

Abstract
Autophagy is a highly conserved cellular process that allows degradation of large macromolecules. p62/SQSTM1 is a key adaptor protein that interacts both with material to be degraded and with LC3 at the autophagosome, enabling degradation of cargos such as protein aggregates, lipid droplets and damaged organelles by selective autophagy. Dysregulation of autophagy contributes to the pathogenesis of many diseases. In this study, we investigated if the interaction of p62/SQSTM1 with LC3B could be regulated. We purified full‐length p62/SQSTM1 and established an in vitro assay that measures the interaction with LC3B. We used the assay to determine the role of the different domains of p62/SQSTM1 in the interaction with LC3B. We identified a mechanism of regulation of p62/SQSTM1 where the ZZ and the PB1 domains regulate the exposure of the LIR‐sequence to enable or inhibit the interaction with LC3B. A mutation to mimic the phosphorylation of a site on the ZZ domain leads to increased interaction with LC3B. Also, a small compound that binds to the ZZ domain enhances interaction with LC3B. Dysregulation of these mechanisms in p62/SQSTM1 could have implications for diseases where autophagy is affected. In conclusion, our study highlights the regulated nature of p62/SQSTM1 and its ability to modulate the interaction with LC3B through a LIR‐sequence Accessibility Mechanism (LAM). Furthermore, our findings suggest the potential for pharmacological modulation of the exposure of LIR, paving the way for future therapeutic strategies.
Keywords: chemical biology, LC3 and GABARAP proteins, LIR, molecular mechanism, p62/SQSTM1, XRK3F2
1. INTRODUCTION
Autophagy is a cellular mechanism that enables the degradation of large cellular particles, intracellular protein aggregates and organelles (Ohsumi, 2014). Ubiquitinated (Ub) proteins would normally be degraded by the proteasome; however, in certain situations, that is, when proteasome cannot cope with the degradation of proteins, Ub‐proteins may also be degraded by autophagy (Mizushima & Komatsu, 2011). Selective autophagy (macroautophagy) requires first the labelling of the material to be degraded and the interaction of the material with p62/sequestosome (p62/SQSTM1 thereafter) and other autophagy receptors (Stolz et al., 2014; Zellner & Behrends, 2021). p62/SQSTM1 was named sequestosome for its ability to participate in the formation of intracellular protein aggregates (Shin, 1998), today described as a liquid–liquid phase separation membraneless organelle termed p62 body (Komatsu, 2022). p62/SQSTM1 is described to participate in multiple cellular functions (Kumar et al., 2022). As part of selected autophagy p62/SQSTM1 is best known as an “adaptor” protein that on one side binds the ubiquitinylated material labeled for degradation and on the other side binds to the autophagosome (Figure 1a).
FIGURE 1.
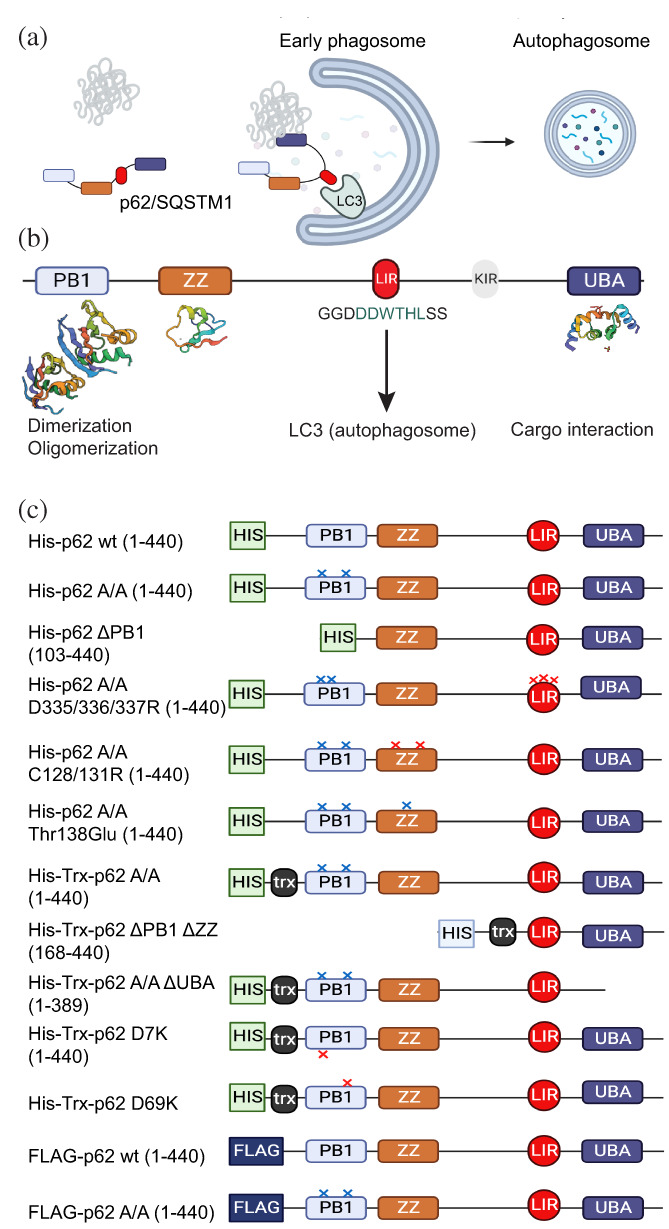
Schematic representation of p62/SQSTM1. (a) Role of autophagy receptor p62/SQSTM1 as an adaptor protein, taking cargo to the autophagosome for degradation via selected autophagy. (b) Representation of p62/SQSTM1 domains and LIR. C, p62/SQSTM1 constructs employed in this study.
The p62/SQSTM1 protein is comprised by a Phox‐BEM1 (PB1) domain, a ZZ‐type zinc finger (ZZ) domain, a ubiquitin‐associated (UBA) domain, a linker sequence containing a Keap1‐interacting region (KIR) that interacts with KEAP and a LC3 Interacting Region (LIR)‐sequence that enables interaction with Atg8‐family proteins LC3A, LC3B, LC3C, GABARAP, GABARAPL1, and GABARAPL2 in humans, reviewed in (Rogov et al., 2023) (Figure 1a,b). Upon interaction of p62/SQSTM1 with LC3/GABARAPs, the autophagosome englobes the material labeled for degradation and the vesicle fuses with lysosomes, which ultimately degrade the material and provide free building blocks to the cell (Kirkin et al., 2009). p62/SQSTM1 contributes to the regulation of multiple cellular processes, including adipogenesis (Lee et al., 2010), NF‐kB signaling (Sanchez et al., 1998; Sanz et al., 1999; Sanz et al., 2000), response to oxidative stress (Komatsu et al., 2010), as well as sensing nutritional status by interacting with key proteins that provide localization and activation of mTORC1 at the lysosome (Duran et al., 2011).
Defective autophagy has been linked to the development of several diseases, including cancer and conformational disorders (Dikic & Elazar, 2018). Some cancers are addicted to autophagy while most other cell types are less affected by the inhibition of autophagy (Mathew et al., 2009). Therefore, it is suggested that inhibiting autophagy may be suitable to specifically target some cancers without producing severe side‐effects. Depletion of p62/SQSTM1 can also inhibit cancer progression (Nguyen et al., 2019). On the other hand, autophagy does not function properly, and intracellular protein aggregates accumulate, in a broad range of diseases grouped with the term “conformational disorders”, such as neurodegenerative disorders (Alzheimer's, Parkinson's diseases) (Bar‐Yosef et al., 2019), and also in metabolic disorders, like type I diabetes (Mukherjee et al., 2015) and Non‐alcoholic steatohepatitis (NASH) (Tan et al., 2023). p62/SQSTM1 is present in the protein aggregates of all conformational disorders studied. Even if p62/SQSTM1 is present in the protein aggregates, these do not get degraded via autophagy.
While p62/SQSTM1 can regulate autophagy, post‐translational modifications can modulate p62/SQSTM1 activity. Activating phosphorylation include modifications by ULK1, Casein kinase 2 (CK2), TBK‐1, and AMPK (Ha et al., 2017; Lim et al., 2015; Matsumoto et al., 2011; Pilli et al., 2012). On the contrary, phosphorylation by protein kinase A (PKA) and MAPK13 inhibit autophagy (Christian et al., 2014; Linares et al., 2015). The Leucine‐Rich Repeat Kinase 2 (LRRK2) indirectly and directly modulates p62/SQSTM1 phosphorylation (Kalogeropulou et al., 2018; Park et al., 2016). Other post translational modifications of p62/SQSTM1, such as oxidation of cysteines (C113, C115), can increase autophagy (Carroll et al., 2018). Finally, it has been shown that arginylated substrates that bind to the ZZ domain of p62/SQSTM1 can enhance autophagy (Zhang et al., 2018).
Targeting p62/SQSTM1 could be a potential therapeutic approach (Chen et al., 2020). However, Full‐length p62/SQSTM1 can form oligomers mediated by the PB1 domain (Jakobi et al., 2020), which results in the formation of large p62/SQSTM1 filaments or “aggregates”. This characteristic has hampered in vitro studies involving full‐length p62/SQSTM1 as well as large screening studies focused on this protein.
To investigate possible mechanisms of p62/SQSTM1 regulation, we developed an in vitro assay to monitor interaction between full‐length p62/SQSTM1 and prototypical human Atg8‐family protein LC3B, and tested the effect on the interaction of mutated and domain‐deleted p62/SQSTM1. Our studies indicate that the ZZ domain and PB1 domain regulate the exposure of the LIR‐sequence, enabling the regulated interaction with LC3B. Furthermore, we found that a known phosphorylation site on p62/SQSTM1 can modulate its interaction with LC3B. Finally, we describe that a small compound that binds to the ZZ domain of p62/SQSTM1 can enhance the interaction with LC3B. These findings shed light on the complex mechanism of regulation of p62/SQSTM1 and provide insights into potential strategies for developing therapeutics that can modulate this process.
2. RESULTS
2.1. Expression, purification and in vitro interaction between full‐length p62/SQSTM1 and LC3B
His‐p62/SQSTM1 comprising an N‐terminal fusion with a 6× His‐tag and in some cases a thioredoxin (Trx) domain was expressed in bacteria from a pDEST59 construct (see Figure 1c for p62/SQSTM1 constructs used in this study). In an attempt to purify His‐p62/SQSTM1, the protein was expressed successfully in bacteria, was bound to Ni‐NTA resin, but could not be eluted using standard protocols. We reasoned that the protein may not have eluted because it could be forming large stable oligomers. The PB1 domain is the most N‐terminal domain of p62/SQSTM1 and has two “faces”, a negatively charged face (represented by Asp69) and a positively charged face (represented by Lys7). In the oligomerization mediated by the PB1 domain, the negatively charged face interacts with the positively charged face. In order to interfere with the oligomerization that inhibited the purification of full‐length p62/SQSTM1, we performed two mutations, one at each side of the PB1 domain, replacing Lys7 and Asp69 by Ala, His‐p62/SQSTM1 (K7A, D69A) (p62/SQSTM1 A/A) (Lamark et al., 2003). The p62/SQSTM1 A/A protein could be eluted from the Ni‐NTA column using a relatively high concentration of imidazol (400–500 mM). In Size Exclusion Chromatography (SEC), His‐p62/SQSTM1 eluted as one peak at a position corresponding approximately with the void volume of the column, indicating that the purified p62/SQSTM1 A/A was present as a large protein oligomer/aggregate (Figure S1). We considered that only monomers or dimers of His‐p62/SQSTM1 would have eluted from the Ni‐NTA resin with imidazole. However, since p62/SQSTM1 eluted from the gel filtration as if they were “large aggregates”, we concluded that the purified form of p62/SQSTM1 A/A still formed large multi‐protein complexes in vitro but that these were in dynamic equilibrium with non‐aggregated forms of p62/SQSTM1 A/A. The finding that the PB1 mutations enabled the purification confirmed that the PB1 domain was indeed involved in the formation of stable large multi‐protein complexes and indicated the A/A mutation was a mild mutation that only diminished the affinity of the PB1 domains to form oligomers.
We then used the AlphaScreen technology to test the interaction of purified full‐length His‐p62/SQSTM1 A/A with purified GST‐LC3B. Previous studies have shown that the fusion to GFP or GST does not affect the interaction of LC3/GABARAPs proteins with p62/SQSTM1 or other interacting partners (Mizushima 2009; Novak et al. 2010; Pankiv et al. 2007). In this assay, each protein is attached to a bead. An exciting light on the donor bead produces a “singlet oxygen”, which travels up to 200 nm and reacts with the acceptor bead to emit light when the two proteins interact (Figure 2a). We clearly observed an interaction between His‐p62/SQSTM1 A/A and GST‐LC3B (Figure 2b). His‐p62/SQSTM1 A/A mutated at the LIR‐sequence did not show binding to GST‐LC3B (Figure 2b), verifying the specificity of the assay. Also, the addition of the polypeptide LIRtide comprising the LIR‐sequence inhibited the interaction between p62/SQSTM1 and LC3B (Figure 2c). Similarly, the addition of novobiocin, identified by us to interact with LC3A and LC3B (Hartmann et al., 2021), displaces the interaction (Figure 2d).
FIGURE 2.
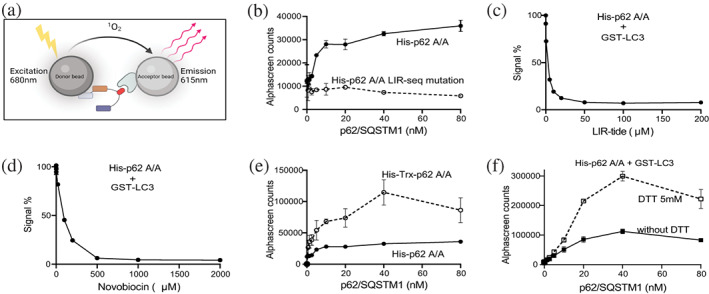
p62/SQSTM1 interaction with LC3B. (a) Graphical representation of the AlphaScreen interaction assay. (b) Purified His‐p62/SQSTM1 A/A and GST‐LC3B interact. His‐p62 K7A K69A did not interact with LC3B, when the LIR‐motif was mutated in p62/SQSTM1. (c) LIRtide inhibits the interaction of p62 and LC3B (IC50 = 1.7 μM). (d) Novobiocin inhibited the interaction of p62 and LC3B (IC50 = 81 μM). Novobiocin is a small compound that binds LC3 and blocks the interaction with LIRtide. (e) Construct containing a thioredoxin domain increased interaction with LC3. (f) DTT increased the interaction of p62/SQSTM1 with LC3B. The data are representative of two independent experiments.
We next compared the interaction of His‐p62/SQSTM1 A/A with an identical construct but fused to a thioredoxin domain. Interestingly, the His‐Trx‐fusion protein consistently showed higher binding to GST‐LC3B (Figure 2f). In addition, pre‐incubation of p62/SQSTM1 A/A with 5 mM DTT enhanced the subsequent binding to LC3B (Figure 2e). The finding suggested that p62/SQSTM1 A/A had a particular sensitivity to oxidation that inhibited the interaction with LC3B. The sensitivity to oxidation is in agreement with reports that p62/SQSTM1 could be regulated by cellular oxidation (Carroll et al., 2018). In practical terms, these findings helped us to set‐up the conditions of the interaction assays and led us to only compare p62/SQSTM1 constructs with the same tags.
2.2. Role of different domains of p62/SQSTM1 on the interaction with LC3B
After establishing a robust p62/SQSTM1 A/A‐LC3B interaction assay with purified full‐length p62/SQSTM1, we aimed to investigate whether domains other than the LIR‐sequence regulate the interaction of p62/SQSTM1 with LC3B. To this end we prepared a set of constructs of p62/SQSTM1 (Figure 1c), expressed them in bacteria, purified them and tested them in the AlphaScreen assay.
We first investigated if the UBA domain participated in the interaction of p62/SQSTM1 with LC3B. Deletion of the UBA domain in p62/SQSTM1 A/A (1‐389) (ΔUBA p62/SQSTM1 A/A) did not affect the maximal binding of p62/SQSTM1 A/A to LC3B, although a close inspection of the concentration dependence of the interaction unveiled a different pattern of the curve, suggesting that the UBA domain could have a modest contribution to binding (Figure 3a).”
FIGURE 3.
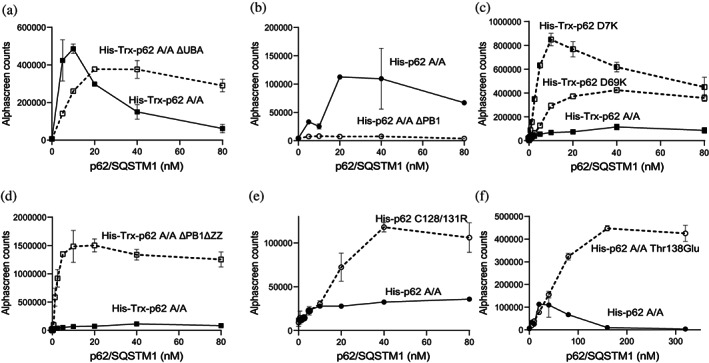
Role of PB1 and ZZ domains of p62/SQSTM1 on the interaction with LC3B using AlphaScreen. (a) Deletion of the UBA domain does not affect the total binding of the protein to LC3B, although the different shape of the curves could indicate a modest contribution to binding. (b) Deletion of the PB1 domain results in decreased interaction with LC3B. (c) Mutations at either side of the PB1 domain renders proteins with increased interaction with LC3B. (d) Deletion of the PB1 and ZZ domains vastly enhances interaction with LC3B. (e) Disruption of the ZZ domain by mutagenesis of key cysteines residues enhances interaction with LC3B. (f) Phosphorylation‐mimicking mutation at Thr138 renders a protein with increased interaction with LC3B. The data are representative of two independent experiments.
We next investigated if the PB1 domain regulated the interaction of p62/SQSTM1 with LC3B. Deletion of the PB1 domain in the p62/SQSTM1 (103‐440) (ΔPB1 p62/SQSTM1) construct decreased the binding, indicating that the PB1 domain supported the binding of p62/SQSTM1 to LC3B (Figure 3b). We also performed charged‐reversal mutations at either side of the PB1 domain to completely disrupt the charge properties of the domain (p62/SQSTM1 Lys7Asp and p62/SQSTM1 Asp69Lys). Interestingly, both mutants of the PB1 domain that completely disrupted the oligomerization of p62/SQSTM1 enhanced the binding to LC3B (Figure 3c). From this we conclude that in our in vitro model, PB1 domain‐mediated oligomerization did not directly participate in the enhancement of the interaction with LC3B. The results are compatible with a dual and therefore more complex involvement of the PB1 domain on the interaction with LC3B.
We showed above that p62/SQSTM1 (103‐440) lacking the PB1 domain (ΔPB1 p62/SQSTM1) had decreased binding to LC3B. Notably, the additional deletion of the ZZ domain of p62/SQSTM1 (168‐440) (ΔPB1 ΔZZ p62/SQSTM1) elicited a vast increase in the ability to interact with LC3B (Figure 3d). Mutations of Cys 128/131 to Ser, predicted to disturb the folding of the ZZ domain, also enhanced binding to LC3B (Figure 3e). The ZZ domain is known to contain a phosphorylation site, Thr138, that is phosphorylated by LRRK2 (Kalogeropulou et al., 2018). Notably, mutation of Thr138 to Glu to mimick the phosphorylation of Thr138 resulted in a protein with increased interaction with LC3B (Figure 3f). These results indicated an important role of the ZZ domain in regulating the interaction of p62/SQSTM1 with LC3B. Such a role could be rationalized if p62/SQSTM1 was a regulated adaptor, where modifications in the ZZ domain would modulate the conformation of the protein.
Using the AlphaScreen assay, we confirmed that the interaction between p62/SQSTM1 and LC3 is mediated by the LIR‐sequence of p62/SQSTM1. Notably, the above findings indicated that the LIR‐sequence is not always available for interaction with LC3 and suggest a model where the interaction between p62/SQSTM1 and LC3B could be modulated by the regulated exposure of the LIR‐sequence. Together, the above results indicated that the PB1 domain in a monomeric form supported the interaction with LC3B and that the ZZ domain exerted an inhibitory effect on the interaction of p62/SQSTM1 with LC3B. Furthermore, the results suggested that the phosphorylation of Thr138 within the ZZ domain could regulate the conformation of p62/SQSTM1, enabling the exposure of the LIR‐sequence and the interaction with LC3B.
2.3. Small compounds can modulate the interaction between p62/SQSTM1 with LC3B
Next, we aimed to investigate if the interaction between p62/SQSTM1 and LC3B could be modulated by small compounds. The AlphaScreen assay, established in vitro using bacterially expressed and purified His‐p62/SQSTM1 A/A, was very robust, with Z′ = 0.87. We tested the possible effect of the small compound XRK3F2, which is described to interact with the ZZ domain of p62/SQSTM1 (Teramachi et al., 2016). XRK3F2 increased the interaction between His‐Trx‐p62/SQSTM1 A/A and LC3B (Figure 4a). Since the above experiments were performed with the purified His‐Trx‐p62/SQSTM1 A/A mutant, we aimed to test if the compounds could also affect the interaction of a non‐mutated form of p62/SQSTM1 with LC3B. To this end we expressed FLAG‐p62/SQSTM1 and used the unpurified tagged protein and appropriate AlphaScreen beads that recognize FLAG‐tagged proteins (Figures 4b and S2). Similarly to the results using the assay with the purified proteins, LIRtide and Novobiocin displaced the interaction between GST‐LC3B and non‐purified FLAG‐p62/SQSTM1 A/A and also displaced the interaction with the non‐mutated p62/SQSTM1 (Figure S2). Using this assay, we further confirmed that the compound XRK3F2 enhanced the interaction of the unpurified wild type FLAG‐p62/SQSTM1 with GST‐LC3B in the context of a cellular lysate (Figure 4c).
FIGURE 4.
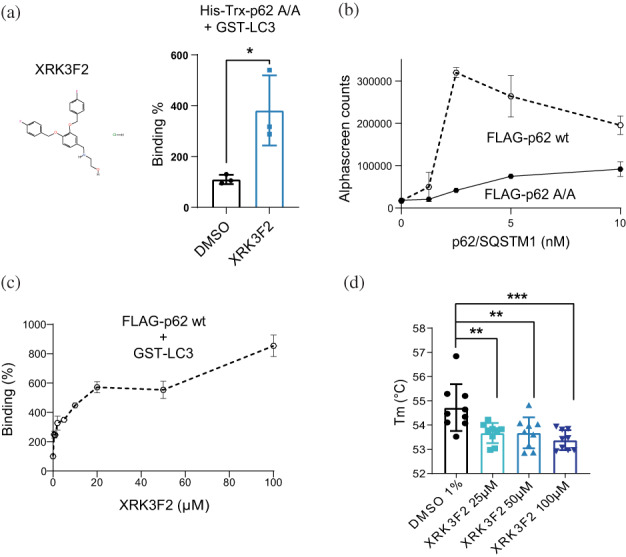
XRK2F2 binds to the ZZ domain and increases the interaction between p62/SQSTM1 and LC3. (a) The compound XRK2F2 increases the interaction between purified His‐Trx‐p62/SQSTM1 A/A (His‐Trx‐p62 A/A) and GST‐LC3. Results expressed as mean ± SD with *p < 0.05 by unpaired t‐test. (b) Non purified FLAG‐p62/SQSTM1 wt (wild type sequence) interacts with LC3B on the modified AlphaScreen assay. Data are representative of two independent experiments. (c) XRK2F2 increases the interaction between FLAG‐p62/SQSTM1 wt and GST‐LC3 in the context of the cell lysate. Data are representative of two independent experiments. (d) Change in the thermal stability of the purified ZZ domain due to the binding of XRK2F2. Graphic depicts three replicates of three independent experiments. Results are expressed as Mean ± SD with **p < 0.01 and ***p = 0.0001 by one‐way ANOVA followed by Bonferroni multiple comparison test. (A, C) The binding (%) was calculated considering the DMSO condition as 100%.
We considered that the ZZ‐domain directly or indirectly sequestered the LIR sequence, inhibiting the interaction with LC3B. In such model, XRK3F2 could bind to the ZZ domain, displace the sequestration of the LIR‐sequence by the ZZ domain, where the release of the LIR‐sequence would enable its binding to LC3B. Such simple model would require that both XRK3F2 and the LIR‐sequence would interact with the ZZ‐domain. In this line, XRK3F2 is indeed claimed to interact with the ZZ domain of p62/SQSTM1 (Teramachi et al., 2016). However, to our knowledge a formal proof for the direct interaction was not published. We established a temperature‐stability assay using a construct that expresses the ZZ domain fused to GST (GST‐ZZ‐domain; Figure 1c). XRK3F2 decreased the melting temperature (Tm), of GST‐ZZ‐domain but not of the construct containing only GST (Figures 4d and S3). Although the binding of a compound to a target protein increases the stability at the binding site, a compound can also decrease the stability of the target protein if the binding produces a larger de‐stabilization of the target at a distant site, allosterically. The de‐stabilization of the ZZ‐domain by XRK3F2 thus indicated to us that the compound binds to the ZZ‐domain and affected its folding.
In the simplest model, the mechanism of sequestration of LIR by the ZZ domain could be direct, where the LIR‐sequence directly interacted with the ZZ domain. We tested this hypothesis by investigating if LIRtide could also directly interact with the ZZ domain. In contrast to the findings with XRK3F2 we could not measure any interaction between LIRtide and LC3B (Figure S2), suggesting that LIR‐sequence does not directly interact with the ZZ domain. Together, our results suggest a general model where p62/SQSTM1 exists in at least two different conformations in equilibrium, a conformation where the PB1 and ZZ domains indirectly sequester the LIR‐sequence and a second conformation that exposes the LIR‐sequence (Figure 5).
FIGURE 5.
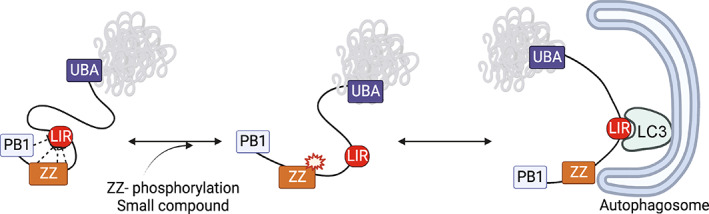
Schematic model of p62/SQSTM1 regulation by a LIR‐accessibility mechanism (LAM). p62/SQSTM1 is represented in equilibrium between populations of conformations hiding the LIR‐sequence (Left) and conformations exposing LIR‐sequence. Post‐translational modification of the ZZ domain or direct interaction with small compounds produce a change in the population of p62/SQSTM1 molecules enhancing the exposure of the LIR‐sequence, in turn favoring the interaction with LC3 at the early phagosome.
3. DISCUSSION
For many years, the p62/SQSTM1 protein was considered an important adaptor protein and a marker for autophagy. More recently, however, there are multiple reports that show that post‐translational modifications of p62/SQSTM1 can regulate different aspects of autophagy. Post‐translational modifications can affect adaptor proteins by modulating their ability to bind to different protein partners. However, in the present work we describe that the adaptor function of p62/SQSTM1 may be regulated by modulation of the exposure of the LIR‐sequence, a mechanism that we term LAM (LIR Accessibility Mechanism). While we still do not know the molecular details of the mechanism, our studies indicate that: (1) the N‐terminal PB1 and ZZ domains play a role in this regulation, while the C‐terminal UBA domain does not, (2) the integrity of the PB1 domain and the ZZ domain are needed to sequester the LIR‐sequence, (3) ZZ domain does not seem to directly sequester the LIR‐sequence and therefore we deduce that the sequestration of the LIR‐sequence should involve other regions of p62/SQSTM1 located C‐terminally to the ZZ domain, (4) a mutation that mimics phosphorylation at the ZZ domain leads to the exposure of the LIR‐sequence, (5) small molecules that bind to the ZZ domain can release the sequestration of the LIR‐sequence and promote binding to LC3B, and (6) the possibility to modulate the system with small molecules further informs about the dynamic in nature of the system where the p62/SQSTM1 conformations are in equilibrium. Together, our results indicate that the essential function of p62/SQSTM1, to bind to LC3 molecules via the LIR‐sequence, may be physiologically regulated, for example, by cellular signaling and phosphorylation and also may be modulated pharmacologically.
p62/SQSTM1 is found in conformational disorders within protein aggregates that are not directed for degradation by autophagy. It is reasonable to speculate that the molecular mechanism regulating p62/SQSTM1 may be affected in diseases, leading to either stabilization of p62/SQSTM1 in conformations that are impaired in binding to LC3B (such as in conformational disorders), or stabilization in conformations that are competent for binding to LC3B (such as in cancers that rely on autophagy). Further research is needed to develop tools that can visualize the actual conformation of p62/SQSTM1 in cellular systems and tissues from patients. The immunological detection of the LIR‐sequence in biological samples, could help deduce if the p62/SQSTM1 conformation is in either the “exposed” or “hidden” conformation. As controls, the total available LIR‐sequences could be obtained by addition of compounds that bind to the ZZ domain with a mechanism similar to that of XRK3F2.
Our results point to a key regulatory role of the ZZ domain of p62/SQSTM1. Interestingly, using an independent approach, the ZZ domain had previously been identified as the domain that can interact with small molecules—arginylated substrates (Zhang et al., 2018), XRK3F2 (Teramachi et al., 2016) and AUTOTACs (Ji et al., 2022) affecting p62/SQSTM1 cellular functions. Here we suggest a molecular mechanism, LAM, whereby interacting proteins or molecules could modulate the conformation of p62/SQSTM1 via the exposure of the LIR‐sequence. The current work provides possible platforms to identify and develop more potent compounds, which could serve as probes for research or to attempt pharmacological intervention for the treatment of diseases that are affected in p62/SQSTM1 function. On one side, the validation of the ZZ domain would strengthen the rational development of drugs directed to this particular domain of p62/SQSTM1. On the other hand, the AlphaScreen interaction assays here described is suitable for HTS and could be used as a means to identify molecules with the desired effect within large libraries of molecules.
4. MATERIALS AND METHODS
4.1. Materials
The peptide LIRtide (SGGDDDWTHLS) was synthesized by Genescript (Hartmann et al., 2021). Primary antibodies were, anti‐p62 (New England Biolab), anti‐His (27E8) (Cell Signaling Technology); Secondary antibody was Fluorescently labeled IRDye 680 (GαM680) (LiCor).
4.2. General methods
Molecular biology techniques were performed using standard protocols. Site‐directed mutagenesis was performed using a QuikChange kit (Stratagene) following the instructions provided by the manufacturer. The oligonucleotides were from Macrogen (Korea). The DNA constructs used for transient transfection were purified from bacteria using a Qiagen plasmid Maxi kit according to the manufacturer's protocol. The DNA sequences were verified by automatic DNA sequencing was performed by Macrogen. Protein concentration was estimated using Coomassie Plus reagent from Thermo Scientific and verified by Coomassie staining using a BSA as a standard. FLAG‐p62/SQSTM1 wt and A/A were transiently transfected and expressed from a pcDNA5 plasmid in HEK293 cells. The levels of FLAG‐p62/SQSTM1 were compared with the amounts of His‐p62/SQSTM1 by using quantitative fluorescent Western‐blotting (anti‐ p62/SQSTM1 primary antibody).
Microbiology procedures were performed using standard protocols. Materials for cell culture were from Greiner and Sarstedt. Human embryonic kidney (HEK) 293T cells (ATCC collection) were cultured in 175 cm2 flasks in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum (Sigma‐Aldrich).
4.3. Protein expression and purification
For expression of HIS constructs, GST‐ZZ and GST control, BL21 DE3 bacteria were transformed according to manufacturer's instructions (Invitrogen). Expression was induced by 1 mM IPTG and incubated over‐night at 18°C. Cells were then lysed by sonication using a Tris‐EDTA lysis buffer containing 50 mM Tris, 100 mM NaCl, a tip of spatula of PMSF (Carl Roth), supplemented with one tablet of protease inhibitor cocktail (Roche) per 50 ml and Benzonase (Sigma). The set‐up of the purification procedure for His‐Trx‐p62/SQSTM1 was done as described in the text using a Ni‐NTA resin (GE Healthcare). For the routine purification of the different His‐tagged p62/SQSTM1 proteins, the cleared lysate was incubated (2 h) with Ni‐IDA resin (Jena BioScience) with addition of NaCl to reach 500 mM and imidazol to reach 10 mM to avoid unspecific binding. This was followed by 10 washes in washing buffer (50 mM Tris, 500 mM NaCl, 10 mM imidazole, one tip of spatula of PMSF, and 0.1 mM ß‐mercaptoethanol. Elution was performed in elution buffer (50 mM Tris; 500 mM NaCl; 400 mM imidazole; one tip of spatula of PMSF and 0.1 mM ß‐mercaptoethanol).
For the expression of FLAG‐p62/SQSTM1, LC3B fused to GST, GST‐ZZ, and GST, plasmids were transfected into 8 × 14.5 cm dishes containing HEK293 cells using the PEI method (125 μg PEI and 12.5 μg plasmid/14.5 cm dish). The cells were lysed after 48 h in a buffer containing 50 mM Tris–HCl pH 7.5, 1 mM EGTA, 1 mM EDTA, 1% (w/v) Triton X‐100, 1 mM sodium orthovanadate, 50 μM sodium fluoride, 5 mM sodium pyrophosphate, 0.27 M sucrose, 0.1% β‐mercaptoethanol, and 1 tablet of protease inhibitor cocktail (Roche) per 50 ml of buffer. Lysates were cleared by centrifugation, frozen in liquid nitrogen and kept at −80°C until required. The cleared lysates of FLAG‐p62/SQSTM1 were diluted and employed in the AlphaScreen interaction assay without purification. Purification of GST‐fusion proteins involved incubation of the cleared lysate with glutathione sepharose 4B (GE Healthcare) followed by two washes with 0.5 M NaCl in lysis buffer, eight washes with a buffer containing 50 mM Tris–HCl, 0.1 mM EGTA and 0.1% β‐mercaptoethanol (buffer A), and a last wash with buffer A supplemented with 0.26 M sucrose. Elution was performed with this last buffer containing 20 mM glutathione, and the GST‐fusion protein was cleared from the resin by filtration through a “SigmaPrep” spin column (Sigma).
In all cases, the purified proteins were aliquoted, snap frozen in liquid nitrogen and kept at −80°C until use. Purity at this stage was above 85% as estimated by SDS‐PAGE and staining with Coomassie Brilliant Blue R250.
4.4. AlphaScreen interaction assay
The AlphaScreen assay was performed according to the manufacturer's protocol (Perkin Elmer). Reactions were performed in a 25 μl final volume in white 384‐well microtiter plates (Greiner). The reaction buffer contained 20 mM Tris–HCl (pH 7.4), 100 mM NaCl, 1 mM dithiotreitol (DTT), 0.05% (v/v) Tween‐20 and 0.1% (w/v) BSA. For the interactions between His‐p62/SQSTM1 and GST‐LC3B the beads were nickel chelate‐coated acceptor beads and GSH‐coated donor beads. For the interactions between FLAG‐p62/SQSTM1 and GST‐LC3B the nickel bead was replaced by the anti‐FLAG conjugated AlphaScreen acceptor beads. The quantification of FLAG‐p62/SQSTM1 in the unpurified fraction was evaluated by quantitative western‐blot using known amount of His‐p62/SQSTM1 as control. For each interaction pair the conditions for the interaction were identified by performing cross‐titration assays. We then fixed the GST‐LC3B concentration in the assay and tested the effect of various concentrations of His‐ or FLAG‐ tagged p62/SQSTM1 constructs. The AlphaScreen assays were read in an EnVision multilabel plate reader equipment (Perkin Elmer). When setting‐up the assay using p62/SQSTM1 A/A and GST‐LC3B in HTS format, 7 nM His‐Trx‐p62/SQSTM1 and 3.5 nM GST‐LC3B produced average binding of (AlphaScreen arbitrary units) 60861 ± 2473; with background values of 1573 ± 108, while the displacement by the control LIRtide rendered 1714 ± 104, with Z′value of 0.87.
4.5. Temperature stability assay
Protein unfolding was monitored by the increase in the fluorescence of the fluorophor SYPRO Orange (Invitrogen) using a real‐time PCR device (StepOnePlus, Applied Biosystems) following the protocol described (Niesen et al., 2007). GST‐ZZ domain/GST control was diluted in 10 mM HEPES (pH 7.5) buffer containing 200 mM NaCl. The reactions were performed in a final volume of 10 μl in 96‐well PCR microtiter plates (Applied Biosystems) and contained 3 μM protein, 10 mM HEPES (pH 7.5), 200 mM NaCl, 1/1000 SYPRO Orange, 1 mM dithiothreitol and 2 mM ZnCl2. XRK3F2 or LIRtide (1% final DMSO concentration) were added to this reaction mixture. The temperature gradient was performed in steps of 0.3°C in the range of 25°C to 85°C. To calculate the T m values, the data were exported to GraphPad Prism and the curves fitted to a Boltzmann sigmoidal equation with all R 2 > 0.998.
4.6. Statistical analysis
All experiments were performed at least twice with similar results. GraphPad Prism was used for the statistical analysis. The values are presented as mean ± SD. Significance was calculated by one‐way analysis of variance followed by Bonferroni post‐hoc testing or unpaired t test. A p value <0.05 was considered statistically significant.
AUTHOR CONTRIBUTIONS
Ricardo M. Biondi: Methodology; conceptualization; investigation; funding acquisition; writing – original draft; writing – review and editing; supervision; project administration. Lucia Alcober‐Boquet: Methodology; investigation; writing – review and editing; writing – original draft. Tabea Zang: Methodology; investigation; writing – review and editing. Larissa Pietsch: Methodology; investigation. Evelyn Suess: Methodology; investigation; supervision. Markus Hartmann: Methodology; investigation. Ewgenij Proschak: Supervision; writing – review and editing. Lissy Zoe Florens Gross: Methodology; investigation. Mariana Sacerdoti: Methodology; investigation. Stefan Zeuzem: Writing – review and editing; funding acquisition. Vladimir V. Rogov: Methodology; investigation; writing – review and editing. Alejandro E. Leroux: Methodology; investigation; writing – review and editing; supervision. Albrecht Piiper: Methodology; investigation; writing – review and editing; project administration; funding acquisition; supervision.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflicts of interest with the contents of this article.
Supporting information
DATA S1. Supporting Information.
ACKNOWLEDGMENTS
We thank the members of the Research Group PhosphoSites and DKTK‐Frankfurt network, specially Ivan Dikic, Christian Behrends, Christian Brands and Stefan Knapp, who participated in the early discussions on the LIR‐LC3B targeting project. The study was supported by the German Cancer Consortium (DKTK) and ANPCyT PICT2018‐737 to Ricardo M. Biondi and the LOEWE‐TMP to Albrecht Piiper. Vladimir V. Rogov is grateful for support by the Structural Genomics Consortium (SGC), a registered charity (no: 1097737) that receives funds from Bayer AG, Boehringer Ingelheim, Bristol Myers Squibb, Genentech, Genome Canada through Ontario Genomics Institute [OGI‐196], EU/EFPIA/OICR/McGill/KTH/Diamond Innovative Medicines Initiative 2 Joint Undertaking [EUbOPEN grant 875510], Janssen, Merck KGaA (aka EMD in Canada and US), Pfizer and Takeda. Figures 1, 2a, and 5 were created with BioRender.com. Open Access funding enabled and organized by Projekt DEAL.
Alcober‐Boquet L, Zang T, Pietsch L, Suess E, Hartmann M, Proschak E, et al. The PB1 and the ZZ domain of the autophagy receptor p62/SQSTM1 regulate the interaction of p62/SQSTM1 with the autophagosome protein LC3B. Protein Science. 2024;33(1):e4840. 10.1002/pro.4840
Lucia Alcober‐Boquet and Tabea Zang have contributed equally to this study.
Reviewing Editor: John Kuriyan
Contributor Information
Albrecht Piiper, Email: piiper@med.uni-frankfurt.de.
Ricardo M. Biondi, Email: dabiondi@yahoo.co.uk.
REFERENCES
- Bar‐Yosef T, Damri O, Agam G. Dual role of autophagy in diseases of the central nervous system. Front Cell Neurosci. 2019;13:196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll B, Otten EG, Manni D, Stefanatos R, Menzies FM, Smith GR, et al. Oxidation of sqstm1/p62 mediates the link between redox state and protein homeostasis. Nat Commun. 2018;9(1):256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li Q, Li Q, Xing S, Liu Y, Liu Y, et al. P62/sqstm1, a central but unexploited target: advances in its physiological/pathogenic functions and small molecular modulators. J Med Chem. 2020;63(18):10135–10157. [DOI] [PubMed] [Google Scholar]
- Christian F, Krause E, Houslay MD, Baillie GS. Pka phosphorylation of p62/sqstm1 regulates pb1 domain interaction partner binding. Biochim Biophys Acta. 2014;1843(11):2765–2774. [DOI] [PubMed] [Google Scholar]
- Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- Duran A, Amanchy R, Linares JF, Joshi J, Abu‐Baker S, Porollo A, et al. P62 is a key regulator of nutrient sensing in the mtorc1 pathway. Mol Cell. 2011;44(1):134–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha S, Jeong SH, Yi K, Chung KM, Hong CJ, Kim SW, et al. Phosphorylation of p62 by amp‐activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J Biol Chem. 2017;292(33):13795–13808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Huber J, Kramer JS, Heering J, Pietsch L, Stark H, et al. Demonstrating ligandability of the lc3a and lc3b adapter interface. J Med Chem. 2021;64(7):3720–3746. [DOI] [PubMed] [Google Scholar]
- Jakobi AJ, Huber ST, Mortensen SA, Schultz SW, Palara A, Kuhm T, et al. Structural basis of p62/sqstm1 helical filaments and their role in cellular cargo uptake. Nat Commun. 2020;11(1):440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji CH, Kim HY, Lee MJ, Heo AJ, Park DY, Lim S, et al. The autotac chemical biology platform for targeted protein degradation via the autophagy‐lysosome system. Nat Commun. 2022;13(1):904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalogeropulou AF, Zhao J, Bolliger MF, Memou A, Narasimha S, Molitor TP, et al. P62/sqstm1 is a novel leucine‐rich repeat kinase 2 (LRRK2) substrate that enhances neuronal toxicity. Biochem J. 2018;475(7):1271–1293. [DOI] [PubMed] [Google Scholar]
- Kirkin V, McEwan DG, Novak I, Dikic I. A role for ubiquitin in selective autophagy. Mol Cell. 2009;34(3):259–269. [DOI] [PubMed] [Google Scholar]
- Komatsu M. P62 bodies: phase separation, nrf2 activation, and selective autophagic degradation. IUBMB Life. 2022;74(12):1200–1208. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor nrf2 through inactivation of keap1. Nat Cell Biol. 2010;12(3):213–223. [DOI] [PubMed] [Google Scholar]
- Kumar AV, Mills J, Lapierre LR. Selective autophagy receptor p62/sqstm1, a pivotal player in stress and aging. Front Cell Dev Biol. 2022;10:793328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Perander M, Outzen H, Kristiansen K, Overvatn A, Michaelsen E, et al. Interaction codes within the family of mammalian phox and bem1p domain‐containing proteins. J Biol Chem. 2003;278(36):34568–34581. [DOI] [PubMed] [Google Scholar]
- Lee SJ, Pfluger PT, Kim JY, Nogueiras R, Duran A, Pages G, et al. A functional role for the p62‐erk1 axis in the control of energy homeostasis and adipogenesis. EMBO Rep. 2010;11(3):226–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Lachenmayer ML, Wu S, Liu W, Kundu M, Wang R, et al. Proteotoxic stress induces phosphorylation of p62/sqstm1 by ulk1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linares JF, Duran A, Reina‐Campos M, Aza‐Blanc P, Campos A, Moscat J, et al. Amino acid activation of mtorc1 by a pb1‐domain‐driven kinase complex cascade. Cell Rep. 2015;12(8):1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/sqstm1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44(2):279–289. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Methods for monitoring autophagy using gfp‐lc3 transgenic mice. Methods Enzymol. 2009;452:13–23 [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Morales‐Scheihing D, Butler PC, Soto C. Type 2 diabetes as a protein misfolding disease. Trends Mol Med. 2015;21(7):439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TD, Shaid S, Vakhrusheva O, Koschade SE, Klann K, Tholken M, et al. Loss of the selective autophagy receptor p62 impairs murine myeloid leukemia progression and mitophagy. Blood. 2019;133(2):168–179. [DOI] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2(9):2212–2221. [DOI] [PubMed] [Google Scholar]
- Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, et al. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep. 2010;11(1):45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohsumi Y. Historical landmarks of autophagy research. Cell Res. 2014;24(1):9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. P62/sqstm1 binds directly to atg8/lc3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145. [DOI] [PubMed] [Google Scholar]
- Park S, Han S, Choi I, Kim B, Park SP, Joe EH, et al. Interplay between leucine‐rich repeat kinase 2 (LRRK2) and p62/SQSTM‐1 in selective autophagy. PLoS One. 2016;11(9):e0163029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilli M, Arko‐Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, et al. Tbk‐1 promotes autophagy‐mediated antimicrobial defense by controlling autophagosome maturation. Immunity. 2012;37(2):223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogov VV, Nezis IP, Tsapras P, Zhang H, Dagdas Y, Noda NN, et al. Atg8 family proteins, LIR/AIM motifs and other interaction modes. Autophagy Rep. 2023;2(1):2188523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez P, de Carcer G, Sandoval IV, Moscat J, Diaz‐Meco MT. Localization of atypical protein kinase c isoforms into lysosome‐targeted endosomes through interaction with p62. Mol Cell Biol. 1998;18(5):3069–3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Diaz‐Meco MT, Nakano H, Moscat J. The atypical pkc‐interacting protein p62 channels nf‐kappab activation by the il‐1‐traf6 pathway. EMBO J. 2000;19(7):1576–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz L, Sanchez P, Lallena MJ, Diaz‐Meco MT, Moscat J. The interaction of p62 with rip links the atypical pkcs to nf‐kappab activation. EMBO J. 1999;18(11):3044–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res. 1998;21(6):629–633. [DOI] [PubMed] [Google Scholar]
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501. [DOI] [PubMed] [Google Scholar]
- Tan CT, Soh NJH, Chang HC, Yu VC. P62/sqstm1 in liver diseases: the usual suspect with multifarious identities. FEBS J. 2023;290(4):892–912. [DOI] [PubMed] [Google Scholar]
- Teramachi J, Silbermann R, Yang P, Zhao W, Mohammad KS, Guo J, et al. Blocking the zz domain of sequestosome1/p62 suppresses myeloma growth and osteoclast formation in vitro and induces dramatic bone formation in myeloma‐bearing bones in vivo. Leukemia. 2016;30(2):390–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zellner S, Behrends C. Autophagosome content profiling reveals receptor‐specific cargo candidates. Autophagy. 2021;17(5):1281–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Mun SR, Linares JF, Ahn J, Towers CG, Ji CH, et al. Zz‐dependent regulation of p62/sqstm1 in autophagy. Nat Commun. 2018;9(1):4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DATA S1. Supporting Information.