

SUMMARY
Misfolded endoplasmic reticulum (ER) proteins are degraded through a process called ER-associated degradation (ERAD). Soluble, lumenal ERAD targets are recognized, retrotranslocated across the ER membrane, ubiquitinated, extracted from the membrane, and degraded by the proteasome using an ERAD pathway containing a ubiquitin ligase called Hrd1. To determine how Hrd1 mediates these processes, we developed a deep mutational scanning approach to identify residues involved in Hrd1 function, including those exclusively required for lumenal degradation. We identify several regions required for different Hrd1 functions. Most surprisingly, we find two cytosolic regions of Hrd1 required for lumenal ERAD substrate degradation. Using in vivo and in vitro approaches, we define roles for disordered regions between structural elements that are required for Hrd1 autoubiquitination and substrate interaction. Our results demonstrate that disordered cytosolic regions promote substrate retrotranslocation by controlling Hrd1 activation and establishing directionality of retrotranslocation for lumenal substrate across the ER membrane.
Graphical abstract
In brief
Peterson et al. report a massively parallelized genetic screen to determine how the Hrd1 E3 ubiquitin ligase functions in ERAD. The genetic and biochemical assays demonstrate that overlooked, disordered cytosolic regions are required for providing directionality in degradation of lumenal ERAD substrates.
INTRODUCTION
Most integral membrane and secretory proteins are translated at the endoplasmic reticulum (ER), where they are folded and undergo protein quality control before distribution to other organelles in the secretory pathway. Newly synthesized proteins that fail to fold are degraded at the proteasome through the conserved pathway called ER-associated degradation (ERAD). In addition to the degradation of misfolded proteins, ERAD regulates biosynthetic pathways and degrades key enzymes in sterol synthesis pathways.1–4 ERAD is critical for maintaining cellular homeostasis, and deletion of the ERAD machinery is embryonically lethal in mice.5–9 When the degradative capacity of the ERAD system is exceeded, unfolded proteins accumulate in the ER and induce the unfolded-protein response (UPR) to restore ER proteostasis.10,11
Over several decades, genetic and biochemical studies have characterized the ERAD machinery and established fundamental principles for ERAD function. Using S. cerevisiae as a model organism, the existence of at least four ERAD pathways has been proposed, based on whether the misfolding lesion is in the lumen, membrane, cytosol, or inner nuclear membrane space (ERAD-L, -M, -C, and -INM, respectively)3,12,13 (for reviews see Christianson and Carvalho14 and Christianson et al.15). Both ERAD-L and ERAD-M substrates are degraded by the Hrd1 complex normally consisting of Hrd1, Hrd3, Yos9, Der1, and Usa1 (Figure S1A). The Hrd1 complex functions by recruiting substrate to the complex, retrotranslocating lumenal substrate (movement from the ER lumen to the cytosol), followed by substrate ubiquitination, extraction by the Cdc48 complex, and degradation by the proteasome.12,16–19 Hrd1 is an integral membrane RING-type ubiquitin ligase with eight transmembrane segments that can directly recognize substrates and is required for retrotranslocation.19–23 Hrd1 forms a heterodimeric retrotranslocation channel with Der1.24,25 Der1 is a rhomboid pseudo-protease that is essential for ERAD-L under normal physiological conditions.24–26 Both Hrd1 and Der1 interact directly with Usa1, which scaffolds the heterodimeric-channel interactions and mediates higher-order oligomerization.24,27 Hrd1 also interacts with Hrd3, a single-pass membrane protein that controls Hrd1 ubiquitination activity, recruits substrates, and bridges an interaction with the substrate-recruiting protein called Yos9.12,28–30 The Hrd1 complex is proposed to be dynamic, with structural studies finding Hrd1 as a ‘‘monomeric’’ (Der1-Hrd1-Hrd3-Usa1) or dimeric (Hrd3-Hrd1-Hrd1-Hrd3) complex state.20,24,31,32
Genetic, biochemical, and structural studies support a central role for Hrd1 in ERAD. Overexpression of Hrd1 bypasses the requirement for other complex components in vivo,21 and Hrd1 can independently recognize, retrotranslocate, and ubiquitinate substrates in vitro.19,22,23 Moreover, Hrd1 controls the activity of the complex by activating itself through autoubiquitination within its RING domain.22 It is currently unclear how Hrd1 autoubiquitination results in Hrd1 activity; proposals include the idea that autoubiquitination drives a conformational change in Hrd1 that permits substrate retrotranslocation,22,23 exposes a cytosolic substrate binding site,23 or alters the oligomeric state from a regulatory to an active stoichiometry.31
To dissect the mechanics of Hrd1 function, we developed fluorescent reporter substrates and used unbiased deep mutational scanning (DMS) to identify residues within Hrd1 required for ERAD. Our approach highlighted two surprising regions on the cytosolic face of Hrd1 that were specifically required for degradation of ERAD-L substrates. These important cytosolic regions are predicted to be disordered but have intrinsic elements that were required for their function in ERAD. Using both in vivo and in vitro assays, we found that the first disordered cytosolic loop between transmembrane segments 6 and 7 is required for Hrd1 autoubiquitination. The second disordered region falls within the C-terminal region and forms a cytosolic substrate-binding domain required to promote retrotranslocation of lumenal substrates across the membrane. Our work unveils mechanics for lumenal substrate selection and retrotranslocation across the ER membrane.
RESULTS
Deep mutational scanning of the Hrd1 ubiquitin ligase
We developed an unbiased deep mutational scanning approach to dissect Hrd1 function. We designed fluorophore-based reporters using model ERAD substrates to allow for high-throughput screening by flow cytometry (Figures 1A and S1B– S1D).1,33–36 For our assay, we selected CPY* (GFP-CPY*) and Hmg2 (Hmg2-RFP) as model ERAD-L and ERAD-M substrates.1,34 We observed clear fluorescence shifts between wild-type Hrd1 (Hrd1(WT)) and the non-functional RING domain mutant Hrd1 (Hrd1(C399S)) (Figures 1A and S1D). Using flow cytometry, we followed substrate degradation by allowing cells to enter stationary phase, which reduces new translation of ERAD substrates (hereafter called a saturated chase) (Figures 1B and S1E).16 We reasoned that this method would give us better recovery of sorted cells compared with the more widely used cycloheximide chase.1,37
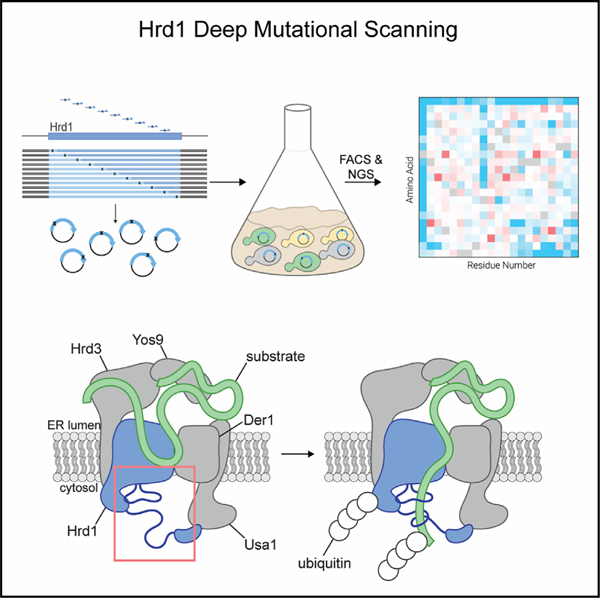
Figure 1. Deep mutational scanning of the Hrd1 ubiquitin ligase.
(A) Degradation of Hmg2-RFP and GFP-CPY* was followed using flow cytometry after the addition of 0.1% ethanol (vehicle control), 10 μg/mL zaragozic acid (for Hmg2 chase), or 50 μg/mL cycloheximide (for CPY* chase). Experiments were performed in hrd1Δ cells expressing either Hrd1(WT) or the non-functional Hrd1(C399S). Histograms were scaled as a percentage of maximum cell count (modal).
(B) Substrate degradation was followed using flow cytometry either during mid-log phase growth treated as in (A) (mid-log chase, top) or with cells grown to saturation and no pharmacological treatment (saturated chase, bottom).
(C) As in (A) except with hrd1Δ cells co-expressing Hmg2-RFP and GFP-CPY*. Cells were complemented either by centromeric Hrd1 plasmids or with Hrd1 integration and subjected to a saturated chase. Centromeric Hrd1(WT) forms two populations of expression, either expressing (‘‘WT:on’’) or low expressing (‘‘WT:off’’), as observed previously.40
(D) Fluorescence-activated cell sorting (FACS) analysis of hrd1Δ cells expressing Hmg2-RFP (y axis) and GFP-CPY* (x axis) that were transformed with centromeric plasmids containing Hrd1(WT), inactive Hrd1(C399S), or one of five different Hrd1 mutant libraries. Wild-type-like (WT) and ERAD-L-defective (L) cells were sorted into bins as indicated and collected for downstream analysis.
(E) Top: topology diagram of Hrd1 with transmembrane segments displayed as TM1–TM8. Colors indicate cytosolic (blue), lumenal (magenta), and cytosolic RING domain (green). Bottom: deep mutational scanning results of cells sorted from the wild-type-like bins in (D) are displayed as a heatmap showing singlecodon changes that were enriched (red) or depleted (blue) compared with the input library. Individual amino acids are on the y axis, and the Hrd1 amino acid position is on the x axis. Dark gray boxes indicate the wild-type amino acids and light gray boxes indicate a lack of sequencing coverage.
See also Figure S1.
We used tiling primer mutagenesis to mutagenize five subregions (approximately 110 amino acids each) that span Hrd1 and are compatible with short-read Illumina sequencing.38 We inserted the mutagenized DNA fragments into a centromeric plasmid backbone using in-cell homologous recombination.39 While expression from centromeric plasmids switches between on- and off-expression states,40 we opted for centromeric plasmids, rather than genomic integration, to increase transformation efficiency (Figure 1C and Table S1). We subjected these cells to a saturated chase and isolated wild-type-like (WT) and ERAD-L-defective (L) populations using fluorescence-activated cell sorting (FACS) (Figure 1D). After outgrowth, we confirmed the phenotype of the sorted populations, followed by DNA extraction, library preparation, Illumina sequencing, and analysis (Figures S1F and S1G).
We began our analysis with WT-sorted cells (WT bin, Figure 1D) with only single amino acid changes (covering 99.7% of the possible substitutions). We compared the relative ratio of mutations in the WT bin to our input libraries and visualized the results with a heatmap (Figure 1E). To validate our screening and analysis pipeline, we considered mutations expected to prevent wild-type function based on prior knowledge. First, we observed that stop codons were strongly depleted (blue in the heatmap) in our WT bin, except within the last 30 amino acids of Hrd1 (Figure 1E and Tables S2–S4). Second, we expected that prolines would disrupt the transmembrane segments and prevent normal function.41,42 Indeed, we observed a strong depletion of prolines in most transmembrane segments. Finally, we expected that mutations in the RING domain of Hrd1 would prevent function. As expected, mutations in the RING-finger cross-brace motifs were highly depleted for most substitutions. Together, these results demonstrated that our screening and analysis pipelines performed as expected.
Transmembrane segments 1 and 2 control complex specificity through distinct mechanics
To specifically identify portions of Hrd1 contributing to ERAD-L, we shifted our attention to cells that were sorted as defective in CPY* degradation (ERAD-L), but functional in Hmg2 degradation (ERAD-M) (L bin, Figures 1D and S1G). We looked for enriched single-point mutations and focused on results with low false discovery rates (FDRs) (Figures 2A and S2A–S2C and Tables S5–S7). We observed clear enrichment of mutations that prevented ERAD-L substrate degradation scattered across transmembrane segments 1 and 2 and in the C-terminal region following the RING domain.
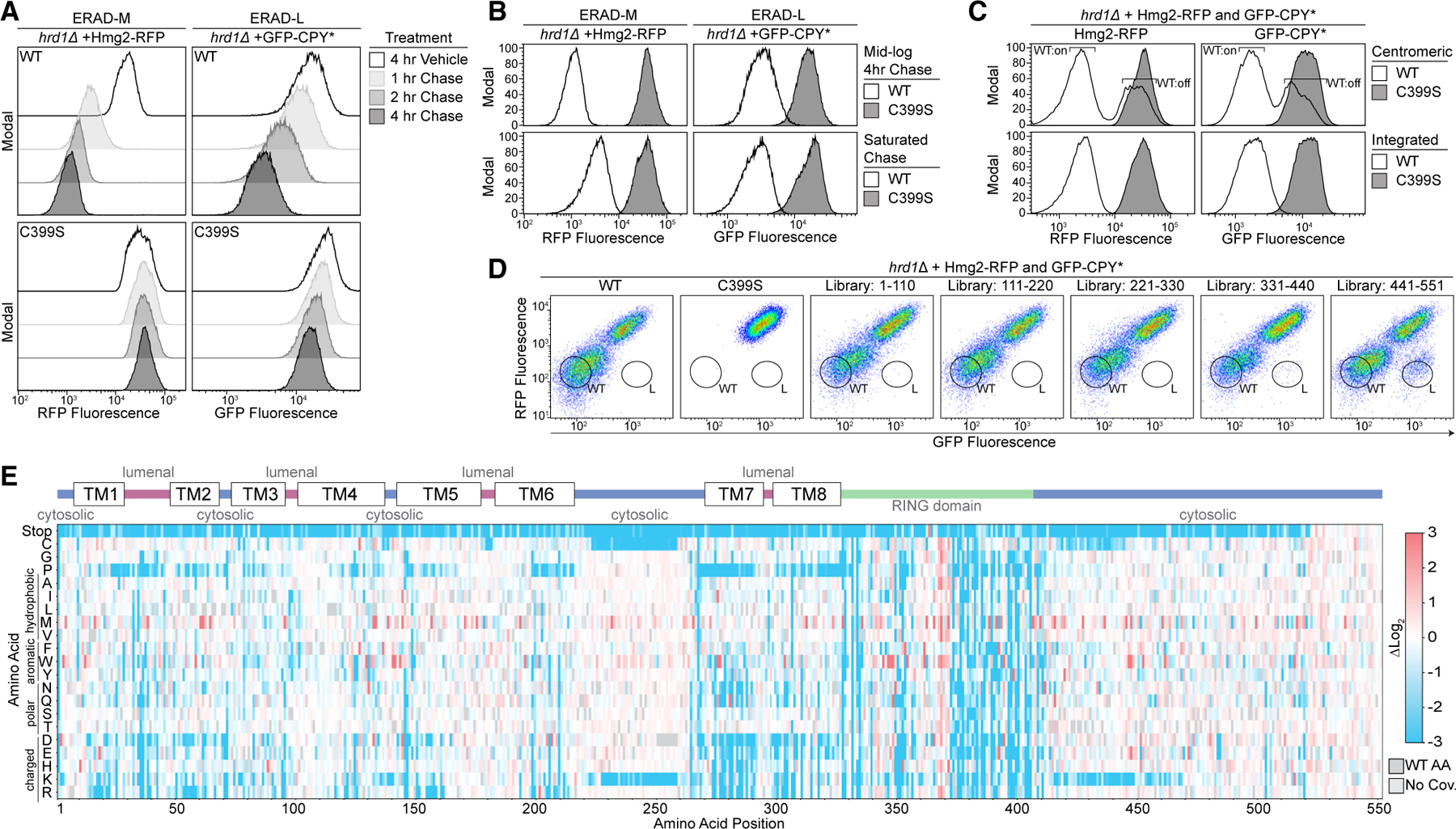
Figure 2. Transmembrane segments 1 and 2 control complex specificity through distinct mechanics.
(A) Top: topology diagram of Hrd1 with transmembrane segments shown as TM1–TM8. Colors indicate cytosolic (blue), lumenal (magenta), and cytosolic RING domain (green). Middle: heatmap showing the highest enrichment value at each amino acid position from cells sorted into the ERAD-L-defective bin. Bottom: deep mutational scanning results of cells sorted into the ERAD-L-defective bins displayed as a heatmap showing single-codon changes that were enriched (red) or depleted (blue) compared with the input library. Color transparency was adjusted based on false discovery rates (FDRs). FDRs below 0.1% were set to 0% transparent, and FDRs between 0.1% and 100% were used to adjust transparency from 0% (opaque) to 90% transparent (see also Figures S2A–S2C). Individual amino acids are on the y axis, and the Hrd1 amino acid position is on the x axis. Dark gray boxes indicate wild-type amino acids and light gray boxes indicate lack of coverage.
(B) As in (A) but with a heatmap representing the cells sorted into the wild-type-like bin between residues Met1 and Gly72. Transparency was not adjusted based on FDR.
(C) As in (A) but showing only amino acids Met1–Gly72.
(D) Topology diagram highlighting the positions of ERAD-L-defective Hrd1 mutations from (C), with the color scheme matching enrichment values. The red circle (F46R) indicates an ERAD-L-defective mutant isolated during screen development.
(E) Flow cytometry analysis of hrd1Δ cells expressing the indicated ERAD substrates and Hrd1(WT), inactive Hrd1(C399S), or Hrd1 variants. Cells were treated for 4 h with 0.1% ethanol (vehicle, solid black line with no fill) or chased for 4 h with cycloheximide or zaragozic acid (solid black line with gray fill). The dashed line highlights the position of Hrd1(C399S) after a 4-h chase.
(F) Quantification of (E). Hrd1(WT) is set to 1 (full function, black) and inactive Hrd1(C399S) is set to 0 (no function, white). Values outside of the range were set to 0 or 1 (see also Figure S2D).
(G) Protein levels of Hrd1 or Hrd1(L20R) expressed from native promoters on centromeric plasmids were determined by quantification of immunoblots normalized to Hrd1(WT) levels displayed as the mean ± SEM.
For this figure, the number of quantified replicates and individual values are shown in Tables S8 and S9. See also Figure S2.
The Hrd1 lumenal segment between transmembrane segments 1 and 2 was previously reported to interact with Hrd3.20,24,28 Our screening data highlight the importance of this interaction interface because mutations to many amino acids, including polar, charged, or proline, were disruptive to Hrd1 function, presumably through loss of Hrd3 association (Figure 2B). Notably, we found specific mutations clustered near or within the Hrd3 interaction site (Ala28, Ser32, Leu42) that reduced ERAD-L function (Figures 2C and 2D).
We also identified mutations clustering within transmembrane segments 1 (Val16, Leu20) and 2 (Phe46, Val50, Ile53) that specifically prevented degradation of ERAD-L substrates (Figures 2C, 2D, and S2D). Based on earlier studies, Phe46, Met49, and Ile53 are likely to be positioned closely to Der1,24 a component required for ERAD-L degradation.26 We expect that mutations in Phe46, Val50, and Ile53 are likely to perturb Hrd1/Der1 interactions, explaining their ERAD-L defects. However, residues within transmembrane segment 1 have not been shown to interact with Der1, so it is unlikely that Val16 and Leu20 substitutions disrupt Hrd1/Der1 interactions.24 To study this region, we selected the highest-enriched variant from transmembrane segment 1, Hrd1(L20R) (Figures 2C and 2D). First, we confirmed that Hrd1(L20R) interacted with Hrd3 and Der1 (Figures S2E and S2F). Next, we confirmed that Hrd1(L20R) was unable to degrade either ERAD-L substrate (GFP-CPY* or GFP-Pep4*) but still degraded all ERAD-M substrates (Hmg2-NR1, 6myc-Hmg2, Hmg2, and Pdr5*), thus demonstrating a strong and specific defect for ERAD-L (Figures 2E, 2F, and S2D). Curiously, Hrd1(L20R) had enhanced degradation activity for Hmg2, which was apparent because of reduced steady-state levels in vehicle-treated cells (Figure 2E). We found Hrd1(L20R) expression to be around 1/5 that of Hrd1(WT), eliminating the possibility that elevated levels of Hrd1(L20R) enhanced Hmg2 degradation (Figure 2G). In summary, we found clusters of mutations across transmembrane segments 1 and 2 that likely disrupt ERAD-L in different ways: perturbing Hrd3 interaction, perturbing Der1 interaction, and altering Hrd1 specificity.
Usa1 interacts with the Hrd1 C-terminal region
Our screening results also demonstrated that the Hrd1 C-terminal region was required for degradation of ERAD-L, but not ERAD-M, substrates (Figure 2A). We observed an enrichment of stop codons beginning at residue Phe404 and ending at Ile521 that prevented ERAD-L degradation (Figures 3A and S2A–S2C and Tables S5–S7). We confirmed that the complete removal of the C-terminal region (Hrd1(Δ408-551)) prevented ERAD-L substrate degradation but maintained the ability to degrade all four ERAD-M substrates (Figures 3B and S3B; see Figure S3A for a schematic of the Hrd1 mutants in Figure 3).
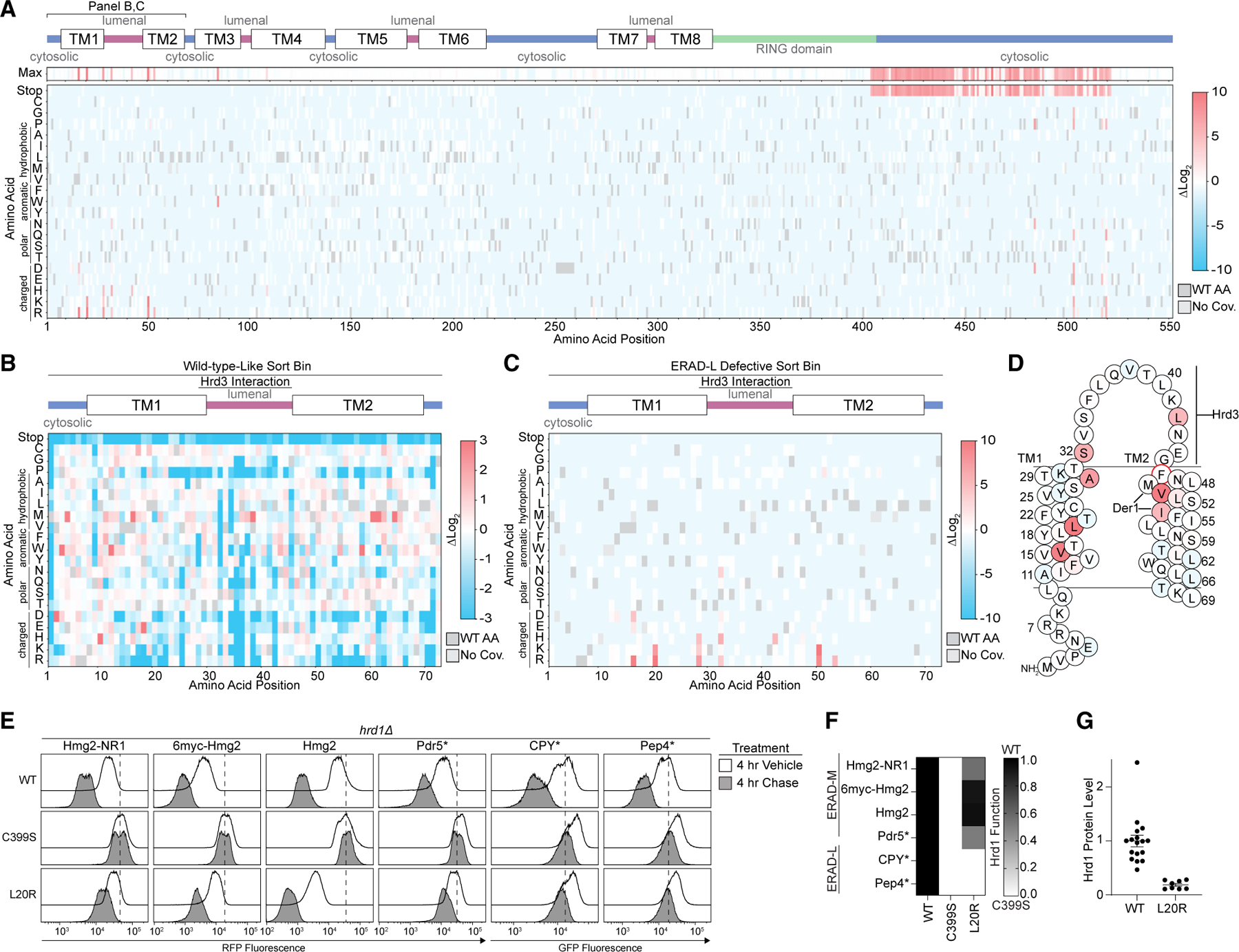
Figure 3. Usa1 interacts with the Hrd1 C-terminal region.
(A) Top: line chart representing predicted disorder (predictors of natural disordered regions [PONDR] VLXT47) of Hrd1 normalized from 0 (ordered) to 1 (disordered). Middle: topology diagram of Hrd1 with transmembrane segments shown as TM1–TM8. Bottom: deep mutational scanning results of stop codon enrichment values within the ERAD-L-defective sorting bin (ERAD-L defective) and wild-type-like sorting bin (WT). Amino acid position is on the x axis. Missing sequencing coverage is shown in light gray. The ERAD-L-defective heatmap’s color transparency was adjusted based on FDR. FDRs below 0.1% were set to 0% transparent, and FDRs between 0.1% and 100% were used to adjust transparency from 0% (opaque) to 90% transparent. Transparency was not adjusted using FDRs for the wild-type-like heatmap.
(B) Degradation of ERAD substrates was followed by flow cytometry and summarized in a heatmap. The indicated Hrd1 variants were integrated into hrd1Δ cells expressing individual ERAD substrates and subjected to a 4-h mid-log chase. Hrd1(WT) is set to 1 (full function, black) and inactive Hrd1(C399S) is set to 0 (no function, white). Values outside of the range were set to 0 or 1 (see also Figure S3B). See Figure S3A for a schematic of Hrd1 mutants used in this figure.
(C) Co-immunoprecipitation of the Hrd1 complex was performed with the indicated Hrd1 variants. 3xHA-Hrd3, 3xV5-Usa1, and Hrd1-3xFLAG were integrated into hrd1Δhrd3Δusa1Δ cells, lysed, and immunoprecipitated with anti-FLAG antibodies. Input represents 5% of the cleared lysate. These immunoblots are representative of three independent replicates.
(D) As in (B) with the indicated Hrd1 variants (see Figure S3C).
(E) As in (B) with the indicated Hrd1 variants.
(F) As in (C) with the indicated Hrd1 variants.
(G) As in (C) with the indicated Hrd1 variants. GSG refers to a poly-Gly-Ser-Gly linker the same length as the indicated deletion.
For this figure, the number of quantified replicates and individual values are shown in Table S8. See also Figure S3.
One possible explanation for this result is that loss of the C-terminal region of Hrd1 prevents an interaction with Usa1. Usa1 is required for ERAD-L substrate degradation, and the C-terminal region of Hrd1 (Lys518–Ile551) was previously demonstrated to interact with Usa1.12,27 Correspondingly, our screen identified a point mutation within the reported Usa1 interaction site at Leu519 that was defective in degradation of ERAD-L substrates (Figures 2A and S2A–S2C and Tables S5– S7). However, in our experiments, truncations beginning at Glu522 retained normal ERAD-L function, meaning that residues after Ile521 were not required for interaction with Usa1 (Figure 3A). Using co-immunoprecipitation we confirmed that Hrd1(Δ522-551) maintained normal Hrd1/Usa1 interaction, refining the Hrd1/Usa1 interaction site to end at Ile521 (Figure 3C).
Based on the deep mutational scanning data, we identified non-truncation point mutations between Phe484 and Leu519 that were enriched in the ERAD-L-defective populations (Figure 2A and Tables S5–S7). We confirmed that these point mutations were defective in ERAD-L, but not ERAD-M, substrate degradation (Figures 3D and S3C). In addition, during screening development, we discovered an ERAD-L-defective triple-point mutant within this region, Hrd1(T416P,W481L,I505T), and confirmed the ERAD-L-defective phenotype (Figure 3E). For each of these mutants, the steady-state levels were similar to those of wild-type Hrd1, indicating the ERAD-L defects were not related to the protein stability (Figure S3D). Based on the proximity of these residues to the reported Hrd1/Usa1 interface, we suspected these point mutations would disrupt Hrd1/Usa1 interactions.27 Co-immunoprecipitation experiments confirmed that Hrd1(T416P,W481L,I505T) and Hrd1(F484D) failed to interact with Usa1, but interacted normally with Hrd3 (Figure 3F).
Given the observed enrichment of stop codons starting at Phe404, we wondered whether the region between the RING domain and Trp481 was also important for Usa1 interaction. To determine whether Gly408–Thr480 was important for Usa1 interaction, we replaced these residues with a poly-Gly-Ser-Gly linker of the same length (Hrd1(Δ408-480_GSG)) and a slightly shorter replacement region from Gly408 to Met469 (Hrd1(Δ408– 469_GSG)). Using co-immunoprecipitation, we found that both Hrd1(Δ408-480_GSG) and Hrd1(Δ408-469_GSG) maintained normal Usa1 and Hrd3 interactions (Figure 3G). These data allowed us to refine the Hrd1/Usa1 interaction interface to Hrd1 Trp481–Ile521, which is required for ERAD-L, but not ERAD-M.
A disordered C-terminal region is required for retrotranslocation
The requirement of a cytosolic region (Gly408–Thr480) for lumenal substrate degradation was surprising because Hrd1(Δ408-480_GSG) appeared to interact with the other ERAD components properly. In addition, this region was predicted to be disordered (Figure 3A), so we considered whether it would function solely as a spacer; in this case, enrichment of stop codons could be a function of removing the subsequent Usa1 interaction interface. We reasoned that if Gly408–Thr480 served strictly as a spacer, any replacement sequence would maintain wild-type-like activity. First, we tested Hrd1(Δ408-469_GSG) and Hrd1(Δ408– 480_GSG), finding that both constructs were completely unable to degrade ERAD-L substrates but were still able to degrade ERAD-M substrates (Figure 4A). Next, we replaced Hrd1 Gly408–Met469 with the corresponding regions of Hrd1 from Saccharomyces kudriavzevii (Hrd1(Sk)) and Lachancea nothofagi (Hrd1(Ln)) (Figure S4A). Each chimera was able to degrade ERAD-L and ERAD-M substrates, although less efficiently than endogenous S. cerevisiae Hrd1(WT) (Figure 4A). We measured steady-state Hrd1 levels and found that each construct exhibited reduced Hrd1 stability that was dependent on Hrd1 catalytic function (Figures 4B, 4C, and S4B). However, stability alone did not explain the activity defects, because Hrd1(Sk), Hrd1(Δ408– 469_GSG), and Hrd1(Δ408-480_GSG) had similar stability but disparate degradation capability (Figures 4A–4C). These data support the idea that the amino acid composition of the disordered Gly408–Thr480 region is important for Hrd1 function in degradation of ERAD-L substrates.
Figure 4. A disordered C-terminal region is required for retrotranslocation.
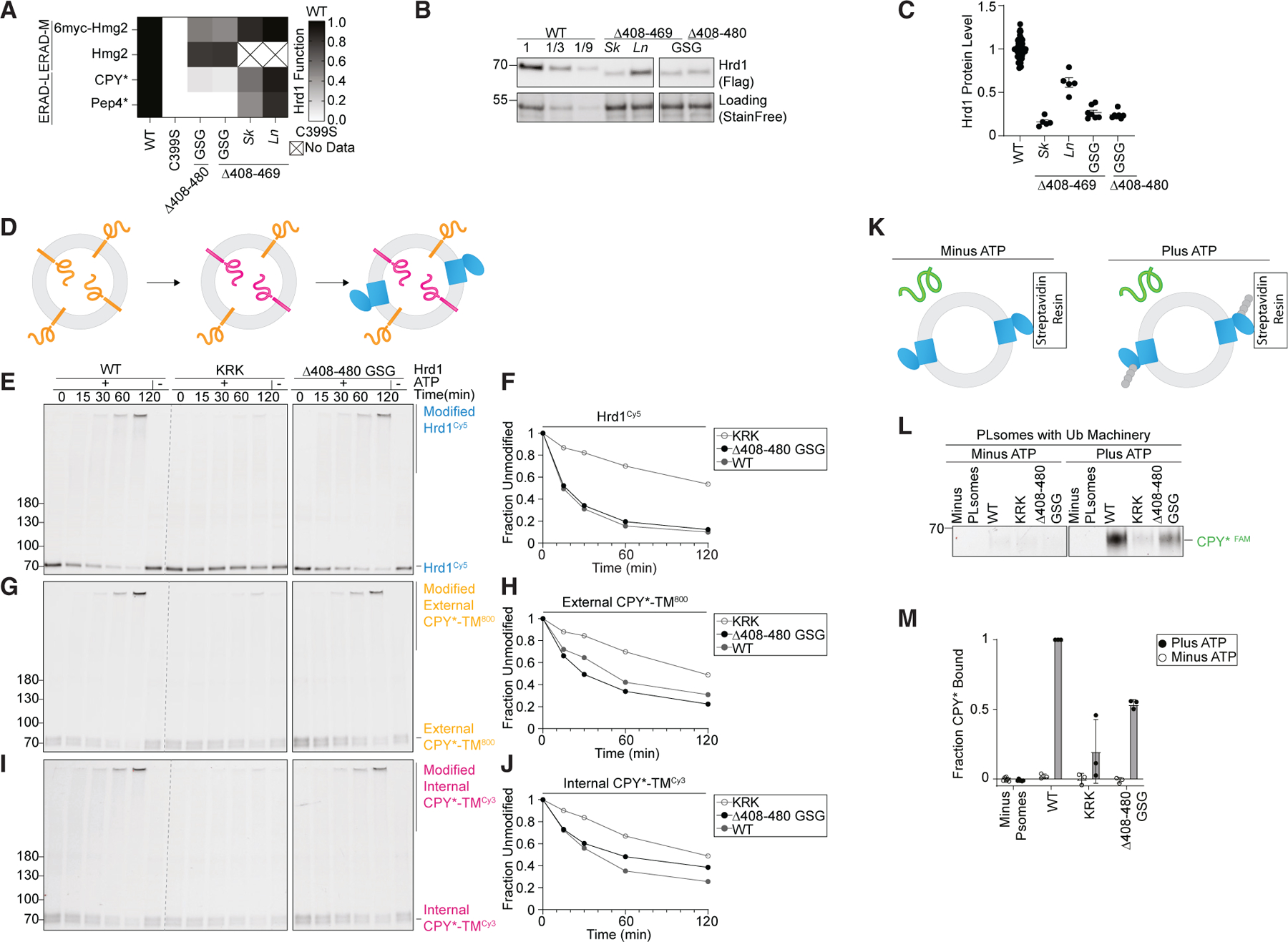
(A) Degradation of ERAD substrates by individual Hrd1 variants was followed by flow cytometry and summarized in a heatmap. The indicated Hrd1 variants were integrated into hrd1Δ cells expressing individual ERAD substrates and subjected to a 4-h mid-log chase. For ‘‘GSG’’ Hrd1 samples, the region indicated was replaced with a poly-Gly-Ser-Gly linker of the same length as the indicated deletion. Sk (S. kudriavzevii) and Ln (L. nothofagi) are chimeras of S. cerevisiae Hrd1 replaced with homologous regions from the indicated species (see Figure S4A). Wild-type Hrd1(WT) is set to 1 (full function, black) and inactive Hrd1(C399S) is set to 0 (no function, white).
(B) Expression levels of Hrd1 variants were determined by immunoblotting. hrd1Δ cells expressing Hrd1-3xFLAG constructs were from (A). The first three lanes are a calibration curve generated with dilutions of the wild-type Hrd1 lysate. Total protein was visualized by stain-free technology as a loading control.
(C) Quantification of (B) normalized to wild-type Hrd1 expression and displayed as the mean ± SEM.
(D) Schematic of the in vitro retrotranslocation assay. CPY*-TM labeled C-terminally with DyLight800 (orange) was reconstituted into proteoliposomes. The C-terminal DyLight800 peptide of internally oriented substrate was exchanged for a new peptide containing Cy3 (magenta) using sortase A followed by incorporation of Hrd1Cy5 (blue).
(E) In vitro autoubiquitination of Hrd1Cy5 (blue) in a reconstituted proteoliposome system with externally oriented CPY*-TM800 (orange) and internally oriented CPY*-TMCy3 (magenta). Wild-type Hrd1 (WT, positive control), a retrotranslocation-defective Hrd1 (Hrd1(KRK), negative control), and Hrd1(Δ408-480 GSG) were reconstituted and incubated with recombinant ubiquitination machinery for the indicated times. Samples were analyzed by SDS-PAGE and in-gel fluorescence scanning to visualize Hrd1. This fluorescence scan is representative of three replicates (see also Figures S4C–S4H).
(F) Quantification of (E), the fraction of unmodified Hrd1Cy5.
(G) As in (E) showing external CPY*-TM800.
(H) Quantification of (G), the unmodified external CPY*-TM800.
(I) As in (E) showing internal CPY*-TMCy3. Red pixels indicate saturation of signal during the imaging.
(J) Quantification of (I), the unmodified internal CPY*-TMCy3.
(K) Schematic for soluble CPY* (green) interaction assay in proteoliposomes. Hrd1 is shown in blue and ubiquitin chains in gray.
(L) Hrd1 proteoliposomes (PLsomes) were immobilized at 1 μM and incubated with recombinant ubiquitination machinery ±ATP, washed, and incubated with 100 nM CPY*FAM (green) (see K). Samples were eluted with biotin and analyzed by SDS-PAGE and in-gel fluorescence scanning. Red pixels indicate image saturation.
(M) Quantification of (L), shown as the mean ± SD, from three independent experiments.
For this figure, the number of quantified replicates and individual values are shown in Tables S8 and S9. See also Figure S4.
To identify the specific function of this disordered region, we turned to an in vitro reconstituted system enabling dissection of the individual steps in the ERAD process. Hrd1 was reconstituted into proteoliposomes with an ERAD-L substrate (CPY*-TM) that was C-terminally labeled with an organic fluorophore to track CPY*-TM orientation (Figure 4D).22 Hrd1 function was followed in different ways: first, by Hrd1 autoubiquitination (Figures 4E and 4F); second, by ubiquitination of externally oriented substrate (active ubiquitin ligase activity) (Figures 4G and 4H); and third, by ubiquitination of internally oriented substrate (demonstration of substrate retrotranslocation) (Figures 4I and 4J). As controls for this assay, we used wild-type Hrd1 and a previously established retrotranslocation-defective Hrd1 (Hrd1(KRK)).22,23 Hrd1 and Hrd1(KRK) performed as expected (Figures 4E–4J). Hrd1(Δ408– 480_GSG) autoubiquitination was similar to wild-type Hrd1 (Figures 4E and 4F) as was the ubiquitination of externally oriented substrate (Figures 4G and 4H). However, this Hrd1 variant showed a notably reduced ability to retrotranslocate and ubiquitinate internally oriented substrate (Figures 4I and 4J). This indicates that Hrd1(Δ408-480_GSG) retrotranslocation function is partially impaired, indicating that the cytosolic-facing Gly408–Thr480 disordered region of Hrd1 plays an important role in substrate retrotranslocation specifically.
Hrd1 was previously proposed to contain a cytosolic substrate binding site that becomes exposed following autoubiquitination.23 To determine whether Gly408–Thr480 directly interacts with substrates, we used a previously described proteoliposome-based substrate interaction assay.23 We reconstituted Hrd1 into proteoliposomes and immobilized the proteoliposomes with an affinity resin. We incubated the proteoliposomes with purified ubiquitination machinery either in the presence or in the absence of ATP. After removing the ubiquitination machinery, we incubated the proteoliposomes with a soluble ERAD-L substrate (CPY*) and followed CPY* interaction with the proteoliposomes (Figure 4K). As expected, Hrd1(WT) efficiently interacted with CPY* after autoubiquitination, while Hrd1(KRK) did not (Figures 4L and 4M).23 Hrd1(Δ408-480_GSG) displayed a reduced capacity to interact with CPY* following autoubiquitination, even though the autoubiquitination activity was comparable to that of wild-type Hrd1 (Figure 4E). Taken together, these results support a role for a previously overlooked disordered C-terminal region in Hrd1 (Gly408–Thr480). This region is necessary for efficient ERAD-L substrate retrotranslocation through direct substrate interaction.
Hrd1 autoubiquitination outside of the RING domain restricts function
We noticed that the C-terminal disordered region was largely devoid of lysine and cysteine residues (Figure S4A). This observation was complemented by our screening data that showed that mutations adding lysine or cysteine residues were largely depleted between Gly408–Met469 in the wild-type-like sorted group (Figures 1E and 5B). We directly tested this observation by substituting lysine across the C-terminal region (Gly408– Ser475) and assaying ERAD function. Consistent with our screening data, lysine substitutions within Gly408–Met469 largely reduced activity against both classes of substrates but demonstrated no change in specificity between ERAD-L and ERAD-M substrates (Figures 5C and S5A).
Figure 5. Disordered cytosolic regions are devoid of lysine and cysteine.
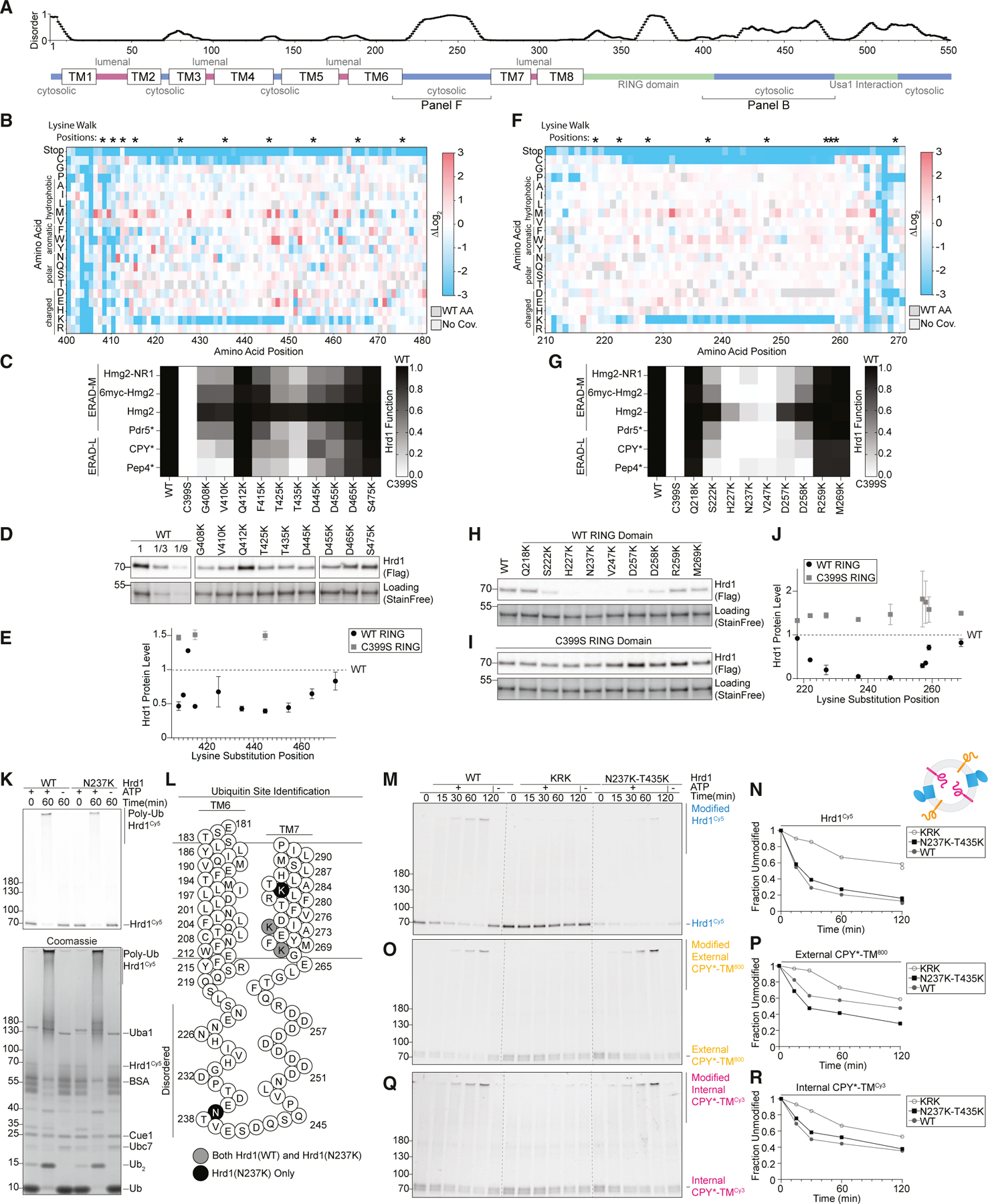
(A) Top: line chart representing predicted disorder (PONDR VLXT47) of Hrd1 normalized from 0 (ordered) to 1 (disordered). Bottom: topology diagram of Hrd1 with transmembrane segments shown as TM1–TM8.
(B) Top: asterisks (*) indicate positions where lysine is substituted in (C), (D), and (E). Bottom: heatmap representing the deep mutational scanning results of Hrd1 variants sorted into the wild-type-like bin. Amino acids from Arg400 to Thr480 are shown. Enrichment values are compared with the input library. Dark gray boxes indicate the wild-type amino acid and light gray boxes indicate lack of coverage.
(c) Degradation of ERAD substrates by individual Hrd1 variants was followed by flow cytometry and summarized in a heatmap. The indicated Hrd1 variants were integrated into hrd1Δ cells expressing individual ERAD substrates and subjected to a 4-h mid-log chase. Hrd1(WT) is set to 1 (full function, black) and inactive Hrd1(C399S) is set to 0 (no function, white). Values outside of this range were set to 0 or 1 (see Figure S5A).
(D) Expression levels of Hrd1 variants were determined by immunoblotting. hrd1Δ cells expressing Hrd1-3xFLAG constructs were from (C). Total protein was visualized by stain-free technology as a loading control. The first three lanes are a calibration curve generated with dilutions of the wild-type Hrd1 lysate.
(E) Quantification of (D) normalized to wild-type Hrd1 expression and displayed as the mean ± SEM. See also Figure S5B.
(F) As in (B) but for amino acid positions Asn210–Tyr270.
(G) As in (C) but with the indicated Hrd1 variants. See also Figure S5E.
(H) As in (D) but with the indicated Hrd1 variants.
(I) As in (H) but containing E3-inactivating C399S RING mutation.
(J) Quantification of (H) as black-filled circles and (I) as gray-filled squares normalized to wild-type Hrd1 (black dashed line). See also Figure S5G.
(K) Purified Hrd1(WT) or Hrd1(N237K) in detergent micelles was incubated with recombinant ubiquitination machinery. Samples were separated by SDS-PAGE, and poly-ubiquitinated Hrd1 bands were excised from the gel and sent for mass spectrometry-based identification of ubiquitination sites. Top: Hrd1Cy5 fluorescence scanning of the in vitro ubiquitination reactions. Bottom: Coomassie staining.
(L) Topology diagram showing TM6 and TM7 and the predicted disordered intervening cytosolic loop. Lysines that were identified as ubiquitinated for both wildtype Hrd1 and Hrd1(N237K) are displayed as gray-filled circles. Lysines ubiquitinated only in Hrd1(N237K) are displayed as a black-filled circles.
(M) In vitro autoubiquitination of Hrd1Cy5 (blue) in a reconstituted proteoliposome system with externally oriented CPY*-TM800 (orange) and internally oriented CPY*-TMCy3 (magenta). Wild-type Hrd1 (WT, positive control), a retrotranslocation-defective Hrd1 (Hrd1(KRK), negative control), and Hrd1(N237K–T435K) were reconstituted and incubated with recombinant ubiquitination machinery for the indicated times. Samples were analyzed by SDS-PAGE and in-gel fluorescence scanning to visualize Hrd1. This fluorescence scan is representative of three replicates (see also Figures S5J–S5U).
((N) Quantification of (M) showing unmodified Hrd1Cy5.
(O) As in (M) showing external CPY*-TM800.
(P) Quantification of (O) showing unmodified external CPY*-TM800.
(Q) As in (M) showing internal CPY*-TMCy3. Red pixels indicate saturation of signal during the imaging.
(R) Quantification (Q) showing unmodified internal CPY*-TMCy3.
For this figure, the number of quantified replicates and individual values are shown in Tables S8 and S9. See also Figure S5.
We suspected that the depletion of lysine residues in proximity to the RING domain could stem from one of three possibilities. First, lysines could reduce Hrd1 function because they are ubiquitinated, resulting in Hrd1 degradation. Alternatively, lysine ubiquitination could sterically obstruct the observed substrate interaction with this C-terminal region. Last, ubiquitination in this C-terminal region could sterically inhibit substrate passage through the retrotranslocon. We determined whether lysine substitutions within Hrd1 residues Gly408–Met469 were destabilizing by measuring Hrd1 steady-state levels. We found that lysine substitutions at all tested residues, except Gln412, resulted in reduced Hrd1 levels (Figures 5D, 5E, and S5B) roughly corresponding to each mutant’s overall ERAD function (Figures 5C– 5E, S5A, S5B, and S5I). We verified that this ubiquitination and destabilization of Hrd1 was caused by Hrd1 itself, rather than another ubiquitin ligase, because with inactive Hrd1(C399S), lysine substitutions at Gly408, Phe415, or Asp445 did not lead to destabilization (Figures 5E and S5B). While wild-type Hrd1 primarily autoubiquitinated on lysine residues, substitution to cysteine residues also resulted in autoubiquitination and destabilization of Hrd1 within the Gly408–Met469 region (Figures S5C and S5D). These data support the hypothesis that lysine and cysteine residues were not tolerated in the Gly408–Met469 region because they were ubiquitinated, resulting in Hrd1 turnover rather than interfering with substrate interaction or retrotranslocation directly.
We observed a second region on the cytosolic face that had characteristics similar to those of the C-terminal region we implicated in direct substrate interaction (Gly408–Thr480). Both regions were predicted to be disordered, devoid of lysine, and devoid of cysteine. This second region falls within a cytosolic loop between transmembrane segments 6 and 7 (Ser222– Asp258, Figures 1E, 5A, and 5F). Similar to the Gly408–Met469 region, our functional screen found that Ser222–Asp258 was tolerant to most substitutions, except for lysine or cysteine (Figure 5F). As with the C-terminal results, substituting lysine or cysteine within Ser222–Asp258 resulted in severe defects degrading ERAD-M and ERAD-L substrates (Figures 5G, S5E, and S5F). Likewise, we found that steady-state levels of Hrd1 variants with lysine or cysteine mutations within Ser222– Asp258 were reduced (Figures 5H, 5J, S5G, and S5H), and this destabilization correlated with the severity of Hrd1 function loss (Figures 5G, 5H, 5J, and S5E–S5I). Similar to the Gly408– Met469 region (Figures 5E, S5B, and S5D), lysine and cysteine substitutions within Ser222–Asp258 destabilized Hrd1 due to Hrd1 autoubiquitination (Figures 5I, 5J, S5G, and S5H). Together, our data suggest that lysine or cysteine substitutions between positions Ser222 and Asp258 destabilized Hrd1 through autoubiquitination and subsequent degradation.
In Hrd1, the Ser222–Asp258 loop is likely in close proximity to the proposed retrotranslocation channel.20,24 Therefore, we tested whether ubiquitination of this cytosolic loop could also directly inhibit Hrd1 function by sterically obstructing substrate passage through the membrane. We confirmed that lysines within Ser222–Asp258 could be ubiquitinated in vitro. We purified wild-type Hrd1 and a destabilizing lysine substitution (Hrd1(N237K)), performed in vitro autoubiquitination, and found that Hrd1(WT) and Hrd1(N237K) both autoubiquitinated efficiently (Figure 5K). To determine whether Hrd1(N237K) was able to ubiquitinate residue 237, we excised polyubiquitinated Hrd1 and identified sites of lysine ubiquitination using mass spectrometry. In wild-type Hrd1, we identified ubiquitination sites near the cytosolic side of transmembrane segment 7 at Lys267 and Lys272 (Figure 5L). With Hrd1(N237K), we also observed ubiquitination at Lys267 and Lys272 but, importantly, observed two additional sites at the introduced N237K site and at Lys282 (Figure 5L). This directly supported the idea that Hrd1 can autoubiquitinate lysines within the cytosolic loop between transmembrane segments 6 and 7.
To test whether ubiquitination within the disordered cytosolic regions sterically inhibits Hrd1-mediated retrotranslocation, we used our in vitro retrotranslocation assay with Hrd1 containing a lysine substitution within each disordered region (Hrd1(N237K–T435K)). We found that Hrd1(N237K–T435K) efficiently autoubiquitinated (Figures 5M and 5N), ubiquitinated externally oriented substrates (Figures 5O and 5P), and retrotranslocated internally oriented substrates, similar to wild-type Hrd1 (Figures 5Q and 5R). These data demonstrate that the ubiquitination of these disordered cytosolic regions does not directly prevent substrate retrotranslocation. Rather, we concluded that the exclusion of lysines and cysteines from the loop between transmembrane segments 6 and 7 and the disordered C-terminal region prevents premature degradation of Hrd1 catalyzed by autoubiquitination.
The cytosolic loop between transmembrane segments 6 and 7 has a unique role in ERAD
Our work has uncovered clear similarities between the disordered C-terminal cytosolic region of Hrd1 and the disordered cytosolic transmembrane 6–7 loop. Furthermore, we defined an important role of the disordered C-terminal region in substrate interaction and retrotranslocation. As such, we hypothesized that the transmembrane 6–7 loop would also have a direct role in ERAD-L retrotranslocation and degradation. We returned to our deep mutational scanning data and found mutations near transmembrane 7 that were highly enriched but had high FDRs (Figures S2A and S2B and Tables S5–S7). Nevertheless, we tested these variants against ERAD-L and ERAD-M substrates and found mutations that specifically inhibited ERAD-L substrate degradation (F268K, F268R, M269P, and I274T) (Figures 6A and S6A) without altered protein stability (Figure S6B).
>Figure 6. Cytosolic loop between transmembrane segments 6 and 7 required for ERAD-L degradation.
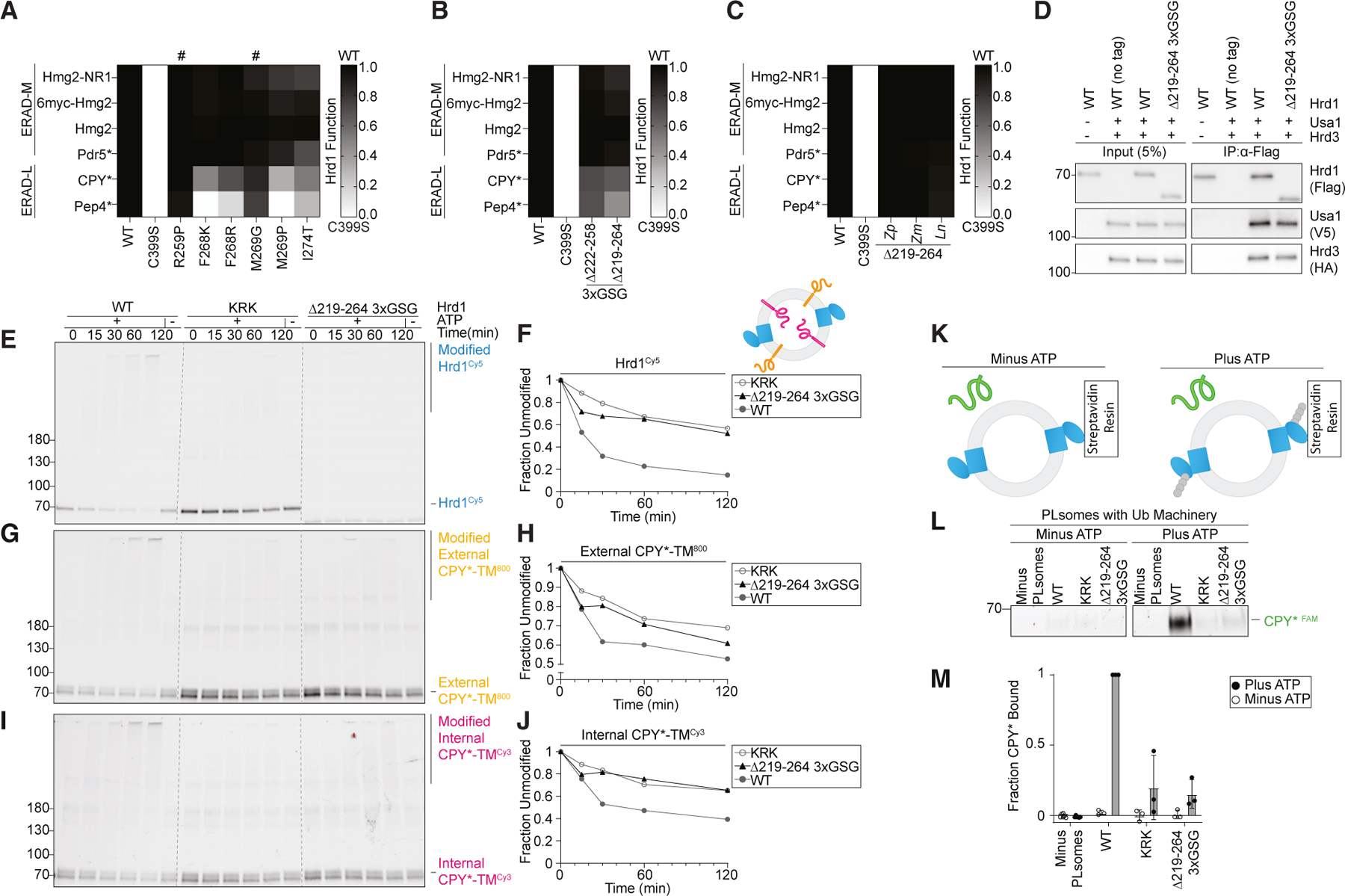
(A) Degradation of ERAD substrates by individual Hrd1 variants was followed by flow cytometry and summarized in a heatmap. Hrd1 variants were integrated into hrd1Δ cells expressing individual ERAD substrates and subjected to a 4-h mid-log chase. Hrd1(WT) is set to 1 (full function, black) and inactive Hrd1(C399S) is set to 0 (no function, white). Values outside of this range were set to 0 or 1 (see Figure S6A). ‘‘#’’ indicates a false-positive hit.
As in (A), but with the indicated Hrd1 regions replaced by a 3xGSG (Gly-Ser-Gly-Gly-Ser-Gly-Gly-Ser-Gly) linker sequence.
(C) As in (A), but with the indicated Hrd1 chimeras. The indicated region of S. cerevisiae Hrd1 was replaced with the homologous region of Zygosaccharomyces parabailii (Zp), Zygotorulaspora mrakii (Zm), or L. nothofagi (Ln). See Figures S6C and S6D.
(D) Co-immunoprecipitation of the Hrd1 complex was performed with the indicated Hrd1 variants. 3xHA-Hrd3, 3xV5-Usa1, and Hrd1-3xFLAG were integrated into hrd1Δhrd3Δusa1Δ cells, lysed, and immunoprecipitated with anti-FLAG antibodies. Input represents 5% of the cleared lysate. These immunoblots are representative of three independent replicates.
(E) In vitro autoubiquitination of Hrd1Cy5 (blue) in a reconstituted proteoliposome system with externally oriented CPY*-TM800 (orange) and internally oriented CPY*-TMCy3 (magenta). Wild-type Hrd1 (WT, positive control), a retrotranslocation-defective Hrd1 (Hrd1(KRK), negative control), and Hrd1(Δ219-264_3xGSG) were reconstituted and incubated with recombinant ubiquitination machinery for the indicated times. Samples were analyzed by SDS-PAGE and in-gel fluorescence scanning to visualize Hrd1. This fluorescence scan is representative of three replicates (see also Figures S6E–S6J).
(F) Quantification of (E) showing unmodified Hrd1Cy5.
(G) As in (E) showing external CPY*-TM800.
(H) Quantification of (G) showing unmodified external CPY*-TM800.
(I) As in (E) showing internal CPY*-TMCy3. Red pixels indicate saturation of signal during the imaging.
(J) Quantification (I) showing unmodified internal CPY*-TMCy3.
(K) Schematic for soluble CPY* (green) interaction assay in proteoliposomes. Hrd1 is shown in blue and ubiquitin chains in gray.
(L) Hrd1 proteoliposomes (PLsomes) were immobilized at 1 μM and incubated with recombinant ubiquitination machinery ±ATP, washed, and incubated with 100 nM CPY*FAM (green) (see K). Samples were eluted with biotin and analyzed by SDS-PAGE and in-gel fluorescence scanning. Red pixels indicate image saturation.
(M) Quantification of (L) displayed as mean ± SD from three independent experiments.
For this figure, the number of quantified replicates and individual values are shown in Table S8. See also Figure S6.
As with the C-terminal disordered region (Figure 3), we generated deletions of two regions (either Ser222–Asp258 or Gln219– Leu264) but only partially replaced the region with a 3xGSG linker (Gly-Ser-Gly-Gly-Ser-Gly-Gly-Ser-Gly). Hrd1(Δ222-258_3xGSG) degraded ERAD-M substrates normally but showed modest defects in ERAD-L substrate degradation (Figure 6B). The slightly expanded deletion (Hrd1(Δ219-264_3xGSG)) also degraded ERAD-M substrates normally but had stronger defects in ERAD-L substrate degradation (Figure 6B). When Gln219– Leu264 was replaced with homologous regions from diverging yeast strains (Figure S6D), we found that each chimera tested maintained a wild-type-like ability to degrade both ERAD-L and ERAD-M substrates (Figures 6C and S6C), even though many of these sequences were significantly shorter (Figure S6D).
We wondered why Hrd1(Δ219-264_3xGSG) was unable to efficiently degrade ERAD-L substrates. First, we confirmed that Hrd1(Δ219-264_3xGSG) interacted normally with Usa1 and Hrd3 (Figure 6D) and had protein levels comparable to wild-type Hrd1 (Figure S6B). Then, we tested Hrd1(Δ219-264_3xGSG) using our in vitro retrotranslocation assay and observed severe defects because Hrd1(Δ219-264_3xGSG) was unable to autoubiquitinate, mirroring the negative control (Hrd1(KRK); Figures 6E and 6F). In addition, this variant barely ubiquitinated the externally oriented substrate (Figures 6G and 6H) and failed to retrotranslocate internally oriented substrate (Figures 6I and 6J). Because this variant could not autoubiquitinate efficiently, we expected that it would be unable to activate and expose the cytosolic substrate binding site that is in part formed by the C-terminal disordered region (Figure 4). To test this, we used the in vitro CPY* binding assay (Figure 6K) and found that Hrd1(Δ219-264_3xGSG) failed to interact with soluble CPY*, similar to Hrd1(KRK) (Figures 6L and 6M). Finally, we tested whether the shorter transmembrane segment 6–7 loop replacement could autoubiquitinate. Similar to Hrd1(Δ219-264_3xGSG), Hrd1(Δ222-258_3xGSG) was unable to efficiently autoubiquitinate (Figure S6K). Taken together, in contrast to the disordered C-terminal region, the cytosolic transmembrane 6–7 loop was required for efficient Hrd1 autoubiquitination and, therefore, each of the subsequent steps in Hrd1 retrotranslocation.
DISCUSSION
In this study, we developed a deep mutational scanning platform to identify critical residues for function of the Hrd1 ubiquitin ligase. This powerful approach allowed us to identify different clusters of mutations that disrupted Hrd1 function through distinct mechanisms (Figure 1). We identified mutations across the first two transmembrane segments that likely perturb Hrd1/Hrd3 interaction, perturb Hrd1/Der1 interaction, and alter the substrate specificity of the complex (Figure 2). We refined the Hrd1/Usa1 interaction site and identified a disordered C-terminal region of Hrd1 that interacts with ERAD-L substrates to facilitate retrotranslocation (Figures 3 and 4). Similarities to the C-terminal region directed us to another disordered cytosolic region between transmembrane segments 6 and 7 that is required for Hrd1 function (Figures 5 and 6). Degradation of ERAD-L substrates, but not ERAD-M substrates, requires both cytosolic disordered regions in vivo, but for different reasons. The disordered cytosolic loop between transmembrane segments 6 and 7 (Gln219–Leu264) promotes Hrd1 autoubiquitination, which is required for retrotranslocation. Following autoubiquitination, the disordered C-terminal region (Gly408– Thr480) promotes retrotranslocation by interacting with the substrate on the cytosolic side of the membrane. Substrate ubiquitination occurs in close proximity to these two regions, explaining why lysine and cysteine substitution are not tolerated there.
Based on our experiments, and previously published data,22–24 we propose the following model (Figure 7). The Hrd1 complex engages substrate on the lumenal side of the membrane. The cytosolic loop between transmembrane segments 6 and 7 coordinates autoubiquitination to activate Hrd1 and open the cytosolic high-affinity substrate binding site(s), providing molecular interactions to enforce directionality and facilitate retrotranslocation prior to substrate ubiquitination. These high-affinity sites ultimately position substrates for ubiquitination, allowing recruitment of the Cdc48 complex to improve the efficiency of retrotranslocation, extraction, and, ultimately, substrate degradation.
Figure 7. Model for retrotranslocation.
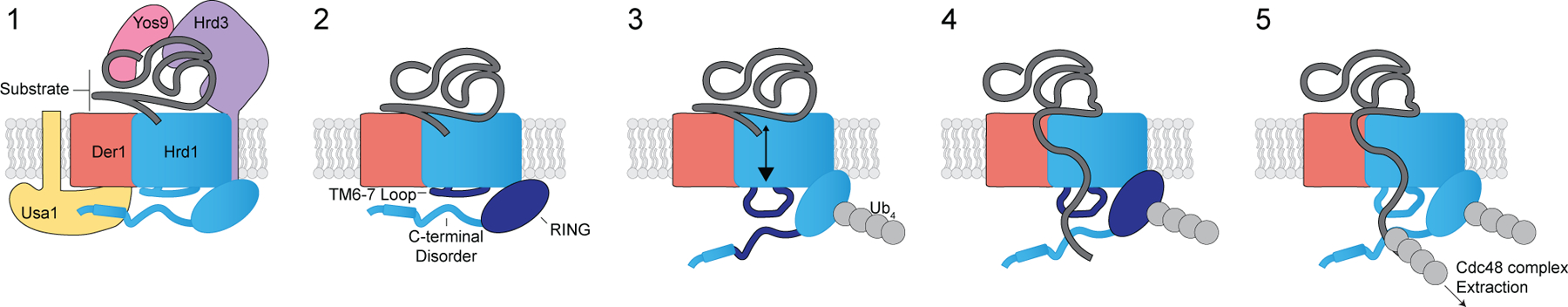
Schematic for Hrd1 complex retrotranslocation. In stage 1, the Hrd1 complex recruits a lumenal substrate. In stage 2, lumenal substrate engages Der1-Hrd1 on the lumenal side of the membrane, and Hrd1 autoubiquitination is driven by the cytosolic loop between transmembrane segments 6 and 7. In stage 3, Hrd1 autoubiquitination exposes the cytosolic substrate-binding sites in the loop between transmembrane segments 6 and 7 and the disordered C-terminal region creating a binding site for substrates on the cytosolic side of the membrane. In stage 4, Hrd1 ubiquitinates the substrate, and in stage 5, ubiquitinated substrate is extracted by the Cdc48 complex for degradation by the proteasome. See also Figure S7.
Initially, we were surprised to find mutations in the cytosolic regions of Hrd1 to be important for retrotranslocation that was independent of Usa1 interaction or RING-finger domain formation, because Hrd1 transmembrane segments can directly recognize substrate.19 Furthermore, structural and biochemical studies have illuminated a proposed retrotranslocon channel formed by the transmembrane segments 3–8.20,22–24 The mutations we found in transmembrane segments that specifically disrupted ERAD-L primarily fell within the first two transmembrane segments. The simplest explanation of these results is that mutations within transmembrane segment 2 would likely disrupt Hrd1/Der1 interaction. Transmembrane segment 1 mutations appear to alter the specificity of the complex, possibly by affecting the entry of substrate into the retrotranslocation channel.20 Within the trans-membrane segments that form the proposed retrotranslocation channel (TM3–TM8), we found relatively few mutations that specifically inhibited lumenal substrate degradation. We suspect that mutations within the channel itself affect both ERAD-L and ERAD-M substrate degradation or destabilize Hrd1.18,43 It is also likely that our method, focused on point mutations, would miss long-range or paired mutations with stronger phenotypes.
The cytosolic regions we have identified as essential for degradation of lumenal substrates are missing from current structural models of Hrd1.20,24 In addition, innovative studies focused on the cytosolic domains of Hrd1 identified highly conserved structured regions that were essential for partner protein interactions, but the role of the surrounding disordered regions themselves remained enigmatic.44 While predicted disordered cytosolic regions devoid of lysine and cysteine are common to many forms of Hrd1, from fungi to humans (Figures S7A–S7D), their amino acid sequences have little conservation. This general trend was observed in our deep mutational scanning because many individual substitutions were tolerated in these regions, except for lysine and cysteine substitution. The apparent lack of sequence conservation and mutational tolerance, while maintaining ERAD function, is another reason these regions may have gone undetected in previous studies. However, there are other ubiquitin ligases that use disordered domains to recognize their substrates.45 Even in ERAD-related proteins, disordered domains appear to interact with integral membrane substrates.46 Moreover, the existence of cytosolic substrate-interaction domains within Hrd1 was previously suggested, but the identity of this substrate interaction domain was unclear.23 We identified a region in the C-terminal region of Hrd1 (Gly408–Thr480) that serves as this cytosolic ubiquitin-activated substrate-binding domain. Disrupting Gly408–Thr480 did not completely inhibit Hrd1’s ability to bind cytosolic substrate, consistent with the idea that another portion of Hrd1 (or even polyubiquitin chains themselves) could contribute additional affinity.23 However, we propose that the cytosolic loop between transmembrane segments 6 and 7 (Gln219–Leu264) is another substrate interaction site because of its similar character to Gly408–Thr480 and the positioning directly adjacent to the retrotranslocation channel.
While our model is the simplest explanation of our results, more complicated models could also be applied to our findings. The Hrd1 complex is increasingly viewed as a dynamic complex with the precise active stoichiometry (or range of active stoichiometries) yet to be determined. Biochemical and structural data have demonstrated that Hrd1 can exist as monomers or dimers in complex with Der1. Moreover, activation of Hrd1 by ubiquitination may drive complex rearrangement.31 The disordered domains of Hrd1 may be required for stabilizing transient complex formations, recruiting additional components like Ubp1,33 or promoting the active Hrd1 stoichiometry.
Here, using deep mutational scanning combined with in vivo assays and in vitro reconstitution, we have demonstrated that S. cerevisiae Hrd1 requires the disordered cytosolic regions for function. It is important to note that many Hrd1 homologs, including mammalian Hrd1, have disordered cytosolic regions largely devoid of lysine and cysteine that vary in length and position (Figure S7). We propose that these disordered cytosolic regions are broadly required for ERAD in all eukaryotes, but future studies will be needed to test this idea.
Limitations of the study
In the deep mutational scan, we analyzed only single-amino-acid substitutions. It is possible that additional regions of interest could be missed because of a requirement for multiple mutations for the most dramatic phenotypes. In addition, our experiments are limited by the fact that our pipeline is unable to completely account for changes in growth rates of individual strains containing Hrd1 mutants. Therefore, it is possible that regions of interest were missed due to slow growth of Hrd1 mutants. In our in vitro assays, we used reconstitution systems containing only Hrd1. These Hrd1-only assays may not be completely reflective of how Hrd1 operates in complex with other components, although the in vivo and in vitro experiments are consistent with each other. Finally, while we used a Hrd1 proteoliposome CPY* binding assay to support that Gly408–Thr480 can bind to CPY*, we do not have direct binding data (i.e., the Gly408–Thr480 fragment directly binding to a misfolded substrate). In this case, it is important to consider that the Hrd1 cytosolic accessible substrate binding site(s) is available only after Hrd1 autoubiquitination. It is still unclear whether structural rearrangements occur in Hrd1 to expose this site. Thus, studying the Gly408–Thr480 region (or any region of Hrd1) on its own may not be reflective of how the region behaves in the context of full-length Hrd1.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Ryan Baldridge (ryanbald@umich.edu).
Materials availability
All materials and reagents generated in this study are available upon request from the lead contact.
Data and code availability
All raw sequencing data are uploaded to the NCBI Short Read Archive: BioProject:PRJNA951752.
Code used to analyze the deep mutational scanning data are available at the following DOI links: https://doi.org/10.5281/zenodo.10038326; https://doi.org/10.5281/zenodo.10034350. Most recent versions of the code are available at the following links: https://github.com/baldridge-lab/hrd1_dms_2023; https://github.com/jwschroeder3/2023_mut_scan_analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Strains
Yeast strains used in this study were purchased from Horizon Discovery Ltd. and are derivatives of BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) or BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) (Table S11). Yeast were cultured in synthetic dropout media (0.17 % (w/v) yeast nitrogen base(Becton, Dickinson and Company), 0.5 % (w/v) ammonium sulfate (Fisher), ~0.1 % (w/v) dropout powder (Teknova and Sigma), 2 % (w/v) glucose (Sigma)) at 30 °C. Drop-out powder for synthetic complete media are at the following concentration: adenine sulfate (20 mg/L), uracil (20 mg/L), L-tryptophan (20 mg/L), L-histidine (20 mg/L), L-arginine (20 mg/L), L-methionine (20 mg/L), L-tyrosine (30 mg/L), L-leucine (60 mg/L), L-isoleucine (30 mg/L), L-lysine (30 mg/L), L-phenylalanine (50 mg/L), L-glutamic Acid (100 mg/L), L-aspartic Acid (100 mg/L), L-valine (150 mg/L), L-threonine (200 mg/L), and L-serine (400 mg/L). Combinational knockout strains were derived from transforming a PCR-amplified antibiotic targeting cassette with the LiAc/PEG methods or by crossing and sporulation48 and appropriate targeting was verified by PCR. All strains available upon request (Table S11).
METHOD DETAILS
Plasmids
Plasmids were constructed using either standard restriction cloning or HiFi DNA assembly (New England Biolabs) and propagated in DH5α E. coli. For most in vivo experiments, we used custom integrating cassettes targeted to the leu2Δ0, his3Δ1, or ura3Δ0 loci in BY4741/BY4742 strains.49 Centromeric plasmids were used only where specified in figure legends.50 Note Pdr5* constructs used in Figures 2E, 2F, S2D, 3E, 6C, and S6C contained a second mutation A1174V that arose spontaneously during restriction cloning between vectors. All plasmids available upon request (Table S12).
Hrd1 steady-state collection
hrd1D cells expressing Hrd1-3xFlag (or the indicated Hrd1 variants) from the Hrd1 native promoter were grown with shaking at 30°C to mid-log phase between an OD600 of 0.35–1.05. Cells were pelleted, had 0.1 mm glass beads (BioSpec) added, frozen on dry ice, and stored at −80 °C until lysis. Samples were resuspended in lysis buffer (10 mM 3-(N-morpholino)propanesulfonic acid (MOPS), pH 6.8, 1 % sodium dodecyl sulfate (SDS), 8 M urea, 10 mM ethylenediaminetetraacetic acid (EDTA), fresh protease inhibitors (1 mM phenylmethylsulfonyl fluoride (PMSF), 1.5 μM pepstatin A)) at 25 OD600/mL. Samples were vortexed for 2 minutes then diluted with an equal volume of urea sample buffer (125mM trisaminomethane (Tris), pH 6.8, 4 % SDS, 8 M urea, 10 % β-mercaptoethanol). Samples were incubated at 65 °C for 5 minutes before separating on SDS-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to a polyvinylidene difluoride (PVDF) membrane, immunoblotted with anti-DYKDDDK (A00187, Genscript) and mouse IgG HRP-linked whole Ab (NA931, Cytiva), then imaged using (ECL Select, RPN2235 Cytiva) with a ChemiDoc MP (Bio-Rad). For gel quantification, band intensities were measured using ImageJ51 (NIH), normalized to total protein in the sample detected using Bio-Rad Stain Free Dye Imaging Technology (StainFree). Hrd1 variant expression levels were normalized to wild-type Hrd1. All individual Hrd1 protein level values can be found in (Table S9). For statistical analysis, GraphPad Prism (Dotmatics) was used to conduct one-way ANOVA tests, and p values were derived using Dunnett’s multiple comparisons test against Hrd1(WT) or a Welch’s t-test.
Co-immunoprecipitation of Hrd1 complex
Hrd1-3xFlag (or Hrd1 variants) were integrated at the his3 locus, 3xHA-Hrd3 at the leu2 locus, and 3xV5-Usa1 at the ura3 locus in hrd1Δhrd3Δusa1Δ cells. Cells were cultured to mid-log phase, pelleted, resuspended in IP buffer (50 mM 2-[4-(2-Hydroxyethyl)-1-piperazine] ethanesulfonic acid (HEPES), pH 7.4, 150 mM potassium chloride (KCl)) with protease inhibitors (1 mM PMSF, 1.5 μM pepstatin A) and flash frozen in liquid nitrogen to form yeast ‘balls’ prior to cryogenic lysis using freezer/mill (SPEX SamplePrep). Cell powders were thawed on ice and centrifuged at 18,000 x g for 10 min to collect the microsomal fraction. The pellets (P18K) were solubilized in IP buffer supplemented with 1 % decyl maltose neopentyl glycol (DMNG) and protease inhibitors by rotating for 1 hour at 4 °C. Following solubilization, we collected the ‘‘input’’ samples, and 10 OD600 of solubilized proteins (180–200 μg proteins) were diluted 1:5 in IP buffer to a final concentration of 0.2 % DMNG. The solubilized proteins were mixed with 20 μL (40 μL slurry) anti-Flag M2 magnetic beads (Sigma) and rolled for 3 hours at 4 °C. The bound proteins were washed 6–7 times with 1 mL of IP buffer (50 mM HEPES, pH 7.4, 150 mM KCl, 0.2 % DMNG) and eluted with 2x SDS-PAGE sample buffer. The samples were analyzed by SDS-PAGE and immunoblotting with anti-Flag (anti-DYKDDDDK, A00187 Genscript), anti-HA (clone 3F10, Roche), and anti-V5 (A01724, Genscript) antibodies with the inputs loaded at 5 %.
Immunoprecipitation of Der1 occurred as described above with the following changes. In hrd1Δder1Δ cells, Hrd1-3xFlag (or Hrd1-3xFlag variants) were integrated at the his3 locus and Der1-HA was expressed from its endogenous promoter on a centromeric plasmid. Following solubilization, either 10 OD600 or 20 OD600 of solubilized proteins were used for the immunoprecipitation. 40 μL (80 μL slurry) of anti-Flag M2 magnetic beads were used for 20 OD600 of solubilized proteins and input was loaded at 2 %.
Flow cytometry-based degradation assays
hrd1Δ cells expressing ERAD substrates were complemented with Hrd1, or Hrd1 variants. Cells were picked from transformation plates and subcultured in synthetic dropout media in 96 deep-well plates until cells entered log-phase (<1.5 OD600/ml). Cells were pelleted and resuspended in fresh synthetic dropout media supplemented with either cycloheximide (at 50 μg/ml), zaragozic acid (at 10 µg/ml, for Hmg2), or ethanol (0.1 % as a vehicle control) for the indicated times. At end of the time course, cells were pelleted, washed with ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM sodium phosphate buffer, and 1.8 mM KH2PO4 (pH 7.4)), resuspended in PBS containing 1 μM SytoxBlue (viability dye; Invitrogen), and placed at 4 °C during acquisition on either a Ze5 (Everest software; Bio-Rad) or a MACSQuant VYB (MACSQuantify software; Miltenyi Biotec). On the Ze5, the 488 nm laser was used for forward/side scatter to identify single cells, and Sytox Blue fluorescence was used to exclude dead cells from the 405 nm laser with a 460nm/22nm bandpass filter. GFP fluorescence was measured from the 488 nm laser with a 509nm/24nm bandpass filter, and mScarlet-I fluorescence was measured from the 561 nm laser with a 615nm/24nm bandpass filter. On the MacsQuant VYB we used the 561 nm laser for forward/side scatter to identify single cells, and SytoxBlue fluorescence was used to exclude dead cells from the 405 nm laser with a 450nm/50nm bandpass filter set. GFP fluorescence was measured from the 488 nm laser with a 525nm/50nm bandpass filter set, and mScarlet-I fluorescence from the 561 nm laser with a 615nm/20nm bandpass filter set. FlowJo V10.7.1 (FlowJo LLC) was used to analyze FCS files (version 3.1) on FSC/SSC to eliminate debris, gate on single cells, and eliminate dead cells. Median GFP, or mScarlet-I, fluorescence values from at least 10,000 cells were exported and used to quantify Hrd1 variant function. For quantification, we determined the fraction of substrate remaining after the treatment, compared to the vehicle control. The fraction of substrate remaining was normalized to wild-type Hrd1 set to ‘‘1’’, and Hrd1(C399S) set to ‘‘0’’. All individual flow cytometry functional values can be found in (Table S8). For statistical analysis, GraphPad Prism (Dotmatics) was used to conduct one-way ANOVA tests, and p values were derived using Dunnett’s multiple comparisons test against Hrd1(WT).
For saturated chases, single cells were inoculated in synthetic dropout media and grown overnight (~14 hours). Cells were diluted 1 to 50, or 1 to 100, in fresh synthetic dropout media and cultured for ~24 hours to enter a ‘‘saturated chase’’ and processed as described above.
Flow cytometry plots were generated using FlowJo V10.7.1, and all flow cytometry data are from at least 10,000 events passing FSC/SSC and viability gates.
Library generation
Tiling primers mutagenesis was performed as previously described.38 We mutagenized Hrd1 in five regions (amino acids: 1–110, 111–220, 221–330, 331–440, 441–551), to enable 2 × 300bp Illumina sequencing across the regions. A linearized PCR template was generated by digesting a wild-type Hrd1 plasmid with restriction enzyme and purified using a QIAquick PCR purification kit (Qiagen). For each region two separate PCR reactions were set up, one for forward tiling primers and one for reverse tiling primers amplifying the linearized wild-type Hrd1 sequence with a flanking primer adding homology for gap-plasmid repair in yeast (Tables S13 and S14). For library generation, the first round of PCR consisted of 7 cycles of the following program using Q5 High-Fidelity DNA Polymerase (New England Biolabs). Step 1: 98 °C for 2 min; step 2: 98 °C for 30 sec; step 3: 72 °C for 1 sec; step 4: 60 °C for 30 sec, cooling at 0.5 °C/sec; step 5: 72 °C for 30 sec; step 6: 7 cycles of return to step 2; step 7: 72 °C for 1 min.
These PCR products were diluted 1 to 4 with water, and each forward and reverse library was combined as the template for a second round of PCR using the flanking primers. Step 1: 98 °C for 2 min; step 2: 98 °C for 30 sec; step 3: 72 °C for 1 sec; step 4: 60 °C for 30 sec, cooling at 0.5 °C/sec; step 5: 72 °C for 30 sec; step 6: 20 cycles of return to step 2; step 7: 72 °C for 1 min. The amplified libraries were cleaned up by a QIAquick PCR purification kit.
For the vector to be used for recombination, centromeric plasmids containing the native promoter and terminator of Hrd1 had partial replacement of coding sequence corresponding to amino acids 1–110, 111–220, 221–330, 331–440, or 441–551 with an EcoRI restriction site. Plasmids were linearized with EcoRI-HF and purified using QIAquick PCR purification kit. This method produced a range of mutations per region (Table S10).
To generate the mutagenized libraries yeast cells expressing integrated substrates were transformed with a 1:5 molar ratio of linearized vector to PCR fragments product using standard LiAc/PEG transformation using homologous recombination for gap-plasmid repair strategy.39 Post transformation, 0.1 % of the cell mixture was plated on synthetic dropout plates to determine transformation efficiency and the rest was inoculated into synthetic dropout media and cultured for two days to reach saturation (Table S1).
Fluorescence-activated cell sorting (FACS)
Two days after the library generation, the cell libraries were diluted 1:100 in fresh synthetic dropout media and grown for 24 hours, allowing cells to enter into a ‘‘saturated chase’’. Prior to FACS, we collected an aliquot of cells as the ‘‘input’’ library, and stored the cells at −80 °C. For FACS, cells were kept at room temperature, pelleted, washed in PBS, resuspended in PBS containing 1 μM SytoxBlue, and sorted using a Bigfoot Spectral Cell Sorter (Thermo Scientific (formally Propel Labs)). Forward and side measurements were analyzed on the 488 nm laser to gate for single cells and SytoxBlue fluorescence was used to exclude dead cells from the 445 nm laser with a 465nm/22nm bandpass filter. GFP fluorescence was measured from the 488 nm laser with a 507nm/19nm bandpass filter set and mScarlet-I fluorescence was measured from the 561 nm laser with a 605nm/15nm bandpass filter set. Cells in the wild-type-like sort bin (WT) and ERAD-L defective sort bin (L) were collected in tubes containing 2x synthetic dropout media to aid cell recovery. Over 3.3 million cells passing FSC/SSC and viability gating were sorted over (see Table S1 for the number of cells sorted into each bin).
Sorted cells were grown in synthetic dropout media to saturation and an aliquot of cells was collected and stored at −80 °C prior to genomic extraction. This frozen aliquot of cells represents the sorted population used for NGS library prep and analysis (wild-type-like or ERAD-L defective populations). The remaining cells were subjected to a saturated chase to confirm phenotype the next day. Some ERAD-L defective populations were subjected to a second round of sorting to enrich for the lumenal defective phenotype (Table S1).
Illumina sequencing and data analysis
DNA was extracted from 30–40 million (3–4 OD600 equivalents) cells using a zymolyase method.52 Briefly, cells were resuspended in 100 μL of buffer Z (50 mM Tris, pH 7.4 and 1–3 mg/ml of zymolyase 100T (Amsbio)) and shaken at 37 °C for 1–2 hours. The solution was heated at 95 °C for 6 minutes before centrifugation to clear insoluble material.
NGS libraries were prepared for amplicon sequencing on an Illumina platform. Libraries were generated from two PCR reactions both using Q5 High-Fidelity DNA Polymerase (NEB). Primers flanking mutated regions amplified, 25 cycles, the extracted DNA from 3–4 million cells (10 μL of extraction solution) while adding on partial R1 and R2 read adaptors (Table S14). The second round of PCR (8–9 cycles) added i5 and i7 indexes using the Index Kit 2 for Illumina (Apexbio Technology LLC). The libraries were cleaned up using AMPure XP beads, normalized, pooled, and submitted for sequencing on a MiSeq 2×300 platform through Genewiz (now Azenta Life Sciences).
For analysis, de-multiplexed FASTQ files were provided from Azenta (reads available at NCBI BioProject PRJNA951752). These files were trimmed with cutadapt (using the following arguments: -a CTGTCTCTTATACACATCT -A CTGTCTCTTATACACATCT -u 8 -U 8 -q 30), 3’ overlapping pair-end reads were merged with fastp (using the default settings), and aligned to a wild-type Hrd1 sequence with bowtie2 (using the ‘‘very-sensitive’’ setting), and resulting SAM files was sorted on name while converting to a BAM file using samtools.53–56 The sorted BAM files were used as input for python scripts that translated and counted Hrd1 mutations for all sequences without insertions or deletions (indels) identified during Bowtie alignment. All code and analysis pipelines are available (https://doi.org/10.5281/zenodo.10038326 ; current version of code: https://github.com/baldridge-lab/hrd1_dms_2023).
To test variant enrichment in the ERAD-L defective Hrd1 sort bin, we only analyzed reads with single amino acid substitutions. We performed rate ratio tests using the test_poisson_2indep function from version 0.13.5 of the python module statsmodels.stats.rates. Arguments to the test_poisson_2indep function were as follows: ‘‘count1’’: the number of reads in the ERAD-L defective phenotype with amino acid ‘a’ at Hrd1 position ‘p’, ‘‘exposure1’’: the total number of reads arising from the ERAD-L defective phenotype, ‘‘count2’’: the number of reads in the input mutant library with amino acid ‘a’ at Hrd1 position ‘p’, ‘‘exposure2’’: the total number of reads in the mutant library, ‘‘alternative’’: ‘‘greater’’. We used stratified jackknife sampling to incorporate unpaired replicates, performing a separate set of hypothesis tests for each jackknife replicate to arrive at our final estimates of p-values and variant enrichment in the ERAD-L defective bin. By this method, the final p-value estimate of a given amino acid substitution at the position was the weighted mean of the jackknife replicate p-values for the given substitution and position. We used the multipletests function in version 0.13.5 of the python module statsmodels.stats.multitest to correct for multiple hypothesis testing using the method of Benjamini and Hochberg.57 Variant enrichment (or depletion) in the wild-type-like bin was analyzed similarly as the ERAD-L defective bin, but the ‘‘alternative’’ argument was set to ‘‘two-sided’’. All variant count information for input libraries, the wild-type-like bin, and the ERAD-L defective phenotype bin, as well as code used for this analysis, are available (https://doi.org/10.5281/zenodo.10034350 ; current version of code: https://github.com/jwschroeder3/2023_mut_scan_analysis).
To identify residues and regions of interest in the ERAD-L defective population, we used a 1 % false discovery rate coupled with an enrichment value greater than 30. When we identified residues near the transmembrane segment 6–7 loop, we only looked for residues with a greater than 50 enrichment value.
Screening optimization mutant isolation
Hrd1(F46R) was isolated with a similar method as the DMS screen above with the following differences. Hrd1 was mutagenized using tiling primers across the first 384 amino acids and transformed into hrd1Δ cells expressing substrates from centromeric plasmids. Cell sorting occurred using a MoFlo Astrios (Beckman). Forward and side measurements were analyzed on the 488 nm laser to gate for single cells. GFP fluorescence was measured from the 488 nm laser with a 513nm/26nm bandpass filter set and mScarlet-I fluorescence was measured from the 561 nm laser with a 614nm/20nm bandpass filter set. Sorted cells were plated and individual colonies were confirmed for ERAD-L defective phenotype before Hrd1 plasmid isolation (Zymoprep Yeast Plasmid Miniprep II) and Sanger-sequencing.
Hrd1(T416P,W481L,I505T) was isolated as follows. Hrd1 was mutagenized across the entire coding sequence and cloned into a centromeric plasmid using HiFi Assembly (New England BioLabs). The centromeric Hrd1 library was transformed into hrd1Δ cells expressing substrates from centromeric plasmids and plated and grown at 30 °C. Once yeast colonies appeared (~2 days), plates were fluorescently imaged using a ChemiDoc MP (Bio-Rad). Individual colonies appearing ERAD-L defective were selected and confirmed using flow cytometry before Hrd1 plasmid isolation (Zymoprep Yeast Plasmid Miniprep II) and Sanger-sequencing.
Strains and plasmids for protein expression
Uba1 was purified from the InvSc1 strain (Invitrogen). Both CPY*-TM and soluble CPY* were purified from a hrd3Δalg3Δ strain derived from BY4741 (yRB129: MATa his3Δ1 leu2Δ0 ura3Δ0 met15Δ0 hrd3Δ::kanRMX4 alg3Δ::hphNT1). Hrd1 was expressed and purified from a hrd1Δubc7Δ diploid (yBGP55B: MATa/α his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 LYS2/lys2Δ0 met15Δ0/MET15 ura3Δ0/ura3Δ0 hrd1::HphNT1/hrd1::HphNT1 ubc7::KanRMX4/ubc7:KanRMX4). Bacterial expression strains were BL21-CodonPlus (DE3) RIPL (Agilent). (See Table S11)
Yeast overexpression for purification were from 2μ plasmids of the pRS42X series driven by the inducible Gal1 promoter.58 Bacterial expression of Cue1 and Ubc7 were expressed from two different fusion proteins. As previously described,19 where Cue1(24–203) (pAS153) and Ubc7 (pAS159) were expressed with an N-terminal His6 tag in a pET28B vector (Novagen). Cue1 and Ubc7 were also expressed from a K27-His14-SUMO vector allowing for the complete removal of affinity and solubility tags.59 (See Table S12)
Yeast protein expression and purification
Yeast strains with plasmids for the expression of proteins of interest were grown at 30°C with shaking in synthetic dropout media. 1:150 dilutions of actively growing cultures were used to inoculate eight 1L cultures in 2.8L Fernbach flasks. The cultures were grown for 24 hours at 30 °C with shaking. To induce expression, 4x yeast extract peptone (YP) broth containing 8 % (w/v) galactose was added to the culture to a final concentration of 1x YP and 2 % galactose. The temperature was shifted to 25 °C, and the culture was grown for 15 to 18 hours. Cells were pelleted, washed with water or 2 mM dithiothreitol (DTT) (to weaken the cell wall), flash-frozen with liquid nitrogen, and stored at −80 °C.
All purification steps were conducted at 4 °C unless otherwise specified. Approximately 150 to 200 grams of yeast cells were resuspended in buffer Y1 (50 mM HEPES, pH 7.4, 300 mM KCl, and 0.5 mM tris(2-carboxyethyl)phosphine (TCEP)) with freshly supplemented protease inhibitors (1 mM PMSF, 1.5 μM pepstatin A, 0.2 mM diisopropyl fluorophosphate (DFP)). 0.5 mm glass beads were added to 1/3 to ½ the volume of the cell suspension and were subjected to bead beating (BioSpec), either split across two bead beater rounds or unbroken cells from the first bead beating round were subjected to a second round of bead beating. Cells were lysed by beat beating for 20 minutes of 20 seconds on/40 seconds off. The lysate was cleared with two low-speed spins (2,000 x g for 10 minutes), and the supernatant was centrifuged at 42,000 rpm for 33 minutes using a 45Ti rotor (Beckham, RCF average 138,000 x g). For Hrd1 purification, the membrane fraction was washed twice by resuspending the membrane fraction in 180 mL of buffer Y1 containing protease inhibitors (1 mM PMSF, 1.5 μM Pepstatin A, 0.1 mM DFP) and pelleting using 45Ti ultracentrifugation.
For Hrd1 purification, the membrane fraction was resuspended in buffer 180 mL of Y1 with freshly supplemented protease inhibitors (1 mM PMSF, 1.5 mM pepstatin A, 0.1 mM DFP) and 1 % (w/v) DMNG. The membrane was rolled for 1 hour, and insoluble material was removed by ultracentrifugation in a 45Ti (42,000 rpm for 33 minutes). The supernatant was incubated with affinity resin (1.5 μL of Streptavidin Agarose (Pierce)) and concurrently sortase-labeled overnight (~12 to 15 hours), using the sortase A (P94R/D160N/D165A/K190E/K196T) pentamutant60 and Sulfo-Cy5-Maleimide (Lumiprobe) coupled to a Gly-Gly-Gly-Cys peptide (Genscript). The next morning, the streptavidin agarose was washed seven times with 50 mL of buffer Y1 containing 1 % (w/v) DMNG, then 1 mM DMNG, and the remaining washes with 120 μM DMNG. The fourth wash was conducted at room temperature with 0.5 mM adenosine triphosphate (ATP) supplemented. Hrd1 was eluted off streptavidin resin in Buffer Y1 supplemented with 120 μM DMNG and 2 mM biotin. Elutions were pooled based on yield and purity, as assessed by SDS-PAGE and Coomassie blue staining. Pooled elutions were concentrated and further purified using size exclusion chromatography (Superose 6 Increase 10/300 GL (Cytiva Life Sciences)) with 25 (or 50) mM HEPES, pH 7.4, 300 mM KCl, 0.25 (or 0.5 mM) TCEP, and 120 μM DMNG. Peak fractions were pooled, concentrated, and flash-frozen in liquid nitrogen.
For soluble CPY* with C-terminal sortase recognition tag, 3C cleavage site, and His14 affinity tag, we collected membranes as described above. The membrane fraction was resuspended under denaturing conditions in buffer Y2 (50 mM HEPES, pH 7.4, 300 mM KCl, 30 mM imidazole, 0.5 mM TCEP, and 6 M urea) with fresh protease inhibitors and rolled for 1 hour. The urea insoluble material was cleared by 45Ti ultracentrifugation (42,000 rpm, 33 minutes). The urea-soluble supernatant was rolled with 10 mL of HisPur Ni-NTA Resin (Thermo Scientific) for 1.5 hours. The nickel resin was washed ten times with 50 mL of buffer Y3 (50 mM HEPES, pH 7.4, 300 mM KCl, 0.5 mM TCEP, 30 mM imidazole) containing the following concentration of urea per wash: wash 1, 6 M urea; wash 2–4, 3 M urea; wash 5–8, 1 M urea; and wash 9–10, no urea. The CPY* material was eluted with 400 mM imidazole under denaturing conditions in buffer Y2. The denatured CPY* was refolded over a 20-hour dialysis. First, the 6 M urea was diluted linearly to 3 M urea using buffer Y1 over a ~6 hour period. Next, 3 M urea was diluted linearly to 0.5 M urea over 10 hours. Finally, the 0.5 M urea solution was dialyzed twice against buffer Y1 bringing the final urea concentration to ~1 mM. The CPY* was labeled overnight (~14 hours) with FAM-maleimide (Lumiprobe) coupled to Gly-Gly-Gly-Cys peptide using Sortase A (P94R/D160N/D165A/K190E/K196T) that replaces the C-terminal 3C cleavage site and His14 affinity tag if labeled. The overnight sortase labeling caused about 1/3 of the CPY* to precipitate out of solution. Insoluble CPY*-FAM was collected by low-speed centrifugation and re-solubilized in buffer Y2 for 1 hour. The solubilized denatured CPY* was depleted of His14 tag or full-length His14 containing species by three successive passes over 2 mL of HisPur Ni-NTA Resin (Thermo Scientific). The urea was dialyzed out over 20 hours bringing the final urea concentration to ~1 mM in buffer Y1, as described above. The protein was concentrated, aliquoted, and flash-frozen.
CPY*-TM was purified as described previously.22 Briefly, the membrane fraction was resuspended in 200 mL of buffer R1 (50 mM HEPES, pH 7.4, 300 mM KCl, 1 mM MgCl2, 1 mM TCEP, 6 M urea, 1 % tridecylphosphocholine (Fos-choline 13, Fos13), 30 mM imidazole) for 60 minutes. Insoluble material was removed by 45Ti ultracentrifugation (42,000 rpm, 30 minutes). Soluble material was incubated with His60 Superflow resin for 60 minutes. The resin was washed with 10 column volumes (CV) of buffer R2 (25 mM HEPES, pH 7.4, 300 mM KCl, 1 mM MgCl2, 0.5 mM TCEP, 2 mM Fos13, 30 mM imidazole) supplemented with 6 M urea. The column was washed with decreasing amounts of urea: 10 CV of R2(5 M Urea), 10 CV of R2(3 M Urea), 10 CV of R2(1 M Urea), and 20 CV of R2(No Urea). The protein was eluted with 400 mM imidazole in buffer R2, labeled with Dylight800 via Sortase A, and purified by gel filtration.
For Uba1 with N-terminal His14 affinity and TEV cleavage site, cell lysis occurred as described above with the following modifications. First, cells were resuspended in buffer Y4 (50 mM HEPES, pH 7.4, 300 mM KCl, 30 mM imidazole) with fresh protease inhibitors (1 mM PMSF, 1.5 μM pepstatin A, 1x protease inhibitor cocktail (PIC: AEBSF (pefabloc) 100 μM, aprotinin 0.6 μM, E-64 1 μM, leupeptin 10 μM, pepstatin A 5 μM, bestatin 5 μM)). Second, the supernatant, not membrane fraction, was taken following ultracentrifugation. The supernatant was rolled with 7.5 mL of HisPur Ni-NTA Resin (Thermo Scientific). The resin was washed with 25 times the bed volume with buffer Y4. Uba1 was eluted with 400 mM imidazole in buffer Y4. 5 mM DTT was immediately added to each elution. Elutions were pooled based on yield and purity, as assessed by SDS-PAGE and Coomassie blue staining. The pooled elutions were supplemented with 5 % glycerol and the His14 tag was removed using TEV protease (1 to 100 molar ratio) overnight (~14 hours). The next morning, cleaved Uba1 was purified by ion-exchange chromatography using a HiTrap MonoQ column (Cytiva). We used a linear gradient of 50 mM KCl to 1000 mM KCl in 50 mM HEPES, pH 7.4; the Uba1 eluted around 350 mM KCl. The peak fractions were pooled, concentrated, and separated by size-exclusion chromatography (HiLoad 16/600 Superdex 200 pg (GE Healthcare, now Cytiva) in 20 mM HEPES, pH 7.4, 150 mM KCl, 300 mM sorbitol, and 0.5 mM TCEP. Peak fractions were pooled, concentrated, aliquoted, and flash-frozen in liquid nitrogen.
Bacterial protein expression and purification
Ubc7 and Cue1(Gln24-Thr203) were expressed and purified in two different ways. The first method was as described previously.1
K27 His14-SUMO Ubc7 was expressed in E. coli BL21-CodonPlus (DE3) RIPL cells (Aligent). Cells were inoculated into starter cultures containing kanamycin (50 μg/ml) and chloramphenicol (25 μg/ml) at 37 °C with shaking and grown overnight (~14 hours). The cultures were diluted 1:150 into 2.8L Fernback flasks with 1 L of terrific broth (TB) containing kanamycin and chloramphenicol at 37 °C. When the cells reached 0.8–1.0 OD600/ml, protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG). The cultures were grown at 18 °C and 220 rpm for 17.5 hours. Cells were collected, washed in water, frozen on dry ice, and stored at −80 °C. K27 His14-SUMO Cue1(Gln24-Thr203) was expressed as above with the following adjustments. Chloramphenicol was at 35 μg/ml, induction occurred at 0.55 OD600/ml, and cells were grown at 18 °C for 18.5 hours.
For K27 His14-SUMO Ubc7, the cell pellet (97 grams) was thawed and resuspended in 180 mL of buffer B1 (50 mM Tris, pH 8, 500 mM NaCl, 0.25 mM TCEP, and 30 mM imidazole) with fresh protease inhibitors (1 mM PMSF, 1.5 μM pepstatin A, 1x PIC). Cells were lysed via sonication, and the cell lysate was subjected to ultracentrifugation using a 45Ti (42,000 rpm, 33 minutes). The supernatant was incubated with 7.5 mL of HisPur Ni-NTA resin for 2.5 hours and washed six times with buffer B1. The resin was washed with an additional 75 mL of buffer B2 (25 mM HEPES pH 7.4, 300 mM KCl, and 0.25 mM TCEP) supplemented with 30 mM imidazole. Ubc7 was eluted in 7.5 mL batches with 400 mM imidazole in buffer B2. An additional 2 mM of TCEP was added to each elution. The peak fractions were pooled, and Ulp1 (SUMO-protease) was added to 4 μM and incubated overnight to cleave the His14-SUMO tag, while dialyzing against buffer B2 (to remove imidazole). Following overnight incubation, precipitated Ubc7 was filtered from solution. The His14-SUMO tag was removed from the solution, by passing the solution over 5 mL of HisPur Ni-NTA resin three successive times. Ubc7 was further purified by ion exchange chromatography using a HiTrap monoQ HP column (Cytiva) with a salt gradient from 50 mM KCl to 530 mM KCl in 25 mM HEPES pH 7.4 containing 0.25 mM TCEP; Ubc7 eluted around 235 mM of KCl. Peak fractions were pooled, concentrated, aliquoted, and flash-frozen in liquid nitrogen.
The K27 His14-SUMO Cue1(Gln24-Thr203) cell pellet (12 grams) was resuspended in 180 mL of buffer B3 (50 mM HEPES, pH 7.4, 300 mM KCl, 6 M urea, 10 mM EDTA) with fresh protease inhibitors (1 mM PMSF, 0.1 mM DFP, 1.5 μM pepstatin A, 0.5 μM bestatin, 1x PIC). The cells were lysed using sonication, and the insoluble material was cleared via ultracentrifugation in a 45Ti (42,000 rpm 33 minutes). The supernatant was collected, supplemented with 5 mM imidazole, and incubated with 4 mL cOmplete His-Tag Purification Resin (Roche) for 1 hour. The resin was washed with 75x bed volume with buffer B3 supplemented with 5 mL imidazole and eluted in batches of 6 mL of B3 supplemented with 400 mM imidazole. 2 mM TCEP was immediately added to the elutions. Cue1 was refolded by removal of the urea using dialysis. 6 M urea was dialyzed linearly to 1.5 M urea over a period of 3 hours using buffer B4 (25 mM HEPES pH 7.4, 300 mM KCl, 0.25 mM TCEP). 1.5 M urea was dialyzed to ~142 mM urea over 14.5 hours. The solution was dialyzed against buffer B4 for 1 hour bringing the final urea to ~4.5 mM. We observed no precipitation of His14-SUMO Cue1. Ulp1 was added at 1:75 (mass:mass) and incubated for 1 hour. To remove the His14-SUMO tag, the solution passed successively over 1.5 mL of HisPur Ni-NTA resin three times. Cue1 was further purified by ion exchange chromatography using a HiTrap monoQ HP column (Cytiva) with a salt gradient from 20 mM to 520 mM in 25 mM HEPES, pH 7.4 with 0.25 mM TCEP; Cue1 eluted around 150 mM KCl. Peak elutions were pooled, concentrated, aliquoted, and flash-frozen in liquid nitrogen.
In vitro ubiquitination
In vitro ubiquitination to assess Hrd1 autoubiquitination activity occurred at 30 °C in 25 mM HEPES, pH 7.4, 150 mM KCl, 1 mM MgCl2, 0.1 mM TCEP, and 120 μM DMNG. The ubiquitination machinery were added at the following concentrations: 0.2 μM Uba1, 2 μM Ubc7, 2 μM Cue1(Gln24-Thr203), 0.2 μM Hrd1, 50 μM ubiquitin (yeast recombinant from bio-techne (R&D Systems)), and 0.6 μM bovine serum albumin (BSA; A3311 from Sigma). 2 mM ATP was added to start the reaction. The samples were analyzed using non-reducing SDS-PAGE and fluorescence scanning (ChemiDoc MP, Bio-Rad).
To assess Hrd1 residues modified by ubiquitin, in vitro ubiquitination of Hrd1(WT) occurred as described above for 60 minutes at 30 °C in presence or absence of ATP. After 60 minutes, the samples were split and treated with either water, Usp2 (final concentration: 10 μM) , BME (final concentration: 5 %) , or sodium hydroxide (NaOH) (final concentration: 200 mM) for 30 minutes at 37 °C. After 30 minutes the NaOH treated sample was neutralized with an equivalent amount of hydrochloric acid (HCl). The samples were analyzed using non-reducing SDS-PAGE and fluorescence scanning (ChemiDoc MP, Bio-Rad).
Ubiquitination site identification
In vitro ubiquitination for ubiquitin site identification occurred at 30°C in 25 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl2, 0.1 mM TCEP, and 120 μM DMNG. The ubiquitination machinery was added at the following concentrations: 0.2 μM Uba1, 2 μM Ubc7, 2 μM Cue1(Gln24-Thr203), 0.5 μM Hrd1, 125 μM ubiquitin (yeast recombinant from bio-techne (R&D Systems)), and 0.6 μM BSA. 2 mM ATP was added to start the reaction. The samples were analyzed using non-reducing SDS-PAGE and fluorescence scanning (ChemiDoc MP, Bio-Rad). The gel section corresponding to poly-ubiquitinated Hrd1 was excised and sent for ubiquitin site identification at the Taplin Mass Spectrometry Facility (Harvard Medical School).
Excised gel bands were reduced with 1 mM DTT for 30 minutes at 60 °C, alkylated with 5 mM iodoacetamide for 15 minutes at room temperature, dehydrated with acetonitrile, and dried in a speed-vac. Gels pieces were rehydrated in 50 mM ammonium bicarbonate, and peptides were generated by in-gel digestion using trypsin (sequencing-grade (Promega)) at 37 °C overnight. Peptides were extracted in 50 % acetonitrile and 1 % formic acid, the peptide solution was dried in a speed-vac for 1 hour and stored at 4 °C until analysis.
On day of analysis, samples were reconstituted in 5–10 μL of HPLC solvent A (2.5 % acetonitrile, 0.1 % formic acid). Samples were separated on a nano-scale reverse-phase HPLC capillary column (100 mm inner diameter x ~25 cm length) packed with 2.6 mm C18 spherical silica beads. Elution occurred over a gradient of increasing HPLC solvent B (97.5 % acetonitrile, 0.1 % formic acid). As peptides eluted off column, they were subjected to electrospray ionization and entered an LTQ Orbitrap Velos Pro ion-trap mass spectrometer (ThermoFischer). Peptides were detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide. Peptide sequences are matched to protein sequences by Sequest (ThermoFinnigan). The modification of 114.0429 mass units to lysine was included in the database searches to determine ubiquitin-modified peptides. Data was filtered with a 1 % false discovery rate.
In vitro retrotranslocation
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) from Avanti Polar Lipids was solubilized in chloroform and aliquoted. The solvent was removed under a nitrogen stream, followed by placing the aliquot into a lyophilizer overnight (~14 hours). DOPC was resuspended at 6.2 mM in buffer R (25 mM HEPES, pH 7.4, 150 mM KCl, 1 mM MgCl2, 100 μM TCEP) by vortexing. Lipids were completely solubilized in 1.2 % (w/v; 18.1 mM) Triton X-100 (Anatrace) for 20 min at room temperature. CPY*-TM substrate labeled with Dylight800 was added to 2.5 μM bringing DOPC to 5.1 mM, and the mixture was incubated at 4 °C with gentle shaking for 60 min. The detergent was removed by three 2-hour incubations with ~33 mg of Bio-Beads SM2 (Bio-Rad) followed by overnight incubation with ~50 mg of Bio-Beads SM2 with all incubations occurring at 4 °C.
With sealed liposomes, color switching internal-CPY* occurred by adding 10 mM CaCl2, 50 μM sulfo-cyanine3-maleimide (Cy3) (Lumiprobe) coupled to a Gly-Gly-Gly-Cys peptide, and 2 mM Sortase A for 1 hour at 4 °C. The proteoliposomes were mixed 1:1 (vol:vol) with 80 % glycerol in buffer R. The proteoliposomes were overlaid with 30 % glycerol, 15 % glycerol, 5 % glycerol, and 0 % glycerol (all prepared in buffer R). The step gradient was centrifuged for 3 hrs at 50,000 rpm in a TLS-55 rotor (RCFavg 166,180). Five fractions were collected, starting from the top of the gradient. Proteoliposomes formed a sharp band between the 15 % and 5 % glycerol layers. Sortase A and GGGC-fluorophore stayed in the 40 % glycerol layer.
To incorporate Hrd1 into the proteoliposomes, the floated samples were partially solubilized by the addition of 0.1 % (w/v) DMNG on ice for 30 min. Hrd1 was added to 2 μM, and the mixture was incubated on ice for 60 min. DMNG was removed by five successive passes over detergent removal resin (Pierce) at a ratio of 2.75x resin to proteoliposome material. The proteoliposomes were then mixed 1:1 with 80 % glycerol in buffer R and subjected to a glycerol step gradient centrifugation consisting of 30 %,15 %, and 0 % glycerol layers (prepared in buffer R).
The proteoliposomes containing CPY*-TM and Hrd1 floated to an interface between 15 % and 0 % glycerol layers. The liposomes were diluted 1 to 2 (or 1 to 4) in buffer R. The liposome solution was brought to room temp for 5 minutes, and then liposomes were added to an equal volume of ubiquitination machinery at the following concentrations: 0.4 μM Uba1, 4 μM Ubc7, 4 μM Cue1(Gln24-Thr203), 50 μM ubiquitin, and 0.6 μM BSA. The retrotranslocation assay was initiated with the addition of 2 mM ATP, and reactions were incubated at 30 °C. The samples were analyzed using non-reducing SDS-PAGE and fluorescence scanning using a ChemiDoc MP (Bio-Rad). Quantification of band intensity occurred using ImageJ,51 and the fraction unmodified was calculated by taking band intensity at indicated time point over band intensity at the zero-minute time point.
Hrd1 CPY* binding
DOPC, as prepared for retrotranslocation assays, was resuspended in buffer C1 (25 mM HEPES, pH 7.4, 150 mM KCl, 5 mM MgCl, 0.1 mM TCEP). Liposomes were extruded through a 100 nm polycarbonate membrane, partially solubilized with 0.1 % DMNG for 30 minutes on ice, and incubated with 2 μM Hrd1 for 1 hour on ice. DMNG was removed by four successive passes over detergent removal resin (Pierce) at a ratio of 3.35x resin to proteoliposome material. The proteoliposomes were then mixed 1:1 with 80 % glycerol prepared in buffer C1 and subjected to glycerol step gradient centrifugation consisting of 30 %, 15 %, 5 %, and 0 % glycerol layers (prepared in buffer C1) in a TLS-55 (50,000 rpm for 3 hours). The liposomes with incorporated Hrd1 floated to the interface between the 15 % and 5 % glycerol layers.
Pierce magnetic streptavidin beads were prewashed with 112.5 μL of buffer C1 supplemented with 2 mg/mL BSA and 2 mM DOPC, followed by 225 μL of buffer C1 supplemented with 2 mg/ml of BSA per 7.5 μL of beads. 20 μL of 1 μM Hrd1 proteoliposomes were bound to 7.5 μL of prewashed beads at 30 °C for 30 min. Background binding controls lacking proteoliposomes were incubated with buffer C2 (25 mM HEPES, pH 7.4, 150 mM potassium chloride, 5 mM magnesium chloride, 0.1 mM TCEP, 0.6 μM BSA). Unbound Hrd1 proteoliposomes were pipetted off, and immobilized Hrd1 proteoliposomes were resuspended in ubiquitin mix consisting of 0.2 μM Uba1, 2 μM Ubc7, 2 μM Cue1(Gln24-Thr203), and 50 μM ubiquitin in buffer C2. Ubiquitination was initiated by the addition of 2 mM ATP and incubated at 30 °C for 1 hr. The beads were subsequently washed with 225 μL of buffer C2 to remove the ubiquitination mix. The substrate, 20 μL of 0.1 μM CPY*-FAM, was incubated with immobilized Hrd1 proteoliposomes at 30 °C for 1 hr. The unbound CPY*-FAM was collected, and the beads were washed with 225 μL of buffer C2. The bound material was eluted with 2 mM biotin in 50 mM MOPS for 5 minutes at 30 °C. The bound samples were analyzed by non-reducing SDS-PAGE and fluorescence scanning with a ChemiDoc MP (Bio-Rad). Substrate binding was quantified from three independent experiments using ImageJ.51 To determine the fraction bound, band intensity was normalized to CPY*-FAM bound to Hrd1(WT) with ATP condition.
QUANTIFICATION AND STATISTICAL ANALYSIS
Details of statistical, software, and quantification methods used were described within the method details of the experiment. For the deep mutational scanning screen, the number of replicates per Hrd1 subregion can be found in Table S1. For flow cytometry-based degradation assays displayed as a heatmap, Hrd1 function was represented as a mean. All replicate values can be found in Table S8. For Hrd1 steady-state levels, data is represented as mean +/− standard error of the mean (SEM). All replicate values can be found in Table S9. Ubiquitination site identification using mass spectrometry was a single replicate. All other experimental data (non-heatmap flow cytometry plots, co-immunoprecipitation assays, retrotranslocation assays, and Figure S5C) were representative of at least three replicates.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| THE DYKDDDDK Tag Antibody, mAb, Mouse | GenScript | Cat#A00187; RRID:AB_1720813 |
| Anti-HA High Affinity; Rat monoclonal antibody (clone 3F10) | Roche | Cat#11867423001; RRID:AB_390918 |
| THE V5 Tag Antibody, mAb, Mouse | GenScript | Cat#A01724; RRID:AB_2622216 |
| Goat Anti-Rat IgG, Whole Ab ECL Antibody, HRP Conjugated | Cytiva | Cat#NA935; RRID:AB_772207 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 800 | Thermo Fisher Scientific | Cat#A32730; RRID:AB_2633279 |
| Sheep Anti-Mouse IgG - Horseradish Peroxidase | Cytiva | Cat#NA931; RRID:AB_772210 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| BL21-CodonPlus (DE3)-RIPL Competent Cells | Agilent | Cat#230280 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| ECL Select Western Blotting Detection Reagent | Cytiva | Cat#RPN2235 |
| Immun-Blot Low Fluorescence PVDF Membrane, Roll, | Bio-Rad | Cat#1620264 |
| 4-20% Criterion TGX Stain-Free Protein Gel, 26 well, 15 μl | Bio-Rad | Cat#5678095 |
| Decyl Maltose Neopentyl Glycol; DMNG; 2,2-dioctylpropane-1,3-bis-β-D-maltopyranoside | Anatrace | Cat#NG322 |
| Fos-Choline-13, Anagrade | Anatrace | Cat#F310 |
| Anti-FLAG M2 Magnetic Beads | Millipore Sigma | Cat#M8823 |
| Zaragozic Acid A | Cayman Chemical | Cat#17452 |
| Zymolyase 100T (Arthrobacter luteus) | Amsbio | Cat#120493 |
| Q5 High-Fidelity DNA Polymerase | New England BioLabs | Cat#M0491S |
| Q5 Hot Start High-Fidelity DNA Polymerase | New England BioLabs | Cat#M0493S |
| Index Kit 2 for Illumina | APExBIO | Cat#K1059 (discontinued) |
| AMPure XP SPRI Reagent | Beckman Coulter | Cat#A63881 |
| Bovine Serum Albumin | Sigma-Aldrich | Cat#A3311 |
| Recombinant Yeast Ubiquitin Protein, CF Bio-Techne Corporation Cat#U-100sc (discontinued) | ||
| 1,2-dioleoyl-sn-glycero-3-phosphocholine ; 18:1 (Δ9-Cis) PC (DOPC) | Avanti | Cat#850375P |
| α-[4-(1,1,3,3-Tetramethylbutyl)phenyl]-ω-hydroxy-poly(oxy-1,2-ethanediyl); TRITON X-100 | Anatrace | Cat#T1001 |
| Pierce High Capacity Streptavidin Agarose | Thermo Fisher Scientific | Cat#20361 |
| Pierce Streptavidin Magnetic Beads | Thermo Fisher Scientific | Cat#88817 |
| Bio-Beads SM-2 Adsorbents | Bio-Rad | Cat#1523920 |
| Fisher BioReagents Yeast Extract (Granulated) | Thermo Fisher Scientific | Cat#BP9727 |
| Peptone, Granulated | Research Products International | Cat#P20250 |
| Phenylmethanesulfonyl fluoride; PMSF Sigma-Aldrich Cat#78830 | ||
| Pepstatin A | AdipoGen Life Sciences or Sigma-Aldrich | Cat#AG-CP3-7001 or Cat#P4265 |
| AEBSF (Pefabloc) | Supelco | Cat#76307 |
| Aprotinin from bovine lung | Sigma-Aldrich | Cat#A1153 |
| E-64 | Sigma-Aldrich | Cat#E3132 |
| Leupeptin | Sigma-Aldrich | Cat#L2884 |
| Bestatin hydrochloride | Sigma-Aldrich | Cat#B8385 |
| Cycloheximide | Sigma-Aldrich | Cat#239763 |
| SYTOX Blue Nucleic Acid Stain - 5 mM Solution in DMSO | Invitrogen | Cat#S11348 |
| Invitrogen UltraPure Salmon Sperm DNA Solution | Invitrogen | Cat#15632011 |
| D(+)-Galactose, 99+%, Thermo Scientific Chemicals | Thermo Fisher Scientific | Cat#15061 |
| Gly-Gly-Gly-Cys (GGGC) Peptide | Genscript | Custom Order |
| DyLight 800 Maleimide | Thermo Fisher Scientific | Cat#46621 |
| sulfo-Cyanine5 maleimide | Lumiprobe | Cat#63380 |
| sulfo-Cyanine3 maleimide | Lumiprobe | Cat#61380 |
| FAM maleimide, 6-isomer | Lumiprobe | Cat#44180 |
| HisPur Ni-NTA Resin | Thermo Fisher Scientific | Cat#88222 |
| cOmplete His-Tag Purification Resin | Roche | Cat#5893682001 |
| Superose 6 Increase 10/300 GL | Cytiva | Cat#29091596 |
| Superdex 200 Increase 10/300 GL | Cytiva | Cat#28990944 |
| HiLoad 16/600 Superdex 200 pg | Cytiva | Cat#28989335 |
| HiTrap Q HP | Cytiva | Cat#17115401 |
| Extruder Set With Holder/Heating | Avanti | Cat#610000 |
| 10mm Filter Supports (100/pk) | Avanti | Cat#610014 |
| Polycarbonate Membranes 0.1μm 19mm (100/pk) | Avanti | Cat#610005 |
| His60 Ni Superflow Resin | Takara | Cat#635660 |
| BD DIFCO Yeast Nitrogen Base without Amino Acids and Ammonium Sulfate | Becton, Dickinson and Company | Cat#233520 |
| TEV, purified in house | This paper | Addgene plasmid # 8827 ; http://n2t.net/addgene:8827 ; RRID:Addgene_8827 |
| Sortase A, purified in house | This paper | Addgene plasmid # 75144 ; http://n2t.net/addgene:75144 ; RRID:Addgene_75144 |
| BeadBeater | BioSpec | Cat#1107900-105 |
|
| ||
| Deposited data | ||
|
| ||
| Raw sequencing data for deep mutational scanning experiment | This paper | NCBI SRA: BioProject: PRJNA951752 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| For strains used in this paper | This paper | See Table S11 |
| INVSc1, S. cerevisiae Yeast Strain | Invitrogen | Cat#C81000 |
| S. cerevisiae: Strain background By4741 | Horizon Discovery | Cat#YSC1048 |
| S. cerevisiae: Strain background By4742 | Horizon Discovery | Cat#YSC1049 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primers for NGS sequencing and Hrd1 library design | This paper | See Table S14 |
|
| ||
| Recombinant DNA | ||
|
| ||
| For plasmids used in this paper | This paper | See Table S12 |
|
| ||
| Software and algorithms | ||
|
| ||
| Custom code | This paper | https://doi.org/10.5281/zenodo.10038326 https://github.com/baldridge-lab/hrd1_dms_2023 |
| Custom code | This paper | https://doi.org/10.5281/zenodo.10034350 https://github.com/jwschroeder3/2023_mut_scan_analysis |
| ImageJ | Schneider et al.51 | https://imagej.net/ij/index.html; RRID:SCR_003070 |
| FlowJo (version 10.7.1) | Becton, Dickinson and Company | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism (version 9 and 10.0.2) | Dotmatics | https://www.graphpad.com/ |
| Python Programming Language | Python Software Foundation | Python.org; RRID:SCR_008394 |
| Cutadapt (version 1.18) | Martin et al.53 | https://cutadapt.readthedocs.io/ |
| Fastp (version 0.20.0) | Chen et al.54 | https://github.com/OpenGene/fastp |
| Bowtie2 (version 2.3.5.1) | Langmead & Salzberg55 | https://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| SAMtools (version 1.10) | Danecek et al.56 | https://www.htslib.org/ |
| CodonTilingPrimers | Bloom38 | https://github.com/jbloomlab/CodonTilingPrimers |
|
| ||
| Other | ||
|
| ||
| ZE5 Cell Analyzer | Bio-Rad | https://www.bio-rad.com/en-us/product/ze5-cell-analyzer |
| MACSQuant® VYB Flow Cytometer | Miltenyi Biotec | https://www.miltenyibiotec.com/US-en/products/macsquant-vyb.html |
| MoFlo Astrios Cell Sorter | Beckman Coulter | https://www.beckman.com/flow-cytometry/cell-sorters/moflo-astrios-eq |
| Bigfoot Spectral Cell Sorter | Thermo Fisher Scientific | https://www.thermofisher.com/us/en/home/life-science/cell-analysis/flow-cytometry/flow-cytometers/bigfoot-spectral-cell-sorter.html |
Highlights.
Deep mutational scanning of the Hrd1 ubiquitin ligase
Clusters of Hrd1 mutations disrupt partner protein interaction sites
Disordered, cytosolic regions of Hrd1 are required for ERAD in vivo and in vitro
ACKNOWLEDGMENTS
We would like to thank Devon Dennison and other members of the Baldridge lab for critical reading of the manuscript and thoughtful discussion. We thank former lab member Morgan Glaser for cloning the Hrd1 libraries used to isolate Hrd1(T416P,W481L,I505T). We thank Dave Adams and other members of the Michigan Flow Cytometry Core for assistance with flow cytometry training and cell sorting. We also thank Ross Tomaino at the Harvard Taplin Mass Spectrometry Facility for assistance in Hrd1 ubiquitination site identification. B.G.P. was supported by NIH/NIGMS Michigan Predoctoral Training in Genetics (T32GM007544) and NSF Graduate Research Fellowship Program (DGE 1841052). J.E.R. was supported by NIH/NIGMS Michigan Predoctoral Training in Genetics (T32GM007544). This work was supported by the University of Michigan Medical School Biological Sciences Scholars Program and by NIH/NIGMS awards (R35GM128592 to R.D.B. and R35GM128637 to P.L.F.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.113451.
DECLARATION OF INTERESTS
The authors declare that they have no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as a member of the LGBTQIA+ community. We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Hampton RY, and Rine J (1994). Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. J. Cell Biol 125, 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, and Carvalho P (2013). Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. Elife 2, e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Foresti O, Rodriguez-Vaello V, Funaya C, and Carvalho P (2014). Quality control of inner nuclear membrane proteins by the Asi complex. Science 346, 751–755. [DOI] [PubMed] [Google Scholar]
- 4.Christiano R, Nagaraj N, Frö hlich, F., and Walther, T.C. (2014). Global proteome turnover analyses of the Yeasts S. cerevisiae and S. pombe. Cell Rep 9, 1959–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yagishita N, Ohneda K, Amano T, Yamasaki S, Sugiura A, Tsuchimochi K, Shin H, Kawahara KI, Ohneda O, Ohta T, et al. (2005). Essential role of synoviolin in embryogenesis. J. Biol. Chem 280, 7909–7916. [DOI] [PubMed] [Google Scholar]
- 6.Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonc a, N., Liu X, Locasale JW, Simpson KW, Duhamel GE, et al. (2014). Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc. Natl. Acad. Sci. USA 111, E582–E591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eura Y, Yanamoto H, Arai Y, Okuda T, Miyata T, and Kokame K (2012). Derlin-1 deficiency is embryonic lethal, Derlin-3 deficiency appears normal, and Herp deficiency is intolerant to glucose load and ischemia in mice. PLoS One 7, e34298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Francisco AB, Singh R, Li S, Vani AK, Yang L, Munroe RJ, Diaferia G, Cardano M, Biunno I, Qi L, et al. (2010). Deficiency of suppressor enhancer Lin12 1 like (SEL1L) in mice leads to systemic endoplasmic reticulum stress and embryonic lethality. J. Biol. Chem 285, 13694–13703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dougan SK, Hu CCA, Paquet M-E, Greenblatt MB, Kim J, Lilley BN, Watson N, and Ploegh HL (2011). Derlin-2-Deficient Mice Reveal an Essential Role for Protein Dislocation in Chondrocytes. Mol. Cell Biol 31, 1145–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cox JS, Shamu CE, and Walter P (1993). Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell 73, 1197–1206. [DOI] [PubMed] [Google Scholar]
- 11.Kozutsumi Y, Segal M, Normington K, Gething M-J, and Sambrook J (1988). The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature 332, 462–464. [DOI] [PubMed] [Google Scholar]
- 12.Carvalho P, Goder V, and Rapoport TA (2006). Distinct ubiquitinligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373. [DOI] [PubMed] [Google Scholar]
- 13.Khmelinskii A, Blaszczak E, Pantazopoulou M, Fischer B, Omnus DJ, Le Dez G, Brossard A, Gunnarsson A, Barry JD, Meurer M, et al. (2014). Protein quality control at the inner nuclear membrane. Nature 516, 410–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christianson JC, and Carvalho P (2022). Order through destruction: how ER-associated protein degradation contributes to organelle homeostasis. EMBO J 41, e109845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christianson JC, Jarosch E, and Sommer T (2023). Mechanisms of substrate processing during ER-associated protein degradation. Nat. Rev. Mol. Cell Biol 24, 777–796. [DOI] [PubMed] [Google Scholar]
- 16.Hampton RY, Gardner RG, and Rine J (1996). Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol. Biol. Cell 7, 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denic V, Quan EM, and Weissman JS (2006). A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359. [DOI] [PubMed] [Google Scholar]
- 18.Sato BK, Schulz D, Do PH, and Hampton RY (2009). Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Mol. Cell 34, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein A, Ruggiano A, Carvalho P, and Rapoport TA (2014). Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell 158, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DiMaio F, Baker D, Chambers MG, Su H, Li D, et al. (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature 548, 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carvalho P, Stanley AM, and Rapoport TA (2010). Retrotranslocation of a misfolded luminal ER protein by the ubiquitin-ligase Hrd1p. Cell 143, 579–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baldridge RD, and Rapoport TA (2016). Autoubiquitination of the Hrd1 Ligase Triggers Protein Retrotranslocation in ERAD. Cell 166, 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vasic V, Denkert N, Schmidt CC, Riedel D, Stein A, and Meinecke M (2020). Hrd1 forms the retrotranslocation pore regulated by auto-ubiquitination and binding of misfolded proteins. Nat. Cell Biol 22, 274–281. [DOI] [PubMed] [Google Scholar]
- 24.Wu X, Siggel M, Ovchinnikov S, Mi W, Svetlov V, Nudler E, Liao M, Hummer G, and Rapoport TA (2020). Structural basis of ER-associated protein degradation mediated by the Hrd1 ubiquitin ligase complex. Science 368, eaaz2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mehnert M, Sommer T, and Jarosch E (2014). Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nat. Cell Biol 16, 77–86. [DOI] [PubMed] [Google Scholar]
- 26.Knop M, Finger A, Braun T, Hellmuth K, and Wolf DH (1996). Der1, a novel protein specifically required for endoplasmic reticulum degradation in yeast. EMBO J 15, 753–763. [PMC free article] [PubMed] [Google Scholar]
- 27.Horn SC, Hanna J, Hirsch C, Volkwein C, Schütz A, Heinemann U, Sommer T, and Jarosch E (2009). Usa1 functions as a scaffold of the HRD-ubiquitin ligase. Mol. Cell 36, 782–793. [DOI] [PubMed] [Google Scholar]
- 28.Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, and Hampton RY (2000). Endoplasmic Reticulum Degradation Requires Lumen to Cytosol Signaling: Transmembrane Control of Hrd1p by Hrd3p. J. Cell Biol 151, 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szathmary R, Bielmann R, Nita-Lazar M, Burda P, and Jakob CA (2005). Yos9 Protein Is Essential for Degradation of Misfolded Glycoproteins and May Function as Lectin in ERAD. Mol. Cell 19, 765–775. [DOI] [PubMed] [Google Scholar]
- 30.Quan EM, Kamiya Y, Kamiya D, Denic V, Weibezahn J, Kato K, and Weissman JS (2008). Defining the glycan destruction signal for endoplasmic reticulum-associated degradation. Mol. Cell 32, 870–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pisa R, and Rapoport TA (2022). Disulfide-crosslink analysis of the ubiquitin ligase Hrd1 complex during endoplasmic reticulum-associated protein degradation. J. Biol. Chem 298, 102373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hwang J, Walczak CP, Shaler TA, Olzmann JA, Zhang L, Elias JE, and Kopito RR (2017). Characterization of protein complexes of the endoplasmic reticulum-associated degradation E3 ubiquitin ligase Hrd1. J. Biol. Chem 292, 9104–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peterson BG, Glaser ML, Rapoport TA, and Baldridge RD (2019). Cycles of autoubiquitination and deubiquitination regulate the ERAD ubiquitin ligase Hrd1. Elife 8, e50903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Finger A, Knop M, and Wolf DH (1993). Analysis of two mutated vacuolar proteins reveals a degradation pathway in the endoplasmic reticulum or a related compartment of yeast. Eur. J. Biochem 218, 565–574. [DOI] [PubMed] [Google Scholar]
- 35.Plemper RK, Egner R, Kuchler K, and Wolf DH (1998). Endoplasmic Reticulum Degradation of a Mutated ATP-binding Cassette Transporter Pdr5 Proceeds in a Concerted Action of Sec61 and the Proteasome. J. Biol. Chem 273, 32848–32856. [DOI] [PubMed] [Google Scholar]
- 36.Shearer AG, and Hampton RY (2005). Lipid-mediated, reversible misfolding of a sterol-sensing domain protein. EMBO J 24, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Basić-Zaninović T, Papes, and Franekić, J. (1991). Cycloheximide genotoxicity in in vitro and in vivo test systems. Mutat. Res 263, 203–210. [DOI] [PubMed] [Google Scholar]
- 38.Bloom JD (2014). An experimentally determined evolutionary model dramatically improves phylogenetic fit. Mol. Biol. Evol 31, 1956–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ma H, Kunes S, Schatz PJ, and Botstein D (1987). Plasmid construction by homologous recombination in yeast. Genes 58, 201–216. [DOI] [PubMed] [Google Scholar]
- 40.Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S, Lee ME, DeLoache W, Dueber JE, Arkin AP, and Cate JHD (2014). Selection of chromosomal DNA libraries using a multiplex CRISPR system. Elife 3, e03703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cordes FS, Bright JN, and Sansom MSP (2002). Proline-induced Distortions of Transmembrane Helices. J. Mol. Biol 323, 951–960. [DOI] [PubMed] [Google Scholar]
- 42.Baker JA, Wong WC, Eisenhaber B, Warwicker J, and Eisenhaber F (2017). Charged residues next to transmembrane regions revisited: “Positive-inside rule” is complemented by the “negative inside depletion/outside enrichment rule. BMC Biol 15, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nakatsukasa K, Wigge S, Takano Y, Kawarasaki T, Kamura T, and Brodsky JL (2022). A positive genetic selection for transmembrane domain mutations in HRD1 underscores the importance of Hrd1 complex integrity during ERAD. Curr. Genet 68, 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schulz J, Avci D, Queisser MA, Gutschmidt A, Dreher L-S, Fenech EJ, Volkmar N, Hayashi Y, Hoppe T, and Christianson JC (2017). Conserved cytoplasmic domains promote Hrd1 ubiquitin ligase complex formation for ER-associated degradation (ERAD). J. Cell Sci 130, 3322–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, and Gardner RG (2011). Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 41, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L, Li J, Wang Q, Ge MX, Ji J, Liu D, Wang Z, Cao Y, Zhang Y, and Zhang ZR (2022). TMUB1 is an endoplasmic reticulum-resident escortase that promotes the p97-mediated extraction of membrane proteins for degradation. Mol. Cell 82, 3453–3467.e14. [DOI] [PubMed] [Google Scholar]
- 47.Romero P, Obradovic Z, Li X, Garner EC, Brown CJ, and Dunker AK (2001). Sequence complexity of disordered protein. Proteins 42, 38–48. [DOI] [PubMed] [Google Scholar]
- 48.Gietz RD, and Schiestl RH (2007). High-efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat. Protoc 2, 31–34. [DOI] [PubMed] [Google Scholar]
- 49.Hwang J, Peterson BG, Knupp J, and Baldridge RD (2023). The ERAD system is restricted by elevated ceramides. Sci. Adv 9, eadd8579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sikorski RS, and Hieter P (1989). A System of Shuttle Vectors and Yeast Host Strains Designed for Efficient Manipulation of DNA in Saccharomyces ceravisiae. Genetics 122, 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fuxman Bass JI, Reece-Hoyes JS, and Walhout AJM (2016). Zymolyase-Treatment and Polymerase Chain Reaction Amplification from Genomic and Plasmid Templates from Yeast. Cold Spring Harb. Protoc 2016. pdb.prot088971. [DOI] [PMC free article] [PubMed]
- 53.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. j 17, 10. Next Generation Sequencing Data Analysis. [Google Scholar]
- 54.Chen S, Zhou Y, Chen Y, and Gu J (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, and Li H (2021). Twelve years of SAMtools and BCFtools. GigaScience 10, giab008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. Roy. Stat. Soc B 57, 289–300. [Google Scholar]
- 58.Mumberg D, Müller R, and Funk M (1994). Regulatable promoters of Saccharomyces cerevisiae: comparison of transcriptional activity and their use for heterologous expression. Nucleic Acids Res 22, 5767–5768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frey S, and Görlich D (2014). A new set of highly efficient, tag-cleaving proteases for purifying recombinant proteins. J. Chromatogr. A 1337, 95–105. [DOI] [PubMed] [Google Scholar]
- 60.Chen I, Dorr BM, and Liu DR (2011). A general strategy for the evolution of bond-forming enzymes using yeast display. Proc. Natl. Acad. Sci. USA 108, 11399–11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw sequencing data are uploaded to the NCBI Short Read Archive: BioProject:PRJNA951752.
Code used to analyze the deep mutational scanning data are available at the following DOI links: https://doi.org/10.5281/zenodo.10038326; https://doi.org/10.5281/zenodo.10034350. Most recent versions of the code are available at the following links: https://github.com/baldridge-lab/hrd1_dms_2023; https://github.com/jwschroeder3/2023_mut_scan_analysis.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.