

Abstract
Background
Declined skeletal muscle mass and function are inevitable consequences of long‐term diabetes and bring about many adverse events. Muscle fibre atrophy and interstitial fibrosis are major pathological manifestations of diabetic sarcopenia. Stimulator of interferon genes (STING) participates in various metabolic diseases. We aimed to explore whether and how STING regulates the above pathological manifestations of diabetic sarcopenia.
Methods
Wild‐type and STINGgt/gt C57BL/6J mice and C2C12 myotubes were used to study the role of STING in the regulation of diabetic sarcopenia and the underlying mechanisms.
Results
STING was abnormally activated in diabetic muscles and in PA‐treated myotubes (P < 0.01 for all parameters). The diabetic mice demonstrated decreased forelimb grip strength, lean mass, muscle weight and hanging impulse, which were improved by STING deficiency due to alleviated muscle fibre atrophy and interstitial fibrosis (P < 0.05 for all parameters). STING deficiency alleviated muscle fibre atrophy through the following mechanisms. Firstly, STING deficiency or inhibition increased the contents of pDRP1Ser616, PINK1, Parkin and LC3‐II, decreased p62 content, and increased the amount of mito‐Keima fluorescent dots at 578 nm in diabetic state (P < 0.05 for all parameters), suggesting improved mitofission and mitophagy. Secondly, STING deficiency or inhibition increased the expression of pAKTSer473 and GLUT4 post‐insulin change in diabetic state (P < 0.05 for all), indicating alleviated insulin resistance (IR). Mechanically, STING deficiency or inhibition increased peroxisome proliferator activated receptors γ (PPARγ) protein content by reducing the degradation of ubiquitinated PPARγ through the proteasome pathway and thus increased the expression of fatty acid oxidation (FAO)‐related proteins in diabetic state (P < 0.05 for all parameters). Decreased expression of FAO‐related proteins caused by PPARγ inhibition abolished the improvements in mitofission, mitophagy and IR achieved by STING inhibition in PA‐treated myotubes and thus promoted muscle fibre atrophy (P < 0.05 for all parameters). STING deficiency alleviated interstitial fibrosis by decreasing TGFβ1 expression in diabetic state and TGFβ1 promoted the fibrogenic differentiation of fibro‐adipogenic progenitors (P < 0.05 for all parameters). PPARγ inhibition abolished the effect of STING inhibition on reducing TGFβ1 content in PA‐treated myotubes (P < 0.01).
Conclusions
STING deficiency exerted protective effects in diabetic sarcopenia by inhibiting the degradation of ubiquitinated PPARγ through the proteasome pathway and enhancing PPARγ‐mediated FAO, which alleviated muscle fibre atrophy by promoting mitophagy and ameliorating IR, and alleviated interstitial fibrosis by reducing TGFβ1 production and suppressing the fibrogenic differentiation of fibro‐adipogenic progenitors.
Keywords: Atrophy, Diabetic sarcopenia, Fibrosis, PPARγ, STING
Introduction
Sarcopenia is a degenerative syndrome characterized by declined muscle mass, muscle strength and physical performance and leads to frailty, falls, physical disability and even mortality. 1 , 2 Diabetes mellitus (DM) has been reported as a common risk factor for sarcopenia. Diabetic patients demonstrated accelerated degeneration of skeletal muscle strength and mass compared with non‐diabetic patients. 3 And this state is termed as diabetic sarcopenia. Muscle fibre atrophy and interstitial fibrosis are major pathological features of diabetic sarcopenia. 4 Muscle fibre atrophy directly leads to declined skeletal muscle mass and strength, whereas interstitial fibrosis limits the contraction and extension of the muscles and triggers muscle loss. 5 , 6 Exploring the underlying mechanisms is the key to prevent the progressive degeneration of muscle function and improve the prognosis of diabetic patients.
Both impaired mitophagy and insulin resistance (IR) are contributing factors for muscle fibre atrophy in the diabetic state. Mitophagy is a form of autophagy that selectively removes damaged mitochondria, and mitofission is an initiation step for mitophagy. 7 Mitophagy initiated by mitofission is an important mechanism to maintain mitochondrial homeostasis. Mitophagy is impaired in long‐term diabetic state, 8 and impaired mitophagy is a promoting factor for muscle fibre atrophy. 9 Methods of improving mitophagy are reported to effectively alleviate muscle fibre atrophy. 10 In addition to improving mitophagy, using insulin sensitizers that alleviate IR could significantly ameliorate muscle atrophy in diabetic mice. 3 Therefore, promoting mitophagy and alleviating IR may be effective targets for the treatment of skeletal muscle atrophy, and it is important to explore the upstream mechanisms.
Increased fibrogenic differentiation of PDGFRα+ fibro‐adipogenic progenitors (FAPs) are the key reason for interstitial fibrosis in the diabetic state. FAPs are a major kind of stem cells in skeletal muscles with both fibrogenic and adipogenic differentiation potentials. 11 They can differentiate into fibroblasts in the pro‐fibrotic environment and promote interstitial fibrosis. 11 As recently reported, the collagens abnormally deposited in the muscle interstitium in the diabetic state mainly derived from the FAPs. 12 Therefore, reducing the fibrogenic differentiation of FAPs may become an effective intervention strategy to alleviate muscle fibrosis in diabetic state.
STING might be the core molecule in promoting diabetic myopathy. STING is an endoplasmic reticulum resident protein involved in regulating apoptosis, pyroptosis, inflammation and metabolic processes. 13 Cytosolic DNA fragments are classic activators of STING. They are recognized by cyclic GMP‐AMP synthase (cGAS) and transformed to cyclic GMP‐AMP (cGAMP), which activates the STING pathway by causing the phosphorylation of STING and interferon regulatory factor 3 (IRF3) with the help of TANK binding kinase‐1 (TBK1). 14 Normally, DNA appears in the nucleus and mitochondria. However, in the state of DM, DNA fragments gradually accumulate in the cytoplasm, 15 forming a microenvironment that promotes the continuous activation of STING. Besides, the mRNA content of STING was highly elevated in serum of pre‐diabetic and diabetic patients. 16 STING deficiency was reported to promote autophagy, 17 alleviate IR 18 and inhibit pathological fibrosis. 19 However, whether and how STING participates in diabetic sarcopenia remain unclear. In this study, we established type 2 diabetes mellitus (T2DM) models in wild‐type and STING‐deficient mice to investigate whether STING participates in diabetic sarcopenia by affecting muscle fibre atrophy and interstitial fibrosis and to analyse the possible mechanisms.
Methods
Animals
STING‐deficient (STINGgt/gt) mice (background: C57BL/6J) were purchased from the Jackson Laboratory. Simply, a missense mutation was chemically induced in exon 6, resulting in an isoleucine‐to‐asparagine change in amino acid 199 of the protein. Wild‐type (WT) and STINGgt/gt C57BL/6J mice at 6 weeks of age were randomized into control and diabetic groups using the random number table method. Mice in the control groups (18 WT mice and 18 STINGgt/gt mice) were raised with normal chow diet containing 10% kcal fat, and mice in the diabetic groups (18 WT mice and 18 STINGgt/gt mice) were raised with high fat diet (HFD) containing 60% kcal fat. After 12 weeks of HFD, the intraperitoneal glucose tolerance test (IPGTT) and intraperitoneal insulin tolerance test (IPITT) were performed. Next, these mice were injected intraperitoneally with citrate buffer or streptozotocin (75 mg/kg) respectively and continuously raised with the former kind of diet for another 12 weeks. After 2 weeks of streptozotocin administration, mice whose random blood glucose were more than 16.7 mmol/L and manifested with polydipsia and polyuria were defined as diabetic mice. The detailed methods of IPGTT, IPITT and streptozotocin administration were shown in the supporting information. All the mice were housed in a specific pathogen‐free and temperature‐controlled environment under a 12/12‐h light–dark cycle with free access to food and water until they were 30 weeks old and were used in various assays described below at the end of the experiment.
Exercise performance
Eight mice in each group were randomly selected for forelimb grip strength test and hanging grid test. The mouse was trained to grasp the horizontal bar connected to a dynamometer (SHSIWI, SK‐10) and was pulled backwards gently. The forelimb grip strength was the maximum value recorded by the dynamometer until the mouse left the bar. The mouse was placed in the centre of a 45 × 45 cm grid (bar thickness, 2 mm; mesh, 18 mm) and turned upside down. The hanging time was recorded as the time from the beginning of the inversion until the mouse fell off, and the hanging impulse was calculated as the product of the hanging time and body weight. Both the tests were repeated thrice for each mouse with a >30‐min interval and the results were averaged.
Dual energy X‐ray absorptiometry body composition analysis
Five mice that were randomly selected from each group were anaesthetized, weighed and attached to the foam board with tape. Lean mass were measured by the dual energy X‐ray absorptiometry equipment (Ge Lunar Prodigy, USA).
In‐vivo metabolic studies
Acute insulin challenge experiments were performed to test the effect of STING on IR. Five mice in each group were randomly selected and fasted for 6 h. Then the quadriceps femoris muscles were collected 15 min post‐intraperitoneal insulin administration (2 U/kg body weight). Proteins were extracted to detect the expression of AKT serine/threonine kinase phosphorylated at Ser473 (pAKTSer473) and insulin‐responsive glucose transporter type 4 (GLUT4).
Serological tests
Five mice in each group were randomly selected and subjected to 12 h starvation, after which the mice were euthanized, and the blood was collected to separate the serum. The serum was analysed for glucose, total cholesterol (TC), low density lipoprotein cholesterol (LDL‐C), high density lipoprotein cholesterol (HDL‐C), and triglyceride (TG) using the Bayer 1650 blood chemistry analyser (Bayer AG, Leverkusen, Germany).
Tissue processing
Considering starvation may affect fatty acid oxidation (FAO), the rest eight mice in each group were normally raised without starvation before tissue acquisition. After being anaesthetized and weighed, the mice were euthanized. Bilateral gastrocnemius muscles and quadriceps femoris muscles were collected and weighed. The tibia length was measured. The quadriceps femoris muscles were stored at −80°C after liquid nitrogen freezing or were fixed with 4% paraformaldehyde, dehydrated by gradient ethanol series and embedded with paraffin to make 5‐μm sections.
Histopathological staining
After being dewaxed and hydrated, the paraffin sections were used for haematoxylin–eosin (HE) staining, Masson staining, Sirius Red staining, immunohistochemical staining of laminin, myosin, fast myosin heavy chain (MyHC), slow MyHC, collagen I, collagen III, and immunofluorescent staining of alpha smooth muscle actin (αSMA) following the corresponding methods. The detailed procedures and antibodies were shown in the supporting information. The images of HE staining, Masson staining, Sirius Red staining and immunohistochemical staining were acquired using an Olympus DP72 digital imaging system (Olympus Corporation, Tokyo, Japan). The images of immunofluorescent staining were acquired using fluorescence microscope (Nikon, DS‐Ri2, Japan). The images were analysed using Image Pro Plus. The size of muscle fibres was calculated from immunohistochemical staining of myosin, fast MyHC and slow MyHC and represented by cross sectional area (CSA, i.e., cell area divided by cell number). Because the quadriceps femoris muscles contain mainly fast muscle fibres, and slow muscle fibres exist mainly in the region adjacent to the femur, the fast and slow MyHC‐stained sections were sampled from different regions where they mainly distributed. For CSA of all muscle fibres and fast muscle fibres, three fields were randomly selected, and the area of 50 fibres was randomly counted in each field. For CSA of slow muscle fibres, the area and number of all slow muscle fibres were calculated. The collagen volume fraction was calculated from Masson staining images and represented as the ratio of green dye area to red dye area. The content of collagen I and collagen III was represented by the integrated optical density values calculated from immunohistochemical staining of collagen I and collagen III. The fibroblasts (αSMA positive cells counter‐stained with DAPI) was counted in immunofluorescent staining images of αSMA.
Cells
C2C12 myoblasts were purchased from Shandong Quanzhen Science and Technology Ltd and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin–streptomycin (Gibco, USA). Myoblasts were differentiated into myotubes by changing the medium to DMEM supplemented with 2% horse serum and 1% penicillin–streptomycin.
Palmitic acid (PA) was applied to mimic the diabetic state. C176 is a strong covalent inhibitor that specifically blocks murine STING. To evaluate the effects of PA treatment and STING inhibition on the myotubes, the fully differentiated myotubes were divided into control group, C176 group, PA group and PA + C176 group. GW9662 is a potent selective antagonist of peroxisome proliferator activated receptors γ (PPARγ). To study whether STING inhibition exerts its effects through PPARγ in the diabetic state, the fully differentiated myotubes were divided into PA group, PA + C176 group and PA + C176 + GW9662 group. Noticeably, because PA was dissolved with 10% FBS free of fatty acids and the other reagents was dissolved with dimethylsulfoxide (DMSO), the control groups were supplemented with the same concentration of corresponding solvents. PA was added after pre‐treatment with C176 or GW9662 for 4 h. The PA stimulation time was 48 h in the studies unless specifically noted. The medium supplemented with C176 or GW9662 was changed every 24 h to ensure the effectiveness of the inhibitors. The dosage, references and other details of the above reagents were listed in Table S1.
CD31−CD45−PDGFRα+ FAPs were separated from the muscles of 1‐month‐old mice using the magnetic activated cell sorting method as previously described. 20 The cells were cultured in DMEM supplemented with 20% FBS, 2 mM l‐glutamine, 1% penicillin–streptomycin and 2.5 ng/mL basic fibroblast growth factor. To verify the effects of transforming growth factor beta 1 (TGFβ1), FAPs were cultured in the medium supplemented with gradient concentration of TGFβ1 (Proteintech, HZ‐1011; 0, 2.5, 5, 7.5 and 10 ng/mL) for 24 h.
Cytosolic DNA assessment
Co‐immunofluorescent staining of mitochondrial marker TOM20 and double‐stranded DNA (dsDNA) in myotubes was used to assess the effect of PA treatment on the cytosolic DNA content. Briefly, after being treated with 4% paraformaldehyde solution, 0.5% TritonX‐100 solution and 5% BSA solution, the cells were incubated with anti‐TOM20 antibody (Proteintech, 11802‐1‐AP) and anti‐dsDNA antibody (Abcam, ab27156) at 4°C overnight, fluorescence secondary antibodies (Abcam, ab150077 and ab150116) at 37°C for 1 h, and DAPI for 5 min. Images were acquired using the microscope. DNA foci that did not co‐localize with nuclei or mitochondria was counted manually. Thirty cells in each group were calculated and averaged.
Pulse‐chase assay and ubiquitination assay
To further explore how STING regulated PPARγ, the following tests were carried out. Firstly, the pulse‐chase assay was performed to test whether STING inhibition prolonged the half‐life of PPARγ protein in the diabetic state. The fully differentiated myotubes were pre‐treated with DMSO or C176 for 4 h before PA was added to the medium. The protein synthesis inhibitor cycloheximide (CHX) was added to the medium at 0, 2, 4, 6 or 8 h post‐PA administration, and the cell lysates were collected at 8 h to detect the PPARγ protein content. Secondly, we inhibited the proteasome on the basis of inhibiting STING to examine whether STING influenced the degradation of PPARγ through the ubiquitin proteasome pathway in the diabetic state. The fully differentiated myotubes were pre‐treated with DMSO or C176 for 4 h and then incubated with PA combined with CHX or proteasome inhibitor MG132 for another 4 h, after which the cell lysates were collected to detect the PPARγ protein content. Thirdly, ubiquitination level of PPARγ was examined to verify how STING regulated the degradation of PPARγ through the proteasome pathway. The fully differentiated myotubes were pre‐treated with DMSO or C176 for 4 h and then incubated with PA for 48 h, after which the cells were collected for immunoprecipitation (IP) analysis. The detailed steps of IP were demonstrated in the supporting information.
Mitophagic flux analysis
Mito‐Keima is a mitochondria‐targeted, pH‐sensitive, dual‐excitation ratiometric fluorescent protein that is resistant to lysosomal degradation. The shorter‐wavelength (440 nm, green) excitation predominates in the mitochondria environment (pH 8.0), whereas the longer‐wavelength (578 nm, red) excitation predominates in the acidic lysosome environment (pH 4.5). Thus, Mito‐Keima is an effective tool used to assess the mitophagic flux. 21 Mito‐Keima overexpression adenovirus was purchased from HanBio biotechnology (Shanghai, China). The fully differentiated myotubes were infected with mito‐Keima overexpression adenovirus for 8 h at a multiplicity of infection of 50, and then cultured in fresh medium supplemented with PA, C176 or GW9662. The cells were observed under a confocal microscope (Leica Microsystems CMS GmbH, Germany) with an excitation wavelength at 440 nm or 578 nm and an emission wavelength at 610 nm.
In‐vitro metabolic studies
Acute insulin challenge experiments were performed to test the effect of STING on IR. After treatment with the corresponding reagents for 48 h, the cells were starved in serum‐free medium for 6 h, and then cultured in previous mediums supplemented with insulin (100 nmol/L) for 15 min. The cell lysis was collected to detect the expression of pAKTSer473 and GLUT4.
Western blotting
The proteins were extracted from quadriceps femoris muscles and C2C12 myotubes, separated in 10% or 12% SDS‐polyacrylamide gels, and transferred onto PVDF membranes (Millipore IPVH304F0 and ISEQ00010, USA), which were soaked in 5% skim milk (room temperature, 1 h), washed, and then incubated at 4°C overnight with primary antibodies: anti‐TMEM173/STING (Proteintech, 19851‐1‐AP), anti‐phospho‐STING (Ser365) (Cell Signalling Technology, 72971), anti‐IRF3 (phospho Ser396) (Immunoway, YP0326), anti‐GAPDH (Proteintech, 66004‐1‐Ig), anti‐Fbx32 (Abcam, ab168372), anti‐fibronectin (Abcam, ab45688), anti‐phospho‐DRP1 (Ser616) (Cell Signalling Technology, 3455), anti‐PINK1 (Proteintech, 23274‐1‐AP), anti‐Parkin (Proteintech, 14060‐1‐AP), anti‐SQSTM1/p62 (Abcam, ab109012), anti‐LC3B (Abcam, ab192890), anti‐PPARγ (Proteintech, 16643‐1‐AP), anti‐ACADM (Abcam, ab92461), anti‐ACADVL (Proteintech, 14527‐1‐AP), anti‐CPT2 (Abcam, ab181114), anti‐CPT1A (Proteintech, 15184‐1‐AP), anti‐ACOX1 (Abclonal, A8091), anti‐ACOX2 (Abclonal, A12796), anti‐CD36 (Abcam, ab252922), anti‐ubiquitin (Proteintech, 10201‐2‐AP), anti‐TGF beta 1 (Abcam, ab215715), anti‐αSMA (Abcam, ab7817), anti‐COL1A1 (Thermo Fisher, PA5‐35379), anti‐phospho‐AKT (Ser473) (Cell Signalling Technology, 4060) and anti‐GLUT4 (Proteintech, 66846‐1‐Ig) antibodies. On the second day, the membranes were washed and incubated with horseradish peroxidase‐labelled anti‐rabbit or anti‐mouse secondary antibodies (ZSGB‐BIO ZB‐2305 or ZB‐2301, Beijing, China) at room temperature for 90 min. After washing, the images were acquired using the enhanced chemiluminescence reagent (Millipore WBKLS0500, USA). The images were quantified using ImageJ.
Statistical analysis
Data are represented as means ± SEM. GraphPad Prism 6.0 was used for statistical analysis and image presentation. Comparisons between two groups were performed using unpaired t tests; comparisons among three or more groups were performed using one‐way or two‐way ANOVA and Tukey's multiple comparisons tests. P < 0.05 was considered significant.
Results
STING deficiency alleviated sarcopenia in diabetic mice
WT and STINGgt/gt mice were raised and the tests were performed accordingly (Figure S1A). Multiple loci sequencing of mouse tail DNA revealed a single base pair change, T596A, in exon 6 of Tmem173 (Figure S1B). Consistent with the Jackson Laboratory, no STING protein was detected by western blotting in control and diabetic STINGgt/gt mice (Figure S1C). Diabetic WT and STINGgt/gt mice had higher fasting blood glucose content compared with control WT and STINGgt/gt mice, respectively (P < 0.001 for both), and no significant differences were discovered between the two control groups and between the two diabetic groups (P > 0.05 for both, Figure S1D). Diabetic WT mice had lower body weight compared with control WT mice (P < 0.05, Figure S1E), which was consistent with the manifestation of weight loss in the late course of T2DM. Control STINGgt/gt mice had lower body weight compared with control WT mice (P < 0.001, Figure S1E), which suggested that STING deficiency influenced body weight. The above results suggested that we had successfully established the diabetic WT and STINGgt/gt animal model.
We examined the effects of DM and PA treatment on the expression and activation of STING in the skeletal muscles and C2C12 myotubes. The results showed that STING expression was higher in diabetic mice compared with control mice (P < 0.001), whereas no significant differences were discovered between the control myotubes and the PA‐treated myotubes (P > 0.05). Diabetic mice showed increased levels of pSTINGSer365 and pIRF3Ser396 compared with control mice (P < 0.001 for both). Similarly, PA‐treated myotubes had higher levels of them compared with control myotubes (P < 0.001 and P < 0.01, respectively, Figure 1A). These results demonstrated abnormal activation of STING in the diabetic skeletal muscles. Considering that dsDNA is an upstream activator of STING and that increased cytosolic dsDNA in diabetic states has been reported, 22 we validated the effect of PA treatment on dsDNA cytosolic exposure in muscle fibres. Compared with control myotubes, the content of cytosolic dsDNA was significantly increased in PA‐treated myotubes (P < 0.001, Figure S2), which might be a key up‐stream factor for the abnormal activation of STING.
Figure 1.
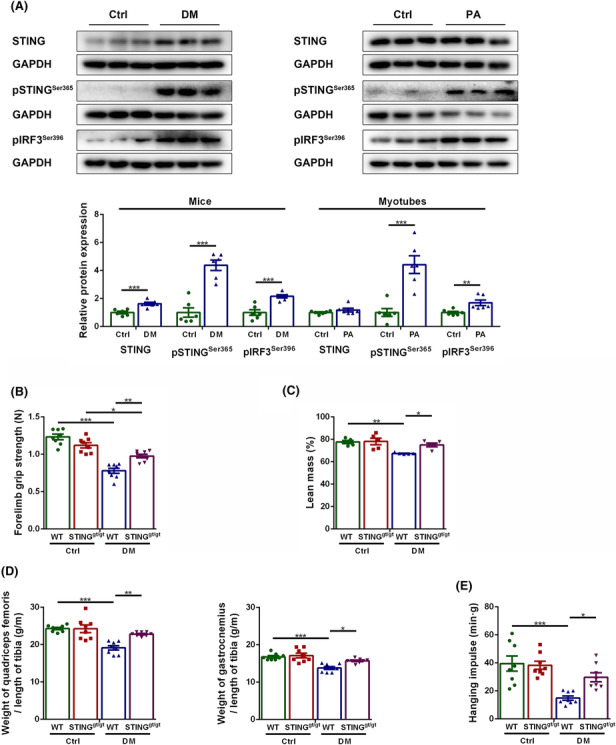
STING deficiency alleviated sarcopenia in diabetic mice. Mice were divided into control group and diabetic group. Myotubes were divided into control group and PA group. (A) Expression of STING, pSTINGSer365 and pIRF3Ser396 in skeletal muscles and C2C12 myotubes detected through western blotting; GAPDH: internal reference (n = 6). Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (B) Forelimb grip strength (N) (n = 8). (C) Lean mass measured through dual energy X‐ray analysis (%) (n = 5). (D) Ratio of quadriceps femoris weight to tibia length (g/m) and ratio of gastrocnemius weight to tibia length (g/m) (n = 8). (E) Hanging impulse (min/g) (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001.
We further evaluated the impact of DM and STING on skeletal muscles by examining the three main manifestations of sarcopenia, that is, declined muscle strength, muscle mass and physical performance. The muscle strength was reflected by the forelimb grip strength, and the muscle mass was reflected by the lean mass measured by dual‐energy X‐ray analysis, and the ratio of muscle mass to tibia length (the muscle mass was corrected by tibia length due to significant differences in body weight). The physical performance was reflected by hanging impulse. Compared with control WT mice, diabetic WT mice showed significantly decreased forelimb grip strength, lean mass, ratios of quadriceps femoris weight and gastrocnemius weight to tibia length, and hanging impulse (P < 0.001, P < 0.01, P < 0.001, P < 0.001 and P < 0.001, respectively), suggesting obvious sarcopenia in diabetic mice; whereas the above parameters were significantly increased in diabetic STINGgt/gt mice compared with diabetic WT mice (P < 0.01, P < 0.05, P < 0.01, P < 0.05 and P < 0.05, respectively, Figure 1B–E), suggesting that STING deficiency alleviated sarcopenia in diabetic mice.
Stimulator of interferon genes deficiency alleviated the pathological manifestations of sarcopenia in diabetic mice
Stimulator of interferon genes deficiency alleviated skeletal muscle fibre atrophy in diabetic mice
Considering that muscle fibre atrophy is a major histopathological feature of sarcopenia, we evaluated the effects of DM and STING on it. HE staining showed that diabetic skeletal muscles exhibited smaller fibre size, which were improved by STING deficiency (Figure 2A). Immunohistochemical staining revealed that the CSA of total muscle fibres, fast muscle fibres and slow muscle fibres declined significantly in diabetic WT mice compared with control WT mice (P < 0.001, P < 0.001 and P < 0.05, respectively) but were increased in diabetic STINGgt/gt mice compared with diabetic WT mice (P < 0.001, P < 0.001 and P < 0.05, respectively). The CSA of total muscle fibres and fast muscle fibres decreased in diabetic STINGgt/gt mice compared with control STINGgt/gt mice (P < 0.05 and P < 0.001, respectively), whereas the CSA of slow muscle fibres showed no obvious changes between these two groups (P > 0.05, Figure 2A,B). Western blotting showed that diabetic WT muscles had higher expression of F‐box protein 32 (Fbx32; a marker of muscle fibre atrophy) compared with control WT muscles (P < 0.001), whereas diabetic STINGgt/gt muscles showed lower expression of this parameter compared with diabetic WT muscles (P < 0.05). No significant differences were shown between the two control groups and between the two STINGgt/gt groups (P > 0.05 for both, Figure 2C). Similarly, the protein content of Fbx32 was higher in PA group compared with control group (P < 0.01) and was lower in PA + C176 group compared with PA group (P < 0.05). No significant differences were shown between the control group and the C176 group, and between the C176 group and the PA + C176 group (P > 0.05 for both, Figure 2D). The above results demonstrated that STING deficiency inhibited the atrophy of skeletal muscle fibres in the diabetic state.
Figure 2.
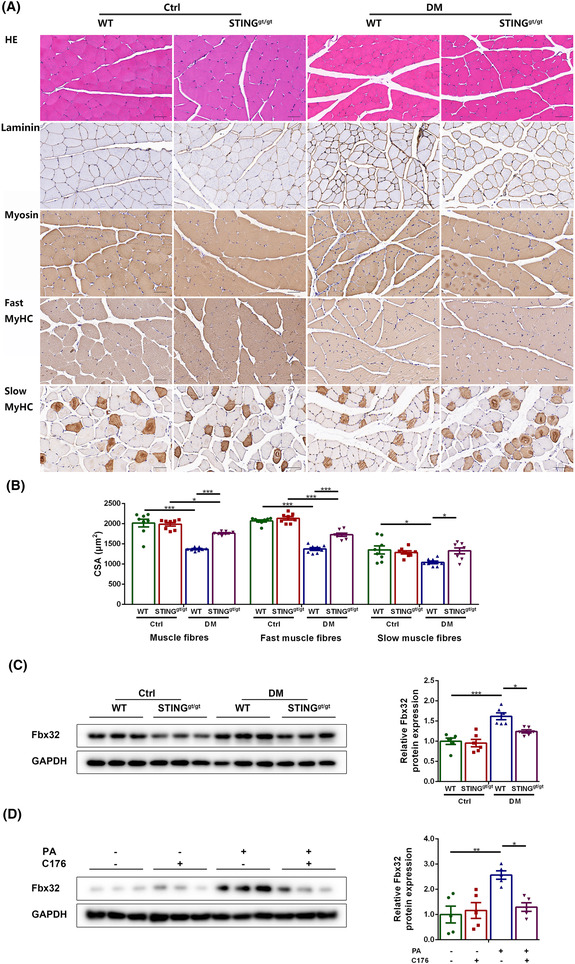
STING deficiency alleviated skeletal muscle fibre atrophy in diabetic mice. Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (A) HE staining and immunohistochemical staining of laminin, myosin, fast MyHC and slow MyHC (scale bar = 50 μm; n = 8). (B) CSA of muscle fibres, fast muscle fibres and slow muscle fibres (μm2) (n = 8). (C) Expression of Fbx32 in skeletal muscles detected through western blotting; GAPDH: internal reference (n = 6). Myotubes were divided into control group, C176 group, PA group and PA + C176 group. (D) Expression of Fbx32 in myotubes detected through western blotting; GAPDH: internal reference (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Stimulator of interferon genes deficiency reduced skeletal muscle interstitial fibrosis in diabetic mice
Interstitial fibrosis is another major histopathological feature of sarcopenia, and we evaluated the effects of DM and STING deficiency on it. Histopathological staining showed that the collagen volume fraction and the contents of collagen I and collagen III were higher in diabetic WT mice as compared with those in control WT mice (P < 0.001 for all) and were lower in diabetic STINGgt/gt mice as compared with those in diabetic WT mice (P < 0.001 for all). The contents of collagen I and collagen III were increased in diabetic STINGgt/gt mice as compared with those in control STINGgt/gt mice (P < 0.001 for both), whereas the collagen volume fraction was non‐significantly increased in diabetic STINGgt/gt mice as compared with that in control STINGgt/gt mice (P > 0.05, Figure 3A–D). Western blotting showed that diabetic WT mice had increased content of fibronectin (a marker of fibrosis) in their muscles compared with control WT mice (P < 0.05), whereas diabetic STINGgt/gt mice had decreased content compared with diabetic WT mice (P < 0.001). No significant differences were discovered between the two control groups and between the two STINGgt/gt groups (P > 0.05 for both, Figure 3E). These results demonstrated that STING deficiency reduced skeletal muscle interstitial fibrosis in diabetic mice.
Figure 3.
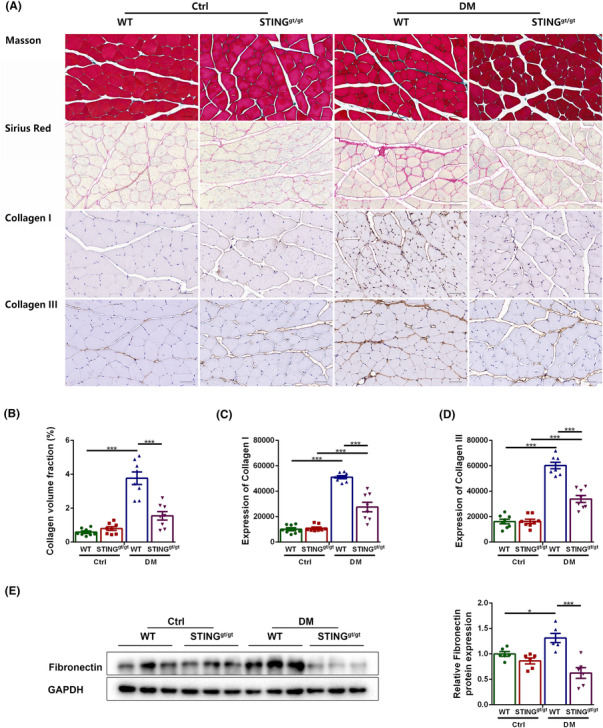
STING deficiency reduced skeletal muscle interstitial fibrosis in diabetic mice. Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (A) Masson staining, Sirius red staining, and immunohistochemical staining of collagen I and collagen III (scale bar = 50 μm; n = 8). (B) Collagen volume fraction (%) (n = 8). (C) Expression of collagen I (n = 8). (D) Expression of collagen III (n = 8). (E) Expression of fibronectin in skeletal muscles detected through western blotting; GAPDH: internal reference (n = 6). *P < 0.05, **P < 0.01, ***P < 0.001.
Mechanisms of stimulator of interferon genes deficiency to alleviate muscle fibre atrophy in diabetic mice
STING deficiency alleviated muscle fibre atrophy by promoting mitofission and mitophagy in skeletal muscles of diabetic mice
Impaired mitophagy is reported to be an important factor for muscle fibre atrophy and PINK1‐Parkin‐dependent mitophagy is a common type of mitophagy. Serine/threonine kinase PTEN induced kinase 1 (PINK1) that accumulate on the outer membrane of impaired mitochondria recruit E3‐ubiquitin ligase Parkin. Then Parkin connects ubiquitin chain (Ub) to mitochondrial surface proteins, which are recognized by autophagy receptor p62 and linked to microtubule‐associated protein 1 light chain 3 (LC3) to initiate the formation of autophagosome and the removal of damaged mitochondria. 7 Mitofission is considered to be the initiation step for mitophagy. Dynamin‐related protein 1 (DRP1) is a key protein mediating mitofission, the function of which is enhanced by phosphorylation at Ser616 site. 23 Thus, we evaluated the effects of STING deficiency or inhibition on mitofission and mitophagy in diabetic muscles and PA‐treated myotubes using the above parameters. In vivo, diabetic WT muscles had lower protein content of pDRP1Ser616, PINK1, Parkin and LC3‐II, and higher content of p62 compared with control WT muscles (P < 0.05, P < 0.01, P < 0.05, P < 0.001 and P < 0.001, respectively), indicating impaired mitofission and mitophagy in the diabetic state. Whereas diabetic STINGgt/gt muscles had higher content of pDRP1Ser616, PINK1, Parkin and LC3‐II, and lower content of p62 compared with diabetic WT muscles (P < 0.05, P < 0.05, P < 0.01, P < 0.01 and P < 0.01, respectively, Figure 4A), indicating improved mitofission and mitophagy. In vitro, PA‐treated myotubes exhibited lower levels of pDRP1Ser616 and LC3‐II, and higher level of p62 relative to control myotubes (P < 0.01, P < 0.05 and P < 0.01, respectively), with no significant differences in the levels of PINK1 and Parkin between the two groups (P > 0.05 for both). STING inhibition significantly increased the levels of pDRP1Ser616, PINK1, Parkin and LC3‐II, and significantly decreased the level of p62 in PA‐treated myotubes (P < 0.05, P < 0.01, P < 0.05, P < 0.05 and P < 0.05, respectively, Figure 4B). The above results suggested that STING deficiency promoted mitofission and mitophagy in skeletal muscles of diabetic mice, which might be one of the key reasons for alleviated muscle fibre atrophy achieved by STING deficiency.
Figure 4.
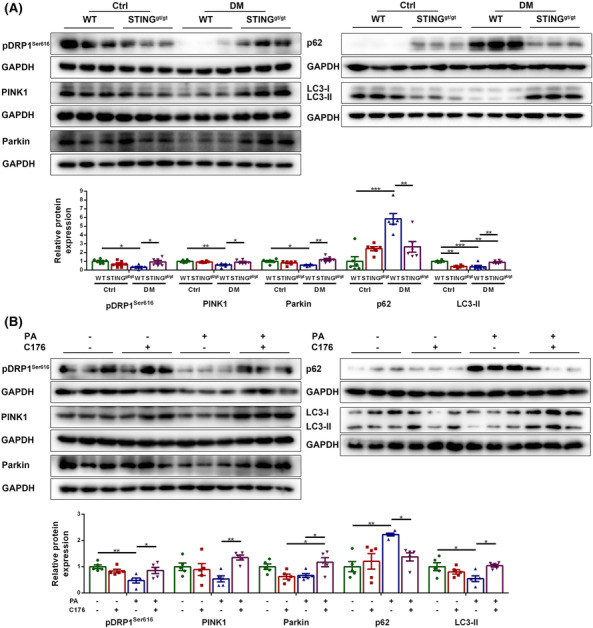
STING deficiency promoted mitofission and mitophagy in skeletal muscles of diabetic mice.
Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (A) Expression of pDRP1Ser616, PINK1, Parkin, p62 and LC3‐II in skeletal muscles detected through western blotting; GAPDH: internal reference (n = 6). Myotubes were divided into control group, C176 group, PA group and PA + C176 group. (B) Expression of pDRP1Ser616, PINK1, Parkin, p62 and LC3‐II in myotubes detected through western blotting; GAPDH: internal reference (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Stimulator of interferon genes deficiency alleviated muscle fibre atrophy by improving insulin resistance in skeletal muscles of diabetic mice
IR has been proved to be a pathogenic factor for muscle fibre atrophy and we also evaluated the effects of STING deficiency or inhibition on IR in diabetic muscles and PA‐treated myotubes. Diabetic WT muscles had lower protein levels of pAKTSer473 and GLUT4 post‐insulin challenge compared with control WT muscles (P < 0.05 and P < 0.01, respectively), whereas diabetic STINGgt/gt muscles had higher content of the above markers compared with diabetic WT muscles (P < 0.05 and P < 0.001, respectively, Figure S3A). PA treatment significantly reduced the content of pAKTSer473 and GLUT4 post‐insulin challenge in myotubes (P < 0.05 and P < 0.01, respectively), both of which were increased by STING inhibition (P < 0.01 and P < 0.05, respectively, Figure S3B). Moreover, IPGTT and IPITT were performed to evaluate the overall effect of STING deficiency on IR. The blood glucose levels at 0, 30, 60, 90 and 120 min post‐glucose or insulin challenge were highest in diabetic WT mice and were lowest in control WT and STINGgt/gt mice. The area under curve of both IPGTT and IPITT was higher in diabetic WT mice compared with control WT mice (P < 0.001 for both) and in diabetic STINGgt/gt mice compared with control STINGgt/gt mice (P < 0.001 and P < 0.01, respectively) but was lower in diabetic STINGgt/gt mice compared with diabetic WT mice (P < 0.001 for both, Figure S3C). The above results suggested that STING deficiency alleviated IR in diabetic muscles, which might be another reason for alleviated muscle fibre atrophy achieved by STING deficiency.
Mechanisms of stimulator of interferon genes deficiency to improve mitofission and mitophagy in skeletal muscles of diabetic mice
FAO is reported to be a promoting factor for mitophagy. 24 Fatty acids in the blood are transported into muscle fibres through the fatty acid transporter CD36 and transformed to acyl‐carnitine, which enters the mitochondrial matrix with the help of carnitine lipoacyltransferases (CPT) and converted to energy under the catalysis of enzymes including acyl coenzyme A dehydrogenase (ACAD) and through the tricarboxylic acid cycle and the respiratory chain. 25 Acyl‐CoA oxidases (ACOX) catalyses the first step of peroxisomal β‐oxidation of very long‐chain fatty acids and branched‐chain fatty acids. 26 PPARγ is an upstream regulator of FAO. Thus, in order to clarify whether the promoting effect of STING inhibition on mitophagy in diabetic muscles is realized by promoting PPARγ‐mediated FAO, the following studies were conducted.
Stimulator of interferon genes deficiency up‐regulated the expression of peroxisome proliferator activated receptors γ and fatty acid oxidation key proteins in skeletal muscles of diabetic mice
Firstly, we examined whether STING intervention affected the content of PPARγ and the levels of FAO key proteins in diabetic muscles and PA‐treated myotubes. Compared with diabetic WT mice, diabetic STINGgt/gt mice had higher levels of PPARγ, acyl coenzyme A dehydrogenase medium chain (ACADM), acyl coenzyme A dehydrogenase very long chain (ACADVL), CPT2, CPT1A, ACOX1, ACOX2, and CD36 (P < 0.05, P < 0.05, P < 0.05, P < 0.01, P < 0.05, P < 0.05, P < 0.05 and P < 0.05, respectively, Figure 5A), which demonstrated that STING deficiency promoted the expression of PPARγ and FAO‐related markers in diabetic state in vivo. Similarly, the levels of the above parameters were higher in PA + C176 group compared with PA group (P < 0.05, P < 0.01, P < 0.05, P < 0.05, P < 0.05, P < 0.001, P < 0.05 and P < 0.01, respectively, Figure 5B), suggesting that STING inhibition up‐regulated the expression of PPARγ and the key proteins of FAO in PA‐treated myotubes.
Figure 5.
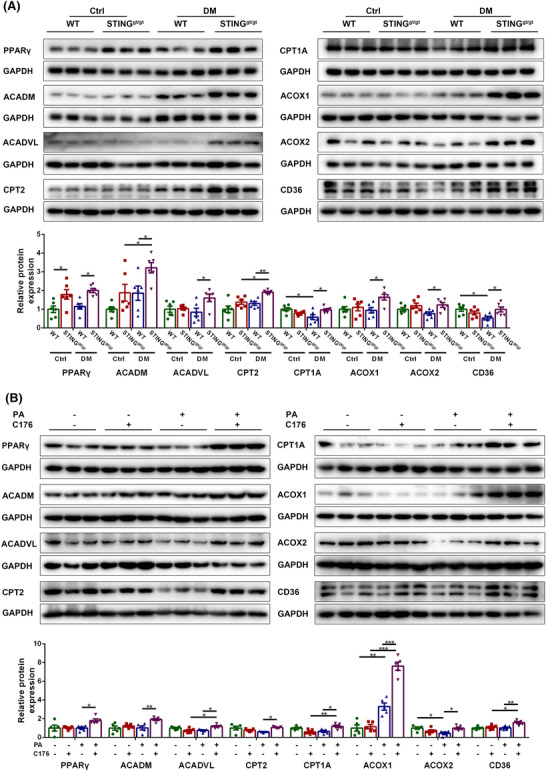
STING deficiency up‐regulated the expression of PPARγ and FAO key proteins in skeletal muscles of diabetic mice. Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (A) Expression of PPARγ, ACADM, ACADVL, CPT2, CPT1A, ACOX1, ACOX2 and CD36 in skeletal muscles detected through western blotting; GAPDH: internal reference (n = 6). Myotubes were divided into control group, C176 group, PA group and PA + C176 group. (B) Expression of PPARγ, ACADM, ACADVL, CPT2, CPT1A, ACOX1, ACOX2 and CD36 in myotubes detected through western blotting; GAPDH: internal reference (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Besides, the content of blood lipids was examined to reflect the overall lipid metabolism. Diabetic WT mice had higher levels of TC, LDL‐C, HDL‐C and TG compared with control WT mice (P < 0.001, P < 0.01, P < 0.001 and P < 0.05, respectively). Whereas diabetic STINGgt/gt mice had lower levels of TC, LDL‐C, and TG compared with diabetic WT mice (P < 0.05, P < 0.01 and P < 0.01, respectively), with no significant difference in the level of HDL‐C between the two groups (P > 0.05, Figure S4A), suggesting that STING deficiency decreased the levels of blood lipids in the diabetic state.
To verify that STING improved the expression of FAO‐related proteins by regulating PPARγ, GW9662 was applied to inhibit PPARγ in the condition of PA and C176 treatment. Western blotting showed that the levels of ACADM, ACADVL, CPT2, CPT1A, ACOX1, ACOX2 and CD36 were lower in PA + C176 + GW9662 group than in PA + C176 group (P < 0.05, P < 0.01, P < 0.01, P < 0.01, P < 0.05, P < 0.05 and P < 0.05, respectively, Figure S5A), demonstrating that the effect of STING inhibition on promoting FAO in PA‐treated myotubes was mediated by PPARγ.
Stimulator of interferon genes inhibition reduced the degradation of peroxisome proliferator activated receptors γthrough the proteasome pathway in the diabetic state
Next, we studied how STING regulated the protein content of PPARγ in the diabetic state. Firstly, a pulse‐chase assay with CHX was performed in myotubes to test whether STING was involved in the degradation of PPARγ. The PPARγ protein contents after 4, 6 and 8 h of CHX treatment were significantly higher in PA + C176 group than in PA + DMSO group (P < 0.05, P < 0.01 and P < 0.01, respectively, Figure 6A), suggesting that STING inhibition prolonged the half‐life of PPARγ protein in PA‐treated myotubes. Secondly, we detected whether STING regulated the degradation of PPARγ through the ubiquitin proteasome pathway. MG132 increased the PPARγ content in PA and CHX‐treated myotubes (P < 0.01), indicating that PPARγ could be degraded through the proteasome pathway. The PPARγ content was significantly increased in the PA + C176 group compared with the PA group (P < 0.01), and in the PA + CHX + C176 group compared with the PA + CHX group (P < 0.001), whereas no significant differences were shown between the PA + CHX + MG132 group and the PA + CHX + MG132 + C176 group (P > 0.05), and between the PA + CHX + C176 group and the PA + CHX + MG132 + C176 group (P > 0.05; Figure 6B). These results indicated that STING inhibition reduced the degradation of PPARγ through the proteasome pathway in the diabetic state. To verify how STING regulated the degradation of PPARγ through the proteasome pathway, we further examined the effect of STING inhibition on the ubiquitination level of PPARγ. STING inhibition significantly increased the ubiquitination degree of PPARγ (P < 0.05, Figure 6C), suggesting that STING inhibition reduced the degradation of ubiquitinated PPARγ through the proteasome pathway instead of decreasing the ubiquitination of PPARγ in the diabetic state.
Figure 6.
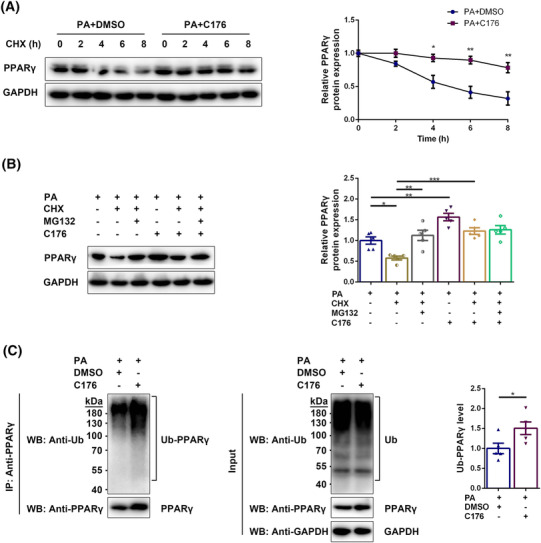
STING inhibition reduced the degradation of PPARγ through the proteasome pathway in the diabetic state. Myotubes were divided into PA + DMSO group and PA + C176 group and treated with CHX for 0, 2, 4, 6, or 8 h. (A) Expression of PPARγ in myotubes detected through western blotting; GAPDH: internal reference (n = 5). Myotubes were divided into PA group, PA + CHX group, PA + CHX + MG132 group, PA + C176 group, PA + CHX + C176 group, and PA + CHX + MG132 + C176 group. (B) Expression of PPARγ in myotubes detected through western blotting; GAPDH: Internal reference (n = 5). Myotubes were divided into PA + DMSO group and PA + C176 group. Immunoprecipitation using anti‐PPARγ antibody was performed and western blotting of IP lysates and whole‐cell lysates was carried out using anti‐Ub antibody and anti‐PPARγ antibody. (C) Detection of ubiquitinated PPARγ in myotubes through western blotting; GAPDH: internal reference (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Peroxisome proliferator activated receptors γ‐inhibition‐mediated fatty acid oxidation impairment abolished the effects of stimulator of interferon genes inhibition on improving mitophagy and alleviating muscle fibre atrophy
Then, we investigated whether STING affected mitofission and mitophagy by regulating PPARγ‐mediated FAO. PPARγ inhibition decreased the expression of pDRP1Ser616, PINK1, Parkin and LC3‐II (P < 0.05, P < 0.05, P < 0.05 and P < 0.01, respectively), and increased the expression of p62 (P < 0.05, Figure 7A) in C176 and PA‐treated myotubes, suggesting that the improvements in mitofission and mitophagy in PA‐treated myotubes achieved by inhibiting STING was abolished by PPARγ inhibition. Moreover, mitophagic flux analysis using mito‐Keima overexpression adenovirus was performed to confirm the effects of PA, C176 and GW9662 on the mitophagy activity of myotubes. Compared with control group, fewer fluorescent dots at 578 nm were observed in PA group (P < 0.05), indicating impaired mitophagic flux in PA‐treated conditions. Compared with PA group, more fluorescent dots at 578 nm were observed in PA + C176 group (P < 0.05), suggesting that STING inhibition improved the mitophagic flux of PA‐treated myotubes. Compared with PA + C176 group, less fluorescent dots at 578 nm were observed in PA + C176 + GW9662 group (P < 0.05, Figure 7B,C), suggesting that PPARγ inhibition abolished the improvements of mitophagic flux in PA‐treated myotubes achieved by STING inhibition. Further, the effect on muscle fibre atrophy was evaluated. The content of Fbx32 was higher in PA + C176 + GW9662 group than in PA + C176 group (P < 0.001, Figure 7D), suggesting that the alleviation of muscle fibre atrophy in PA‐treated myotubes by inhibiting STING was abolished by PPARγ inhibition. The above results indicated that STING inhibition promoted mitophagy and alleviated skeletal muscle fibre atrophy by increasing the PPARγ protein content and subsequently promoting FAO in the diabetic state.
Figure 7.
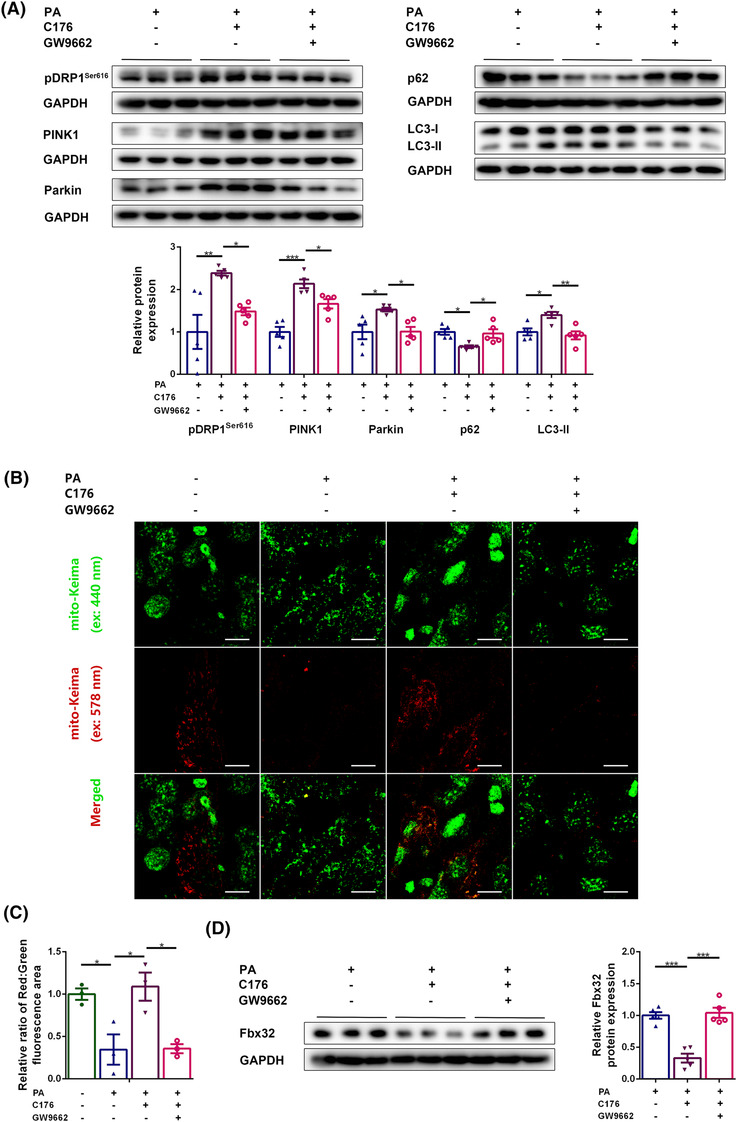
PPARγ‐inhibition‐mediated FAO impairment abolished the effects of STING inhibition on improving mitophagy and alleviating muscle fibre atrophy. Myotubes were divided into PA group, PA + C176 group, and PA + C176 + GW9662 group. (A) Expression of pDRP1Ser616, PINK1, Parkin, p62, and LC3‐II in myotubes detected through western blotting; GAPDH: internal reference (n = 5). Myotubes were transfected with the Mito‐Keima overexpression adenovirus and divided into control group, PA group, PA + C176 group, and PA + C176 + GW9662 group. (B) Fluorescent dots of Mito‐Keima at 440 nm (green) and 578 nm (red) (scale bar = 10 μm; n = 3). (C) The relative ratio of red to green fluorescence area per cell (n = 3). Myotubes were divided into PA group, PA + C176 group, and PA + C176 + GW9662 group. (D) Expression of Fbx32 in myotubes detected through western blotting; GAPDH: internal reference (n = 5). *P < 0.05, **P < 0.01, ***P < 0.001.
Mechanisms of stimulator of interferon genes deficiency to improve insulin resistance in skeletal muscles of diabetic mice
In addition to mitophagy, we also explored the possible mechanisms of STING inhibition in alleviating IR. Considering that promoting FAO has been shown to effectively alleviate IR in chronic metabolic diseases, we evaluated whether STING regulated IR by influencing PPARγ‐mediated FAO. PPARγ inhibition significantly decreased the content of pAKTSer473 and GLUT4 in PA and C176‐treated myotubes (P < 0.001 and P < 0.05, respectively, Figure S6A), suggesting that STING inhibition alleviated IR by increasing the PPARγ protein content in the diabetic state.
Mechanisms of stimulator of interferon genes deficiency in reducing interstitial fibrosis in skeletal muscles of diabetic mice
Finally, the mechanisms of STING deficiency in reducing interstitial fibrosis in diabetic muscles were explored. Considering that FAPs are a major source of intermuscular fibroblasts and fibrous tissue, we examined whether there was a fibrosis‐promoting microenvironment in diabetic muscles, and whether STING deficiency reduced the fibrogenic differentiation of FAPs by alleviating such fibrosis‐promoting microenvironment. Western blotting showed that the content of secreted form of TGFβ1 (molecular weight ≈ 13 kDa) was significantly increased in diabetic WT muscles relative to control WT muscles (P < 0.001) and in PA‐treated myotubes compared with control myotubes (P < 0.05), which was reversed by STING deficiency or inhibition (P < 0.01 and P < 0.05, respectively, Figure 8A,B), suggesting that STING inhibition decreased the production of TGFβ1 in muscle fibres in the diabetic state. PPARγ inhibition increased the secreted TGFβ1 content in C176 and PA‐treated myotubes (P < 0.01, Figure 8C), suggesting that the inhibitory effect of STING inhibition on TGFβ1 production was abolished by PPARγ inhibition. Further, the effect of TGFβ1 on FAPs was evaluated and the results demonstrated that treatment with TGFβ1 at concentrations of 7.5 and 10 ng/mL significantly increased the expression of fibrogenic markers including αSMA (P < 0.05 for both) and collagen type I alpha 1 chain (COL1A1) (P < 0.05 and P < 0.01, respectively, Figure 8D), suggesting that TGFβ1 promoted the fibrogenic differentiation of FAPs. Finally, we detected the effects of STING deficiency on the content of fibroblasts in diabetic muscles. The content of αSMA positive cells was higher in diabetic WT group compared with control WT group (P < 0.001) and was lower in diabetic STINGgt/gt group compared with diabetic WT group (P < 0.001, Figure 8E,F). These results demonstrated that STING inhibition reduced the fibrogenic differentiation of FAPs by increasing PPARγ and subsequently reducing the production of secreted TGFβ1 in muscle fibres and thus alleviated interstitial fibrosis in the diabetic state.
Figure 8.
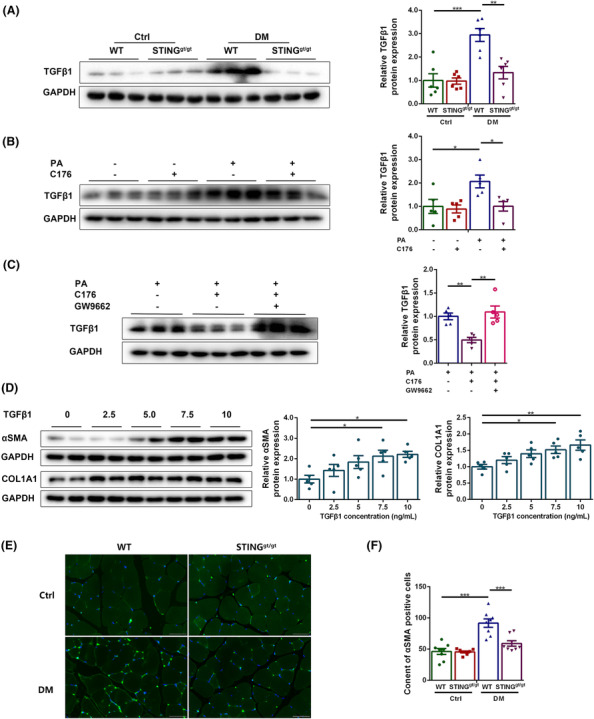
STING inhibition reduced the fibrogenic differentiation of FAPs by reducing the production of secreted TGFβ1 in muscle fibres in the diabetic state. Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group, and diabetic STINGgt/gt group. (A) Expression of TGFβ1 in skeletal muscles detected through western blotting; GAPDH: internal reference (n = 6). Myotubes were divided into control group, C176 group, PA group, and PA + C176 group. (B) Expression of TGFβ1 in myotubes detected through western blotting; GAPDH: internal reference (n = 5). Myotubes were divided into PA group, PA + C176 group, and PA + C176 + GW9662 group. (C) Expression of TGFβ1 in myotubes detected through western blotting; GAPDH: internal reference (n = 5). FAPs were treated with TGFβ1 in gradient concentration (0, 2.5, 5, 7.5 and 10 ng/mL). (D) Expression of αSMA and COL1A1 in FAPs detected through western blotting; GAPDH: internal reference (n = 5). Mice were divided into control WT group, control STINGgt/gt group, diabetic WT group and diabetic STINGgt/gt group. (E) Immunofluorescent staining of αSMA (scale bar = 50 μm; n = 8). (F) Content of αSMA positive cells (n = 8). *P < 0.05, **P < 0.01, ***P < 0.001.
Discussion
Diabetic sarcopenia is an inevitable complication of T2DM, characterized by three major features, that is, decreased muscle mass, weakened muscle strength and reduced physical performance. 1 In our study, the diabetic mice demonstrated the above features, suggesting obvious sarcopenia. Also, we found that cytosolic dsDNA was significantly increased under metabolic stress in PA‐treated C2C12 myotubes, and STING was significantly activated in the diabetic state. STING has been reported to be activated by ectopic DNA that is abnormally leaked to the cytoplasm under metabolic stress and promote the pathological process of metabolic diseases. 27 , 28 STING deficiency or inhibition effectively protected the organs, including the heart, kidney, retina and skin from structural and functional degeneration in the diabetic state. 29 , 30 , 31 , 32 In our study, STING deficiency significantly improved the muscle mass, muscle strength and exercise capacity in diabetic mice, indicating the protective effects of STING deficiency on diabetic sarcopenia.
Skeletal muscle fibre atrophy is one of the major pathological manifestations of diabetic sarcopenia. In our study, muscle fibre atrophy in diabetic mice was significantly alleviated by STING deficiency. The possible mechanisms lied in improved mitofission and mitophagy and alleviated IR in the diabetic state. Mitophagy selectively removes damaged mitochondria and maintains mitochondrial quality, and mitofission is an initiation step for mitophagy. 7 PINK1‐Parkin‐dependent mitophagy is the most comprehensively studied type of mitophagy in mammals. In our study, both mitofission and PINK1‐Parkin‐dependent mitophagy were impaired under diabetic conditions. Impaired mitophagy was reported to increase the content of dysfunctional mitochondria and reactive oxygen species, up‐regulate the expression of Fbx32 and MuRF1, and lead to excessive protein degradation and cell apoptosis, 33 which might be the reasons for muscle atrophy. Parkin deficiency caused severe impairment in mitochondrial respiration and decreased muscle strength. 34 Whereas Parkin overexpression significantly reduced oxidative stress and increased muscle fibre size. 10 Muscle atrophy was also alleviated via improving PINK1‐mediated mitophagy. 35 Therefore, impaired mitophagy may be an important cause for skeletal muscle fibre atrophy in the diabetic state. In our study, STING deficiency or inhibition significantly increased mitofission and mitophagy both in diabetic skeletal muscles and in PA‐treated myotubes, which may be an important mechanism of STING intervention to alleviate muscle fibre atrophy in diabetic state.
Alleviated IR achieved by STING deficiency is another reason for ameliorated muscle fibre atrophy in diabetic state. Significant loss of lean mass was shown in individuals with IR as compared with individuals without IR. 36 The application of insulin sensitizers was reported to increase muscle mass in T2DM mice. 37 Hence, IR is an important promoter of skeletal muscle fibre atrophy in diabetic state. It caused muscle fibre atrophy by accelerating protein degradation, inhibiting protein synthesis, and affecting mitochondrial function. 36 , 38 In our study, STING deficiency improved IR both in diabetic muscles and in PA‐treated myotubes, suggesting that STING deficiency ameliorated skeletal muscle fibre atrophy in diabetic state possibly by alleviating IR.
Impaired FAO is an important factor leading to disordered mitophagy and IR in the diabetic state. PPARγ is an upstream regulator of FAO. In our study, STING deficiency or inhibition significantly up‐regulated the protein levels of PPARγ and FAO‐related proteins in diabetic skeletal muscles and PA‐treated myotubes, and these effects were attributed to reduced degradation of ubiquitinated PPARγ through the proteasome pathway. Elevated CD36 demonstrated increased transport of fatty acids into muscle fibres, while elevated CPT2, CPT1A, ACADM, ACADVL, ACOX1 and ACOX2 indicated up‐regulated FAO. The increased intracellular transport and oxidation of fatty acids might be the reason for decreased blood lipids in STING‐deficient diabetic mice. As recently reported, increasing FAO prevented HFD‐induced cardiomyopathy through promoting Parkin‐mediated mitophagy. 24 Similarly, our study showed that the improvements in mitofission and mitophagy achieved by STING deficiency were abolished by PPARγ inhibition, indicating a promoting effect of FAO on mitofission and mitophagy. In addition to mitofission and mitophagy, PPARγ‐mediated FAO also influences IR. As reported, PPARγ inhibition promoted the development of IR in response to metabolic stress. 39 Promoting FAO by overexpressing CPT1 alleviated IR in skeletal muscles under HFD conditions, 40 whereas inhibiting FAO by knocking out CPT2 potentiated PA‐induced IR in C2C12 myotubes. 41 Although transient inhibition of mitochondrial FAO can increase glucose utilization by peripheral tissue and reduce fasting blood glucose, long‐term FAO impairment can lead to systemic IR. 42 Similarly, our study showed that the improvements in IR achieved by STING deficiency were abolished by PPARγ inhibition, demonstrating that increasing PPARγ‐mediated FAO by STING deficiency alleviated IR in the diabetic state. Furthermore, impaired FAO was reported to be an independent predictor of reduced muscle endurance in heart failure patients. 43 Enhancing FAO in aging skeletal muscles partially alleviated the pathological phenotype of sarcopenia. 44 The above reports and results suggested that STING inhibition improved mitofission and mitophagy and reduced IR by improving PPARγ‐mediated FAO, and thus alleviated muscle fibre atrophy.
Interstitial fibrosis is another major histopathological feature of diabetic sarcopenia. Our study showed that STING deficiency alleviated interstitial fibrosis in diabetic muscles. To figure out the mechanisms, PDGFRα+ FAPs were studied. FAPs are a kind of stem cells in skeletal muscles and possess both fibrogenic and adipogenic differentiation potentials. 11 Increased differentiation of FAPs to fibroblasts was reported as an important source of interstitial fibrous tissue in diabetic muscles. 12 In our study, the secreted form of TGFβ1 protein was highly expressed in diabetic muscles and PA‐treated myotubes, and the recombinant TGβ1 protein significantly promoted the differentiation of FAPs to fibroblasts. STING deficiency reduced the level of secreted TGFβ1 protein and decreased the content of fibroblasts in diabetic muscles. These results suggested that STING deficiency reduced the fibrogenic differentiation of FAPs by decreasing the production of secreted TGFβ1 proteins in muscle fibres, which might be a key mechanism for decreasing interstitial fibrosis in diabetic muscles. Further, recent studies reported the protective effect of PPARγ on reducing tissue fibrosis. 45 In our study, the down‐regulatory effect of STING inhibition on the production of secreted TGFβ1 in PA‐treated myotubes was counteracted by PPARγ inhibition, indicating the regulatory effect of PPARγ on fibrosis in the diabetic state. Besides, promoting FAO by overexpressing CPT1A was also reported to reduce TGFβ1 expression and organ fibrosis, 46 suggesting that the anti‐fibrosis effect of STING inhibition might be attributed to its up‐regulatory effect on PPARγ‐mediated FAO.
In conclusion, our results suggested that STING deficiency promoted mitofission and mitophagy and decreased IR by inhibiting the degradation of PPARγ through the proteasome pathway and promoting PPARγ‐mediated FAO, and thus alleviated muscle fibre atrophy. STING deficiency decreased the production of secreted TGFβ1 and reduced the fibrogenic differentiation of FAPs by increasing the PPARγ protein content and possibly by promoting PPARγ‐mediated FAO, and thus reduced the interstitial fibrosis. The above mechanisms jointly improved the muscle mass and muscle strength in T2DM and exerted protective effects in diabetic sarcopenia. STING is expected to be an important therapeutic target to alleviate diabetic sarcopenia.
Conflict of interests
The authors declare that they have no conflict of interests.
Supporting information
Table S1. Detailed information of reagents for in‐vivo and in‐vitro experiments.
Figure S1. Establishment of the diabetic animal model.
Figure S2. PA treatment increased cytosolic ectopic dsDNA in myotubes Myotubes were divided into control group and PA group. (A) Immunofluorescent staining of mtDNA and TOM20. (B) The number of cytosolic dsDNA foci per cell (n = 30 cells/group). *P < 0.05, ** P < 0.01, *** P < 0.001.
Figure S3. STING deficiency alleviated IR in skeletal muscles of diabetic mice.
Figure S4. STING deficiency decreased the levels of blood lipids in the diabetic state.
Figure S5. The effect of STING inhibition on promoting FAO in PA‐treated myotubes was mediated by PPARγ.
Figure S6. STING inhibition alleviated IR by increasing the PPARγ protein content in the diabetic state.
Acknowledgements
This work was supported by the research grants from the Natural Science Foundation of Shandong Province (ZR202111180226). No human data was included in this manuscript. Animal studies have been approved by the appropriate ethics committee and have therefore been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments. All authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle.
Yan S.‐b., Liang H., Zhan P., Zheng H., Zhao Q.‐x., Zheng Z.‐j., et al (2023) Stimulator of interferon genes promotes diabetic sarcopenia by targeting peroxisome proliferator activated receptors γ degradation and inhibiting fatty acid oxidation, Journal of Cachexia, Sarcopenia and Muscle, 14, 2623–2641, doi: 10.1002/jcsm.13336
Contributor Information
Guo‐kai Shang, Email: beijiyibeisgk@163.com.
Xiao‐ping Ji, Email: jixiaoping@sdu.edu.cn.
References
- 1. Jimenez‐Gutierrez GE, Martínez‐Gómez LE, Martínez‐Armenta C, Pineda C, Martínez‐Nava GA, Lopez‐Reyes A. Molecular mechanisms of inflammation in sarcopenia: diagnosis and therapeutic update. Cell 2022;11:2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Izzo A, Massimino E, Riccardi G, Della PG. A narrative review on sarcopenia in type 2 diabetes mellitus: prevalence and associated factors. Nutrients 2021;13:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang X, Zhao Y, Chen S, Shao H. Anti‐diabetic drugs and sarcopenia: emerging links, mechanistic insights, and clinical implications. J Cachexia Sarcopenia Muscle 2021;12:1368–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giha HA, Alamin OAO, Sater MS. Diabetic sarcopenia: metabolic and molecular appraisal. Acta Diabetol 2022;59:989–1000. [DOI] [PubMed] [Google Scholar]
- 5. Honda Y, Tanaka M, Tanaka N, Sasabe R, Goto K, Kataoka H, et al. Relationship between extensibility and collagen expression in immobilized rat skeletal muscle. Muscle Nerve 2018;57:672–678. [DOI] [PubMed] [Google Scholar]
- 6. Chen H, Qian Z, Zhang S, Tang J, Fang L, Jiang F, et al. Silencing COX‐2 blocks PDK1/TRAF4‐induced AKT activation to inhibit fibrogenesis during skeletal muscle atrophy. Redox Biol 2021;38:101774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahuja P, Ng CF, Pang BPS, Chan WS, Tse MCL, Bi X, et al. Muscle‐generated BDNF (brain derived neurotrophic factor) maintains mitochondrial quality control in female mice. Autophagy 2022;18:1367–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Mitophagy is essential for maintaining cardiac function during high fat diet‐induced diabetic cardiomyopathy. Circ Res 2019;124:1360–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gambarotto L, Metti S, Chrisam M, Cerqua C, Sabatelli P, Armani A, et al. Ambra1 deficiency impairs mitophagy in skeletal muscle. J Cachexia Sarcopenia Muscle 2022;13:2211–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Leduc‐Gaudet JP, Reynaud O, Hussain SN, Gouspillou G. Parkin overexpression protects from ageing‐related loss of muscle mass and strength. J Physiol 2019;597:1975–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen W, You W, Valencak TG, Shan T. Bidirectional roles of skeletal muscle fibro‐adipogenic progenitors in homeostasis and disease. Ageing Res Rev 2022;80:101682. [DOI] [PubMed] [Google Scholar]
- 12. Farup J, Just J, de Paoli F, Lin L, Jensen JB, Billeskov T, et al. Human skeletal muscle CD90(+) fibro‐adipogenic progenitors are associated with muscle degeneration in type 2 diabetic patients. Cell Metab 2021;33:2201–2214.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS‐STING signalling. Nat Rev Mol Cell Biol 2020;21:501–521. [DOI] [PubMed] [Google Scholar]
- 14. Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H, et al. The cGAS‐STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B 2022;12:50–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zang N, Cui C, Guo X, Song J, Hu H, Yang M, et al. cGAS‐STING activation contributes to podocyte injury in diabetic kidney disease. iScience 2022;25:105145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ali HS, Boshra MS, Agwa SHA, Hakeem MSA, Meteini MSE, Matboli M. Identification of a multi‐messenger RNA signature as type 2 diabetes mellitus candidate genes involved in crosstalk between inflammation and insulin resistance. Biomolecules 2022;12:1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rong Y, Zhang S. STING controls energy stress‐induced autophagy and energy metabolism via STX17. J Cell Biol 2022;221:e202202060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiao J, Zhang Z. A distinct role of STING in regulating glucose homeostasis through insulin sensitivity and insulin secretion. Proc Natl Acad Sci U S A 2022;119:e2101848119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang D, Liu Y, Zhu Y, Zhang Q, Guan H, Liu S, et al. A non‐canonical cGAS‐STING‐PERK pathway facilitates the translational program critical for senescence and organ fibrosis. Nat Cell Biol 2022;24:766–782. [DOI] [PubMed] [Google Scholar]
- 20. Shang GK, Han L, Wang ZH, Song M, Wang D, Tan YM, et al. Pim1 knockout alleviates sarcopenia in aging mice via reducing adipogenic differentiation of PDGFRα(+) mesenchymal progenitors. J Cachexia Sarcopenia Muscle 2021;12:1741–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sun N, Malide D, Liu J, Rovira II, Combs CA, Finkel T. A fluorescence‐based imaging method to measure in vitro and in vivo mitophagy using mt‐Keima. Nat Protoc 2017;12:1576–1587. [DOI] [PubMed] [Google Scholar]
- 22. Yan M, Li Y, Luo Q, Zeng W, Shao X, Li L, et al. Mitochondrial damage and activation of the cytosolic DNA sensor cGAS‐STING pathway lead to cardiac pyroptosis and hypertrophy in diabetic cardiomyopathy mice. Cell Death Dis 2022;8:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hu Q, Zhang H, Gutiérrez Cortés N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res 2020;126:456–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shao D, Kolwicz SC Jr, Wang P, Roe ND, Villet O, Nishi K, et al. Increasing fatty acid oxidation prevents high‐fat diet‐induced cardiomyopathy through regulating Parkin‐mediated mitophagy. Circulation 2020;142:983–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sithara T, Drosatos K. Metabolic complications in cardiac aging. Front Physiol 2021;12:669497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ferdinandusse S, Denis S, van Roermund CWT, Preece MA, Koster J, Ebberink MS, et al. A novel case of ACOX2 deficiency leads to recognition of a third human peroxisomal acyl‐CoA oxidase. Biochim Biophys Acta Mol Basis Dis 2018;1864:952–958. [DOI] [PubMed] [Google Scholar]
- 27. Mao Y, Luo W, Zhang L, Wu W, Yuan L, Xu H, et al. STING‐IRF3 triggers endothelial inflammation in response to free fatty acid‐induced mitochondrial damage in diet‐induced obesity. Arterioscler Thromb Vasc Biol 2017;37:920–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pham PT, Fukuda D, Nishimoto S, Kim‐Kaneyama JR, Lei XF, Takahashi Y, et al. STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle‐related diseases. Eur Heart J 2021;42:4336–4348. [DOI] [PubMed] [Google Scholar]
- 29. Ma XM, Geng K, Law BY, Wang P, Pu YL, Chen Q, et al. Lipotoxicity‐induced mtDNA release promotes diabetic cardiomyopathy by activating the cGAS‐STING pathway in obesity‐related diabetes. Cell Biol Toxicol 2023;39:277–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang X, Chen Z, Luo Z, Yang D, Hao Y, Hu J, et al. STING deletion alleviates podocyte injury through suppressing inflammation by targeting NLRP3 in diabetic kidney disease. Cell Signal 2023;109:110777. [DOI] [PubMed] [Google Scholar]
- 31. Liu H, Ghosh S, Vaidya T, Bammidi S, Huang C, Shang P, et al. Activated cGAS/STING signaling elicits endothelial cell senescence in early diabetic retinopathy. JCI Insight 2023;8:e168945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Geng K, Ma X, Jiang Z, Huang W, Gu J, Wang P, et al. High glucose‐induced STING activation inhibits diabetic wound healing through promoting M1 polarization of macrophages. Cell Death Dis 2023;9:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marzetti E, Calvani R, Cesari M, Buford TW, Lorenzi M, Behnke BJ, et al. Mitochondrial dysfunction and sarcopenia of aging: from signaling pathways to clinical trials. Int J Biochem Cell Biol 2013;45:2288–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gouspillou G, Godin R, Piquereau J, Picard M, Mofarrahi M, Mathew J, et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J Physiol 2018;596:2565–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dantas WS, Zunica ERM, Heintz EC, Vandanmagsar B, Floyd ZE, Yu Y, et al. Mitochondrial uncoupling attenuates sarcopenic obesity by enhancing skeletal muscle mitophagy and quality control. J Cachexia Sarcopenia Muscle 2022;13:1821–1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee CG, Boyko EJ, Strotmeyer ES, Lewis CE, Cawthon PM, Hoffman AR, et al. Association between insulin resistance and lean mass loss and fat mass gain in older men without diabetes mellitus. J Am Geriatr Soc 2011;59:1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin‐proteasome pathway by defects in muscle cell signaling. Endocrinology 2006;147:4160–4168. [DOI] [PubMed] [Google Scholar]
- 38. Levine ME, Crimmins EM. The impact of insulin resistance and inflammation on the association between sarcopenic obesity and physical functioning. Obesity (Silver Spring) 2012;20:2101–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guo F, Xu S, Zhu Y, Zheng X, Lu Y, Tu J, et al. PPARγ transcription deficiency exacerbates high‐fat diet‐induced adipocyte hypertrophy and insulin resistance in mice. Front Pharmacol 2020;11:1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bruce CR, Hoy AJ, Turner N, Watt MJ, Allen TL, Carpenter K, et al. Overexpression of carnitine palmitoyltransferase‐1 in skeletal muscle is sufficient to enhance fatty acid oxidation and improve high‐fat diet‐induced insulin resistance. Diabetes 2009;58:550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Blackburn ML, Ono‐Moore KD, Sobhi HF, Adams SH. Carnitine palmitoyltransferase 2 knockout potentiates palmitate‐induced insulin resistance in C(2)C(12) myotubes. Am J Physiol Endocrinol Metab 2020;319:E265–E275. [DOI] [PubMed] [Google Scholar]
- 42. Fritzen AM, Lundsgaard AM. Tuning fatty acid oxidation in skeletal muscle with dietary fat and exercise. Nat Rev Endocrinol 2020;16:683–696. [DOI] [PubMed] [Google Scholar]
- 43. Bekfani T, Bekhite Elsaied M, Derlien S, Nisser J, Westermann M, Nietzsche S, et al. Skeletal muscle function, structure, and metabolism in patients with heart failure with reduced ejection fraction and heart failure with preserved ejection fraction. Circ Heart Fail 2020;13:e007198. [DOI] [PubMed] [Google Scholar]
- 44. Hénique C, Mansouri A, Vavrova E, Lenoir V, Ferry A, Esnous C, et al. Increasing mitochondrial muscle fatty acid oxidation induces skeletal muscle remodeling toward an oxidative phenotype. FASEB J 2015;29:2473–2483. [DOI] [PubMed] [Google Scholar]
- 45. Legchenko E, Chouvarine P, Borchert P, Fernandez‐Gonzalez A, Snay E. PPARγ agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med 2018;10:eaao0303. [DOI] [PubMed] [Google Scholar]
- 46. Miguel V, Tituaña J, Herrero JI, Herrero L, Serra D, Cuevas P, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest 2021;131:e140695. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Detailed information of reagents for in‐vivo and in‐vitro experiments.
Figure S1. Establishment of the diabetic animal model.
Figure S2. PA treatment increased cytosolic ectopic dsDNA in myotubes Myotubes were divided into control group and PA group. (A) Immunofluorescent staining of mtDNA and TOM20. (B) The number of cytosolic dsDNA foci per cell (n = 30 cells/group). *P < 0.05, ** P < 0.01, *** P < 0.001.
Figure S3. STING deficiency alleviated IR in skeletal muscles of diabetic mice.
Figure S4. STING deficiency decreased the levels of blood lipids in the diabetic state.
Figure S5. The effect of STING inhibition on promoting FAO in PA‐treated myotubes was mediated by PPARγ.
Figure S6. STING inhibition alleviated IR by increasing the PPARγ protein content in the diabetic state.