

Abstract

The catalytic translocation of a metal catalyst along a saturated hydrocarbon side chain constitutes a powerful strategy for enabling bond-forming reactions at remote, yet previously unfunctionalized, sp3 C–H sites. In recent years, Ni-catalyzed chain-walking reactions have offered counterintuitive strategies for forging sp3 architectures that would be difficult to accomplish otherwise. Although these strategies have evolved into mature tools for advanced organic synthesis, it was only recently that chemists showed the ability to control the motion at which the catalyst “walks” throughout the alkyl chain. Specialized ligand backbones, additives and a judicious choice of noninnocent functional groups on the side chain have allowed the design of “a la carte” protocols that enable regiodivergent bond-forming scenarios at different sp3 C–H sites with distinct topological surface areas. Given the inherent interest in increasing the fraction of sp3 hybridized carbons in medicinal chemistry, Ni-catalyzed regiodivergent chain-walking reactions might expedite the access to target leads in drug discovery campaigns.
Keywords: Chain-Walking, Ni-Catalysis, C(sp3)−H Functionalization, Regiodivergency, Site-Selectivity, Cross-Coupling
Introduction
Saturated hydrocarbons are the major constituents of natural gas and crude oil. In addition, these compounds constitute the main skeleton of a myriad of natural products and compounds that display important biological activities. Indeed, compounds with increased number of saturated sp3 hybridized carbons have showed recently to improve several molecular attributes that contribute to clinical success, including molecular shape (3D-structure), enhanced solubility or bioavailability, among others.1 Driven by these observations, chemists have been challenged to design de novo catalytic techniques that harness the potential of C(sp3)–H linkages as molecular handles for enabling further bond-forming reactions, including late-stage functionalization of advanced synthetic intermediates.2−4 However, the catalytic functionalization of C(sp3)–H bonds is not particularly straightforward compared to their parent C(sp2)–H analogues. Indeed, the former are considerably less acidic than their corresponding C(sp2)–H congeners, and they lack empty low-energy or filled high energy orbitals that interact with the d electrons of the transition metal. In addition, multiple, yet similar, C(sp3)–H bonds are present in an alkyl side chain, thus raising a reasonable doubt on whether a metal catalyst might be able to discriminate between C(sp3)–H bonds with similar steric or electronic considerations (Scheme 1).5 If successful, however, the ability to enable a mild and site-selective functionalization of C(sp3)–H bonds might offer the advantage of carrying orthogonally reactive motifs through a chemical synthesis while providing access to chemical libraries of utmost significance in medicinal chemistry settings.
Scheme 1. Metal-Catalyzed C(sp3)–H Functionalization.

As judged by the wealth of literature data, recent years have witnessed considerable advances in the C(sp3)–H functionalization arena. At present, the majority of these processes rely primarily on the utilization of proximal directing groups that allow exquisite control of both chemical reactivity and site-selectivity via the intermediacy of cyclometalated structures (Scheme 2).6−9 Despite the advances realized with transient, hemilabile and native directing groups, these approaches tend to favor five-membered metallacycles, hence limiting the number of C(sp3)–H sites amenable for functionalization (Scheme 2, left).10 Although other elegant alternatives have been designed, particularly in the context of metallacarbene-mediated strategies,11,12 open-shell species13,14 or enzymatic C(sp3)–H functionalization (Scheme 2, right),15,16 reactivity and selectivity are typically dictated by bond-dissociation energies or by the presence of specific substituents, thus reinforcing the need for more flexible approaches for functionalizing remote C(sp3)–H linkages.
Scheme 2. Site-Selectivity in C(sp3)–H Functionalization.

While originally designed to control the topology of polymers, chain walking reactions have recently offered new vistas for the functionalization of remote C(sp3)–H sites.17−19 Although a variety of transition metals have been shown to be competent for promoting such processes, nickel catalysts have gained considerable momentum as an enabling new technique for C(sp3)–H functionalization. Its popularity is likely ascribed to the versatility of nickel to forge a variety of sp3 architectures with exceptional ease and its redox promiscuity that allows operation via either one- or two-electron manifolds. Ni-catalyzed chain walking is typically orchestrated by a dynamic and reversible series of β-hydride elimination and migratory insertion events, thus allowing formal translocation of the nickel center ranging from one carbon to distal positions along the saturated backbone (Figure 1). Given that site-selectivity in chain walking is formally controlled by the motion at which the nickel center is displaced throughout the alkyl side-chain, it comes as no surprise that recent years have witnessed impressive developments on the ability to promote regiodivergent C(sp3)–H functionalization events by means of chain walking scenarios, with proposed mechanisms operating via either Ni(I) or Ni(II) centers. This perspective aims at summarizing the recent efforts toward this goal by utilizing different strategies, with bond-formation occurring (a) adjacent to a stabilizing functional group, (b) at terminal C(sp3)–H sites on steric grounds or (c) at methylene C(sp3)–H linkages via “interrupted” chain-walking events aided by proximal directing groups. The regiodivergent chain-walking strategies delineated in this perspective have been classified as either ligand- or substrate-controlled. These techniques appear in chronological order, and can selectively target multiple C(sp3)–H sites by changing the reaction parameters from the same precursor.
Figure 1.
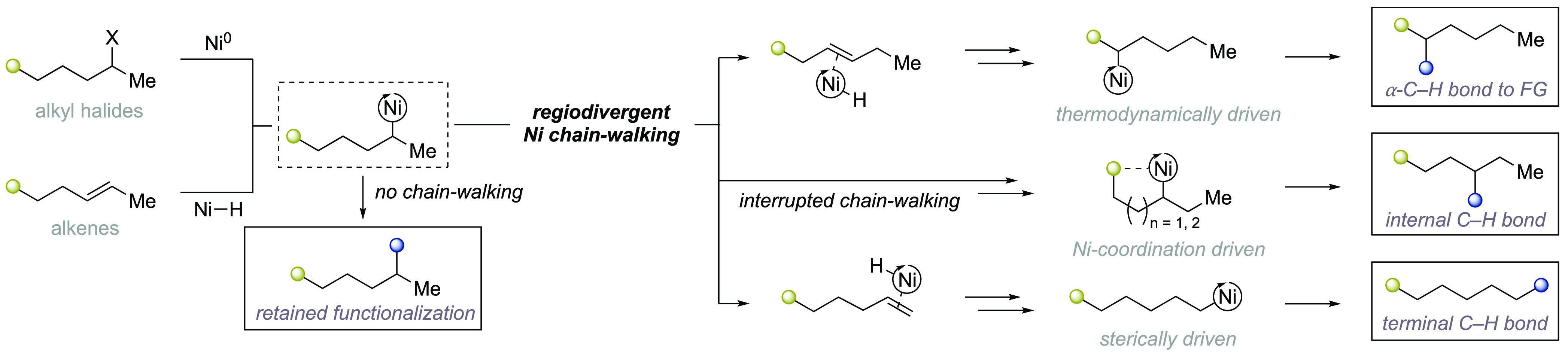
Regiodivergent Ni-catalyzed chain-walking C(sp3)–H functionalizations by utilizing alkyl halides or olefins.
Ligand-Controlled Ni-Catalyzed Chain-Walking
In 2013, Ong described an intriguing Ni-catalyzed regiodivergent scenario by reacting allyl benzene derivatives with a variety of heteroarene architectures, resulting in compounds arising from either benzyl C(sp3)–H functionalization or with a canonical anti-Markovnikov selectivity across the double bond (Scheme 3).20 While the majority of chain walking reactions with olefins typically rely on external hydride sources, this technique relied on the utilization of particularly acidic C(sp2)–H sites at the heteroarene to form the propagating Ni–H species.
Scheme 3. Site-Selective Heteroarylation of Allyl Benzene.

While the utilization of 2,2′-bipyridines and 1,10-phenanthrolines as ligands proved to be inefficient, regiodivergency could be effected by the nature of the N-heterocyclic carbene utilized. Specifically, anti-Markovnikov selectivity was illustrated by the combination of IPr and AlMe3 with benzimidazole as substrates (2a–d) whereas benzylic C(sp3)–H functionalization was found to be competent with otherwise related IMes instead (3a–d). The crucial role of AlMe3 dictating linear selectivity could be rationalized in the context of cooperative Lewis-acid/transition metal catalysis, whereas Lewis-acid coordination to the N-heteroaromatic likely outcompetes chain walking due to steric repulsion, thus guiding the Ni-complex at terminal positions.21 Subsequently, Ong described an otherwise similar behavior with differently substituted pyridines at coupling partner, resulting in functionalization at C4 by an intriguing combination of aluminum Lewis acids and low-valent Ni(0) complexes supported by N-heterocyclic carbene ligands (Scheme 4).22 As shown, the utilization of MAD [methylaluminum bis(2,6-di-tert-butyl-4-methylphenoxide] or AlMe3, respectively, led to the para-selective C(sp2)–H functionalization of pyridines. Although regioselectivity was not altered regardless of the Lewis acid utilized, its nature was critical to achieve a high para-selectivity. However, site-selectivity could be controlled by subtle ligand modulation; indeed, IMes delivered 4 with anti-Markovnikov selectivity whereas the inclusion of sterically less encumbered L1 resulted in benzylic C(sp3)–H functionalization 5. Even 3-substituted pyridines could be utilized as coupling partners, delivering the corresponding linear (4c,d) or branched (5c,d) products with excellent regioselectivities.
Scheme 4. Hydroarylation of Allyl Benzene with Pyridine.

Recently, Sun showed that regiodivergency can be enabled beyond allyl benzene derivatives (Scheme 5),23 with substrates possessing distal unsaturation from the appendant arene being competent as precursors, albeit in lower regioselectivity patterns (6b vs 6c). In addition, the reaction could be applied to heteroarenes other than benzimidazole, such as pyrazoles (6d, 7c) or furans (6e, 7d) with similar selectivity. Regiodivergency could be rationalized by steric considerations; IMes triggered rapid reversible alkene isomerization leading to functionalization of benzylic C(sp3)–H bonds whereas the utilization of bulky IPr*OMe prevented the isomerization process, thus resulting in anti-Markovnikov selectivity across the double bond.
Scheme 5. Hydroarylation of Arene-Containing Olefins.

In 2017, Martin described a regiodivergent Ni-catalyzed chain walking strategy of unactivated alkyl bromides with CO2 as coupling partner, resulting in either linear or α-branched carboxylic acids (Scheme 6).24 1,10-Phenanthroline ligands bearing substituents adjacent to the nitrogen atom (L2) were found to be critical for reactivity whereas site-selectivity could be easily modulated and controlled by a subtle modulation of the reaction temperature and the nature of the substituents on the alkyl side chain. Indeed, linear carboxylic acids could be obtained at 10 °C whereas branched-selectivity, likely the thermodynamic C–H site, was observed at 42 °C if carbonyl derivatives were utilized as coupling partners (9a vs 9e). It is worth noting that CO2 insertion was not observed in any case at the internal methylene positions of the alkyl-side chain. Notably, chain walking could be utilized for forging all-carbon quaternary centers (10d). Interestingly, control experiments with enantioenriched 8a indicated a significant preservation of the chiral stereogenic center, thus arguing against a dissociative-type mechanism in which the nickel is disengaged from the olefin backbone prior to migratory insertion. The ability of Ni/L2 to promote chain-walking can tentatively be ascribed to the intermediacy of cationic alkyl-Ni(II) species that result from a bromide dissociation at the nickel center imposed by the steric bulk around the 1,10-phenanthroline backbone, thus facilitating the agostic interaction required for enabling β–H elimination events. Moreover, the authors showed that regioconvergent carboxylation of statistical mixtures of olefins with CO2 could be realized via a tandem hydrobromination/carboxylation event. Later on, the authors described a related chain walking scenario by combining olefins feedstocks, CO2 and water, with the latter being formally employed as a hydride source, thus representing a bonus from a synthetic standpoint (bottom).25
Scheme 6. Regiodivergent Carboxylation of Alkyl-Br.

The reactivity shown in Scheme 6 could be expanded beyond carboxylation events, as illustrated by the development of a regiodivergent amidation with isocyanates as coupling partners (Scheme 7).26 Exquisite site-selectivity was achieved by a subtle modulation of the substituents at the 2,2′-bipyridine backbone; while a Ni/L4 regime resulted in linear selectivity, the utilization of a protocol based on otherwise related L5 prevented the metal translocation throughout the alkyl side-chain, resulting in α-branched amide backbones instead. The authors showed that differently substituted isocyanates could participate in the regiodivergent scenario, even in the presence of different functional groups that might a priori compromise the selectivity of the process. In addition, the utilization of tert-butyl isocyanate allowed the authors to prepare a series of mono-, di-, and trisubstituted amides by simple homologation procedures. Although speculative, the higher steric bulk around the nickel center with L4 as the ligand might facilitate the access to cationic intermediates that would facilitate the key β-hydride elimination event.
Scheme 7. Site-Selective Amidation of Alkyl Bromides.

In 2020, Zhu and Wang reported an elegant site-selective hydroamination of alkenes utilizing hydroxylamine esters as coupling partners (Scheme 8).27 The choice of the ligand backbone was critical for site-selectivity, with monodentate phosphine ligands such as SPhos leading to α-branched amines 16 whereas linear selectivity (17) was accomplished with L6 instead. Careful optimization revealed 2-methylpyridine as a key additive for boosting yield and chemoselectivity, probably due to its beneficial role as an ancillary ligand in the oxidative addition step. Unfortunately, however, a single substrate was described to undergo regiodivergent chain-walking en route to either 16a or 17a. Regioisomeric furans of types 16b and 17b could be obtained in high regioselectivities by utilizing different olefins; while 16b was prepared from a terminal alkene, the utilization of an internal olefin resulted in 17b in high regioselectivity. Unfortunately, no explanation or mechanistic experiments were given to understand the difference in reactivity between phosphine and bipyridine ligands.
Scheme 8. Regiodivergent Hydroamination of Alkenes.

In addition, Zhu showed the viability for enabling a Ni-catalyzed regiodivergent hydrothiolation of alkenes (Scheme 9).28 The authors observed an intriguing dichotomy exerted by the nature of the ligand backbone. While the utilization of bathocuproine (L3) resulted in benzyl sulfides (18a–c) when combined with HBpin in THF/MeCN, the utilization of a protocol based on 2,2′-bipyridine L6 and (MeO)2MeSiH in DMSO resulted in anti-Markovnikov products 19a–c in high yields and selectivities. While a single example demonstrated the viability for enabling such a regiodivergent scenario starting from the same precursor (18a–19a), it is worth noting that the benzylic C(sp3)–H functionalization could accommodate a broad range of functional groups, including unprotected alcohols, amines, and N-heterocycles, among others. Unfortunately, however, it is not yet clear how the ligand backbone exerts influence on site-selectivity.
Scheme 9. Regiodivergent Hydrothiolation of Alkenes.

Later on, Fang showed that a Ni-catalyzed hydrocyanation of internal alkenes could be within reach with phosphite ligands (Scheme 10).29 Intriguingly, regioselectivity was controlled by the utilization of different diastereomers of BINOL-derived ligand L7, differing in the axial chirality of the biaryl backbone (S-enantiomer illustrated). DFT calculations indicated that chain-walking was facile and reversible (ΔΔG# < 13 kcal/mol), whereas reductive elimination was found to be irreversible. Site-selectivity could be interpreted on the differences in the coordination sphere of the two Ni-complexes; while Ni/(S)-L7 features a large pocket, allowing for rapid chain-waking en route to the thermodynamic benzylic C(sp3)–H bond, Ni/(R)-L7 experiences steric repulsions between the substrate and the ligand, disrupting the otherwise beneficial coordination of the aryl group with Ni. Consequently, the less sterically encumbered transition state leads to cyanation at the terminal C(sp3)–H bond. As shown in Scheme 11, regiodivergent hydrocyanation occurred with a broad scope and high regioselectivities. In line with other disclosures in the area of chain-walking, regioselectivity can also be accomplished with statistical mixtures of olefins, leading to either branched (21c) or linear nitriles (22c). It is worth noting that the utilization of (S)-L7 resulted in 21b with a rather promising 77% ee, thus representing a significant step forward for the development of highly enantioselective chain-walking events.
Scheme 10. Regiodivergent Hydrocyanation of Alkenes.

Scheme 11. Coupling of Allyl Benzenes, BnCl and B2pin2.

Recently, Yin described an interesting three-component Ni-catalyzed chain-walking aided by Co(II) porphyrin Co-1 (Scheme 11).30 Specifically, the authors showed that allyl benzenes underwent carboboration with B2pin2 and benzyl chlorides. In line with Weix31 and Reisman’s reports,32 the use of cobalt additives was believed to stabilize the transient radical species. Strikingly, the nature of the solvent exerted a profound influence on site-selectivity; 1,4-dioxane was found to be suited for benzylic C(sp3)–H functionalization, whereas DMA or NMP resulted in a 1,1-difunctionalization event. While lower yields and regioselectivities were found for the former protocol (23a–c), excellent yields and site-selectivities were obtained in the latter (24a–c).
Recently, Yin demonstrated the crucial role of lithium bases such as MeOLi for chain-walking scenarios with cyclic alkenes; interestingly, the corresponding sodium counterparts predominantly led to the formation of 1,2-difunctionalization products.33 Subsequent studies by Yin extended this regiodivergent method to alkenyl bromides, delivering 1,1- or 1,3-difuncitonalized products with excellent regioselectivities, even without cobalt porphyrin additives (Scheme 12).34 Neither the arene electron-density of the substrate (25a–c, 26a–c) nor the presence of ortho-substituents (25d, 26d) altered the regioselectivity, evidencing the robustness of this nickel chain-walking regime.
Scheme 12. Site-Selective Carboboration of Allyl Benzenes.

The authors conducted DFT studies to elucidate the striking differences in the reactivity exerted by two similar ligand backbones. Theoretical calculations on β–H elimination enabled by Ni(II) intermediates concluded that the α-boronate products (26) are thermodynamically favored, while benzylic C(sp3)–H functionalization is kinetically driven (25). The authors claimed that a kinetic equilibrium is established during Ni-chain-walking. However, deuterium experiments elucidated little deuterium scrambling along the hydrocarbon fragment, but rather a very selective 1,2-Ni shift for benzylic alkylation (27, Scheme 13).
Scheme 13. Deuterium-Labeling Studies on the Regiodivergent Ni-Catalyzed Carboboration with Alkenyl Bromides.

Recently, photoredox-catalysis has offered an entry point to enable regiodivergent alkylation of alkyl bromides with α-silyl amines counterparts (Scheme 14).35 Specifically, the utilization of 1,10-phenantroline L11 under a dual Ir/Ni metallaphotoredox scenario led to retentive functionalization. Such a protocol tolerated a wide number of functional groups, including esters, amides or nitriles (30a–c) or cyclic backbones (30d) in excellent site-selectivities (>20:1). Intriguingly, the utilization of L3 in combination with 4-CzIPN resulted in a chain walking adjacent to stabilizing functional groups (31a–c). Notably, cyclobutane 30d was within reach, and the method could be extended beyond cyclic amines with similar ease and site-selectivities. Experimental and theoretical studies supported a mechanistic scenario initiated by single-electron transfer oxidation of the α-silyl amine followed by reduction of Ni(II) to Ni(0). Oxidative addition of the alkyl bromide to the latter followed by chain walking and interception of the α-amino radical sets the stage for a reductive elimination, leading to the targeted product while recovering back the propagating Ni species.
Scheme 14. Regiodivergent Ni-Catalyzed Alkylation with α-Silyl Amines under Photoredox-Catalytic Conditions.

Chelation-Controlled Ni-Catalyzed Chain-Walking
Prompted by an example described by Martin on the coupling of N-Boc 2,5-dihydropyrrole with haloboranes,36 Hu described a regiodivergent hydroalkylation of similar substrates aided by carbamate protecting groups (Scheme 15).37 Interestingly, subtle differences at the pyrox backbone led to either olefin functionalization (34) or nickel translocation at C2 (33). In line with recent developments in chain-walking reactions, the inclusion of substituents adjacent to the nitrogen atom at the pyridine core was found to be critical for enabling the nickel translocation. Secondary (33a,b, 34a,b) and primary alkyl iodides (33c, 34c) could be used as electrophiles for forging sp3 C–C linkages. Unfortunately, no mechanistic investigations were conducted to shed light on the role of the N-protecting group, thus questioning whether C2 stabilization arises from chelation of the carbamate fragment or from electronic considerations of the α-C–Ni bond to nitrogen.
Scheme 15. Hydroalkylation of N-Protected Pyrrolines.

Despite the existing precedents from Martin and Hu on chain-walking events at internal C(sp3)–H bonds in cyclic, N-protected heteroarenes, the means to promote nickel translocation in aliphatic side-chains at internal C(sp3)–H bonds was an unmet synthetic challenge. In 2021, Yu described a Ni-catalyzed interrupted chain-walking by utilizing thioethers as chelating groups (Scheme 16).38 As anticipated, subtle differences at the 1,10-phenanthroline backbone were critical for dictating the regioselectivity. The utilization of L14 facilitated a C(sp3)–N bond-formation at the γ-position, whereas less-sterically encumbered ligand L15 resulted in a 1,2-hydroamination across the olefin backbone. The authors argued that site-selectivity might arise by forming five- or six-membered nickelacycles facilitated by the different steric environments at L14 and L15. Regiodivergency proved to be compatible with a range of protected thioether scaffolds (36d, 37b). However, a significant loss in regioselectivity was observed when placing the reactive alkene at long-range (36a vs 36b-c), which was rationalized due to diminished chelation in the hydrometalation step.
Scheme 16. Hydroamidation of Thioethers.

Driven by seminal contributions from Koh39 and Hong40 on β- and γ-selective chain-walking events, Lu and Fu reported in 2022 an alternative strategy for interrupted nickel chain-walking using 8-aminoquinolinamides as directing groups (38, Scheme 17).41 While Markovnikov hydroalkylation (40) occurred in the presence of ethanolamine, a Ni translocation occurred with the Ni(PPh3)2Cl2 precatalyst to obtain β-functionalized products (39), albeit in lower yields. Alkyl iodides bearing esters, nitriles, protected amines, indoles, aryl bromides, and free alcohols could be tolerated without affecting the γ-regioselectivity (40a–c). However, only n-butyl iodide was used to show the potential of a regiodivergent β-alkylation (39a–c). Interestingly, the utilization of ethylenediamine, butylamine, or aniline in lieu of ethanolamine delivered the targeted products in good yields but moderate regioselectivities. Further ligand optimization revealed that 80% ee could be achieved with L16, which is a significant achievement in the area of asymmetric hydroalkylation events.
Scheme 17. Hydroalkylation of 8-Aminoquinolinamides.

While the previous examples constituted a proof-of-concept for enabling Ni-catalyzed interrupted chain-walking at internal C(sp3)–H bonds, the utilization of protected thiols and engineered amides as chelating moieties might hamper the applicability of these strategies. Indeed, chemists have been challenged to design alternatives with native directing groups for enabling interrupted chain-walking scenarios. For example, Lu described a temperature-controlled regiodivergent hydroalkylation using bisoxazoline ligand L17 (Scheme 18).42 Thus, nickel translocation could be promoted either at the α or β-position by promoting the reaction at 10 or 100 °C, with site-selectivity arising from the formation of five- or six-membered nickelacycle intermediates. Notably, good reactivity could be accomplished when the olefin was located at distal positions from the amine moiety (42a,b). Note, however, that selectivity was highly dependent on the protecting group utilized at the amine backbone; while a 12:1 ratio was observed for N-pivalates (43a,b), otherwise related benzoates eroded the targeted β-selectivity (40c). In addition, N-Boc, N-acetate, or N-phthalimide derivatives resulted in an α-functionalization. Good reactivity was achieved with geminal and internal olefins, even in the presence of benzylic C(sp3)–H sites (42e, 43e). In addition, high enantioselectivities, regioselectivities, and yields were obtained when utilizing chiral ligands (45a,b). Note, however, that these results could only be obtained for a Markovnikov alkylation rather than a chain-walking scenario.
Scheme 18. Lu’s Hydroalkylation of N-Protected Amines.

Wang independently reported an otherwise identical transformation using a ligand-controlled approach at room temperature (Scheme 19).43 With very similar regioisomeric ratios obtained for α- and β-alkylated products, the reported substrate scope proved to tolerate a wide range of functional groups in the electrophile backbone and included the successful reactivity of secondary alkyl iodides. Nevertheless, the selectivity for β-alkylation was only demonstrated in a discrete chain-walking regime using homoallylic amine substrates with L18.
Scheme 19. Wang’s Hydroalkylation of N-Protected Amines.

Mechanistic experiments with L18 showed that alkene isomerization occurred en route to protected enamines (41a′), thus setting the basis for promoting α-alkylation (Scheme 20). On the contrary, L19 delivered a slower kinetic rate of Ni-migration comparable to the overall cross-coupling, with low amounts (<10%) of other alkene regioisomers detected, thus leading to β-alkylation, likely via the six-membered ring nickelacycles. In line with the initial results reported by Hu and Martin,33,34 α-alkylation was obtained with pyrroline 41b.
Scheme 20. Mechanistic Studies for the Ni-Catalyzed Hydroalkylation of N-Protected Amines.

Shortly after, Shu described the α- and β-alkylation of protected allyl amines by utilizing alkyl iodides as electrophiles (45 and 46, Scheme 21).44 Interestingly, α- and β-selectivity was dictated by subtle differences at the bis-oxazoline backbone, whereas the inclusion of cobalt catalysts enabled an anti-Markovnikov hydroalkylation of the pending olefin (47a–c). The observed site-selectivity was computationally investigated, revealing the preferred formation of a five-membered nickelacycle when using L20 (ΔG# = −3.2 kcal/mol compared to its six-membered analogue). As for L21, the six-membered metallacycle is thermodynamically favored by ΔG# = +1.3 kcal/mol, preventing further chain walking and affording the β-alkylated product.
Scheme 21. Shu’s Hydroalkylation of N-Protected Amines.

More recently, Shu described that a similar methodology could be applicable to functionalize amides containing an appendant C–Br bond (Scheme 22).45 Importantly, the reaction allowed for dictating ipso-, γ-, or β-functionalization by subtle control of the reaction conditions and the ligand backbone. Box-type ligands proved to be suited for β-alkylation (49), whereas bipyridine ligands were particularly efficient for γ-functionalization (50). Furthermore, tridentate pybox ligands delivered ipso-functionalized products (51), likely due to the absence of vacant sites in the nickel complex that are amenable to β-hydride elimination. Notably, the directing amide can be mono- or disubstituted with either aromatic rings or alkyl fragments without affecting the regioselectivity of the process. It is worth noting that all amide substrates displayed an internal C(sp2)–Br bond. Therefore, it remains uncertain whether the ligands employed might be capable of overcoming the facile ipso-reactivity of nickel complexes when utilizing terminal, primary C(sp2)–Br bonds. As judged by the data provided by the authors, both primary and secondary alkyl bromides could be utilized for the other counterpart, resulting in some regioselectivity issues only when targeting γ-functionalization (50c-d).
Scheme 22. Regiodivergent Cross-Electrophile Coupling of Bromine-Containing Amides.

In parallel, Martin leveraged unsaturated amides as directing groups to promote a regiodivergent hydroalkylation of olefins with redox-active pyridinium salts as electrophiles, resulting in the formation of three different products arising from functionalization at the β-, γ-, and terminal position (Scheme 23).46 As shown, subtle differences at the pyrox-type ligand were critical for success. While β-alkylation (49) was found with L26 possessing a methyl substituent adjacent to the pyridyl ring, a γ-alkylation (50) was found instead for 2-oxazoquinoline L27. It is worth noting that a wide variety of native amides could participate in the targeted reaction, including compounds possessing N-aryl substituents, ring-strained heterocycles, or Weinreb amides. Similarly, terminal, internal, or geminal alkenes all delivered the targeted products with similar site-selectivities, even at long-range. The reaction could be extended beyond pyridiniums as alkyl iodides could also be employed as electrophilic partners (49h). It is worth noting that compounds possessing a free NH group such as natural capsaicin could be selectively functionalized at the β- or γ-positions by selecting the adequate ligand (49i, 50i).
Scheme 23. Martin’s Hydroalkylation of Unsaturated Amides.

While β-selectivity might a priori be achieved via the intermediacy of α,β-unsaturated amides arising from olefin isomerization followed by 1,4-addition of in situ generated alkyl radicals, the lack of deuterium scrambling observed for 49e-d2 indicates otherwise (Scheme 24). In line with this observation, enantioenriched amide 52b (85% ee) possessing a α-methyl substituent could be coupled with primary alkyl iodides to furnish compound 49j with preservation of the chiral integrity. Careful monitoring of the reaction revealed no evidence for acrylamide derivatives or intermediate olefins, an observation that is in contrast to previously developed chain-walking scenarios (Scheme 20). Taking these results into consideration, the authors argued that site-selectivity arises from the intermediacy of five- or six-membered nickelacycle intermediates by coordination of the native amide backbone to the nickel center.
Scheme 24. Mechanistic Experiments.

A recent example by Cao and Wang described the development of a regiodivergent nickel-catalyzed chain-walking hydroamination reaction with protected amines and N–O electrophiles (Scheme 25).47 Interestingly, two a priori similar ligands L3 and L6 were responsible for dictating the site-selectivity switch. With N-benzoyl protected amines affording better regioisomeric control (53a,b, 54a,b), the reaction was compatible with both acyclic and N-heterocycles. However, a direct comparison was elusive as only dibenzyl amines delivered excellent regioselectivities for β-amination, whereas α-amination predominantly used morpholine as the N–O electrophile.
Scheme 25. Hydroamination with N–O Electrophiles.

Interestingly, mechanistic experiments showed that exposure of 41a to the standard reaction conditions without electrophile delivered 1-carbon olefin isomerization for ligand L6, whereas a 1:1 ratio of protected enamine 41c and allylamine 41d was observed with L3, likely suggesting the intervention of nickelacycles, respectively (Scheme 26). This was further evidenced by reacting enamine 41c under both catalytic regimes; L6 delivered a 3:1 rr favoring β-amination, whereas L3 afforded α-functionalization almost exclusively.
Scheme 26. Ligand Effects in Regiodivergent Hydroamination.

In addition, the authors reported the asymmetric β-functionalization of protected amines (55) and directed Markovnikov olefin hydroamination (56) (Scheme 27). Importantly, seemingly similar hindered box-type ligands were responsible for dictating the selectivity of these reactions, with the inclusion of PMe3 being essential for obtaining β-aminated products in high enantiomeric excesses. While poor yields were obtained with internal olefins (55c), it is worth noting that the development of asymmetric transformations within the context of nickel chain-walking is extremely rare. Therefore, the ability to obtain products 55a-c stands as a testament to the impact that chiral ligands might have when controlling olefin isomerization in asymmetric Ni-catalyzed chain-walking reactions.
Scheme 27. Regiodivergent Asymmetric Hydroamination.

Conclusion
Ni-catalyzed chain walking reactions have recently offered new vistas for enabling functionalization at remote C(sp3)–H sites prior to carbon-carbon or carbon-heteroatom bond-formation. The dynamic and reversible metal translocation throughout the alkyl side chain enables functionalization (i) adjacent to a stabilizing functional group, (ii) at the terminal site of an alkyl chain based on steric grounds, or (iii) at internal C(sp3)–H sites via an interrupted chain-walking event. Site-selectivity can be modulated by the ligand backbone or by the fine-tuning of the reaction conditions, including the utilization of additives or a subtle temperature control. While it is evident that remarkable advances have been realized in the context of Ni-catalyzed chain-walking reactions, these technologies are in the midst of a transition that might impact the practice of organic synthesis. Indeed, a number of daunting challenges remain in these scenarios: (i) chain-walking regimes with polyolefin backbones still constitutes “terra incognita”; (ii) the majority of chain-walking reactions with olefin counterparts are initiated by nickel hydride intermediates. Therefore, the potential applicability of these reactions beyond olefin hydrofunctionalization has not yet been fully assessed; (iii) the inclusion of branched substituents typically hinders the nickel translocation, whereas the utilization of densely substituted olefins still remains particularly problematic; (iv) despite the enormous potential that catalytic asymmetric chain-walking has for forging enantioenriched sp3 architectures, a limited number of these methods have been described in the literature.
There is ample consensus, however, that the major challenge that chain-walking techniques should overcome lies in the existing ambiguity behind the nature of the nickel species that enable the dynamic displacement throughout the alkyl side-chain. Indeed, speculation remains on whether Ni(I), Ni(II), or even Ni(III) might be responsible for the motion that ultimately results in bond-formation at remote C(sp3)–H sites. This is largely due to the fleeting nature of short-lived, yet exceptionally sensitive, nickel entities, the presence of low-coordinate species, and the redox promiscuity of nickel complexes supported by nitrogen-containing ligands, thus constituting serious barriers for studying the intricacies of these processes. In addition, a non-negligible number of chain-walking reactions require specific substitution patterns at the ligand backbone and enigmatic additives for these processes to occur efficiently. Taking into consideration the recent efforts made at the molecular level for understanding the nickel speciation in a myriad of catalytic processes, it is fair to assume that these challenges will be eventually met in the not so distant future, thus setting the basis to design even more powerful techniques that might forge exceedingly complex sp3 architectures via remote functionalization.
Acknowledgments
We thank ICIQ, FEDER/MCI PID2021-123801NB-I00 and European Research Council (ERC) under European Union’s Horizon 2020 research and innovation program (grant agreement 883756) for financial support. J.R. and F.-L.H. thank the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement 101105032 and Alexander von Humboldt Foundation for a Feodor Lynen Research Fellowship.
Author Contributions
# J.R. and F.-L.H. contributed equally to this work.
The authors declare no competing financial interest.
References
- Lovering F.; Bikker J.; Humblet C. Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem. 2009, 52, 6752–6756. 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- Guillemard L.; Kaplaneris N.; Ackermann L.; Johansson M. J. Late-stage C–H functionalization offers new opportunities in drug discovery. Nat. Rev. Chem. 2021, 5, 522–545. 10.1038/s41570-021-00300-6. [DOI] [PubMed] [Google Scholar]
- Docherty J. H.; Lister T. M.; Mcarthur G.; Findlay M. T.; Domingo-Legarda; Kenyon J.; Choudhary S.; Larrosa I. Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. 10.1021/acs.chemrev.2c00888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellotti P.; Huang H.-M.; Faber T.; Glorius F. Photocatalytic Late-Stage C–H Functionalization. Chem. Rev. 2023, 123, 4237–4352. 10.1021/acs.chemrev.2c00478. [DOI] [PubMed] [Google Scholar]
- Bergman R. G. C–H Activation. Nature 2007, 446, 391–393. 10.1038/446391a. [DOI] [PubMed] [Google Scholar]
- Huang Z.; Lim H. N.; Mo F.; Young M. C.; Dong G. Transition Metal-Catalyzed Ketone-Directed or Mediated C–H Functionalization. Chem. Soc. Rev. 2015, 44, 7764–7786. 10.1039/C5CS00272A. [DOI] [PubMed] [Google Scholar]
- He J.; Wasa M.; Chan K. S. L.; Shao Q.; Yu J.-Q. Palladium-Catalyzed Transformations of Alkyl C–H Bonds. Chem. Rev. 2017, 117, 8754–8786. 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Dong G. Sp3 C–H Activation via Exo-Type Directing Groups. Chem. Sci. 2018, 9, 1424–1432. 10.1039/C7SC04768A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rej S.; Ano Y.; Chatani N. Bidentate Directing Groups: An Efficient Tool in C–H Bond Functionalization Chemistry for the Expedient Construction of C–C Bonds. Chem. Rev. 2020, 120, 1788–1887. 10.1021/acs.chemrev.9b00495. [DOI] [PubMed] [Google Scholar]
- Mingo M. M.; Rodríguez N.; Gómez Arrayás R.; Carretero J. C. Remote C(sp3)–H functionalization via catalytic cyclometallation: beyond five-membered ring metallacycle intermediates. Org. Chem. Front. 2021, 8, 4914–4946. 10.1039/D1QO00389E. [DOI] [Google Scholar]
- Davies H. M. L.; Morton D. Recent Advances in C–H Functionalization. J. Org. Chem. 2016, 81, 343–350. 10.1021/acs.joc.5b02818. [DOI] [PubMed] [Google Scholar]
- He Y.; Huang Z.; Wu K.; Ma J.; Zhou Y.-G.; Yu Z. Recent Advances in Transition-Metal-Catalyzed Carbene Insertion to C–H Bonds. Chem. Soc. Rev. 2022, 51, 2759–2852. 10.1039/D1CS00895A. [DOI] [PubMed] [Google Scholar]
- Yi H.; Zhang G.; Wang H.; Huang Z.; Wang J.; Singh A. K.; Lei A. Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev. 2017, 117, 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- Sarkar S.; Cheung K. P. S.; Gevorgyan V. C–H functionalization reactions enabled by hydrogen atom transfer to carbon-centered radicals. Chem. Sci. 2020, 11, 12974–12993. 10.1039/D0SC04881J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. C.; Coelho P. S.; Arnold F. H. Enzymatic Functionalization of Carbon–Hydrogen Bonds. Chem. Soc. Rev. 2011, 40, 2003–2021. 10.1039/C0CS00067A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham N. P.; Arnold F. H. Nature’s Machinery, Repurposed: Expanding the Repertoire of Iron-Dependent Oxygenases. ACS Catal. 2020, 10, 12239–12255. 10.1021/acscatal.0c03606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasseur A.; Bruffaerts J.; Marek I. Remote Functionalization through Alkene Isomerization. Nat. Chem. 2016, 8, 209–219. 10.1038/nchem.2445. [DOI] [PubMed] [Google Scholar]
- Sommer H.; Juliá-Hernández F.; Martin R.; Marek I. Walking Metals for Remote Functionalization. ACS Cent. Sci. 2018, 4, 153–165. 10.1021/acscentsci.8b00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S.; Patel S.; Chatterjee I. Chain-walking reactions of transition metals for remote C–H bond functionalization of olefinic substrates. Chem. Commun 2021, 57, 11110–11130. 10.1039/D1CC04370F. [DOI] [PubMed] [Google Scholar]
- Lee W.-C.; Wang C.-H.; Lin Y.-H.; Shih W.-C.; Ong T.-G. Tandem Isomerization and C–H Activation: Regioselective Hydroheteroarylation of Allylarenes. Org. Lett. 2013, 15, 5358–5361. 10.1021/ol402644y. [DOI] [PubMed] [Google Scholar]
- For a selected example of pyridine functionalization aided by Lewis acids:; Nakao Y.; Yamada Y.; Kashihara N.; Hiyama T. Selective C-4 Alkylation of Pyridine by Nickel/Lewis Acid Catalysis. J. Am. Chem. Soc. 2010, 132, 13666–13668. 10.1021/ja106514b. [DOI] [PubMed] [Google Scholar]
- Lee W.-C.; Chen C.-H.; Liu C.-Y.; Yu M.-S.; Lin Y.-H.; Ong T.-G. Nickel-Catalysed Para C-H Activation of Pyridine with Switchable Regioselective Hydroheteroarylation of Allylarenes. Chem. Commun. 2015, 51, 17104–17107. 10.1039/C5CC07455J. [DOI] [PubMed] [Google Scholar]
- Imran S.; Jin W.-H.; Li R.-P.; Ismaeel N.; Sun H.-M. Ligand-Controlled Nickel-Catalyzed Tandem Isomerization/Regiodivergent Hydroheteroarylation of α-Alkenes with Heteroarenes. Org. Lett. 2022, 24, 8875–8879. 10.1021/acs.orglett.2c03689. [DOI] [PubMed] [Google Scholar]
- Juliá-Hernández F.; Moragas T.; Cornella J.; Martin R. Remote Carboxylation of Halogenated Aliphatic Hydrocarbons with Carbon Dioxide. Nature 2017, 545, 84–88. 10.1038/nature22316. [DOI] [PubMed] [Google Scholar]
- Gaydou M.; Moragas T.; Juliá-Hernández F.; Martin R. Site-Selective Catalytic Carboxylation of Unsaturated Hydrocarbons with CO2 and Water. J. Am. Chem. Soc. 2017, 139, 12161–12164. 10.1021/jacs.7b07637. [DOI] [PubMed] [Google Scholar]
- Tortajada A.; Menezes Correia J. T.; Serrano E.; Monleón A.; Tampieri A.; Day C. S.; Juliá-Hernández F.; Martin R. Ligand-Controlled Regiodivergent Catalytic Amidation of Unactivated Secondary Alkyl Bromides. ACS Catal. 2021, 11, 10223–10227. 10.1021/acscatal.1c02913. [DOI] [Google Scholar]
- Zhang Y.; He J.; Song P.; Wang Y.; Zhu S. Ligand-Enabled NiH-Catalyzed Migratory Hydroamination: Chain Walking as a Strategy for Regiodivergent/Regioconvergent Remote sp3 C–H Amination. CCS Chem. 2021, 3, 2259–2268. 10.31635/ccschem.020.202000490. [DOI] [Google Scholar]
- Zhang Y.; Xu X.; Zhu S. Nickel-Catalysed Selective Migratory Hydrothiolation of Alkenes and Alkynes with Thiols. Nat. Commun. 2019, 10, 1752. 10.1038/s41467-019-09783-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J.; Jiao M.; Ni J.; Yu R.; Cheng G.-J.; Fang X. Nickel-Catalyzed Migratory Hydrocyanation of Internal Alkenes: Unexpected Diastereomeric-Ligand-Controlled Regiodivergence. Angew. Chem., Int. Ed. 2021, 60, 1883–1890. 10.1002/anie.202011231. [DOI] [PubMed] [Google Scholar]
- Li Y.; Wei H.; Yin G. Nickel-Catalyzed Migratory Benzylboration of Allylbenzenes. Tetrahedron Lett. 2022, 100, 153889 10.1016/j.tetlet.2022.153889. [DOI] [Google Scholar]
- Ackerman L. K. G.; Anka-Lufford L. L.; Naodovic M.; Weix D. J. Cobalt Co-Catalysis for Cross-Electrophile Coupling: Diarylmethanes from Benzyl Mesylates and Aryl Halides. Chem. Sci. 2015, 6, 1115–1119. 10.1039/C4SC03106G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstra J. L.; Cherney A. H.; Ordner C. M.; Reisman S. E. Synthesis of Enantioenriched Allylic Silanes via Nickel-Catalyzed Reductive Cross-Coupling. J. Am. Chem. Soc. 2018, 140, 139–142. 10.1021/jacs.7b11707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W.; Bao Y.; Lu L.; Han Z.; Zhong Y.; Zhang R.; Li Y.; Yin G. Base-Modulated 1,3-Regio- and Stereoselective Carboboration of Cyclohexenes. Angew. Chem., Int. Ed. 2023, 62, e202308041 10.1002/anie.202308041. [DOI] [PubMed] [Google Scholar]
- Sun C.; Ding C.; Yu Y.; Li Y.; Yin G. Ligand-Modulated Regiodivergent Alkenylboration of Allylarenes: Reaction Development and Mechanistic Study. Fundamental Research 2023, 10.1016/j.fmre.2023.03.016. [DOI] [Google Scholar]
- Wang W.; Yan X.; Ye F.; Zheng S.; Huang G.; Yuan W. Nickel/Photoredox Dual-Catalyzed Regiodivergent Aminoalkylation of Unactivated Alkyl Halides. J. Am. Chem. Soc. 2023, 145, 23385–23394. 10.1021/jacs.3c09705. [DOI] [PubMed] [Google Scholar]
- Sun S-Z.; Börjesson M.; Martin-Montero R.; Martin R. Site-Selective Ni-Catalyzed Reductive Coupling of α-Haloboranes with Unactivated Olefins. J. Am. Chem. Soc. 2018, 140, 12765–12769. 10.1021/jacs.8b09425. [DOI] [PubMed] [Google Scholar]
- Qian D.; Hu X. Ligand-Controlled Regiodivergent Hydroalkylation of Pyrrolines. Angew. Chem., Int. Ed. 2019, 58, 18519–18523. 10.1002/anie.201912629. [DOI] [PubMed] [Google Scholar]
- Du B.; Ouyang Y.; Chen Q.; Yu W.-Y. Thioether-Directed NiH-Catalyzed Remote γ-C(sp3)–H Hydroamidation of Alkenes by 1,4,2-Dioxazol-5-Ones. J. Am. Chem. Soc. 2021, 143, 14962–14968. 10.1021/jacs.1c05834. [DOI] [PubMed] [Google Scholar]
- Chen X.; Rao W.; Yang T.; Koh M. J. Alkyl halides as both hydride and alkyl sources in catalytic regioselective reductive olefin hydroalkylation. Nat. Commun. 2020, 11, 5857. 10.1038/s41467-020-19717-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C.; Seo H.; Jeon J.; Hong S. γ-Selective C(sp3)–H amination via controlled migratory hydroamination. Nat. Commun. 2021, 12, 5657. 10.1038/s41467-021-25696-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.-X.; Xu Y.-T.; Zhang Z.-L.; Lu X.; Fu Y. NiH-Catalysed Proximal-Selective Hydroalkylation of Unactivated Alkenes and the Ligand Effects on Regioselectivity. Nat. Commun. 2022, 13, 1890. 10.1038/s41467-022-29554-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-W.; Liu D.-G.; Chang Z.; Li Z.; Fu Y.; Lu X. Nickel-Catalyzed Switchable Site-Selective Alkene Hydroalkylation by Temperature Regulation. Angew. Chem., Int. Ed. 2022, 61, e202205537 10.1002/anie.202205537. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Zhu Y.; Liu M.; Xie L.; Liang J.; Shi H.; Meng X.; Chen Z.; Han J.; Wang C. Ligand-Controlled NiH-Catalyzed Regiodivergent Chain-Walking Hydroalkylation of Alkenes. Angew. Chem., Int. Ed. 2022, 61, e202204716 10.1002/anie.202204716. [DOI] [PubMed] [Google Scholar]
- Yang P.-F.; Shu W. Orthogonal Access to α-/β-Branched/Linear Aliphatic Amines by Catalyst-Tuned Regiodivergent Hydroalkylations. Angew. Chem., Int. Ed. 2022, 61, e202208018 10.1002/anie.202208018. [DOI] [PubMed] [Google Scholar]
- Zhao W-T.; Meng H.; Lin J-N.; Shu W. Ligand-Controlled Nickel-Catalyzed Regiodivergent Cross-Electrophile Alkyl-Alkyl Couplings of Alkyl Halides. Angew. Chem., Int. Ed. 2023, 62, e202215779 10.1002/anie.202215779. [DOI] [PubMed] [Google Scholar]
- Rodrigalvarez J.; Wang H.; Martin R. Native Amides as Enabling Vehicles for Forging sp3-sp3 Architectures via Interrupted Deaminative Ni-Catalyzed Chain-Walking. J. Am. Chem. Soc. 2023, 145, 3869–3874. 10.1021/jacs.2c12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L.; Liang J.; Bai H.; Liu X.; Meng X.; Xu Y-Q.; Cao Z-Y.; Wang C. Ligand-Controlled NiH–Catalyzed Regiodivergent and Enantioselective Hydroamination of Alkenyl Amides. ACS Catal. 2023, 13, 10041–10047. 10.1021/acscatal.3c01845. [DOI] [Google Scholar]