

Abstract
Background
Duchenne muscular dystrophy (DMD) is a progressive muscle degenerative disorder, culminating in a complete loss of ambulation, hypertrophic cardiomyopathy and a fatal cardiorespiratory failure.
Necroptosis is the form of necrosis that is dependent upon the receptor‐interacting protein kinase (RIPK) 3; it is involved in several inflammatory and neurodegenerative conditions. We previously identified RIPK3 as a key player in the acute myonecrosis affecting the hindlimb muscles of the mdx dystrophic mouse model. Whether necroptosis also mediates respiratory and heart disorders in DMD is currently unknown.
Methods
Evidence of activation of the necroptotic axis was examined in dystrophic tissues from Golden retriever muscular dystrophy (GRMD) dogs and R‐DMDdel52 rats. A functional assessment of the involvement of necroptosis in dystrophic animals was performed on mdx mice that were genetically depleted for RIPK3. Dystrophic mice aged from 12 to 18 months were analysed by histology and molecular biology to compare the phenotype of muscles from mdxRipk3 +/+ and mdxRipk3 −/− mice. Heart function was also examined by echocardiography in 40‐week‐old mice.
Results
RIPK3 expression in sartorius and biceps femoris muscles from GRMD dogs positively correlated to myonecrosis levels (r = 0.81; P = 0.0076). RIPK3 was also found elevated in the diaphragm (P ≤ 0.05). In the slow‐progressing heart phenotype of GRMD dogs, the phosphorylated form of RIPK1 at the Serine 161 site was dramatically increased in cardiomyocytes. A similar p‐RIPK1 upregulation characterized the cardiomyocytes of the severe DMDdel52 rat model, associated with a marked overexpression of Ripk1 (P = 0.007) and Ripk3 (P = 0.008), indicating primed activation of the necroptotic pathway in the dystrophic heart. MdxRipk3 −/− mice displayed decreased compensatory hypertrophy of the heart (P = 0.014), and echocardiography showed a 19% increase in the relative wall thickness (P < 0.05) and 29% reduction in the left ventricle mass (P = 0.0144). Besides, mdxRipk3 −/− mice presented no evidence of a regenerative default or sarcopenia in skeletal muscles, moreover around 50% less affected by fibrosis (P < 0.05).
Conclusions
Our data highlight molecular and histological evidence that the necroptotic pathway is activated in degenerative tissues from dystrophic animal models, including the diaphragm and the heart. We also provide the genetic proof of concept that selective inhibition of necroptosis in dystrophic condition improves both histological features of muscles and cardiac function, suggesting that prevention of necroptosis is susceptible to providing multiorgan beneficial effects for DMD.
Keywords: Animal model, Cardiac failure, Duchenne muscular dystrophy, Fibrosis, Myogenesis, Myonecrosis, Necroptosis, Programmed cell death
Introduction
Duchenne muscular dystrophy (DMD) is an X‐linked genetic disorder, affecting 1/5000 male birth worldwide. Loss‐of‐function mutations in the dystrophin gene primarily lead to chronic myofibre necrosis (myonecrosis) and severe locomotor muscle disability starting in early childhood. Notably, the absence of dystrophin at the sarcolemma renders the myofibre more susceptible to mechanical stress, which favours contraction‐induced myonecrosis. Following injury, muscle repair requires myogenesis thanks to the contribution of PAX7‐expressing resident muscle stem cells (MuSC). 1 However, in disabling neuromuscular disorders involving chronic myofibre degeneration such as DMD, regeneration does not fully compensate for chronic myofibre demise. The loss of a sustained homeostatic balance between the two mechanisms leads to the increasing replacement of contractile myofibres by fibrotic or adipose tissue. Biopsies with interstitial fibrosis positively correlate with a poor outcome in DMD boys. 2 Thus, identifying the molecular pathways involved in fibrosis formation is relevant for improving therapeutic perspectives. Dystrophin deficiency also challenges the survival of respiratory and cardiac muscle cells, leading to the premature death of DMD patients from cardiorespiratory weakness or failure. 3 , 4
Recently, we reported that necroptosis, a receptor‐interacting protein kinase (RIPK)1‐, RIPK3‐, and mixed lineage domain kinase‐like (MLKL)‐dependent cell death pathway, is activated in the locomotor musculature of murine dystrophin‐deficient muscles. 5 Necroptosis inhibition dampens the peak of myonecrosis in the locomotor muscles of three‐week‐old mdx mice and improves muscle function in young adults. 5 However, two recent studies reported a link between necroptosis inhibition and a deficit in early myogenesis and myofibre atrophy. 6 , 7 One of them reported that RIPK3 depletion in mdx MuSCs dramatically impairs muscle function. Repressing necroptosis might therefore lead to two putative conflictual outcomes: a beneficial decrease in myofibre degeneration, together with a default in muscle regeneration leading to deleterious muscle atrophy and sarcopenia. Consequently, the long‐term effects of necroptosis inhibition in dystrophic skeletal and heart muscle remodelling need to be determined, before further envisaging the necroptotic pathway as a putative therapeutic target for DMD.
To this end, we first examined the diaphragm and heart for evidence of necroptosis activation from Golden retriever muscular dystrophy (GRMD) dogs. The functional assessment of necroptosis in the cardiorespiratory system was achieved using mdxRipk3 −/− mice. Compared with mdxRipk3 +/+ , locomotor muscles from mdxRipk3 −/− mice displayed a significant reduction of fibrosis. Despite its supposed involvement in early myogenesis, RIPK3 deficiency did not generate sarcopenia in this model. In the diaphragm, fibrosis reduction indicates that necroptosis not only participates in the pathology of limbs, but also of respiratory muscles. Importantly, we found that RIPK3 depletion in the heart improves cardiomyopathy by reducing myocardium hypertrophy and fibrosis deposition. Together, our data demonstrate that in dystrophic mdx mice, RIPK3 is dispensable for post‐lesional myogenesis and suggests that necroptosis inhibition is beneficial on the hindlimb, respiratory, and heart muscles in dystrophinopathies. This work opens new avenues for further research exploring the improvement of patient's health status by reducing necroptosis, in complement to other proposed therapies.
Methods
Animals
Mice and rats were bred in the Institut Mondor de Recherche biomédicale, Créteil, France in a pathogen‐free facility with 12‐h light and 12‐h dark cycles in accordance with European Directive 2010/63/EU. Only male mice and rats were used for the experiments. Both wild‐type (WT) and DMD rats and mdx mice were born from the same litter. The GRMD dogs included in this study were housed in the facilities of the CARE‐NMD platform of the Veterinary School of Alfort. Experiments were approved by the Anses/EnvA/Upec Ethics Committee. All care and manipulations were performed by national and European legislation on animal experimentation. Ripk3 −/+ mice were kindly provided by Genentech (San Francisco, CA, USA). mdxRipk3 −/− mice were generated by crossing Ripk3 −/− with mdx mice. The resulting mice were backcrossed for at least four generations to obtain final mdxRipk3 +/− breeders.
Treadmill tests
The running capacity was evaluated on a treadmill (Tecmachine, Medical Development) at a + 5% slope. Males of 70‐ to 76‐week‐old mice were initially acclimatized to the treadmill for 5 days (10 min/day and 12 m/min). Mice were challenged to an exercise of progressively increased intensity. Speed was increased every 90 to 120 s/min and the speed of the last completed running step before exhaustion was considered as the individual maximal aerobic speed.
Molecular biology
Total skeletal muscle RNA was extracted from muscle and heart samples using TRIzol (Thermo Fisher Scientific) following the manufacturer's instructions. SuperScript III Reverse Transcriptase from the Invitrogen kit converted RNA into cDNA using the Veriti 96‐Well Fast Thermal Cycler (Applied Biosystems). Gene expression was quantified by real‐time qPCR with the StepOnePlus real‐time PCR system (Applied Biosystems) using SYBR Green detection tools (Applied Biosystems). Gene expression for the mouse transcripts in skeletal muscles was normalized to expression of Tbp (encoding TATA‐binding protein). Results are reported as relative gene expression (2‐DDCT). In the heart, from 500 ng of extracted RNA, the first‐strand cDNA was then synthesized using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with random hexamers according to the manufacturer's instructions. Using the Light Cycler® 480 system (Roche Diagnostics), the reaction was carried out in duplicate for each sample in a 6‐μL reaction volume containing 3 μL of SYBR Green Master Mix, 500 nM of the forward and reverse primers each and 3 μL of diluted (1:25) cDNA. The thermal profile for the SYBR Green qPCR was 95°C for 8 min, followed by 40 cycles at 95°C for 15 s, 60°C for 15 s and 72°C for 30 s. Gene expression the mouse transcripts in the heart was normalized to expression of Hprt1 and Rpl32. In dogs, RNA was extracted from frozen muscle slides using the NucleoSpin RNA extraction kit (Macherey Nagel). RNA was quantified by Nanodrop and the Maxima first strand cDNA master kit (Thermo Fisher) was used to convert 300 ng of RNA into cDNA. Gene expression was quantified by real‐time qPCR with the QuantStudio™ 3 Real‐Time PCR System (Applied Biosystems) using SYBR Green detection kit (Thermo Fisher). The reaction was carried out in triplicates for each sample of diluted (1:10) cDNA. Gene expression of transcripts in dog biopsies was normalized to the expression of ACTB (encoding ß‐actin). Results are reported as relative gene expression (2‐DDCT).
Primers used: Mouse: forward Ripk3 5′‐CGGGCACACCACAGAACAT‐3′, Ripk3 and reverse 5′‐GTAGCACATCCCCAGCACCAC‐3′, forward Ripk1 5′‐AGAAGAAGGGAACTATTCGC‐3′ and reverse Ripk1 5′‐TTCTATGGCCTCCACGAT‐3′, forward Bnp 5′‐CAGCTCTTGAAGGACCAAGG‐3′, reverse 5′‐AGACCCAGGCAGAGTCAGAA‐3′, forward Myh7 5′‐AGGTGTGCTCTCCAGAATGG‐3′, reverse 5′‐CAGCGGCTTGATCTTGAAGT‐3′, Rpl32 ‐ forward 5′‐TGGTGAAGCCCAAGATCGTC‐3′, reverse 5′‐GGATCTGGCCCTTGAACCTT‐3′, Hprt1 ‐ forward 5′‐CAGGCCAGACTTTGTTGGAT‐3′, reverse 5′‐TTGCGCTCATCTTAGGCTTT‐3′. Rat: forward Ripk3 5′‐CGTACACGTAGTCCCACTCG‐3′, reverse 5′‐AGGGAGGTGAAGGCTATGGT‐3′, forward Ripk1 5′‐GGTCTCCCATGACCCCTTTG‐3′, reverse 5′‐GGTAGGTTGGTCTCAGGCAC‐3′. Dog: Forward RIPK1 5′‐GAATGAGTTCAGCCCTGCTC‐3′, reverse 5′‐CTCGCTCATAGTCGTGGTCA‐3′, forward RIPK3 5′‐CAGAGAGGCTCAAGGTCAGG‐3′, reverse 5′‐CGATGTCTGGGCCACTATCT‐3′ ACTB forward 5′‐CCATCTACGAGGGGTACGCCC‐3′ reverse TGCTCGAAGTCC AGGGCGACGTA.
Histology
Fibrosis was labelled using Epredia Varistain Gemini ES Automated Slide Stainer. For immunofluorescence: rat antibody to CD68 (clone FA‐11, 137001, 1/50), rabbit antibody to mouse pan‐Laminin (Sigma, L9393, 1/1000), Phospho‐RIPK1 (Ser161) antibody (1/100 Invitrogen 105640).
Biopsies from GRMD dogs were processed and analysed as previously described. 8 The dried sections of biceps femoris, sartorius cranialis, diaphragm, and heart were stained for 10 min in Hematein and 5 min in 1% Eosin, dehydrated in four consecutive baths of ethanol, one bath of xylene and mounted in Canada balsam. One entire section per biopsy was photographed using an AxioObserver Z1 linked to its ICC1 camera (Zeiss), and the MosaiX application of the software AxioVision (Zeiss). The image was then analysed using the software Visilog 6.4 (Noesis). A grid of 10 000 mm2 squares was superimposed onto the image of the entire section. At each intercept of the grid (i.e., every 100 mm) the histological aspect of the underlying tissue was manually captioned using predefined annotations. The percentage of each type of histological event was calculated, defined as the percentage of events not corresponding to normal shape fibres. Inflammatory infiltration in dog biopsies was quantified using CD4, CD8, and CD11b immunostainings; 7 μm sections from each biopsy were fixed in cold acetone‐methanol. After having blocked the endogenous peroxidase activity, the primary antibody (either a rat anti‐canine CD4, Serotec H, 1/50, or a rat anti‐canine CD8, Serotec H, 1/50, or a mouse anti‐canine CD11b, Serotec H, 1/50) was used. The amount of inflammatory cells was normalized to the area of the section. 8
Myonecrosis levels in mdx biopsies were determined by measuring the area corresponding to mouse IgG uptake in myofibres (and not in interstitial areas) and expressed as the percentage of the cross‐sectional area. Muscle fibre minimal Feret diameter, corresponding to the minimum distance between the two parallel tangents of myofibres, was determined. Images analyses were performed with ImageJ by a specific self‐developed macro that recognizes muscle fibres as previously described. 9
Cardiac phenotyping
Mice were trained to be grasped because transthoracic echocardiography (TTE) was performed in conscious, non‐sedated mice to avoid any cardiac depressor effect of sedative or anaesthetic agents. Data acquisition was performed every month by a single operator (JT) as previously described. 10 Images were acquired from a parasternal position at the level of the papillary muscles using a 13‐MHz linear‐array transducer with a digital ultrasound system (Vivid 7, GE Medical System, Horton, Norway). Left ventricular diameters and ejection fraction, anterior and posterior wall thicknesses were serially obtained from M‐mode acquisition. Relative LV wall thickness (RWT) was defined as the sum of septal and posterior wall thickness over LV end‐diastolic diameter, and LV mass was determined using the uncorrected cube assumption formula (LV mass = (IVSd + LVIDd + PWTd)3 − LVIDd3). Peak systolic values of radial SR in the anterior and posterior wall were obtained using tissue Doppler imaging (TDI) as previously described. 10 TDI loops were acquired from the same parasternal view at a mean frame rate of 514 fps and a depth of 1 cm. The Nyquist velocity limit was set at 12 cm/s. Radial SR analysis was performed offline using the EchoPac Software (GE Medical Systems) by an observer (GD) blinded to the diet of the animals. The peak systolic of radial SR was computed from a region of interest positioned in the mid anterior wall and was measured over an axial distance of 0.6 mm. The temporal smoothing filters were turned off for all measurements. Because slight respiratory variations exist, we averaged peak systolic of radial SR on 8 consecutive cardiac cycles.
Results
Firstly, we addressed the relevance of necroptosis in dystrophin deficiency as a potential cell demise involved in locomotor and respiratory muscles and the heart. Because heart or diaphragm muscle biopsies cannot be performed in patients, we aimed at examining tissues in different animal models for DMD. We first investigated degenerating tissues from golden retriever muscular dystrophy (GRMD) dogs that represent a gold standard relevant animal model for DMD. 11 Compared with age‐matched wild‐type (WT) dogs, 6‐month‐old‐GRMD dogs were characterized by typical high and variable blood creatine kinase levels (P = 0.0167) (Figure S1A), assessing a significant ongoing, global myofibre demise. Histological features of myonecrosis in the sartorius and biceps femoris muscles confirmed the presence of recently injured area (Figure 1A). Because myofibre necroptosis comes with an increase of Ripk3 transcripts in mdx TA muscles, and that RIPK3 upregulation sensitizes muscle cells to TNF‐elicited necroptosis, 5 we examined whether there was a correlation between muscles presenting high histological necrotic extent and RIPK3 expression in muscles of GRMD dogs. Sartorius muscles from 6‐ and 12‐month‐old GRMD dogs were together quantified by qPCR for RIPK3 transcripts and quantified for evidence of necrotic events and inflammation in haematoxylin & eosin staining. Interestingly, RIPK3 expression was highly correlated with the extent of myonecrosis (Pearson test, P = 0.0076, r = 0.8138, n = 9 GRMD muscles) (Figure 1A), but not with the inflammatory area (P = 0.1409) (Figure S1B), suggesting that RIPK3 elevation is specific to necrotic demise, rather than post‐necrotic events such as myeloid infiltrate. Similarly, RIPK3 transcripts increased in the GRMD diaphragm compared with age‐matched WT dogs, and RIPK3 expression was also positively correlated with RIPK1 expression in these muscles (P = 0.0307) (Figure 1B,C). Thus, RIPK3‐dependent cell death does not only involve ambulatory, but also the main respiratory musculature in the DMD pathogenic course. In the heart, while we observed no significant increase in the necrosome transcripts in the left ventricle of 6‐month‐old GRMD dogs (Figure 1E), we found a dramatic increase of immunoreactivity of GRMD dog's cardiomyocytes to antibodies directed against phospho‐RIPK1 (Ser161), indicating that RIPK1 is primed for either extrinsic apoptosis or canonical necroptosis (Figure 1F). 12 , 13
Figure 1.
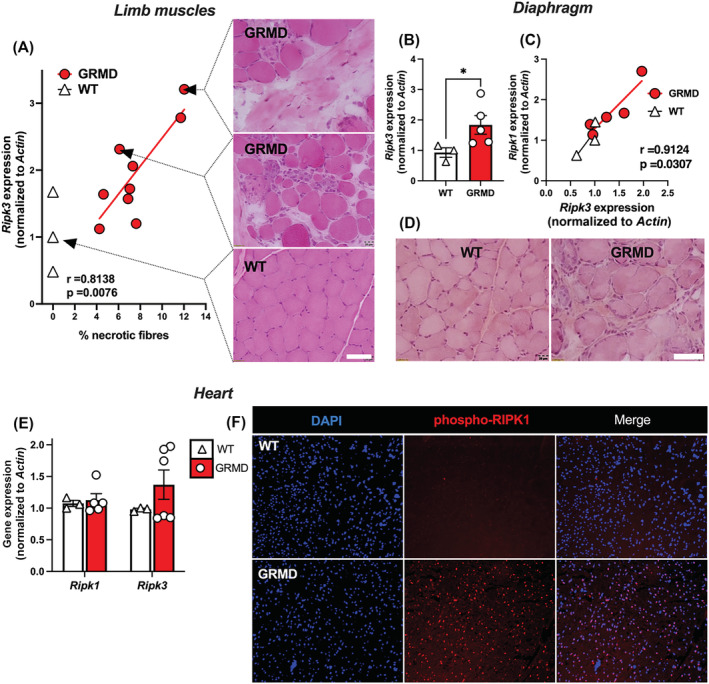
The RIPK1‐RIPK3 axis is dysregulated in the skeletal muscles and the heart of dystrophic dogs. (A) Pearson correlation coefficient (r) and P‐value (P) between necrotic fibre percentage and RIPK3 transcripts expression normalized to ACTIN. Representative H&E staining of the sartorius muscle corresponding to related biopsied muscle. Scale bar: 20 μm. (B) Quantification of RIPK3 expression in the diaphragms of 6‐month‐old WT and GRMD dogs. (C) Pearson correlation between RIPK1 and RIPK3 expression in the diaphragm. (D) H&E staining of the diaphragm muscle. (E) in the heart left ventricle of GRMD and WT dogs, quantification of RIPK1 and RIPK3 transcripts normalized to ACTIN. (F) Representative image of a left ventricle from WT and GRMD dogs labelled with an antibody directed against the phosphorylated form of RIPK1 (S161). Data are given as means ± SEM. ns: Not significant, *P‐value < 0.05.
Thus, to determine whether necroptosis has a functional role in progressive cardiomyopathy or in myonecrosis affecting the diaphragm of dystrophin‐deficient mice, we generated mdxRipk3 −/− mice by crossing C57BL/10ScSn‐Dmd mdx (hereafter named mdx) mice with Ripk3‐KO mice (originally generated in a C57BL/6 N strain), which compromise dystrophic mice for RIPK3‐dependent necroptosis. 14 MdxRipk3 +/+ (hereafter named mdxRipk3 +/+ or mdx) and mdxRipk3 −/− littermates were kept for ageing to reach the maximum mdx pathogenic state in striated muscles. We first examined whether the long‐term genetic inhibition of necroptosis would promote sarcopenia by repressing MuSCs density and the regenerative capacity of skeletal muscle. Indeed, a recent article indicated that necroptosis inhibition generates a MuSCs depletion by inducing the death of PAX7‐positive cells, with a subsequent deficit in myogenesis associated with a dramatically reduced muscle strength in adult mdx mice. 6 In biceps from 18‐month‐old mdxRipk3 −/− mice, Ripk1 transcripts were downregulated compared with Ripk3 +/+ littermates (Figure 2A), indicating that canonical necroptosis is repressed in mdxRipk3 −/− mice. To determine whether the defect of necroptosis signalling compromises muscle function, mdx and mdxRipk3 −/− littermates underwent forced treadmill exercise with progressive increase of speed, until exhaustion. Control mdx mice were characterized by an important variability in running performance, expressed in the running distance or the maximum aerobic speed before exhaustion (Figure 2B). mdxRipk3 −/− mice showed more homogenous capacities (coefficient of variation: mdx: 20.99% vs. mdxRipk3 −/− 12.66%). On average, mdxRipk3 −/− ran ~10% longer than mdx littermates. However, no significant differences were observed in running performance between mdx and mdxRipk3 −/− mice. The putative atrophy of Ripk3 −/− dystrophic muscles was examined using muscle mass normalized to the individual size of the animals, assessed by the tibia length (TL). TA, EDL and biceps muscle mass remained unchanged in aged mdxRipk3 −/− compared with mdxRipk3 +/+ mice (Figure 2C). Although young mdx muscles show extensive necrotic events leading to the accumulation of fibrotic tissue, muscles from old mice are typically spared. In the rare post‐necrotic areas in TAs from old mdx and mdxRipk3 −/− mice, we found an equal density of Ki67‐positive cells (Figure 2D), suggesting that necroptosis inhibition does not affect MuSC proliferation in mdx muscle at this age. The mean of pooled minimum Feret of TA myofibres (Figure S2A), as well as the relative distribution of myofibre (Figure S2B), were strictly unchanged. Interestingly, we found that RIPK3 depletion reduced fibrosis in the TA (P = 0.0252) (Figure 2E), suggesting a cumulative cytoprotective effect regarding ancient degenerative events. To further examine any possible evidence of regenerative default in mdxRipk3 −/− muscles, we analysed biceps muscles for Pax7 and myogenin (Figure 2F) expression but observed no difference. Of note, we found a significant downregulation of myostatin expression (P = 0.0082), a biomarker of neuromuscular disorders while follistatin was not significantly repressed (P = 0.4452) (Figure 2F).
Figure 2.
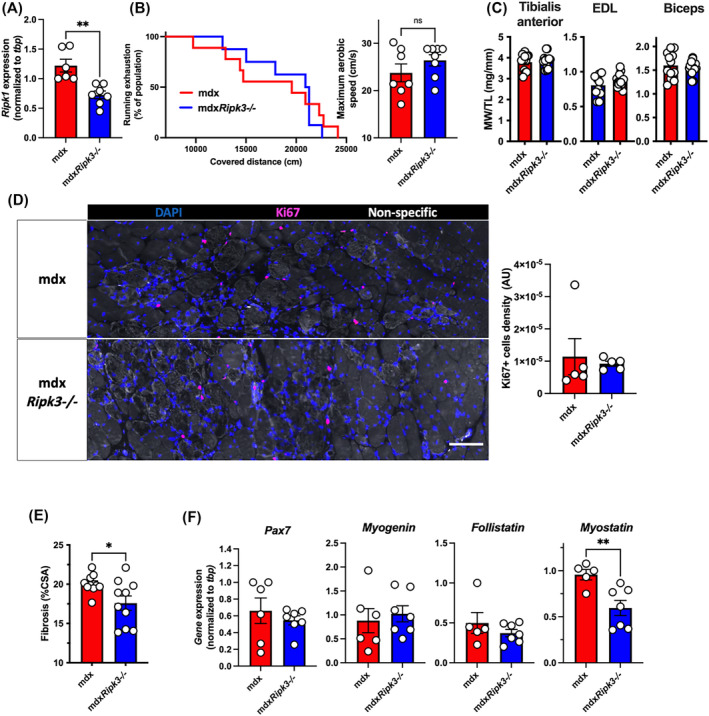
The genetic ablation of RIPK3 does not promote sarcopenia in 18‐month‐old mdx mice. (A) Quantification of Ripk1 transcripts in the 18‐month‐old biceps muscles. Seventeen‐month‐old mdxRipk3 +/+ (i.e., mdx) and mdxRipk3 −/− male mice were submitted to forced treadmill running with a progressive increase of speed. (B) Endurance curve of running mice against covered distance expressed in cm and maximum aerobic speed before mice exhaustion. Data are given as means ± SEM. mdxRipk3 +/+, n = 7, mdxRipk3 −/−, n = 8. Shapiro–Wilk test followed by a Mann–Whitney test. (C) Tibialis anterior (TA), EDL and biceps muscles were harvested, and muscle weight (MW) was determined (and normalized to tibial length (MW/TL). (D) TA immunolabelling using antibodies directed against Ki67 (red) and quantification of the density of Ki67‐positive cells. (E) Quantification of collagen deposition using Sirius red dye. The normal distribution of data was tested using the Kolmogorov–Smirnov test (passing normality test with α = 0.05), leading to either t‐tests or Mann–Whitney tests. (F) Quantification of the transcript levels of Pax7, myogenin, follistatin, and myostatin in the biceps, by quantitative PCR. Data are given as means ± SEM. ns: Not significant, *P‐value < 0.05, **P‐value < 0.01. Scale bar: 100 μm.
We then examined whether the pathogenic features of the mdx diaphragm are improved by RIPK3 depletion (Figure 3). In histology, the localization of blood proteins such as albumin or immunoglobulins within the sarcoplasm is a reliable marker for the necrotic fate of myofibre. 15 , 16 Muscles from 18‐month‐old mice showed IgG immunoreactivity almost only localized at the extracellular compartment (Figure 3A), and rarely within myofibres, indicating no/little ongoing myonecrosis. No difference was found in both genotypes (unpaired t‐test, P = 0.1914). The density of myofibres with leaky sarcolemma was extremely low in both genotypes (2.3 fibres/mm2 in mdx vs. 2.6 fibres/mm2 in mdxRipk3 −/− ). Only a low percentage of centrally nucleated fibres was found in the mdx diaphragm (5–10% of myofibres) (Figure 3B). In both genotypes, this was not associated with infiltrations of CD68‐positive cells (Figure 3C), suggesting that the diaphragm from 18‐month‐old mdx mice stand at relative homeostasis with little/no ongoing myonecrosis, regardless of RIPK3 expression. Dystrophin‐deficient muscles are characterized by an increased heterogeneity in myofibre size, illustrated by an increase of the variance coefficient. 17 RIPK3‐deficiency decreased the mean–variance coefficient of myofibres in mdx mice (Figure 3D, unpaired t‐test P = 0.0431). Examining the profile of myofibre size, we observed a moderate decrease of myofibre size in mdxRipk3 −/− diaphragm compared with mdx littermates (16.6 in mdx vs. 16.1 in mdxRipk3 −/− , P < 0.0001, Mann–Whitney test, n > 16 000 myofibres in each group, included in six mdx muscles, and eight mdxRipk3 −/− muscles) (Figure 3E). Fibrosis deposition in respiratory muscles, such as the diaphragm, is a key pathogenic event in DMD pathogenesis and correlates with a bad prognosis in patients. To determine whether RIPK3 also mediates fibrosis deposition in the diaphragm, muscle sections were stained with Sirius red (Figure 3F). We observed less collagen deposition in mdxRipk3 −/− diaphragms compared with mdx littermates (unpaired t‐test, P = 0.0414). Together, our data demonstrate that RIPK3 participates in diaphragm pathogenesis in mdx mice by promoting interstitial fibrosis.
Figure 3.
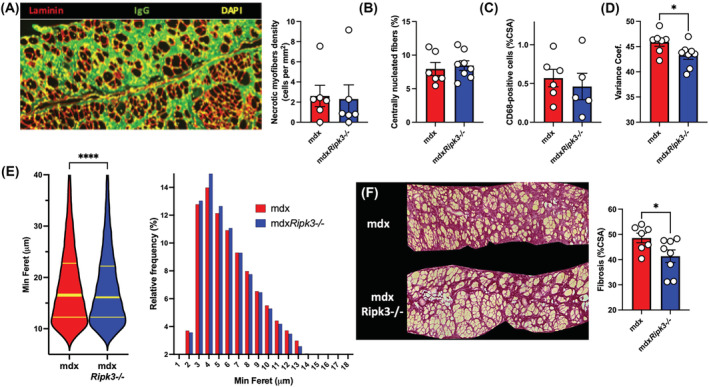
Necroptosis participates in the diaphragm fibrosis in mdx mice. (A) Representative image of IgG uptake in diaphragms of 18‐month‐old mdx (mdxRipk3 +/+) and mdxRipk3 −/− mice and quantification of myonecrosis extent. Myonecrosis is defined as the area occupied by leaky myofibres (i.e., immunoreactive for mouse IgG uptake). (B) Quantification of the percentage of centrally nucleated myofibres. (C) CD68‐positive macrophage infiltrates expressed in percentage of CSA. (D) Quantification of the variance coefficient of the minimum Feret of myofibres. (E) Violin plot of the minimum Ferets of diaphragm myofibres and relative frequency of minimum Ferets from diaphragm myofibres (mdx, n = 16 873 including seven distinct muscles, mean: 18.61 ± 0.06555; mdxRipk3 −/−, n = 31 666 including eight distinct muscles, mean: 18.21 ± 0.04554, two‐tailed Mann–Whitney test P < 0.0001). Data are expressed as a percentage. (F) Representative picture of Sirius red staining and quantification of diaphragm fibrosis. The normal distribution of data was tested using the Kolmogorov–Smirnov test (passing normality test with α = 0.05), leading to either t‐tests or Mann–Whitney tests. Data are given as means ± SEM. ns: Not significant, *P‐value < 0.05, ****P‐value < 0.0001.
We next addressed the role of necroptosis in the development of cardiomyopathy upon dystrophin deficiency. Importantly, end‐stage cardiomyopathy is a major concern in the survival of boys and is associated with hallmarks of cardiomyoblast necrosis and fibrosis deposition. In GRMD dogs, RIPK1 was found phosphorylated at the Ser161 site in the heart (Figure 1F), indicating an early step of activation of RIPK1‐dependant apoptosis or necroptosis. 13 R‐DMDdel52 rats develop an early cardiomyopathy 18 and the analysis of hearts by H&E staining showed evidence of cardiomyoblast degeneration in 6‐month‐old rats (Figure 4A). Cell death was associated with a significant increase of Ripk1 (P = 0.0070, t‐test) and Ripk3 (P = 0.0080, t‐test) transcripts compared with age‐matched WT rats (Figure 4A), suggesting advanced necroptosis activation in the heart from R‐DMDdel52 rats at this age. Similar p‐RIPK1 immunoreactivity to GRMD dogs was observed in R‐DMDdel52 rats (Figure 4B). This contrasted with dogs that displayed no significant elevation of RIPK1 and RIPK3 expression (Figure 1F), in agreement with rare fresh necrotic events observed in dystrophic hearts of GRMD dogs. To determine the role of necroptosis in the progressive cardiomyopathy of dystrophin‐deficient hearts, we further analysed the heart phenotype of 18‐month‐old mdxRipk3 −/− mice (Figure 4C–F). RIPK3 depletion decreased cardiac compensatory hypertrophy in mdx mice (Figure 4C). At the investigated age, we found no significant levels of ongoing degeneration in the heart, displaying little CD68‐positive cell infiltration whatever the genotype, which suggested an accumulation of past degenerative events with little evolution over time at this age. Furthermore, the hearts of mdxRipk3 −/− mice showed significantly reduced expression of Bnp and Myf7, two sensitive markers of cardiac hypertrophy (Figure 4D). The minimum Feret of mdxRipk3 −/− cardiomyocytes was slightly reduced compared with Ripk3 +/+ littermates (two‐tailed Mann–Whitney test P < 0.05), likely due to a relative over‐representation of cardiomyocytes (Figure 4E). Interstitial collagen deposition was reduced by almost 50% in the right ventricle of mdxRipk3 −/− mice compared with their mdx control littermates (Figure 4F).
Figure 4.
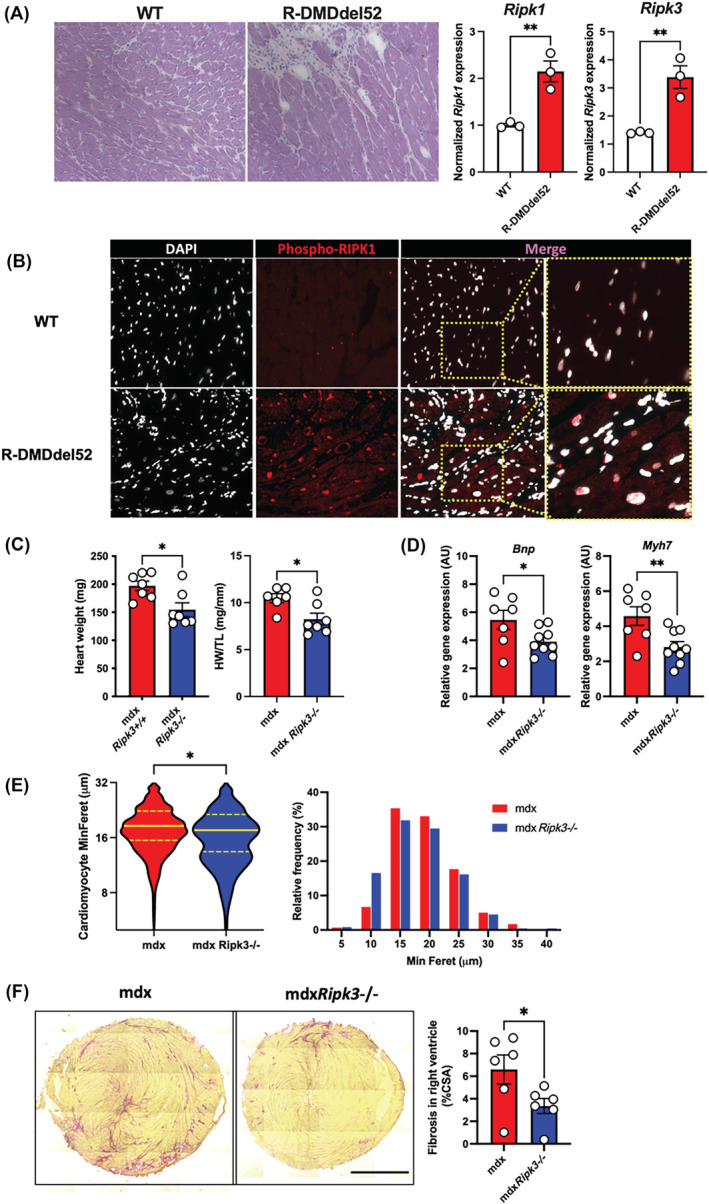
RIPK3 mediates the compensatory hypertrophy of the dystrophin‐deficient heart. (A) Representative pictures of H&E labelling from 6‐month‐old wild‐type (WT) and dystrophin‐deficient R‐DMDdel52 rats and quantification of the rat Ripk1 and Ripk3 transcripts in hearts from WT and R‐DMDdel52 rats. (B) Representative pictures of heart labelling using an antibody directed against the phosphorylated form of RIPK1 at S161 from 6‐month‐old rats. (C) Heart weights of 18‐month‐old mdx and mdxRipk3−/− mice respectively without with normalization to the tibial length (TL). (D) Hearts from 18‐month‐old mdxRipk3 +/+ and −/− were analysed for Bnp and Myh7 mRNA levels by quantitative PCR. (E) Violin plot of the minimum Ferets of mouse cardiomyocytes and relative frequency of minimum Ferets from cardiomyocytes. Data are expressed as a percentage (fifty cardiomyocytes measured per mouse including n = 6 mdx and 5 mdxRipk3 −/− hearts, two‐tailed Mann–Whitney test P < 0.05). (F) Representative picture of Sirius red staining of mdx and mdxRipk3 −/−, and quantification of RV fibrosis. Scale bar: 1700 μm. The normal distribution of data was tested using the Kolmogorov–Smirnov test (passing normality test with α = 0.05), leading to either t‐tests or Mann–Whitney tests. Data are given as means ± SEM. ns: Not significant, *P‐value < 0.05, **P‐value < 0.01.
We thus examined whether the improvement of the deleterious phenotype of cardiomyopathy conferred by RIPK3 depletion had a positive impact at the 2D and functional levels. Routine echocardiographic parameters were measured using the M (otion)‐mode (Figure 5A). Compared with wild‐type mice, aged mdx mice are generally characterized by eccentric hypertrophy characterized by a transiently increased LV mass with increased left ventricle (LV) end‐diastolic and end‐systolic diameter. This is followed by a decompensated dilated cardiomyopathy associated with a reduction in LV fractional shortening and a decreased LV mass. 19 We confirmed that our 18‐month‐old mdx mice depleted for RIPK3 had an improved phenotype, with reduced LV internal diameter in diastole (LVIDd) and systole (LVIDs) (Figure 5B and Table S1). Of note, compared with mdx mice, mdxRipk3 −/− littermates displayed a 30% reduction of the LV mass linked to a decrease in LVIDd that contributed to the restoration of the LV relative wall thickness value (Figure 5D). Fractional shortening of the LV, strain rate of cardiomyocytes from the anterior wall (Figures 5E–F) and heart rate remained unchanged (Table S1). Altogether, these data suggested that in the mdx model, RIPK3‐dependent necroptosis participates in the long‐term development of cardiac hypertrophy and fibrosis and that in these conditions, RIPK3 deficiency has overall cytoprotective effects against dystrophic‐associated cardiac deleterious remodelling in the mouse.
Figure 5.
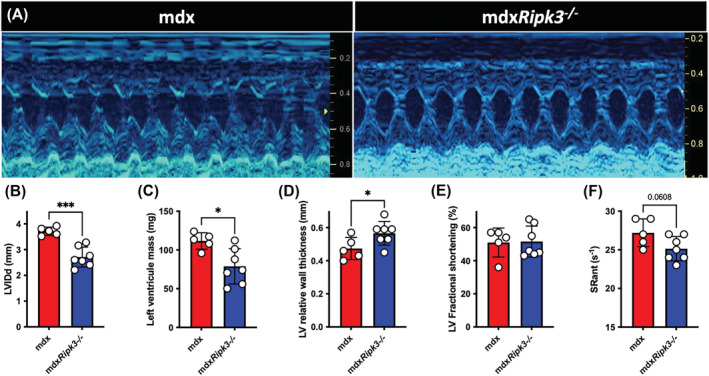
The genetic ablation of RIPK3 reduces cardiomyopathy in mdx mice. (A) Representative M‐mode echocardiogram traces in mdx and mdxRipk3 −/− 40‐week‐old mice showing a reduced left ventricle (LV) dilation in mdxRipk3 −/− mice. (B) Quantification of LVIDd, (C) LV mass, (D) LV relative wall thickness, (E) LV fractional shortening, and (F) strain rate (SR) of the anterior LV wall. The normal distribution of data was tested using the Kolmogorov–Smirnov test (passing normality test with α = 0.05), leading to t‐tests. Data are given as means ± SEM. *P‐value < 0.05, ***P‐value < 0.001.
Discussion
Necroptosis is now considered a new therapeutic target for several degenerating disorders, including cardiovascular neurodegenerative and autoimmune diseases. 20 Dystrophin absence and DUX4 overexpression can lead to myofibre necroptosis in vivo. 5 , 21 , 22 Translating these discoveries into the clinic for neuromuscular disorders requires an understanding of (1) the long‐term consequences of necroptosis inhibition in dystrophic conditions and (2) the role of necroptosis in the DMD cardiorespiratory phenotype.
While locomotor tissues can be harvested in DMD patients for diagnostic purposes and could be used for research purposes, respiratory or heart biopsies were not available. We then used dystrophic dogs and rats which are highly relevant animal models for DMD and reproduce the main aspects of DMD pathogenesis. 11 Transient RIPK3 overexpression is a biomarker for necroptosis in multiple animal models in vivo, 23 including for myofibre in mdx mice, and sensitizes myoblasts to TNF‐elicited necroptosis. 5 Using sartorius and femoral biceps, diaphragm and cardiac tissues from 6‐month‐old GRMD dogs, we first assessed different levels of activation of the early steps of the RIPK1‐RIPK3 canonical necroptotic pathway. RIPK3 upregulation in limb muscles, and diaphragm, and its correlation with the myonecrosis extent (Figure 1A,B), suggest a role for RIPK3 in the myonecrosis affecting GRMD dogs, especially in cells expressing p‐RIPK1, which prime cells to programmed necrosis. 13 RIPK3 typically recruits MLKL during final necroptosis induction, and p‐MLKL causes membrane permeability and necrotic morphology. Interestingly, dogs and other species belonging to the Carnivora order express crucial proteins required for necroptosis activation such as RIPK1, RIPK3, DNA‐dependant activator of interferon regulatory factors (DAI) and TIR‐domain‐containing adaptor‐inducing IFNβ (TRIF), however, they do not express MLK. 24 The molecular mechanisms of cell death execution in RIPK3‐dependant but MLKL‐independent necrosis, involvement remains an open question.
Notably, necrotic demise affecting the diaphragm and heart are key events leading to the short life expectancy of DMD boys. Herein, we provide evidence that beyond its role in the degeneration of locomotor muscles, necroptosis also takes part in the respiratory and cardiac phenotypes in the mdx model of DMD, notably by promoting tissue fibrosis. In skeletal muscle as in other tissues, necrotic cells generate a living cell‐free area that is rapidly infiltrated by myeloid cells, in charge of phagocyting cell debris and promoting tissue repair. In the regenerating muscle, transient overexpression of the extracellular matrix supports myogenesis in many ways, including as an anchorage structure and controlling cell polarity. 25 Muscles affected by chronic injuries and necroinflammation experience prolonged production of extracellular matrix components and adversely affect myogenesis. Post‐lesional area, therefore, progressively becomes colonized by scar/fibrotic tissue. 26 Accumulation of extracellular matrix deposition following myonecrosis is a central pathogenic process contributing to the loss of function of dystrophic muscles, by progressively replacing functional muscle tissue in both the myocardium and skeletal muscles. 2 Therefore, understanding the molecular pathways directly or indirectly leading to myofibre or cardiomyocyte death or fibrosis is highly relevant in the context of long‐term muscle repair.
In dystrophin‐deficient mice, investigating the phenotypes of the diaphragm and heart requires the generation of dystrophic and necroptosis‐incompetent mice that would reach sufficient age to progressively develop a significant phenotype. 19 , 27 Compared with young dystrophic TA, the diaphragm from 18‐month‐old mdx mice showed IgG immunoreactivity mainly trapped in the large fibrotic area (Figure 3A) and barely detected within the myofibre compartment, suggesting little/no ongoing myonecrosis. The observed low percentage of central nucleation together with low cell infiltration and high fibrosis indicates that this phenotype is the outcome of accumulating necrotic and regenerative events over time. Fibrosis decrease was observed in the diaphragm and was associated with a decrease in the mean myofibres size and size variability (Figure 3D–F). These results are consistent with a previous report assessing significant myofibre cytoprotection in the diaphragm. 28 We also found a significant reduction of fibrosis in the TA of old mdxRipk3 −/− (Figure 2E) compared with mdx littermates, suggesting that the effects of necroptosis inhibition on muscle histology benefit both locomotor and respiratory muscles.
Two recent studies reported that necroptosis is involved in post‐lesional myogenesis. 6 , 29 However, the underlying mechanisms of action of necroptosis on regeneration seem unclear: Sreenivasan and colleagues observed exclusive expression of RIPK3 in mdx MuSCs and not in myofibres. MuSCs depleted for RIPK3 led to a dramatic loss of PAX7‐expressing cells and severely impaired muscle function. 6 On another hand, Zhou and colleagues found that myofibres express RIPK3 and MLKL and are susceptible to necroptotic demise. They demonstrated that myofibre necroptosis indirectly modulates muscle regeneration by releasing Tenascine‐C, which is required for MuSCs proliferation. An effect of myofibre necroptosis on Pax7 expression and myofibre size was observed in vivo at 7 and 15 days post‐injury. 29 As the full effects on regeneration are generally observed at a later stage, after 4 weeks post‐injury, it is unclear in this study whether the depletion of necroptosis irreversibly stops myogenesis or only delays it.
Herein, we found no impairment of running performance in dystrophic Ripk3 −/− mice, and no/little effect on muscle weight, while confirming the downregulation of Ripk1 in muscle extracts. The weight of limb muscles was unchanged, as myofibre size (Figures 2C and S2). Pax7 and myogenin expression were not reduced in mdxRipk3−/− mice, suggesting there is no depletion of MuSCs. Together, our results demonstrate that in mdx mice, a long‐term necroptosis inhibition does not lead to sarcopenia of dystrophic muscle, but rather has beneficial effects because it dampens fibrosis. The lack of observed sarcopenia in our mdxRipk3 −/− dystrophic mice could for instance be explained by a delay of myogenesis when necroptosis is prevented. Indeed, slowing down the kinetics of differentiation does not necessarily impair regeneration. 30 It is also possible that any effect, positive or negative of RIPK3‐deficiency on regeneration could be mitigated by the cytoprotective effects of necroptosis inhibition. Not to be forgotten that the regenerative process remains a consequence of previous degenerative events. Hence, the less degeneration, the less resulting regeneration. The hypothesis that long‐term inhibition of necroptosis may have a more beneficial than harmful impact will be extensively examined in the severe DMD rat model. This model is marked by continuous and active cycles of myonecrosis/regeneration in the first 6 months of the disease's progression, which are eventually halted by the animals' depleted repair capacity towards the end of their lifespan. 31
In mdx mice, myofibre necrosis does not lead to a dramatic lack of function of the limb, respiratory or cardiac muscles. 32 On another hand, pathology is dramatically exacerbated in R‐DMDdel52 rats with a reduced life expectancy and cardiomyopathy functionally assessed in young adults. 18 Phospho‐Ser161 RIPK1 is strongly upregulated in nuclei from cardiomyocytes, and the RIPK1/3 axis is largely overexpressed, suggesting ongoing necroptosis activation. In mdx mice, the heart pathogenic phenotype becomes detectable using M‐mode echocardiography only after 10–12 months of age compared with wild‐type mice. Heart weight, histological features, and analysis can discriminate healthy from dystrophic aged hearts. 19 , 33 , 34 Necroptosis depletion reduced heart weight, and biomarkers of hypertrophic cardiomyopathy such as Bnp and Myh7 transcripts 35 , 36 and fibrosis (Figure 4). BNP is upregulated in mdx compared with C57BL/10 control mice and restored to baseline in mdx mice treated by rAAV9‐mediated microdystrophin. 37 The RIPK1/3 axis may therefore represent an interesting therapeutic target to counteract the development of cardiomyopathy in DMD. The role of canonical necroptosis has been found in cardiovascular diseases, such as atherosclerosis, ischemia–reperfusion injuries, myocardial infarction, myocarditis and chemically‐induced cardiomyopathy. 38 We failed to observe a significant decrease in cardiomyocyte demise and necroinflammation. This is likely due to rare cell death events affecting the cardiomyocytes of mdx mice at this age. Indeed, only a minority of mdx hearts showed ongoing active necroinflammation area. However, a reduction of cardiac hypertrophy linked with a reduction of fibrosis deposition suggests, over time, less accumulation of myocardial cell death susceptible to be replaced by collagen and overall improved tissue homeostasis.
In conclusion, using dogs, rats and mice models of DMD pathogenesis, we found that RIPK3 participates in the pathogenesis of mdx mice at different levels, mediating necrosis and fibrosis accumulation in locomotor, respiratory, and cardiac muscles without evidence of a detrimental effect on skeletal muscle regeneration. This study, therefore, suggests that necroptosis prevention can be beneficial both to skeletal muscle and heart phenotype in DMD conditions and paves the way for considering necroptosis as a promising therapeutic target for DMD.
Conflict of interest statement
The authors declare no conflict of interest.
Supporting information
Figure S1: CK levels and Pearson correlation coefficient and p‐value between the percentage of inflammation and RIPK3 transcripts expression. (A) Quantification of serum CK in six‐month‐old WT and GRMD dogs. (B) Pearson correlation coefficient and p‐value between the percentage of inflammation and RIPK3 transcripts expression. Data are given as means ± s.e.m. Ns: Not significant, *: p‐value < 0.05.
Figure S2: Distribution of myofibre in mdx and mdxRipk3−/− skeletal muscle. (A) Violin plot of the pooled minimum Feret of TA myofibres (mdx: n = 33,142 myofibres out of 7 distinct muscles; mdxRipk3−/−, n = 26,469 myofibres out of 9 distinct muscles, Mann–Whitney test p = 0.1526). (B) Relative frequencies of minimum Feret from TA myofibres are expressed in percentage.
Table S1: M‐Mode echocardiographic values.
Acknowledgements
We thank Dr V. M. Dixit for Ripk3 knockout mice. We also thank J. Ternacle, B. Drayton Libotte, L. Guillaud and X. Decrouy for technical assistance and C. Laisne and D. Gelperowic for taking good care of animals, and the VOUSH group for fruitful discussions. This work was supported by the Association Française contre les Myopathies (AFM‐Téléthon) through the Translamuscle I (#19507) and Translamuscle II (#22946) programmes.
Bencze M., Periou B., Punzón I., Barthélémy I., Taglietti V., Hou C., et al (2023) Receptor interacting protein kinase‐3 mediates both myopathy and cardiomyopathy in preclinical animal models of Duchenne muscular dystrophy, Journal of Cachexia, Sarcopenia and Muscle, 14, 2520–2531, doi: 10.1002/jcsm.13265
References
- 1. Relaix F, Bencze M, Borok MJ, der Vartanian A, Gattazzo F, Mademtzoglou D, et al. Perspectives on skeletal muscle stem cells. Nat Commun 2021;12:692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desguerre I, Mayer M, Leturcq F, Barbet JP, Gherardi RK, Christov C. Endomysial fibrosis in Duchenne muscular dystrophy: a marker of poor outcome associated with macrophage alternative activation. J Neuropathol Exp Neurol 2009;68:762–773. [DOI] [PubMed] [Google Scholar]
- 3. Khirani S, Ramirez A, Aubertin G, Boulé M, Chemouny C, Forin V, et al. Respiratory muscle decline in Duchenne muscular dystrophy. Pediatr Pulmonol 2014;49:473–481. [DOI] [PubMed] [Google Scholar]
- 4. Fayssoil A, Abasse S, Silverston K. Cardiac involvement classification and therapeutic management in patients with Duchenne muscular dystrophy. J Neuromuscul Dis 2017;4:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morgan JE, Prola A, Mariot V, Pini V, Meng J, Hourde C, et al. Necroptosis mediates myofibre death in dystrophin‐deficient mice. Nat Commun 2018;9:3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sreenivasan K, Ianni A, Künne C, Strilic B, Günther S, Perdiguero E, et al. Attenuated epigenetic suppression of muscle stem cell necroptosis is required for efficient regeneration of dystrophic muscles. Cell Rep 2020;31:107652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhou S, Zhang W, Cai G, Ding Y, Wei C, Li S, et al. Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin‐C during regeneration. Cell Res 2020;30:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Barthélémy I, Uriarte A, Drougard C, Unterfinger Y, Thibaud JL, Blot S. Effects of an immunosuppressive treatment in the GRMD dog model of Duchenne muscular dystrophy. PLoS ONE 2012;7:e48478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Reyes‐Fernandez PC, Periou B, Decrouy X, Relaix F, Authier FJ. Automated image‐analysis method for the quantification of fiber morphometry and fiber type population in human skeletal muscle. Skelet Muscle 2019;9:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Czibik G, Mezdari Z, Murat Altintas D, Bréhat J, Pini M, d’Humières T, et al. Dysregulated phenylalanine catabolism plays a key role in the trajectory of cardiac aging. Circulation 2021;144:559–574. [DOI] [PubMed] [Google Scholar]
- 11. Wells DJ. Tracking progress: an update on animal models for Duchenne muscular dystrophy. Dis Model Mech 2018;11:dmm035774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McQuade T, Cho Y, Chan FK‐M. Positive and negative phosphorylation regulates RIP1 and RIP3‐induced programmed necrosis. Biochem J 2013;456:409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang Y, Su SS, Zhao S, Yang Z, Zhong CQ, Chen X, et al. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat Commun 2017;8:14329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF‐kappa Bs, signaling by the B‐cell and T‐cell receptors, tumor necrosis factor receptor 1, and Toll‐like receptors 2 and 4. Mol Cell Biol 2004;24:1464–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Straub V, Rafael JA, Chamberlain JS, Campbell KP. Animal models for muscular dystrophy show different patterns of sarcolemmal disruption. J Cell Biol 1997;139:375–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bencze M, Periou B, Baba‐Amer Y, Authier FJ. Immunolabelling myofiber degeneration in muscle biopsies. JoVE (Journal of Visualized Experiments) 2019;e59754. 10.3791/59754 [DOI] [PubMed] [Google Scholar]
- 17. Briguet A, Courdier‐Fruh I, Foster M, Meier T, Magyar JP. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx‐mouse. Neuromuscul Disord 2004;14:675–682. [DOI] [PubMed] [Google Scholar]
- 18. Taglietti V, Kefi K, Bronisz‐Budzyńska I, Mirciloglu B, Rodrigues M, Cardone N, et al. Duchenne muscular dystrophy trajectory in R‐DMDdel52 preclinical rat model identifies COMP as biomarker of fibrosis. Acta Neuropathol Commun 2022;10:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Quinlan JG, Hahn HS, Wong BL, Lorenz JN, Wenisch AS, Levin LS. Evolution of the mdx mouse cardiomyopathy: physiological and morphological findings. Neuromuscul Disord 2004;14:491–496. [DOI] [PubMed] [Google Scholar]
- 20. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight 2021;4:e128834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bencze M. Mechanisms of myofibre death in muscular dystrophies: the emergence of the regulated forms of necrosis in myology. Int J Mol Sci 2023;24:362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mariot V, Joubert R, le Gall L, Sidlauskaite E, Hourde C, Duddy W, et al. RIPK3‐mediated cell death is involved in DUX4‐mediated toxicity in facioscapulohumeral dystrophy. J Cachexia Sarcopenia Muscle 2021;12:2079–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jouan‐Lanhouet S, Riquet F, Duprez L, vanden Berghe T, Takahashi N, Vandenabeele P. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol 2014;35:2–13. [DOI] [PubMed] [Google Scholar]
- 24. Dondelinger Y, Hulpiau P, Saeys Y, Bertrand MJM, Vandenabeele P. An evolutionary perspective on the necroptotic pathway. Trends Cell Biol 2016;26:721–732. [DOI] [PubMed] [Google Scholar]
- 25. Lu P, Takai K, Weaver VM, Werb Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb Perspect Biol 2011;3:a005058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, et al. Aberrant repair and fibrosis development in skeletal muscle. Skeletal Muscle 2011;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stedman HH, Sweeney HL, Shrager JB, Maguire HC, Panettieri RA, Petrof B, et al. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991;352:536–539. [DOI] [PubMed] [Google Scholar]
- 28. Stupka N, Schertzer JD, Bassel‐Duby R, Olson EN, Lynch GS. Stimulation of calcineurin Aα activity attenuates muscle pathophysiology in mdx dystrophic mice. Am J Physiol‐Regul Integr Comparat Physiol 2008;294:R983–R992. [DOI] [PubMed] [Google Scholar]
- 29. Zhou S, Zhang W, Cai G, Ding Y, Wei C, Li S, et al. Myofiber necroptosis promotes muscle stem cell proliferation via releasing Tenascin‐C during regeneration. Cell Res 2020;30:1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bencze M, Negroni E, Vallese D, Yacoub‐Youssef H, Chaouch S, Wolff A, et al. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol Ther 2012;20:2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Taglietti V, Kefi K, Rivera L, Bergiers O, Cardone N, Coulpier F, et al. Thyroid‐stimulating hormone receptor signaling restores skeletal muscle stem cell regeneration in rats with muscular dystrophy. Sci Transl Med 2023;15:eadd5275. [DOI] [PubMed] [Google Scholar]
- 32. Partridge TA. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J 2013;280:4177–4186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang W, ten Hove M, Schneider JE, Stuckey DJ, Sebag‐Montefiore L, Bia BL, et al. Abnormal cardiac morphology, function and energy metabolism in the dystrophic mdx mouse: an MRI and MRS study. J Mol Cell Cardiol 2008;45:754–760. [DOI] [PubMed] [Google Scholar]
- 34. Li W, Liu W, Zhong J, Yu X. Early manifestation of alteration in cardiac function in dystrophin deficient mdx mouse using 3D CMR tagging. J Cardiovasc Magn Reson 2009;11:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hasegawa K, Fujiwara H, Doyama K, Miyamae M, Fujiwara T, Suga S, et al. Ventricular expression of brain natriuretic peptide in hypertrophic cardiomyopathy. Circulation 1993;88:372–380. [DOI] [PubMed] [Google Scholar]
- 36. Fananapazir L, Dalakas MC, Cyran F, Cohn G, Epstein ND. Missense mutations in the beta‐myosin heavy‐chain gene cause central core disease in hypertrophic cardiomyopathy. PNAS 1993;90:3993–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shin J‐H, Nitahara‐Kasahara Y, Hayashita‐Kinoh H, Ohshima‐Hosoyama S, Kinoshita K, Chiyo T, et al. Improvement of cardiac fibrosis in dystrophic mice by rAAV9‐mediated microdystrophin transduction. Gene Ther 2011;18:910–919. [DOI] [PubMed] [Google Scholar]
- 38. Zhe‐Wei S, Li‐Sha G, Yue‐Chun L. The role of necroptosis in cardiovascular disease. Front Pharmacol 2018;9:721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: CK levels and Pearson correlation coefficient and p‐value between the percentage of inflammation and RIPK3 transcripts expression. (A) Quantification of serum CK in six‐month‐old WT and GRMD dogs. (B) Pearson correlation coefficient and p‐value between the percentage of inflammation and RIPK3 transcripts expression. Data are given as means ± s.e.m. Ns: Not significant, *: p‐value < 0.05.
Figure S2: Distribution of myofibre in mdx and mdxRipk3−/− skeletal muscle. (A) Violin plot of the pooled minimum Feret of TA myofibres (mdx: n = 33,142 myofibres out of 7 distinct muscles; mdxRipk3−/−, n = 26,469 myofibres out of 9 distinct muscles, Mann–Whitney test p = 0.1526). (B) Relative frequencies of minimum Feret from TA myofibres are expressed in percentage.
Table S1: M‐Mode echocardiographic values.