan M, 2004. Finding and evaluating community structure in networks. Phys. Rev. E 69 (2), 026113. [DOI] [PubMed] [Google Scholar]
- Ping L, et al. 2018. Global quantitative analysis of the human brain proteome in Alzheimer’s and Parkinson’s disease. Sci Data 5, 180036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping L, et al. 2020. Global quantitative analysis of the human brain proteome and phosphoproteome in Alzheimer’s disease. Sci Data 7 (1), 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayaprolu S, et al. 2021. Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology 46 (1), 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santarpia L, Lippman SM, El-Naggar AK, 2012. Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin. Ther. Targets 16 (1), 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders AM, et al. 1993. Association of apolipoprotein E allele ϵ4 with late-onset familial and sporadic Alzheimer’s disease. Neurology 43 (8), 1467. [DOI] [PubMed] [Google Scholar]
- Schneider JA, et al. 2007. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology 69 (24), 2197–2204. [DOI] [PubMed] [Google Scholar]
- Seyfried NT, et al. 2017. A multi-network approach identifies protein-specific co-expression in asymptomatic and symptomatic Alzheimer’s disease. Cell Syst 4 (1), 60–72 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara M, et al. 2017. Role of LRP1 in the pathogenesis of Alzheimer’s disease: evidence from clinical and preclinical studies. J. Lipid Res 58 (7), 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, et al. 1993. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. U. S. A 90 (5), 1977–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swarup V, et al. 2020. Identification of conserved proteomic networks in neurodegenerative dementia. Cell Rep 31 (12), 107807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki S, et al. 2022. Inferring protein expression changes from mRNA in Alzheimer’s dementia using deep neural networks. Nat. Commun 13 (1), 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tijms BM, et al. 2020. Pathophysiological subtypes of Alzheimer’s disease based on cerebrospinal fluid proteomics. Brain 143 (12), 3776–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasoni M, et al. 2020. MONET: a toolbox integrating top-performing methods for network modularization. Bioinformatics 36 (12), 3920–3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tubi MA, et al. 2021. Regional relationships between CSF VEGF levels and Alzheimer’s disease brain biomarkers and cognition. Neurobiol. Aging 105, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umoh ME, et al. 2018. A proteomic network approach across the ALS-FTD disease spectrum resolves clinical phenotypes and genetic vulnerability in human brain. EMBO Mol. Med 10 (1), 48–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM, 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet 13 (4), 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RS, et al. 2015. Temporal course and pathologic basis of unawareness of memory loss in dementia. Neurology 85 (11), 984–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingo AP, et al. 2019. Large-scale proteomic analysis of human brain identifies proteins associated with cognitive trajectory in advanced age. Nat. Commun 10 (1), 1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao MF, et al. 2017. NPTX2 and cognitive dysfunction in Alzheimer’s disease. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, et al. 2021. Dimensionality reduction by UMAP reinforces sample heterogeneity analysis in bulk transcriptomic data. Cell Rep 36 (4), 109442. [DOI] [PubMed] [Google Scholar]
- Yu L, et al. 2018. Targeted brain proteomics uncover multiple pathways to Alzheimer’s dementia. Ann. Neurol 84 (1), 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, et al. 2020. Cortical proteins associated with cognitive resilience in community-dwelling older persons. JAMA Psychiatry 77 (11), 1172–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zacchigna S, Lambrechts D, Carmeliet P, 2008. Neurovascular signalling defects in neurodegeneration. Nat. Rev. Neurosci 9 (3), 169–181. [DOI] [PubMed] [Google Scholar]
- Zheng C, Xu R, 2021. Molecular subtyping of Alzheimer’s disease with consensus non-negative matrix factorization. PLoS One 16 (5), e0250278. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw mass spectrometry data from the ROSMAP dorsolateral prefrontal cortex tissues can be found at https://www.synapse.org/#!Synapse:syn17015098. Pre- and post-processed protein expression data and case traits related to this manuscript are available at https://www.synapse.org/ADsubtype. The algorithm used for batch correction is fully documented and available as an R function, which can be downloaded from https://github.com/edammer/TAMPOR. The results published here are in whole or in part based on data obtained from the AMP-AD Knowledge Portal (https://adknowledgeportal.synapse.org). The AMP-AD Knowledge Portal is a platform for accessing data, analyses, and tools generated by the AMP-AD Target Discovery Program and other programs supported by the National Institute on Aging to enable open-science practices and accelerate translational learning. The data, analyses, and tools are shared early in the research cycle without a publication embargo on secondary use. Data are available for general research use according to the following requirements for data access and data attribution (https://adknowledgeportal.synapse.org/#/DataAccess/Instructions). Additional ROSMAP resources can be requested at www.radc.rush.edu.