

Summary
Prospective isolation of defined cell types is critical for the functional study of stem cells, especially in primary human tissues. Here, we present a protocol for purifying 10 transcriptomically and functionally distinct neural stem and progenitor cell types from the developing human brain using fluorescence-activated cell sorting. We describe steps for tissue dissociation, staining, and cell sorting as well as downstream functional experiments for measuring clonogenicity, differentiation, and engraftment potential of purified populations.
For complete details on the use and execution of this protocol, please refer to Liu et al. (2023).1
Subject areas: Classification Description, Cell culture, Cell isolation, Cell-based Assays, Flow Cytometry, Developmental biology, Neuroscience, Stem Cells
Graphical abstract
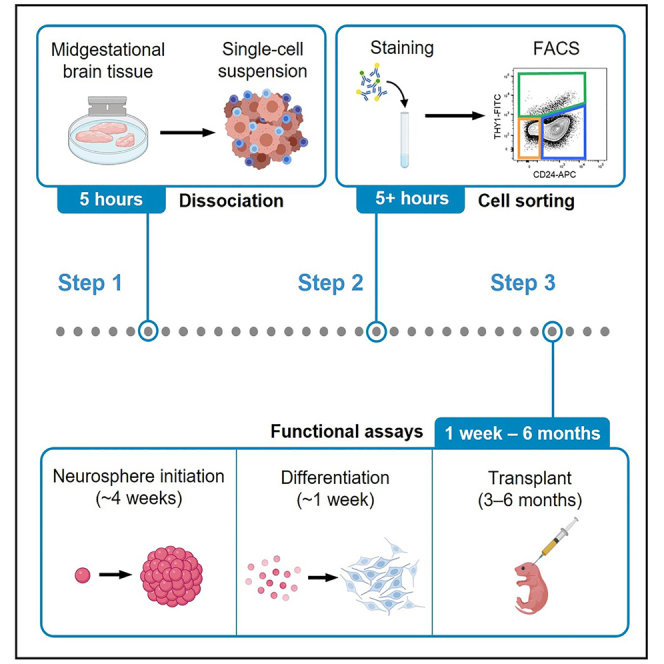
Highlights
-
•
Optimized dissociation and staining protocol for midgestational human brain tissue
-
•
Validated FACS gating strategy for 10 distinct neural stem and progenitor cell types
-
•
Guide for clonogenicity, differentiation, and engraftment assays with purified cells
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Prospective isolation of defined cell types is critical for the functional study of stem cells, especially in primary human tissues. Here, we present a protocol for purifying 10 transcriptomically and functionally distinct neural stem and progenitor cell types from the developing human brain using fluorescence-activated cell sorting. We describe steps for tissue dissociation, staining, and cell sorting as well as downstream functional experiments for measuring clonogenicity, differentiation, and engraftment potential of purified populations.
Before you begin
Institutional permissions
All experiments were performed strictly per pre-approved guidelines set by Stem Cell Research Oversight (SCRO protocol #735) and Laboratory Animal Care (APLAC protocol #26209) at Stanford University.
Researchers seeking to use to this protocol for their own work would need to acquire permission from their respective institutions for the procurement of fetal human brain tissue, as well as all downstream experiments using purified cell populations, and follow proper ethical and legal guidelines as per their local, state, and federal laws.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-human CD24-APC, clone 32D12 (1:25 dilution) | Miltenyi | Cat#130-095-954; RRID: AB_10829613 |
| Mouse anti-human CD90-FITC, clone 5E10 (1:50 dilution) | BioLegend | Cat#328108; RRID: AB_893429 |
| Mouse anti-human CXCR4-APC/Cy7, clone 12G5 (1:50 dilution) | BioLegend | Cat#306528; RRID: AB_2565994 |
| Mouse anti-human EGFR-PE-Cy7, clone AY13 (1:50 dilution) | BioLegend | Cat#352910; RRID: AB_2562159 |
| Mouse anti-human PDGFRA-biotin, clone 16A1 (1:50 dilution) | BioLegend | Cat#323504; RRID: AB_2162487 |
| Mouse anti-human CD31-PE, clone WM59 (1:50 dilution) | BD Biosciences | Cat#563652; RRID: AB_2738349 |
| Mouse anti-human CD34-PE, clone 581 (1:50 dilution) | BD Biosciences | Cat#562383; RRID: AB_11154586 |
| Mouse anti-human CD45-PE, clone HI30 (1:50 dilution) | BioLegend | Cat#304010; RRID: AB_314398 |
| Mouse anti-human CD105-PE, clone 266 (1:50 dilution) | BD Biosciences | Cat#562380; RRID: AB_11154054 |
| Mouse anti-human CD235a-PE, clone HIR2 (1:50 dilution) | BioLegend | Cat#306606; RRID: AB_314624 |
| BUV395 streptavidin (1:50 dilution) | BD Biosciences | Cat#564176; RRID: AB_2869553 |
| Chemicals, peptides, and recombinant proteins | ||
| Liberase | Roche | Cat#5401119001 |
| DNase I | Worthington Biochemical | Cat#LS002007 |
| Accutase | Innovative Cell Technologies | Cat#AT104 |
| Polyvinyl alcohol | Sigma | Cat#P8136 |
| Poly-L-ornithine hydrobromide | Thermo Fisher Scientific | Cat#P3655 |
| Laminin | Thermo Fisher Scientific | Cat#23017015 |
| HBSS without Ca2+/Mg2+ | Thermo Fisher Scientific | Cat#14175103 |
| HBSS with Ca2+/Mg2+ | Thermo Fisher Scientific | Cat#24020117 |
| X-VIVO 15 media | Lonza | Cat#04-744Q |
| DMEM/F12 | Thermo Fisher Scientific | Cat#11320082 |
| Hibernate-A medium | Thermo Fisher Scientific | Cat#A1247501 |
| HEPES buffer | Thermo Fisher Scientific | Cat#15630080 |
| N-2 | Thermo Fisher Scientific | Cat#17502048 |
| Heparin | STEMCELL Technologies | Cat#07980 |
| N-acetylcysteine | VWR | Cat#E-3710 |
| Fibroblast growth factor 2 (FGF) | Shenandoah Biotechnology | Cat#100-146 |
| Epidermal growth factor (EGF) | Shenandoah Biotechnology | Cat#100-26 |
| Leukemia inhibitory factor (LIF) | Sigma | Cat#LIF1010 |
| B27 | Thermo Fisher Scientific | Cat#17504044 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat#15710 |
| Fast green dye | Sigma | Cat#F7252 |
| Propidium iodide | Sigma | Cat#P4170 |
| Anti-rat and anti-hamster Ig κ compensation particles | BD Biosciences | Cat#552845 |
| Anti-mouse Ig κ compensation particles | BD Biosciences | Cat#552843 |
| Experimental models: Organisms/strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG); neonatal | The Jackson Laboratory | JAX: 005557 |
| Human: human fetal brain tissue; gestational week 16–20 | Advanced Bioscience Resources | N/A |
| Software and algorithms | ||
| ImageJ | NIH | http://wsr.imagej.net/distros/ |
| GraphPad Prism 9.0 | GraphPad Software | http://www.graphpad.com/scientific-software/prism |
| BD FACSDiva | BD Biosciences | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsdiva-software |
| FlowJo | FlowJo LLC | https://www.flowjo.com/ |
| Extreme limiting dilution analysis | Hu and Smyth2 | https://bioinf.wehi.edu.au/software/elda/ |
| Other | ||
| Stereotaxic frame | Harvard apparatus | Cat#75-1808 |
| Neonate adaptor | Stoelting | Cat#51625 |
| Microsyringe pump | World Precision Instruments | Cat#UMP3 |
| Syringe (10 μL, model 701 RN) | Hamilton | Cat#7635 |
| Needle (34 gauge, small hub RN needle, 0.375 in, point style 4 at 12°) | Hamilton | Cat#207434 |
Materials and equipment
Wash Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HBSS without Ca2+/Mg2+ | 1× | 450 mL |
| PVA 1% | 0.1% | 50 mL |
| Total | N/A | 500 mL |
Storage: Store at 4°C indefinitely.
Mincing Buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| HBSS with Ca2+/Mg2+ | 1× | 49 mL |
| HEPES (1 M) | 10 mM | 500 μL |
| DNase I (20 mg/mL) | 200 μg/mL | 500 μL |
| Total | N/A | 50 mL |
Storage: Freshly prepared. Keep on ice during experiment.
Liberase Enzyme Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| HBSS with Ca2+/Mg2+ | 1× | 48.5 mL |
| HEPES (1 M) | 10 mM | 500 μL |
| Liberase (1 mg/mL) | 10 μg/mL | 500 μL |
| DNase I (20 mg/mL) | 200 μg/mL | 500 μL |
| Total | N/A | 50 mL |
Storage: Freshly prepared. Keep on ice until ready to use.
Accutase Enzyme Solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Accutase | 1× | 49.5 mL |
| DNase I (20 mg/mL) | 200 μg/mL | 500 μL |
| Total | N/A | 50 mL |
Storage: Freshly prepared. Keep on ice until ready to use.
Primary Staining Cocktail
| Reagent | Final concentration | Amount |
|---|---|---|
| Wash Buffer | 1× | 340 μL |
| Brilliant Stain Buffer | N/A | 50 μL |
| Mouse anti-human CD24-APC, Clone 32D12 | 1:25 | 20 μL |
| Mouse anti-human CD90-FITC, Clone 5E10 | 1:50 | 10 μL |
| Mouse anti-human CXCR4-APC/Cy7, Clone 12G5 | 1:50 | 10 μL |
| Mouse anti-human EGFR-PE-Cy7, Clone AY13 | 1:50 | 10 μL |
| Mouse anti-human PDGFRA-biotin, Clone 16A1 | 1:50 | 10 μL |
| Mouse anti-human CD31-PE, Clone WM59 | 1:50 | 10 μL |
| Mouse anti-human CD34-PE, Clone 581 | 1:50 | 10 μL |
| Mouse anti-human CD45-PE, Clone HI30 | 1:50 | 10 μL |
| Mouse anti-human CD105-PE, Clone 266 | 1:50 | 10 μL |
| Mouse anti-human CD235a-PE, Clone HIR2 | 1:50 | 10 μL |
| Total | N/A | 500 μL |
Storage: Freshly prepared. Keep on ice shielded from light.
Secondary Staining Cocktail
| Reagent | Final concentration | Amount |
|---|---|---|
| Wash Buffer | 1× | 490 μL |
| BUV395 Streptavidin | 1:50 | 10 μL |
| Total | N/A | 500 μL |
Storage: Freshly prepared. Keep on ice shielded from light.
Fetal Growth Media
| Reagent | Final concentration | Amount |
|---|---|---|
| X-VIVO 15 Media | 1× | 488 mL |
| N-2 | 1× | 5 mL |
| EGF (20 μg/mL) | 20 ng/mL | 500 μL |
| FGF (20 μg/mL) | 20 ng/mL | 500 μL |
| LIF (10 μg/mL) | 10 ng/mL | 500 μL |
| Heparin (2 mg/mL) | 2 μg/mL | 500 μL |
| N-acetylcysteine (6 mg/mL) | 60 μg/mL | 5 mL |
| Total | N/A | 500 mL |
Storage: Store at 4°C for up to 1 month. Keep shielded from light.
Differentiation Media
| Reagent | Final concentration | Amount |
|---|---|---|
| DMEM/F12 | 1× | 47.95 mL |
| N-2 | 1× | 0.5 mL |
| B-27 (50×) | 1× | 1 mL |
| Heparin (2 mg/mL) | 2 μg/mL | 50 μL |
| N-acetylcysteine (6 mg/mL) | 60 μg/mL | 500 μL |
| Total | N/A | 50 mL |
Storage: Store at 4°C for up to 1 month.
Alternatives: Fluorophores in the primary and secondary staining cocktails can be swapped out to accommodate the user’s cell sorter configuration. In this case, it is important to still use the same antibody clone, just conjugated to a different fluorophore (see troubleshooting 1).
Step-by-step method details
Step 1: Tissue dissociation
Timing: 5 h
In this step, fetal human brain tissue is dissociated into a single-cell suspension using a combination of mechanical and enzymatic digestion. Red blood cells are depleted using a density gradient (Figure 1A).
Optional: If the tissue is intact, perform any dissection of specific brain regions. Our protocol was optimized on gestational week (GW) 16–20 cortical tissue but is applicable to any brain region.
Note: Fetal human brain tissue can be kept overnight on ice with no significant impact on viability or cell type makeup. Our tissue is generally shipped overnight in Hibernate-A medium.
-
1.
Transfer tissue to a 50 mL tube. Spin down the sample at 300 × g for 3 min. Remove supernatant. Note the packed volume of the tissue.
Note: In our experience, every 10 mL packed tissue volume typically yields ∼2×108 cells. We have used this protocol successfully for as little as 1 cm3 and as much as 25 cm3 of tissue.
-
2.Mechanical dissociation.
-
a.Transfer the tissue into a sterile petri dish with 5 mL cold Mincing Buffer. Chop the sample with sterile razor blades, aiming for 1–2 mm sized chunks (Figure 1B).Note: Do not mince the tissue too finely at this stage, as it may adversely affect cell survival.
-
b.Transfer the minced sample into 50 mL tubes, washing the petri dish with additional Mincing Buffer as needed.Note: In our experience, one tube per 5 mL packed tissue volume works well.
-
c.Spin down the minced tissue at 300 × g for 3 min. Remove supernatant.
-
a.
-
3.Enzymatic dissociation in Liberase.
-
a.Resuspend the minced tissue in pre-warmed Liberase Enzyme Solution, 30 mL per tube.
-
b.Shake the tubes on a thermomixer at 37°C and 750 RPM for 30 min.
-
c.Spin down the cell suspension at 300 × g for 3 min. Remove 20 mL supernatant per tube, leaving behind 10 mL volume for trituration.
-
d.Triturate (i.e., pipette up and down) the cell suspension with progressively smaller-aperture pipettes as detailed below.
-
i.Pipette the cell suspension up and down with a 10 mL serological pipette until it goes through smoothly (typically 10–15 strokes).Note: If there are still large chunks that have a hard time passing through the pipette, consider shaking in Liberase Enzyme Solution for another 15 minutes.
-
ii.Attach a P1000 pipette tip (no filter) to the end of a 10 mL serological pipette, and cut the end off with a sterile razor blade (∼5 mm). Pipette the cell suspension up and down until it goes through smoothly (typically 10–15 strokes).
-
iii.Attach a P1000 pipette tip to the end of a 10 mL serological pipette, without cutting off the tip. Pipette the cell suspension up and down until it goes through smoothly (typically 10–15 strokes).
-
i.
-
e.Fill the tube with Wash Buffer, spin down the cell suspension at 300 × g for 3 min, and remove the supernatant.
-
a.
-
4.Enzymatic dissociation in Accutase.
-
a.Resuspend the cell pellet in Accutase Enzyme Solution, 25 mL per tube.Note: Accutase quickly loses its activity at high temperatures, so avoid prewarming it too early in advance. Do not forget to add DNase to the enzyme solution, as it helps significantly with cell viability.
-
b.Shake the tubes on a Thermomixer at 25°C and 750 RPM for 30 min.
-
c.Spin down the cell suspension at 300 × g for 3 min, and remove the supernatant.
-
d.Wash the cells with 10 mL Wash Buffer. Spin down the cell suspension at 300 × g for 3 min, and remove the supernatant.
-
e.Resuspend the cell pellet in 9 mL Wash Buffer per tube.
-
a.
-
5.Red blood cell depletion using Ficoll (Figure 1C).Note: RBC depletion is optional but recommended as it speeds up cell sorting (for some samples, we have observed up to 30% of cells to be RBCs at this step, though this varies widely) (Figure 1D). Our Ficoll step will not decrease the yield of neural cells. Alternative RBC depletion methods such as ACK lysis or MACS depletion are feasible, but their effects on cell viability and function have not been tested.
-
a.Prepare one FACS tube per 3 mL of cell suspension.Note: We find that skinnier tubes (such as FACS tubes or 15 mL tubes) make it easier to recover cells following Ficoll density centrifugation. Other tube formats will also work.
-
b.Add 1 mL Ficoll to each tube.
-
c.Gently layer 3 mL cell suspension on top of Ficoll for each tube.
-
d.Spin the tubes at 400 × g for 30 min at 25°C. Set the acceleration of the centrifuge to 4 out of 10, and turn the brakes off.
 CRITICAL: For successful separation, is important for the centrifuge to be at room temperature, and for the brakes to be off.
CRITICAL: For successful separation, is important for the centrifuge to be at room temperature, and for the brakes to be off. -
e.After centrifugation, the red blood cells will have pelleted to the bottom, and the remainder of the cells will be found in a buffy coat at the interface of the two layers (Figure 1C).
-
f.Carefully pipette the upper layer and buffy coat and transfer into a new 50 mL tube.Note: Be liberal in taking cells from the buffy coat. It is better to recover more cells at the cost of some Ficoll contamination, which will be washed away in the next step anyways.
-
g.Fill the tube with Wash Buffer and spin down at 400 × g for 3 min. Remove the supernatant and resuspend in 10 mL Wash Buffer.
-
a.
-
6.
Determine the cell concentration and viability on a hemocytometer or other automated cell counting machine.
Note: In our experience, every 10 mL packed tissue volume yields around ∼2×108 cells, though this varies. We reliably obtain 85%–95% cell viability with this protocol.
Figure 1.

Dissociation of fetal human brain tissue
(A) Outline of tissue dissociation workflow.
(B) Left: typical appearance of non-intact fetal human brain tissue as received. Right: appearance of tissue after mincing.
(C) Left: Ficoll layering prior to centrifugation: upper layer contains cell suspension, and bottom layer contains Ficoll. Right: Ficoll layering after centrifugation: cells of interest are at the interface, and red blood cells have been pelleted to the bottom.
(D) Histograms of cell size distribution before and after Ficoll, on two separate donors.
Step 2: Fluorescence-activated cell sorting (FACS)
In this step, the single cell suspension will be stained with a cocktail of fluorescently-labeled antibodies. Cells will then be analyzed on a flow cytometer, and specific cell types will be gated and sorted.
-
7.Stain the cell suspension with fluorescently-labeled antibodies.
-
a.Set aside a small number of cells for an unstained control (1% of total cells).Optional: For new markers or initial troubleshooting, it is helpful to include fluorescence-minus-one (FMO) controls, in which a small number of cells is stained for all but one marker, in order to determine the cutoff for positive staining. The gating scheme described here, however, can be drawn based on contours alone.Note: We perform staining in FACS tubes, though this is not critical.
-
b.Stain the cell suspension by resuspending it in Primary Staining Solution.
-
i.We have achieved good results with as much as 2×108 cells/mL Primary Staining Solution. For example, 108 cells should be resuspended in 500 μL Primary Staining Solution.Note: While the exact fluorophores can be switched around to accommodate different cell sorters (see troubleshooting 1), we highly recommend using the same clone of antibodies. For example, we found that the 16A1 but not the αR1 clone of the PDGFRA antibody shows staining.
-
i.
-
c.Incubate the cells on ice for 20 min. Protect from light.
-
d.Wash the cells with 2 mL Wash Buffer. Spin down at 300 × g for 3 min. Remove supernatant.
-
e.Resuspend the cell pellet in Secondary Staining Solution.
-
f.Incubate the cells on ice for 20 min. Protect from light.
-
g.Wash the cells with 2 mL Wash Buffer. Spin down at 300 × g for 3 min. Remove supernatant. Perform this step twice prior to analysis.
-
h.Propidium iodide is added to the cells immediately prior to analysis.
-
a.
-
8.Prepare compensation controls.
-
a.Prepare one FACS tube for each fluorophore on the panel, as well as a negative control.
-
i.For the specific panel described here, this would entail 7 tubes: unstained control, APC, FITC, PE/Cy7, APC/Cy7, BUV395, and PE.
-
i.
-
b.In the unstained control tube, add one drop of negative control compensation particles.
-
c.In each stained tube, add one drop each of (1) Anti-Rat/Anti-Hamster Ig, κ compensation particles, and (2) Anti-Mouse Ig, κ compensation particles.
-
d.In each stained tube, add 1 μL of any antibody conjugated to the corresponding fluorophore.
-
e.Add 300 μL Wash Buffer to each tube.
-
a.
Pause point: If FACS machines are not immediately available, stained cells may be kept on ice for several hours.
-
9.Set up the cell sorting instrument.Note: The instructions here apply for BD FACSAria II/III/Fusion instruments, and most BD instruments running FACSDiva software. The equivalent steps should be taken for other cell sorting instruments and software.
-
a.Run Cytometer Setup and Tracking (CST) to set voltages.
-
b.Select all fluorophores used in the staining panel, in addition to PI, forward scatter, and side scatter. Forward and side scatter should be recorded on a linear scale and all other channels on a log scale. Troubleshooting 1.
-
c.Run compensation controls and apply the compensation matrix.CRITICAL: Proper compensation is essential for high-dimensional flow experiments. This setup should be carefully optimized for the investigator’s specific flow cytometer and optical configuration.
-
d.Set the Drop Delay using Auto Delay.
-
e.Aim each of the four streams to ensure they hit the bottom of the collection tube.
-
a.
-
10.Draw gates.Note: When initially learning how to draw gates, we find it helpful to set the plot display type to “Contour” with outliers instead of “Pseudocolor,” as it allows for better visualization of the cutoffs.
-
a.Load the stained cells and record 105 events for gating.
-
b.Draw the following gates as depicted in Figure 2.
-
c.From All Events, plot FSC-A against SSC-A. Gate for Cells (Figure 2A).
-
d.From Cells, plot FSC-H against FSC-W. Gate for Singlets (FSC) (Figure 2B).
-
e.From Singlets (FSC), plot SSC-H against SSC-W. Gate for Singlets (SSC) (Figure 2C).Note: We use a stringent two-step doublet discrimination strategy. Other methods (such as gating FSC-A against FSC-H) will also work.
-
f.From Singlets (SSC), plot Lineage-PE against PI. Gate for Live Lineage− (Figure 2D).Note: The “Lineage” channel includes markers against all vascular (CD31, CD34, CD105) and hematopoietic (CD45, CD235a) lineages. It is used together with PI to select all live cells of neuroectodermal origin.
-
g.From Live Lineage−, plot CD24-APC against THY1-FITC. Gate for the following (Figure 2E):
-
i.CD24−THY1−/lo, enriched for radial glia and astrocytes.
-
ii.CD24+THY1−/lo, enriched for neuron precursors.
-
iii.THY1hi, enriched for oligodendrocyte lineages.
-
i.
-
h.From CD24−THY1−/lo, plot CXCR4-APC/Cy7 against EGFR-PE/Cy7. Gate for the following (Figure 2F):
-
i.EGFR+CXCR4−, enriched for ventricular radial glia (vRG).
-
ii.EGFR−CXCR4−, enriched for outer radial glia (oRG).
-
iii.EGFR+CXCR4+, enriched for astrocytes (AC).
-
i.
-
i.From THY1hi, plot PDGFRA-BUV395 against EGFR-PE/Cy7. Gate for the following (Figure 2G):
-
i.EGFRhiPDGFRA−, enriched for glial progenitor cells (GPCs).
-
ii.EGFR+PDGFRA+, enriched for pre-oligodendrocyte precursor cells (pre-OPCs).
-
iii.EGFR−PDGFRA+, enriched for oligodendrocyte precursor cells (OPCs).
-
iv.EGFR−PDGFRA−, enriched for oligodendrocytes (OL).
-
i.
-
j.From CD24+THY1−/lo, plot CXCR4-APC/Cy7 against EGFR-PE/Cy7. Gate for the following (Figure 2H):
-
i.EGFR+CXCR4−, enriched for early excitatory neurons (ExN).
-
ii.EGFR−CXCR4−, enriched for late excitatory neurons.
-
iii.EGFR−CXCR4+, enriched for inhibitory neurons (InN).
-
i.
-
a.
-
11.Sort cells.
-
a.Choose which cell populations to sort.Note: FACS machines can generally only sort four populations in parallel. Strategies for sorting more than four populations can be found in troubleshooting 2
-
b.Load collection tubes with 0.5 mL Wash Buffer added. Vortex prior to loading.
-
c.Sort cells on four-way purity (for bulk sorts) or single-cell purity (for clonal assays) until the desired number of cells is obtained.
-
a.
Figure 2.

FACS gating strategy for human NSPCs
(A) Plot of side scatter area (SSC-A) against forward scatter area (FSC-A), gated on all events.
(B) Plot of forward scatter height (FSC-H) against forward scatter width (FSC-W), gated on “Cells.”
(C) Plot of side scatter height (SSC-H) against side scatter width (SSC-W), gated on “Singlets (FSC).”
(D) Plot of Lineage-PE against Viability-PI, gated on “Singlets (SSC).” Lineage channel consists of CD31, CD34, CD45, CD105, and CD235a.
(E) Plot of CD24-APC against THY1-FITC, gated on “Live Lineage−.”
(F) Plot of CXCR4-APC/Cy7 against EGFR-PE/Cy7, gated on “CD24−THY1−/lo.”
(G) Plot of PDGFRA-BUV395 against EGFR-PE/Cy7, gated on “THY1hi.”
(H) Plot of CXCR4-APC/Cy7 against EGFR-PE/Cy7, gated on “CD24+THY1−/lo.”
(I) List of numbered gate names, along with the enriched cell type for that gate.
(J) Graphical summary of purified neural stem and progenitor cell types, their immunophenotypes, and simplified hierarchical organization. Adapted from Liu et al., 2023.1
Step 3: Functional assays
Following purification of NSPCs, various downstream functional assays can be performed. A few examples are listed here.
-
12.Neurosphere initiation assay.
-
a.Prepare 96 well plates filled with 100 μL Fetal Growth Media.
-
i.Antibiotic/antimycotic should be added to the media for freshly isolated cells. It is no longer needed in subsequent media changes.Note: Even with proper humidification, we find that wells on the edge of the plate tend to dry up over the several weeks needed for clonal outgrowth. We recommend only using the non-edge wells, and filling the edge wells with 200 μL PBS.
-
i.
-
b.During sorting, set purity to “single-cell” mode.
-
c.Sort limiting dilutions of cells into each row of the plate.
-
d.Every week, replace half the volume of each well with fresh Fetal Growth Media.Note: We maintain our neurosphere cultures in hypoxic conditions (5% O2), as it improves neurosphere initiation. This is not strictly required, but culture conditions should be clearly reported to ensure cross-study comparability.
-
e.After 3 weeks, score the wells for the presence of neurospheres (i.e., a well with at least one neurosphere is scored as positive). Examples of neurospheres can be found in Figure 3B.
- f.
-
a.
-
13.In vitro differentiation.
-
a.Coat 96 well plates with polyornithine and laminin.
-
i.Add 50 μL polyornithine solution (20 μg/mL) to each well. Incubate at 37°C for at least 1 h.
-
ii.Aspirate the polyornithine solution.
-
iii.Add 50 μL laminin solution (50 μg/mL) to each well. Incubate at 37°C for at least 1 h.
-
iv.Aspirate the laminin solution.
-
i.
-
b.Bulk sort the cell population of interest as described above.
-
c.Spin down the sorted cells at 300 × g for 3 min. Resuspend in Differentiation Media at a concentration of 5×104 cells/mL.Note: The given seeding density works well for the NSPC types described here, but may have to be optimized for new populations.
-
d.Add 100 μL of the cell suspension per 96 well, which gives 5×103 cells per well.
-
e.After 5 days, fix the cells by adding 100 μL 4% PFA on top of the media. Fix for 15 min at 4°C.
-
f.Remove the PFA and wash three times with 200 μL PBS.
-
g.Perform immunofluorescent staining for lineage markers of interest.Note: We have provided some representative images of purified cell populations stained with markers for astrocytes (GFAP), oligodendrocytes (O4), and neurons (DCX, MAP2) in Figures 3C–3E, though the specific markers used will depend on the goals of the experiment.
-
h.Image stained cells on a fluorescent microscope.
-
a.
-
14.In vivo transplantation.
-
a.Bulk sort the cell population of interest as described above.Pause point: At this point it will have typically been 16+ h since the beginning of tissue processing. It is okay to keep the sorted cells at 4°C overnight in a 50/50 mix of Wash Buffer and Fetal Growth Media, and transplant the cells the following day.
-
b.Spin down the sorted cells at 300 × g for 3 min. Resuspend in Fast Green dye at a concentration of 105 cells/μL.Note: Our standard is to inject 2×105 neural stem cells per mouse (105 cells into each lateral ventricle). It is perfectly fine, however, to inject fewer cells, especially for rare populations where it is not even possible to sort so many cells. We have observed engraftment with as few as 2×104 transplanted cells. We do, however, recommend injecting as many cells as possible below 2×105, as there is a direct correlation between the number of transplanted cells and the level of subsequent engraftment.
-
c.Anesthetize neonatal NSG pups by wrapping them in a tissue and covering them with ice for 2 min.CRITICAL: It is required to use an immunocompromised mouse strain for xenotransplantation. It is always a logistical challenge to procure neonatal pups on the same day that brain samples arrive, but we have achieved good engraftment on pups anywhere from P1-P4 in age.Note: Alternative methods for anesthesia, such as isoflurane, would also be suitable.
-
d.Once the pup is anesthetized, place them on a stereotaxic device fitted with a mouse neonate adaptor. Secure the pup’s head in place using the adaptor.Note: Keep the platform of the stereotaxic device cold so that the pup remains anesthetized. This can be accomplished by laying a tissue over the pup’s body and covering with ice.
-
e.Load the cell suspension into a Hamilton syringe with a 33-gauge needle.
-
f.Load the syringe onto a microsyringe pump controller.
-
g.Make a burr hole at the injection site using a 30-gauge needle.
-
i.For injection into the lateral ventricles, use the following coordinates: anteroposterior from midline (A), lateral from midline (L), ventral from surface of brain (V). Lateral ventricles (A, L, V) = (0.8, ±1.5, 2.0) mm with reference to lambda
-
i.
- h.
-
i.Retract the needle and release the pup from the mouse neonate adaptor.Optional: If transplanting several different populations, it will be necessary to distinguish pups. This can be done by injecting India ink into the foot pads of the mouse (Figure 4B). Record which tattoo patterns correspond to which transplanted population. We do not recommend tattooing more than 2 of the foot pads per mouse.
-
j.Warm the pup on a 37°C heating pad or in your palm until movement is observed.
-
k.Reintroduce the pup to its mother. Ensure the mother accepts the pup.Note: We recommend placing the pup in the opposite corner of the cage as its littermates. Wait for the mother to pick up the pup and return it to its littermates.
-
l.Wait between 3–6 months for the mouse to mature.Note: Transplanted human neural stem cells will engraft in the mouse subventricular zone and give rise to interneurons that migrate through the rostral migratory stream into the olfactory bulb. We find that 6 months of maturation is required for human interneurons to migrate to the olfactory bulb. For other cell types, a shorter engraftment period would be acceptable.
-
m.Sacrifice mouse.
-
i.Anesthetize the mouse using isoflurane.
-
ii.Open the thoracic cavity to expose the heart.
-
iii.Cut the right atrium.
-
iv.Inject 20 mL PBS with 5 mM EDTA into the left ventricle to perfuse the mouse.Note: Successful perfusion can be visually observed by the liver lightening in color. If the entire brain is to be fixed, then perfusion with 4% PFA is recommended instead.
-
v.Decapitate the mouse and dissect out the brain.
-
vi.Brains can either be fixed in 4% PFA for histology, or dissociated to recover engrafted human cells.Note: For histology, we recommend staining with a human-specific antigen (e.g., STEM101 for human nuclear antigen, STEM121 for human cytoplasmic antigen, STEM123 for human-specific GFAP), along with lineage-specific markers (e.g., SOX2, OLIG2, NeuN). We have provided representative images of engrafted human astrocytes, oligodendrocytes, and neurons in Figures 4C–4E.
-
i.
-
a.
Figure 3.

In vitro assays for human NSPCs
(A) Typical sort layout for limiting dilution assays to measure neurosphere initiation frequency.
(B) Representative images of neurospheres, indicative of a positive well. Scale bar 100 μm.
(C) Immunofluorescence (IF) images of CD24−THY1−/lo populations differentiated in vitro for 5 days, then stained for GFAP (red), DCX (green), O4 (magenta), or DAPI (blue).
(D) IF images of THY1hi populations differentiated in vitro for 5 days, then stained for GFAP (red), O4 (green), or DAPI (blue). Abbreviations: E, EGFR; P, PDGFRA.
(E) IF images of CD24+THY1−/lo populations differentiated in vitro for 5 days, then stained for DCX (green) or MAP2 (red). Scale bar 50 μm. Adapted from Liu et al., 2023.1
Table 1.
Expected frequencies and clonogenicity of immunophenotypic NSPC populations
| Population | Enriched population | Percent of live lineage− gate | Percent of parent gate | 1 / neurosphere initiation frequency |
|---|---|---|---|---|
| CD24−THY1−/lo | Radial glia, astrocytes | 13 | 13 | |
| EGFR− | Outer radial glia (oRG) | 4.3 | 33 | 104 |
| EGFRmid | Ventricular radial glia (vRG) | 4.0 | 31 | 11.7 |
| EGFRhi | 4.0 | 31 | 4.5 | |
| EGFR+CXCR4+ | Astrocytes | 0.6 | 4.7 | 38 |
| CD24+THY1−/lo | Neurons | 86 | 86 | |
| CXCR4−EGFR+ | Early excitatory neurons | 5.2 | 6.3 | 61 |
| CXCR4−EGFR− | Late excitatory neurons | 59 | 69 | ∞ |
| CXCR4+ | Inhibitory neurons | 22 | 25 | ∞ |
| THY1hi | Oligodendrocyte lineages | 1.2 | 1.2 | |
| EGFR+PDGFRA− | Glial progenitor cells (GPCs) | 0.19 | 16 | 9.8 |
| EGFR+PDGFRA+ | Pre-OPCs | 0.12 | 10 | 5.6 |
| EGFR−PDGFRA+ | OPCs | 0.49 | 41 | 872 |
| EGFR−PDGFRA− | Oligodendrocytes | 0.40 | 33 | 1634 |
Values are approximate.
Figure 4.

In vivo assays for human NSPCs
(A) Positioning of pup on stereotaxic device and positioning of needle for intracerebroventricular injection (left), and visualization of successful ventricular injection by fast green dye (right).
(B) Pup before and after injection of tattoo ink back right (BR) foot (left), and appearance of back right foot tattoo after 2 months.
(C–E) Confocal immunofluorescence (IF) images of mouse brains engrafted with CD24−THY1−/lo human fetal radial glia. 40 μm thick sections were stained with anti-human GFAP (C) or human cytoplasmic antigen-specific STEM121 antibody (D, E), in addition to species cross-reactive antibodies against SOX2, OLIG2, or NeuN and MAP2 (C, D, E, respectively). Abbreviations: SVZ, subventricular zone. Scale bar 25 μm. Adapted from Liu et al., 2023.1
Expected outcomes
Tissue processing
The number of cells that can be obtained follow dissociation is on the order of ∼2×108 cells for 10 mL initial tissue volume, though this can vary from sample to sample. The viability of our single cell suspension following dissociation is consistently in the range of 85%–95%. The expected proportion of each immunophenotypic gate is tabulated in Table 1, though it should be noted that these frequencies depend on the brain region under investigation, as detailed in Liu et al.1 The surface marker profile shown in Figure 2 is highly consistent across donors.
In vitro assays
The expected neurosphere initiation frequencies for each population are listed in Table 1. We note that clonogenicity does not necessarily map onto stemness, but rather reflects some combination of self-renewal potential, proliferation rate, and culture condition effects.
The expected results from the in vitro differentiation assay are shown in Figures 3C–3E. We find that radial glia are tripotent, giving rise to neurons, astrocytes, and rare oligodendrocytes; glial progenitor cells are bipotent, giving rise to astrocytes and oligodendrocytes; astrocytes, OPCs, and neurons are unipotent, giving rise exclusively to their respective committed lineages. We were unsuccessful in culturing inhibitory neurons, which may require additional growth factors for survival.
In vivo transplant
Following transplant into the lateral ventricles of neonatal NSG mice, we observe robust engraftment from radial glia, glial progenitor cells, pre-OPCs, and OPCs (Table 2). We did not observe engraftment from oligodendrocytes or neurons; these populations may be too mature to engraft, or may require direct transplant into the brain parenchyma rather than the ventricles. Consistent with the in vitro differentiation, radial glia gave rise to all three neural lineages (astrocytes, oligodendrocytes, neurons), glial progenitor cells gave rise only to astrocytes and oligodendrocytes, while pre-OPCs and OPCs gave rise exclusively to oligodendrocytes. The relative proportions of the engrafted cells are documented in Table 2. We note that the lineage output differs considerably between in vitro and in vivo assays, reflecting the difference in microenvironmental signals between the two systems. In our hands, the engraftment is very robust—we have not yet observed a sample that failed to engraft.
Table 2.
Expected results of in vivo engraftment of bulk sorted populations
| Population | Enriched population | Engraft? | Percent GFAP + | Percent OLIG2+ | Percent DCX+ |
|---|---|---|---|---|---|
| CD24−THY1−/lo | Radial glia, astrocytes | ||||
| EGFR− | Outer radial glia (oRG) | Yes | 87 | 13 | <1 |
| EGFR+ | Ventricular radial glia (vRG) | Yes | 82 | 18 | <1 |
| EGFR+CXCR4+ | Astrocytes | Yes | 100 | 0 | 0 |
| CD24+THY1−/lo | Neurons | ||||
| CXCR4−EGFR+ | Early excitatory neurons | Not tested | |||
| CXCR4−EGFR− | Late excitatory neurons | No | — | — | — |
| CXCR4+ | Inhibitory neurons | No | — | — | — |
| THY1hi | Oligodendrocyte lineages | ||||
| EGFRhiPDGFRA− | Glial progenitor cells (GPCs) | Yes | 33 | 67 | 0 |
| EGFR+PDGFRA+ | Pre-OPCs | Yes | 0 | 100 | 0 |
| EGFR−PDGFRA+ | OPCs | Yes | 0 | 100 | 0 |
| EGFR−PDGFRA− | Oligodendrocytes | No | — | — | — |
Values represent percentage of human cells (marked by anti-human cytoplasmic antigen) that also stained positive for each lineage marker.
The purpose of this protocol is not merely to reproduce our results, but moreover to iterate and build upon them. Our surface marker panel is easily adaptable, and new markers may be added to further stratify NSPC subtypes. These candidate subtypes may be functionally interrogated using methods including but not limited to those detailed here, to explore the full breadth of stem cell heterogeneity in human brain development.
Limitations
Our study used brain tissue from gestational week (GW) 16–20. We have profiled brain tissue from as early as GW9 to as late as GW21, and have found that the staining pattern is remarkably consistent to that shown in Figure 2. However, we have not validated the gating strategy with functional assays at early gestational ages. Functional validation should be performed prior to applying our gating strategy to other systems (e.g., earlier gestational ages, neural organoids, other species, postnatal tissue).
Troubleshooting
Problem 1
Fluorophore panel design (related to step 2).
Description: Available FACS machine does not accommodate all fluorophores in panel.
Potential solution
-
•Remove markers that are not of interest.
-
○For example, if the investigator is not interested in oligodendroglial lineages, PDGFRA can be safely removed. If the investigator is only interested in oligodendroglial lineages, then CXCR4 can be removed.
-
○
-
•Switch antibodies into a fluorophore than can be accommodated.
-
○While fluorophores can often be switched, the clone of the antibody should not be switched without prior validation that both show the same staining pattern.
-
○The lineage dump channel can be put in a channel that has some spectral overlap with the viability channel, as both will be gated out anyways. Good combinations of dump and viability channels include PE/Cy5 and PI, BUV737 and Sytox Red, or PacBlue and DAPI.
-
○
Problem 2
Sorting more than four populations (related to step 2).
Description: Only four populations can be sorted in parallel on most FACS machines, but investigators may be interested in sorting more than four populations.
Potential solution
-
•We have successfully used magnetic-activated cell sorting (MACS) prior to FACS to separate out THY1-enriched and THY1-depleted fractions. Each fraction can then be run in parallel on separate FACS machines, where the THY1-enriched fraction was used to sort GPCs, pre-OPCs, OPCs, and OLs, while the THY1-depleted fraction was used to sort radial glia, astrocytes, and neurons.
-
○Following antibody staining, the cells are further stained with anti-FITC microbeads, which bind to the anti-THY1-FITC antibody.
-
○Cells are then put through an LS column for magnetic separation. The cells that flow through are THY1-depleted, and the cells that are flushed out of the column are THY1-enriched.
-
○
-
•Though more wasteful, populations can be sorted in series (i.e., sort 4 populations, and once enough has been collected, sort another 4 populations).
-
○In this scenario, we recommend sorting the most common populations first, and once enough cells have been collected, sort the rarer populations.
-
○
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rahul Sinha (sinhar@stanford.edu).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
We thank A. McCarty for mouse colony management, L. Quinn and T. Naik for laboratory management, C. Carswell-Crumpton and C. Pan for FACS support, and T.T.H. Wu for microscopy support. D.D.L. and J.Q.H. were supported by Stanford University Medical Scientist Training Program grants T32-GM007365 and T32-GM145402. This work was supported by NIH/NCI Outstanding Investigator Award R35-CA220434 to I.L.W., the Virginia and D.K. Ludwig Fund for Cancer Research to I.L.W., and the Chan Zuckerberg Biohub.
Author contributions
N.U., R.S., J.Q.H., and D.D.L. optimized the dissociation protocol. N.U., D.D.L., J.Q.H., and R.S. developed the antibody panel. D.D.L., R.S., and N.U. developed the FACS gating strategy. D.D.L. wrote the manuscript. R.S., N.U., and I.L.W. supervised the study.
Declaration of interests
D.D.L., J.Q.H., R.S., N.U., and I.L.W. are listed as inventors on a pending patent related to this protocol. I.L.W. is a co-founder of Bitterroot Bio, Inc. and Pheast, Inc., neither of which are related to the current protocol. I.L.W. was an initial co-founder and N.U. a former employee of StemCells, Inc., but they are not currently consultants or employees at the company or its successor.
Contributor Information
Irving L. Weissman, Email: irv@stanford.edu.
Rahul Sinha, Email: sinhar@stanford.edu.
Data and code availability
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Liu D.D., He J.Q., Sinha R., Eastman A.E., Toland A.M., Morri M., Neff N.F., Vogel H., Uchida N., Weissman I.L. Purification and characterization of human neural stem and progenitor cells. Cell. 2023;186:1179–1194.e15. doi: 10.1016/j.cell.2023.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 3.Uchida N., Buck D.W., He D., Reitsma M.J., Masek M., Phan T.V., Tsukamoto A.S., Gage F.H., Weissman I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.