

Abstract

The article shows that the definition of the HOMA index of geometrical aromaticity satisfies the axioms of a similarity function between the examined and benzene ring. Consequently, for purely mathematical reasons, the index works exceptionally well as an index of aromaticity: it expresses a geometric similarity to the archetypal aromatic benzene. Thus, if the molecule is geometrically similar to benzene, then it is also chemically similar, and therefore, it is aromatic. However, the similarity property legitimizes using the HOMA-like indices to express similarity to molecules other than benzene, whether cyclic or linear and existing or hypothetical. The paper demonstrates an example of HOMA-similarity to cyclohexane, which expresses a (relaxed)-saturicity property not accompanied by strong structural strains or steric hindrances. Further, it is also shown that the HOMA index can evaluate the properties of whole molecules, such as 25 unbranched catacondensed isomers of hexacene. The index exhibits a significant quadratic correlation with the total energy differences of planar isomers from which the nonplanar ones deviate. Moreover, the HOMA index of hexacene isomers significantly correlates with the Kekulé count connected to the resonance energy in the Hückel approximation. As a result, the study shows that the HOMA index can be used not only for aromaticity analyses but also as a general chemical descriptor applicable to rings, chains, composed molecular moieties, or even whole molecules.
1. Introduction
Aromaticity is not an observable molecular property, although it is essential in modern chemistry. It manifests itself in multiple molecular characteristics and is thus defined in various ways, highlighting its unique features. Different definitions expose the specifics of molecular geometry, reactivity, additional energy stabilization, magnetic currents, and some other features and sometimes disagree. Therefore, it is imperative to understand every aspect of the aromaticity definition. This study is focused on analyzing the definition of the Harmonic Oscillator Model of Aromaticity, HOMA, and geometrical aromaticity index.1 It is one of the most successful and widely used measures of aromaticity2−4 despite its foundation being grounded on a simple approximative quantum chemical model. Nevertheless, at the very outset, the authors stated that the index “is quite independent/in principle/of quantum chemical models”.1 Hereafter, we demonstrate why it is the absolutely correct statement.
For hydrocarbon rings, HOMA is defined as follows
| 1 |
where Ri and Ropt (Å) stand for the i-th CC bond length in the analyzed ring and the reference benzene ring (Ropt = RB) for which HOMA = 1, respectively, n is number of CC bonds in the ring, and α = 257.7 Å–2 normalizes the index to be unitless and equal to 0 for a hypothetical perfectly alternating Kekulé cyclohexatriene ring.
HOMA can be reformulated in terms of two destabilizing factors: energetic (EN) and geometric (GEO), disrupting the perfect aromaticity, for which HOMA = 1.5−7
| 2 |
| 3 |
| 4 |
where Rav is the averaged bond length in the examined ring, EN is a normalized squared difference of the bond length arithmetic means, and GEO is a normalized bond variance in the examined molecule.
First defined for aromatic hydrocarbons, HOMA was then parametrized in several ways to evaluate the geometrical aromaticity of heterocyclic structures:3,7−11 The HOMA index has also been defined as a function of ring bond properties other than bond lengths (R) like parameters of the electron density in bond critical points.12−14 Interestingly, the HOMA index asymptotically converges as the number n of molecular units increases. For example, for alkanes, spiro cyclopropanes, polyynes, and allenes, the index decreases with the increasing n and converges to negative values from −4 to −2.13 This can suggest an increase in the degree of insulation with an increase in the number of units. For alkenes and polythiophenes, the index increases and converges to values above 0.6, while for polypyrrole, it reaches ca. 0.8.13,15 This can suggest the increase of π-electron delocalization with an increase in the number of units.13,16 The HOMA indices were also used to study linear and branched alkanes, and for n-alkanes, a good correlation with the boiling points was found. On the other hand, for n-alkane constitutional isomers, the HOMA index tends to increase with the boiling points from the most branched to the most extended isomer.17
The above data indicate that the HOMA index is more than just a geometrical aromaticity index. However, why it may be so is known.16 First, the HOMA index is connected to a distance in the abstract n-dimensional molecular space and, second, to a statistical value such as a variance.17 Third, the association of HOMA with aromaticity is only through Ropt, taken from the aromaticity standard: benzene. The α factor establishes HOMA = 0 for a nonaromatic, hypothetical cyclohexatriene molecule.
This study shows that HOMA expresses a similarity between the studied and reference moiety. If a molecule is geometrically similar to benzene, HOMA-similar, then it also is chemically similar and, thus, aromatic. By replacing benzene with a different reference molecule, the HOMA index with this new reference provides geometrical similarity to the new reference. Consequently, the similarity of the chemical properties of the new reference is also satisfied. Thus, the HOMA index referred to benzene measures aromaticity, but when referred to cyclohexane, it provides similarity to an unstrained, saturated hydrocarbon. We demonstrated that HOMA can also be used to evaluate the properties of entire molecules, such as isomers of hexacene. Ultimately, we argue that the HOMA index can describe a general geometric similarity between chemical moieties and can be shaped according to the unconstrained will of a researcher rather than the form intended by its inventors.
2. Computations
All structure optimizations were done with the DFT calculations using the B3LYP functional,18,19 along with the D3 Grimme correction for dispersion forces,20 the 6-31G** Pople-type basis set,21 and Gaussian 09 software.22 The 6-31G** basis set was chosen to enable the reproduction of the calculations in any laboratory and because this small basis set yields sufficiently good geometry, energy, and electron density data23 to build organic physical chemistry indices. It was confirmed that each structure exhibits proper symmetry and is at the energy minimum, owning all positive harmonic frequencies. The XYZ coordinates as well as the original input and output Gaussian 16 files of all molecules calculated are given in the Supporting Information.
3. Results and Discussion
3.1. EN and GEO Factors
Let us denote by x1,x2,···,xn and y1,y2,···,yn the CC bonds in the analyzed and reference rings x and y, respectively. Then, EN factor (3) can be redrawn as the square of the difference between the CC bond lengths arithmetic means x̅ = Rav and y̅ = Ropt normalized by α
| 5 |
The GEO factor (4) is then a normalized variance of the CC bond lengths in the examined moiety x
| 6 |
Thus
| 7 |
So, to calculate the HOMA index, it is enough to know the appropriate arithmetical means and the variation of the CC bond lengths in the single moiety x. Note that only the GEO = α·Var(x) term contains information about the ring size hidden in the summation operation. For the rings other than six-membered, the summation stops at n ≠ 6, but still, Ropt = RB is used. So, it is tacitly assumed that in every reference ring, the CC distance equals that in benzene, even though the matching n-membered hydrocarbon ring does not exist. Hence, HOMA = 1 for every reference ring consisting of k = 3, 4, 5, 7,··· bonds, and it is as ideally aromatic as benzene. However, if arithmetical means of comparing moieties and variation of the ring bond lengths in one of them are sufficient to determine HOMA, then even a single distance can play a role in the reference moiety. It can be present in a selected molecule but can also be equal to the arithmetic mean of the bond lengths in an existing or a hypothetical moiety.
3.2. HOMA Establishes a Similarity between Moieties
A similarity between two elements x and y of a set X is a function 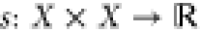 which for any x,y ∈ X satisfies the following axioms24
which for any x,y ∈ X satisfies the following axioms24
 |
8 |
Notice that if α = 1, then HOMA becomes a similarity function sHOMA(x,y)
| 9 |
The non-negativity is satisfied since
for hydrocarbons 0 < (xi – y̅)2 <
1, so 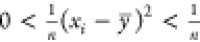 ; thus,
; thus, 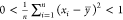 . The symmetricity is given by the symmetricity
of the (xi – y̅)2 factors. Eventually, if xi = y̅ for any i, then HOMA = sHOMA (y,y) = 1 is the maximum. Notice that HOMA
truncated to a single EN factor, 1 – EN, for α = 1 is
the similarity function for analogous reasons. Notably, HOMA is connected
also to a distance in abstract molecular space.17
. The symmetricity is given by the symmetricity
of the (xi – y̅)2 factors. Eventually, if xi = y̅ for any i, then HOMA = sHOMA (y,y) = 1 is the maximum. Notice that HOMA
truncated to a single EN factor, 1 – EN, for α = 1 is
the similarity function for analogous reasons. Notably, HOMA is connected
also to a distance in abstract molecular space.17
However, the HOMA non-negativity is not essential in studying the aromaticity of molecules. Notice that α ≈ 250 > 1 provides most figures between 1 and ca. −10, which have been convenient so far. Besides, returning to the strict similarity function is trivial. Thus, for chemical purposes, let us use a similarity-like function satisfying only symmetricity and having maximum axioms (8). Then, the HOMA and HOMA shortened to the single EN factor functions are just such “chemical” similarity functions.
Therefore, for the hexahydroxy and hexafluoro benzene, C6(OH)6 and C6F6, the rings are geometrically nearly identical to that of benzene, HOMA ≈ 1.17 The side ring in triphenylene is quite similar to benzene (HOMA = 0.93, Table 1). The off-center coronene and C6Li6 rings are less similar to benzene. Indeed, the CC bonds in the former vary, and though the latter has D6h symmetry, the CC bonds are longer than those in benzene. Hence, they have HOMA ≈ 0.8,17 meaning smaller aromaticity. HOMA of the ring in cycloheptatriene or pentalene dianions, C7H72– and C8H62–, equals ca. 0.5 (Table 1). Although nothing seems similar between the five-membered pentalene dianion and benzene rings, in terms of similarity established by the HOMA function, they are somehow aromatic, which for C8H62 is confirmed by the NICS index.25 Again, according to (7), HOMA-similarity requires similar CC bond arithmetic means in the studied and reference rings and slight variation of the CC bonds in the former. It is difficult to judge whether the central ring in triphenylene (HOMA = 0.28) is still similar to benzene or not. Unsurprisingly, ethene and cyclooctatetraene are HOMA-dissimilar to benzene and are not aromatic: HOMA ≈ 0.1 and −0.1, respectively (Table 1). The CC bond in the former is double and radically different from that in aromatic benzene, whereas the double and single CC bonds in the latter alter, and the molecule has a significant GEO factor. In agreement with the intention of the HOMA index founders,1 similarity with benzene ends at HOMA = 0, reflecting the nonaromaticity of the perfectly alternating Kekulé cyclohexatriene ring. However, according to HOMA, chair cyclohexane and archetypically antiaromatic cyclobutadiene rings have similar HOMA indices around −4.4 (Table 1). Moreover, the HOMA of the ring in antiaromatic pentalene is close to 0. Thus, the HOMA-similarity with benzene does not allow for distinguishing between cycloalkane, nonaromatic, and antiaromatic hydrocarbons. We shall check whether such differentiation could be possible by going beyond the primary definition in Section 3.3.2.
Table 1. HOMAB and HOMAc Indices Calculated Using, Respectively, Benzene (B) and Chair Cyclohexane (C) Reference Moleculesa.
| molecule/moiety | # | HOMAB | HOMAC | molecule/moiety | # | HOMAC | HOMAB |
|---|---|---|---|---|---|---|---|
| benzene | 1 | 1.00 | –4.53 | chair cyclohexane | 41 | 1.00 | –4.53 |
| cyclooctatetraene dianion | 2 | 0.94 | –3.46 | adamantane | 42 | 1.00 | –4.58 |
| side ring in triphenylene | 3 | 0.93 | –4.14 | neopentane | 46 | 1.00 | –4.74 |
| cyclooctatetraene dication | 4 | 0.89 | –3.07 | cycloheptane | 38 | 1.00 | –4.32 |
| cyclopentadienyl anion | 5 | 0.85 | –2.88 | boat cyclohexane | 44 | 0.99 | –4.61 |
| side ring in coronene | 6 | 0.81 | –3.30 | cyclooctane | 48 | 0.99 | –4.90 |
| central ring in coronene | 7 | 0.75 | –2.43 | cyclopentane | 47 | 0.98 | –4.81 |
| benzene anion radical | 8 | 0.61 | –3.15 | cyclobutane | 49 | 0.97 | –5.39 |
| ring in pentalene dianion | 9 | 0.57 | –2.00 | cyclopropane | 32 | 0.86 | –2.90 |
| cycloheptatriene dianion | 10 | 0.52 | –2.37 | ring in bicyclo[1.1.0]butane | 29 | 0.36 | –2.21 |
| central ring in triphenylene | 11 | 0.28 | –1.65 | central ring in 12H-coronene | 23 | 0.20 | –1.13 |
| cycloheptatriene anion | 12 | 0.17 | –3.76 | cyclobutadiene dianion | 21 | 0.10 | –1.04 |
| ethene | 13 | 0.07 | –9.99 | cyclohexene | 43 | –0.91 | –4.59 |
| benzyne | 14 | 0.06 | –7.03 | cyclopentene | 35 | –1.33 | –3.08 |
| cyclooctatetraene | 15 | –0.15 | –4.82 | cyclobutene | 37 | –1.59 | –3.79 |
| C6 cyclic molecule | 16 | –0.20 | –10.89 | central ring in triphenylene | 11 | –1.65 | 0.28 |
| ring in pentalene | 17 | –0.22 | –3.32 | cyclobuta-1-yn-3-ene | 28 | –1.70 | –1.97 |
| cyclohexa-1,3-diyn-5-ene | 18 | –0.45 | –8.94 | ring in pentalene dianion | 9 | –2.00 | 0.57 |
| ring in 1,4-dihydropentalene | 19 | –0.66 | –3.01 | off-center ring in 12H-coronene | 25 | –2.25 | –1.25 |
| cyclopentadiene | 20 | –0.93 | –2.97 | cycloheptatriene dianion | 10 | –2.37 | 0.52 |
| cyclobutadiene dianion | 21 | –1.04 | 0.10 | central ring in coronene | 7 | –2.43 | 0.75 |
| allene | 22 | –1.08 | –13.39 | cyclohexa-1,4-diene | 27 | –2.72 | –1.53 |
| central ring in 12H-coronene | 23 | –1.13 | 0.20 | cyclohexa-1,3-diene | 26 | –2.75 | –1.48 |
| cyclopentadienyl cation | 24 | –1.16 | –3.79 | cyclopentadienyl anion | 5 | –2.88 | 0.85 |
| off-center ring in 12H-coronene | 25 | –1.25 | –2.25 | cyclopentadiene | 20 | –2.97 | –0.93 |
| cyclohexa-1,3-diene | 26 | –1.48 | –2.75 | ring in 1,4-dihydropentalene | 19 | –3.01 | –0.66 |
| cyclohexa-1,4-diene | 27 | –1.53 | –2.72 | cyclooctatetraene dication | 4 | –3.07 | 0.89 |
| cyclobuta-1-yn-3-ene | 28 | –1.97 | –1.70 | benzene anion radical | 8 | –3.15 | 0.61 |
| ring in bicyclo[1.1.0]butane | 29 | –2.21 | 0.36 | side ring in coronene | 6 | –3.30 | 0.81 |
| cyclopropene | 30 | –2.70 | –3.91 | ring in pentalene | 17 | –3.32 | –0.22 |
| cyclopenta-1-yn-3-ene | 31 | –2.78 | –5.49 | cyclooctatetraene dianion | 2 | –3.46 | 0.94 |
| cyclopropane | 32 | –2.90 | 0.86 | cycloheptatriene anion | 12 | –3.76 | 0.17 |
| cyclopentadiyne | 33 | –2.98 | –5.51 | cyclopentadienyl cation | 24 | –3.79 | –1.16 |
| cyclohexa-1-yn-3-ene | 34 | –2.98 | –5.47 | cyclohexyne | 45 | –3.90 | –4.68 |
| cyclopentene | 35 | –3.08 | –1.33 | cyclopropene | 30 | –3.91 | –2.70 |
| cyclohexa-1-yn-4-ene | 36 | –3.12 | –5.34 | side ring in triphenylene | 3 | –4.14 | 0.93 |
| cyclobutene | 37 | –3.79 | –1.59 | cyclobutadiene | 40 | –4.43 | –4.33 |
| cycloheptane | 38 | –4.32 | 1.00 | cyclopentyne | 50 | –4.52 | –5.66 |
| cyclohexa-1,4-diyne | 39 | –4.32 | –8.02 | benzene | 1 | –4.53 | 1.00 |
| cyclobutadiene | 40 | –4.33 | –4.43 | cyclooctatetraene | 15 | –4.82 | –0.15 |
| chair cyclohexane | 41 | –4.53 | 1.00 | cyclohexa-1-yn-4-ene | 36 | –5.34 | –3.12 |
| adamantane | 42 | –4.58 | 1.00 | cyclohexa-1-yn-3-ene | 34 | –5.47 | –2.98 |
| cyclohexene | 43 | –4.59 | –0.91 | cyclopenta-1-yn-3-ene | 31 | –5.49 | –2.78 |
| boat cyclohexane | 44 | –4.61 | 0.99 | cyclopentdiyne | 33 | –5.51 | –2.98 |
| cyclohexyne | 45 | –4.68 | –3.90 | benzyne | 14 | –7.03 | 0.06 |
| neopentane | 46 | –4.74 | 1.00 | cyclobutyne | 53 | –7.43 | –10.69 |
| cyclopentane | 47 | –4.81 | 0.98 | cyclohexa-1,4-diyne | 39 | –8.02 | –4.32 |
| cyclooctane | 48 | –4.90 | 0.99 | cyclohexa-1,3-diyn-5-ene | 18 | –8.94 | –0.45 |
| cyclobutane | 49 | –5.39 | 0.97 | ethene | 13 | –9.99 | 0.07 |
| cyclopentyne | 50 | –5.66 | –4.52 | cyclohexa-1,3-diyne | 51 | –10.51 | –7.75 |
| cyclohexa-1,3-diyne | 51 | –7.75 | –10.51 | C6 cyclic molecule | 16 | –10.89 | –0.20 |
| ethyne | 52 | –7.82 | –27.33 | allene | 22 | –13.39 | –1.08 |
| cyclobutyne | 53 | –10.69 | –7.43 | ethyne | 52 | –27.33 | –7.82 |
Finally, let us emphasize that the HOMA-similarity criterion is not always intuitive and should be operated deliberately. Equation 7 states that a molecule must have similar average bond lengths and as slight bond length variation as possible to be similar to the reference one. An unreflective HOMA analysis of the CC distances in tetracyano, tetrathiocyanato, and 1,1-difluoro-2,2-dichloro ethylene, C2(CN)4, C2(NCS)4, and F2C=CCl2, with the CC distances at the B3LYP/D3/6-31G** level equal to 1.3719, 1.3773, and 1.3351 Å, respectively, would lead to the conclusion that the first two molecules are aromatic, while the third is nonaromatic, as HOMA equals 0.85, 0.91, and 0.04, respectively. Therefore, the HOMA analysis must always be placed in a narrowly defined chemical context.
3.3. HOMA as a General Chemical Index
3.3.1. Similarity to a Molecule Other Than Benzene
Several hydrocarbons other than benzene have all identical CC bonds and could serve as a reference for the HOMA analysis: chair cyclohexane (D3d symmetry), adamantane (Td), cyclobutane (D2d), cyclopropane (D3h), cyclopentadienyl anion (C5H5–, D5h), cyclooctatetraenyl anion and cation (C8H8–, C8H8+, D5h), and cycloheptatrienyl cation (C7H7+, D7h), while the anion has slightly lowered symmetry by the Jahn–Teller effect.26 The persubstituted molecules listed above, like, e.g., hexamethylbenzene,27 or fragments of a larger symmetric structure, such as the central ring in coronene, also have all identical CC bonds. Nevertheless, one-CC-bond molecules are the limit case eq 7 allows. They would set the HOMA-similarity to the most basic organic molecules such as ethene or acetylene.
Now, let us replace Ropt = RB = 1.3962 Å with RC = 1.5428 Å but keep α = 257.7 Å–1 (B and C denote benzene and cyclohexane, respectively). Then, HOMAC(C) = 1.0000 and HOMAC(B) = −4.5338, and HOMAC(B) = HOMAB(C) = −4.5. An intuition on what the HOMAC-similarity to cyclohexane means can be shaped to some extent based on Table 1. Tricyclic adamantane, acyclic and branched neopentane, and unstrained cycloheptane are exceptionally similar to cyclohexane. Indeed, they are even more HOMAC-similar than the boat cyclohexane. The more strained the cyclic alkane (cyclopentane, cyclobutane, cyclopropane), the less HOMAC-similar it is. After all, the three-membered ring of bicyclo[1.1.0]butane (HOMAC ≈ 0.36), the central ring in coronene (HOMAC ≈ 0.20), and the cyclobutadiene dianion (HOMAC ≈ 0.10) already seem to be not similar to cyclohexane.
Once a molecule has an unsaturated bond, it becomes utterly dissimilar from cyclohexane (Table 1, Figure 1). The most dissimilar are acetylene and allene, C6 ring molecule, cyclohexa-1,3-diyne, and ethene molecules (HOMAC between −27.5 and −10.00). HOMAC of cyclooctatetraene dianion, cyclooctatetraene dication, cyclopentadienyl anion, side ring in coronene, ..., and pentalene anion ring is from ca. −4.5 to −2.3. However, nonaromatic structures such as side rings in 12H-coronene, cyclohexa-1,3-diene, cyclohexa-1,4-diene, cyclopropene, and cyclohexyne also fall into the same HOMAC range.
Figure 1.

Structures for which the HOMAB and HOMAC indices were calculated (Table 1).
Remark that except for saturated not-strained cyclic adamantane and cycloheptane, HOMAC of neopentane equals 1.00, and HOMAC of isopentane, isohexane, hexane, pentane, and butane exceeds 0.97 (Table 1). Thus, for a moiety to be HOMAC-similar, saturation instead of molecular cyclicity is necessary. Such a similarity allows for a moderate strain as in cyclobutane (HOMAC = 0.96) or a much larger one as in cyclopropane (HOMAC = 0.85) or even in cubane (HOMAC = 0.80). Still, a three-membered ring in bicyclo[1.1.0]butane exhibits HOMAC = 0.36, and if the six-membered ring is flat as in 12H-coronene, in which each C atom is double-bonded with the external rim, HOMAC = 0.20. The index of very strained, elusive tetrahedrane is equal to −0.81. Thus, the HOMAC index expresses a (relaxed)-saturicity not accompanied by strong structural strains or steric hindrances.
To better see the difference between using the HOMAB and HOMAC indices, let us look at the HOMAC(A) = f(HOMAB(A)) function, where A stands for an arbitrary moiety (Figure 2a). At first glance, the points are spread erratically. Nevertheless, at closer inspection, a regular, nonlinear boundary can be seen (Figure 2a). One boundary branch goes through ethyne, allene, and benzene, and the other through cyclohexane and similar compounds. The former seems composed of primarily unsaturated compounds, while the other is composed of nonstrained structures, irrespectively saturated or not. All interior points seem to correspond to strained and simultaneously unsaturated hydrocarbons.
Figure 2.

Relationships between indices calculated using benzene (B) and chair cyclohexane (C) reference molecules. The HOMA indices (a) and the HOMA indices truncated to single EN (black points) and GEO factors (red points) (b).
Notwithstanding partial chaos present in Figure 2a, the HOMA(A) decomposition into the EN(A) and GEO(A) factors reveals where the irregularities come from. The plot of the HOMA(A) function truncated to the 1-EN(A) term forms the border curve where all of the moieties are positioned (black points, Figure 2b). The HOMA(A) function truncated to the 1-GEO(A) term establishes a half straight line (red points, Figure 2b). All of the moieties with GEO = 0 are in the point (1,1) - the beginning of the half straight line. The benzene and cyclohexane molecules and all of the moieties are located at the border curve in Figure 2a. Thus, the border is formed by moieties that have all CC bonds of equal lengths and thus GEO = 0, whereas the interior is formed by the moieties that have (significant) bond length alteration. Seemingly, the curve in Figure 2b is a deformed parabola, but the rotation of this curve by 45° using a pair of parametric equations shows that it is nothing but a parabola (Figure S1 in the Supporting Information).
3.3.2. Similarity to a Molecule with a Nonzero GEO Factor
Now, the feasibility of discrimination between nonaromatic and antiaromatic hydrocarbons is addressed using the HOMA-similarity function. The former compounds should exhibit single and double bond alternation in the ring, as in the elusive cyclohexatriene, while the latter should have 4 instead of 6 π-electrons per ring, as Hückel’s rule for the aromatic systems predicted. The HOMAB index does not allow for distinguishing these species classes (Section 3.2). However, the HOMAC index enables an additional qualification: the nonaromatic and antiaromatic rings have a negative HOMAC index, and they are dissimilar to cyclohexane (Table 1). Hence, the necessary, though insufficient, condition of being a non- or antiaromatic ring is to be HOMAB and HOMAC dissimilar. Immediately a question appears of whether a sufficient condition cannot be formulated using the HOMA index. The answer would be positive if we could find good references for these hydrocarbons.
The first problem in finding such references is the presence of the GEO factor in the archetypical cyclohexatriene and cyclobutadiene rings. In such a case, the condition of the HOMA-similarity should be supplemented by comparing the GEO factors. Hence, expression 7 has to be rewritten in a new HOMAΔ form as follows
| 10 |
where module of variances |·| is necessary to conserve the having a maximum axiom (8) for HOMAΔ.
To calculate the HOMACHTΔ-similarity to cyclohexatriene (CHT), it is necessary first to know the averaged CC distances and their variation in the cyclohexatriene moiety. From Krygowski’s original paper addressing the parameters of the HOMA index for cyclohexatriene,28 the following relationship between the bond distances and the parameter α emerges17
 |
11 |
In the case of the B3LYP/D3/6-31G** calculations used throughout this study: y̅ = Ropt = RB = 1.39621 Å and α = 257.7 Å–2, R(C=C)CHT = 1.35681 Å, R(C–C)CHT = 1.47501 Å, and Var(x)CHT= 0.00349 Å2.
According to HOMACHTΔ, the following moieties from Table 1 are the most similar to cyclohexatriene: ring in pentalene, cycloheptatriene anion, cyclooctatetraene, ring in 1,4-dihydropentalene, butadiene, and benzyne: their HOMACHTΔ is equal to 0.91, 0.81, 0.77, 0.57, 0. 54, and 0.52, respectively. This sequence shows that the HOMACHTΔ similarity criterion is not discriminative. Indeed, pentalene, 1,4-dihydropentalene, and the cycloheptatriene anion in the singlet state are antiaromatic for their 4n π electrons per ring29 and the NICS index criterion for the anion.30 Nonplanar cyclooctatetraene with alternating single and double bonds is nonaromatic. Butadiene has bond length alternation but is not cyclic, and we are not inclined to call it nonaromatic in the sense that the cyclohexatriene and cyclooctatetraene rings are. The structural formula of benzyne (cyclohexa-1-yn-3,5-diene) is formally similar to cyclohexatriene’s but, according to the NICS criterion, is more aromatic than benzene.31
Let us therefore apply the HOMACHTΔ index to rings very close to cyclohexatriene. A search for rings similar to “Kekulé benzene” among a series of triply fused hexasubstituted benzenes with the C3 axis using HOMAB = 0 as a criterion demonstrated that only some molecules fulfilling HOMAB ≈ 0 met another necessary condition such that R(C–C) > RB > R(C=C).17 However, most of the molecules satisfying the two necessary conditions contained the reactive (antiaromatic) pentalene system fused to the central ring. Such compounds are inappropriate as model reference molecules.
Application of the HOMACHTΔ criterion to the molecules exhibiting HOMAB ≈ 0 considered before17 shows that HOMACHTΔ exhibits similarity to cyclohexatriene only for those molecules that satisfied R(C–C) > RB > R(C=C), whereas for the others, HOMACHTΔ is close to 0 or negative (Table 2). This demonstrates that eq 10 can help search for moieties similar to rings with significant GEO factors and that the search can be more effective than using the classical HOMA index supplemented by additional criteria (Figure 3).17
Table 2. HOMAB and HOMACHTΔ Indices Calculated for the Molecules Shown in Figure 3, Using, Respectively, Benzene (B) and Cyclohexatriene (CHT) Reference Molecules (See Table 1 and Eqs 10 and 11, Respectively)a.
| # | HOMAB | ENB | GEOB | HOMACHTΔ | ENCHT | GEOCHT | mean CC | variance |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.06 | 0.87 | 0.07 | –0.21 | 0.38 | 0.83 | 1.4542 | 0.00028 |
| 2 | 0.04 | 0.26 | 0.71 | 0.77 | 0.04 | 0.19 | 1.4279 | 0.00274 |
| 3 | 0.03 | 0.18 | 0.80 | 0.88 | 0.01 | 0.10 | 1.4226 | 0.00309 |
| 4 | 0.02 | 0.89 | 0.10 | –0.19 | 0.39 | 0.80 | 1.4549 | 0.00037 |
| 5 | 0.01 | 0.89 | 0.10 | –0.20 | 0.40 | 0.80 | 1.4551 | 0.00038 |
| 6 | 0.00 | 0.20 | 0.80 | 0.89 | 0.02 | 0.10 | 1.4238 | 0.00311 |
| 7 | –0.01 | 0.92 | 0.09 | –0.22 | 0.41 | 0.81 | 1.4558 | 0.00035 |
| 8 | –0.01 | 0.92 | 0.09 | –0.22 | 0.41 | 0.81 | 1.4559 | 0.00035 |
| 9 | –0.04 | 0.27 | 0.77 | 0.83 | 0.04 | 0.13 | 1.4285 | 0.00299 |
| 10 | –0.05 | 0.26 | 0.78 | 0.84 | 0.04 | 0.12 | 1.4283 | 0.00303 |
Figure 3.

Structures in which the central 6-membered ring has HOMAB ≈ 0, but only 2, 3, 6, 9, and 10 have this ring are similar to cyclohexatriene according to the HOMACHTΔ criterion (see Table 2).
A moderate success of the HOMACHTΔ index in examining the similarity to cyclohexatriene prompts the supposition that a good index can also be constructed for antiaromatic compounds. However, the issue of a good reference system is returned. The strain of the ring of the antiaromatic cyclobutadiene determines its HOMACBDΔ index. Therefore, it is inadequate as a reference for antiaromaticity. On the other hand, the similarity of less strained antiaromatic pentalene to cyclohexatriene makes discrimination between nonaromatic and antiaromatic hydrocarbons using sole HOMAΔ similarity function doubtful. The sole representation of the 4n π-electron requirement in the bond lengths and their variations may not be sufficiently specific. However, further study of this issue goes beyond this project.
At the end of this section, let us mention that the HOMAΔ index can also be constructed based on a reference free of any symmetry constraints. If one assumes the averaged bond length y̅ and Var(y) in eq 10 are taken from an arbitrary asymmetric reference, then the HOMAΔ value shows the HOMAΔ-similarity to such a symmetryless compound. This can help assess the similarity to asymmetric compounds such as isooctane, on which the octane number is based, but perhaps also to important natural products such as terpenes. Although, for an asymmetric reference, the analysis could become fuzzier, without further detailed research, it is impossible to assess the benefits of such an approach. However, detailed research on this issue again goes beyond this project.
3.3.3. HOMA as an Index of the Entire Molecule
Consider all 25 unbranched catacondensed isomers of hexacene.32 Let us calculate the HOMAB index of the entire structures rather than of the individual rings (Figure 4). This can be done using expressions 1 or 7, in which now n denotes the number of all CC bonds in the structure instead of only in the single ring. The HOMAB values range from 0.76 for benzo[c]picene to 0.60 for hexacene (Figure 4). Notably, the former is the most stable, and the latter is the least stable. Also, the former has the largest, while the latter has the smallest Kekulé count33 (Figure 4).
Figure 4.

Structural formulas of all unbranched catacondensed hexacene isomers. Total energy differences vs benzo[c]picene are in blue and parentheses (kcal/mol), HOMAB of the entire structure are in red, and the Kekulé count are in black and brackets. Point group symmetry and the PAH 3-digit code34 are in the second row in black.
The stability of the unbranched catacondensed isomers of hexacene is biased by a steric hindrance, overcrowding, associated with a specific annelation leading to nonplanarity of the structure.32 Their symmetry and planarity are reflected in the PAH 3-digit code.34 The code used here assigns numbers 0, 1, and 2 to the nonterminal rings depending on annelation (number of H atoms in the edge, where the “clockwise” and “counterclockwise” angular annelations are differentiated by numbers 0 and 2, respectively). For instance, hexacene, benzo[c]picene, and hexahelicene are coded by 1111, 0202, and 0000, respectively (Figure 4). Notice that the original Balaban’s 3-digit code34 defines differently: 0 for linear annulations, and 1 or 2 for angular annulation. Still, the two 3-digit codes are equivalent.
A large number of nonplanar structures of the C2 or C1 point group symmetry (00 or 22 sequence present in the code) cause that correlation between the stabilization energy and molecular descriptors such as the Kekulé count occurs only after the addition of a term associated with the structure deviation from planarity.32 The Kekulé count K is the number of different perfect matchings of the structure with alternating single and double bonds (as in the Kekulé formula of benzene). Within the resonance theory stemming from the Hückel approximation of the molecular orbitals theory, the Kekulé count allows calculating the resonance energy (RE): RE = A·ln(K), where coefficient A = 0.1185 (eV).33 Still, the resonance theory of the conjugated polyhex hydrocarbons refers to planar structures.35,36 Consequently, the correlation of the total energy difference and the logarithm of the Kekulé count occurs only for planar hexacene isomers, while the nonplanar ones deviate substantially (Figure 5a).
Figure 5.

(a) Linear correlation between the total energy difference and the natural logarithm of the Kekulé count ln(K) for 25 unbranched catacondensed isomers of hexacene calculated at the B3LYP/D3/6-31G** level. (b) Logarithmic correlation between the total energy difference and the HOMAB index calculated for entire molecules and (c) the linear correlations between the HOMAB index calculated for entire molecules and the Kekulé count for all 25 structures (in blue) and only for the planar ones (in red). Blue empty cycles correspond to the nonplanar molecules (Figure 4).
However, the HOMAB index calculated for the entire molecules of the unbranched catacondensed isomers of hexacene performs as well as the Kekulé count (Figure 5b). Indeed, there is a significant logarithmic correlation between HOMAB and the total energy differences for planar structures (R2 = 0.948, Figure 5b). As for the Kekulé count, energies of the nonplanar ones deviate from it. Moreover, the linear correlation between the calculated HOMAB index and the Kekulé count for all 25 structures is satisfactorily significant (R2 = 0.906, Figure 5c), and if only planar ones are considered, the correlation coefficient increases to R2 = 0.966 (Figure 5c). Finally, let us suggest that the HOMAB index calculated for the entire molecules of the hexacene isomers can be understood as a similarity to a hypothetical isomer that would consist of six perfectly aromatic benzene rings. Let us also stress that, similarly to the classical HOMA index, the index can be applied to molecules composed of different numbers of rings and fused differently. So, it can be applied to how the topological indices are used in the Chemical Graph Theory. In fact, we have already demonstrated that the HOMA index is a topological index of the Structural Formula version of the Graph Theory.37
4. Final Remarks
4.1. HOMA Index for Molecules Containing Heteroatoms
A significant strength of the HOMA index is its ability to be generalized and parametrized for heteroatoms3,7−11 as follows
| 12 |
where j = CC, CN or NC, CO or ON, ..., NN, etc., and thus k denotes the number of different (unordered) pairs of atoms in the cycle, αj is a normalization factor for the j-th type of bonds, nj is the number of the j-th kind of bonds, Rji is the i-th bond of j-th type, and Rjopt is the optimal bond for the j-th type. The αj and Rjopt parameters are found in the parametrization procedures referred to the experimental technique or computational level at which distances in a given ring are found or calculated.
Expression 12 can be rewritten to a more convenient form as follows
| 13 |
where j indexes the kind of bond, k is the number of differentiated bonds, αj is the j-th normalization factor, x̅j and y̅j are the means of all bonds of the j-th type and the appropriate j-th reference, respectively, and in the variance symbol, the nj number of bonds of j-th-type is hidden.
The HOMA index defined in eqs 12 and 13 satisfies symmetricity and has a maximum axioms (8) and thus is a “chemical” similarity function as the primary HOMA index does.
4.2. Use of the HOMA Formula to Express Similarity Other Than Geometrical
The HOMA index was already studied as a function of the electron density properties in bond critical points (BCPs).12,13 In such a case, HOMA expressed similarity between electron density properties (electron density, potential, and kinetic forms of energy) measured in BCPs and was not a geometrical parameter anymore. However, the HOMA expression allows an even further deviation from the original intention. We can assume that x is an atom property such as partial charge, spin, or chemical shift. Then, y would be the appropriate property in the reference atom, and Var(x) would be the variance of this parameter in the examined moiety such as a ring. Since, for years, the 13C NMR spectra have been used for determining the aromaticity of the compounds, the comparison between the NICS class of indices and the HOMA-based NMR with the benzene C atom chemical shift as a reference would be especially intriguing. Such comparison is even more important because the chemical shift is observable. In contrast, most of the NICS indices are not, and only recently was it proven that the integral NICS index introduced by Stanger,38 INICS,39,40 was physically justified through its relation to the ring current via Ampère–Maxwell’s law as demonstrated by Berger et al.41,42
4.3. Use of the HOMA Formula to Study Acyclic Molecules
The HOMA index revealed both an increase in delocalization in polyene-like structures and an increase in insulation in unsaturated hydrocarbons,12,13,16 and a correlation with the boiling point in n-alkanes.17 However, juxtaposing the HOMA indices taken against ethane and ethene or acetylene references in analogy to that presented in Section 3.3.1 could uncover interesting new molecular features.
5. Conclusions
A close inspection of the definition of the HOMA aromaticity index revealed that it has the mathematical property of a similarity function. This property explains why the index, derived based on simple quantum chemical approximation, is such a good measure of geometrical aromaticity. It expresses a similarity to perfectly aromatic benzene.
An expression based on the transformation of the EN and GEO components of HOMA displays that to calculate the index, it is enough to know the arithmetical means of the CC bond lengths in the examined and reference rings and the variation of the CC bond lengths only in the examined moiety.
The similarity property of the HOMA function enables applying the index to evaluate similarity to other molecules, like cyclohexane. The HOMA-similarity to cyclohexane appeared to express a (relaxed)-saturicity not accompanied by strong structural strains or steric hindrances. A slight reformulation of the HOMA definition to allow the reference moiety to have some variation of the bond lengths, denoting a nonzero GEO factor, showed that similarity to the archetypical elusive cyclohexatriene ring can be better discriminated than using the classical HOMA index.
We demonstrated that HOMA can also be used to evaluate the properties of entire molecules, such as isomers of hexacene. The index calculated for all 25 unbranched catacondensed isomers of hexacene shows a significant quadratic correlation with the total energy differences of planar isomers from which the nonplanar ones deviate. Such an index could be interpreted as a similarity to a hypothetical isomer consisting of six perfectly aromatic benzene rings. Moreover, the HOMA index is significantly correlating with the Kekulé count (connected to the resonance energy within the frame of the Hückel approximation of the molecular orbitals theory) for all 25 isomers of hexacene (R2 > 0.9), but if only planar ones are considered, the correlation is much stronger (R2 > 0.96).
Acknowledgments
The article is dedicated to Professor Tadeusz Marek Krygowski, founder of the HOMA index and one of the most creative Polish organic physical chemists. This project was financially supported by the Polish National Science Centre Grant No. 2020/39/B/ST4/01670. The computational grant from the Świerk Computing Centre (CIŚ) for the J.Cz.D. group is gratefully acknowledged. The authors are grateful to the anonymous reviewers of this article for their insightful and constructive comments and suggestions.
Data Availability Statement
All quantum chemical DFT calculations were done using commercially available Gaussian 09 software.22 The B3LYP functional,18,19 along with the D3 Grimme correction for dispersion forces,20 the 6-31G** Pople-type basis set,21 are accessible directly in the Gaussian program. Calculations of the HOMA indices and their components were done using commercial Microsoft Excel program, while correlations were performed using commercial SigmaPlot for Windows ver. 14.43
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.3c01551.
The XYZ coordinates of all molecules used, as well as six zipped files with original input and output Gaussian 09 files of all molecules calculated for Figures 1, 3, and 4. After decompression, the files can be automatically read by commercial Gaussian or GaussView software44 (PDF)
ad-Fig1-Gaussian16-input (ZIP)
ad-Fig1-Gaussian16-output (ZIP)
ad-Fig3-Central 6-membered-ring-Gaussian16-input (ZIP)
ad-Fig3-Central 6-membered-ring-Gaussian16-output (ZIP)
ad-Fig4-hexacene-Gaussian16-input (ZIP)
ad-Fig4-hexacene-Gaussian16-output (ZIP)
The authors declare no competing financial interest.
Supplementary Material
References
- Kruszewski J.; Krygowski T. M. Definition of aromaticity basing on the harmonic oscillator model. Tetrahedron Lett. 1972, 13, 3839–3842. 10.1016/S0040-4039(01)94175-9. [DOI] [Google Scholar]
- Krygowski T. M.; Cyrański M. K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1419. 10.1021/cr990326u. [DOI] [PubMed] [Google Scholar]
- Krygowski T. M.; Szatylowicz H.; Stasyuk O. A.; Dominikowska J.; Palusiak M. Aromaticity from the Viewpoint of Molecular Geometry: Application to Planar Systems. Chem. Rev. 2014, 114, 6383–6422. 10.1021/cr400252h. [DOI] [PubMed] [Google Scholar]
- Szatylowicz H.; Wieczorkiewicz P. A.; Krygowski T. M.. Molecular geometry as a source of electronic structure of π-electron systems and their physicochemical properties. In Aromaticity. Modern Computational Methods and Applications; Fernández I., Ed.; Elsevier Inc., 2021; Chapter 3, pp 71–98. [Google Scholar]
- Krygowski T. M.; Cyrański M. K. Separation of the Energetic and Geometric Contributions to the Aromaticity of π-electron Carbocyclics. Tetrahedron 1996, 52, 1713–1722. 10.1016/0040-4020(95)01007-6. [DOI] [Google Scholar]
- Krygowski T. M.; Cyrański M. K. Separation of the Energetic and Geometric Contribution to Aromaticity. Part X. The Case of Benzene Rings in Fused Polycyclic Benzenoid Hydrocarbons. Tetrahedron 1998, 54, 14919–14924. 10.1016/S0040-4020(98)00934-X. [DOI] [Google Scholar]
- Krygowski T. M.; Cyrański M. K. Separation of the Energetic and Geometric Contributions to the Aromaticity. Part IV. A General Model for the π-Electron Systems. Tetrahedron 1996, 52, 10255–10264. 10.1016/0040-4020(96)00560-1. [DOI] [Google Scholar]
- Zborowski K. K.; Proniewicz L. M. HOMA Model Extension for the Compounds Containing the Carbon-Selenium Bond. Polym. J. Chem. 2009, 83, 477–484. [Google Scholar]
- Raczyńska E. D.; Hallman M.; Kolczyńska K.; Stępniewski T. M. On the Harmonic Oscillator Model of Electron Delocalization (HOMED) Index and its Application to Heteroatomic π-Electron Systems. Symmetry 2010, 2, 1485–1509. 10.3390/sym2031485. [DOI] [Google Scholar]
- Frizzo C. P.; Martins M. A. P. Aromaticity in heterocycles: new HOMA index parametrization. Struct. Chem. 2012, 23, 375–380. 10.1007/s11224-011-9883-z. [DOI] [Google Scholar]
- Arpa E. M.; Durbeej B. HOMER: a reparameterization of the harmonic oscillator model of aromaticity (HOMA) for excited states. Phys. Chem. Chem. Phys. 2023, 25, 16763–16771. 10.1039/D3CP00842H. [DOI] [PubMed] [Google Scholar]
- Ostrowski S.; Dobrowolski J. Cz. What does the HOMA index really measure?. RSC Adv. 2014, 4, 44158–44161. 10.1039/C4RA06652A. [DOI] [Google Scholar]
- Dobrowolski J. Cz.; Ostrowski S. On the HOMA index of some acyclic and conducting systems. RSC Adv. 2015, 5, 9467–9471. 10.1039/C4RA15311A. [DOI] [Google Scholar]
- Tokatlı A.; Ucun F. CTED: the new aromaticity index based on corrected total electron density at bond critical points. J. Phys. Org. Chem. 2014, 27, 380–386. 10.1002/poc.3274. [DOI] [Google Scholar]
- Dobrowolski J. Cz.; Jamróz M. E. On the variation of the belt and chiral screw and spring conformations of substituted regioregular HT undecathiophenes. RSC Adv. 2018, 8, 2116–2122. 10.1039/C7RA12777D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avramopoulos A.; Reis H.; Tzeli D.; Zaleśny R.; Papadopoulos M. G. Photoswitchable Molecular Units with Tunable Nonlinear Optical Activity: A Theoretical Investigation. Molecules 2023, 28, 5646 10.3390/molecules28155646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolski J. Cz. Three Queries about the HOMA Index. ACS Omega 2019, 4, 18699–18710. 10.1021/acsomega.9b02628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A. Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Keith T.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2013.
- Islam S. M.; Huelin S. D.; Dawe M.; Poirier R. A. Comparison of the Standard 6-31G and Binning-Curtiss Basis Sets for Third Row Elements. J. Chem. Theory Comput. 2008, 4, 86–100. 10.1021/ct700224j. [DOI] [PubMed] [Google Scholar]
- Deza M. M.; Deza E.. Encyclopedia of Distances; Springer-Verlag: Berlin, Heidelberg, 2009. [Google Scholar]
- Wannere C. S.; Schleyer P. v. R.; Schaefer H. F. III The Design of “Neutral” Carbanions with Intramolecular Charge Compensation. J. Org. Chem. 2016, 81, 1885–1898. 10.1021/acs.joc.5b02695. [DOI] [PubMed] [Google Scholar]
- Zwaard A. W.; Brouwer A. M.; Mulder J. J. C. Semi-empirical calculations concerning the geometry of the cycloheptatrienyl anion. Recl. Trav. Chim. Pays-Bas 1982, 101, 137–141. 10.1002/recl.19821010402. [DOI] [Google Scholar]
- Lonsdale K. Y. The Structure of the Benzene Ring. Nature 1928, 122, 810. 10.1038/122810c0. [DOI] [Google Scholar]
- Krygowski T. M. Crystallographic Studies of Inter- and Intramolecular Interactions Reflected in Aromatic Character of π-Electron Systems. J. Chem. Inf. Comput. Sci. 1993, 33, 70–78. 10.1021/ci00011a011. [DOI] [Google Scholar]
- Konishi A.; Okada Y.; Kishi R.; Nakano M.; Yasuda M. Enhancement of Antiaromatic Character via Additional Benzoannulation into Dibenzo[a,f]pentalene: Syntheses and Properties of are reactive even at low temperatures. Benzo[a]naphtho[2,1-f]pentalene and Dinaphtho[2,1-a,f]pentalene. J. Am. Chem. Soc. 2019, 141, 560–571. 10.1021/jacs.8b11530. [DOI] [PubMed] [Google Scholar]
- Allen A. D.; Tidwell T. T. Antiaromaticity in Open-Shell Cyclopropenyl to Cycloheptatrienyl Cations, Anions, Free Radicals, and Radical Ions. Chem. Rev. 2001, 101, 1333–1348. 10.1021/cr990316t. [DOI] [PubMed] [Google Scholar]
- De Proft F.; von Rague-Schleyer P.; van Lenthe J. H.; Stahl F.; Geerlings P. Magnetic Properties and Aromaticity of o-, m-, and p-Benzyne. Chem.—Eur. J. 2002, 8, 3402–3410. [DOI] [PubMed] [Google Scholar]
- Cioslowski J.; Dobrowolski J. Cz. Structural Dependence of Thermodynamic Stability of Unbranched Catacondensed Benzenoid Hydrocarbons. Chem. Phys. Lett. 2003, 371, 317–320. 10.1016/S0009-2614(03)00276-8. [DOI] [Google Scholar]
- Graovac A.; Gutman I.; Trinajstić N.. Topological Approach to the Chemistry of Conjugated Molecules; Springer-Verlag, Berlin, 1977. [Google Scholar]
- Balaban A. T. Applications of graph theory in chemistry. J. Chem. Inf. Comput. Sci. 1985, 25, 334–343. 10.1021/ci00047a033. [DOI] [Google Scholar]
- Trinajstić N. On the classification of polyhex hydrocarbons. J. Math. Chem. 1990, 5, 171–175. 10.1007/BF01166426. [DOI] [Google Scholar]
- Dias J. R. Nonplanarity Index for Fused Benzenoid Hydrocarbons. Polycycl. Aromat. Comp. 2014, 34, 161–176. 10.1080/10406638.2013.867515. [DOI] [Google Scholar]
- Dobrowolski J. Cz. The Structural Formula Version of Graph Theory. MATCH Commun. Math. Comput. Chem. 2019, 81, 527–555. [Google Scholar]
- Stanger A. Reexamination of NICSπ,zz: Height Dependence, Off-Center Values, and Integration. J. Phys. Chem. A 2019, 123, 3922–3927. 10.1021/acs.jpca.9b02083. [DOI] [PubMed] [Google Scholar]
- Dudek W. M.; Ostrowski S.; Dobrowolski J. Cz. On Aromaticity of the Aromatic Amino Acids and Tuning of the NICS Indices to Find the Aromaticity Order. J. Phys. Chem. A 2022, 126, 3433–3444. 10.1021/acs.jpca.2c00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarończyk M.; Ostrowski S.; Dobrowolski J. Cz. On Integral INICS Aromaticity of Pyridodiazepine Constitutional Isomers and Tautomers. Molecules 2023, 28, 5684. 10.3390/molecules28155684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger R. J. F.; Dimitrova M.; Nasibullin R. T.; Valiev R. R.; Sundholm D. Integration of global ring currents using the Ampère–Maxwell law. Phys. Chem. Chem. Phys. 2022, 24, 624–628. 10.1039/D1CP05061C. [DOI] [PubMed] [Google Scholar]
- Berger R. J. F.; Dimitrova M. A natural scheme for the quantitative analysis of the magnetically induced molecular current density using an oriented flux-weighted stagnation graph. I. A minimal example for LiH. Phys. Chem. Chem. Phys. 2022, 24, 23089–23095. 10.1039/D2CP02262A. [DOI] [PubMed] [Google Scholar]
- SigmaPlot for Windows version 14, Copywright © 2017 Systat Software, Inc.
- GaussView 6.0 software; Gaussian Inc.: Wallingford, CT, 2009.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All quantum chemical DFT calculations were done using commercially available Gaussian 09 software.22 The B3LYP functional,18,19 along with the D3 Grimme correction for dispersion forces,20 the 6-31G** Pople-type basis set,21 are accessible directly in the Gaussian program. Calculations of the HOMA indices and their components were done using commercial Microsoft Excel program, while correlations were performed using commercial SigmaPlot for Windows ver. 14.43