

Abstract
The past two decades have witnessed a rapid evolution in our ability to measure RNA and protein from biological systems. As a result, new principles have arisen regarding how information is processed in cells, how decisions are made, and the role of networks in biology. This essay examines this technological evolution, reviewing (and critiquing) the conceptual framework that has emerged to explain how RNA and protein networks control cellular function. We identify how future investigations into transcriptomes, proteomes, and other cellular networks will enable development of more robust, quantitative models of cellular behavior whilst also providing new avenues to use knowledge of biological networks to improve human health.
You are not a beautiful and unique snowflake.
Tyler Durden (119)
In both biological and manmade systems, reducing the frequency of failure often requires an enormous increase in the complexity of circuits.
Dr. Leland Hartwell and others (56)
Introduction
Humans and other multicellular organisms are extraordinarily complex systems exhibiting features of both emergence and engineering. Because humans are not studied by an engineer who made them—rather, by scientists behaving like someone trying to reverse engineer a satellite fallen to earth from a more advanced civilization in a far away galaxy—one of the basic efforts of biology for the last century has necessarily been categorization of a molecular parts list. First, however, we figured out many of the basic principles for how the major classes of molecules in the cell interact with each other. DNA is the genetic material and carrier of information transgenerationally. RNA performs many important roles in all cellular processes and encodes proteins, which in turn make up molecular machines and function within networks that carry out subcellular and intercellular processes. Lipids, carbohydrates, and small molecules are building blocks for the cell’s organelles, and energy sources for, and regulators of, its activities. With these principles understood for some reactions, technological and conceptual convergence at the end of the last century enabled exploration of the vastness of biological molecules. With the human genome (as well as the genomes of many other species) in the hard drive, transcriptomes and proteomes are now being explored with confidence.
But what are the objectives of omics studies, particularly—to be addressed in this essay—transcriptomics and proteomics? Should we be attempting to determine the rate constants for every extant biological reaction in a complex multicellular eukaryote, such that each reaction could be recapitulated in a test tube with recombinant proteins? In this article, we examine the natural history of transcriptome and proteome analysis, attempting to discern patterns in how the methods and thinking evolved. Next, we endeavor to answer the question of what comes after the parts lists are generated. Bioinformatics and gene ontology are recipes for obfuscation in understanding biological systems. Rather, genes, transcripts, and proteins should be treated as nameless entities related only by their observed abundance, interactions, location, thermodynamics, and the central dogma. Bioinformatics is needed on some level to compile and organize the data, but computational modeling is needed to reveal the principles that govern the networks. This is not a new principle in physiology: some of the greatest leaps in our understanding of physiology have come when mathematical modeling reveals fundamental rules that govern a system [e.g., physiological control of breathing (103) or biochemical feedback circuits in the cell cycle (41)]. It is clear that individual molecules can operate in different modules, enabling the same proteins to participate in distinct biological functions (56, 165). Furthermore, these modules act within networks, influencing basic cellular functions (8) as well as complex phenotypes like disease progression (12).
Finally, we see the field as poised to answer two interrelated questions: What are the laws that govern the interactions amongst proteins and RNA at a network level? And how can a theory of network function, one that goes beyond its structure and the annotation of transcripts and proteins into pathways, enable a more perfect description of cellular function and identify new avenues for treatment of human disease? We address these questions by undertaking an analysis of the progression of research in omics, with an emphasis on the transcriptome and proteome, highlighting the yin and yang of technique and theory. We analyze emerging areas of interaction among different types of biological networks, including the realm of genetic control over RNA and protein expression and the relationship between computational modeling and omics measurement in systems biology.
Arrays to RNA Sequencing: The Many Types of Transcriptome Variation
In 1995, Patrick Brown and colleagues used an automated method to print 45 cDNAs from Arabidopsis thaliana onto a glass slide (144) (Fig. 1A), arguably launching the era of transcriptome analysis. Since the intensity of the fluorescence, detection method was directly proportional to the amount of transcript, this innovation allowed for the simultaneous analysis of large numbers of transcripts in a quantitative manner. Soon thereafter, the microarray field rapidly evolved new and more sophisticated platforms, all of which were based on the attachment of probes representing various expanses of the genome to a solid surface, which was in turn incubated with fluorescent cDNA libraries from a sample of interest. The resulting hybridization pattern enabled profiling of a transcriptome (180). In the ensuing 10 to 15 years, this technique was widely used in biology, fundamentally altering the basic question asked about transcription in a given system: rather than sufficing to measure a few genes, the turn of the century saw measurement of large numbers of genes and even transcriptome-wide analysis (including multiple types of RNA species) become commonplace across biological research (Fig. 1B).
Figure 1.
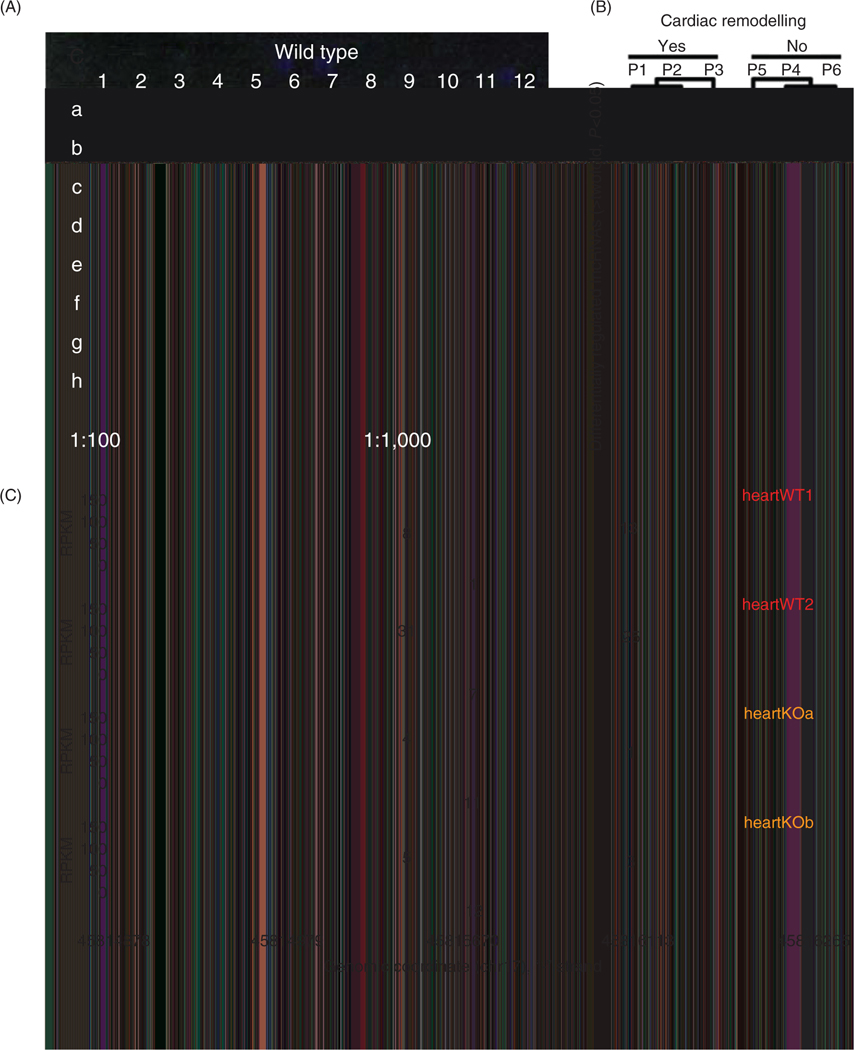
Evolution of RNA quantitation techniques toward a more comprehensive catalogue of RNA species. (A) The first microarray was published in 1995 by Patrick Brown and quantified 45 mRNA species simultaneously using hybridization to DNA probes [reprinted with permission (144)]. (B) Microarrays have since advanced to measure tens of thousands of RNAs, including noncoding RNA. Shown is a heatmap of 768 lncRNAs found by array to exhibit altered abundances in the blood between patients with and without left ventricular remodeling following myocardial infarction [reprinted with permission (89)]. In this case, transcriptome measurements enabled unbiased identification of disease progression biomarkers. (C) Due to the development of RNA-sequencing and subsequent advances in the library preparation, sequencing and data analysis, quantification a greater diversity of RNA species on a transcriptome-wide scale is now routine. Shown is a Sashimi plot displaying the relative abundances of different exons in an example measured from the hearts of wild-type mice (red) and mice with a knockout of a splicing factor. y-axis represents normalized RNA-seq reads (expression), x-axis represents genomic coordinates. The arcs are numbered to indicate the raw number of junction reads. Arcs with greater values bridge two exons that are more often spliced next to each other [reprinted with permission (77)]. While these data were acquired from mice that were experimentally manipulated to disrupt splicing, many studies find exon usage is an important component to the transcriptome regulation of cell-type specificity, development, and disease.
The microarray had one obvious limitation, however: it can only measure genes on the array. Enter the RNA sequencing (RNA-seq; Table 1) revolution of the mid-2000s. RNA-seq is based on the high-throughput sequencing of a double strand cDNA library generated from an RNA population. After sequencing, the reads are aligned with the specific reference genome to generate the expression profile for a given transcript or gene (depending on how the experimenter chooses to circumscribe the data). RNA-seq has clear advantages over the preexisting hybridization-based microarrays (Fig. 1C): (i) there is no requirement for transcript-specific probes and thus the analyses are not limited to the detection of transcripts previously detected (including noncoding RNAs, further discussed below, and splice variants) (185); (ii) since DNA sequences can be precisely mapped to unique regions of the genome, RNA-seq experiments present lower background levels, avoiding the common microarray cross- and nonspecific-hybridization problems (94); and (iii) RNA-seq quantifies gene expression based on the number of reads generated during sequencing, allowing the quantification of gene expression in absolute rather than relative values. This feature facilitates the detection of RNAs with very low expression and provides a more accurate measurement of extremely abundant transcripts (101). Depending on the biological question, RNA-seq applications can be tailored to cover more of the transcriptome, with greater coverage requiring deeper sequencing.
Table 1.
Techniques for Measuring the Transcriptome, Proteome, Transcription, and Translation
| Technique | Pros | |
|---|---|---|
|
| ||
| RNA abundance | Microarray | Inexpensive standardized |
| RNA-seq | Measure unknown RNAs absolute quantitation | |
| SAGE | RNA-seq for 3′-UTR | |
| CAGE | RNA-seq for 5′-UTR | |
| Single-cell RNA-seq | Capture intercellular heterogeneity | |
| RNA splicing | RNA-Pet TIF-seq |
Identify splice-junctions and allele differences |
| Long-read single-molecule Real-time sequencing | Capture splicing and allele data | |
| MapSplice SpliceMap |
Identify splice junctions from RNA-seq data | |
| HMMsplicer | ||
| MISO MATS |
Quantify alternatively spliced genes | |
| SpliceR | ||
| Transcription | GRO-seq | Quantify nascent RNAs |
| PAR-CLIP iCUP |
Snapshot of transcribed RNAs (protein bound) | |
| Translation | Ribo-seq | Snapshot of translated RNAs (ribosome bound) |
| Protein species and abundance | 2D-PAGE | Provide visual display |
| Shotgun, bottom-up LC/MS/MS | Easy to implement Measure many proteins | |
| MRM | Quantify known subset of proteins | |
| SWATH | Combine accurate quantitation with depth | |
| SILAC | Precise quantitation | |
| iTRAQ | Isobaric label Greater multiplexing | |
| Dimethyl-labeling | Least expensive, most amenable label | |
| Label-free quantitation | Amenable to many experimental workflows | |
Sequencing depth in turn depends on different factors. First, one can sequence either one or both ends of a DNA molecule, respectively called “single” or “paired” end sequencing, determined by the adaptors used for selection. Since paired-end sequencing provides a superior alignment across the reference genome, it is the method of choice to sequence repetitive elements or to detect novel transcripts or genomic rearrangements (e.g., insertions, deletions, and inversions) (45). By contrast, single-end sequencing enables less accurate alignment than paired-end sequencing; however, it is cheaper and the library prep and data analysis faster. Thus, single-end is normally used in experiments where annotating the genome is not the goal, such as in simple transcriptome profiling experiments where alternative splicing or ncRNA will not be analyzed (52). A second consideration is the length of the sequencing reads: usually 50 or 100 bp is analyzed, but longer reads can be sequenced (52). The more nucleotides sequenced on each cDNA fragment of the library, the more reliably the data are aligned to the proper location in the genome. Longer reads not only provide a better alignment, but they also increase the cost of the experiment. Next, different RNA-seq approaches require different RNA library preparation protocols to address the individual research needs. For the analysis of polyadenylated RNA molecules, including not only mRNA, but also some long noncoding RNA (lncRNA) and small nucleolar RNA (snoRNA), a PolyA selection is performed. This methodology separates mRNA from ribosomal RNA (rRNA) very efficiently, enriching the former such that greater sequencing depth can be achieved—toward a targeted subset of RNA—with fewer reads (186). In the analysis of prokaryotic RNA or in experiments that analyze nonpolyadenylated RNAs, an rRNA depletion step has to be included in the protocol. This step uses magnetic beads that contain capture probes for depletion of both cytoplasmic and mitochondrial rRNAs, thereby enriching the remaining RNAs of interest (186). This approach is more expensive than PolyA selection but necessary for sequencing non-polyA RNA transcripts and/or noncoding RNA (159). Lastly, depth of coverage is a variable for which one must independently account. The depth of coverage is a measure of the number of times that a specific genomic site is sequenced with a certain number of reads, assuming that reads are randomly distributed across the genome (152). In general the higher the number of times that a base is sequenced, the better the quality of the data. Coverage can be modulated by different factors like the length of the reads, the number of sequenced ends and the number of samples runs in a given lane of the sequencer. It is widely accepted that garden variety transcriptome profiling experiments require 20 million reads per sample, whereas more detailed analyses, such as alternative splicing, allele-specific expression or expression of low-abundant transcripts may require 40, 60, or even 100 million reads per sample, respectively (152).
In addition to increasing sequencing depth, increasing thenumber of biological replicates can also improve identification of differentially expressed genes. Statistical analysis using a t test requires a minimum of three biological replicates, whereas the Fisher’s exact test can be used on fewer samples, because this method is dependent on the total number of reads mapped across biological replicates. Beyond the obvious benefits to ensure the effect of a treatment is greater than the effect of biological and technical variability, increasing read number through addition of biological replicates as opposed to deeper sequencing is more effective at identifying additional differentially regulated genes (179) [one study finds this is true only for additional reads beyond 10 million per sample (98)].
RNA-seq applications
In the relatively short time since its emergence, RNA-seq has transformed analyses and understanding of the transcriptome (Table 1). As with many large-scaled omics techniques before and since, RNA-seq was first applied in yeast and plants (97, 108), but the reduction of sequencing costs and the versatility of the technique have enabled its application in virtually all eukaryotic cell types (27, 82) and tissues (86, 107). Emergent areas of investigation now focus on transcript functionality, transcriptome diversity, and the role of transcripts in disease by measuring alternative splicing, polyadenylation and/or new transcription start sites (TSS), in addition to abundance, on a genome-wide scale. In this transcript-centric view, the gene annotation or expression level is only partially helpful in determining the in vivo functionality of the RNA because multiple RNA forms usually exist for a single gene. As a result, novel hypotheses about the biology of RNA in eukaryotes have led to new technologies to study functional cohorts of these molecules distinguished by the physical features of the RNA species (e.g., circular RNAs, micro RNAs, and long noncoding RNAs).
Alternative polyadenylation and TSS analyses investigate how the same gene template gives rise to various protein-coding mRNAs depending on cellular conditions or identity. Two methods in particular have been used in this space: serial analysis of gene expression (SAGE)-like and cap analysis of gene expression (CAGE)-like sequencing, both of which were originally developed to be used with Sanger sequencing (160), but which have been repurposed for use with next generation sequencing technology (59). SAGE methodologies are focused on the study of the 3′ untranslated regions (3′UTR), whose length and sequence regulate alternative polyadenylation. On the other hand, CAGE technology studies processing of the 5′ ends where the appearance of alternative TSS and the length of the 5′UTR, respectively, regulate the formation of new isoforms and the efficiency of the translation. SAGE (187) includes a step to capture polyadenylated transcripts and CAGE uses a 5′-cap isolation step (158) prior to the generation of the cDNA library. The captured RNAs are converted to cDNA and then, depending on the chosen methodology, the samples are subjected to enzymatic digestion and adapter ligation, to generate short sequences of 21 to 27 nucleotides called tags. The final sequencing step generates the reads from these tags that directly depend on the amount of a specific mRNA molecule.
Alternative splicing is an important layer of gene regulation that dramatically increases the complexity of the transcriptome. No longer a niche field of investigation into a few genes, RNA-seq-driven exploration of eukaryotic transcriptomes have revealed that >90% of genes undergo alternative splicing (167). Detection of alternative splicing events requires high sequencing coverage, in the realm of 40 to 60 million reads per sample, and intense, specialized computational efforts (96). The development of new algorithms to address this goal is still under active research but specific aligners that identify splice junctions like MapSplice (168), SpliceMap (7), or HMMsplicer (32) as well as alternative expression tools designed to quantify the expression level of alternatively spliced genes like MISO (76), MATS (149), or SpliceR (163) have been reported. A related but separate challenge emergent with RNA-seq is the goal of determining complete transcript structures. A key limitation of RNA-seq methodologies is the short length of the reads (84) which limits the reconstruction of the transcripts, and therefore the identification of splicing variants, fusion transcripts, and the discrimination between different alleles. To circumvent these problems, different methods have been invented toward the goal of sequencing the entire transcript. One approach is based on the detection of both 3′ and 5′ ends of each transcript using pair-end sequencing, and include techniques like RNA-PET (139) and TIF-seq (121) which are based on the formation of a circular template that is digested or sonicated to generate a single molecule with the information from both ends. An alternative methodology is the sequencing of a full-length cDNA library using long-read single-molecule real-time sequencing technology (142). This new technology can generate 15 to 20 kb reads and thus has emerged as a useful tool to complement the short-read sequencing experiments.
The aforementioned RNA-seq approaches provide information on the abundance of different RNA species—they provide a snapshot of the transcriptome at a given moment, but can reveal the dynamics of neither real-time transcription nor protein synthesis. To address these aims, three different methodologies have been developed: methods based on the immunoprecipitation of RNA-protein complexes (PAR-CLIP, iCLIP) (55,83), global run-on sequencing (GRO-seq) (30), and ribosome profiling (Ribo-seq) (67). The first group of techniques is based on the ability of ultraviolet irradiation to crosslink RNA and proteins, in the process forming complexes that can be immunoprecipitated using antibodies. The immunoprecipitated RNA is sequenced by conventional approaches, representing the portion of the transcriptome bound to a given target. These techniques have been used to identify the binding sites of cellular RNA-binding proteins and microRNA-containing ribonucleoprotein complexes (54), but the low efficiency of crosslinking using UV has prompted the invention of alternative methodologies. GRO-seq is a technique used to map, orient and quantify nascent RNAs that are associated with transcriptionally engaged polymerases, providing a genome-wide readout of active transcription (30). This protocol is based in the use of a ribonucleotide analog to BrU-tag (BrUTP) that is added to the sample and incorporated to the nascent RNA during the run-on step. After a brief pulse period (to keep the labeled mRNA short, allowing better resolution), the RNA is hydrolyzed and the nuclear run-on RNA (NRO-RNA) is captured using an anti-BrU anti-body conjugated to magnetic beads. An NRO-cDNA library is then generated for sequencing, thereby enabling measurement of the number and identity of transcripts synthesized during the pulse period. In one example, this technique was used to compare the complete transcriptional profiles of RNA polymerases in mouse embryonic stem cells and mouse embryonic fibroblasts, showing that 40% of genes have peaks of paused Pol II upstream of their promoters (104). Importantly, GRO-seq captures nascent transcripts of approximately 100 nucleotides (30); however, the majority of initiated transcriptional events abort after ∼10 nucleotides (9), a size too small to be caught by GRO-seq or aligned to the genome. At present, these aborted transcripts are usually thought of as byproducts of an imperfect transcriptional system (i.e., “not real,” in a biological sense); however, some investigators have suggested that some aborted transcripts may have regulatory function, such as the case of a bacteriophage aborted transcript regula ing antitermination activity of a terminator (93).
What about regulation of the transcriptome at the translational machinery? A convergence of technologies measuring inputs and outputs of translational machinery has revealed some interesting observations (Fig. 2). Studies of the so-called translatome, a sequencing based proxy for protein abundance (80), have emerged in which RNAs bound to ribosomal proteins are quantified. Because myriad factors conspire to make the connection between mRNA levels and protein levels nonlinear, including mRNA processing (17) or RNA modulation via miRNAs (173) among others, direct measurement of mRNAs undergoing translation is necessary to understand this process, rather than total mRNA or protein measurements. Ribosome profiling (Ribo-seq) is a technique to sequence the mRNAs that are being actively translated, which entails inhibition of ribosomal activity (various methods have been described) (68, 92), cell lysis and RNaseI digestion to generate single monosomes from the original polysomes (complexes of mRNA with two or more ribosomes). During this step, all the RNA is digested except for the fragments of RNA that are protected by the ribosomes, called nuclease-resistant ribosome-protected fragments or footprints (RPFs). The monosomes are then isolated using sucrose gradients or size-exclusion chromatography and the RPFs are released from the ribosome to generate a cDNA library that is sequenced (92). This technique has limitations in that it only provides data from mRNAs (since it is the only RNA protected by the ribosome during the nuclease digestion) and the small size of the RPFs (around 27 nucleotides) complicates the subsequent alignment tasks (46). One application of this technique has been to better understand the correlation between mRNA and protein abundance by capturing the level of translation. However, the actual level of translation in the cell may be much greater than is measured with this approach (an issue separate from the abundance of proteins) due to translation that is terminated after only several nucleotides, and the peptide quickly (<1 min) degraded (9). Some investigators have proposed that this extra translation serves a proofreading step for inappropriate stop-codons, performed by the ribosome (9).
Figure 2.

RNA abundance and protein abundance both correlate better with ribosome occupancy than they do with each other. Expression analysis was performed on lymphoblastoid cell lines of diverse genetic backgrounds taken from the HapMap project. Genes were clustered into modules or neurons (hexagon, right panels) within a self-organizing map based on similar expression profiles across four different measurements (protein abundance, translation efficiency, RNA abundance, and ribosome occupancy; left panel). The right panel displays the same self-organizing map colored to portray the mean expression of the genes within the module based on the four different datasets. The authors ask if hexagons with similar mean expression by one measurement (either both colored red or both colored blue) also show similar expression when using an alternate measure of expression. Ribosome occupancy correlates with RNA expression and protein level better than RNA expression and protein level correlate with each other. Note, ribosome occupancy is defined by the total read counts for an RNA after ribosome profiling, while translation efficiency takes into account the total pool of RNA (RNA-seq) in addition to the ribosome occupancy [reprinted with permission (23)].
The rapid improvement of RNA-seq technologies in terms of limit of detection and sample size has enabled researchers to tackle a previously unassailable question in biology: cell-to-cell variability within a complex tissue. It has been established that cells with high expression of a given gene in a heterogeneous population can significantly bias measurements across the rest of the tissue (16, 143), and it was for this reason, and to address the analyses of specific rare cells or specific subpopulations for which the amount of RNA is not sufficient to perform conventional RNA-seq, that single-cell RNA sequencing was developed (34, 57). This protocol is similar to conventional RNA-seq, but it includes two nontrivial innovations, which are the isolation of individual cells of interest (the methods for which must be optimized depending on the tissue of interest) and the conversion of small amount of RNA to cDNA. Single cell isolation has been accomplished by a suite of techniques including flow cytometry followed by sorting, micromanipulation, optical tweezers, microfluidics-based techniques, or laser-capture microdissection. A variety of single cell commercial kits are now available and have been applied to examine conclusions made from whole tissue studies (176). This methodology has been used to study transcriptome features in specific cells whose expression profiles were otherwise masked in a tissue level experiment, and single-cell RNA-seq has also emerged as an effective protocol to study splicing events (147), different allelic expression (31), SNPs, and mutations (78, 127). Accordingly, the specific methods for single-cell RNA-seq are married to technical challenges. RNA loss during library generation is a problem and can reach up to 50%, having the greatest impact on transcripts with low expression. Related to this, limited starting material can impair sensitivity, making it difficult to distinguish between the noise—especially that generated during cDNA amplification—and real biological variation. These two factors make single-cell profiling difficult, since the expression of key regulatory transcripts like lncRNAs and microRNAs are often expressed at the lowest level.
Of course, genomic DNA sequencing is progressing in technological lock step with RNA sequencing and, not reviewed here, is quickening the arrival of a time in which genome sequences will be available for entire populations of humans. Indeed, the vast majority of RNA-seq experiments—as well as all epigenomics experiments—are reliant on a reference genome, although some interesting approaches have been reported that enable direct measurement of RNA sequence (183) without reference genome, enabling more accurate testing of still very controversial fields like RNA editing.
Building Protein Networks: Identification and Quantitation for Discerning Biological Interactions
What sequencing is to genomics, transcriptomics and epigenomics, mass spectrometry is to proteomics (Table 1). Discovery-driven proteomics experiments enable unbiased identification and quantification of protein isoforms in complex samples (i.e., several thousand proteins in a cell lysate). Before mass spectrometry for proteins came into its own, however, in 1975, two-dimensional polyacrylamide gel electrophoresis (2D-PAGE) was used to separate large groups of proteins (114). The principle was to separate proteins by two features (commonly mass and isoelectric focusing point, pI) followed by total protein staining allowing visual comparison, aided by software, to detect differences between samples. Figure 3A is an example of such a comparison (29). Several 2D-PAGE databases were developed (122), such as the multispecies database of cardiac 2D gels (37). The Internet was becoming mainstream at this time, making possible the comparison of a gel you ran in your lab against another from anywhere in the world. This 2D-PAGE plus limited mass spectrometry technique was the primary method for proteome level studies through the mid/end of the 1990s.
Figure 3.

Evolution of proteomics toward network analysis. (A) Two-dimensional protein gels were published in 1975 (top panel, [reprinted with permission (113)]), and remained a common tool for identifying proteome-level quantitative differences between samples up into the late 1990s (bottom panel [reprinted with permission (29)]). Bottom panel is a computer-processed image of a silver-stained 2D gel from a human dilated cardiomyopathy sample. Spots represent protein isoforms identifiable by their position in the gel (number indicates database protein ID). Note that PTM can shift a protein’s location in the gel, providing additional information. Red spots indicate isoforms which were less abundant (weaker signal; similar to Western blot analysis) across the dilated cardiomyopathy patients as compared to ischemic cardiomyopathy samples run in a separate gel, and analyzed together using computer software. Note that this analysis reveals on average 1282 spots per sample, in the same general scale as LC/MS/MS analyses; however, the identification of the individual spots, when not coupled to mass spectrometry, remains imprecise. (B) By contrast, advances in mass spectrometry and sample preparation pipeline have enabled quantification of PTMs across entire signaling cascades from multiple conditions. Shown here is the known insulin signaling pathway curated from multiple databases, overlaid with phosphorylation quantitation (expressed as fold-change) from a mass spectrometry analysis performed on liver samples from mice treated with PBS or insulin at two time points [reprinted with permission (64)]. These techniques are optimized for a focused subproteome, thus enabled thorough, dynamic measurements of the system, which go beyond identifying proteins into the realm of mapping biological processes within a network. (C) Shown is a protein-protein interaction network from HeLa cells generated through combining coimmunoprecipitation followed by mass spectrometry for 1125 different proteins [reprinted with permission (58)]. Red indicates edges previously annotated in CORUM. On its own, this network represents a database to inform other protein interaction studies. However, the authors took this study a step further to compare their interaction network with the relative abundance of the proteins to infer complex stability. Thus, by comparing across networks, the omics datasets are able to generate new understanding of properties of the proteome.
There were three major drawbacks to this method, however. First, the gel separation needed to be highly reproducible to capitalize on the existing databases of protein identifications and/or to compare between different samples analyzed in the same lab, let alone between different labs (with different gel apparatuses, buffers, and so forth). Second, analyses were biased toward the most abundant proteins, which could be visualized in the gel, as well as against high molecular weight and hydrophobic proteins that were poorly resolved by 2D-PAGE. Finally, in addition to 2D-PAGE databases, proteins could be characterized by peptide mass fingerprinting mass spectrometry. With this method, 2D-PAGE spots were excised, enzymatically digested, and analyzed by MALDI (matrix-assisted laser desorption/ionization) to generate the peptide-mass fingerprint which, briefly, can be thought of as a low resolution protein identification that lacks information on single amino acid differences and posttranslational modifications (PTMs), while also having limited quantitative capacity and high false identification rates. When working with complex samples containing multiple proteins, there was the possibility for multiple peptides to generate a similar-enough fingerprint spectra that precise identification was impossible. However, tandem mass spectrometry, which involves scanning the original peptide (MS1) followed by fragmentation and a second scan of the product ions (MS2) overcame this problem (see (181) for a review on the history of mass spectrometry innovations).
In 2001, the Yates lab published MudPIT (multidimensional protein identification technology) (169), a shotgun proteomics technique that served as an alternative to 2D gels and MALDI. By coupling tandem mass spectrometry [which had recently emerged as a powerful technique for peptide identification (36)] to 2D liquid chromatography, they overcame the protein-level bias of 2D gels, since the LC separation occurs on digested peptides. Enzymatic digestion also meant it was easier to get reproducible separation. The workflow entailed passing the sample through a strong cation exchange column, and eluting one fraction at a specific salt concentration. The elutant passed over a reversed phase column that allowed further fractionation with an organic solvent gradient that was then eluted directly onto an electrospray setup to introduce peptide ions into the gas phase in the mass spectrometer (an innovation itself made possible a decade earlier (40) and which has since become the most widely used ionization technique in proteomic mass spectrometry). There were then iterative increases in the salt concentration followed by subsequent rounds of increasing organic solvent to analyze the entire sample. When published, the MudPIT technique dramatically increased the number of protein IDs from a single experiment (the first paper reported 1484) and increased detection of low abundant proteins through the multiple rounds of fractionation, which decreased the complexity of the sample entering the mass spectrometer at any given moment, thus increasing the likelihood that a given peptide would be detected. In subsequent years, most investigators have eschewed the strong cation exchange step and performed tandem mass spectrometry after reverse phase LC separation of tryptic peptides, so-called LC/MS/MS, a technique that has been used in probably thousands of papers focused on protein identification in the last decade and a half (4, 184).
Most LC/MS/MS analyses of proteins and peptides operate in data-dependent mode, wherein the mass spectrometer selects the most abundant ions from the MS1 scan for fragmentation and analysis in the MS2 scan. Complementary to this type of discovery proteomics, however, is multiple reaction monitoring (MRM)-based mass spectrometry. MRM also employs data-dependent, tandem mass spectrometry in an initial experiment; however in a subsequent experiment, and based on the observations from the first analysis, the user specifies a set of peptides by the m/z of the parent and product ions (known as a “transition”) in a data-independent manner. The mass spectrometer only detects and fragments these prespecified peptides, each constituting a transition (which is counted by the computer as a quantitative measurement) which increases the accuracy of quantifying the peptide by biasing the mass spectrometer to analyze the total of the peptide signal, even when it is not the most abundant peptide eluting from the column at a given time in the LC gradient. This approach has been especially important for biomarker analysis in blood samples, which have a large dynamic range due to highly abundant serum proteins, and has other applications that are complementary with data-dependent analyses (33).
Nonetheless, many labs perform quantitation using LC/MS/MS-based proteomics to quantify a group of proteins with differential abundance between samples (43). While less accurate than MRM, these techniques do not require the laborious multiple experiment format to establish MRM assays and can be implemented on multiple types of tandem mass spectrometers. Label-free quantitation is the least accurate but cheapest of the shotgun quantitation methods (109). With this approach, the two samples to be compared are run separately, in successive LC/MS/MS runs. Run-to-run variability can introduce bias in the detection of peptides; however, this is overcome with technical replicates. Peptides are then quantified and the ratio of signal for a given peptide between the two samples is compared. Ideally, multiple peptides for a given protein are detected and the ratios for each peptide converge on a consensus fold difference value. As for all quantitation techniques, sample-specific PTM can lead to inaccuracy in quantification if the modifications lead to aberrant identification of the peptide (or failure to identify) due to deviation from the expected mass. Search algorithms allow for flexibility for specified modifications, but peptides with a large amount of modification can still escape identification. Ideally, individual proteins contain several tryptic peptides that are not modified and can thus be relied upon for quantification. Quantitation can be done on the MS2 scan, wherein the number of MS2 scans is counted and compared between samples, an approach called spectral counting. Most data-dependent experiments set a several-second delay (called dynamic exclusion) such that a peptide cannot be sent for MS2 multiple times within a prescribed time frame to allow for deeper sampling of less abundant peptides. Thus, differences between samples in the abundance of other peptides eluting from the column at the same time can lead to differences in the number of MS2 scans that are not reflective of differences in the abundance of the peptide of interest. This problem is not only present in label-based quantification experiments, but also in other omics such as RNA-seq which starts with a fixed amount of RNA between all samples and counts changes in a specific transcript between samples. Alternatively, label-free quantitation can be performed on the MS1 data. In this case, the extracted ion chromatogram for a peptide is isolated with the retention time on the x-axis and the intensity as detected by the mass spectrometer on the y-axis. Integration of the area under the curve is used to estimate the abundance of the peptide throughout its entire time of elution, thereby obviating errors in spectral counting due to dynamic exclusion. However, like all shotgun quantitation methods, it requires the peptide be among the top-most abundant peptides such that it is selected for MS2 and fragmented.
These same techniques can be applied to quantify samples that have been multiplexed. In this case, protein samples are covalently labeled and then combined so that a single LC/MS/MS run contains multiple samples, thereby mitigating run-to-run variability. Among the labeling methods, SILAC (stable isotope labeling by amino acids in cell culture) (116) controls for the most variability in sample preparation because it incorporates the label at the earliest time point in the experimental workflow. In this way, the proteins themselves are labeled and can be combined before the majority of preparation steps that lead to sample loss and error in measurement are performed. The SILAC approach introduces a metabolic label into growing cells through heavy isotope containing amino acids introduced into the culture media. This method is expensive and very challenging to apply in animals, which requires feeding several generations with heavy isotopes. Chemical labeling can circumvent some of these limitations, by introducing the label to the peptides after tryptic digest but before sample fractionation. Dimethyl labeling (19) is the cheapest but can only be multiplexed up to three samples (light, medium, and heavy isotopes), while iTRAQ (145) is more costly but can multiplex up to eight samples. For large-scale proteome level analyses, label-free quantitation is still used and has the benefit that by not combining samples into a single run, sample-specific, low-abundant proteins are less diluted and so have a greater chance of being identified. However, with low-cost, easy techniques like dimethyl-labeling, label-based quantitation is a good option for proteomic experiments that start unbiased, but plan to identify and focus on a small subset of proteins for which higher accuracy in quantitation is desired. Furthermore, label-based quantification requires greater investment in sample preparation time and cost, but is often faster to analyze due to shrink-wrapped proteomic software solutions from reagent and instrument manufacturers that automate quantification, while still enabling the investigator to manually investigate peptides of interest. By contrast, label-free quantitation often requires greater investment in homemade informatic tools as well as more extensive knowledge of mass spectrometry in the data analysis and validation phase.
In 2012, a new proteomic mass spectrometry method was published by the Aebersold lab that sought to combine the advantages of data-dependent LC/MS/MS and MRM. SWATH (sequential window acquisition of all theoretical mass spectra) (51) improves upon data-independent acquisition methods to allow simultaneous fragmentation and analysis of a larger number of peptides (this group also provided a new data analysis platform known as OpenSWATH (136) for interrogating the datasets from this distinct proteomic workflow). MRM has remained the gold standard for quantitation despite the sophisticated labeling and analysis pipelines developed for shotgun proteomics due to the persistent irreproducibility in precursor ion selection for fragmentation due to the biases already discussed (33). Furthermore, while shotgun proteomics enables identification of thousands of proteins, as compared to MRM, there is still consistent under sampling of complex mixtures due to the requirement for a peptide to be in the top ∼10 most abundant peptides to be fragmented for MS2 and thus privileged for identification (33). SWATH overcomes these two major disadvantages of MRM and shotgun proteomics by performing unbiased fragmentation (in MS1) of all peptides, independent of their abundance (i.e., data-independent acquisition). Following the MS1 scan, there are a series of MSn scans. In a typical shotgun proteomics experiment, this would consist of 10 MSn scans, one for each of the top 10 most abundant peptides from the MS1. In SWATH, there are 32 MSn scans (with each scan covering a proportion of the m/z range of the instrument), and in each one, multiple peptides are fragmented (Fig. 4A). The major challenge with this technique is matching the product ion spectra to the correct parent ion in the MS1 spectra, since there are product ions from many parent ions in the same MSn scan. Borrowing from MRM analyses, OpenSWATH relies on searching for known transitions (m/z product and parent ion pairs) from already existing spectral libraries to identify peptides in the sample—this is the opposite of how traditional shotgun proteomics data are analyzed, in which experimental spectra are matched against theoretical spectra generated from a reference protein sequence database and a search algorithm. Currently, implementation of SWATH remains restricted primarily to labs specializing in mass spectrometry, but may become more broadly implemented with time.
Figure 4.

Mass spectrometry techniques for building protein networks. (A) Peptides (circles; size indicates relative abundance) elute from the LC column into the mass spectrometer. In shotgun/bottom-up proteomics, peptides are scanned in the MS1 and the most abundant ions selected for fragmentation and identification via multiple MS2 scans. In MRM, both the MS1 and MS2 scan are performed on predetermined m/z ratios set by the user to precisely quantify peptides of interest, including low abundant peptides. SWATH by contrast fragments all ions from the MS1 scan, resulting in many more MS2 scans, each containing spectra from many parent ions. (B) Upstream techniques can be used in conjunction with mass spectrometry to enable protein and PTM identification, quantitation, and spatial localization information used to build protein networks.
Some consider a goal for proteomic mass spectrometry to be identification of all proteins in the cell. Unlike other systems biology projects, this specific goal would not fundamentally advance the understanding of molecular networks or their interactions, but rather, would provide a foundational parts list of protein players. In 1997, around the time tandem mass spectrometry was being implemented, it was thought that the human genome contained 60,000 to 80,000 genes, and at the time there were only 3719 human proteins in the SWISS-PROT database (122). We now know there are closer to 20,000 genes, but many more proteoforms (154) due to alternative splicing and PTM. As of November 2015, there were 26,133 entries in the SWISS-PROT database for human proteins. Two papers were published in May 2014 in the same issue of Nature that attempted to catalogue the entire human proteome by combining their own mass spectrometry data with published reports. The first claimed to have amassed protein-level evidence for 84% of annotated protein coding genes, which they compiled into humanproteomemap.org (79). The second organized their dataset into ProteomicsDB (174), incorporating data from 47 human tissues and body fluid samples, and observing that coverage of the proteome plateaued at 16,000 to 17,000 proteins. This observation suggested to some observers a limit of current data-dependent MS approaches that could be surpassed only by transformative technical advancements in how proteins are identified (it remains to be seen whether SWATH fits this bill).
At present, identification of all proteins in an unbiased manner in a single experiment (or even with a single experimental workflow in multiple attempts) is impossible, given issues of protein chemistry, abundance, PTM and other issues (including the inability to amplify proteins in a cell free environment, akin to PCR, which transformed the genomics field). However, there are important questions that can be asked with the existing data alone such as quantifying protein complex stoichiometry and comparing mRNA to protein abundance (a discipline that requires unbiased use of techniques from transcriptional analysis and protein synthesis/degradation). Furthermore, recent studies have shown that some lncRNAs, contrary to the implication from their names, contain open reading frames (9, 28). These large-scale studies to identify all human proteins also raise the question as to the coverage necessary for designing a more focused mass spectrometry experiment that a nonproteomics lab may conduct. If the most specialized mass spectrometry labs, working with a huge number of datasets, saturate at 16,000 proteins, and the typical shotgun experiment identifies ∼1000 proteins, how does an experimenter know that her or his dataset is complete enough to make conclusions about the system, especially if the goal is to ask systems-level questions? We propose that a practical solution to generate meaningful data that is not only thorough, but also reasonably attainable, is to focus on subproteomes as informed by a specific biological hypothesis (106).
Large-scale coverage of the human proteome remains prohibitively costly and time consuming to be carried out in a comprehensive manner (i.e., to measure the totality of proteins expressed in multiple individuals). For instance in the aforementioned studies in humans, the authors find that ∼70% of the top 100 most abundant proteins in each of the 47 tissues and body fluids tested are expressed in all of the samples, though the abundance varies by up to 5 orders of magnitude (174). One of the current salient questions for translational research and system biology is the influence of genetics on intermediate molecular networks (such as transcripts, metabolites, or proteins) that bridge differences in disease susceptibility from the genetic perturbations to the phenotypic manifestation (172). Such approaches in the proteomic field have been rare, although a few large mass spectrometry studies on the HapMap project have been successful (177). Current practice is to conduct proteome-level, unbiased quantitation in a small mouse or human cohort to identify a specific candidate protein, which can then be measured in many more samples in a focused manner, thenceforth interfacing with known protein interaction networks and pathways. Alternatively, studies that aim to measure networks and not candidate molecules rely on RNA-based network modeling with only focused proteome-level validation, if any. However, recent studies describe protein quantitative trait loci (pQTL), which match SNPs to differences in the expression of individual proteins (see forthcoming section on the role of genetic variation in RNA and protein function). A frontier in this field is the development of a quantitative molecular understanding (and the resultant mathematical tools) to scale these analyses up to cohorts of proteins across various genetic backgrounds.
For most labs that utilize, as opposed to develop, mass spectrometry technology, their interaction with these mass spectrometry advancements is through access to instruments with greater resolution and sensitivity to identify more proteins, the affordability of certain of the labeling approaches for quantitation, and the ever-growing size of the UniProt database of human and model-organism proteins which shotgun experiments are searched against and which, for most experiments, define the totality of what is possible to be identified by the protein identification software. As RNA-seq refines genome annotation, large-scale studies can use these new data to build additional protein databases, but these databases must balance being inclusive while maintaining a reasonable scale of multiple hypothesis testing to preserve laudable false discovery rates (110) (123).
In addition to measuring protein expression, technical advances have also enabled large-scale measurements of PTMs (115) (Fig. 3B). Databases of PTMs and the specific spectral changes associated with each functional group have been compiled (175). For studies of a single specific modification, investigators often perform an enrichment step for modified peptides to achieve greater coverage, with the most successful methods for enrichment existing for phosphorylation (115). As a result, a good enrichment for phosphorylation can lead to identification of 10,000 modified residues in a single mass spectrometry run, enabling dissection of entire signaling pathways (115). Further aiding phosphoproteomics was the development of electron transfer dissociation (ETD) (155). An alternative to the more common collision induced dissociation (used for virtually all of the aforementioned LC/MS/MS shotgun experiments), ETD was developed from electron capture dissociation and utilizes nonkinetic fragmentation of the peptide in a manner that better preserves the phosphorylation. However, dissecting the combinations of modifications that concurrently decorate a protein remains a great challenge. Top-down mass spectrometry (in which peptides are measured and identified without fragmentation and/or undigested proteins themselves are subjected to gas phase fragmentation) is a technique employed by far fewer labs than the bottom-up, peptide-based mass spectrometry described above, primarily due to the difficulty in optimizing a single method that will equally fractionate, ionize, and detect proteins with different chemical properties. However, top-down mass spectrometry offers the opportunity to measure the entire set of modifications occurring on a single protein (153,157). The other major challenge for PTMs is uncovering the biological significance for the large number of modifications being discovered to dissect cellular processes. Once discovered and characterized, PTMs can also be used in the clinic as biomarkers, although rigorous quantitation by ELISA or MRM is necessary (3,118). Special considerations are necessary for measuring, validating and implementing clinically relevant biomarkers, but progress is being made, especially in the area of cancer (118).
Like RNA-seq, single-cell technologies are also being developed for proteomics. The sensitivity of the mass spectrometer is not suited for unbiased proteomic measurements of a single cell; however, enrichment and labeling strategies have enabled single-cell measurements for panels of proteins. Mass cytometry is similar in concept to flow cytometry; however, the protein is detected by measuring the tagged mass as opposed to the fluorescence. This can be performed using GFP-tagged cell lines (111) or antibodies labeling over 30 proteins in the same cell (15). The advancement of these studies lies in overcoming the spectral overlap that limits the number of fluorescently tagged protein species that can be detected by flow cytometry, by relying instead on the significant resolving power of the mass spectrometer to discern subtle mass differences. These studies have revealed principles for protein noise in a population, finding much of it can be explained by mRNA abundance, buffered by the longer half-life of proteins (111). Using antibodies for PTMs has also led to analysis of signaling pathways (88), and this technology is also being applied to understand the heterogeneity of patients samples as well as screen drug compounds (87). Other antibody-based methods have also been developed, including a modified DNA microarray chip to an antibody-based chip (150), as well as single-cell western blotting allowing measurements of up to 11 proteins per cell (63). These examples remain niche technologies: at present and for the foreseeable future, unbiased analysis of whole proteomes will be reliant on mass spectrometry and a fine tuned biochemical workflow that is cell type dependent.
Principles for Interaction Across Molecular Scales in Biology
Role of genetic variation in RNA and protein function
The steady-state abundances of proteins are determined by rates of transcription, mRNA degradation (and modulation by other factors, like miRNAs), translation, protein stability/ modification, and protein degradation. An underlying assumption in many biological studies for decades has been a concordance of transcript and protein levels, due to the flow of information from DNA to phenotype. In recent years, systemwide relationships between transcript and protein levels have been studied in yeast, plants, and mice and have yielded unexpected results: the agreement between transcript and protein abundances is often surprisingly low.
A comparative study in a yeast segregating population showed that there is a significant, but modest correlation between transcript and protein levels (42). A molecular phenotype mapping study in Arabidopsis reported similar findings (44). In both of these studies, in addition to relatively modest correlation between protein and mRNA abundance, genetic loci that influence protein abundance are different from those affecting transcript abundance. Investigating the commonality of hotspot loci (defined as loci affecting a large number of traits within each biological class) across various biological scales, the investigators identified fewer pQTL (defined here as genetic variation that influences the expression of a protein) compared to expression quantitative trait loci (eQTL; genetic variation that affects the expression of an mRNA), leading to the conclusion that phenotypic buffering of perturbations affects molecular phenotypes as one looks to scales further away from the DNA variation (e.g., proteome vs. transcriptome). As moderate to low correlation between protein and mRNA abundance data (coefficient of determination R2 ≤ 0.4) was found, it was concluded that no more than 40% of the variance in protein levels is explained by variance in the rates of transcription and mRNA degradation; the remaining variance in protein expression (≥60%) is explained by translation and protein degradation.
Because most biological functions occur at the protein level, protein abundances are more direct determinants of cellular function than transcript abundances. A logical step forward is the mapping of pQTLs to determine genetic control of protein expression. Using isobaric tag-based quantitative mass spectrometry, Wu et al. quantified relative protein levels of 5953 genes in lymphoblastoid cell lines from 95 individuals from the HapMap Project and found that protein levels, like expression levels, were heritable molecular phenotypes (177). In addition to proteins varying based on ethnic background and gender, sets of proteins involved in the same biological process covaried, suggesting tight regulation at the protein level. Mapping for pQTLs revealed overlaps between eQTLs and pQTLs. In addition, the group identified novel cis-pQTLs that were not previously detected by eQTL analysis. The authors showed that IMPA1 protein, which has a poor correlation with its mRNA (r = 0.04, p = 0.76), demonstrated a strong pQTL (p = 3 × 10−7), indicating that distinct genetic mechanisms control gene expression at different levels and the importance of the complementary knowledge provided by systematically characterizing the human proteome.
Schwanhäusser et al. estimated that transcription explains 34% of the variance in protein abundance, mRNA degradation 6%, translation 55%, and protein degradation 5% (146). These early studies explained the weak correlation between transcript and protein levels by claiming that mechanisms of posttranscriptional regulation buffered changes in transcript abundance so that they either do not lead to changes in protein abundance, or they do lead to changes in protein abundance, but in the absence of a corresponding effect on transcripts (53, 99). In addition, comparative studies suggest that protein levels are under greater evolutionary constraint than transcript levels, an observation consistent with buffering of protein abundance vis-à-vis variation introduced at the transcript level (164). These findings are consistent with the concept that translational control makes a larger contribution in protein abundance than transcriptional control, although computational efforts to reexamine transcriptome and proteome data have questioned this interpretation. Recent findings suggest that the high-throughput methods used in these early studies suffered several systematic biases, highlighting a number of relevant and important technical and biological considerations for system-wide transcriptome and proteome investigations. Several of these early studies used label-free mass spectrometry that may have underestimated the amounts of lower abundance proteins by as much as a factor of 10. Also, guanine-cytosine base pair content has been suggested to bias mRNA-seq data by a factor of up to 3. Both biases introduced errors in the estimates that would lower the apparent correlation between transcript and protein levels (95). Subsequent studies using statistical efforts to estimate and reduce the impact of errors resulted in a higher correlation between true protein and true mRNA levels (14, 73, 95). Correction for errors allowed Jovanovic et al. to calculate that at steady state, mRNA levels explain 68% of the variance in protein expression, translation rate 26%, and protein degradation rates 8%. Furthermore, Li et al. found that by correcting for a nonlinear scaling error in protein abundance estimates and accounting for error estimates using replica and control data, the variance in true mRNA levels explained a minimum of 56% of the variance in true protein levels. Finally, by measuring translation rates directly by ribosome footprinting, true mRNA levels were found to explain 84% of the variance in true protein expression, with transcription accounting for 73%, RNA degradation 11%, and translation and protein degradation each only 8% of variance in protein abundance.
Battle et al. performed ribosome profiling to measure changes in translational regulation in addition to transcriptome and proteome assessment (14). Mapping of genetic association with each of the regulatory phenotypes detected 2355 eQTLs, 939 rQTLs (ribosomal QTL), and 278 pQTLs. There is significant overlap among the detected QTLs. Of the 4322 genes quantified for all three phenotypes, 54% of the genes with pQTLs also have significant rQTL and/or eQTL. In addition, most (90%) genetic variants associated with ribosome occupancy are also associated with transcript levels. In contrast, eQTLs showed the lowest overlap with pQTLs (35%). The fact that many eQTL SNPs are not associated with differences in protein levels is consistent with either incomplete mapping power in protein levels due to higher measurement error or buffering. It can be concluded that the majority of genetic variants affecting transcript levels also alter ribosomal occupancy but many eQTLs have attenuated effects on steady-state protein levels. Furthermore, comparison of expression-specific QTLs (esQTLs) and protein-specific QTLs (psQTLs) showed that ribosome data usually tracked with levels of RNA. These results allowed the identification of loci which affect protein levels that are not mediated by transcription or translation but rather protein degradation. Enrichment analysis revealed that exonic and UTR SNPs are enriched for more significant psQTL effects, compared with intergenic or intronic SNPs. Finally, psQTLs are further enriched for nonsynonymous sites (compared with all exonic SNPs), especially near acetylation sites, reflecting possible functional role of lysine acetylation in modulating protein degradation. In addition to discordance between mRNA and protein abundance, we have found indirect relationships between genetic variation and mRNA abundance as well as between the transcriptome and organ-level phenotypes (Fig. 5). These analyses suggest that the one SNP to one gene’s expression comparisons used in QTL analysis are insufficient to explain the transcriptome due to regulatory interaction amongst and between SNPs, mRNAs, and proteins, which ultimately dictate biological processes.
Figure 5.

The role of genetics in gene expression is organ specific. To test the relationship between genetics, gene expression, and phenotype, we examined data from a panel of 37 genetically diverse, inbred mouse strains with microarray data from multiple organs: Macrophages with and without LPS stimulation (unpublished), striatum (120), hippocampus (120), bone marrow (38), and heart with and without isoproterenol (ISO) stimulation to induce heart failure (128). Strains were clustered based on expression of all genes on the microarray (All) or a class of genes known as the “fetal gene program” (Fetal), whose cardiac expression are considered to be biomarkers of heart failure. The relatedness between each strain-by-strain comparison (Euclidean) was compared across organs. If the relative similarity in expression between two strains is similar across two organs, those two organs cluster closer together on the dendrogram. We also incorporated genetic relatedness based on kinship matrix derived from SNPs (Genetics). Macrophages cluster according to genetics, suggesting that strains with similar genetics also show similar expression patterns in macrophages regardless of if we examine all genes, or the cardiac fetal genes, and even when examining expression after LPS stimulation. By contrast, other organs, such as bone marrow, have expression relationships that less closely match genetic relationships. For context, we compared the relationships between genetics versus mRNA expression to that of genetics versus cardiac phenotype [ejection fraction (EF) and heart weight/body weight (HW/BW), two indices which change in heart failure]. In some cases, the genetic relationship more closely matched the phenotype than the expression (basal EF), but in other cases it did not (EF after ISO). We hypothesized that the “fetal gene program” was an intermediate between genetics and phenotype, but found that it no more closely matched the phenotypic relationships than when we examined all genes together. These analyses indicate that the relationship between genetic variation, mRNA expression, and ultimately phenotype is buffered at each level. For example, complex SNP interactions and chromatin features may buffer the relationship between genetic variation and mRNA expression, while posttranscriptional and posttranslational processing as well as compartmentalization may buffer the relationship between mRNA and protein levels, with the relationship between protein and phenotype in turn buffered by protein network properties and interaction with other classes of molecules.
In summary, multiple QTL-based analyses have been deployed to reveal relationships between genetic variability, transcriptional variability, and protein expression. Disagreements in the correlation between these measurements result from technical variability (principally in the design of the proteomics experiment and subsequent analysis of mass spectrometry data, in our view) as well as biological differences between cell types and species (particularly yeast contrasted with multicellular eukaryotes).
Genetic control of gene expression is often mediated through chromatin
Epigenomic regulation of gene expression, protein expression and cellular phenotype is an exploding field that is conceptually interrelated to the topics of transcriptome and proteome in this review, but which we will not endeavor to cover in great detail as several timely reviews exist on this matter (72, 134, 156). An interesting observation regarding the interaction of genetic variation with transcriptome and proteome regulation presages as-yet unknown mechanisms of chromatin regulation. The majority of SNPs associated with disease lie in noncoding regions (introns and intergenic regions) (60), with the assumed functional significance being to modify gene expression. But what is the molecular basis for such modification? The simplest mechanism is when a SNP acts in cis to change the chromatin features in a nearby gene, which in turn alters gene expression. The ENCODE project (Encyclopedia of DNA Elements) is a collaborative effort by many labs to measure multiple features of chromatin using consistent protocols on a shared panel of cell lines and tissues, allowing integration of datasets across experiments. Collectively, the project has mapped regulatory regions across many human tissues (1, 49), which can be used to annotate SNPs. Furthermore, ENCODE makes all of their data easily downloadable in multiple file formats for other researchers to scrutinize in their own studies. However, while the technology to define regulatory regions enables fast and scalable data collection, the next step of discerning how they regulate a specific gene is usually still an intensive, single DNA locus effort. A recent effort to annotate SNPs associated with autoimmune diseases found that 90% fell in noncoding regions with 60% specifically in enhancer regions (39). However, the majority of these SNPs did not disrupt known DNA consensus motifs for chromatin proteins (39), that is, the aforementioned behavior to regulate chromatin proteins in cis is not supported as the mechanism of action. Despite major advances in uncovering the relationship between expression and chromatin at genes and regulatory regions, this ongoing study highlights our lack of understanding for the genetic control of chromatin structure. As the cost of next-generation sequencing continues to decrease, the aim of developing thorough and consistent datasets like that of ENCODE from diverse genetic backgrounds becomes possible. While it may not be organized under a single umbrella like ENCODE, datasets from multiple labs can be compared. To foster these analyses, labs should provide more of their intermediate processed data (between raw sequencing files and an excel sheet of target loci) that other labs could incorporate into their data analysis.
Importantly, the relationship between chromatin and disease phenotype can also be directly probed, bypassing gene expression, in what is known as an epigenome-wide association study (EWAS) as opposed to genome-wide association study (GWAS) (117, 126). In fact, the effect size of causal CpGs (cytosines followed by guanines, whose DNA methylation status is correlated with a trait) tend to be larger than SNPs, despite only small differences in the percent methylation between cases and controls (126). Recent studies have also found that DNA methylation correlates with complex phenotypes in an ostensibly heritable manner that is independent of, although it may be influenced by, SNP (24), yet the mechanisms for how these epigenetic features control phenotype remain to be determined.
Features of Protein and RNA Networks
As the foregoing discussion of transcriptome and proteome analyses have described, our ability to measure large groups of biological molecules has rapidly advanced over the last two decades. If RNA, protein and other molecular species function in networks, then it is in networks they must be studied. How exactly does that work? Imperfectly executed, omics studies become list generators, but properly matched with network theory and mathematical biology, omics investigations can reveal fundamentally new principles of biology.
Several different modeling approaches have been employed to examine how large groups of molecules enable the structures and behaviors of a cell. Many epigenomic studies turn to hidden Markov models, to define genomic domains populated by similar chromatin features. However, metabolomics, transcriptomics, and proteomics often use networks to model molecular interactions based on coexpression, physical interaction, shared domains, substrate/product, or epistatic/signaling/regulatory relationships. This higher order analysis can also be integrated across multiple tiers of molecules (from genetic variants to RNA to protein, for instance). Biological networks have been shown to exhibit scale-free properties: most nodes (molecules) have few edges (connections), whereas select hub nodes have many edges and tend to be older evolutionarily (130). These features make biological networks both robust (the network can sustain loss of the majority of its nonhub nodes) and well connected (exhibiting small world effects wherein any two nodes are separated by only a few links) (13). An added feature to the scalefree topology, cellular networks are disassortative, in that hub nodes tend not to interact directly with other hub nodes (13). Within a network are modules of nodes (61) that exhibit higher connectivity amongst themselves than with nonmodule nodes and that together contribute to a specific cellular function (13). Hubs not only exist within modules but also serve as bridges between modules (13). Depending on the nature of the network (e.g., protein interaction or signaling) links can be directional and the local topology of several interconnecting nodes can form functional motifs, indicative of the prevailing relationship(s) in that region network (e.g., positive feedback) (13). Several resources exist for visualizing networks including Cytoscape (148), VisANT (62) and NetGestalt (151).
Coexpression of RNAs and proteins is one property that can be used to assign the links in a network, operating on the premise that coexpression is indicative of shared functionality (21, 35, 112, 178) and/or shared regulation (5). Weighted gene coexpression network analysis (WGCNA) (182) is a method for building coexpression networks from RNA expression data. The tools for WGCNA are available in an R package (91) that enables the user to first identify modules and hub genes, and then designate eigengenes, fictional genes whose expression is representative of the module’s members. Eigengenes or hub genes can then be used to probe statistical relationships between module behavior and a given phenotype, bypassing the multiple hypothesis-testing problem that arises from examining every gene on the microarray (90). Furthermore, differential network analysis and consensus module analysis on networks from different physiological states can be used to identify network properties that are conserved or altered under different circumstances. In addition to exploring system-level differences, coexpression analysis can also be used to identify new candidate genes for subsequent single molecule analyses. Such identification can be done using network neighborhoods, where genes of known biological significance are used to pull out novel genes that are found to directly interact with the significant genes in the network, so-called Guilt-By-Association, (166) an approach that can also be taken on coexpression networks built without WGCNA (10). As RNA-seq replaces microarray data, new issues have been identified. Namely, hub nodes differ depending on whether the network was built with microarray or RNA-seq data, due to differences in the noise of each technology (10). The WGCNA methodology has also been adapted for protein expression data, showing that peptide modules of coexpression also enrich for overlapping functionality and protein-protein interactions (50). In addition to WGCNA, other algorithms have been optimized for particular situations. For example, Maximal Information Component Analysis performs better on expression data with many nonlinear relationships by incorporating Module Identification in Networks and allowing genes to exist in multiple modules (129). Networks can also be built from PTM abundance, as was done for mapping the phosphotyrosine signaling cascades in HeLa cells (18) and the insulin signaling pathway in mouse liver (Fig. 3B).
Networks can also be built from protein-protein interaction data, traditionally from yeast two-hybrid screens or affinity purification mass spectrometry (Fig. 4B). Seminal work came in 2006 from two separate studies of the yeast interactome (47, 85), with mammalian studies following. CORUM is a database of curated mammalian protein complexes (140). While not strictly comprised of protein-protein interactions, the Kyoto Encyclopedia of Genes and Genomes database is a major curator of signaling and metabolic pathways (74) as well as drug/target interactions and disease specific pathways and molecules (75). Mass spectrometry experiments can be optimized to detect specific subsets of interactions (124), including identification of stable versus dynamic complexes (81) and interactions that are direct, as determined by cross linking (132). A human network, BioPlex, built on HEK293T cells reveals that protein-interaction modules also enrich for shared functionality, subcellular localization, and protein domains (65). The BioPlex interactome is extensive (7668 proteins); however, like most proteomics-based datasets, it is noncomprehensive. Guidelines for estimating data quality exist (161). Fortunately, a recent collaboration between mass spectrometry labs showed that standardized protocols can dramatically increase reproducibility of affinity purification mass spectrometry, suggesting a human interactome could be attainable (20). The largest human binary interaction map to date is HI-II-14, made by testing ∼13,000 genes pairwise by yeast two hybrid finding 14,000 interactions (133), while the alternative approach of next-generation interaction survey using coimmunoprecipitation in HeLa has recently identified 28,000 interactions, in addition to quantifying stoichiometry, allowing authors to infer the stable and dynamic components of protein complexes (58) (Fig. 3C).
Importantly, several studies have examined the overlap between networks built on coexpression data versus those built with physical interaction data (48), specifically noting that permanent protein complexes (stable under most cellular conditions) have highly correlated mRNA expression, while transient complexes have less-correlated expression (70). Furthermore, coexpression coupled with gene function annotation can be used to predict protein expression in yeast (71), which not only demonstrates the convergence of the two measurements, but also offers a tool to overcome the proteomic limitations of building complete interactomes by incorporating other types of datasets. Networks can also be built on other combinations of datasets, for example regulatory networks that incorporate coexpression and shared transcription factor occupancy (11), coexpression networks that derive directionality by incorporating eQTL data (6), or protein interaction networks that derive directionality from genephenotype interactions gleaned from RNAi screens (162).
In addition to the more common protein-protein interaction networks based on physical binding, spatial interaction networks also exist, which define proteins found in the same organelle or cytoplasmic space. High spatial resolution can be achieved using a spatially restricted enzymatic tag to mark proteins for purification before mass spectrometry analysis, such as BirA*,a modified biotin protein ligase which was used to identify nuclear proteins when fused to the nuclear lamin A via proximity-dependent biotin identification (137, 138). Peroxidase enzymes have also been used for a similar purpose. Engineered ascorbate peroxidase (APEX) targeted to the mitochondrial matrix was used to biotinylate mitochondrial proteins when the cell was exposed to 1 mmol/L hydrogen peroxide and biotin-phenol which reacts with electron rich amino acids in an APEX- and hydrogen peroxide-dependent manner (131). However, it is also now possible to interrogate many organelles at once from the same cell sample, without developing specialized fractionation protocols to enrich each organelle into a pure population. Localization of Organelle Proteins by Isotope Tagging relies on a gentle lysis which breaks the cell membrane while leaving organelle membranes intact followed by centrifugation for subcellular fractionation (25). Individual fractions are labeled with isobaric tags (by definition they possess the same mass) after which all fractions are combined and analyzed by shotgun proteomics (25). Because of the isobaric tag, the same peptide will produce the same MS1 scan from all samples, but the MS2 will reveal the relative contributions from the different fractions based on the different fragmentation patterns of the individual tags. The crux of this method is that it does not require pure fractions for each organelle, only the presence of several known biomarker proteins restricted to each organelle (25) which are used to calibrate the relative abundance profile of each organelle across the fractions. Next, the remaining proteins are matched to the organelle with which they share a similar elution profile and plotted in a 2D space based on coelution (25). An exciting finding to come out of this work is the appreciation that many proteins reside in multiple subcellular locations where they may be carrying out different functional roles, highlighting that measuring expression or PTM in a whole cell lysate may be insufficient to determine the abundance of specific pools of proteins.
The power of networks comes from their ability to predict physiological outcomes. A recent paper screened 2890 disease-causing human missense mutations and 1140 nondisease causing mutations for protein interactions, revealing that the majority of disease-causing missense mutations do not alter overall protein stability (inferred by interaction with chaperones); however they disrupt protein-protein interactions seven times more than nondisease causing mutations (141). This study highlights the potential for networks to predict consequences of genetic perturbations. Work in Escherichia coli using phenotype phase plane analysis of the metabolic network predicted all possible network solutions by which the bacterium could use a given substrate to achieve growth, with some being deemed some more suitable than others (66). Similar to the “good enough solutions” concept (171), the investigators found that for some substrates the E. coli population used a suboptimal solution, that could be adapted to the environment over ∼700 generations to become optimal (66). A challenge now is to integrate human genetic diversity with organ-specific and disease-specific networks to predict patient response to physiological insults and identify the critical nodes for modulation of the network toward a different “solution.”
Moving Forward
“Many molecular details are simply not needed to describe phenomena on the desired functional level.”
Dr. Leland Hartwell and others (56)
The aforementioned quote from Dr. Hartwell and coauthors identified one of the paradoxes of the omics era that is as true now as when they wrote it in 1999: although development in the instrumentation, computation, and concepts associated with transcriptomics and proteomics has been remarkable, and has fundamentally enabled an ever more granular understanding of the molecular basis of physiology, one of the principal lessons from the last 20 years of these studies has been that, as just discussed in detail, the function of molecules can only be fully understood in the context of modules or networks. The terminology of modules and networks must be sufficiently precise to account for a key biological process and yet sufficiently fluid to allow multifunctionality (56, 165), over the time scale of instants to generations: natural selection can act on modules and networks, not individual molecules. Moreover, reducing the behavior of complex systems to component parts precludes quantitative measurement and real world modeling. Systems analysis is most successful when it evokes a hypothesis … not for the purpose of biasing the experiment in a reductionist manner, but to focus the data-acquisition step to a finite realm wherein it can be thorough, after which, modeling coupled with hypothesis can be used transcend cataloging changes to uncovering novel properties of the system (Fig. 6). We need new tools, but moreover new thinking, in particular a structure that removes genetically engineered gain/loss of function mouse models from their privileged position in the analysis of biological systems (100, 102, 105). One of the new salient questions in biology, then, is: at what scale must biological processes be investigated to make new discoveries and/or to use our knowledge of biological networks to improve the human condition? We endeavor to answer this question with a few predictions.
Figure 6.

Spectrum of cognitive bias in basic and translational research. Implementation of discovery science and hypothesis-driven research comprise a spectrum analogous to the “opportunity cost” principle. Points along the curve represent experiments where the opportunity cost is minimized, because some perfect balance between discovery and hypothesis is struck. Point A defines a species of research with very high uncertainty and little or no theoretical underpinning, but with the potential to be very novel. Point B defines another type in which highly focused and inherently biased research reaches full potential by maximizing prior knowledge. Studies that lie under the curve, due to shoddy or underexplored data or an experimental design that builds only incrementally on precedent, fail to meet the ideal balance of discovery and hypothesis [reprinted with permission (106)].
A promising area of development is to use patterns within transcriptome and proteome networks, perhaps in combination with genetics, to predict disease incidence, and tailor personalized therapy. Oncology is one area where patients are stratified by the presence or absence of a particular genetic lesion and clinical treatment administered differentially as a result. There are now several mouse models and human cell line studies showing that despite the multitude of genetic mutations acquired successively during the course of oncogenesis, cancer cells remain particularly dependent on maintaining the initiating oncogenic lesions (either overexpression of oncogenes or downregulation of tumor-suppressor genes) such that losing only one of these key changes is enough to cause growth inhibition, apoptosis, or differentiation into normal tissue (170). This area offers a promising application for network biology to integrate gene expression networks and DNA sequencing for genetic mutations to determine a panel of candidates specific to a patient, such that if any of the candidates are targetable by drugs or gene therapy, the treatment could be tailored accordingly.
Furthermore, oncology research has also incorporated networks into expression analysis for patient stratification. One group found that they could increase accuracy of predicting metastasis in breast cancer patients by combining protein interaction networks with mRNA expression versus using mRNA expression alone to identify predictive genes (26). The group used existing protein interaction networks to identify subnetworks and then calculated an overall expression value for all the genes in the subnetwork, identifying groups of genes which together had greater predictive value compared with individual genes as well as greater conservation when applied across patient cohorts (26). The conclusion was that incorporating interaction data allowed the investigators to capture genes known to play a major role in driving the disease, but whose expression changes were subtle and thus not identified as significant by rote expression profiling. Other groups are also incorporating distinct types of omics data including chromatin modifications, DNA mutations, and analysis of noncoding RNAs (2).
Another fledgling application of network biology is drug repurposing. By using existing gene-drug and disease-drug networks, researchers can predict novel diseases for which a drug may be useful based on the genes it targets, with the effect being to dramatically decrease drug-development cost by starting only with existing, approved drugs, and removing the first stage of unbiased small molecule screening (69).
Interpretation of how genetic variation impacts phenotype on a clinical scale will depend on reliable in silico prediction based on dynamical network modeling across biological scales of genomic variation, transcript dynamics, protein turnover, and metabolite dynamics (22,125). Existing network modeling methods are frameworks of convenience, based on coexpression or physical interaction, which are properties that are accessible based on measurements from current technology. However, these frameworks are rough and static models of the biological system in question that can be improved upon by further experimental and modeling. As dynamical measures under different in vivo conditions become ascertainable in the future, the reaction constants for individual processes will become available. With ever expanding computational power, the opportunity will exist to model on a cellular level the predicted metabolic readout of an individual genetic variant under different cell culture conditions, as has been pioneered for simpler systems where such time series and stoichiometric data are already available (8).
Proteomic experiments are unique amongst the omics measurements in that they can target a physiologically relevant process in a single tier of information, unlike genomics or transriptomics where the resident networks or modules are not connected by physical means (transcripts and genes must transfer information to other molecules to affect each other, with the exception of some RNA-RNA interactions). In part because of this, some investigators have promulgated the idea of the proteotype (135), or a mass spectrometry equivalent of a genotype, in which a set of proteomic markers is proven to be connected to a physiological outcome and subsequently assayed in large populations. The expertise, instrumentation, and computational infrastructure now exists to implement such proteotypes across institutions and on a population scale for the purposes of better molecular stratification of patients. Moving forward, interconnected challenges for basic and translational science require us to utilize transcriptome and proteome networks as discrete molecular phenotypes. Data integration and modeling will enable new cellular principles to be defined—rules through which transcriptomes, proteomes and other networks of molecules underpin cellular physiology. Combined with insights from genetic variability across human populations, a critical translational task will then be to make these principles actionable in the clinical setting, such that omics measurements can become part of the electronic medical record, informing physician and patient alike about health and disease.
Acknowledgements
The authors thank colleagues at UCLA for discussions about networks and systems biology. Research in the Vondriska laboratory is supported by the National Institutes of Health, the Department of Anesthesiology in the David Geffen School of Medicine, and the American Heart Association.
References
- 1.An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Integrated genomic analyses of ovarian carcinoma. Nature 474: 609–615, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Addona TA, Shi X, Keshishian H, Mani DR, Burgess M, Gillette MA, Clauser KR, Shen D, Lewis GD, Farrell LA, Fifer MA, Sabatine MS, Gerszten RE, Carr SA. A pipeline that integrates the discovery and verification of plasma protein biomarkers reveals candidate markers for cardiovascular disease. Nat Biotechnol 29: 635–643, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature 422: 198–207, 2003. [DOI] [PubMed] [Google Scholar]
- 5.Allocco DJ, Kohane IS, Butte AJ. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinformatics 5: 18, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aten JE, Fuller TF, Lusis AJ, Horvath S. Using genetic markers to orient the edges in quantitative trait networks: The NEO software. BMC Syst Biol 2: 34, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Au KF, Jiang H, Lin L, Xing Y, Wong WH. Detection of splice junctions from paired-end RNA-seq data by SpliceMap. Nucleic Acids Res 38: 4570–4578, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azeloglu EU, Iyengar R. Signaling networks: Information flow, computation, and decision making. Cold Spring Harb Perspect Biol 7: a005934, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baboo S, Cook PR. “Dark matter” worlds of unstable RNA and protein. Nucleus 5: 281–286, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ballouz S, Verleyen W, Gillis J. Guidance for RNA-seq co-expression network construction and analysis: Safety in numbers. Bioinformatics 31: 2123–2130, 2015. [DOI] [PubMed] [Google Scholar]
- 11.Bar-Joseph Z, Gerber GK, Lee TI, Rinaldi NJ, Yoo JY, Robert F, Gordon DB, Fraenkel E, Jaakkola TS, Young RA, Gifford DK. Computational discovery of gene modules and regulatory networks. Nat Biotechnol 21: 1337–1342, 2003. [DOI] [PubMed] [Google Scholar]
- 12.Barabasi AL, Gulbahce N, Loscalzo J. Network medicine: A network-based approach to human disease. Nat Genet 12: 56–68, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barabasi AL, Oltvai ZN. Network biology: Understanding the cell’s functional organization. Nat Rev Genet 5: 101–113, 2004. [DOI] [PubMed] [Google Scholar]
- 14.Battle A, Khan Z, Wang SH, Mitrano A, Ford MJ, Pritchard JK, Gilad Y. Genomic variation. Impact of regulatory variation from RNA to protein. Science 347: 664–667, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bendall SC, Simonds EF, Qiu P, Amir el AD, Krutzik PO, Finck R, Bruggner RV, Melamed R, Trejo A, Ornatsky OI, Balderas RS, Plevritis SK, Sachs K, Pe’er D, Tanner SD, Nolan GP. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332: 687–696, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Gene expression profiling in single cells from the pancreatic islets of Langerhans reveals lognormal distribution of mRNA levels. Genome Res 15: 1388–1392, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blagoev B, Ong SE, Kratchmarova I, Mann M. Temporal analysis of phosphotyrosine-dependent signaling networks by quantitative proteomics. Nat Biotechnol 22: 1139–1145, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Boersema PJ, Aye TT, van Veen TA, Heck AJ, Mohammed S. Triplex protein quantification based on stable isotope labeling by peptide dimethylation applied to cell and tissue lysates. Proteomics 8: 4624–4632, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Braun P Reproducibility restored—On toward the human interactome. Nat Methods 10: 301, 303, 2013. [DOI] [PubMed] [Google Scholar]
- 21.Brown MP, Grundy WN, Lin D, Cristianini N, Sugnet CW, Furey TS, Ares M Jr.,, Haussler D. Knowledge-based analysis of microarray gene expression data by using support vector machines. Proc Natl Acad Sci U S A 97: 262–267, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carter H, Hofree M, Ideker T. Genotype to phenotype via network analysis. Curr Opin Genet Dev 23: 611–621, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cenik C, Cenik ES, Byeon GW, Grubert F, Candille SI, Spacek D, Alsallakh B, Tilgner H, Araya CL, Tang H, Ricci E, Snyder MP. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res 25: 1610–1621, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Orozco L, Wang J, Rau CD, Rubbi L, Ren S, Wang Y, Pellegrini M, Lusis AJ, Vondriska TM. DNA methylation indicates susceptibility to isoproterenol-inducd cardiac pathology and is associated with chromatin states. Circ Res 118: 786–797, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christoforou A, Arias AM, Lilley KS. Determining protein subcellular localization in mammalian cell culture with biochemical fractionation and iTRAQ 8-plex quantification. Methods Mol Biol 1156: 157–174, 2014. [DOI] [PubMed] [Google Scholar]
- 26.Chuang HY, Lee E, Liu YT, Lee D, Ideker T. Network-based classification of breast cancer metastasis. Mol Syst Biol 3: 140, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cloonan N, Forrest AR, Kolle G, Gardiner BB, Faulkner GJ, Brown MK, Taylor DF, Steptoe AL, Wani S, Bethel G, Robertson AJ, Perkins AC, Bruce SJ, Lee CC, Ranade SS, Peckham HE, Manning JM, McKernan KJ, Grimmond SM. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nat Methods 5: 613–619, 2008. [DOI] [PubMed] [Google Scholar]
- 28.Cohen SM. Everything old is new again: (linc)RNAs make proteins! Embo J 33: 937–938, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corbett JM, Why HJ, Wheeler CH, Richardson PJ, Archard LC, Yacoub MH, Dunn MJ. Cardiac protein abnormalities in dilated cardiomyopathy detected by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis 19: 2031–2042, 1998. [DOI] [PubMed] [Google Scholar]
- 30.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deng Q, Ramskold D, Reinius B, Sandberg R. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science 343: 193–196, 2014. [DOI] [PubMed] [Google Scholar]
- 32.Dimon MT, Sorber K, DeRisi JL. HMMSplicer: A tool for efficient and sensitive discovery of known and novel splice junctions in RNA-Seq data. PLoS One 5: e13875, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Domon B, Aebersold R. Options and considerations when selecting a quantitative proteomics strategy. Nat Biotechnol 28: 710–721, 2010. [DOI] [PubMed] [Google Scholar]
- 34.Eberwine J, Sul JY, Bartfai T, Kim J. The promise of single-cell sequencing. Nat Methods 11: 25–27, 2014. [DOI] [PubMed] [Google Scholar]
- 35.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci U S A 95: 14863–14868, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom 5: 976–989, 1994. [DOI] [PubMed] [Google Scholar]
- 37.Evans G, Wheeler CH, Corbett JM, Dunn MJ. Construction of HSC-2DPAGE: A two-dimensional gel electrophoresis database of heart proteins. Electrophoresis 18: 471–479, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Farber CR, Bennett BJ, Orozco L, Zou W, Lira A, Kostem E, Kang HM, Furlotte N, Berberyan A, Ghazalpour A, Suwanwela J, Drake TA, Eskin E, Wang QT, Teitelbaum SL, Lusis AJ. Mouse genome-wide association and systems genetics identify Asxl2 as a regulator of bone mineral density and osteoclastogenesis. PLoS genetics 7: e1002038, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farh KK, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJ, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ, Hafler DA, Bernstein BE. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518: 337–343, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Electrospray ionization for mass spectrometry of large biomolecules. Science 246: 64–71, 1989. [DOI] [PubMed] [Google Scholar]
- 41.Ferrell JE Jr., Self-perpetuating states in signal transduction: Positive feedback, double-negative feedback and bistability. Curr Opin Cell Biol 14: 140–148, 2002. [DOI] [PubMed] [Google Scholar]
- 42.Foss EJ, Radulovic D, Shaffer SA, Ruderfer DM, Bedalov A, Goodlett DR, Kruglyak L. Genetic basis of proteome variation in yeast. Nat Genet 39: 1369–1375, 2007. [DOI] [PubMed] [Google Scholar]
- 43.Franklin S, Chen H, Mitchell-Jordan S, Ren S, Wang Y, Vondriska TM. Quantitative analysis of the chromatin proteome in disease reveals remodeling principles and identifies high mobility group protein B2 as a regulator of hypertrophic growth. Mol Cell Proteomics 11: M111, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fu J, Keurentjes JJ, Bouwmeester H, America T, Verstappen FW, Ward JL, Beale MH, de Vos RC, Dijkstra M, Scheltema RA, Johannes F, Koornneef M, Vreugdenhil D, Breitling R, Jansen RC. System-wide molecular evidence for phenotypic buffering in Arabidopsis. Nat Genet 41: 166–167, 2009. [DOI] [PubMed] [Google Scholar]
- 45.Fullwood MJ, Wei CL, Liu ET, Ruan Y. Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses. Genome Res 19: 521–532, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao X, Wan J, Liu B, Ma M, Shen B, Qian SB. Quantitative profiling of initiating ribosomes in vivo. Nat Methods 12: 147–153, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, Rau C, Jensen LJ, Bastuck S, Dumpelfeld B, Edelmann A, Heurtier MA, Hoffman V, Hoefert C, Klein K, Hudak M, Michon AM, Schelder M, Schirle M, Remor M, Rudi T, Hooper S, Bauer A, Bouwmeester T, Casari G, Drewes G, Neubauer G, Rick JM, Kuster B, Bork P, Russell RB, Superti-Furga G. Proteome survey reveals modularity of the yeast cell machinery. Nature 440: 631–636, 2006. [DOI] [PubMed] [Google Scholar]
- 48.Ge H, Liu Z, Church GM, Vidal M. Correlation between transcriptome and interactome mapping data from Saccharomyces cerevisiae. Nat Genet 29: 482–486, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, Min R, Alves P, Abyzov A, Addleman N, Bhardwaj N, Boyle AP, Cayting P, Charos A, Chen DZ, Cheng Y, Clarke D, Eastman C, Euskirchen G, Frietze S, Fu Y, Gertz J, Grubert F, Harmanci A, Jain P, Kasowski M, Lacroute P, Leng J, Lian J, Monahan H, O’Geen H, Ouyang Z, Partridge EC, Patacsil D, Pauli F, Raha D, Ramirez L, Reddy TE, Reed B, Shi M, Slifer T, Wang J, Wu L, Yang X, Yip KY, Zilberman-Schapira G, Batzoglou S, Sidow A, Farnham PJ, Myers RM, Weissman SM, Snyder M. Architecture of the human regulatory network derived from ENCODE data. Nature 489: 91–100, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gibbs DL, Baratt A, Baric RS, Kawaoka Y, Smith RD, Orwoll ES, Katze MG, McWeeney SK. Protein co-expression network analysis (ProCoNA). J Clin Bioinforma 3: 11, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gillet LC, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: A new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: O111 016717, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gonzalez E, Joly S. Impact of RNA-seq attributes on false positive rates in differential expression analysis of de novo assembled transcriptomes. BMC Res Notes 6: 503, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19: 1720–1730, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr.,, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp AC, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. PAR-CliP–a method to identify transcriptome-wide the binding sites of RNA binding proteins. J Vis Exp 41: e2034, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature 402: C47–52, 1999. [DOI] [PubMed] [Google Scholar]
- 57.Methods Hebenstreit D., challenges and potentials of single cell RNA-seq. Biology (Basel) 1: 658–667, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 163: 712–723, 2015. [DOI] [PubMed] [Google Scholar]
- 59.Hestand MS, Klingenhoff A, Scherf M, Ariyurek Y, Ramos Y, van Workum W, Suzuki M, Werner T, van Ommen GJ, den Dunnen JT, Harbers M, t Hoen PA. Tissue-specific transcript annotation and expression profiling with complementary next-generation sequencing technologies. Nucleic Acids Res 38: e165, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hindorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, Manolio TA. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc Natl Acad Sci U S A 106: 9362–9367, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Holme P, Huss M, Jeong H. Subnetwork hierarchies of biochemical pathways. Bioinformatics 19: 532–538, 2003. [DOI] [PubMed] [Google Scholar]
- 62.Hu Z, Hung JH, Wang Y, Chang YC, Huang CL, Huyck M, DeLisi C. VisANT 3.5: Multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res 37: W115–121, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hughes AJ, Spelke DP, Xu Z, Kang CC, Schaffer DV, Herr AE. Single-cell western blotting. Nat Methods 11: 749–755, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humphrey SJ, Azimifar SB, Mann M. High-throughput phosphoproteomics reveals in vivo insulin signaling dynamics. Nat Biotechnol 33: 990–995, 2015. [DOI] [PubMed] [Google Scholar]
- 65.Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP. The BioPlex network: A systematic exploration of the human interactome. Cell 162: 425–440, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ibarra RU, Edwards JS, Palsson BO. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420: 186–189, 2002. [DOI] [PubMed] [Google Scholar]
- 67.Ingolia NT. Ribosome profiling: New views of translation, from single codons to genome scale. Nat Rev Genet 15: 205–213, 2014. [DOI] [PubMed] [Google Scholar]
- 68.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genomewide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Issa NT, Kruger J, Byers SW, Dakshanamurthy S. Drug repurposing a reality: From computers to the clinic. Expert Rev Clin Pharmacol 6: 95–97, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jansen R, Greenbaum D, Gerstein M. Relating whole-genome expression data with protein-protein interactions. Genome Res 12: 37–46, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jansen R, Yu H, Greenbaum D, Kluger Y, Krogan NJ, Chung S, Emili A, Snyder M, Greenblatt JF, Gerstein M. A Bayesian networks approach for predicting protein-protein interactions from genomic data. Science 302: 449–453, 2003. [DOI] [PubMed] [Google Scholar]
- 72.Jones PA. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492, 2012. [DOI] [PubMed] [Google Scholar]
- 73.Jovanovic M, Rooney MS, Mertins P, Przybylski D, Chevrier N, Satija R, Rodriguez EH, Fields AP, Schwartz S, Raychowdhury R, Mumbach MR, Eisenhaure T, Rabani M, Gennert D, Lu D, Delorey T, Weissman JS, Carr SA, Hacohen N, Regev A. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science 347: 1259038, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kanehisa M A database for post-genome analysis. Trends Genet 13: 375–376, 1997. [DOI] [PubMed] [Google Scholar]
- 75.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 38: D355–360, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods 7: 1009–1015, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Katz Y, Wang ET, Silterra J, Schwartz S, Wong B, Thorvaldsdottir H, Robinson JT, Mesirov JP, Airoldi EM, Burge CB. Quantitative visualization of alternative exon expression from RNA-seq data. Bioinformatics 31: 2400–2402, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kim KT, Lee HW, Lee HO, Kim SC, Seo YJ, Chung W, Eum HH, Nam DH, Kim J, Joo KM, Park WY. Single-cell mRNA sequencing identifies subclonal heterogeneity in anti-cancer drug responses of lung adenocarcinoma cells. Genome Biol 16: 127, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, Madugundu AK, Kelkar DS, Isserlin R, Jain S, Thomas JK, Muthusamy B, Leal-Rojas P, Kumar P, Sahasrabuddhe NA, Balakrishnan L, Advani J, George B, Renuse S, Selvan LD, Patil AH, Nanjappa V, Radhakrishnan A, Prasad S, Subbannayya T, Raju R, Kumar M, Sreenivasamurthy SK, Marimuthu A, Sathe GJ, Chavan S, Datta KK, Subbannayya Y, Sahu A, Yelamanchi SD, Jayaram S, Rajagopalan P, Sharma J, Murthy KR, Syed N, Goel R, Khan AA, Ahmad S, Dey G, Mudgal K, Chatterjee A, Huang TC, Zhong J, Wu X, Shaw PG, Freed D, Zahari MS, Mukherjee KK, Shankar S, Mahadevan A, Lam H, Mitchell CJ, Shankar SK, Satishchandra P, Schroeder JT, Sirdeshmukh R, Maitra A, Leach SD, Drake CG, Halushka MK, Prasad TS, Hruban RH, Kerr CL, Bader GD, Iacobuzio-Donahue CA, Gowda H, Pandey A. A draft map of the human proteome. Nature 509: 575–581, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.King HA, Gerber AP. Translatome profiling: Methods for genomescale analysis of mRNA translation. Brief Funct Genomics 15: 22–31, 2014. [DOI] [PubMed] [Google Scholar]
- 81.Kito K, Kawaguchi N, Okada S, Ito T. Discrimination between stable and dynamic components of protein complexes by means of quantitative proteomics. Proteomics 8: 2366–2370, 2008. [DOI] [PubMed] [Google Scholar]
- 82.Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J, Pau G, Reeder J, Cao Y, Mukhyala K, Selvaraj SK, Yu M, Zynda GJ, Brauer MJ, Wu TD, Gentleman RC, Manning G, Yauch RL, Bourgon R, Stokoe D, Modrusan Z, Neve RM, de Sauvage FJ, Settleman J, Seshagiri S, Zhang Z. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 33: 306–312, 2015. [DOI] [PubMed] [Google Scholar]
- 83.Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol 17: 909–915, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kratz A, Carninci P. The devil in the details of RNA-seq. Nat Biotechnol 32: 882–884, 2014. [DOI] [PubMed] [Google Scholar]
- 85.Krogan NJ, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis AP, Punna T, Peregrin-Alvarez JM, Shales M, Zhang X, Davey M, Robinson MD, Paccanaro A, Bray JE, Sheung A, Beattie B, Richards DP, Canadien V, Lalev A, Mena F, Wong P, Starostine A, Canete MM, Vlasblom J, Wu S, Orsi C, Collins SR, Chandran S, Haw R, Rilstone JJ, Gandi K, Thompson NJ, Musso G, St Onge P, Ghanny S, Lam MH, Butland G, Altaf-Ul AM, Kanaya S, Shilatifard A, O’Shea E, Weissman JS, Ingles CJ, Hughes TR, Parkinson J, Gerstein M, Wodak SJ, Emili A, Greenblatt JF. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature 440: 637–643, 2006. [DOI] [PubMed] [Google Scholar]
- 86.Krupp M, Marquardt JU, Sahin U, Galle PR, Castle J, Teufel A. RNA-Seq Atlas—A reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics 28: 1184–1185, 2012. [DOI] [PubMed] [Google Scholar]
- 87.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Methods 3: 361–368, 2006. [DOI] [PubMed] [Google Scholar]
- 88.Krutzik PO, Trejo A, Schulz KR, Nolan GP. Phospho flow cytometry methods for the analysis of kinase signaling in cell lines and primary human blood samples. Methods Mol Biol 699: 179–202, 2011. [DOI] [PubMed] [Google Scholar]
- 89.Kumarswamy R, Bauters C, Volkmann I, Maury F, Fetisch J, Holzmann A, Lemesle G, de Groote P, Pinet F, Thum T. Circulating long noncoding RNA, LIPCAR, predicts survival in patients with heart failure. Circ Res 114: 1569–1575, 2014. [DOI] [PubMed] [Google Scholar]
- 90.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol 1: 54, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Langfelder P, Horvath S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9: 559, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee S, Liu B, Huang SX, Shen B, Qian SB. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci U S A 109: E2424–2432, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lee S, Nguyen HM, Kang C. Tiny abortive initiation transcripts exert antitermination activity on an RNA hairpin-dependent intrinsic terminator. Nucleic Acids Res 38: 6045–6053, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Li B, Ruotti V, Stewart RM, Thomson JA, Dewey CN. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 26: 493–500, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li JJ, Biggin MD. Gene expression. Statistics requantitates the central dogma. Science 347: 1066–1067, 2015. [DOI] [PubMed] [Google Scholar]
- 96.Li W, Dai C, Kang S, Zhou XJ. Integrative analysis of many RNA-seq datasets to study alternative splicing. Methods 67: 313–324, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lister R, O’Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133: 523–536, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu Y, Zhou J, White KP. RNA-seq differential expression studies: More sequence or more replication? Bioinformatics 30: 301–304, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lu P, Vogel C, Wang R, Yao X, Marcotte EM. Absolute protein expression profiling estimates the relative contributions of transcriptional and translational regulation. Nat Biotechnol 25: 117–124, 2007. [DOI] [PubMed] [Google Scholar]
- 100.Marian AJ. Modeling human disease phenotype in model organisms: “It’s only a model!”. Circ Res 109: 356–359, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res 18: 1509–1517, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mestas J, Hughes CC. Of mice and not men: Differences between mouse and human immunology. J Immunol 172: 2731–2738, 2004. [DOI] [PubMed] [Google Scholar]
- 103.Milhorn HT Jr, Benton R, Ross R, Guyton AC. A Mathematical Model of the Human Respiratory Control System. Biophys J 5: 27–46, 1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Min IM, Waterfall JJ, Core LJ, Munroe RJ, Schimenti J, Lis JT. Regulating RNA polymerase pausing and transcription elongation in embryonic stem cells. Genes Dev 25: 742–754, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Molkentin JD, Robbins J. With great power comes great responsibility: Using mouse genetics to study cardiac hypertrophy and failure. J Mol Cell Cardiol 46: 130–136, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Monte E, Lopez R, Vondriska TM. Not low hanging but still sweet: Metabolic proteomes in cardiovascular disease. J Mol Cell Cardiol 90: 70–73, 2015. [DOI] [PubMed] [Google Scholar]
- 107.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods 5: 621–628, 2008. [DOI] [PubMed] [Google Scholar]
- 108.Nagalakshmi U, Wang Z, Waern K, Shou C, Raha D, Gerstein M, Snyder M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320: 1344–1349, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA. Less label, more free: Approaches in label-free quantitative mass spectrometry. Proteomics 11: 535–553, 2011. [DOI] [PubMed] [Google Scholar]
- 110.Nesvizhskii AI. Proteogenomics: Concepts, applications and computational strategies. Nat Methods 11: 1114–1125, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441: 840–846, 2006. [DOI] [PubMed] [Google Scholar]
- 112.Niehrs C, Pollet N. Synexpression groups in eukaryotes. Nature 402: 483–487, 1999. [DOI] [PubMed] [Google Scholar]
- 113.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250: 4007–4021, 1975. [PMC free article] [PubMed] [Google Scholar]
- 114.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem 250: 4007–4021, 1975. [PMC free article] [PubMed] [Google Scholar]
- 115.Olsen JV, Mann M. Status of large-scale analysis of post-translational modifications by mass spectrometry. Mol Cell Proteomics 12: 3444–3452, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386, 2002. [DOI] [PubMed] [Google Scholar]
- 117.Orozco LD, Morselli M, Rubbi L, Guo W, Go J, Shi H, Lopez D, Furlotte NA, Bennett BJ, Farber CR, Ghazalpour A, Zhang MQ, Bahous R, Rozen R, Lusis AJ, Pellegrini M. Epigenome-wide association of liver methylation patterns and complex metabolic traits in mice. Cell Metabolism 21: 905–917, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Pagel O, Loroch S, Sickmann A, Zahedi RP. Current strategies and findings in clinically relevant post-translational modification-specific proteomics. Expert Rev Proteomics 12: 235–253, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palahniuk C Fight club. New York, NY: W.W. Norton, 1996. [Google Scholar]
- 120.Park CC, Gale GD, de Jong S, Ghazalpour A, Bennett BJ, Farber CR, Langfelder P, Lin A, Khan AH, Eskin E, Horvath S, Lusis AJ, Ophoff RA, Smith DJ. Gene networks associated with conditional fear in mice identified using a systems genetics approach. BMC Syst Biol 5: 43, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497: 127–131, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pennington SR, Wilkins MR, Hochstrasser DF, Dunn MJ. Proteome analysis: From protein characterization to biological function. Trends Cell Biol 7: 168–173, 1997. [DOI] [PubMed] [Google Scholar]
- 123.Perez-Riverol Y, Alpi E, Wang R, Hermjakob H, Vizcaino JA. Making proteomics data accessible and reusable: Current state of proteomics databases and repositories. Proteomics 15: 930–949, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pflieger D, Gonnet F, de la Fuente van Bentem S, Hirt H, de la Fuente A. Linking the proteins–elucidation of proteome-scale networks using mass spectrometry. Mass Spectrom Rev 30: 268–297, 2011. [DOI] [PubMed] [Google Scholar]
- 125.Qu Z, Garfinkel A, Weiss JN, Nivala M. Multi-scale modeling in biology: How to bridge the gaps between scales? Prog Biophys Mol Biol 107: 21–31, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet 12: 529–541, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ramskold D, Luo S, Wang YC, Li R, Deng Q, Faridani OR, Daniels GA, Khrebtukova I, Loring JF, Laurent LC, Schroth GP, Sandberg R. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol 30: 777–782, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rau CD, Wang J, Avetisyan R, Romay M, Ren S, Wang Y, Lusis AJ. Mapping genetic contributions to cardiac pathology induced by beta-adrenergic stimulation in mice. Circ Cardiovasc Gene 8: 40–49, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Rau CD, Wisniewski N, Orozco LD, Bennett B, Weiss J, Lusis AJ. Maximal information component analysis: A novel non-linear network analysis method. Front Genet 4: 28, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ravasz E, Somera AL, Mongru DA, Oltvai ZN, Barabasi AL. Hierarchical organization of modularity in metabolic networks. Science 297: 1551–1555, 2002. [DOI] [PubMed] [Google Scholar]
- 131.Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339: 1328–1331, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R. Identification of cross-linked peptides from large sequence databases. Nat Methods 5: 315–318, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Rolland T, Tasan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, Yi S, Lemmens I, Fontanillo C, Mosca R, Kamburov A, Ghiassian SD, Yang X, Ghamsari L, Balcha D, Begg BE, Braun P, Brehme M, Broly MP, Carvunis AR, Convery-Zupan D, Corominas R, Coulombe-Huntington J, Dann E, Dreze M, Dricot A, Fan C, Franzosa E, Gebreab F, Gutierrez BJ, Hardy MF, Jin M, Kang S, Kiros R, Lin GN, Luck K, MacWilliams A, Menche J, Murray RR, Palagi A, Poulin MM, Rambout X, Rasla J, Reichert P, Romero V, Ruyssinck E, Sahalie JM, Scholz A, Shah AA, Sharma A, Shen Y, Spirohn K, Tam S, Tejeda AO, Trigg SA, Twizere JC, Vega K, Walsh J, Cusick ME, Xia Y, Barabasi AL, Iakoucheva LM, Aloy P, De Las Rivas J, Tavernier J, Calderwood MA, Hill DE, Hao T, Roth FP, Vidal M. A proteome-scale map of the human interactome network. Cell 159: 1212–1226, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Rosa-Garrido M, Karbassi E, Monte E, Vondriska TM. Regulation of chromatin structure in the cardiovascular system. Circ J 77: 1389–1398, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rost HL, Malmstrom L, Aebersold R. Reproducible quantitative proteotype data matrices for systems biology. Mol Biol Cell 26: 3926–3931, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rost HL, Rosenberger G, Navarro P, Gillet L, Miladinovic SM, Schubert OT, Wolski W, Collins BC, Malmstrom J, Malmstrom L, Aebersold R. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nat Biotechnol 32: 219–223, 2014. [DOI] [PubMed] [Google Scholar]
- 137.Roux KJ, Kim DI, Burke B. BioID: A screen for protein-protein interactions. Curr Protoc Protein Sci 74: 23, 2013. [DOI] [PubMed] [Google Scholar]
- 138.Roux KJ, Kim DI, Raida M, Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196: 801–810, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ruan X, Ruan Y. Genome wide full-length transcript analysis using 5′ and 3′ paired-end-tag next generation sequencing (RNA-PET). Methods Mol Biol 809: 535–562, 2012. [DOI] [PubMed] [Google Scholar]
- 140.Ruepp A, Waegele B, Lechner M, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Mewes HW. CORUM: The comprehensive resource of mammalian protein complexes–2009. Nucleic Acids Res 38: D497–D501, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Sahni N, Yi S, Taipale M, Fuxman Bass JI, Coulombe-Huntington J, Yang F, Peng J, Weile J, Karras GI, Wang Y, Kovacs IA, Kamburov A, Krykbaeva I, Lam MH, Tucker G, Khurana V, Sharma A, Liu YY, Yachie N, Zhong Q, Shen Y, Palagi A, San-Miguel A, Fan C, Balcha D, Dricot A, Jordan DM, Walsh JM, Shah AA, Yang X, Stoyanova AK, Leighton A, Calderwood MA, Jacob Y, Cusick ME, Salehi-Ashtiani K, Whitesell LJ, Sunyaev S, Berger B, Barabasi AL, Charloteaux B, Hill DE, Hao T, Roth FP, Xia Y, Walhout AJ, Lindquist S, Vidal M. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 161: 647–660, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sakai H, Naito K, Ogiso-Tanaka E, Takahashi Y, Iseki K, Muto C, Satou K, Teruya K, Shiroma A, Shimoji M, Hirano T, Itoh T, Kaga A, Tomooka N. The power of single molecule real-time sequencing technology in the de novo assembly of a eukaryotic genome. Sci Rep 5: 16780, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sanchez A, Golding I. Genetic determinants and cellular constraints in noisy gene expression. Science 342: 1188–1193, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Schena M, Shalon D, Davis RW, Brown PO. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270: 467–470, 1995. [DOI] [PubMed] [Google Scholar]
- 145.Schmidt C, Urlaub H. iTRAQ-labeling of in-gel digested proteins for relative quantification. Methods Mol Biol 564: 207–226, 2009. [DOI] [PubMed] [Google Scholar]
- 146.Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature 473: 337–342, 2011. [DOI] [PubMed] [Google Scholar]
- 147.Shalek AK, Satija R, Adiconis X, Gertner RS, Gaublomme JT, Ray-chowdhury R, Schwartz S, Yosef N, Malboeuf C, Lu D, Trombetta JJ, Gennert D, Gnirke A, Goren A, Hacohen N, Levin JZ, Park H, Regev A. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature 498: 236–240, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. MATS: A Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res 40: e61, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shi Q, Qin L, Wei W, Geng F, Fan R, Shin YS, Guo D, Hood L, Mischel PS, Heath JR. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc Natl Acad Sci U S A 109: 419–424, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shi Z, Wang J, Zhang B. NetGestalt: Integrating multidimensional omics data over biological networks. Nat Methods 10: 597–598, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: Key considerations in genomic analyses. Nat Rev Genet 15: 121–132, 2014. [DOI] [PubMed] [Google Scholar]
- 153.Siuti N, Kelleher NL. Decoding protein modifications using top-down mass spectrometry. Nat Methods 4: 817–821, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Smith LM, Kelleher NL. Proteoform: A single term describing protein complexity. Nat Methods 10: 186–187, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A 101: 9528–9533, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tessarz P, Kouzarides T. Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15: 703–708, 2014. [DOI] [PubMed] [Google Scholar]
- 157.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Mapping intact protein isoforms in discovery mode using top-down proteomics. Nature 480: 254–258, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Valen E, Pascarella G, Chalk A, Maeda N, Kojima M, Kawazu C, Murata M, Nishiyori H, Lazarevic D, Motti D, Marstrand TT, Tang MH, Zhao X, Krogh A, Winther O, Arakawa T, Kawai J, Wells C, Daub C, Harbers M, Hayashizaki Y, Gustincich S, Sandelin A, Carninci P. Genome-wide detection and analysis of hippocampus core promoters using DeepCAGE. Genome Res 19: 255–265, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.van Heesch S, van Iterson M, Jacobi J, Boymans S, Essers PB, de Bruijn E, Hao W, MacInnes AW, Cuppen E, Simonis M. Extensive localization of long noncoding RNAs to the cytosol and mono- and polyribosomal complexes. Genome Biol 15: R6, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Velculescu VE, Zhang L, Vogelstein B, Kinzler KW. Serial analysis of gene expression. Science 270: 484–487, 1995. [DOI] [PubMed] [Google Scholar]
- 161.Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI,Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A,Vinayagam A,Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M. An empirical framework for binary interactome mapping. Nat Methods 6: 83–90, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Vinayagam A, Zirin J, Roesel C, Hu Y, Yilmazel B, Samsonova AA, Neumuller RA,Mohr SE, Perrimon N. Integrating protein-protein interaction networks with phenotypes reveals signs of interactions. Nat Methods 11: 94–99, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Vitting-Seerup K, Porse BT, Sandelin A,Waage J. spliceR: An R package for classification of alternative splicing and prediction of coding potential from RNA-seq data. BMC Bioinformatics 15: 81, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Vogel C,Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227–232, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Vondriska TM, Klein JB, Ping P. Use of functional proteomics to investigate PKC epsilon-mediated cardioprotection: The signaling module hypothesis. Am J Physiol Heart Circ Physiol 280: H1434–1441, 2001. [DOI] [PubMed] [Google Scholar]
- 166.Walker MG, Volkmuth W, Sprinzak E, Hodgson D, Klingler T. Prediction of gene function by genome-scale expression analysis: Prostate cancer-associated genes. Genome Res 9: 1198–1203, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wang K, Singh D, Zeng Z, Coleman SJ, Huang Y, Savich GL, He X, Mieczkowski P, Grimm SA, Perou CM, MacLeod JN, Chiang DY, Prins JF, Liu J. MapSplice: Accurate mapping of RNA-seq reads for splice junction discovery. Nucleic Acids Res 38: e178, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Washburn MP, Wolters D, Yates JR, III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 19: 242–247, 2001. [DOI] [PubMed] [Google Scholar]
- 170.Weinstein IB. Cancer. Addiction to oncogenes—The Achilles heal of cancer. Science 297: 63–64, 2002. [DOI] [PubMed] [Google Scholar]
- 171.Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ. “Good enough solutions” and the genetics of complex diseases. Circ Res 111: 493–504, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Weiss JN, Karma A, MacLellan WR, Deng M, Rau CD, Rees CM, Wang J, Wisniewski N, Eskin E, Horvath S, Qu Z, Wang Y, Lusis AJ. “Good enough solutions” and the genetics of complex diseases. Circ Res 111: 493–504, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Wilczynska A, Bushell M. The complexity of miRNA-mediated repression. Cell Death Differ 22: 22–33, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wilhelm M, Schlegl J, Hahne H, Moghaddas Gholami A, Lieberenz M, Savitski MM, Ziegler E, Butzmann L, Gessulat S, Marx H, Mathieson T, Lemeer S, Schnatbaum K, Reimer U, Wenschuh H, Mollenhauer M, Slotta-Huspenina J, Boese JH, Bantscheff M, Gerstmair A, Faerber F, Kuster B. Mass-spectrometry-based draft of the human proteome. Nature 509: 582–587, 2014. [DOI] [PubMed] [Google Scholar]
- 175.Witze ES, Old WM, Resing KA, Ahn NG. Mapping protein post-translational modifications with mass spectrometry. Nat Methods 4: 798–806, 2007. [DOI] [PubMed] [Google Scholar]
- 176.Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, Quake SR. Quantitative assessment of single-cell RNA-sequencing methods. Nat Methods 11: 41–46, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wu L, Candille SI, Choi Y, Xie D, Jiang L, Li-Pook-Than J, Tang H, Snyder M. Variation and genetic control of protein abundance in humans. Nature 499: 79–82, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wu LF, Hughes TR, Davierwala AP, Robinson MD, Stoughton R, Altschuler SJ. Large-scale prediction of Saccharomyces cerevisiae gene function using overlapping transcriptional clusters. Nat Genet 31: 255–265, 2002. [DOI] [PubMed] [Google Scholar]
- 179.Wu Z,Wu H. Experimental Design and Power Calculation for RNA-seq Experiments. Methods Mol Biol 1418: 379–390, 2016. [DOI] [PubMed] [Google Scholar]
- 180.Xiang CC, Chen Y. cDNA microarray technology and its applications. Biotechnol Adv 18: 35–46, 2000. [DOI] [PubMed] [Google Scholar]
- 181.Yates J III. A century of mass spectrometry: From atoms to proteomes. Nat Methods 8: 5, 2011. [Google Scholar]
- 182.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol 4: Article17, 2005. [DOI] [PubMed] [Google Scholar]
- 183.Zhang Q,Xiao X. Genome sequence-independent identification of RNA editing sites. Nat Methods 12: 347–350, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.ZhangY FonslowBR, Shan B Baek MC,Yates JR III. Protein analysis by shotgun/bottom-up proteomics. Chem Rev 113: 2343–2394, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhao S, Fung-Leung WP, Bittner A, Ngo K, Liu X. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells. PLoS One 9: e78644, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhao W, He X, Hoadley KA, Parker JS, Hayes DN, Perou CM. Comparison of RNA-Seq by poly (A) capture, ribosomal RNA depletion, and DNA microarray for expression profiling. BMC Genomics 15: 419, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Zhernakova DV, de Klerk E, Westra HJ, Mastrokolias A, Amini S, Ariyurek Y, Jansen R, Penninx BW, Hottenga JJ, Willemsen G, de Geus EJ, Boomsma DI,Veldink JH, van den Berg LH,Wijmenga C, den Dunnen JT, van Ommen GJ, t Hoen PA, Franke L. DeepSAGE reveals genetic variants associated with alternative polyadenylation and expression of coding and non-coding transcripts. PLoS Genet 9: e1003594, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]